Спосіб одержання інгібітору трипсину
Номер патенту: 54993
Опубліковано: 17.03.2003
Автори: Діхтярьов Сергій Іванович, Чушенко Валентина Миколаївна, Сухінін Валентин Миколайович, Маслова Наталія Федорівна, Кузнєцова Ірина Володимирівна, Січкар Лілія Анатоліївна








Формула / Реферат
1. Спосіб одержання інгібітору трипсину з насіння сої, що включає підготовку сировини шляхом її подрібнення, екстракцію, відділення екстракту від шроту, очистку екстракту від супутніх білків, адсорбцію інгібітору трипсину та промивку його на сорбенті, елюювання інгібітору трипсину розчинником, відмивання елюату від домішок, фільтрацію очищеного елюату, висушування цільового продукту, який відрізняється тим, що при підготовці сировини перед подрібненням здійснюють замочування насіння сої у воді для набухання протягом 10-12 год, екстракцію сировини здійснюють водою очищеною одночасно з подрібненням, відділення екстракту від шроту проводять за допомогою центрифугування або фільтрації під вакуумом, очистка екстракту від супутніх білків включає їх осадження шляхом додавання до екстракту лимонної кислоти або оцтової кислоти, або трихлороцтової кислоти, або сульфату кальцію, або хлориду кальцію з подальшим відділенням осаджених продуктів центрифугуванням або фільтрацію під вакуумом, обробку одержаного екстракту інгібітору трипсину на ультрафільтраційній установці шляхом його діафільтрації та концентрації, для адсорбції інгібітору трипсину використовують як біоспецифічні сорбенти трипсин-сефарозу або трипсин-целюлозу, або ензайт-трипсин, або трипсин-тойоперл та буфер 0,045-0,05 М трис-НСl з рН 7,0-8,0, а промивку його на сорбенті проводять спочатку буфером 0,045-0,05 М трис-НСl з рН 7,0-8,0, потім 0,5-0,7 М натрію хлориду та водою очищеною, елюювання інгібітору трипсину проводять підкисленою до рН 2,0-2,5 водою, або підкисленою водою з додаванням 0,15-0,20 М натрію хлориду, або 0,20-0,25 М калію хлориду, відмивання елюату від домішок здійснюють електродіалізом або діафільтрацією з подальшим його концентруванням, а висушування продукту здійснюють шляхом ліофілізації.
2. Спосіб згідно з п. 1, який відрізняється тим, що після відмивання елюату від домішок та перед висушуванням його піддають стерильній фільтрації у випадку створення стерильних лікарських засобів, таких як ін'єкційні та інфузійні розчини або стерильні мазі та інші стерильні місцеві засоби.
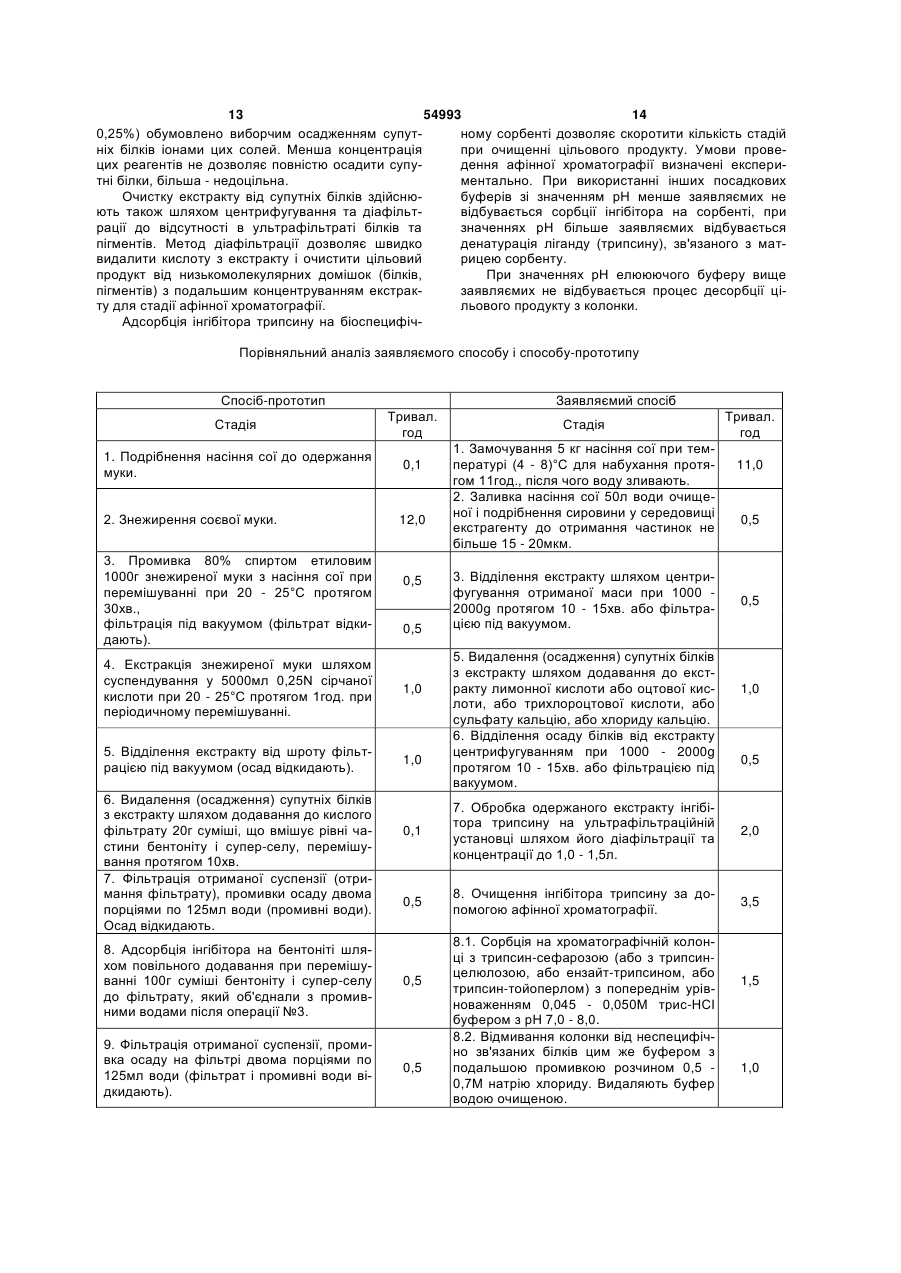
Текст
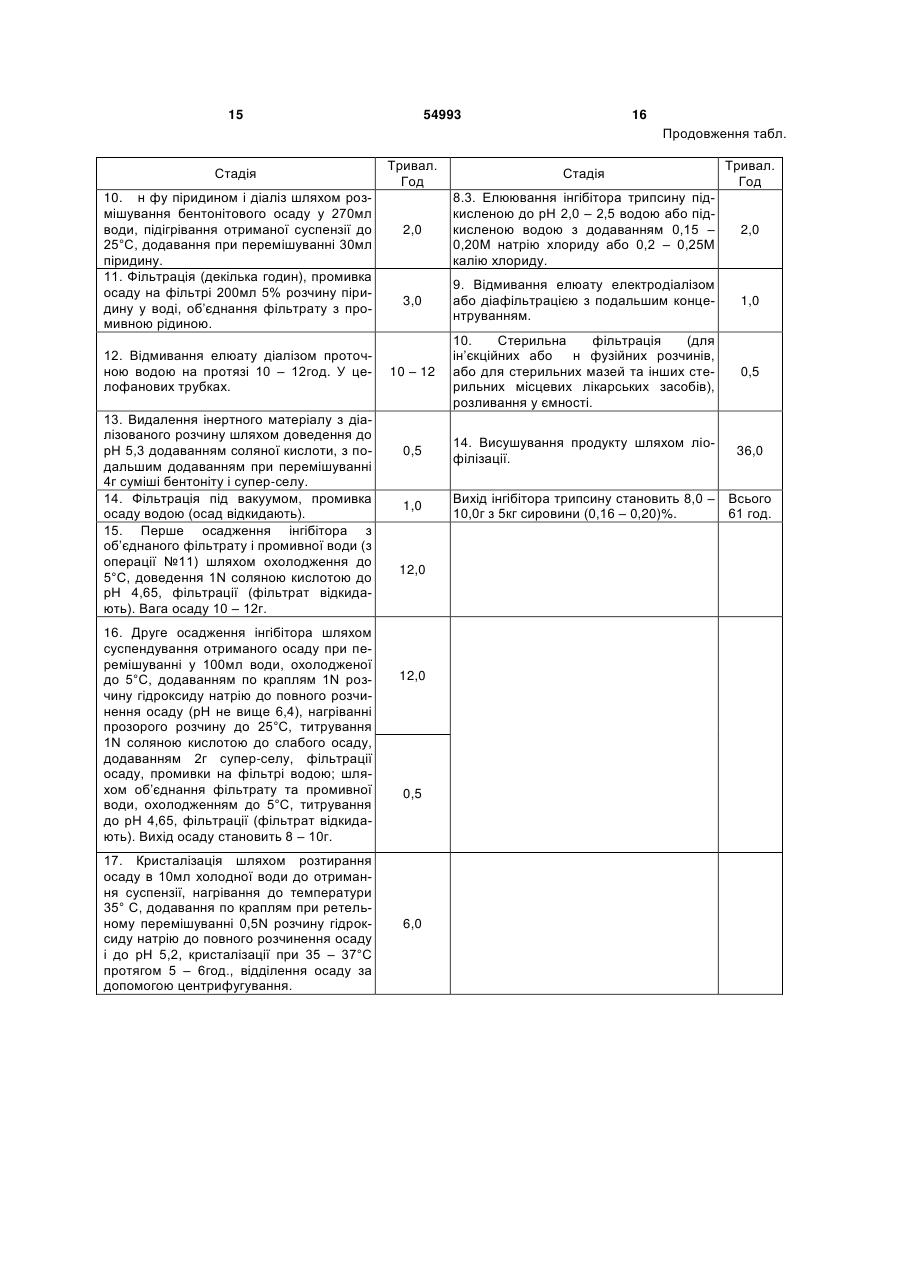
1. Спосіб одержання інгібітору трипсину з насіння сої, що включає підготовку сировини шляхом її подрібнення, екстракцію, відділення екстракту від шроту, очистку екстракту від супутніх білків, адсорбцію інгібітору трипсину та промивку його на сорбенті, елюювання інгібітору трипсину розчинником, відмивання елюату від домішок, фільтрацію очищеного елюату, висушування цільового продукту, який відрізняється тим, що при підготовці сировини перед подрібненням здійснюють замочування насіння сої у воді для набухання протягом 10-12 год, екстракцію сировини здійснюють водою очищеною одночасно з подрібненням, відділення екстракту від шроту проводять за допомогою центрифугування або фільтрації під вакуумом, очистка екстракту від супутніх білків включає їх C2 2 (19) 1 3 54993 4 люють, діазолюють, ретентат ліофілізують. Всі таким чином. Зелену масу люцерни подрібнюють, операції з білками проводять на холоді (4 - 5°С), а під пресом віджимають сік, підкислюють 1N. НСІ хроматографію - при кімнатній температурі (17 до рН 4,2 і відділяють коагульовані білки центри20°С). Отримують 35мг інгібітора трипсину з питофугуванням. Отриманий сік підкислюють 20% тримою активністю білка 1050мІЄ/мг (1). хлороцтовою кислотою до кінцевої концентрації Відомий спосіб одержання інгібітора трипсину 2%, центрифугують, осад відкидають, надосадну шляхом подрібнення скошеної люцерни (у стадії рідину діалізують на холоду проти 0,1Μ ацетатнобутонізації) з подальшим віджиманням соку. Сік го буферу рН 5,0. Хроматографію на колонці з КМ3 підігрівають до 85°С. Коагульовані білки осаджуцелюлозою (3,5 x 20)см проводять в 0,1Μ ацетатють центрифугуванням, супернатант (4,2л) виконому буфері, рН 5,0. Білок елююють, використористовують для біоспецифічного хроматографувуючи безперервний градієнт концентрації розчину 3 вання на трипсин-сефарозі. Колонку (150 x 15)см натрію хлориду 0,1 - 0,5М. Афінну хроматографію з сорбентом заповнюють порцією супер-натанту, інгібітора трипсину здійснюють на трипсинщо дорівнює 250мл (зовнішній об'єм, сорбенту). сефарозі 4В. Сорбцію інгібітора проводять при рН Кожну порцію (всього 17 порцій) витримують у ко5,0 в 0,1М ацетатном буфері, що містить 0,22Μ лонці у відповідності з терміном зв’язування інгібінатрію хлориду і 0,01Μ кальцію хлориду. Елюютора з трипсином. Пігменти видаляють 8 об'ємами вання інгібітора здійснюють розчином, що містить води, неспецифічно зв’язані білки десорбують 10 0,01N соляну кислоту, рН 2,0, 1Μ натрію хлориду і об'ємами 0,15Μ ацетатного буферного розчину з 15% (по об'єму) ізопропілового спирту. Гель0,3Μ хлориду натрію, рН 7,0. Десорбцію здійснюхроматографію очищеного інгібітора трипсину ють 0,15Μ ацетатним буферним розчином з 0,3Μ проводять на колонці з сефадексом G-100, урівнохлориду натрію при рН 7,0. Хлорид натрію віддіваженій 0,05Μ цитратним буфером, рН 2,7 і кололяють промивкою 6 об'ємами води. Елююють нці з акрилексом П-60, урівноваженій 0,1Μ ацетат0,08Μ уксусною кислотою. Отримують 53мг інгібіним буфером, рН 5,0. Диск-електрофорез тора трипсину, електрофоретичне гомогенного, препаратів ферменту проводять у системі гелів: нетоксичного: так, внутрішньовенне введення пре7,5% поліакриамідний розподіляючий гель, лужна парату в дозі 50мг, що є 20-кратною лікувальною система буферів, 1 - 5мА на 1 стовпчик гелю. Акдозою, не визиває загибелі щурів (2). тивність інгібітора трипсину виражають у кількості Відомий спосіб виділення інгібітора трипсину з трипсину (мг), інгібуємого 1мг білка препарату інгікурячих яєць, який здійснюють таким чином. З бітора (5). 13мл курячого білку механічно відділяють 6мл рідВідомий спосіб одержання інгібіторів цистеінокого білка, екстрагують його у 20мл 2N розчину вих протеїназ із соєвих бобів сорту "Приморська хлориду натрію протягом 30хв. при перемішуванні 491", який здійснюють таким чином. при 4 - 5°С. Отриманий екстракт підкислюють 0,5N 1. Отримання препарату спирторозчинних білсоляною кислотою до рН 1,5, потім центрифугують ків. Подрібнену сировину знежирюють ацетоном, 45хв. при 5500g. Надосадочну рідину відділяють і білки екстрагують 60% спиртом етиловим при витримують 28год при 4 - 5°С, потім знову 55°С. Після доведення рН екстракту до 5,3 (за доцентрифугують. Отриману надосадочну рідину помогою концентрованої НСІ) білки висаджують діалізують у целофанових мішках роти дистильодвома об’ємами охолодженого ацетону при 0°С, ваної води, підлуженої до рН 10,0 (до отримання відстоюють 2,0 - 2,5год. при 0°С, осад відокремнегативної реакції на сульфатіон). Вміст целофалюють. Отриманий осад розчиняють у мінімальній нових мішків центрифугують, а надосадкову рідину кількості води, діалізують, висушують ліофільно. ліофілізують. Отримують 115мг препарату інгібіто2. Виділення інгібіторів типу Баумана-Бірк. Для ра трипсину, який має питому активність отримання очищених інгібіторів препарат спирто290,2мІО/мг білка (3). розчинних білків розчинюють у 0,1Μ фосфатному Відомий спосіб одержання Інгібітора трипсину буфері (рН 8,0), наносять на колонку з хімотрипз зерен ячменю, який здійснюють таким чином: син-сефарозой 4В. Колонку промивають тим же 300г зерен ярового ячменю сорту „Чорноморець" буфером, який містить 0,5Μ натрію хлориду, потім подрібнюють і екстрагують 1,8л 0,1Μ соляної киспроводять елюцію інгібітора 0,2Μ калію хлоридом лоти. Після діалізу проти 20-кратного надлишку при рН 2,0. дистильованої води на протязі 20год. збирають 3. Виділення інгібіторів цистеїнових протеїназ. осад шляхом центрифугування. Із сухого осаду Препарат фіцину активують у розчині, що містить інгібітор здобувають 0,1Μ триетаноламіновим бу0,1Μ фосфатний буфер (рН 6,0), 2мМ цистеїн та фером рН 8,0 трикратними порціями по 75; 50 і 0,1мМ ЕДТА, протягом 1год. при 20°С і обробля25мл. Об'єднані екстракти прогрівають на киплячій ють іодацетамідом до кінцевої концентрації 10мМ. водяній бані 15хв., після чого денатуровані білки Отримане неактивне похідне фіцину приєднують відділяють центрифугуванням. До надосадкової до CNBr-активованої сефарози 4В у стандартних рідини додають 31г сульфату амонію (до 40% наумовах. Препарат спирторозчинних білків розчисичення) і після відстоювання на холоду на протязі няють у 0,1Μ фосфатному буфері рН 8,0 і переміночі відділяють осад центрифугуванням. До осаду шують з суспензією фіцин-сефарози протягом додають 100мл 0,1Μ амоній-ацетатного буферу 18год. при 4°С. Потім сорбент переносять на хрорН 6,0, перемішують 45хв., центрифугують, суперматографічну колонку і промивають 0,1Μ фосфатнатант ліофільно висушують. Отримують 132г інгіним буфером (рН 8,0), який містить 0,5Μ NaCl. бітора трипсину з питомою активністю 0,61 (4). Елюцію інгібіторів проводять 0,1Μ NaCl при рН Відомий спосіб одержання інгібітора трипсину 12,0. Елюат доводять до рН 7 - 8 за допомогою з вегетативних органів люцерни, який здійснюють 0,24Μ КН2РО4 (6). 5 54993 6 Відомий спосіб одержання інгібітора трипсиФільтрат і промивні води (операція №6) ’ ноподібних протеаз, який здійснюють таким чином: об єднують, охолоджують до 5°С, титрують 1N легені білих мишей промивають від крові фосфатсоляною кислотою до рН 4,65. Відфільтровують ним буфером (рН 7,5) при температурі 4°С, потім осад, що утворився. Фільтрат відкидають. Вага подрібнюють, додають фосфатний буфер, гомогеосаду складає 10 - 12г. нізують ультразвуком протягом 30 - 60сек. і 8. Друге осадження інгібітора при рН 4,65. центрифугують. Супернатант заморожують до Отриманий осад суспендують при переміщутемператури -18 -20°С, до осаду додають 1% розванні у 100мл води, охолодженої до 5°С, додають чин тритону Х-100, перемішують і поміщають на по краплям 1N розчин гідроксиду натрію до повнохолод при температурі 4 - 6°С на 18 - 20год. Після го розчинення осаду (рН не вище 6,4). Прозорий цього осад гомогенізують і центрифугують при розчин нагрівають до 25°С, титрують до утворення вказаних вище параметрах. І і II супернатанти осаду, додають 2г супер-селу. Осад відфільтрооб'єднують і піддають розподілу протеази та інгібівують, промивають на фільтрі декільками мл води. тора за допомогою іонообмінної хроматографії. Фільтрат і промивні води об'єднують, охолоджують Очистку інгібітора протеаз до гомогенного стану до 5°С, титрують до рН 4,65. Одержаний осад віпроводять за допомогою гельфільтрації на сефадфільтровують, вихід становить 8 - 10г. Фільтрат дексах з подальшою афінною хроматографією на відкидають. трипсин-сефарозі 4В. Десорбцію білків проводять 9. Кристалізація. фосфатним буфером і діалізом проти води з поОтриманий від операції №8 осад (приблизно дальшою ліофільною сушкою та ампулюванням 10г) розтирають у 10мл холодної води до отри(7). мання суспензії, потім нагрівають до 35°С. ДодаНайбільш близьким до заявляємого є спосіб ють по краплям при ретельному перемішуванні одержання інгібітора трипсину з соєвих бобів, який 0,5N розчин гідроксиду натрію до повного розчиздійснюють таким чином. нення осаду і до рН 5,2, залишають для кристалі1. Промивка 80% спиртом етиловим. 1000г обзації при 35 - 37°С протягом 5 - 6год. Осад віддіробленої на холоді знежиреної муки з соєвих бобів ляють за допомогою центрифугування. додають до суміші з 2400мл 95% спирту етилово10. Перекристалізація. го, охолодженого до 5°С та 450мл води дистильоОтриманий кристалічний осад розмішують у ваної. Суспензію ретельно перемішують і залишаподвійній кількості холодної води та титрують 0,5N ють при 20 - 25°С на 30хв. Потім фільтрують під розчином гідроксиду натрію до одержання прозовакуумом, фільтрат відкидають. рого розчину і до рН 6,0. Прозорий розчин нагрі2. Екстракція 0,25Ν сірчаною кислотою. Напіввають до 35°С і титрують 0,5N розчином соляної суху муку знову суспендують у 5000мл 0,25N сіркислоти до рН 5,1 до отримання слабкого осаду, чаної кислоти при 20 - 25°С і залишають на 1год. додають 2г супер-селу і фільтрують до тих пір, при кімнатній температурі, періодично перемішуюпоки розчин не стане прозорим. У фільтрат додачи. Суспензію знову фільтрують під вакуумом, ють затравку і залишають при 36 - 37°С. Поступоосад відкидають. во утворюється густа суспензія кристалів, яку че3. Видалення інертного білка. До кислого фірез 5 - 6год. відділяють центрифугуванням. Осад льтрату додають 20г суміші, що вміщує рівні часзберігають при 5°С. Додаванням 1 - 2 крапель ротини бентоніту і супер-селу, і перемішують 10хв. зчину 0,2N соляної кислоти доводять рН відстою Одержану суспензію фільтрують, отримують фільдо рН 5,1, вводять затравку і розчин залишають трат. Осад промивають двома порціями по 125мл ще на декілька годин при 36 - 37°С для отримання води (промивні води). Осад відкидають. додаткової порції кристалів, яку відділяють таким 4. Адсорбція інгібітора на бентоніті. же чином, як описано вище. Можна ще отримати До фільтрату, який об'єднали з промивними кристали з кінцевого відстою, якщо його охолодити водами після операції №3, повільно додають при до 5°С і додати 0,25 об'єму холодного 95% спирту перемішуванні 100г суміші бентоніту і супер-селу, етилового, як описано у наступному розділі. продовжують перемішувати ще 10хв., після чого 11. Кристалізація з розбавленого спирту. суспензію фільтрують, а осад на фільтрі промиваОтримані кристали розмішують у п'ятикратноють двома порціями води по 125мл. Фільтрат і му об'ємі холодної води і додають по краплям розпромивні води відкидають. чин 0,5N гідроксиду натрію до повного розчинення 5. Елюція піридином та діаліз. кристалів до рН розчину не вище 6,6. Прозорий Бентонітовий осад розмішують у 270мл води, розчин титрують 0,2N соляної кислоти до рН 5,2, суспензію нагрівають до 25°С, додають при передо утвореного осаду додають супер-сел з розрамішуванні 30мл піридину, фільтрують, осад на хунку 4г на 100мл розчину, осад відділяють і профільтрі промивають 200мл 5% розчину піридину у мивають на фільтрі незначною кількістю води. Фіводі. Фільтрат об'єднують з промивною рідиною, льтрат охолоджують до 5°С, повільно додають діалізують проточною водою протягом 10 - 12год. у чверть об’єму охолодженого 95% спирту етиловоцелофанових трубках довжиною 30см. го для утворення осаду. Додаванням розчину 0,2N 6. Видалення інертного матеріалу при рН 5,3. соляної кислоти доводять рН суміші до 5,0 і залиДіалізований розчин, вільний від клейкого осашають її при 30°С. Аморфний осад перетворюєтьду, доводять до рН 5,3 додаванням приблизно 2мл ся на протязі 2год. у кристалічний, який швидко 1N соляної кислоти. У розчині перемішують 4г суосідає на дно ємності. Відстій розчину кожну годиміші бентоніту і супер-селу, фільтрують під вакууну декантують, доводять розчином соляної кислот мом, осад промивають водою і відкидають. до рН 5,0 і повертають до основної ємності. Так 7. Перше осадження інгібітора при рН 4,65. повторюють протягом декількох годин до повного 7 54993 8 припинення появи осаду при рН 5,0. Після цього сушування продукту здійснюють шляхом ліофілісуміш, яка кристалізується, витримують ще 30хв. зації, причому після відмивання елюату від доміпри 30°С. Утворений осад відфільтровують і висушок та перед висушуванням його піддають стеришують при кімнатній температурі протягом 24год. льній фільтрації у випадку створення стерильних 12. Перекристалізація із спирту етилового. лікарських засобів, таких, як ін'єкційні та інфузійні Сухі кристали суспендують у тридцятикратнорозчини або стерильні мазі та інші стерильні місму (по відношенню до їх ваги) об'ємі холодної воцеві засоби. ди, витримують 5 - 10хв., а потім обробляють таТехнічний результат, якого досягають при ким же чином, як і в операції №11. здійсненні винаходу, полягає у розробці способу Вихід кристалів інгібітора трипсину змінюється одержання інгібітора трипсину з рослинної сироу залежності від сорту муки і, в середньому, ставини, який, за рахунок підбору технологічних опеновить приблизно 1г з 1000г сировини - 0,1% (8). рацій у такій послідовності та взаємозв'язку і з таДо недоліків способу-прототипу слід віднести кими режимами та параметрами, забезпечує багатостадійність, значну тривалість процесу, непідвищення виходу цільового продукту, спрощення високий вихід цільового продукту, великі енерготехнологічного процесу шляхом зменшення кільта трудовитрати, екологічну забрудненість виробкості стадій і їх тривалості у часі, скорочення енерництва. го- і трудовитрат, екологічну чистоту виробництва. В основу винаходу поставлено завдання ствоНаводимо конкретні приклади здійснення вирення способу одержання з рослинної сировини находу. інгібітора трипсину шляхом підбору технологічних Приклад 1. 5кг промитого насіння сої замочуоперацій у такій послідовності та взаємозв'язку і з ють у воді очищеній при температурі 4°С для натакими режимами та параметрами, які б забезпебухання протягом 10год. Воду зливають, а розбухчили підвищення виходу цільового продукту, ле насіння сої заливають 50л води очищеної і спрощення технологічного процесу шляхом зменекстрагують, одночасно подрібнюючи сировину у шення кількості стадій і їх тривалості у часі, скоросередовищі екстрагенту. Отриману суміш центричення енерго- і трудовитрат, екологічну чистоту фугують або фільтрують під вакуумом для віддівиробництва. лення екстракту від шроту, після чого отриманий Поставлене завдання вирішується тим, що у екстракт інгібітора трипсину передають на стадію способі одержання інгібітора трипсину з рослинної очистки, яку здійснюють у декілька етапів. Спочатсировини, що включає підготовку насіння сої шляку для осадження супутніх білків до екстракту дохом його подрібнення, екстракцію, відділення ексдають при перемішуванні кислоту лимонну до рН тракту від шроту, очистку екстракту від супутніх 3,5 - 4,0, після чого осаджені білки відділяють на білків, адсорбцію інгібітора трипсину та промивку центрифузі при 1000g протягом 15хв. або фільтйого на сорбенті, елюювання інгібітора трипсину рують під вакуумом. Одержаний прозорий екстракт розчинником, відмивання елюату від домішок, фіінгібітора трипсину на ультрафільтраційній устальтрацію очищеного елюату, висушування цільоновці діафільтрують до відсутності білків і пігменвого продукту, згідно з винаходом, при підготовці тів і концентрують до 1,0л. Подальшу очистку одесировини перед подрібненням здійснюють замочуржаного концентрату інгібітора трипсину вання насіння сої у воді для набухання протягом здійснюють методом афінної хроматографії на 10 - 12год., екстракцію сировини здійснюють вохроматографічній колонці, що заповнена біоспедою очищеною одночасно з подрібненням, віддіцифічним сорбентом трипсин-сефорозою з попелення екстракту від шроту проводять за допомореднім урівноваженням 0,045Μ трис-НСІ буфером гою центрифугування або фільтрації під вакуумом, з рН 7,0. Колонку відмивають від неспецифічно очистка екстракту від супутніх білків включає їх зв'язаних білків цим же буфером з подальшою осадження шляхом додавання до екстракту, липромивкою розчином 0,5Μ натрію хлориду. Видамонної кислоти або оцтової кислоти, або трихлоляють буфер водою очищеною і елююють інгібітор роцтової кислоти, або сульфату кальцію, або хлотрипсину підкисленою до рН 2,0 водою. Одержариду кальцію з подальшим відділенням осаджених ний елюат відмивають від кислоти електродіалізом продуктів центрифугуванням або фільтрацієй під або діафільтрацією, концентрують, розливають у вакуумом, обробку одержаного екстракту інгібітора ємності і ліофілізують. Вихід інгібітора трипсину трипсину здійснюють на ультрафільтраційній становить 8,0. установці шляхом його діафільтрації та концентПриклад 2. 5кг промитого насіння сої замочурації, для адсорбції інгібітора трипсину використоють у воді очищеній при температурі 6°С для навують як біоспецифічні сорбенти трипсинбухання протягом 11год. Воду зливають, а розбухсефарозу або трипсин-целюлозу, або ензайтле насіння сої заливають 50л води очищеної і трипсин, або трипсин-тойоперл та буфер 0,045 екстрагують, одночасно подрібнюючи сировину у 0,05Μ трис-НСІ з рН 7,0 - 8,0, а промивку його на середовищі екстрагенту. Отриману суміш центрисорбенті проводять спочатку буфером 0,045 фугують при 1500g протягом 12хв. або фільтрують 0,05Μ трис-НСІ з рН 7,0 - 8,0, потім 0,5 - 0,7Μ напід вакуумом для відділення екстракту від шроту, трію хлориду та водою очищеною, елюювання після чого отриманий екстракт інгібітора трипсину інгібітора трипсину проводять підкисленою до рН передають на стадію очистки, яку здійснюють у 2,0 - 2,5 водою, або підкисленою водою з додадекілька етапів. Спочатку для осадження супутніх ванням 0,15 - 0,20Μ натрію хлориду, або 0,20 білків до екстракту додають при перемішуванні 0,25Μ калію хлориду, відмивання елюату від докислоту оцтову до рН 2,0 - 4,0, після чого осаджені мішок здійснюють електродіалізом або діафільтбілки відділяють на центрифузі або фільтрують під рацією з подальшим його концентруванням, а вивакуумом. Одержаний прозорий екстракт інгібітора 9 54993 10 трипсину на ультрафільтраційній установці діафімом. Одержаний прозорий екстракт інгібітора трильтрують до відсутності білків і пігментів і концентпсину на ультрафільтраційній установці діафільтрують до 1,1л. Подальшу очистку одержаного конрують до відсутності білків і пігментів і концентруцентрату інгібітора трипсину здійснюють методом ють до 1,4л. Подальшу очистку одержаного афінної хроматографії на хроматографічній колоконцентрату інгібітора трипсину здійснюють метонці, що заповнена біоспецифічним сорбентом тридом афінної хроматографії на хроматографічній псин-сефарозою з попереднім урівноваженням колонці, що заповнена біоспецифічним сорбентом 0,045Μ трис-НСІ буфером з рН 7,5. Колонку відензайт-трипсином з попереднім урівноваженням мивають від неспецифічно зв'язаних білків цим же 0,05Μ трис-НСІ буфером з рН 8,0. Колонку відмибуфером з подальшою промивкою розчином 0,5Μ вають від неспецифічно зв'язаних білків цим же натрію хлориду. Видаляють буфер водою очищебуфером з подальшою промивкою розчином 0,7Μ ною і елюють інгібітор трипсину підкисленою до рН натрію хлориду. Видаляють буфер водою очище2,0 водою. Одержаний елюат відмивають від кисною і елююють інгібітор трипсину підкисленою до лоти електродіалізом або діафільтрацією, конценрН 2,0 - 2,5 водою з додаванням 0,2Μ натрію хлотрують, розливають у ємності і ліофілізують. Вихід риду. Одержаний елюат відмивають від кислоти інгібітора трипсину становить 8,5г. електродіалізом або діафільтрацією, концентруПриклад 3. 5кг промитого насіння сої замочують, стерильно фільтрують (для ін'єкційних або ють у воді очищеній при температурі 8°С для наінфузійних розчинів, або для стерильних мазей та бухання протягом 12год. Воду зливають, а розбухінших стерильних місцевих лікарських засобів), ле насіння сої заливають 50л води очищеної і розливають у ємності і ліофілізують. Вихід інгібітоекстрагують, одночасно подрібнюючи сировину у ра трипсину становить 9,5г. середовищі екстрагенту. Отриману суміш центриПриклад 5. 5кг промитого насіння сої замочуфугують при 2000g протягом 10хв. або фільтрують ють у воді очищеній при температурі 8°С для напід вакуумом для відділення екстракту від шроту, бухання протягом 12год. Воду зливають, а розбухпісля чого отриманий екстракт інгібітора трипсину ле насіння сої заливають 50л води очищеної і передають на стадію очистки, яку здійснюють у екстрагують, одночасно подрібнюючи сировину у декілька етапів. Спочатку для осадження супутніх середовищі екстрагенту. Отриману суміш центрибілків до екстракту додають при перемішуванні фугують при 1000g протягом 15хв. або фільтрують кислоту трихлороцтову до рН 2,0 - 3,0, після чого під вакуумом для відділення екстракту від шроту, осаджені білки відділяють на центрифузі або фільпісля чого отриманий екстракт інгібітора трипсину трують під вакуумом. Одержаний прозорий екстпередають на стадію очистки, яку здійснюють у ракт інгібітора трипсину на ультрафільтраційній декілька етапів. Спочатку для осадження супутніх установці діафільтрують до відсутності білків і пігбілків до екстракту додають при перемішуванні ментів і концентрують до 1,2л. Подальшу очистку кальцію хлорид, після чого осаджені білки відділяодержаного концентрату інгібітора трипсину здійсють на центрифузі або фільтрують під вакуумом. нюють методом афінної хроматографії на хромаОдержаний прозорий екстракт інгібітора трипсину тографічній колонці, що заповнена біоспецифічна ультрафільтраційній установці діафільтрують ним сорбентом трипсин-целюлозою з попереднім до відсутності білків і пігментів і концентрують до урівноваженням 0,05Μ трис-НСІ буфером з рН 7,8. 1,5л. Подальшу очистку одержаного концентрату Колонку відмивають від неспецифічно зв'язаних інгібітора трипсину здійснюють методом афінної білків цим же буфером з подальшою промивкою хроматографії на хроматографічній колонці, що розчином 0,6Μ натрію хлориду. Видаляють буфер заповнена біоспецифічним сорбентом трипсинводою очищеною і елююють інгібітор трипсину тойоперлом з попереднім урівноваженням 0,05Μ підкисленою до рН 2,0 - 2,5 водою з додаванням трис-НСІ буфером з рН 8,0. Колонку відмивають 0,15Μ натрію хлориду. Одержаний елюат відмивід неспецифічно зв'язаних білків цим же буфером вають від кислоти електродіалізом або діафільтз подальшою промивкою розчином 0,5Μ натрію рацією, концентрують, стерильно фільтрують (для хлориду. Видаляють буфер водою очищеною і ін'єкційних або інфузійних розчинів, або для стеелююють інгібітор трипсину підкисленою до рН 2,0 рильних мазей та інших стерильних місцевих ліводою з додаванням 0,20 - 0,25М калію хлориду. карських засобів), розливають у ємності і ліофіліОдержаний елюат відмивають від кислоти електзують. родіалізом або діафільтрацією, концентрують, Вихід інгібітора трипсину становить 9,0г. стерильно фільтрують (для ін'єкційних розчинів Приклад 4. 5кг промитого насіння сої замочуабо стерильних мазей та інших місцевих засобів), ють у воді очищеній при температурі 4°С для нарозливають у ємності і ліофілІзують. бухання протягом 10год. Воду зливають, а розбухле насіння сої заливають 50л води очищеної і Вихід інгібітора трипсину становить 10,0г. У екстрагують, одночасно подрібнюючи сировину у значній мірі вихід інгібітора залежить від якості середовищі екстрагенту. Отриману суміш центрисировини, тобто вмісту інгібітора трипсину в ній. фугують при 1000g протягом 15хв. або фільтрують Заявляємим способом отримують цільовий під вакуумом для відділення екстракту від шроту, продукт (інгібітор трипсину) - порошок білого або після чого отриманий екстракт інгібітора трипсину білого з жовтуватим відтінком кольору. передають на стадію очистки, яку здійснюють у Інгібітор трипсину, одержаний по заявляємому декілька етапів. Спочатку для осадження супутніх способу, проявляє інгібуючу активність по віднобілків до екстракту додають при перемішуванні шенню до таких ферментів, як трипсин, хімотрипкальцію сульфат, після чого осаджені білки віддісин і фібринолізин (плазмін) і може бути викорисляють на центрифузі або фільтрують під вакуутаний у різних лікарських формах: у формі 11 54993 12 ін'єкційних та інфузійних розчинів, у таблетованій ся найбільш біологічно придатною складовою часформі, у капсулах, супозиторіях, мазях та ін. Фартиною рослинної сировини, завдяки чому швидко макологічними дослідженнями встановлено, що відбувається її дифузія у клітини і міжклітинний отриманий інгібітор трипсину є нетоксичним. простір. Термін процесу замочування насіння сої, Білкові інгібітори протеолітичних ферментів що становить 10 - 12год., необхідний для повнограють важливу роль у підтримці гомеостазу. Вони цінного набухання сировини з метою оптимізації беруть участь у регулюванні функцій протеаз шлупроцесу подрібнення, а також підвищення виходу нково-кишкового тракту, кровоносної системи, кліцільового продукту. При тривалості замочування тин шкіри та інших органів і тканин. Численними менше заявляємих значень не досягається необдоклінічними і клінічними дослідженнями доведехідний рівень набухання насіння, при значеннях но, що активація протеаз є основним фактором у більше заявляємих - у воду для замочування відланцюзі патогенезу таких тяжких захворювань любувається небажане виділення (екстракція) цільодини, як панкреатити різної етіології, захворюванвого продукту. ня системи згортання крові, шокові та алергічні Використання води для проведення екстракції стани, різні запальні процеси та ін. Уведення до обумовлено належною розчинністю цільового просхеми лікування цих захворювань препаратів на дукта у воді, а також необхідністю збереження основі інгібіторів протеолітичних ферментів дозвовисокого рівня біологічної активності інгібітора ляє швидко купірувати біль і знизити розвиток патрипсину, економією сировини, енерго- і трудовиттологічного процесу. З цією метою застосовують рат. При цьому зберігається необхідний фармакоінгібітори синтетичного і тваринного походження. терапевтичний ефект цільового продукту. Зараз у клініках використовують такі препарати, як Подрібнення сировини у середовищі екстрагеконтрикал, трасілол, гордокс, амінокапронову киснту скорочує тривалість технологічного процесу і лоту та ін. спрощує його за рахунок об'єднання стадій подріПотреба в інгібіторах протеаз задовольняєтьбнення і екстрагування. ся, в основному, за рахунок препаратів тваринного Екстрагування цільового продукту водою з попоходження, що імпортуються, і які часто є недодальшим осадженням супутніх білків харчовими ступними для населення через високу вартість, а кислотами (наприклад, оцтової, лимонної) дозвотакож можуть ставати джерелом алергізації оргаляє використовувати відходи виробництва (шрот, нізму через присутність у їх складі супутніх білків, білки після осадження) як харчові продукти (окара, пігментів та інших домішок. У зв'язку з цим актуатофу). льною є задача створення вітчизняних антипротеДля успішного здійснення заявляємого спосоазних препаратів, особливо, на основі рослинної бу велика увага була приділена умовам подрібсировини, а також розробки ефективних способів нення рослинної сировини, водної екстракції та їх одержання. очищення цільової речовини, як найвразливішим Заявляємий спосіб одержання інгібітора трипстадіям процесу, при яких можуть спостерігатися сину має такі переваги перед прототипом і аналовтрати цільового продукту. гами: Подрібнення і екстракція за заявляємим спозбільшення виходу цільового продукту за расобом здійснювалися у "щадящому" режимі із захунок оптимізації технологічного процесу; стосуванням інтенсивної технології - подрібнення спрощення процесу одержання цільового пропроводилося одночасно з екстракцією при оптидукту за рахунок зменшення кількості технологічмальному співвідношенні екстрагент-сировина, у них стадій, скорочення термінів їх проведення (напевних температурних і часових умовах екстрагуприклад, відсутність стадії знежирення сировини вання, завдяки чому зберігається біологічна актиорганічними розчинниками, які негативно впливавність цільового продукту. Таким чином, в заявляють на організм людини); ємому способі не застосовували подрібнення комплексна переробка рослинної сировини; рослинної сировини загальноприйнятими спосоекологічна чистота технології (наприклад, за бами, при яких спостерігається місцеве перегрірахунок підібраних для проведення процесу умов і вання, окислення і осмолювання сировини, внареактивів, які не проявляють негативного впливу слідок чого відбувається розклад діючої сполуки і на організм людини), що дозволяє використовувазменшується її біологічна активність, а отже, погіти відходи виробництва (шрот, білки після стадії ршується якість цільового продукту. осадження) як харчові продукти (окара, тофу - соВідділення екстракту від шроту проводять за євий сир); допомогою центрифугування у зв'язку з тим, що отримання цільового продукту високої якості при цьому зменшуються втрати екстракту, отримушляхом використання для його виробництва обється більш очищений екстракт, скорочуються ладнання, що вже є у наявності на вітчизняних терміни проведення стадії фільтрації. хіміко-фармацевтичних підприємствах. Очистку екстракту від супутніх білків здійснюВзаємозв'язок і послідовність технологічних ють шляхом додавання лимонної або оцтової, або операцій заявляємого способу, підбір режимів і трихлороцтової кислот, або сульфату кальцію, або параметрів повністю забезпечують виконання посхлориду кальцію, концентрації яких підібрані екставленого завдання. периментальним шляхом. Додавання кислот, наПопереднє набухання насіння сої у середовиприклад, лимонної або оцтової (до рН 2,0 - 4,0) щі води інтенсифікує технологічний процес одеробумовлено вибірковою денатурацією супутніх жання інгібітора трипсину. білків при очищенні інгібітора трипсину, які осаВода є найбільш прийнятним розчинником для джуються у кислому середовищі, а додавання супроведення процесу набухання, тому що являєтьльфату чи хлориду кальцію (до конценпрації 0,20 13 54993 14 0,25%) обумовлено виборчим осадженням супутному сорбенті дозволяє скоротити кількість стадій ніх білків іонами цих солей. Менша концентрація при очищенні цільового продукту. Умови провецих реагентів не дозволяє повністю осадити супудення афінної хроматографії визначені експеритні білки, більша - недоцільна. ментально. При використанні інших посадкових Очистку екстракту від супутніх білків здійснюбуферів зі значенням рН менше заявляємих не ють також шляхом центрифугування та діафільтвідбувається сорбції інгібітора на сорбенті, при рації до відсутності в ультрафільтраті білків та значеннях рН більше заявляємих відбувається пігментів. Метод діафільтрації дозволяє швидко денатурація ліганду (трипсину), зв'язаного з матвидалити кислоту з екстракту і очистити цільовий рицею сорбенту. продукт від низькомолекулярних домішок (білків, При значеннях рН елююючого буферу вище пігментів) з подальшим концентруванням екстракзаявляємих не відбувається процес десорбції ціту для стадії афінної хроматографії. льового продукту з колонки. Адсорбція інгібітора трипсину на біоспецифічПорівняльний аналіз заявляємого способу і способу-прототипу Спосіб-прототип Заявляємий спосіб Стадія Тривал. год 1. Подрібнення насіння сої до одержання муки. 0,1 2. Знежирення соєвої муки. 12,0 3. Промивка 80% спиртом етиловим 1000г знежиреної муки з насіння сої при перемішуванні при 20 - 25°С протягом 30хв., фільтрація під вакуумом (фільтрат відкидають). 0,5 0,5 4. Екстракція знежиреної муки шляхом суспендування у 5000мл 0,25N сірчаної кислоти при 20 - 25°С протягом 1год. при періодичному перемішуванні. 1,0 5. Відділення екстракту від шроту фільтрацією під вакуумом (осад відкидають). 1,0 6. Видалення (осадження) супутніх білків з екстракту шляхом додавання до кислого фільтрату 20г суміші, що вмішує рівні частини бентоніту і супер-селу, перемішування протягом 10хв. 7. Фільтрація отриманої суспензії (отримання фільтрату), промивки осаду двома порціями по 125мл води (промивні води). Осад відкидають. Стадія 1. Замочування 5 кг насіння сої при температурі (4 - 8)°С для набухання протягом 11год., після чого воду зливають. 2. Заливка насіння сої 50л води очищеної і подрібнення сировини у середовищі екстрагенту до отримання частинок не більше 15 - 20мкм. 3. Відділення екстракту шляхом центрифугування отриманої маси при 1000 2000g протягом 10 - 15хв. або фільтрацією під вакуумом. 5. Видалення (осадження) супутніх білків з екстракту шляхом додавання до екстракту лимонної кислоти або оцтової кислоти, або трихлороцтової кислоти, або сульфату кальцію, або хлориду кальцію. 6. Відділення осаду білків від екстракту центрифугуванням при 1000 - 2000g протягом 10 - 15хв. або фільтрацією під вакуумом. Тривал. год 11,0 0,5 0,5 1,0 0,5 0,1 7. Обробка одержаного екстракту інгібітора трипсину на ультрафільтраційній установці шляхом його діафільтрації та концентрації до 1,0 - 1,5л. 2,0 0,5 8. Очищення інгібітора трипсину за допомогою афінної хроматографії. 3,5 8. Адсорбція інгібітора на бентоніті шляхом повільного додавання при перемішуванні 100г суміші бентоніту і супер-селу до фільтрату, який об'єднали з промивними водами після операції №3. 0,5 9. Фільтрація отриманої суспензії, промивка осаду на фільтрі двома порціями по 125мл води (фільтрат і промивні води відкидають). 0,5 8.1. Сорбція на хроматографічній колонці з трипсин-сефарозою (або з трипсинцелюлозою, або ензайт-трипсином, або трипсин-тойоперлом) з попереднім урівноваженням 0,045 - 0,050Μ трис-НСІ буфером з рН 7,0 - 8,0. 8.2. Відмивання колонки від неспецифічно зв'язаних білків цим же буфером з подальшою промивкою розчином 0,5 0,7М натрію хлориду. Видаляють буфер водою очищеною. 1,5 1,0 15 54993 16 Продовження табл. Стадія 10. н фу піридином і діаліз шляхом розмішування бентонітового осаду у 270мл води, підігрівання отриманої суспензії до 25°С, додавання при перемішуванні 30мл піридину. 11. Фільтрація (декілька годин), промивка осаду на фільтрі 200мл 5% розчину піридину у воді, об’єднання фільтрату з промивною рідиною. 12. Відмивання елюату діалізом проточною водою на протязі 10 – 12год. У целофанових трубках. 13. Видалення інертного матеріалу з діалізованого розчину шляхом доведення до рН 5,3 додаванням соляної кислоти, з подальшим додаванням при перемішуванні 4г суміші бентоніту і супер-селу. 14. Фільтрація під вакуумом, промивка осаду водою (осад відкидають). 15. Перше осадження інгібітора з об’єднаного фільтрату і промивної води (з операції №11) шляхом охолодження до 5°С, доведення 1N соляною кислотою до рН 4,65, фільтрації (фільтрат відкидають). Вага осаду 10 – 12г. 16. Друге осадження інгібітора шляхом суспендування отриманого осаду при перемішуванні у 100мл води, охолодженої до 5°С, додаванням по краплям 1N розчину гідроксиду натрію до повного розчинення осаду (рН не вище 6,4), нагріванні прозорого розчину до 25°С, титрування 1N соляною кислотою до слабого осаду, додаванням 2г супер-селу, фільтрації осаду, промивки на фільтрі водою; шляхом об’єднання фільтрату та промивної води, охолодженням до 5°С, титрування до рН 4,65, фільтрації (фільтрат відкидають). Вихід осаду становить 8 – 10г. 17. Кристалізація шляхом розтирання осаду в 10мл холодної води до отримання суспензії, нагрівання до температури 35° С, додавання по краплям при ретельному перемішуванні 0,5N розчину гідроксиду натрію до повного розчинення осаду і до pH 5,2, кристалізації при 35 – 37°С протягом 5 – 6год., відділення осаду за допомогою центрифугування. Тривал. Год Стадія Тривал. Год 2,0 8.3. Елюювання інгібітора трипсину підкисленою до рН 2,0 – 2,5 водою або підкисленою водою з додаванням 0,15 – 0,20Μ натрію хлориду або 0,2 – 0,25Μ калію хлориду. 2,0 3,0 9. Відмивання елюату електродіалізом або діафільтрацією з подальшим концентруванням. 1,0 10 – 12 10. Стерильна фільтрація (для ін’єкційних або н фузійних розчинів, або для стерильних мазей та інших стерильних місцевих лікарських засобів), розливання у ємності. 0,5 0,5 14. Висушування продукту шляхом ліофілізації. 36,0 1,0 Вихід інгібітора трипсину становить 8,0 – Всього 10,0г з 5кг сировини (0,16 – 0,20)%. 61 год. 12,0 12,0 0,5 6,0 17 54993 18 Продовження табл. Стадія Тривал. Год 18. Перекристалізація отриманого кристалічного осаду при розмішуванні у подвійній кількості холодної води і титруванні 0,5N розчином гідроксиду натрію (рН 6,0) до отримання прозорого розчину, який нагрівають до 35°С і титрують 0,5N розчином соляної кислоти (рН 5,1) до отримання слабого осаду, додавання 2г супер-селу, фільтрація; додавання до фільтрату затравки, витримування при температурі 36 - 37°С на протязі 5 - 6год. (поступово утворюється густа суспензія кристалів); центрифугування; додавання розчину 0,2N соляної кислоти до рН 5,1, вводять затравку і розчин залишають ще на декілька годин при 36 - 37°С для отримання додаткової порції кристалів, яку відділяють так, як описано вище (можна ще отримати кристали з кінцевого відстою, якщо його охолодити до 5°С і додати 0,25 об'єму холодного 95% спирту етилового, як описано у наступному розділі). 12,0 19. Кристалізація з розбавленого спирту розмішуванням отриманих кристалів у п'ятикратному об'ємі холодної води і додаванням по краплям розчину 0,5N гідроксиду натрію до повного розчинення кристалів (рН не вище 6,6). Титрування прозорого розчину 0,2N соляної кислоти (рН 5,2), додавання до утвореного осаду супер-селу з розрахунку 4г на 100мл розчину, відділення осаду, промивання його на фільтрі водою; охолодження фільтрату до 5°С, додавання 1/4 об'єму охолодженого 95% спирту етилового для утворення осаду; додавання розчину 0,2N HCІ до рН 5,0 і витримування суміші при 30°С (аморфний осад перетворюється на протязі 2год. у кристалічний). Відстій розчину кожну годину декантують, доводять розчином соляної кислоти до рН 5,0 і повертають до основної ємності. Так повторюють на протязі декількох годин до повного припинення появи осаду при рН 5,0; після цього суміш, яка кристалізується, витримують ще 30хв. при 30°С). Стадія Тривал. Год 0,5 0,5 1,0 2,0 4,0 0,5 20. Фільтрація осаду, висушування при 24,0 кімнатній температурі протягом 24год. 21. Перекристалізація із спирту етилового сухих кристалів шляхом суспендування у тридцятикратному об'ємі холодної води, 26 витримування 5 - 10хв і кристалізація з розбавленого спирту по операції №19. Вихід інгібітора трипсину становить 1,0г з Всього 1000г сировини (0,1%). 136,2год. Виходячи з даних порівняльної таблиці, завдяки заявляємому способу одержання інгібітора трипсину вихід цільового продукту підвищується у 1,5 - 2,0 рази, кількість стадій технологічного процесу значно зменшується, тривалість у часі скорочується більше, ніж у 2 рази. Крім того, у заявляємому способі відсутні екологічно- та пожежонебезпечні розчинники, які є необхідними для здійснення спо 19 54993 20 собу-прототипу, наприклад, піридин, етиловий 3. Авторское свидетельство №1439782, кл. А 61 К спирт, розчинники, що застосовуються для знежи37/64. Опубл. офиц. бюл. „Открытия, изобретерення соєвої муки. ния", 1988, №43. Результати доклінічних досліджень свідчать, 4. Авторское свидетельство СССР №1412063, кл що специфічна активність цільового продукту, А 61 К 37/64. Опубл. офиц. бюл. „Открытия, изободержаного по заявляємому способу з рослинної ретения", 1988, №27. сировини, співвідносна з активністю препарату 5. Сухинин В.Н., Березин В.А., Новиков Ю.Ф. и др. порівняння "Контрикал" на основі сировини тваОчистка и некоторые свойства ингибитора трипсиринного походження. на из вегетативных органов люцерны. // Ж. БиохиТаким чином, у заявляємому винаході повнісмия, 1981, Т. 46, вып. 7, с. 1183 - 1187. тю виконується задача по створенню високоефек6. Зимачева А.В., Мосолов В.В. Ингибиторы цистивного способу одержання інгібітора трипсину з теиновых протеиназ из семян сои. Ж. Биохимия, рослинної сировини. 1995, Т. 60, вып. 1, с. 118 - 123. ЛІТЕРАТУРА 7. Патент України №23548, кл. А 61 К 35/00. 1. Авторское свидетельство СССР Ν 698623, кл. А Опубл. офіц. бюл. "Промислова власність", 1998, 61 К 37/64. Опубл. 25.11.79. Бюл. "Открытия, изо№4. бретения, промышленные образцы, товарные зна8. Нортрон Д., Кунитц М., Хорриотт P.M. Кристалки", N43. лический ингибитор трипсина из соевых бобов. // 2. Авторское свидетельство СССР №1162081, кл. Кн. Кристаллические ферменты, - М.; Изд. "ИностА 61 К 35/78. Опубл. офиц. бюл, „Открытия, изобранная литература", 1950. - С. 260 - 265 (проторетения", 1985, №22. тип). Комп’ютерна верстка М. Клюкін Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for producing trypsin inhibitor
Автори англійськоюDihtiariov Serhii Ivanovych, Maslova Nataliia Fedorivna, Sukhinin Valentyn Mykolaiovych, Chushenko Valentyna Mykolaivna
Назва патенту російськоюСпособ получения ингибитора трипсина
Автори російськоюДихтярев Сергей Иванович, Маслова Наталья Федоровна, Сухинин Валентин Николаевич, Чушенко Валентина Николаевна
МПК / Мітки
МПК: A61K 38/56, A61K 38/55, A61K 36/00
Мітки: одержання, трипсину, спосіб, інгібітору
Код посилання
<a href="https://ua.patents.su/10-54993-sposib-oderzhannya-ingibitoru-tripsinu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання інгібітору трипсину</a>
Спосіб одержання інгібітору протеази віл з 2(s)-4-піколіл-2-піперазин-трет-бутилкарбоксаміду
Номер патенту: 49826
Опубліковано: 15.10.2002
Автори: Аскін Девід, Єнг Кан К., Рідер Пол, Волант Ральф П.
МПК: C07D 401/06
Мітки: інгібітору, спосіб, 2(s)-4-піколіл-2-піперазин-трет-бутилкарбоксаміду, віл, одержання, протеази
Формула / Реферат:
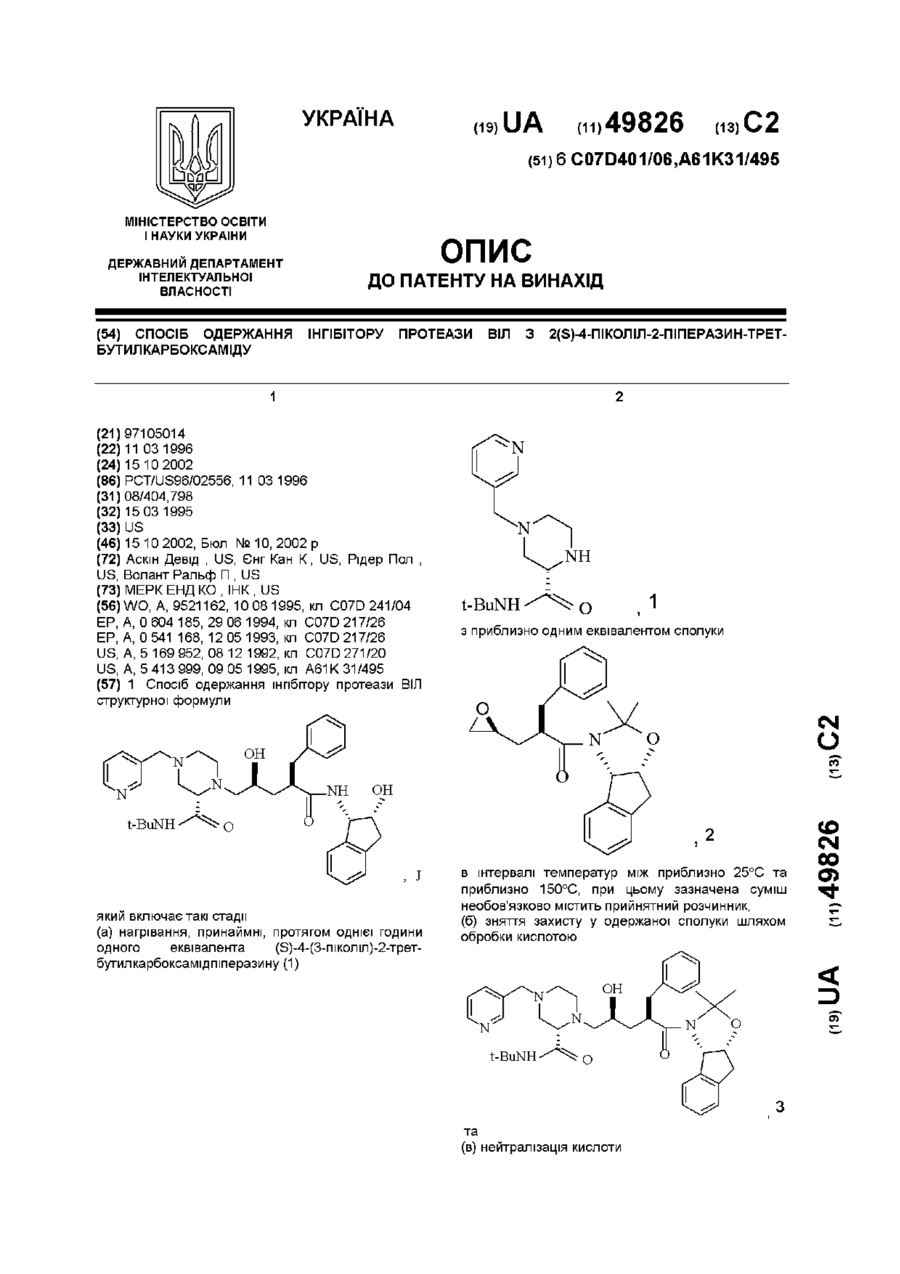
1. Спосіб одержання інгібітору протеази ВІЛ структурної формулиякий включає такі стадії:(а) нагрівання, принаймні, протягом однієї години одного еквівалента (S)-4-(3-піколіл)-2-трет-бутилкарбоксамідпіперазину (1)з приблизно одним еквівалентом сполуки
Спосіб одержання інгібітору трипсіноподібних протеаз

Номер патенту: 23548
Опубліковано: 02.06.1998
Автор: Дівоча Валентина Афанасівна
МПК: A61K 35/00
Мітки: протеаз, трипсіноподібних, інгібітору, одержання, спосіб
Формула / Реферат:
Способ получения ингибитора трипсиноподобных протеаз, включающий афинную хроматографию фракций, содержащих ингибитор протеаз, использование в качестве сорбента трипсин - сефарозы в буферном состоянии, элюирование неспецифически связанных белков и диализ против воды для удаления солей, отличающийся тем, что легкое животного отмывают от крови в фосфатном буфере pH 7,5 при температуре +4°С, затем растирают в стерильной охлажденной посуде...
Спосіб одержання інгібітору нітріфікації азотних добрив

Номер патенту: 3531
Опубліковано: 27.12.1994
Автори: Калашников Сергій Григорович, Орлов Євген Петрович, Чупринко Віталій Георгійович, Пархоменко Володимир Дмитрович, Новак Анатолій Федорович, Стеба Володимир Костянтинович, Водоп'янов Віталій Григорович
МПК: C05G 3/08
Мітки: нітрифікації, інгібітору, азотних, спосіб, одержання, добрив
Формула / Реферат:
Способ получения ингибитора нитрификации азотных удобрений, включающий обработку органического носителя на основе агримуса или гидролизного лигнина водным раствором 3/5/-метилпиразола и сушку продукта, отличающийся тем, что органический носитель предварительно смешивают с фосфорной кислотой или фурфуролом или смесью фосфорной кислоты и фурфурола в количестве 0,5-5 % от массы носителя, при массовом отношении фосфорной кислоты к фурфуролу...
Спосіб склеропластики з використанням штучного біоінертного синтетичного трансплантату з панкреатичним інгібітором протеїназ (трипсину)

Номер патенту: 50968
Опубліковано: 15.11.2002
Автори: Чарковський Олександр Володимирович, Бушуєва Наталія Миколаївна, Абу-Афіфі Шаріф
МПК: A61F 9/00
Мітки: панкреатичним, склеропластики, протеїназ, інгібітором, трансплантату, біоінертного, трипсину, синтетичного, використанням, спосіб, штучного
Формула / Реферат:
Спосіб склеропластики, який полягає у імплантації сітчастого біоінертного експланту із стрічки, яка має форму кульового поясу згідно з кривизною склери у задньому відділі очного яблука, який відрізняється тим, що в експлант додатково вводиться панкреатичний інгібітор протеїназ (трипсину) в кількості 2 мг/г.
Спосіб визначення рівня активності та стабільності інгібіторів трипсину в сиворотці крові і пристрій для його здійснення

Номер патенту: 17562
Опубліковано: 06.05.1997
Автори: Ващук Василь Васильович, Ващук Всеволод Васильович
МПК: C12M 1/00, G01N 33/68
Мітки: трипсину, пристрій, визначення, здійснення, стабільності, крові, інгібіторів, сиворотці, активності, рівня, спосіб
Формула / Реферат:
1. Спосіб визначення рівня активності та стабільності інгібіторів трипсину в сироватці крові, який включає отримання сироватки крові, обробку різної концентрації трипсину сироваткою крові, визначення ступеня протеолітичної активності, який відрізняється тим, що обробляють трипсин різної концентрації сироваткою крові в лунках пристрою, промокнути вмістом лунок диски з фільтрувального паперу накладають на молочно-агарове середовище в чашках...
Попередній патент: Спосіб зміцнення вибухом
Наступний патент: Спосіб очищення внутрішньої поверхні труб нафтових і газових свердловин від гідратних і парафінових відкладень і пробок
Випадковий патент: Дріт для легування рідкої сталі молібденом