Спосіб одержання заміщених 2-[2-(3,5-діарил-4,5-дигідро-1н-піразол-1-іл)-4-оксо-4,5-дигідро-1,3-тіазол-5-іл]-n-арилацетамідів
Номер патенту: 94597
Опубліковано: 25.05.2011
Автори: Десенко Сергій Михайлович, Комихов Сергій Олександрович, Руденко Роман Володимирович, Афанасіаді Людмила Михайлівна



Формула / Реферат
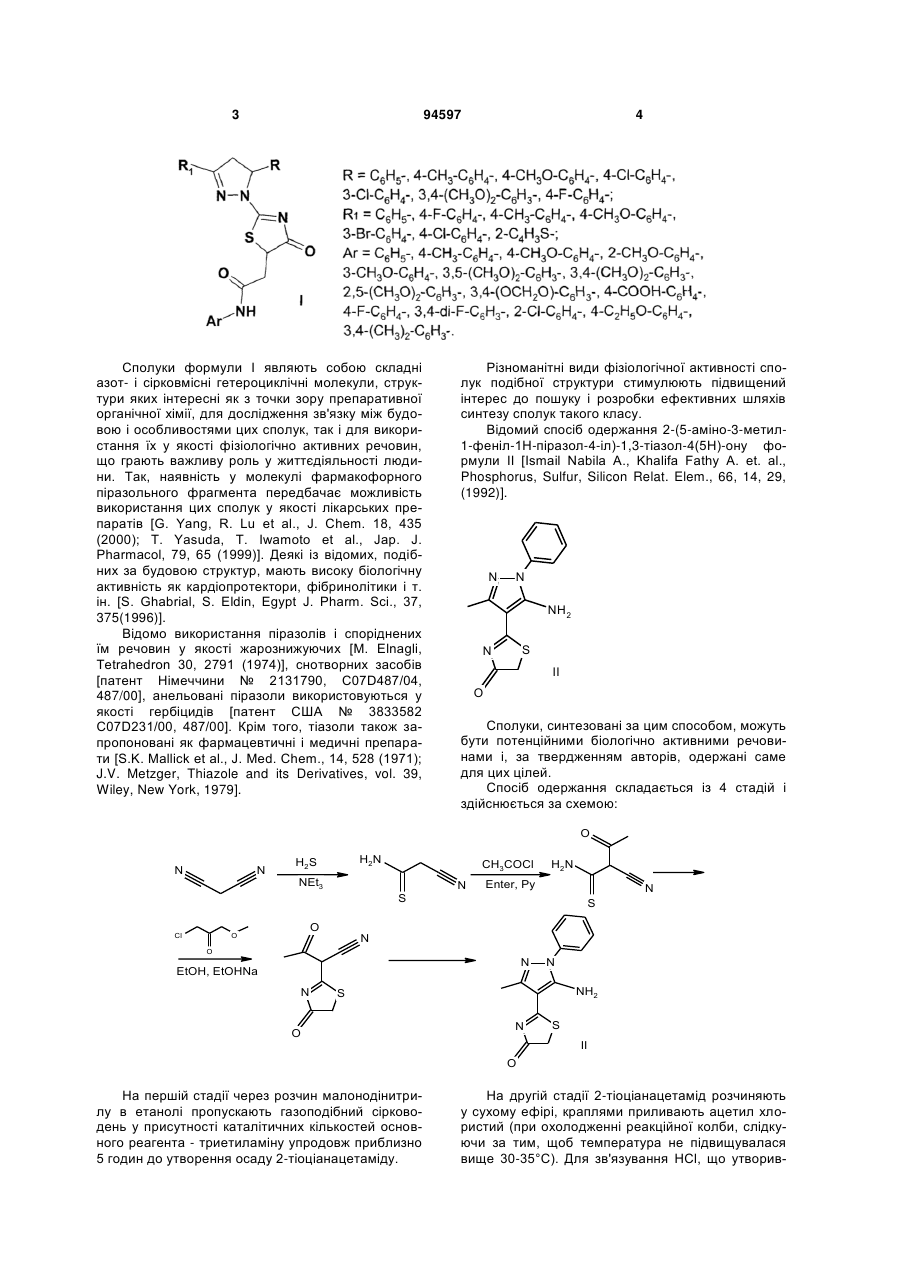
Спосіб одержання 2-[2-(3,5-діарил-4,5-дигідро-1Н-піразол-1-іл)-4-оксо-4,5-дигідро-1,3-тіазол-5-іл]-N-арилацетамідів формули І
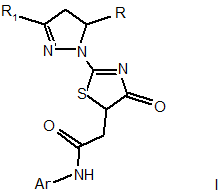 ,
,
де:
R = C6H5-, 4-CH3-C6H4-, 4-CH3O-C6H4-, 4-Cl-C6H4-, 3-Cl-C6H4-, 3,4-(CH3O)2-C6H3-, 4-F-C6H4-;
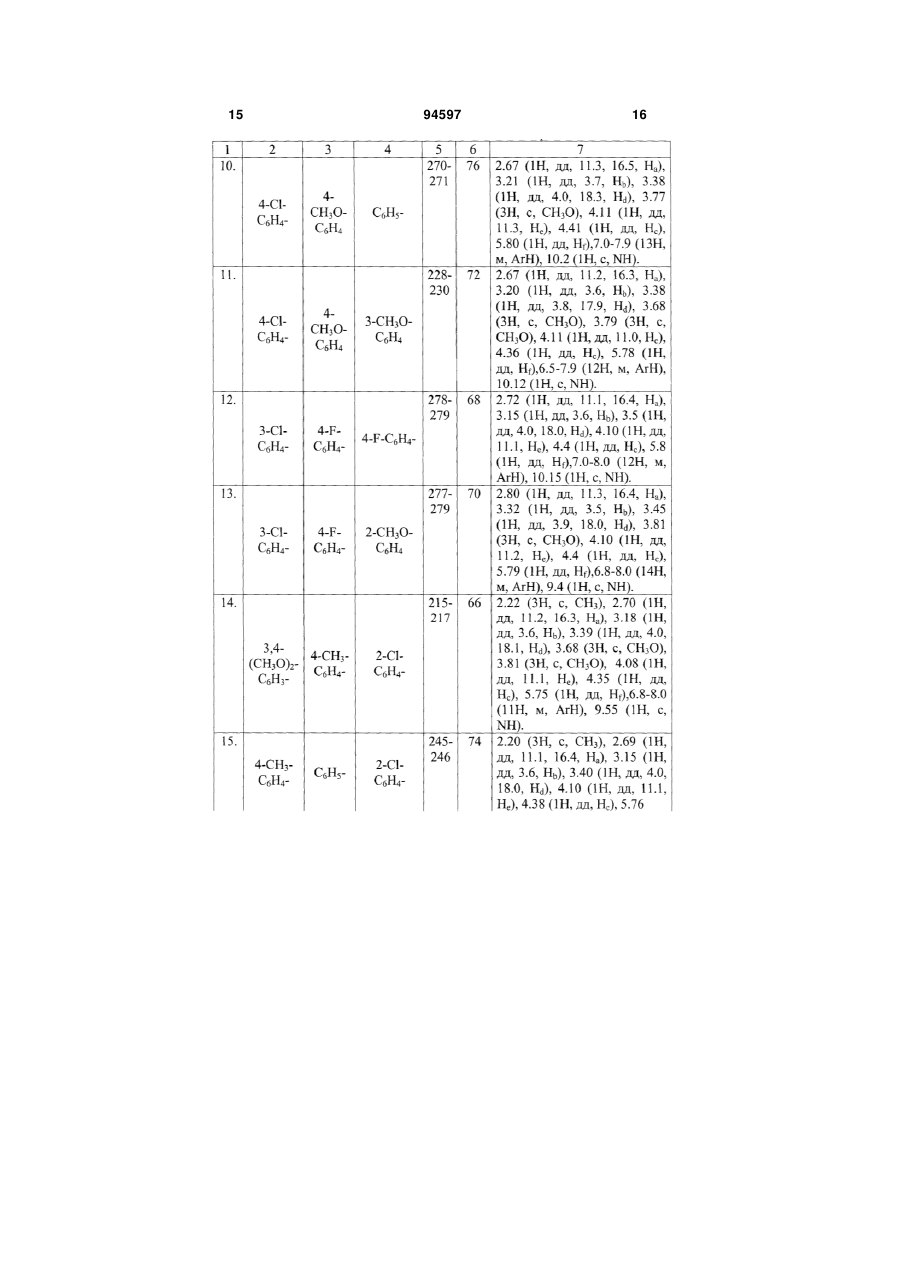
R1 = C6H5-, 4-F-C6H4-, 4-CH3-C6H4-, 4-CH3O-C6H4-, 3-Br-C6H4-, 4-Cl-C6H4-, 2-C4H3S-;
Ar = C6H5-, 4-CH3-C6H4-, 4-CH3O-C6H4-, 2-CH3O-C6H4-, 3-CH3O-C6H4-, 3,5-(CH3O)2C6H3-, 3,4-(CH3O)2-C6H3-, 2,5-(CH3O)2-C6H3-, 3,4-(OCH2O)-C6H3-, 4-COOH-C6H4-, 4-F-C6H4-, 3,4-ди-F-C6H3-, 2-Cl-C6H4-, 4-C2H5O-C6H4-, 3,4-(CH3)2-C6H3-,
що включає взаємодію рівномолярних кількостей тіосемікарбазиду з карбонільною сполукою у середовищі етилового спирту і конденсацію одержаної заміщеної гетероциклічної сполуки з малеімідом у середовищі оцтової кислоти при кипінні, який відрізняється тим, що як карбонільну сполуку використовують R, R1 - заміщені халкони, як заміщену гетероциклічну сполуку - одержаний при взаємодії тіосемікарбазиду і карбонільної сполуки - 3-R-5-R1-4,5-дигідро-1Н-піразол-1-карботіамід, як малеімід - арилзаміщені малеіміди, реакцію конденсації проводять упродовж 5-10 хвилин до утворення цільового продукту.
Текст
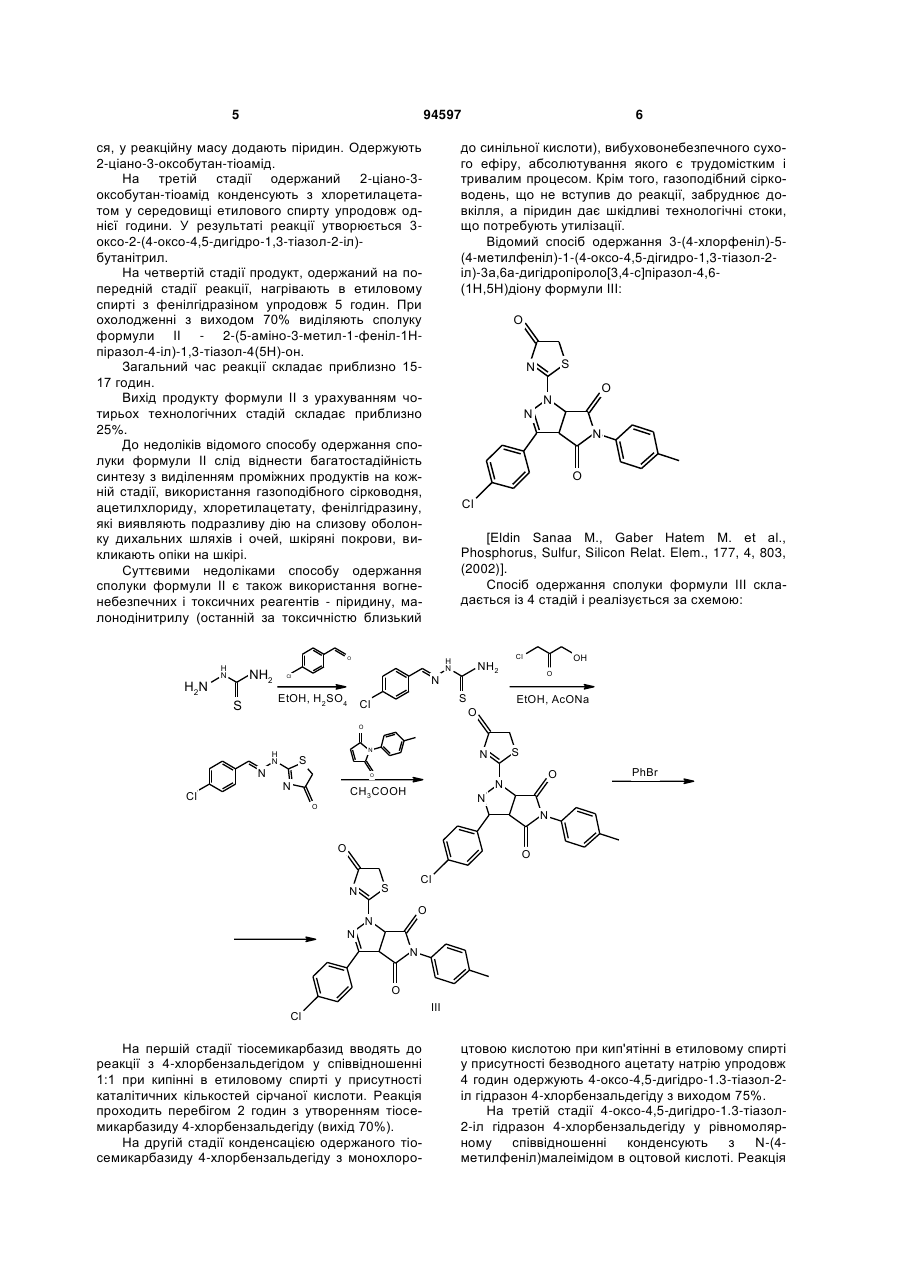
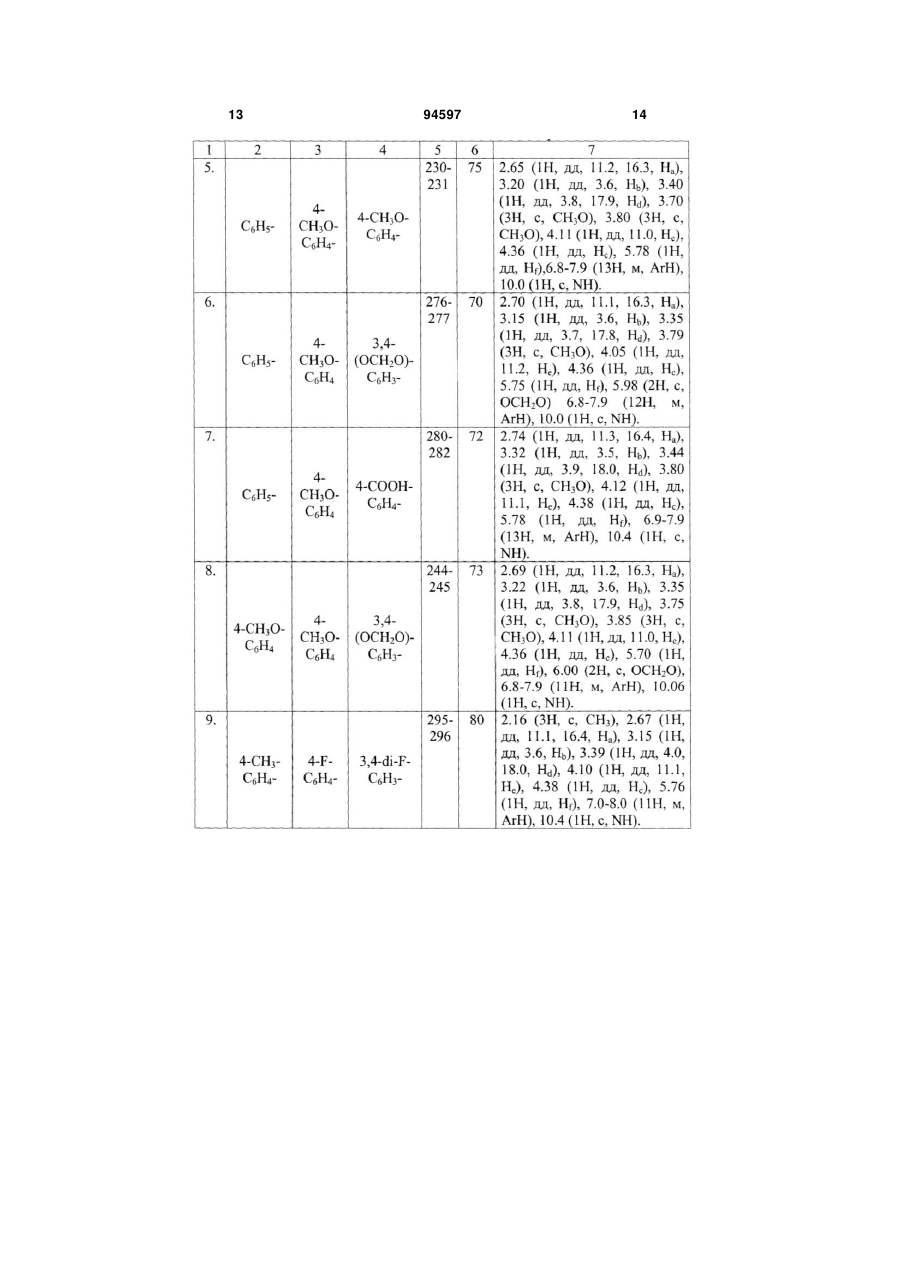
Спосіб одержання 2-[2-(3,5-діарил-4,5-дигідро1Н-піразол-1-іл)-4-оксо-4,5-дигідро-1,3-тіазол-5-іл]N-арилацетамідів формули І 2 Винахід відноситься до області органічної хімії, а саме до способу одержання заміщених 2-[2-(3,5діарил-4,5-дигідро-1Н-піразол-1-іл)-4-оксо-4,5 дигідро-1,3-тіазол-5-іл]-N-арилацетамідів загальної формули І: 3 94597 Сполуки формули І являють собою складні азот- і сірковмісні гетероциклічні молекули, структури яких інтересні як з точки зору препаративної органічної хімії, для дослідження зв'язку між будовою і особливостями цих сполук, так і для використання їх у якості фізіологічно активних речовин, що грають важливу роль у життєдіяльності людини. Так, наявність у молекулі фармакофорного піразольного фрагмента передбачає можливість використання цих сполук у якості лікарських препаратів [G. Yang, R. Lu et al., J. Chem. 18, 435 (2000); Т. Yasuda, Т. Iwamoto et al., Jap. J. Pharmacol, 79, 65 (1999)]. Деякі із відомих, подібних за будовою структур, мають високу біологічну активність як кардіопротектори, фібринолітики і т. ін. [S. Ghabrial, S. Eldin, Egypt J. Pharm. Sci., 37, 375(1996)]. Відомо використання піразолів і споріднених їм речовин у якості жарознижуючих [М. Elnagli, Tetrahedron 30, 2791 (1974)], снотворних засобів [патент Німеччини № 2131790, C07D487/04, 487/00], анельовані піразоли використовуються у якості гербіцидів [патент США № 3833582 C07D231/00, 487/00]. Крім того, тіазоли також запропоновані як фармацевтичні і медичні препарати [S.K. Mallick et al., J. Med. Chem., 14, 528 (1971); J.V. Metzger, Thiazole and its Derivatives, vol. 39, Wiley, New York, 1979]. 4 Різноманітні види фізіологічної активності сполук подібної структури стимулюють підвищений інтерес до пошуку і розробки ефективних шляхів синтезу сполук такого класу. Відомий спосіб одержання 2-(5-аміно-3-метил1-феніл-1Н-піразол-4-іл)-1,3-тіазол-4(5Н)-ону формули II [Ismail Nabila Α., Khalifa Fathy A. et. al., Phosphorus, Sulfur, Silicon Relat. Elem., 66, 14, 29, (1992)]. N N NH2 S N II O Сполуки, синтезовані за цим способом, можуть бути потенційними біологічно активними речовинами і, за твердженням авторів, одержані саме для цих цілей. Спосіб одержання складається із 4 стадій і здійснюється за схемою: O N N H2N H2 S CH3COCl NEt3 N H2N Enter, Py N S Cl O O S N O N EtOH, EtOHNa N N NH2 S O N S II O На першій стадії через розчин малонодінитрилу в етанолі пропускають газоподібний сірководень у присутності каталітичних кількостей основного реагента - триетиламіну упродовж приблизно 5 годин до утворення осаду 2-тіоціанацетаміду. На другій стадії 2-тіоціанацетамід розчиняють у сухому ефірі, краплями приливають ацетил хлористий (при охолодженні реакційної колби, слідкуючи за тим, щоб температура не підвищувалася вище 30-35°С). Для зв'язування НСl, що утворив 5 94597 ся, у реакційну масу додають піридин. Одержують 2-ціано-3-оксобутан-тіоамід. На третій стадії одержаний 2-ціано-3оксобутан-тіоамід конденсують з хлоретилацетатом у середовищі етилового спирту упродовж однієї години. У результаті реакції утворюється 3оксо-2-(4-оксо-4,5-дигідро-1,3-тіазол-2-іл)бутанітрил. На четвертій стадії продукт, одержаний на попередній стадії реакції, нагрівають в етиловому спирті з фенілгідразіном упродовж 5 годин. При охолодженні з виходом 70% виділяють сполуку формули II - 2-(5-аміно-3-метил-1-феніл-1Нпіразол-4-іл)-1,3-тіазол-4(5Н)-он. Загальний час реакції складає приблизно 1517 годин. Вихід продукту формули II з урахуванням чотирьох технологічних стадій складає приблизно 25%. До недоліків відомого способу одержання сполуки формули II слід віднести багатостадійність синтезу з виділенням проміжних продуктів на кожній стадії, використання газоподібного сірководня, ацетилхлориду, хлоретилацетату, фенілгідразину, які виявляють подразливу дію на слизову оболонку дихальних шляхів і очей, шкіряні покрови, викликають опіки на шкірі. Суттєвими недоліками способу одержання сполуки формули II є також використання вогненебезпечних і токсичних реагентів - піридину, малонодінитрилу (останній за токсичністю близький до синільної кислоти), вибуховонебезпечного сухого ефіру, абсолютування якого є трудомістким і тривалим процесом. Крім того, газоподібний сірководень, що не вступив до реакції, забруднює довкілля, а піридин дає шкідливі технологічні стоки, що потребують утилізації. Відомий спосіб одержання 3-(4-хлорфеніл)-5(4-метилфеніл)-1-(4-оксо-4,5-дігидро-1,3-тіазол-2іл)-3а,6а-дигідропіроло[3,4-с]піразол-4,6(1Н,5Н)діону формули III: O H2N NH2 N O Cl [Eldin Sanaa Μ., Gaber Hatem Μ. et al., Phosphorus, Sulfur, Silicon Relat. Elem., 177, 4, 803, (2002)]. Спосіб одержання сполуки формули III складається із 4 стадій і реалізується за схемою: S NH2 N EtOH, H2SO4 O N N H N Cl S N O H N 6 S Cl Cl OH O EtOH, AcONa O O N H N N Cl S N S O O N CH3COOH N O N PhBr N O O N Cl S N O N N O Cl На першій стадії тіосемикарбазид вводять до реакції з 4-хлорбензальдегідом у співвідношенні 1:1 при кипінні в етиловому спирті у присутності каталітичних кількостей сірчаної кислоти. Реакція проходить перебігом 2 годин з утворенням тіосемикарбазиду 4-хлорбензальдегіду (вихід 70%). На другій стадії конденсацією одержаного тіосемикарбазиду 4-хлорбензальдегіду з монохлоро III цтовою кислотою при кип'ятінні в етиловому спирті у присутності безводного ацетату натрію упродовж 4 годин одержують 4-оксо-4,5-дигідро-1.3-тіазол-2іл гідразон 4-хлорбензальдегіду з виходом 75%. На третій стадії 4-оксо-4,5-дигідро-1.3-тіазол2-іл гідразон 4-хлорбензальдегіду у рівномолярному співвідношенні конденсують з N-(4метилфеніл)малеімідом в оцтовой кислоті. Реакція 7 94597 8 проходить при нагріванні упродовж 4 годин з утворенням 3-(4-хлорфеніл)-5-(4-метилфеніл)-1-(4оксо-4,5-дігидро-1,3-тіазол-2-іл)тетрагідропіроло[3,4-с]піразол-4,6-(1H,5Н)діону, вихід якого складає 88%. Продукт очищають перекристалізацією із льодяної оцтової кислоти, одержують жовті кристали з т.пл. 268-269°С. На четвертій стадії для отримання похідного піразоліну одержаний на попередній стадії тетрагідропродукт нагрівають з бромбензолом вперебіг 3 годин. Продукт формули III, що утворився після окислення з виходом 90%, хроматографують на колонці з силікагелем (адсорбент) із толуолу і очищають перекристалізацією із льодяної оцтової кислоти. Загальний час реакції складає приблизно 1820 годин. Сумарний вихід аналога формули III з урахуванням всіх технологічних стадій складає приблизно 40-42%. До недоліків способу одержання відомої сполуки формули III слід віднести багатостадійність синтезу, необхідність виділення проміжних продуктів на кожній технологічній стадії і їх очищення, а також використання ароматичних альдегідів, які легко окислюються у повітрі, що відбивається на чистоті кінцевих продуктів. Хроматографування кінцевого продукту з подальшою перекристалізацією приводить до додаткових витрат реактивів, збільшенню часу синтезу і зменшенню виходу кінцевого продукту. Хлор- і бромвмістні реагенти (монохлороцтова кислота, бромбензол) виявляють подразнювальну дію на слизову оболонку очей, органи дихання і шкіру; бромбензол крім того, є дуже токсичною речовиною - кров'яною отрутою. Використання свіжовиготовленого безводного ацетату натрію для циклодегідратації продукту на другій стадії збільшує час проведення синтезу, бо процес зневоднення його достатньо тривалий. Все це також слід вважати недоліками указаного способу одержання аналога формули III. У зв'язку з наведеними недоліками спосіб має обмежене використання. В якості найближчого аналогу як найбільш близький за технічною суттю обрано останній із наведених аналогів. В основу винаходу поставлено задачу розробки простого і доступного способу одержання заміщених тіазолонопіразолів, що дозволяє зменшити кількість технологічних стадій і тривалість процесу, підвищити вихід кінцевих продуктів реакції і їх якість. Рішення поставленої задачі забезпечується тим, що у способі одержання заміщених 2-[2-(3,5діарил-4,5-дигідро-1Н-піразол-1-іл)-4-оксо-4,5дигідро-1,3-тіазол-5-іл]-N-арилацетамідів загальної формули І що включає взаємодію рівномолярних кількостей тіосемикарбазиду з карбонільною сполукою у середовищі етилового спирту і конденсацію одержаної заміщеної гетероциклічної сполуки з малеімідом у середовищі оцтової кислоти при кипінні, згідно винаходу, в якості карбонільної сполуки використовують R, R1 - заміщені халкони, в якості заміщеної гетероциклічної сполуки - одержаний при взаємодії тіосемикарбазиду і карбонільної сполуки 3-R-5-R1-4,5-дигідро-1Н-піразол-1карботіамід, в якості малеіміду - арилзаміщені малеіміди, реакцію конденсації проводять упродовж 5-10 хвилин до утворення цільового продукту. Взаємодія тіосемикарбазиду з R, R1 - халконами дозволяє одразу одержувати циклічний продукт 1-(3-R-5-R1-4,5-дигідро-1Н-піразоло)карботіамід, який потім конденсують з Arзаміщеними малеімідами, спосіб не потребує використання циклодегідратуючих агентів. Такий вибір компонентів суміші, що реагує (використання R, R1 - заміщених халконів замість ароматичних альдегідів), приводить до зменшення тривалості синтезу; реакція проходить швидко, у дві стадії упродовж 2-2,5 годин (у способінайближчому аналогу - 18-20 годин). Крім того, вибір компонентів суміші, що реагує (халкони, що використовуються, - стійкі у повітрі сполуки) суттєво впливає і на чистоту кінцевих сполук, після виділення із реакційної маси і промивки ацетоном сполуки формули І достатньо чисті, що підтвер1 джено даними H ЯМР спектроскопії. Спосіб, що заявляється, дає можливість відразу одержувати дигідрозаміщені тіазолонопіразолінів, в той час як у способі-прототипі пропонується утворення спочатку тетрагідропохідного, який потім окислюють у середовищі бромбензолу упродовж 3 годин. Таким чином, спосіб, що заявляється, виключає ще одну тривалу стадію процесу і тим самим збільшує вихід і чистоту кінцевих продуктів. Експериментальним шляхом було підібрано часовий режим реакції конденсації: кип'ятіння продуктів суміші, що реагує, менш 5 хвилин не приво1 дить до одержання цільових продуктів. Спектр H ЯМР показав наявність вихідних 3,5-диарил-4,5дигідро-1Н-піразол-1-карботіамідів і заміщених 9 94597 10 1 малеімідів, що не прореагували, реакція проходить приблизно з виходом 15-25% в залежності від складу компонентів суміші, що реагує. Збільшення часу реакції більш 10 хвилин недоцільно, тому що реакція за цей відрізок часу проходить повністю, про що свідчать дані Н ЯМР спектра, наведені у таблиці 2. Спосіб, що заявляється, здійснюється за схемою: Ar H2N H N + R NH2 R1 C2H5OH R N N S O H2N R1 Розроблені експериментальним шляхом умови синтезу сполук формули І, що заявляються, дозволяють зменшити кількість технологічних стадій до 2 (у способі-прототипі 4 стадії). Виходи чистих сполук формули І складають 66-80%, за способом-прототипом вихід 40-42%. Для повного проходження реакції у необхідному напрямку компоненти реакції беруть у рівномолярному співвідношенні (1:1:1). При використанні одного із будь-яких компонентів у меншому або більшому співвідношенні, ніж потребує реакція, у реакційній масі можна виявити реагенти, що не вступили до реакції, які впливають на чистоту цільових продуктів. Можливість модифікації хімічної структури сполук, що одержані за способом, який заявляється, шляхом вільного варіювання R, R1 - замісників у карбонільному компоненті (халконі) і арильного радікала (Ar) у малеіміді відкриває перспективу синтезу широкого асортименту сполук структури І і, таким чином, нових потенційних біологічно активних речовин. У таблиці 1 наведено порівняльні характеристики способу-прототипу і способу, що заявляється. У таблиці 2 дано виходи сполук формули І, що заявляються, їх температури плавлення і спектри 1 ЯМР Н. Нижче приведено приклади конкретного виконання. Приклад 1. Одержання 2-[2-(3,5-діфеніл-4,5дигідро-1Н-піразол-1-іл)-4-оксо-4,5-дигідро-1,3тіазол-5-іл]-N-фенілацетаміду (див. табл. 2, поз. 1). 2,1 г (0,01 моль) халкона і 0,9 (0,01 моль) тіосемикарбазиду розчиняють у 20 мл етилового спирту. До цього розчину додають 0,6 г (0,01 моль) твердого КОН. Реакційну суміш нагрівають упродовж однієї години, охолоджують. Випадає кристалічний осад, що відфільтровують, промивають двічі порціями по 5 мл етилового спирту і сушать. Т.пл. 202-204°С. Суміш 0,28 г (0,001 моль) 3,5-дифеніл-4,5дигідро-1Н-піразол-1-карботіаміду і 0,17 г (0,001 моль) N-фенілмалеіміду нагрівають у 0,8 мл оцтової кислоти упродовж 10 хвилин. Реакційну суміш охолоджують, розбавляють 10 мл ацетону. Осад, що випав, відфільтровують, промивають ацетоном, і сушать. O N R R1 O N N N S S CH3COOH O O Ar NH Вихідні дані: т.пл. 259-260°С, вихід 0,42 г (75%). Структуру 2-[2-(3,5-діфеніл-4,5-дигідро-1Нпіразол-1-іл)-4-оксо-4,5-дигідро-1,3-тіазол-5-іл]-N1 фенілацетаміду підтверджено спектроскопією H ЯМР. Спектри ядерного магнітного резонансу виміряно на спектрометрі VARIAN Mercury VX-200 (200 МГц) у розчинах дейтеродиметилсульфоксиду (DMSO-d6), внутрішній стандарт - тетраметилсилан; (м.ч.): 2.65 (1Н, дд, 11.3, 16.5, На), 3.21 (1Н, дд, 3.7, Нb), 3.38 (1Н, дд, 4.0, 18.3, Hd), 4.11 (1Н, дд, 11.3, Не), 4.39 (1Н, дд, Нс), 5.80 (1Н, дд, Hf),7.0-7.9 (15Н, м, ArН), 10.1 (1Н, с, NH). Приклад 2. Одержання 2-[2-(3-(4-фторфеніл)5-(4-метилфеніл)-4,5-дигідро-1Н-піразол-1-іл)-4оксо-4,5-дигідро-1,3-тіазол-5-іл]-N-(3,4-диметоксифеніл)-ацетаміду (див. табл. 2, поз. 2). Одержують аналогічно прикладу 1. Для реакції беруть: 2,4 г (0,01М) 4-метилфеніл-4'фторфенілхалкона, 0,9 г (0,01М) тіосемикарбазиду, 0,6 г (0,01М) КОН, 2,3 г (0,01М) N-(3,5диметоксифеніл)малеіміду, 20 мл етилового спирту, 5 мл оцтової кислоти. Вихідні дані: т.пл. 260-261°С, вихід 4,26 г (78 %). 1 Спектр Н ЯМР: 2.23 (3H, с, СН3), 2.70 (1Н, дд, 11.2, 16.3, На), 3.18 (1Н, дд, 3.6, Нb), 3.39 (1Н, дд, 4.0, 18.1, Hd), 3.70 (3H, с, СН3О), 3.80 (3H, с, СН3О), 4.08 (1Н, дд, 11.1, Не), 4.37 (1Н, дд, Нс), 5.75 (1Н, дд, Hf), 6.8-8.0 (11Н, м, ArН), 10.1 (1Н, с, NH). Решту прикладів зведено у таблиці 2. Спосіб, що заявляється, дозволяє: - зменшити кількість технологічних стадій до 2 (у способі-прототипі 4 стадії); - зменшити тривалість процесу одержання сполук формули І до 2-2,5 годин (у способіпрототипі 18-20 годин); - виключити перекристалізації кінцевих продуктів (у способі-прототипі використовують очистку як проміжних, так і кінцевого продукту, і хроматографування кінцевого продукту); - підвищити вихід чистих кінцевих продуктів до 66-80% (у способі-прототипі вихід складає приблизно 40-42%); забезпечити відсутність вогненебезпечних і токсичних розчинників. Спосіб одержання сполук формули І простий у технологічному відношенні, пройшов апробацію у 11 лабораторних умовах. Можливість широкої модифікації структури заміщених 2-[2-(3,5-діарил-4,5дигідро-1Н-піразол-1-іл)-4-оксо-4,5-дигідро-1,3тіазол-5-іл]-N-арил-ацетамідів і синтезу великої 94597 12 кількості сполук за заявляємим способом дозволяє передбачити, що спосіб знайде широке застосування як у лабораторній практиці, так і в умовах виробництва. Таблиця 1 Спосіб, що заявляєтьСпосіб за прототипом ся Кількість стадій реакції 2 4 Тривалість всього процесу, годин 2-2,5 18-20 Вихід кінцевих продуктів, % 66-80 40-42 Очищення проміжних і кінцевих потребуються перекристалізації на кожній стадії і не потребується продуктів хроматографування кінцевого продукту Застосування токсичних речовин не використовуються бромбензол, монохлороцтова кислота 13 94597 14 15 94597 16 17 Комп’ютерна верстка О.Гапоненко 94597 Підписне 18 Тираж 24 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for the preparation of 2-[2-(3,5-diaryl-4,5-dihydro-1h-pyrazole-1-yl)-4-oxo-4,5-dihydro-1,3-tiazole-5-yl]-n-arylacetamides
Автори англійськоюRudenko Roman Volodymyrovych, Komikhov Serhii Oleksandrovych, Desenko Serhii Mykhailovych, Afanasiadi Liudmyla Mykhailivna
Назва патенту російськоюСпособ получения замещенных 2-[2-(3,5-диарил-4,5-дигидро-1н-пиразол-1-ил)-4-оксо-4,5-дигидро-1,3-тиазол-5-ил]-n-арилацетамидов
Автори російськоюРуденко Роман Владимирович, Комихов Сергей Александрович, Десенко Сергей Михайлович, Афанасиади Людмила Михайловна
МПК / Мітки
МПК: C07D 417/00
Мітки: заміщених, 2-[2-(3,5-діарил-4,5-дигідро-1н-піразол-1-іл)-4-оксо-4,5-дигідро-1,3-тіазол-5-іл]-n-арилацетамідів, спосіб, одержання
Код посилання
<a href="https://ua.patents.su/9-94597-sposib-oderzhannya-zamishhenikh-2-2-35-diaril-45-digidro-1n-pirazol-1-il-4-okso-45-digidro-13-tiazol-5-il-n-arilacetamidiv.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання заміщених 2-[2-(3,5-діарил-4,5-дигідро-1н-піразол-1-іл)-4-оксо-4,5-дигідро-1,3-тіазол-5-іл]-n-арилацетамідів</a>
N-(1,5-диметил-3-оксо-2-феніл-2,3-дигідро-1н-піразол-4-іл)-4-(5-ариліден-4-оксо-2-тіоксотіазолідин-3-іл)алканаміди, що проявляють антиексудативну дію

Номер патенту: 28396
Опубліковано: 10.12.2007
Автори: Нєктєгаєв Ігор Олексійович, Лесик Роман Богданович, Горішній Володимир Ярославович, Зіменковський Борис Семенович, Демчук Інна Леонідівна
МПК: C07D 277/08
Мітки: проявляють, дію, n-(1,5-диметил-3-оксо-2-феніл-2,3-дигідро-1н-піразол-4-іл)-4-(5-ариліден-4-оксо-2-тіоксотіазолідин-3-іл)алканаміди, антиексудативну
Формула / Реферат:
N-(1,5-диметил-3-оксо-2-феніл-2,3-Дигідро-1Н-піразол-4-іл)-4-(5-ариліден-4-оксо-2-тіоксотіазолідин-3-іл)алканаміди загальної формули,де R = H, OCH3; n = 1,3, які проявляють антиексудативну дію.
Спосіб одержання заміщених 7-гідрокси-7-метил-2-(алкілтіо)n,5-діарил-4,5,6,7-тетрагідро[1,2,4]триазоло[1,5-a] піримідин -6-карбоксамідів

Номер патенту: 50266
Опубліковано: 25.05.2010
Автори: Десенко Сергій Михайлович, Чебанов Валентин Анатолійович, Афанасіаді Людмила Михайлівна, Муравйова Олена Олександрівна
МПК: C07D 239/00
Мітки: одержання, 6-карбоксамідів, 7-гідрокси-7-метил-2-(алкілтіо)n,5-діарил-4,5,6,7-тетрагідро[1,2,4]триазоло[1,5-a, спосіб, заміщених, піримідин
Формула / Реферат:
Спосіб одержання заміщених 7-гідрокси-7-метил-2-(алкілтіо)-N,5-діарил-4,5,6,7-тетрагідро[1,2,4]триазоло[1,5-а]піримідин-6-карбоксамідів формули І, Iдe Аr = С6Н5; 2-СН3О-С6Н4; 4-СН3О-С6Н4; 2,4-диCH3-C6H3; 2-CH3-C6H4; 4-СlС6Н4Alk = СН3; СН2СН3; СН(СН3)2; СН2С6Н5; СН2(3-СН3-С6Н4); СН2(3-Сl-С6Н4); СН2(4-Сl-С6Н4); CH2(4-F-C6H4);...
Спосіб одержання 5-[2-етокси-5-(4-метилпіперазин-1-іл-сульфоніл)-феніл]-1-метил-3-н-пропіл-1,6-дигідро-7н-піразол-[4,3-d]-піримідин-7-ону та способи одержання проміжних продуктів

Номер патенту: 78692
Опубліковано: 25.04.2007
Автори: Моді Шіріш Бхагванлал, Шріканде Атул Анант, Доші Мадхукант Мансукхлал
МПК: C07D 237/00
Мітки: способи, продуктів, одержання, спосіб, 5-[2-етокси-5-(4-метилпіперазин-1-іл-сульфоніл)-феніл]-1-метил-3-н-пропіл-1,6-дигідро-7н-піразол-[4,3-d]-піримідин-7-ону, проміжних
Формула / Реферат:
1. Cпосіб одержання 5-[2-етокси-5-(4-метилпіперазин-1-ілсульфоніл)-феніл]-1-метил-3-н-пропіл-1,6-дигідро-7Н-піразол-[4,3-d]-піримідин-7-ону (V) та його фармацевтично прийнятних солей, який включає такі стадії:a) взаємодія 1-метил-4-аміно-3-н-пропілпіразол-5-карбоксаміду (І) із хлористим воднем у розчиннику, такому як ізопропанол, з одержанням 1-метил-4-аміно-3-н-пропілпіразол-5-карбоксамідгідрохлориду (II);b) взаємодія сполуки...
Спосіб одержання заміщених 2-метилсульфоніл-1,3-діарил(гетерил)-проп-2-ен-1-онів

Номер патенту: 57633
Опубліковано: 10.03.2011
Автори: Гладков Євгеній Станіславович, Десенко Сергій Михайлович, Сірко Світлана Миколаївна, Афанасіаді Людмила Михайлівна
МПК: C07C 317/08
Мітки: заміщених, одержання, спосіб, 2-метилсульфоніл-1,3-діарил(гетерил)-проп-2-ен-1-онів
Формула / Реферат:
Спосіб одержання заміщених 2-метилсульфоніл-1,3-діарил(гетерил)-проп-2-ен-1-онів загальної формули І:I,де R=С6Н5, 4-СН3-С6Н4;R1=С6Н5, 4-СН3-С6Н4, 4-Сl-С6Н4,4-ОСН3-С6Н4, 4-Вr-С6Н4, 4-F-С6Н4, 3-СН3-С6Н4, 2-СН3-С6H4,,що включає взаємодію відповідної...
Спосіб одержання необов’язково 2-заміщених 1,6-дигідро-6-оксо-4-піримідинкарбонових кислот

Номер патенту: 92171
Опубліковано: 11.10.2010
Автор: Шапіро Рафаель
МПК: C07D 239/28, A61K 31/505
Мітки: одержання, необов'язково, спосіб, 1,6-дигідро-6-оксо-4-піримідинкарбонових, 2-заміщених, кислот
Формула / Реферат:
1. Спосіб одержання сполуки Формули 1, 1 де R1 являє собою Н або необов'язково заміщений вуглецевий компонент;який відрізняється тим, що здійснюють наступні стадії: (1) реакцію суміші, що містить (а) сполуку Формули 2а,...
Попередній патент: N-сульфамоїл-n’-бензопіранпіперидини, призначені для застосування у медицині
Наступний патент: Діарилсечовини для лікування легеневої гіпертензії
Випадковий патент: Бур для відбору зразків ґрунту та підкарантинної продукції для виявлення карантинних організмів