Спосіб оптичного поділу піранобензоксадіазольної сполуки






Формула / Реферат
(57) 1. Способ оптического разделения пиранобензоксадиазольного соединения формулы ( ±1)
отличающийся тем, что осуществляют реакцию взаимодействия соединения фор-
мулы (±)1 с оптически активной карбоновой кислотой формулы ll:
с последующей обработкой полученной кристаллической диастереомерной соли или ее сольвата основанием.
2. Способ по п. 1, отличающийся тем, что соединение формулы (±)1 подвергают взаимодействию с соединением формулы (-)ll и полученную кристаллическую диастереомерную соль формулы (+)І, (-)ІІ илиее сольват обрабатывают основанием и получают соединение формулы (+)І.
3. Способ по п. 2, отличающийся тем, что процесс проводят при - 20...-100°С.
4. Способ по п. 2, отличающийся тем, что диастереомерная соль (+)І, (-)ІІ, или ее сольват кристаллизуется при - 20...+50°С.
Текст
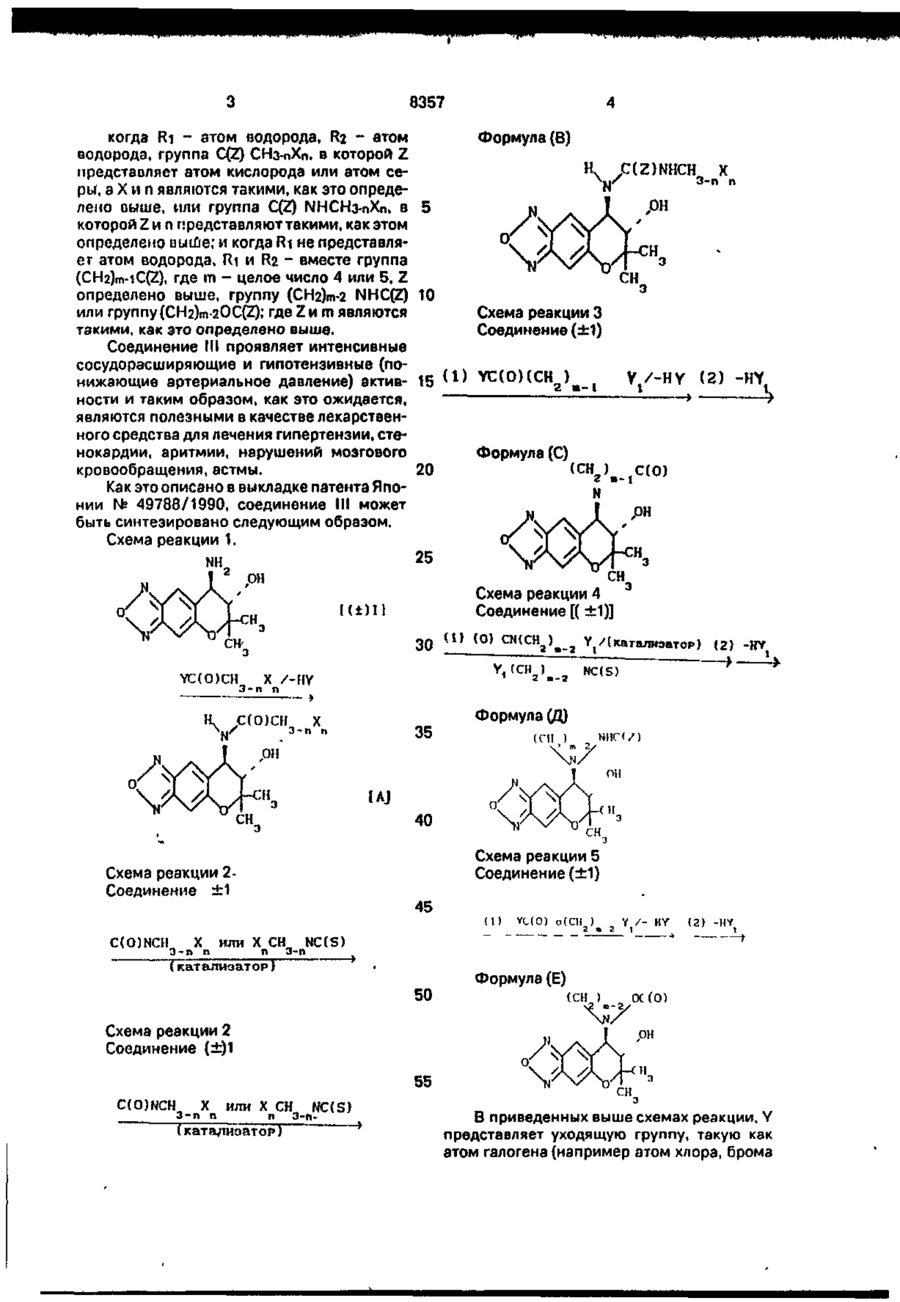
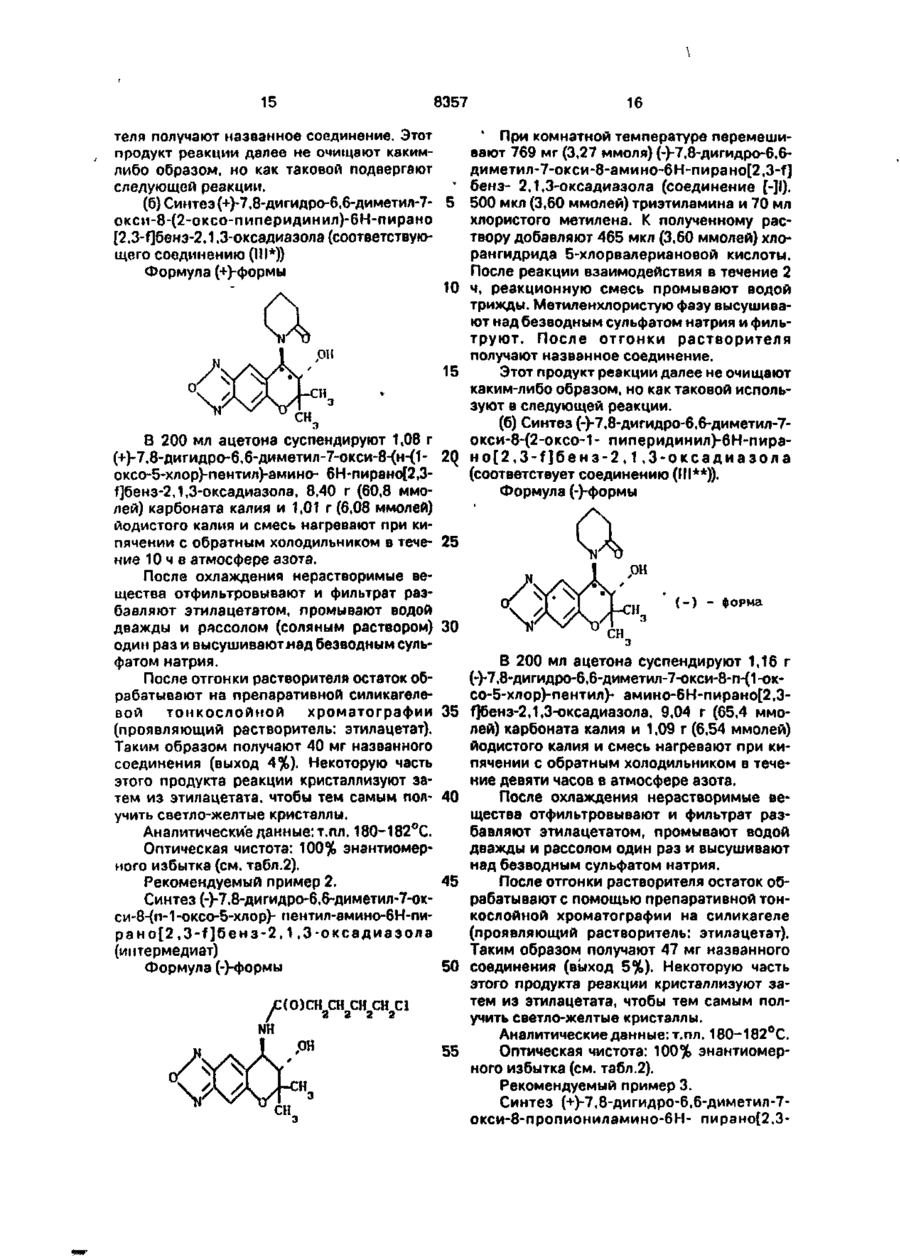
1. Способ оптического разделения пиранобензоксадиазольного соединения формулы ( ±1) мулы (±)1 с оптически активной карбоновой кислотой формулы И: с последующей обработкой полученной кристаллической диастереомерной соли или ее сольвата основанием. 2. Способ по п. 1 . о т л и ч а ю щ и й с я тем, что соединение формулы (±)1 подвергают взаимодействию с соединением формулы (-)!! и полученную кристаллическую диастереомерную соль формулы (+)1, (-)11 или ее сольват обрабатывают основанием и получают соединение формулы (+)!. 3. Способ по п.2, о т л и ч а ю щ и й с я тем, что процесс проводят при -2О...-ИОО°С. о т л и ч а ю щ и й с я тем, что осуществляют реакцию взаимодействия соединения фор Изобретение относится к способам оптческого разделения пиранобензоксадиазольного соединения, которые являются важным промежуточным продуктом при синтезе оптически активного производного пиранобензоксадиазола, полезного при лечении гипертензии (артериальной гипертонии) и астмы. В форме рацемической смеси [1] получается производное пирамобензоксадиазола формулы III 4. Способ по п.2, о т л и ч а ю щ и й с я тем, что диастереомерная соль (+)1, (-)Н, или ее сольват кристаллизуется при -2О...+5О°С. 00 со ел s О где А - гидроксиальная группа или группа ОС(О) СНз-пХп, где X - атом фтора, атом хлора, атом брома, метильная группа или метоксигруппа, а п - 0 или целое число от 1 доЗ; 8357 когда Ri - атом водорода, R2 - атом водорода, группа C(Z) СНз-nXn, в которой Z представляет атом кислорода или атом серь*, а X и п являются такими, как это определено выше, или группа C(Z) ІМНСНз-пХп, в 5 которой Z и п гфедставляют такими, как этом определено виїде; и когда Ri не представляет атом водорода, Ri и R2 - вместе группа (CH2)m-iC(Z), где m - целое число 4 или 5, Z определено выше, группу (СНг)т-2 NHC(Z) 10 или группу (CH2)m-2OC(Z)«' где Z и т являются такими, как это определено выше. Соединение III проявляет интенсивные сосудорасширяющие и гипотензивные (понижающие артериальное давление) актив- 15 ( 1 ) ности и таким образом, как это ожидается, являются полезными в качестве лекарственного средства для лечения гипертензии, стенокардии, аритмии, нарушений мозгового кровообращения, астмы. 20 Как это описано в выкладке патента Японии № 49788/1990, соединение III может быть синтезировано следующим образом. Схема реакции 1. Формула (В) Н C(Z)NHCH X 3-П П ^xtS Схема реакции З Соединение(±1) YC(O)(CH ) У /-HY (2) -HY Формула (С) (СІ У.-іС(0) 25 NH .ОН Схема реакции 4 Соединение [(±1)] 30 YC(O)CH Ii \N/ 3-n (О) CN(CH a ) e 2 Y t /(катализатор) (2) -HY NC(S) X /-HY n C(O)CH X 3-n n 35 Формула (Д) (ГЦ 40 Схема реакции 5 Соединение(±1) Схема реакции 2Соединение ± 1 C(O)NCH X 3-n n 45 (1) YL(O) «{ Y / - HY ( 2 ) -HY или X CH NC(S) n 3-n (катализатор) 50 Схема реакции 2 Соединение (±)1 Формула (Е) (СН ) V7 ОС(О) 55 C(O)NCH X 3-n n или X CH NC(S) n 3-n» В приведенных выше схемах реакции, Y представляет уходящую группу, такую как атом галогена (например атом хлора, брома 8357 или йода), ацетокси-группа или трифторацетокси-группа; Yi представляет атом хлора, атом брома, атом йода, орто- или пара-толуолсульфонатную группу или метансульфонатную 5 группу; и m, n и X являются такими, как это определено выше. Соединение (А), которое является соединением III, в котором Rt представляет атом 10 водорода, может быть получено путем взаимодействия пиранобензоксадиаэольного соединения, которое получается в форме рацемической смеси (вкратце называется как соединение (±1) в дальнейшем), с ацилиру- 15 ющим реагентом УС(О)-СНз-пХп, в котором X,Y и п являются такими, как это определено выше, необязательно в присутствии основания (относится к схеме реакции 1). Соединение (В), которое является соеди- 20 нением (III), в котором Ri представляет атом водорода, может быть получено путем взаимодействия соединения ( ±1) с из оцианатом C(O)NCH3-nXn или изотиоцианатом XnCH3-nNC(S), в котором X, Z и п являются 25 такими, как это определено выше (относится к схеме реакции 2). Соединение (С), которое является соединением (III), в котором Ri и R2 вместе представляют группу (СНг)т-іС(О), может быть 30 получено путем взаимодействия соединения (±1) с ацилирующим реагентом YC(OXCH2)m-iYi, в котором Y, Yi и m являются такими, как это определено выше, необязательно в присутствии основания, а затем 35 циклизацией продукта реакции необязательно в присутствии основания (относится к схеме реакции 3). Соединение (Д), которое является соединением III, где Ri и R2 вместе представля- 40 ют группу (CH2)m-2NHC(Z), где Х и т являются такими, как это определено выше, может быть получено путем взаимодействия соединения (±1) с изоцианатом (O)CN(CH2)m-2Yi или изотиоцианатом 45 (S)CN(CH2)m-2Yi, в которой Yi и m являются такими, как это определено выше, а затем циклизацией продукта реакции необязательно в присутствии основания (относится к схеме реакции 4). 50 Соединение (Е). которое является соединением (III), в котором Ri и R2 вместе представляют Группу (СН2)т-2ОС(О). В КОТОрОЙ Ш является таким, как это определено выше, может быть получено путем взаимодействия 55 соединения (±1) с галогенкарбонатом YC(O)O(CH2)m-2Yi, в котором Y, Yi и m являются такими, как это определено выше, необязательно в присутствии основания, а затем циклизацией продукта реакции необязательно в присутствии основания (относится к схеме реакции 5). В указанных схемах реакции, соединение III, в котором Z является атомом серы, может быть получено сульфирирование соответствующего соединения, в котором Z является атомом кислорода, с помощью реагента Ловиссона. Как описывается в выкладке патента Японии № 49788/1990, соединение (±1) может быть получено следующим образом. Соединение (±1) может быть получено путем обработки известного соединения (F) с гипохлоритом натрия, восстановлением Nоксидной группы соединения (С), полученного таким образом, с помощью восстановителя, такого как триэтилфосфит, а затем взаимодействием полученного таким образом соединения (Н) с аммиаком в инертном растворителе. Однако нигде не сообщено об оптическом разделении соединения (±1). Кроме того, упомянутое рацемическое соединение III, которое обладает асимметрическими атомами углерода в положениях 3 и 4 пиранового цикла, имеет два оптических изомера (соединения (III*) и (Ш**)). Однако упомянутая выкладка патента Японии № 49788/1990 не описывает ни эти оптиче 8357 ски активные производные пиранобензокездиазола, пи какой-либо способ получения этих оптически активных производных. В медицине часто наблюдается, что оптические изомеры отличаются друг от друга 5 по фармакологической активности и безопасности (безвредности). Поэтому является желательным оптически разделять эти изоглеры дня того, чтобы разрабатывать лучшие лекарственные средства. 10 Обнаружено, что оптически активное производное пиранобензоксадиазола [соответствующее соединению (III*)], которое синтезируется через оптически активное пиранобензоксадиазольное соединение, пока- 15 зывающес правое вращение в этаноле [соответствующее соединению (±1), которое будет описано ниже], заметно является превосходным по сравнению с оптически активным производным пиранобензоксадиазола 20 [соответствующим соединению (III*)], которое синтезируется через энантиомер [соответствующий соединению (-)l, которое будет описано ниже], с точки зрения лекарственной активности, определяя таким образом 25 цельность изобретения. Таким образом, изобретение относится к способу оптического разделения соединения (±1), который включает реакцию взаимодействия пиранобензоксадиазольного 30 соединения формулы (±1) К±) \ с оптически активной карбоновой кислотой формулы (И). с последующим разделением полученной таким образом диастереомерной соли или ее сольвата основанием. Соединение И, которое является оптически активным разделяющим реагентом и встречается а виде двух оптических изомеров соединения Q+jll) и ([-]11), может быть синтезировано по методу, описанному в выкладке патента Японии № 83144/1986. Теперь способ оптического разделения соединения (±1) ня соединение ([+]!) и его антипод, а именно, соединение ([-]!), будет описан подробно. 8 Схема реакции 1 Стадия А \ lit) I) ? ((•нч-ип СНСО Н кристаллы 2 К-)] -> [ ( - И - ( - ) Ш маточник 1С) І.(-)1І] -°-смова"ие NH \ В стадии А, соединение ( ±1) подвергается взаимодействию с соединением ([-]l), которое используется в качестве оптически активного разделяющего реагента, и кристаллизуется. Таким образом диастереомерная соль ([+]1 •[-]!!) может быть легко получена в форме кристаллов. Однако должны здесь отметить, что полученные кристаллы могут быть сольвагированы в зависимости от используемого 35 растворителя (см. примеры). Подобным образом диастереомерная соль ([-]l •[+]!!) может быть легко получена при использовании соединения ([+]!!) в качестве оптически активного разделяющего ре40 агента. Таким образом соответствующий оптический изомер соединения (±1) может быть легко получен, выбирая соответствующим образом разделяющий реагент. 45 В качестве растворителя, который используется в стадии А, предпочтительными являются кетоны, такие как ацетон и метилизобутилкетон, хотя настоящее изобретение не ограничивает выбора растворителя. 50 В этом случае диастереомерная соль кристаллизуется в форме сольвата. Температури реакции обычно может Базировать от-20 до 100°С, предпочтительно от 10доЗО°С. 55 Температура кристаллизации обычно может варьировать в пределах от -20 до 50°С> предпочтительно от -10 до 20°С. Выкристаллизованная таким образом диастереомерная соль может быть дополни 8357 тельно перекристаллизована, например из ацетона, тем самым получая кристаллическую диастереомерную соль с высокой чистотой. В стадии Б, кристаллическая диастереомерная соль ([+]| •[-] II) или ее сольват подвергается реакции с* ^основанием, выбираемым из следующих - бикарбоната натрия, бикарбоната калия, карбоната натрия, карбоната калия, гидроокиси натрия и гидроокиси калия. Таким образом целевое соединение ([+]!), показывающее правое вращение в этаноле, может быть легко получено. Подобным образом, соединение ([-]1) может быть легко получено из диастереомерной соли ([-]1 [+]!!) или ее сольвата. Оптическая чистота соединения ([+]!) может быть определена путем взаимодействия указанного соединения с метил-изоцианатом. получая тем самым соединение мочевины формулы ([+JIV) NHCONHCH .ОН 5 10 15 20 25 30 а затем анализируя полученное соединение с использованием оптически активной жидкостной хроматографической колонки (Чиралсел ОС, производимой фирмой Дэйсел Кемикал Индастриз Лимитед). 35 Оптическая чистота соединения ([-]1) может быть определена подобным путем. Как будет видно в примере испытаний, соединение (III*), полученное из соединения d+]l) в соответствии с методом, описанным в 40 патенте Японии № 49788/1990, показывает исключительно высокую активность при понижении кровяного давления, по сравнению с энантиомером (соединение (III**)), полученным из соединения ([-]!). 45 Поэтому очевидно, что применение соединения (III*) при лечении, например, гипертензии является более эффективным, чем применение соединения (III). Анализ с использованием оптически ак- 50 тивной жидкостной хроматографической колонки (Чиралсел ОС, производимый фирмой Дэйсел Кемикал Индастриз Лимитед) доказывает, что не происходит рацемизации во время получения соединения (III*) из соеди- 55 нения ([+]!) или получения соединения (III**) из соединения ([-]1). (1) Пример испытаний (гипотензивная активность). 10 Соединения (III*) и (III**) были каждый по отдельности растворены или суспендированы в 0,5%-ном водном растворе метилцєллюлозы и принудительно внесены пероральным путем трем самцам спонтанно гипертензивных крыс 11-недельного возраста с использованием желудочного зонда. Животные были предварительно обогреты в обогреваемом ящике при 50°С в течение 3-5 мин, а затем перенесены в ограничительную клетку при 37°С, чтобы измерить систолитическое кровяное давление методом хвостовой манжетки (К11-210-1, производимой Натсуме Сейсакушко Ко., Лимитед). В табл.1 приведены проценты понижения кровяного давления спустя 1 ч после введения лекарственного средства. Каждая величина представляет среднее значение от трех животных, подвергнутых испытанию. В табл.2 приведены аналитические данные соединений (III*) и (III**). П р и м е р 1. Стадия А: разделение диастереомерной соли (ацетонового сольвата ([+]! *[-]ll) и диастереомерной соли (ацетонового сольвата ([-]l *[+3ll)В 100 г ацетона растворяют 117,6 г (0,500 ммоля) [±]-7,8-дигиДро-6,6-диметил-7окси-8-амино-6Н-пирано[2,3-т]бенз- 2,1,3оксадиазола (соединение ([±]l)) и 92,9 г (0,510 ммоля) (-)-2-(4-оксифенокси-пропионовой кислоты (соединение (НИ)) и смесь перемешивают в течение 3 ч при охлаждении со льдом. Выпадающие таким образом в осадок кристаллы фильтруют путем отсасывания, промывают с помощью 500 мл охлажденного льдом ацетона и высушивают при пониженном давлении. Таким образом получают 64,6 г диастереомерной соли (ацетоновый сольват ([+]!• [-JII)) в форме светло-желтых кристаллов (выход: 27,2%, оптическая чистота: 95,7% энантиомерного избытка). Эту диастереомерную соль (ацетоновый сольватflf+JI*[-]!!)) нагревают при кипячении с обратным холодильником в 270 г ацетона, а затем отгоняют 77 г ацетона. Остаток кристаллизуют при охлаждении со льдом в течение 2 ч. Таким образом оптическая чистота дизстереомерной соли (ацетонового сольвата Q+]l #[-]И)) повышается до 100% энантиомерного избытка (выход 80%). При измерении т.пл. этого вещества оно начинает медленно разлагаться примерно при 102°С. В результате неводного титрования с помощью хлорной кислоты в уксусной кислоте в качестве растворителя было подтверждено, что одна молекула ацетона находится в сольватной форме. 11 8357 С другой стороны, фильтраты объединяют и ацетон удаляют из фильтратов. Затем к остатку добавляют 1500 мл этилацетата, 1000 мл воды, 32,0 г (0,39 ммоля) бикарбоната натрия и 200 г хлористого натрия и смесь встряхивают. Полуночный раствор оставляют стоять, чтобы тем самым вызвать разделение фаз. Этилацетатную фазу собирают и к ней добавляют 200 мл воды, 6,56 г (0.078 ммоля) бикарбоната натрия и 40 г хлористого натрия. Полученную смесь снова встряхивают и оставляют стоять, чтобы тем самым вызвать разделение фаз. Полученную таким образом этилацетатную фазу высушивают путем добавления безводного сульфата натрия и фильтруют, а затем из нее отгоняют этилацетат. Таким образом получают 94,7 г коричневого твердого вещества. Это коричневое твердое вещество и 73,4 г (0,403 ммоля) [+]-2-(4-оксифенокси)пропионовой кислоты (соединение ([+•]! I) растворяют в 700 г ацетона и перемешивают в течение 3 ч при охлаждении со льдом. Выпадающие в осадок таким образом кристаллы фильтруют путем отсасывания, промывают с помощью 280 мл охлажденного льдом ацетона, а затем высушивают при пониженном давлении. Таким образом получают 75,79 г диастереомерной соли (HI *[+]'О ацетоновой сольват) в фор^е светло-желтых кристаллов (выход 31,9%, оптическая чистота: 100% энантомерногоизбытка). При измерении т.пл. этого продукта реакции он начинает медленно разлагаться примерно при 102°С. В результате неводного титрования с помощью хлорной кислоты в уксусной кислоте подтверждено, что одна молекула цетона сольватирована. Стадия Б: разделение соединений fl+]l) и 12 ммоля) карбоната натрия и 40 г хлористого натрия. Кроме того, эту фазу промывают водным раствором хлористого натрия (40 г/200 мл 5 воды). Затем этилацетатную фазу высушивают добавлением безводного сульфата нат р и я и фильтруют. После о т г о н к и этилацетата получают 32,85 г соединения (1+]1) (выход 96%). 10 Отдельно, диастереомерную соль (ацетоновый сольват ([-]І •[+]!) обрабатывают подобным образом, как это описано выше. Таким образом получают соединение ([-]!), аналитичные данные): т.пл.: 145-146°С (для 15 обоих соединений ([+]!) и (НО). 20 25 30 35 40 (НО Формула Q+]l) 45 Оптическое вращение: соединение ([+]!): [а]д 2 5 + 189° (С - 0,050, этанол соединение Ц-]\: [а]д 2 5 - 189° (С - 0,50, этанол) Оптическая чистота определяется при условиях, указанных в табл.2. Каждое испытуемое соединение подвергают взаимодействию с метилизоцианатом. Мочевиновое соединение, полученное таким образом, анализируют с использованием оптически активной жидкостной хроматографической колонки (Чиралсел ОС, производимой фирмой Дэйсел Кемикал Индастриз Лимитед), Каждое из соединений ([+]!) и (НО показывает оптическую чистоту 100% энантомерного избытка. ЯМР спектр: каждое из обоих соединений ([+]!) и (НО показывает спектр, идентичный со спектром соединения (±1), т.е. рацемической смеси. ЯМР (CDCI3) + DMCO-de) м.д.: 1,26 (ЗН). 1,49 (ЗН), 2,80-3,30 (ЗН), 3,33 (1Н), 3,76 (1Н), 6,82(1Н)и7,98(1Н). (1 р и м е р 2. Стадия А: разделение диастереомерной соли (метил-изобутил кетоновый сольват ([+]1*[-]11) и диастереомерной соли (метил-изобутилкетоновый сольват Є -МИ). В 27,8 гметилизобутилкетона растворяют 4,70 г (20 ммолей) (±)-7,8-дигидро-6,6-диметил-7-оксо-8-амино-6Н-пирано[2,3-т}-бе 50 из- 2,1,3-оксадиазола (соединение [ ±]1)) и К 66,7 г (0,140 ммоля) диастереомерной 3,70 г (20,3 ммолей) (-)-2-(4-оксифенокси)соли (ацетоновый сольват ([+]l •[-]!!) добавляпропионовой кислоты (соединение (НИ) и ют 1000 мл этилацетата, 700 г воды, 17,0 г раствор перемешивают при 21 °С в течение (0,160 ммоля) карбоната натрия и 140 г хло15 мин. ристого натрия, за которым следует встряхи- 55 К полученному таким образом раствору вание, и оставляют стоять, чтобы тем самым добавляют 10 мг затравочного кристалла (мевызывать разделение фаз. тилизобутилкетонового сольвата [+]l *[-]И). ПеЭтилацетатную фазу собирают и промыр е м е ш и в а н и е п р о д о л ж а ю т в течение вают с помощью 200 мл воды, 2,1 (0,020 дополнительных 2 ч, чтобы тем самым вы N 13 8357 14 кристаллизовать продукт реакции.Затем Стадия Б: разделение соединений ([+]!) и прекращают перемешивание и реакцион(НО. ную смесь оставляют стоять в холодильнике К 4,26 г (8,23 ммолей) диастереомерной в течение ночи. соли (метилизобутилкетонового сольвата Выпадающие таким образом кристаллы 5 ([+]! •[-]!!), полученной в стадии А, добавляют собирают отсасыванием, промывают с по53,4 г этилацетата. 42,7 г воды, 0,873 г (8,23 мощью 7,1 г холодного метилизобутилкетоммолей) карбоната натрия и 10,7 г хлористона и высушивают при пониженном го натрия, за которым следует встряхивание давлении. Таким образом получают 4,59 г и оставление стоять, чтобы тем самым выдиастереомерной соли (метилизобутилкето- 10 звать разделение фаз. Зтилацетатную фазу нового сольвата [+]І*[-]ІІ) в форме светлособирают и промывают с 14,2 г воды и 3.6 г желтых кристаллов (выход 44,4%). хлористого натрия. При измерении т.пл. этого продукта реакции он наминает медленно разлагаться примерно при 95°С. В результате неводного титрования с помощью хлорной кислоты в уксусной кислоте подтверждено, что одна молекула метилизобутилкетона сольватирована. С другой стороны, фильтраты объединяют и к нему добавляют 28,2 г 20%-ного водного раствора хлористого натрия и 1,22 г (11,5 ммолей) карбоната натрия. Полученную смесь встряхивают, оставляют стоять, чтобы тем самым вызвать разделение фаз. Метилизобутилкетоновую фазу собирают и встряхивают снова вместе с 9,4 г 20%-ного водного раствора хлористого натрия. Затем ее оставляют стоять, чтобы тем самым вызвать разделение фаз. К полученной таким образом метилизобутилкетоновой фазе добавляют 2,07 г (11,4 ммолей) (+)-2-(4-оксифенокси)-пропионовой кислоты (соединениеtt+]ll))-После растворения соединения путем перемешивания при комнатной температуре к этому раствору добавляют 10 мг затравочного кристалла (метилизобутилкетонового сольвата (HK+jll). чтобы тем самым выкристаллизовать продукт реакции. Затем реакционную смесь оставляют стоять в холодильнике в течение ночи. Выпадающие таким образом кристаллы фильтруют путем отсасывания, промывают с помощью 7,1 г охлажденного метилизобутилкетона и высушивают при пониженном давлении. Таким образом получают 4,25 г диастереомерной соли (метилизобутилкетонового сольвата d+]l Н'О в виде светло-желтых кристаллов (выход 41,1%). Зтилацетатную фазу высушивают до15 бавлением в нее безводного сульфата натрия и фильтруют. После отгонки 48,6 г этилацетата к остатку добавляют 7,3 г гексана, за которым следует кристаллизация при охлаждении со льдом в течение 3 ч. Затем 20 выпадающие таким образом кристаллы собирают. Таким образом получают 1,84 г соединения ([+]!) (выход 95%). Диастереомерную соль (метил изобутилкетоновый сольват ([-]І *[+]И) образуют таким 25 же образом, что и описанный выше. Таким образом получают соединение (МО. ' Соединения d+]l и ([-]!) показывают, каждое из них, оптическую чистоту 100% энан30 тиомерного избытка: Рекомендуемый пример 1. Синтез (+)-7,8-дигидро-6,6-диметил-7окси-8-(н-(1-оксо-5-хлорп)-пентил)-амино6Н-пирано[2,3-т]бенз-2,1,3-оксадиаэола (ин35 термедиат) Формула (+}-формы £(о)сн сн сн сн сі 40 / 2 2 2 2 NH .он 45 715 мг (3,04 ммоля) (+)-7,8-дигидро-6,6диметил-7-окси-8-амино- 6Н-пирано[2,350 т]бенз-2,1,3-оксадиазола (соединение [+]!), 470 мкл триэтиламина и 70 мл хлористого метилена перемешивают при комнатной температуре. К полученному раствору доПри измерении т.пл. этого продукта ребавляют 430 мкл (3,34 ммоля) хлорангидрида акции он начинает медленно разлагаться примерно 95°С. В результате неводного тит- 55 5~хлорвапериановой кислоты. После реакции е течение 2 ч реакционную смесь промырования с помощью хлорной кислоты в уквают водой трижды. Метилхлористую фазу сусной кислоте подтверждено, что одна высушивают над безводным сульфатом намолекула метилизобутилкетона сольватиротрия и фильтруют. После отгонки растворивана. 15 8357 16 теля получают названное соединение. Этот ' При комнатной температуре перемешипродукт реакции далее не очищают какимвают 769 мг (3,27 ммоля) (-)-7,8-дигидро-6,6либо образом, но как таковой подвергают диметил-7-окси-8-амино-6Н-пирано[2,3-т1 следующей реакции. бенз- 2,1,3-оксадиазола (соединение [-]!). (б) Синтез (+}-7,8-дигидро-6,6-диметил-7- 5 500 мкл (3,60 ммолей) триэтиламина и 70 мл окси-8-(2-оксо-пиперидинил)-6Н-пирано хлористого метилена. К полученному рас[2,3-т]бенз-2,1,3-оксадиазола (соответствуютвору добавляют 465 мкл (3,60 ммолей) хлощего соединению (111*)) рангидрида 5-хлорвалериановой кислоты. После реакции взаимодействия в течение 2 Формула (+)-формы 10 ч, реакционную смесь промывают водой трижды. Метиленхлористую фазу высушивают над безводным сульфатом натрия и фильтруют. После отгонки растворителя получают названное соединение. 15 Этот продукт реакции далее не очищают каким-либо образом, но как таковой используют в следующей реакции. (б) Синтез (-)-7,8-дигидро-6,6-диметил-7В 200 мл ацетона суспендируют 1,08 г окси-8-(2-оксо-1- пиперидинил)-6Н-пира(+)-7,8-дигидро-6,6-диметил-7-окси-8-{н-{1- 20 н о [ 2 , 3 - П б е н з - 2 , 1 , 3 - о к с а д и а з о л а оксо-5-хлор)-пентил)-амино- 6Н-пирано[2,3(соответствует соединению (III**)). Г)бенз-2,1,3-оксадиазола, 8,40 г (60,8 ммоФормула (-)-формы лей) карбоната калия и 1,01 г (6,08 ммолей) йодистого калия и смесь нагревают при кипячении с обратным холодильником в тече- 25 ниє 10 ч в атмосфере азота. После охлаждения нерастворимые вещества отфильтровывают и фильтрат раз(-) - форма бавляют этилацетатом, промывают водой дважды и рассолом (соляным раствором) 30 один раз и высушивают лад безводным сульфатом натрия. В 200 мл ацетона суспендируют 1,16 г После отгонки растворителя остаток об(-)-7,8-дигидро-6,6-диметил-7-окси-8-п-{1-окрабатывают на препаративной силикагелесо-5-хлор)-пентил)- амино-6Н-пирано[2,3вой тонкослойной хроматографии 35 т]бенз-2,1,3-оксадиазола, 9,04 г (65,4 ммо(проявляющий растворитель: этилацетат). лей) карбоната калия и 1,09 г (6,54 ммолей) Таким образом получают 40 мг названного йодистого калия и смесь нагревают при кисоединения (выход 4%). Некоторую часть пячении с обратным холодильником в течеэтого продукта реакции кристаллизуют зание девяти часов в атмосфере азота. тем из этилацетата. чтобы тем самым пол- 40 После охлаждения нерастворимые веучить светло-желтые кристаллы. щества отфильтровывают и фильтрат разбавляют этилацетатом, промывают водой Аналитические данные: т.пл. 180-182°С. дважды и рассолом один раз и высушивают Оптическая чистота: 100% знантиомернад безводным сульфатом натрия. ного избытка (см. табл.2). После отгонки растворителя остаток обРекомендуемый пример 2. 45 рабатывают с помощью препаративной тонСинтез (-)-7,8-дигидро~6,6~диметил-7-оккослойной хроматографии на силикагеле си-8-(п-1-оксо-5-хлор)- пентил-амино-6Н-пи(проявляющий растворитель: этилацетат). paHo[2,3-f]6eH3-2,1,3-OKcaflna3Ofla Таким образом получают 47 мг названного (интермедиат) Формула (-)-формы 50 соединения (выход 5%). Некоторую часть этого продукта реакции кристаллизуют затем из этилацетата, чтобы тем самым полуС(О)СН СН СН СН С1 / 2 2 2 2 учить светло-желтые кристаллы. Аналитические данные: т.пл. 180-182°С. Оптическая чистота: 100% энантиомер55 ного избытка (см. табл.2). Рекомендуемый пример 3. Синтез (+)-7,8-дигидро-6,6-диметил-7окси-8-пропиониламино-6Н- пирано[2.3 8357 17 18 f]6eH3-2.1,3-оксадиазола (соответствует соединению (III*)). Формула (+)-формы ют при комнатной температуре в течение 6 ч. После завершения реакции реакционную смесь промывают водой трижды и высуNHC(O)CH CH шивают над безводным сульфатом магния. 2 з После отгонки растворителя остаток переОН' кристаллизовывают из этанола, чтобы тем самым получить 15 мг названного чистого соединения (выход 23%). ( + ) - форма 10 Аналитические данные: т.пл. 179-180°С. Оптическая чистота: 100% э.и. (см. При комнатной температуре перемешитабл.2). вают 1,29 г (5,48 ммолей) (+)-7,8-дигидро-6,2Рекомендуемый пример 5. диметил-7-окси-8-амино-6Н-пирано-[2,3-1] Синтез (+)-7,8-дигидро-6,6-диметил-7бенз-2,1,3-оксадиазола (соединение [+]l), 15 окси-8-метилуреидо-6Н- пирано-[2,3-т*|бенз690 мг (6,8 ммолей) триэтиламина и 40 мл 2,1,3-оксадиазола (соответствующего хлористого метилена, добавляя при этом к соединению (+HV). смеси 610 мг (6,6 ммолей) пропчонилхлориФормула (+)-формы да (хлорангидрид пропионовой кислоты). Смесь перемешивают при комнатной темпе- 20 NHCONHCH ратуре в течение 4 ч. Реакционную смесь экстрагируют с помощью 600 мл этилацетата и 300 мл воды. Органическую фазу собирают и высушивают над безводным сульфатом натрия. Фильтрат и остаток, пол- 25 ученный после отгонки растворителя, кристаллизуют из смеси растворителей, При комнатной температуре перемешисодержащей 10 г этилацетата и 5 г гексана, вают 300 мг (1,29 ммоля) (+)-7,8-дигидро~6,6оставляют стоять в холодильнике в течение диметил-7-окси-амино-6Н-пираноночи, а затем фильтруют путем отсасывания. 30 {2,3-f]6eH3- 2,1,3-оксадиазола (соединение Полученные кристаллы промывают с по(+) и 1) и 15 мл хлористого метилена. К полмощью 3 мл смеси этилацетата/гексана (2:1) ученному раствору добавляют 120 мг (2,10 дважды и высушивают при пониженном давммоля) метилизоцианата. Смесь перемешилении, чтобы тем самым получить названное вают при комнатной температуре (20°С) в соединение в виде бесцветного вещества. 35 течение 5 ч. Аналитические данные: т.пл. 170-180°С. Реакционную смесь кристаллизуют в хоОптическая чистота: 100% э.т. (см. лодильнике и кристаллы, выпадающие в осатабл.2). док таким образом, фильтруют. Таким Рекомендуемый пример 4. образом получают 214 мг названного соедиСинтез (-)-7,8-дигидро*6,6-диметил-7-ок- 40 нения в виде бесцветных кристаллов (выход си-8-пропиониламино-6Н- пирано-[2,358%). f]6eH3-2,1,3-оксадиазола (соответствующего Аналитические данные: т.пл. 165-167°С. соединению (III**) Оптическая чистота: 100% э.и. (см. Формула (-)-формы табл.2). 45 Рекомендуемый пример 6. Синтез (-)-7,8-дигидро-6,6-диметил-7-окNHC(O)CH CH си-метилуреидо-6Н-пирано- [2,3-f]6eH32,1,3-оксадиазола (соответствующего соединению (2) IV). 50 Формула (-)-формы 2 3 NHCONHCH При комнатной температуре перемешивают 52 мг (0,22 ммоля) (-)-7,8-дигидро-6,6диметил-7-окси-8-{н-(1 -оксо-5-хлор)-пентил)- 55 амино~6Н-пирано-[2,3-т]бенз-2.1,3-оксадиазола (соединение) (-)l), 34 мкл (0,24 ммоля) триэтиламина и 5 мл хлористого метилена, При комнатной температуре перемешидобавляя при этом к смеси 21 мкл (0,24 ммовают 300 мг (1,28 ммоля) (-)-7,8-дигидро-6,6ля) пропионил-хлорида. Смесь перемешива 8357 19 диметил-7-окси-8-амино-6Н-пирано-[2,3-т]бенз-2,1,3-оксадиазола (соединение (2) 1) и 20 мл хлористого метилена. К полученному раствору добавляют 120 мг(2,10 ммоля) метилизоцианата. Смесь перемешивают при комнатной температуре (20°С) в течение 5 ч. Рекомендуемую смесь кристаллизуют в холодильнике и выпадающие таким образом кристаллы фильтруют. Таким образом получают 195 мг названного соединения в виде бесцветных кристаллов (выход 52%). Аналитические данные: т.пл. 165-167°С. Оптическая чистота: 100% э.и. (см. табл.2). Рекомендуемый пример 7. Синтез 7,8-дигидро-6,6-диметил-7,8эпокси-6Н-пирано-{2,3-т]- бенз-2,1,3-оксадиазола-3-оксида (соединение (G)). СН При комнатной температуре перемешивают 4,41 г (18,9 ммолей) 6-амино-3,4-дигидро-2,2-диметил-3,4-эпокси-7-нитро-2Н-бензо-[Ь] пирана(соединение (F))i 1.29 г (32 ммолей) гидроокиси натрия, 400 мл этанола и 40 мл воды, добавляя медленно по каплям в эту смесь 32,£ г (26 ммолей) 6%-ного водного раствора гипохлорита натрия. Затем полученную смесь перемешивают в течение 1 ч. После завершения реакции добавляют 1 л водного раствора обычной соли и смесь экстрагируют этилацетатом трижды. Этилацетатные фазы объединяют, промывают рассолом и высушивают над безводным сульфатом натрия. После отгонки растворителя остаток обрабатывают для очистки с помощью колоночной хроматографии на силикагеле (проявляющий растворитель: этилацетат/гексан 1:2 по объему). Таким образом получают 4,00 г названного соединения в виде желтых кристаллов (выход 92%). Аналитические данные: т.пл. 144-145°С. Рекомендуемый пример 8. Синтез 7,8-дигидро-6,6-диметил-7,7эпокси-6Н-пирано[2,3-т>бенз- 2.1,3-оксадиазола (соединение (Н)). 20 При 60°С перемешивают 1,00 г (4,27 ммоля) 7,8-дигидро-6,6-диметил-7,8-эпокси6Н-пирано-2[3-т}-бенз- 2,1,3-оксадиазол-Зоксида (соединение (G)) и 6 мл бензола, добавляя при этом по каплям в пределах 15 мин 0,80 мл (4,70 ммоля) триэтилфосфита. Затем полученную смесь перемешивают в течение 3 ч. После отгонки растворителя остаток подвергают очистке с помощью хроматографии на силикагелевой колонке (проявляющий растворитель: этилацетат/гексан 1:1 по объему). Таким образом получают 0,82 г названного соединения (выход 88%). Некоторую часть этого вещества перекристаллизовывают из гексайа, чтобы тем самым получить желтые кристаллы. Аналитические данные: т.пл. 97-99°С. Рекомендуемый пример 9. Синтез 7,8-дигидро-6,6-диметил-7-окси8-амино-6Н-пирано-{2,3-т> бенз-2,1,3-оксадиазола (соединение [+]!). Формула В 25 мл 16,7%-ного раствора аммиака в этаноле растворяют 0,82 г(3,8 ммоля) 7,8-дигидро-6,8-диметил-7,8-эпокси-6Н-пирано-{2, З-fJ- бенз-2,1,3-оксадиазола (соединение (Н)), а затем смеси позволяют реагировать в толстостенной стеклянной трубке под давлением при 60°С в течение 48 ч. Растворитель реакции отгоняют и остаток подвергают хроматографии на силикагелевой колонке (проявляющий растворитель: смесь этилацетата и метанола 5:1 по объему), чтобы тем самым получить 0,77 г названного соединения в виде коричневого твердого вещества (выход 87%). Некоторую часть этого вещества перекристаллизовывают из этанола, чтобы тем самым получить чистое названное соединение s виде бесцветных кристаллов. Аналитические данные: т.пл. 159-162°С. ЯМР (CDCI3 + DMCO-de) м.д.: 1,26 (ЗН), 1.49 (ЗН). 2,80-3,30 (ЗН). 3.33 (1Н). 3,78 (1Н), 6.82 (1Н) и 7,98 (1Н). Масс-спектр: 133(50%), 163(100%)и235 + * 21 22 8357 Таблица 1 Ri R2 I CH3NHCO NH 37 23 17 Доза, мг/кг Понижение кровяного давления, % 9 о о со со CH3CH2CONH Понижение кровяного давления, % со со о о I о со Доза, мг/кг «о \ / N Соединение (III*) Соединение (III*) (энантиомер из соединения d+] I) (энантиомер из соединения ([-] I) 1 4 I Таблица 2 Ri R2 \ / N і і Соединение (III*) Соединение (III*) (энантиомер из соединения ([+] I) (энантиомер из соединения ([-] 1 ) Т. пл., °С Время удержания при оптически активной жидкостной храматографии, мин Т. пл., °С Время удержания при оптически активной жидкостной храматографии, мин 180-182 15.4 180-182 14,3 CH3CH2CONH 179-180 9,7 170-180 9,0 CH3NHCO I 165-167 13.8 165-167 12,0 П р и м е ч а н и е . Оптически активная жидкостная хроматография: колонка: Чиралсел ОС (Дэйсел Кемикал Индастриз Лимитед) элюенттексан/этанол (4:1 по объему) температура колонки: 40°С скорость потока: 0,5 мл/мин детектор: по УФ-поглощению при 254 нм. Каждый образец, используемый при испытании гипотензивной активности, имеет оптическую чистоту 100% энантиомерного избытка, определенную активной жидкостной хроматографии. 8357 Упорядник Замовлення 4532 Техред М.Моргентал Коректор М.Самборська Тираж Підписне Державне патентне відомство УкраТни, 254655, ГСП, Київ-53, Львівська пл., 8 Відкрите акціонерне товариство "Патент", м. Ужгород, вул.ГагарІна, 101
ДивитисяДодаткова інформація
Автори англійськоюRiozo Sakoda
Автори російськоюРиозо Сакода
МПК / Мітки
МПК: C07D 311/70, C07D 311/62, C07D 498/04, C07D 489/00, C07D 271/12, C07D 311/68, A61K 31/35, A61K 31/42
Мітки: спосіб, оптичного, піранобензоксадіазольної, сполуки, поділу
Код посилання
<a href="https://ua.patents.su/12-8357-sposib-optichnogo-podilu-piranobenzoksadiazolno-spoluki.html" target="_blank" rel="follow" title="База патентів України">Спосіб оптичного поділу піранобензоксадіазольної сполуки</a>
Спосіб отримання 4-ацил-2,3-дигідро-1,4-бензоксазинів чи бензтиазинів

Номер патенту: 4761
Опубліковано: 28.12.1994
Автор: Ханс Мозер
МПК: C07D 265/28, A01N 25/32, C07D 279/00
Мітки: бензтиазинів, отримання, спосіб, 4-ацил-2,3-дигідро-1,4-бензоксазинів
Формула / Реферат:
Способ получения 4-ацил-2,3-дигидро-1,4-бензаксазинов или -бензтиазинов формулыR1- С1-С2-алкил, замещенный одним, двумя или тремя атомами галогена, или С2-алкенил, замещенный тремя атомами галогена, R2 и R3 независимо друг от друга - водород или метил;R4 и R5 независимо друг от друга - водород или хлор, или метил, отличающийся тем, что соединение формулыгде Х, R2, R3, R4 и R5 имеют указанные...
Спосіб одержання діетилового ефіра /е/-4-[2-(3-третбутоксі-3-оксо-1-пропеніл) феніл]-1,4-дігідро-2,6-діметіл-3,5-пірідіндікарбонової кислоти

Номер патенту: 6323
Опубліковано: 29.12.1994
Автори: Алан Девід Бортвік, Даніеле П'єраккіолі, Джованні Гавірагі, Діно Мікелі, Клаудіо Семераро
МПК: C07D 211/84
Мітки: одержання, спосіб, ефіра, кислоти, е/-4-[2-(3-третбутоксі-3-оксо-1-пропеніл, діетилового, феніл]-1,4-дігідро-2,6-діметіл-3,5-пірідіндікарбонової
Формула / Реферат:
Способ получения диэтилового эфира (Е) -4-[2- (3-трет-бутокси-3-оксо-1-пропенил) фенил ]-1, 4-дигидро-2,6-диметил-3,5-пиридиндикарбоновой кислоты формулыотличающийся тем, что трет-бутиловый эфир (Е)-3-(2-формилфенил)-2-пропеновой кислоты формулыподвергают взаимодействию с этиловым эфиром 3-аминокротоновой кислоты формулыпри температуре от -70 до +30 °С в присутствии кислотного катализатора,...
Спосіб одержання сполуки, що терморозширюється, на основі графіту

Номер патенту: 3865
Опубліковано: 27.12.1994
Автори: Хабарова Тетяна Вікторівна, Рудаков Єлісей Сергійович, Савоськін Михайло Віталійович
МПК: C01B 31/04
Мітки: одержання, основі, графіту, терморозширюється, спосіб, сполуки
Формула / Реферат:
1. Способ получения терморасширяющегося соединения на основе графита, включающий последовательную обработку, исходного графита азотной и уксусной кислотами, промывку водой и сушку, отличающийся тем, что исходный графит обрабатывают парами азотной кислоты с концентрацией не менее 85 мас.% или их смесью с газом-носителем при расходе азотной кислоты 0,4-4,0 г на 1 г графита при температуре 20-50°С в течение 1-4 часов.2. Способ по п. 1,...
Спосіб одержання комплексної сполуки платини /іі/з н-днк, що має протипухлинну активність

Номер патенту: 3801
Опубліковано: 27.12.1994
Автори: Волченскова Іліма Іліодорівна, Бударін Лев Іванович, Шалімов Сергій Олександрович, Кейсевич Людвіг Владіславович, Майданевич Надія Миколаївна, Трохименко Олена Петрівна
МПК: C07F 15/00, A61K 31/295, A61P 35/00
Мітки: н-днк, одержання, має, активність, протипухлинну, іі/з, сполуки, платини, комплексної, спосіб
Формула / Реферат:
Способ получения комплексного соединения платины (II) с н-ДНК, обладающего противоопухолевой активностью, взаимодействием цис-дихлордиаминплатины с дезоксирибонуклеиновой кислотой, выделенной из селезенки крупного рогатого скота, марки А, при нагревании до 78,0±0,5°С в водной среде, отличающийся тем, что процесс проводят в присутствии хлористого натрия и цитрата натрия при мольном соотношении цис-дихлордиаминплатины, фосфора, н-ДНК,...
Спосіб біологічної очистки стічних вод, що містять неорганічні сполуки азоту

Номер патенту: 4679
Опубліковано: 28.12.1994
Автори: Жанталай Ольга Борисівна, Шукайло Борис Миколайович, Глікін Марат Аронович, Жанталай Володимир Борисович, Жанталай Валентина Олександрівна, Войтова Ольга Дмитрівна
МПК: C02F 3/02
Мітки: азоту, стічних, містять, спосіб, вод, очистки, біологічно, сполуки, неорганічні
Формула / Реферат:
Способ биологической очистки сточных вод, содержащих неорганические соединения азота, включающий обработку в аэробных условиях нитрифицирующими микроорганизмами, отличающийся тем, что в сточные воды перед обработкой нитрифицирующими микроорганизмами вводят источник сульфид-иона - соль сероводородной кислоты.
Попередній патент: Спосіб активації цітотоксичних клітин
Наступний патент: Безалкогольний напій “таврійський”
Випадковий патент: Пристрій для розбризкування рідких мінеральних добрив та отрутохімікатів