Бета-лактам або його енантіомер для синтезу таксолів та спосіб одержання таксолів






Текст
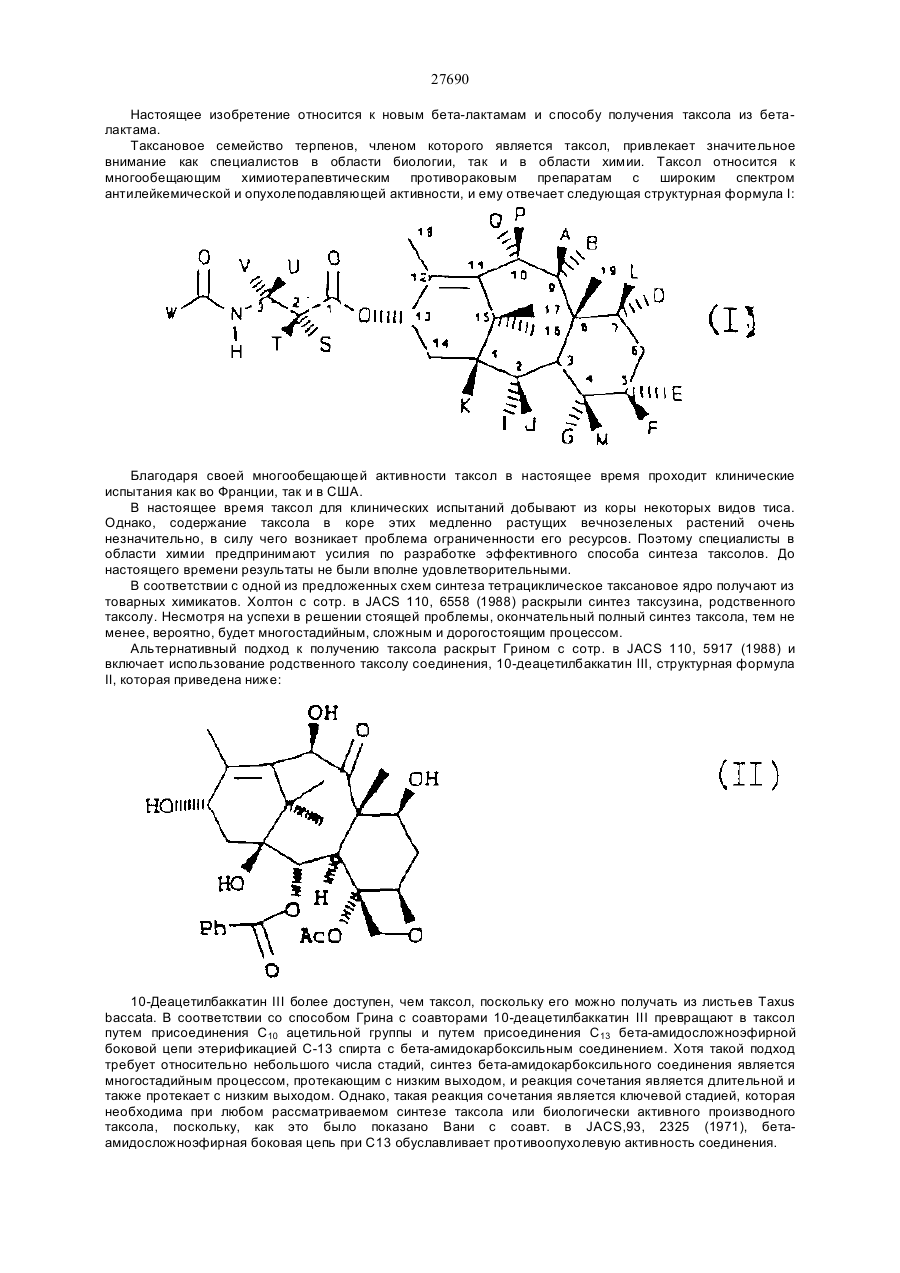
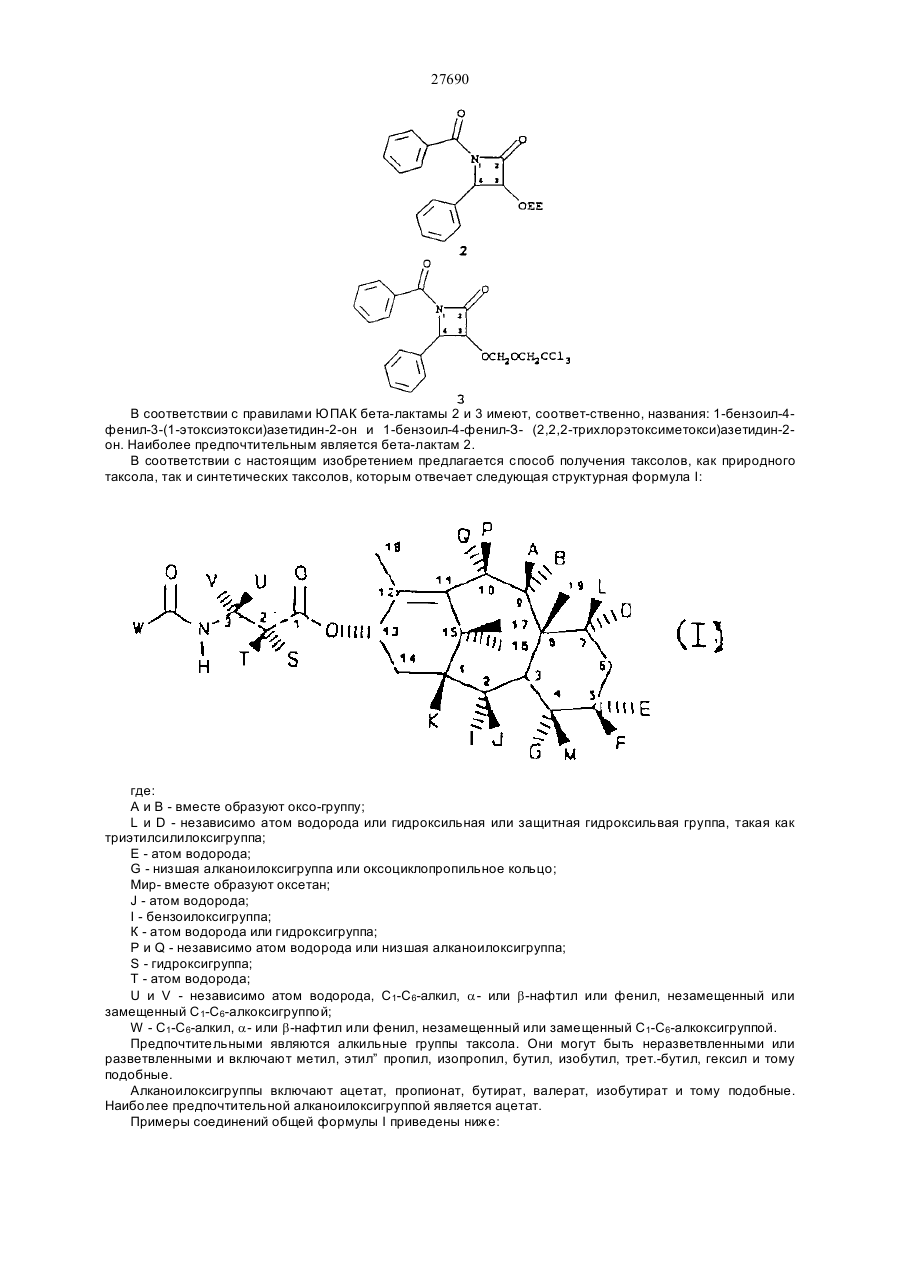
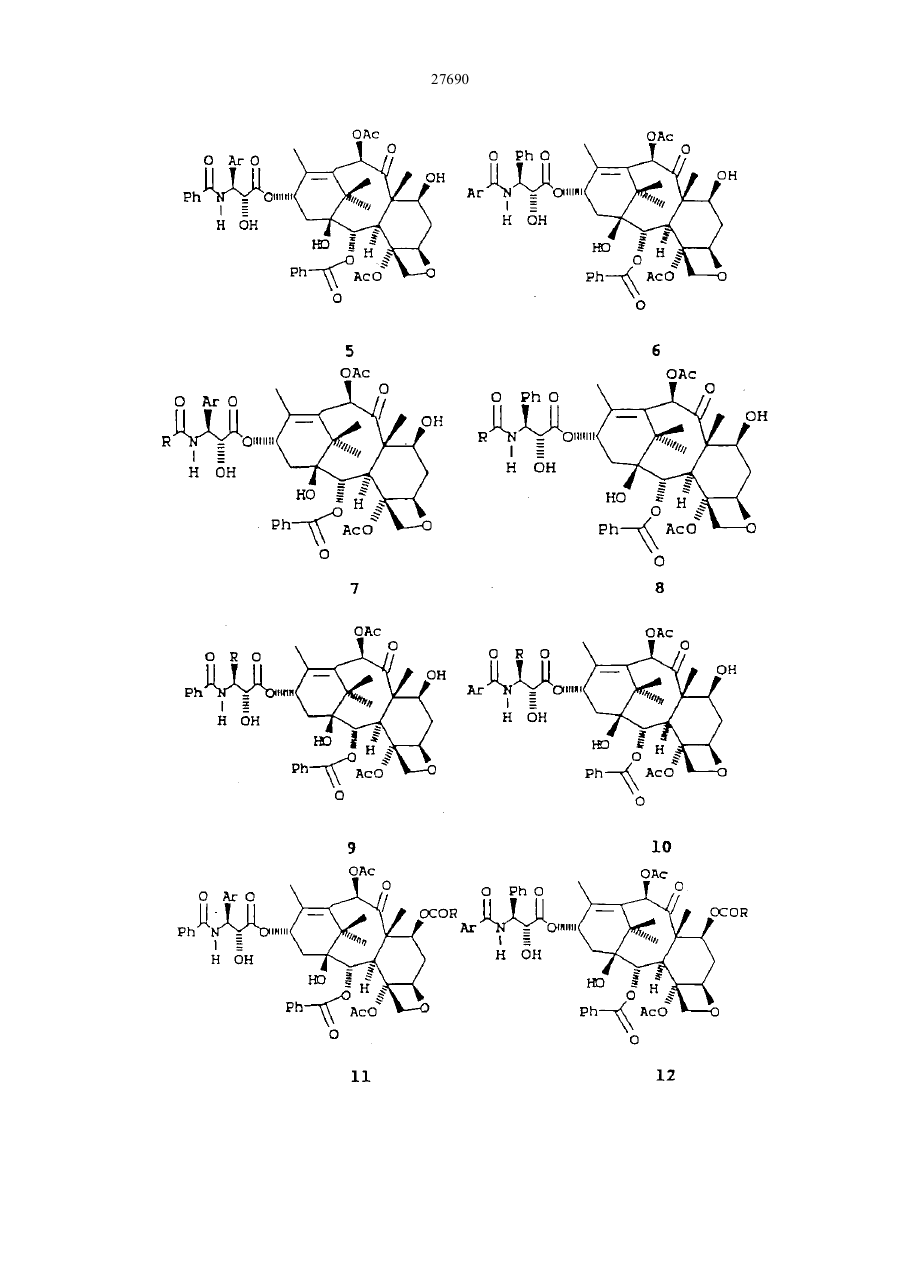
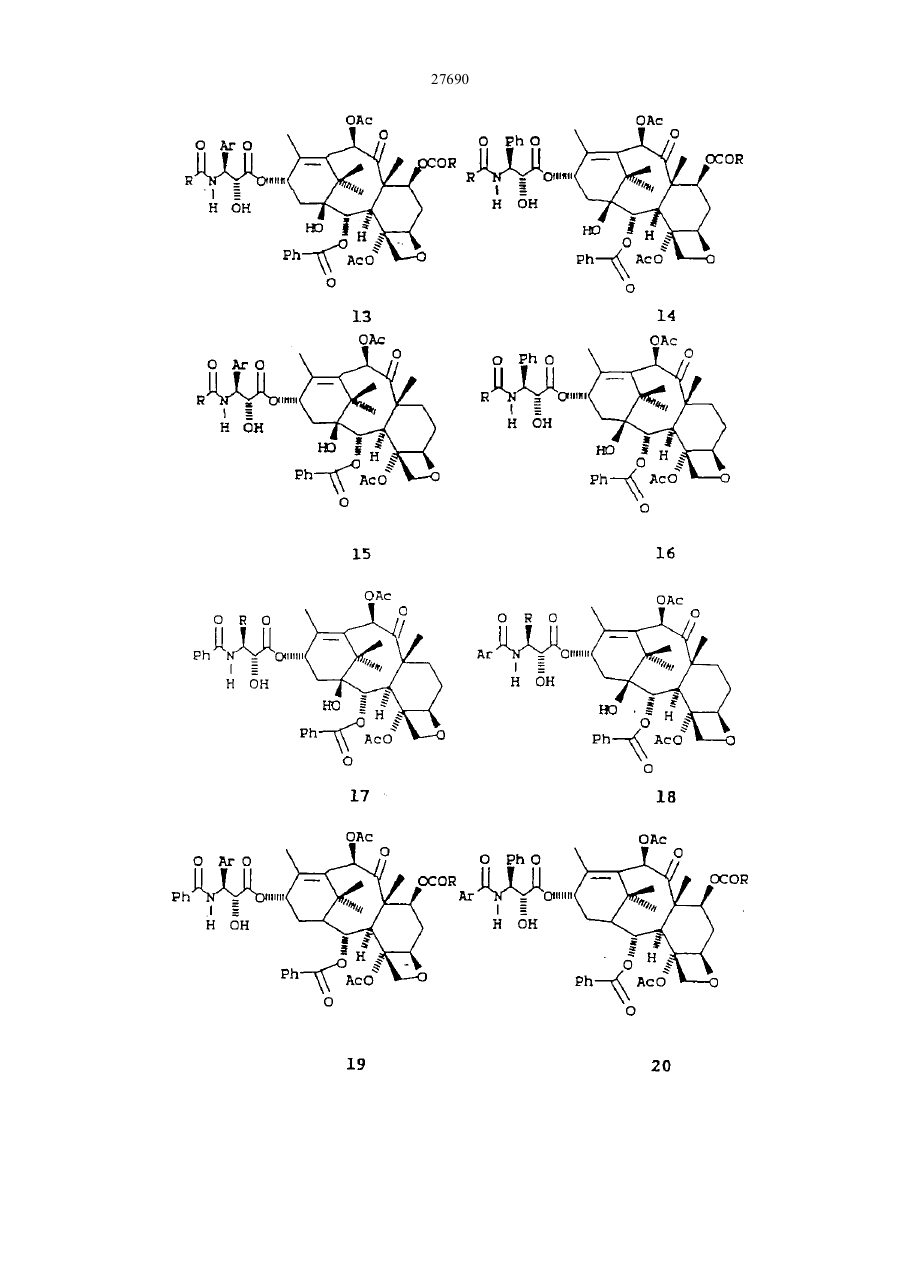
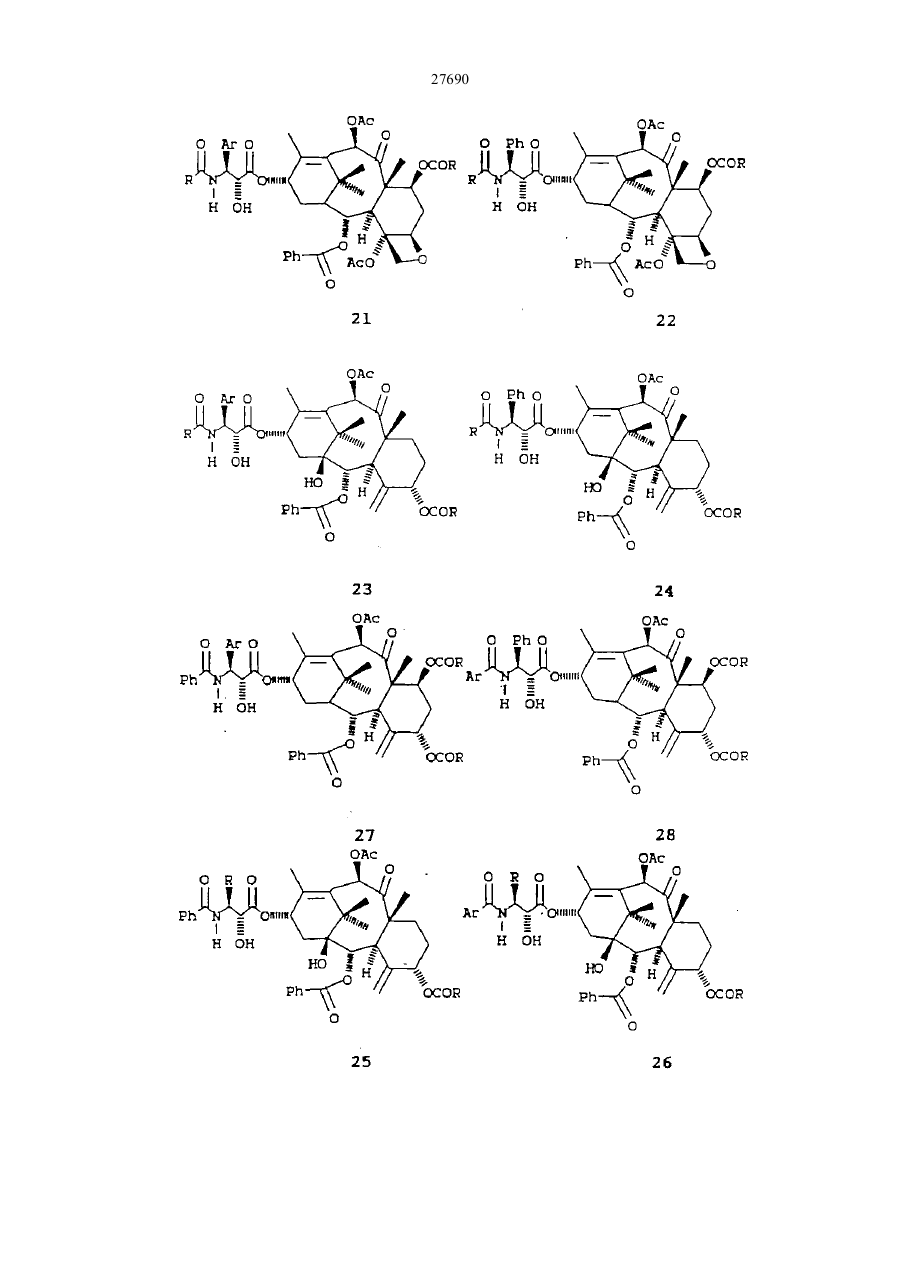
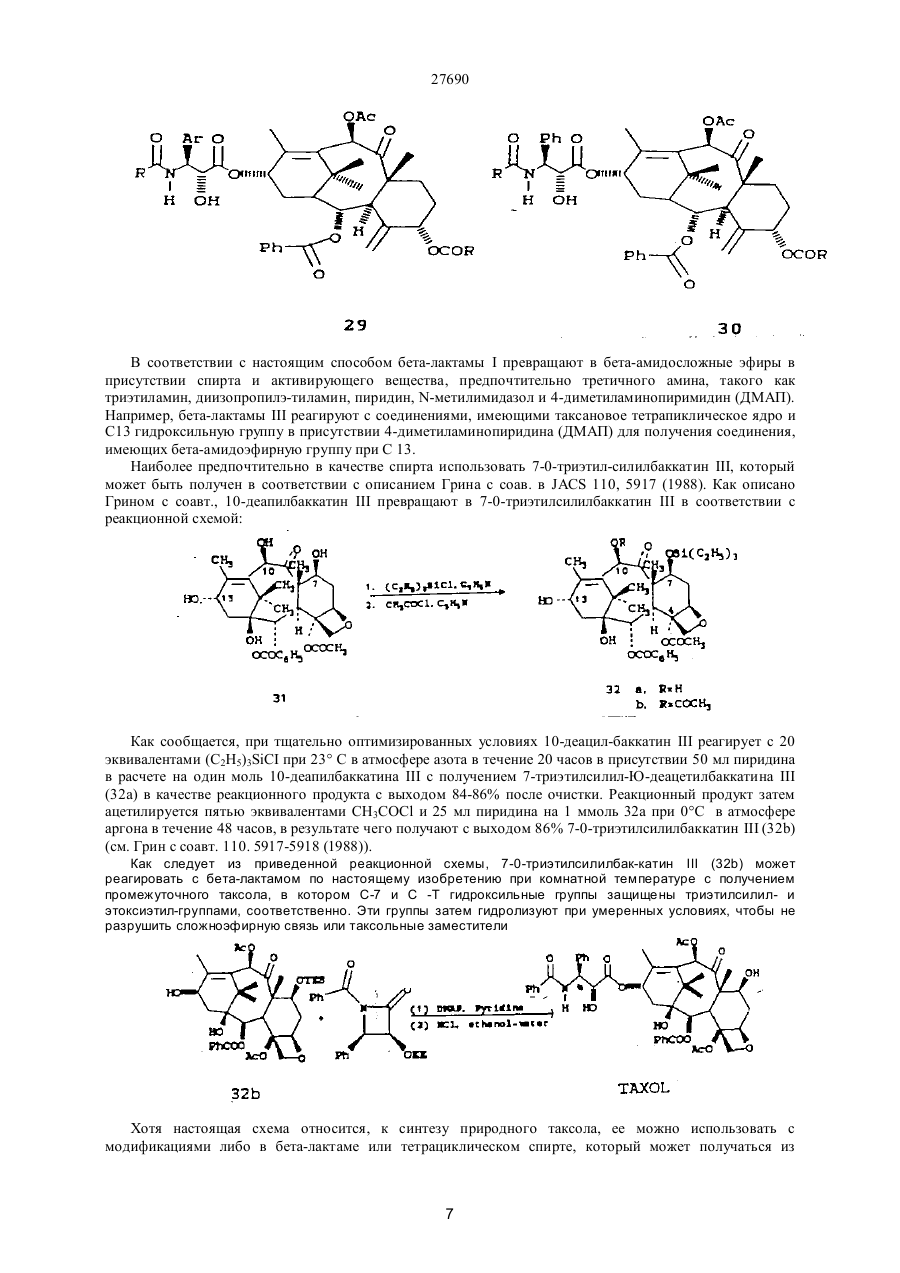
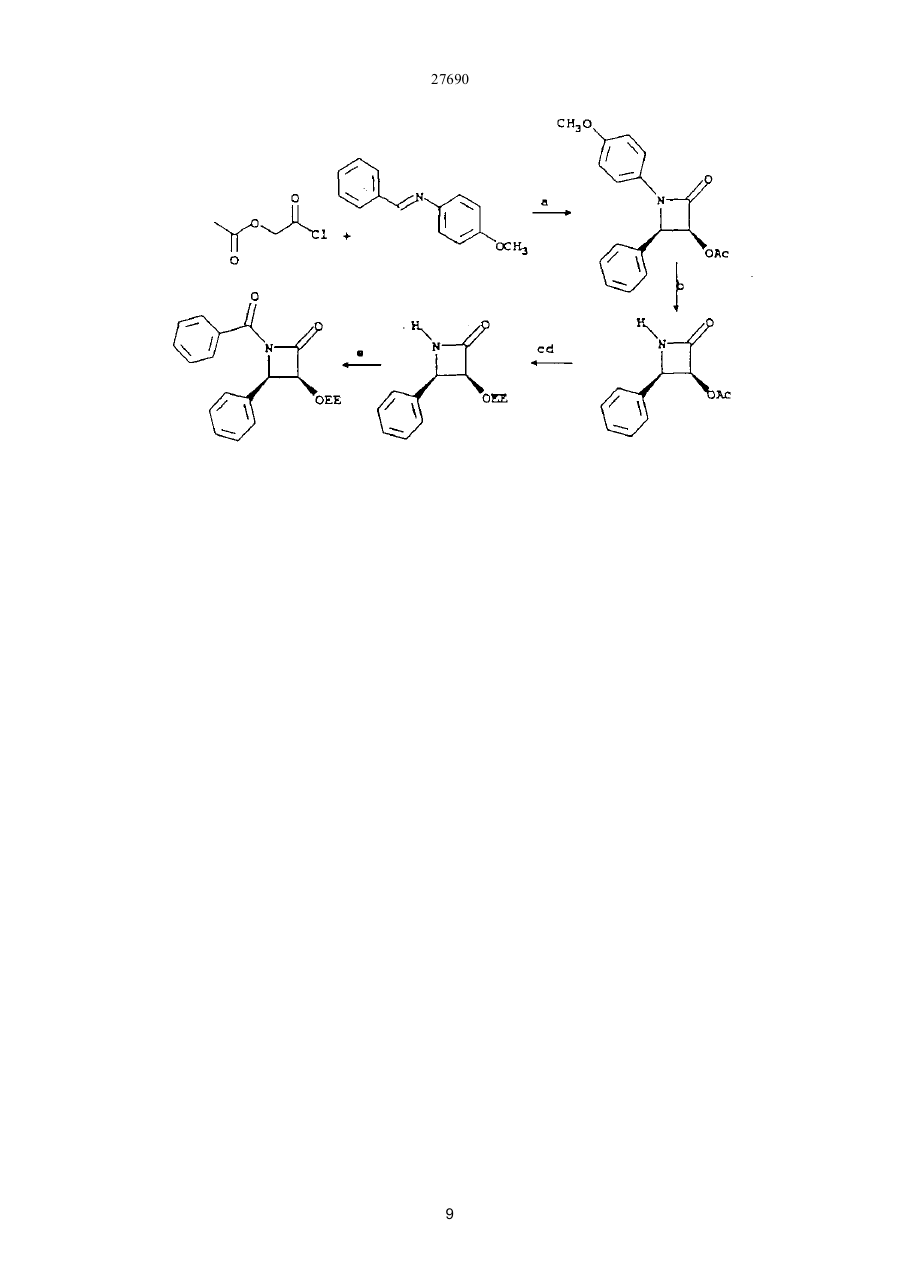
27690 Настоящее изобретение относится к новым бета-лактамам и способу получения таксола из беталактама. Таксановое семейство терпенов, членом которого является таксол, привлекает значительное внимание как специалистов в области биологии, так и в области химии. Таксол относится к многообещающим химиотерапевтическим противораковым препаратам с широким спектром антилейкемической и опухолеподавляющей активности, и ему отвечает следующая структурная формула I: Благодаря своей многообещающей активности таксол в настоящее время проходит клинические испытания как во Франции, так и в США. В настоящее время таксол для клинических испытаний добывают из коры некоторых видов тиса. Однако, содержание таксола в коре этих медленно растущих вечнозеленых растений очень незначительно, в силу чего возникает проблема ограниченности его ресурсов. Поэтому специалисты в области химии предпринимают усилия по разработке эффективного способа синтеза таксолов. До настоящего времени результаты не были вполне удовлетворительными. В соответствии с одной из предложенных схем синтеза тетрациклическое таксановое ядро получают из товарных химикатов. Холтон с сотр. в JACS 110, 6558 (1988) раскрыли синтез таксузина, родственного таксолу. Несмотря на успехи в решении стоящей проблемы, окончательный полный синтез таксола, тем не менее, вероятно, будет многостадийным, сложным и дорогостоящим процессом. Альтернативный подход к получению таксола раскрыт Грином с сотр. в JACS 110, 5917 (1988) и включает использование родственного таксолу соединения, 10-деацетилбаккатин III, структурная формула II, которая приведена ниже: 10-Деацетилбаккатин III более доступен, чем таксол, поскольку его можно получать из листьев Taxus baccata. В соответствии со способом Грина с соавторами 10-деацетилбаккатин III превращают в таксол путем присоединения С10 ацетильной группы и путем присоединения С13 бета-амидосложноэфирной боковой цепи этерификацией С-13 спирта с бета-амидокарбоксильным соединением. Хотя такой подход требует относительно небольшого числа стадий, синтез бета-амидокарбоксильного соединения является многостадийным процессом, протекающим с низким выходом, и реакция сочетания является длительной и также протекает с низким выходом. Однако, такая реакция сочетания является ключевой стадией, которая необходима при любом рассматриваемом синтезе таксола или биологически активного производного таксола, поскольку, как это было показано Вани с соавт. в JACS,93, 2325 (1971), бетаамидосложноэфирная боковая цепь при С13 обуславливает противоопухолевую активность соединения. 27690 Существенным затруднением при синтезе таксола и других активных противоопухолевых препаратов является недостаток легко доступных соединений, которые могут легко присоединяться к С13 кислороду, для создания бета-амидосложноэфирной боковой цепи. Разработка таких соединений и способа их присоединения с высоким выходом облегчает синтез таксола, а также родственных противоопухолевых препаратов, содержащих модифицированный ряд циклических заместителей или модифицированную С13 боковую цепь. Такая потребность была удовлетворена открытием нового, легко доступного соединения, являющегося предшественником боковой цепи, и эффективного способа их присоединения к С13 кислороду. Таким образом, объектами настоящего изобретения являются предшественник боковой цепи для синтеза таксолов и способ присоединения предшественника боковой цепи с относительно высоким выходом. Настоящее изобретение относится к предшественнику боковой цепи -новому бета-лактаму формулы III: или его энантиомеру, где R1 – С1-С6-алкил, a- или b-нафтил или фенил, незамещенный или замещенный С1-С6алкоксигруппой; R2 - гидроксизащитная группа, такая как этоксиэтил; а R3- С1-С6-алкил, a- или b-нафтил или фенил, незамещенныи или замещенный С 1-С6- алкоксигруппой, используемого для синтеза таксолов, как природного, так и таксолов, не имеющихся в природе. Настоящее изобретение относится также к способу получения таксолов, включающий осуществление контакта спирта с бета-лактамом в присутствия достаточного количества активирующего вещества при эффективных для такого процесса условиях с получением бета-амидосложноэфирного промежуточного соединения таксола, из которого в дальнейшем получают таксолы. Для получения таксолов используют бета-лактам или его производные структурной формулы III или его энантиомер. Как указывалось выше, R1 – С1-С6-алкил, a- или b-нафтил или фенил, незамещенный или замещенный С1-С6-алкоксигруппой; R2 -гидроксизащитная группа, и R3 – С1-С6-алкил, a- или b-нафтил или фенил, незамещенный или замещенный С 1-С6-алкоксигруппой. Предпочтительно R1 -фенил, незамещенный или замещенный С1-С6-алкоксигруппой, R2 - этоксиэтил, 2,2,2-трихлорэтоксиметил или другая ацеталь гидроксилзащищающая группа; и R3 - фенил, незамещенный или замещенный С1-С6-алкоксигруппой. Структурные формулы двух предпочтительных бета-лактамов, в которых R1 и R3 - фенил, приведены ниже: Как указывалось выше, R1 – С1-С6-алкил, a- или b-нафтил или фенил, незамещенный или замещенный С1-С6-алкоксигруппой; R2-гидроксизащитная группа, и R3 - С1-С6-алкил, а- или р-нафтил или фенил, незамещенный или замещенный С 1-С6-алкоксигруппой. Предпочтительно R1-фенил, незамещенный или замещенный С1-С6-алкоксигруппой, R2- этоксиэтил, 2,2,2-трихлорэтоксиметил или другая ацеталь гидроксилзащищающая группа; и R3 - фенил, незамещенный или замещенный С1-С6-алкоксигруппой. Структурные формулы двух предпочтительных бета-лактамов, в которых R1 и R3 - фенил, приведены ниже: 27690 В соответствии с правилами ЮПАК бета-лактамы 2 и 3 имеют, соответ-ственно, названия: 1-бензоил-4фенил-3-(1-этоксиэтокси)азетидин-2-он и 1-бензоил-4-фенил-З- (2,2,2-трихлорэтоксиметокси)азетидин-2он. Наиболее предпочтительным является бета-лактам 2. В соответствии с настоящим изобретением предлагается способ получения таксолов, как природного таксола, так и синтетических таксолов, которым отвечает следующая структурная формула I: где: А и В - вместе образуют оксо-группу; L и D - независимо атом водорода или гидроксильная или защитная гидроксильвая группа, такая как триэтилсилилоксигруппа; Е - атом водорода; G - низшая алканоилоксигруппа или оксоциклопропильное кольцо; Мир- вместе образуют оксетан; J - атом водорода; I - бензоилоксигруппа; К - атом водорода или гидроксигруппа; Р и Q - независимо атом водорода или низшая алканоилоксигруппа; S - гидроксигруппа; Т - атом водорода; U и V - независимо атом водорода, С1-С6-алкил, a- или b-нафтил или фенил, незамещенный или замещенный С1-С6-алкоксигруппой; W - С1-С6-алкил, a- или b-нафтил или фенил, незамещенный или замещенный С1-С6-алкоксигруппой. Предпочтительными являются алкильные группы таксола. Они могут быть неразветвленными или разветвленными и включают метил, этил” пропил, изопропил, бутил, изобутил, трет.-бутил, гексил и тому подобные. Алканоилоксигруппы включают ацетат, пропионат, бутират, валерат, изобутират и тому подобные. Наиболее предпочтительной алканоилоксигруппой является ацетат. Примеры соединений общей формулы I приведены ниже: 27690 27690 27690 27690 В соответствии с настоящим способом бета-лактамы I превращают в бета-амидосложные эфиры в присутствии спирта и активирующего вещества, предпочтительно третичного амина, такого как триэтиламин, диизопропилэ-тиламин, пиридин, N-метилимидазол и 4-диметиламинопиримидин (ДМАП). Например, бета-лактамы III реагируют с соединениями, имеющими таксановое тетрапиклическое ядро и С13 гидроксильную группу в присутствии 4-диметиламинопиридина (ДМАП) для получения соединения, имеющих бета-амидоэфирную группу при С 13. Наиболее предпочтительно в качестве спирта использовать 7-0-триэтил-силилбаккатин III, который может быть получен в соответствии с описанием Грина с соав. в JACS 110, 5917 (1988). Как описано Грином с соавт., 10-деапилбаккатин III превращают в 7-0-триэтилсилилбаккатин III в соответствии с реакционной схемой: Как сообщается, при тщательно оптимизированных условиях 10-деацил-баккатин III реагирует с 20 эквивалентами (C2H5)3SiCI при 23° С в атмосфере азота в течение 20 часов в присутствии 50 мл пиридина в расчете на один моль 10-деапилбаккатина III с получением 7-триэтилсилил-Ю-деацетилбаккатина III (32а) в качестве реакционного продукта с выходом 84-86% после очистки. Реакционный продукт затем ацетилируется пятью эквивалентами СН3СОСl и 25 мл пиридина на 1 ммоль 32а при 0°С в атмосфере аргона в течение 48 часов, в результате чего получают с выходом 86% 7-0-триэтилсилилбаккатин III (32b) (см. Грин с соавт. 110. 5917-5918 (1988)). Как следует из приведенной реакционной схемы, 7-0-триэтилсилилбак-катин III (32b) может реагировать с бета-лактамом по настоящему изобретению при комнатной температуре с получением промежуточного таксола, в котором С-7 и С -Т гидроксильные группы защищены триэтилсилил- и этоксиэтил-группами, соответственно. Эти группы затем гидролизуют при умеренных условиях, чтобы не разрушить сложноэфирную связь или таксольные заместители Хотя настоящая схема относится, к синтезу природного таксола, ее можно использовать с модификациями либо в бета-лактаме или тетрациклическом спирте, который может получаться из 7 27690 природных или искусственных источил -ков, для приготовления других синтетических таксолов, относящихся к настоящему изобретению. Альтернативно, бета-лактам I может превращаться в бета-амидосложный эфир в присутствии активирующего вещества и спирта, иного нежели 7-0-трийэтилсилилбаккатин III, с получением промежуточного таксола. Синтез таксола может затем осуществляться с использованием промежуточного соединения таксола в соответствии с реакционной схемой. Алкильные группы бета-лактама могут быть неразветвленными или разветвленными и включают метил, этил, пропил, изопропил, бутил, изобутил, трет.-бутил, гексил и тому подобные. Примерами алканоилоксигрупп бета-лактама служат ацетат, пропионат, бутират, валерат, изобутират и тому подобные. Наиболее предпочтительным -алканоилокси является ацетат. Бета-лактамовые арильные группы либо одни, либо с различными заместителями, содержат от б до 15 атомов углерода и включают фенил, незамещенный или замещенный C1-C6-алкоксигруппой, a- или bнафтил. Как отмечалось выше, R2 бета-лактама (III) может быть этоксиэтилом (ОЕЕ), 2,2,2трихлорэтоксиметокси или другой гидроксилзапщщающей группой, такой как ацетали или простые эфирные группы, то есть метоксиметил, бензилоксиметил; сложные эфирные, такие как ацетаты; карбонаты, такие как метилкарбонаты и тому подобные. Перечень защищающих групп для гидроксильной группы и их синтез описан в книге "Защитные группы в органическом синтезе" Т.В.Грина, Джон Вилли и Сане, 1981. Гидроксилза-щищающая группа должна легко отщепляться при условиях, достаточно умеренных, чтобы не разрушать сложноэфирную связь или другие заместители промежуточного таксола. Однако, R2 предпочтительно выбирают из числа таких групп, как этоксиэтил или 2,2,2-трихлорэтоксиметил, и наиболее предпочтительно этоксиэтил. Предпочтительные значения заместителей бета-лактама R1 R2 и R3 перечислены ниже: R1 = Ph, R1 = p-MeOPh, R1 = С1-С6 - алкил R2 = ЕЕ, R2 = Si R3, R3 = С1-С6 -алкил, R2 = OCOR R3 = Ph, R3 = p-MeOPh, R3 == С1-С6 -алкил. Ниже приведены общие структурные формулы иллюстрируемых соединений: Поскольку бета-лактам I имеет несколько асимметричных атомов углерода, специалистам в данной области очевидно, что соединения с асимметрическими атомами углерода могут существовать в виде диастереоизомеров, рацематов или в оптически активных формах. Все указанные формы находятся в объеме патентных притязаний по настоящему изобретению. Более конкретно, настоящее изобретение включает энантиомеры, диастереоизомеры, рацемические смеси и другие их смеси. Бета-лактамы I могут быть приготовлены из легко доступных материалов, как это показано для беталактама 2 в схеме ниже: 8 27690 9 27690 Реагенты: (а) триэтиламин, дихлорметан, 25° С, 18 часов; (б) 4-эквивалента нитрата церийаммония, ацетонитрил, -10° С, 10 минут; (в) едкое кали, тетрагидрофуран, вода, 0°С, 30 минут; (г) этилвиниловый простой эфир, тетрагидрофуран, толуолсульфокислота (катализатор), 0°С, 1,5 часа; (д) метиллитий, эфир, -78° С, 10 минут, бензоилхлорид, -78-С, 1 час. Исходные вещества легко доступны. Альфа -ацилоксиацетилхлорид получают из гликолевой кислоты и в присутствии третичного амина ее подвер-гают циклоконденсации с иминами, приготовленными из альдегидов и параметоксианилина, получая 1-параметоксифенил-3-ацилокси-4-арилазетидин-2-оны. Пара-метоксифенильную группу легко можно удалить окислением нитратом церийаммония, и ацилокси-группу можно гидролизовать при - стандартных условиях, аналогичных тем, которые используются в технике для приготовления 3-окси-4-арилазетидин-2-онов. 3-Гидроксильная группа может защищаться разными защитными группами, такими как 1-этоксиэтил. Предпочтительно рацемический З-окси-4-арилазетидин-2-он расщепляют на чистые энантиомеры, перед защитой, используя для этого способ перекристаллизации соответствующих 2-метокси-2(трифторметил)фенилуксусных сложных эфиров, и при приготовлении таксола используется лишь правовращаюпщй энантиомер. В любом случае 3-(1-этоксиэтокси)-4-фенил-азетидин-2-он можно превратить в бета-пактам 2 путем обработки основанием, предпочтительно н-бутиллитием, и ароилхлоридом при температуре -78° С или ниже. Приведенные ниже примеры иллюстрируют настоящее изобретение. Пример 1 Синтез цис-1-бензоил-3-(1-этоксиэтокси)-4-фенилазетидин-2-она Цис -1 - пара - метоксифенил - 3 ацетокси - 4 - фенилаз етидин - 2 - он К раствору 962 мг (4,56 ммоля) имина, полученного из бензальдегида и пара-метоксианилина, и 0,85 мл (6,07 ммоля) триэтиламина в 15 мл хлористого метилена при -20° С по каплям добавляют раствор 413 мг (3.04 ммоля) альфа-ацетоксиацетилхлорида в 15 мл хлористого метилена.Реакционную смесь самопроизвольно нагревают до 25° С в течение 18 часов. Затем смесь разбавляют 100 мл хлористого метилена и раствор экстрагируют 30 мл 10% водной соляной кислоты. Органический слой промывают 30 мл воды и 30 мл насыщенного водного бикарбоната натрия, сушат над сульфатом натрия и концентрируют, получая твердую массу. Последнюю растирают с 50 мл гексана и смесь фильтруют. Оставшийся твердый продукт подвергают пере-кристаллизации из смеси этилацетата и гексана, получая 645 мг (68% выход) цис1-пара-метоксифенил-3-ацетокси-4-фенилазетидин-2-она в виде белых кристаллов с точкой плавления 163° С. Цис-3-ацетокси-4-фенилазетидин-2-он К раствору 20,2 г цис-1-пара-метоксифенил-3-ацетокси-4-фенилазе-тидин-2-она в 700 мл ацетонитрила при -10° С медленно добавляют раствор нитрата церийаммония в 450 мл воды в течение одного часа. Смесь перемешивают в течение 30 минут при -10° С и разбавляют 500 мл серного эфира. Водный слой дважды экстрагируют 100 мл порциями серного эфира и объеди-ненный органический слой промывают дважды 100 мл порциями воды, двумя порциями по 100 мл насыщенного водного бикарбоната натрия и концентрируют, получая 18,5 г твердого вещества. Перекристаллизацией его в смеси ацетона -гексана получают 12,3 г (92%) цис-3-ацетокси-4-фенилазетидин-2-она в виде белых кристаллов с точкой плавления 152-154° С. Цис-3-окси-4-фенилазетидин-2-он К смеси 200 мл тетрагидрофурана и 280 мл 1М водного раствора едкого кали при 0°С добавляют раствор 4,59 г (22,4 ммоля) цис-З-ацетокси-4-фенилазетидин-2-она в 265 мл тетрагидрофурана через капельную воронку в течение 40 минут. Раствор перемешивают при 0°С в течение часа и добавляют 100 мл воды и 100 мл насыщенного бикарбоната натрия. Смесь экстрагируют четырежды порциями по 200 мл этилацетата, и объединенные органические слои сушат над сульфатом натрия и концентрируют, получая 3,54 г (97%) рацемического цис-3-окси-4-фенилазетидин-2-она в виде белых кристаллов, точка плавления 147-149° С. Этот продукт расщепляют на его энантиомеры путем перекристаллизации его 2-метокси-2(трифторметил)фенилуксусного сложного эфира в смеси ацетона-гексана с последующим гидролизом. [a]25 Hg 177°С. Цис-3-(1-этоксиэтокси)-4-Фенилазетидин-2-он К раствору 3,41 г (20,9 ммоля) цис-3-окси-4-фенилазетидин-2-она в 15 мл тетрагидрофурана при 0° С добавляют 5 мл этилвинилового простого эфира и 20 мг (0,2 ммоля) метансульфокислоты. Смесь перемешивают при 0°С 20 минут, разбавляют 20 мл насыщенного водного раствора бикарбоната натрия и экстрагируют трижды 40-мл порциями этилацетата. Объединенные этилацетат-ные слои сушат над сульфатом натрия и концентрируют, получая 4,87 г (99%) цис-3-(1-этоксиэтокси)-4-фенилазетидин-2-она в виде бесцветного масла. Цис-1 -бензоил-3 - (1 -этоксиэтокси) -4 -фенилазетидин-2-он К раствору 2,35 г (10 ммоля) цис-3-0 -этоксиэтокси) -4-фенилазетидин-2-она в 40 мл тетрагидрофурана при -78° С добавляют 6,1 мл (10,07 ммоля) 1,65М раствора н-бутиллития в гексане. Смесь перемешивают 10 минут при 78° С и добавляют раствор 1,42 г (10,1 ммоля) бензоилхлорида в 10 мл тетрагидрофурана. Смесь перемешивают при -78-С 1 час и разбавляют 70 мл насыщенного водного бикарбоната натрия и экстрагируют трижды 50-мл порциями этилацетата. Объединенные этилацетатные экстракты сушат над сульфатом натрия и концентрируют, получая 3,45 г масла. Хроматографией масла на силикагеле, с использованием в качестве элюирующего растворителя смеси этилацетата-гексан, получают 3,22 г (95%) цис-1-бензоил-3-(1 10 27690 этоксиэтокси)-4-фенилазетидин-2-она (2) в виде бесцветного масла. Пример 2 Синтез цис-бета-амидо сложных эфиров из цис-1-бензоил-3-(1-этоксиэтокси)-4-фенилазетидин-2-она 2 Бензил-3-бензамидо-З-фенил-оксипропионат К раствору 88 мг (0,26 ммоля) цис-1-бензоил-3-(1-этоксиэтокси)-4-фенилазетидин-2-она в 0,3 мл тетрагидрофурана добавляют 28 мг (0,26 ммоля) бензилового спирта и 32 мг (0,26 ммоля) 4диметиламинопиридина (ДМАП). Через 5 часов при 25° С смесь разбавляют 10 мл насыщенного водного раствора бикарбоната натрия и трижды экстрагируют 20-мл порциями этилацетата. Объединенные этилацетатные слои экстрагируют 10 мл 5% водной соляной кислоты и 10 мл насыщенного бикарбоната натрия, сушат над сульфатом натрия и концентрируют, получая 112 мг (100%) бензилового сложного эфира в виде масла, которое по данным ЯМР-анализа имеет чистоту более 97%. К раствору этого масла в 4 мл тетрагидрофурана добавляют 1 мл 10% водного раствора соляной кислоты. Смесь перемешивают при 25° С 30 минут, разбавляют 20 мл насыщенного раствора бикарбоната натрия в воде и четырежды экстрагируют 30-мл порциями этилацетата. Объединенные этилацетатные экстракты сушат над сульфатом натрия и концентрируют, получая твердый продукт. Перекристаллизацией твердого продукта из хлоро-форма получают 92 мг (95%) белого кристаллического бензил-З-бензамидо-3-фенил-2-оксипропионата с точкой плавления 129-131° С. Таксол. В маленький реакционный сосуд добавляют 109 мг (0,320 ммоля) (+)-цис-1-бензоил-3-(1-этоксиэтокси)-4-фенилазетидин-2-она, 45 мг (0,064 ммоля) 7-0триэтилсилилбаккатина III, 7,8 мг (0,064 ммоля) 4-диметиламинопиридина (ДМАП) и 0,032 мл пиридина. Смесь перемешивают при 25° С 12 часов и разбавляют 100 мл этилацетата. Этилацетатный раствор экстрагируют 20 мл 10%-ного водного раствора сульфата меди, сушат над сульфатом натрия и концентрируют. Остаток фильтруют через силикагельную насадку, используя для элюирования этилацетат. Хроматографией на силикагеле с использованием в качестве элюирующего растворителя смесь этилацетатгексан с последующей перекристаллизацией из смеси гексана и этилацетата получают 61 мг (92%) Т-(1этоксиэтокси)-7-0-триэтилсилилтаксол в виде смеси 2:1 диастереоизомеров. 5 мг пробы указанного таксола растворяют в 2 мл этанола и добавляют 0,5 мл 0,5%-ного водного раствора соляной кислоты. Смесь перемешивают при 0°С в течение 30 часов и разбавляют 50 мл этилацетата. Раствор экстра-гируют 20 мл насыщенного водного раствора бикарбоната натрия, сушат над сульфатом натрия и концентрируют. Остаток очищают хроматографически на силикагеле, используя для элюирования смесь этилацетат-гексан с получением 4,5 мг (примерно 90%) таксола, который во всех отношениях идентичен аутентичной пробе. N-дебензоил-N- (1 -нафтоил)таксол. В маленький реакционный сосуд добавляют 125мг (0,320 ммоль) (+)цис-1-(1-нафтоил)-3-(1-этоксиэтокси)-4-фенилацетидин-2-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (ДМАП) и 0,032 мл пиридина. Смесь перемешивают при 25° С в течение 12 часов и разбавляют 100 мл этилацетата. Этилацетатный раствор экстрагируют 20 мл 10%ного раствора сульфата меди, сушат над сульфатом натрия и концентрируют. Остаток отфильтровывают через силикагельную насадку, используя для элюирования этилацетат. Флешхроматографией на силикагеле с использованием в качестве элюирующего растворителя смесь этилацетата-гексан с последующей перекристаллизацией из смеси гексана и этилацетата получают 62 мг (90%) N-дебензоил-N-(1-нафтоил)-2'-(1этоксиэтокси)-7-0-триэтилсилил таксола. 5 мг пробы N-дебензоил-N-(1-нафтоил)-2'-(1-этоксиэтокси)-7-0-триэтилсилил таксола растворяют в 2 мл этанола и добавляют 0,5 мл 0,5%-ного водного раствора соляной кислоты. Смесь перемешивают при 0° С в течение 30 часов и разбавляют 50 мл этилацетата. Раствор экстрагируют 20 мл насыщенного водного раствора бикарбоната натрия, сушат над сульфатом натрия и концентрируют. Остаток очищают хроматографически на силикагеле, используя для элюирования смесь этилацетата-гексан с получением 3,9 мг (примерно 95%) N-дебензоил-N-(1-нафтоил)таксола. N-дебензоил-N-(2-нафтоил)таксол. В маленький реакционный сосуд добавляют 125 мг (0,320 ммоль) (+)цис-1-(2-нафтоил)-3-(1-этоксиэтокси)-4-фенилацетидин-2-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (ДМАП) и 0,032 мл пиридина. Смесь перемешивают при 25° С в течение 12 часов и разбавляют 100 мл этилацетата. Этилацетатный раствор экстрагируют 20 мл 10%ного раствора сульфата меди, сушат над сульфатом натрия и концентрируют. Остаток отфильтровывают через силикагельную насадку, используя для элюирования этилацетат. Флешхроматографией на силикагеле с использованием в качестве элюирующего растворителя смесь этилацетата-гексан с последующей перекристаллизацией из смеси гексана и этилацетата получают 64 мг (93%) N-дебензоил-N(2-нафтоил)-2'-(1этоксиэтокси)-7-0-триэтилсилил таксола. 5 мг пробы N-дебензоил-N-(2-нафтойл)-2'-(1-этоксиэтокси)-7-0-триэтилсилил таксола растворяют в 2 мл этанола и добавляют 0,5 мл 0,5%-ного водного раствора соляной кислоты. Смесь перемешивают при 0° С в течение 30 часов и разбавляют 50 мл этилацетата. Раствор экстрагируют 20 мл насыщенного водного раствора бикарбоната натрия, сушат над сульфатом натрия и концентрируют. Остаток очищают хроматографически на силикагеле, используя для элюирования смесь этилацетата-гексана с получением 3,5 мг (примерно 90%) N-дебензоил-N-(2-нафтоил)таксола. N-дебензоил-N-пивалоил таксол. В маленький реакционный сосуд добавляют 102 мг (0,320 ммоль) (+)-цис1-пивалоил-3-(1-этоксиэтокси)-4-фенилацетидин-2-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (ДМАП) и 0,032 мл пиридина. Смесь перемешивают при 25° С в течение 12 часов и разбавляют 100 мл этилацетата. Этилацетатный раствор экстрагируют 20 мл 10%-ного раствора сульфата меди, сушат над сульфатом натрия и концентрируют. Остаток отфильтровывают через силикагельную насадку, используя для элюирования этилацетат. Флешхроматографией на силикагеле с использованием в качестве элюирующего растворителя смесь этилацетата-гексана с последующей перекристаллизацией из смеси гексана и этилацетата получают 55мг(85%) N-дебензоил-N-пивалоил-2'-(1 11 27690 этоксиэтокси)-7-0-триэтилсилил таксола. 5 мг пробы N-дебензоил-N-пивалоил-2'-(1-этоксиэтокси)-7-0-триэтил-силил таксола растворяют в 2 мл этанола и добавляют 0,5 мл 0,5%-ного водного раствора соляной кислоты. Смесь перемешивают при 0° С в течение 30 часов и разбавляют 50 мл этилацетата. Раствор экстрагируют 20 мл насыщенного водного раствора бикарбоната натрия, сушат над сульфатом натрия и концентрируют. Остаток очищают хроматографически на силикагеле, используя для элюирования смесь этилацетата-гексана с получением 3,6 мг (примерно 92%) N-дебензоил-N-пивалоилтаксола. N-дебензоил-N-пентаноил таксол. В маленький реакционный сосуд добавляют 102 мг (0,320 ммоль) (+)-цис1-пентаноил-3-(1-этоксиэтокси)-4-фенилацетидин-2-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (ДМАП) и 0,032 мл пиридина. Смесь перемешивают при 25° С в течение 12 часов и разбавляют 100 мл этилацетата. Этилацетатный раствор экстрагируют 20 мл 10%-ного раствора сульфата меди, сушат над сульфатом натрия и концентрируют. Остаток отфильтровывают через силикагельную насадку, используя для элюирования этилацетат. Флешхроматографией на силикагеле с использованием в качестве элюирующего растворителя смесь этилацетата-гексана с последующей перекристаллизацией из смеси гексана и этилацетата получают 58 мг (89%) N-дебензоил-N-пентаноил-2'-(1этоксиэтокси)-7-0-триэтилсилил таксола. 5 мг пробы N-дебензоил-N-пентаноил-2'--(1-этоксиэтокси)-7-0-триэтил-силил таксола растворяют в 2 мл этанола и добавляют 0,5 мл 0,5%-ного водного раствора соляной кислоты. Смесь перемешивают при 0°С в течение 30 часов и разбавляют 50 мл этилацетата. Раствор экстрагируют 20 мл насыщенного водного раствора бикарбоната натрия, сушат над сульфатом натрия и концентрируют. Остаток очищают хроматографически на силикагеле, используя для элюирования смесь этилацетата-гексана с получением 3,6 мг (примерно 92%) М-дебензоил-М-пентаноил таксола. 3'-десфенил-3'-(1-нафтил) таксол. В маленький реакционный сосуд добавляют 125 мг (0,320 ммоль) (+)циc-l-бeнзoил-3-(l-этoкcиэтoкcи)-4т-(l-нaфтил)-ацетидин-2-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (ДМАП) и 0,032 мл пиридина. Смесь перемешивают при 25° С в течение 12 часов и разбавляют 100 мл этилацетата. Этилацетатный раствор экстрагируют 20 мл 10%ного раствора сульфата меди, сушат над сульфатом натрия и концентрируют. Остаток отфильтровывают через силикагельную насадку, используя для элюирования этилацетат. Флешхроматог-рафией на силикагеле с использованием в качестве элюирующего растворителя смесь этилацетата-гексана с последующей перекристаллизацией из смеси гексана и этилацетата получают 62 мг (90%) 2'-(1-этоксиэтокси)-3'-десфенил3'-(1-нафтил)-7 - О-триэтилсилил .таксола. 5 мг пробы 2`-(l-этoкcиэтoкcи)-3'-дecфeнил-3`-(l-нaфтил)-7-0-тpиэ-тилсилил таксола растворяют в 2 мл этанола и добавляют 0,5 мл 0,5%-него водного раствора соляной кислоты. Смесь перемешивают при 0°С в течение 30 часов и разбавляют 50 мл этилацетата. Раствор экстрагируют 20 мл насыщенного водного раствора бикарбоната натрия, сушат над сульфатом натрия и концентрируют. Остаток очищают хроматографически на силикагеле, используя для элюирования смесь этилацетата-гексана с получением 3,9 мг (примерно 95%) 3'-десфенил-3'-(1-нафтил) таксола. 3'-десфенил-3'-(2-нафтил) таксол. В маленький реакционный сосуд добавляют 125 мг (0,320 ммоль) (+)цис-1-бензоил-3-(1-эгоксиэтокси)-4'-(2-нафтил)-ацетидин-2-она, 45 мг (0,064 ммоль) 7-0триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (ДМАП) и 0,032 мл пиридина. Смесь перемешивают при 25° С в течение 12 часов и разбавляют 100 мл этилацетата. Этилацетатный раствор экстрагируют 20 мл 10%-ного раствора сульфата меди, сушат над сульфатом натрия и концентрируют. Остаток отфильтровывают через силикагельную насадку, используя для элюирования этилацетат. Флешхроматог-рафией на силикагеле с использованием в качестве элюирующего растворителя смесь этилацетата-гексана с последующей перекристаллизацией из смеси гексана и этилацетата получают 62 мг (90%) 2`-(l-этoкcиэтoкcи)-3`-дecфeнил-3'-(2-нафтил) -7-0-триэтилсилил-таксола. 5 мг пробы 2'-(1-этоксиэтокси)-3'-десфенил-3'-(2-нафтил)-7-0-триэ-тилсилил-таксола растворяют в 2 мл этанола и добавляют 0,5 мл 0,5%-ного водного раствора соляной кислоты. Смесь перемешивают при 0° С в течение 30 часов и разбавляют 50 мл этилацетата. Раствор экстрагируют 20 мл насыщенного водного раствора бикарбоната натрия, сушат над сульфатом натрия и концентрируют. Остаток очищают хроматографически на силикагеле, используя для элюирования смесь этилацетата-гексана с получением 3,9 мг (примерно 95%) 3'-десфенил-3'-(2-нафтил) таксола. 3'-десфенил-3'-трет.-бутил таксол. В маленький реакционный сосуд добавляют 125 мг (0,320 ммолъ) (+)цис-1-бензоил-3-(1-этоксиэтокси)-4-трет.-бутил-ацетидин-2-она, 45 мг (0,064 ммоль) 7-0триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (ДМАП) и 0,032 мл пиридина. Смесь перемешивают при 25° С в течение 12 часов и разбавляют 100 мл этилацетата. Этилацетатный раствор экстрагируют 20 мл 10%-ного раствора сульфата меди, сушат над сульфатом натрия и концентрируют. Остаток отфильтровывают через силикагельную насадку, используя для элюирования этилацетат. Флешхроматог-рафией на силикагеле с использованием в качестве элюирующего растворителя смесь этилацетата-гексана с последующей перекристаллизацией из смеси гексана и этилацетата получают 58 мг (89%) 2'-(1-этоксиэтокси)-3'-десфенил-3'-трет.-бутил-7-0-триэтилсилил таксола. 5 мг пробы 2'-(1-этоксиэтокси)-3'-десфенил-3'-трет.-бутил-7-0-триэ-тилсилил таксола растворяют в 2 мл этанола и добавляют 0,5 мл 0,5%-ного водного раствора соляной кислоты. Смесь перемешивают при 0° С в течение 30 часов и разбавляют 50 мл этилацетата. Раствор экстрагируют 20 мл насыщенного водного раствора бикарбоната натрия, сушат над сульфатом натрия и концентрируют. Остаток очищают хроматографически на силикагеле, используя для элюирования смесь этилацетата-гексана с получением 3,6 мг (примерно 92%) 3'-десфенил-3' -трет. -бутил, таксола. 3' -десфенил-3' -п-метоксифенил таксол. В маленький реакционный сосуд добавляют 125 мг (0,320 ммоль) (+)-цис-1-бензоил-3-(1-этоксиэтокси)-4'-п-метоксифенил-ацетидин-2-она, 45 мг (0,064 ммоль) 7-0триэтилсилилбак-катина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (ДМАП) и 0,032 мл пиридина. 12 27690 Смесь перемешивают при 25° С в течение 12 часов и разбавляют 100 мл этилацетата. Этилацетатный раствор экстрагируют 20 мл 10%-ного раствора сульфата меди, сушат над сульфатом натрия и концентрируют. Остаток отфильтровывают через силикагельную насадку, используя для элюирования этилацетат. Флешхроматографией на силикагеле с использованием в качестве элюирующего растворителя смесь, этилацетата-гексана с последующей перекристаллизацией из смеси гексана и этилацетата получают 58 мг (87%) 2'-(1 -этоксиэтокси) -3' -десфенил-3'-п-метоксифенил- 7 -О -триэтилсилил таксола. 5 мг пробы 2` -(1-этоксиэтокси)-3` -десфенил-3'-п-метоксифенил-7-0-триэтилсилил таксола растворяют в 2 мл этанола и добавляют 0,5 мл 0,5%-ного водного раствора соляной кислоты. Смесь перемешивают при 0° С в течение 30 часов и разбавляют 50 мл этилацетата. Раствор экстрагируют 20 мл насыщенного водного раствора бикарбоната натрия, сушат над сульфатом натрия и концентрируют. Остаток очищают хроматографически на силикагеле, используя для элюирования смесь этилацетата-гексана с получением 3,6 мг (примерно 92%) 3'-десфенил-3`-п-метоксифенил таксола. 3'-дебензоил-N-(2-нафтоил)-3' -десфенил-3 '-(1-нафтил) таксол. В маленький реакционный сосуд добавляют 140 мг (0,320 ммоль) (+)-цис-1-(2-нафтоил)-3-(1 -этоксиэтокси) -4- (1-нафтил)-ацетидин-2-она, 45 мг (0,064 ммолъ) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (ДМАП) и 0,032 мл пиридина. Смесь перемешивают при 25° С в течение 12 часов и разбавляют 100 мл этилацетата. Этилацетатный раствор экстрагируют 20 мл 10%-ного раствора сульфата меди, сушат над сульфатом натрия и концентри-руют. Остаток отфильтровывают через силикагельную насадку, используя для элюирования этилацетат. Флешхроматографией на силикагеле с использованием в качестве элюирующего растворителя смесь этилацетата-гексана с последующей перекристаллизацией из смеси гексана и этилацетата получают 51 мг (70%) N-дебензоил- N - (2-нафтоил) –2`- (1-этоксиэтокси) –3` -десфенил-3' - (1 -нафтил) -7 -0триэтилсилил таксола. 5 мг пробы N-дебензоил-N-(2-нафтоил)-2'-(1-этоксиэтокси)-3`-десфенил-3'-(1-нафтил)-7-0-триэтилсилил таксола растворяют в 2 мл этанола и добавляют 0,5 мл 0,5%-ного водного раствора соляной кислоты. Смесь перемешивают при 0° С в течение 30 часов и разбавляют 50 мл этилацетата. Раствор экстрагируют 20 мл насыщенного водного раствора бикарбоната натрия, сушат над сульфатом натрия и концентрируют. Остаток очищают хроматографи-чески на силикагеле, используя для элюирования смесь этилацетата-гексана с получением 3,3 мг (примерно 80%) N-дебензоил-N-(2-нафтоил)-3'-десфенил-3'-(1-нафтил) таксола. 13
ДивитисяДодаткова інформація
Назва патенту англійськоюBeta-lactam or enethiomer thereof for taxols synthesis and process for the preparation of taxol
Автори англійськоюHOLTON Robert A.
Назва патенту російськоюB-лактам или его энантиомер для синтеза таксолов и способ получения таксолов
Автори російськоюХолтон Роберт А.
МПК / Мітки
МПК: C07D 305/00, C07C 39/00, A61K 31/395, C07C 233/45, C07C 231/00, A61P 35/02, C07F 7/18, A61P 35/00, A61K 31/397, C07D 205/00
Мітки: синтезу, спосіб, бета-лактам, одержання, енантіомер, таксолів
Код посилання
<a href="https://ua.patents.su/13-27690-beta-laktam-abo-jjogo-enantiomer-dlya-sintezu-taksoliv-ta-sposib-oderzhannya-taksoliv.html" target="_blank" rel="follow" title="База патентів України">Бета-лактам або його енантіомер для синтезу таксолів та спосіб одержання таксолів</a>
Спосіб одержання похідних ціс-4-феніл-1,2,3,4-тетрагідра-1-нафтіламіна або його солей
Номер патенту: 6301
Опубліковано: 29.12.1994
Автори: Біллі Кеннет Кое, Віллард Маккован, Чарльз Армон Херберт, Аллен Річард Краска
МПК: C07C 255/58, C07C 217/74, C07C 49/697, A61K 31/165, C07C 211/41, C07C 45/28, A61P 25/26, C07C 67/00, A61K 31/135, C07C 213/00, C07C 45/00, A61K 31/13, A61K 31/137, C07C 211/42, A61P 25/24, C07C 45/46, C07C 209/00, C07C 57/00, C07C 43/21
Мітки: похідних, солей, ціс-4-феніл-1,2,3,4-тетрагідра-1-нафтіламіна, спосіб, одержання
Формула / Реферат:
Способ получения производных цис-4-фенил-1,2,3,4-тетрагидро-1-нафтиламина общей формулы Ігде R1 - атом водорода или С1-С3-н-алкил; R2-С1-С3-н-алкил; R3 - атом водорода, хлора или С1-С3 - алкоксигруппа; R4 и R5 - атомы водорода, фтора, хлора, брома или трифторметильные группы, причем одновременно R4 и R5 не являются атомами водорода, или их солей, отличающийся тем, что соединение общей формулы IIгде R3, R4 и R5...
Спосіб одержання l-етилового ефіру n-ацетил 3,5-дійод-4-п-метоксифеноксифенілаланіну-реагента для синтезу l-трийодтироніну та l-тироксину

Номер патенту: 8285
Опубліковано: 29.03.1996
Автори: Бальон Ярослав Григорович, Корпачьов Вадим Валерійович, Рабштина Михайло Васильович, Тронько Микола Дмитрович
МПК: A61P 5/18, A61P 5/16, C07C 219/00, A61P 5/00
Мітки: ефіру, l-тироксину, 3,5-дійод-4-п-метоксифеноксифенілаланіну-реагента, одержання, синтезу, l-трийодтироніну, l-етилового, n-ацетил, спосіб
Формула / Реферат:
Способ получения L-этилового эфира N-ацетил-3,5-дийод-4-п-метоксифеноксифенилаланина - реагента для синтеза L-трийодтиронина и L-тироксина путем взаимодействия этилового эфира N-ацетил-3,5-дийодтирозина с йодониевыми солями в присутствии целевых добавок в среде органического растворителя, отличающийся тем, что используют 4,4-диметоксидифенилйодоний сульфат или трифторацетат, в присутствии оснований и краун эфиров при молярном соотношении...
Спосіб отримання складного 1-етоксікарбонілоксіетілового ефіру 6-[d-(-)-2-аміно-2-фенілацетамідо]-пеніциланової кислоти у вигляді його адітивної солі з галоідводневою кислотою

Номер патенту: 3638
Опубліковано: 27.12.1994
Автори: Дерек Регінальд Пальмер, Луіджі Ратті, Роберт Грахем Тисон
МПК: C07D 501/00, A61P 31/04, C07D 499/00, A61K 31/43, C07C 69/96
Мітки: 1-етоксікарбонілоксіетілового, вигляді, кислотою, 6-[d-(-)-2-аміно-2-фенілацетамідо]-пеніциланової, галоідводневою, адітивної, складного, солі, ефіру, отримання, спосіб, кислоти
Формула / Реферат:
Способ получения сложного 1-этоксикарбонилоксиэтилового эфира 6-[D-(-)-2-амино-2-фенилацетамидо]-пенициллановой кислоты в виде его аддитивной соли с галоидводородной кислотой путем взаимодействия соли щелочного металла 6-[D-(-)-2-N-производного 2-фенилацетамино]-пенициллановой кислоты с а-галоиддиэтилкарбонатом формулы I где Наl - хлор, бром или под, в апротронном полярном растворителе...
Спосіб одержання 2-бром-alрha-ергокріптіну або його кислотно-адітивних солей

Номер патенту: 5571
Опубліковано: 28.12.1994
Автори: Габор Медьєрі, Ерік Богш, Ана Колошаі, Ференц Трішлер, Лайош Ковач, Габор Сепеші, Тибор Кєва, Бела Штефко, Марія Газдаг
МПК: C07C 67/00, C07B 39/00, C07D 519/00, C07B 31/00, C07D 457/00
Мітки: спосіб, 2-бром-alрha-ергокріптіну, солей, одержання, кислотно-адітивних
Формула / Реферат:
Способ получения 2-бром-а-эргокриптина формулыили его кислотно-аддитивных солей из а-эргокриптина путем его бромирования, отличающийся тем, что, с целью повышения селективности процесса и увеличения выхода целевого продукта, бромированию подвергают а-эргокриптин или смесь оснований, содержащих наряду с а-эргокриптином такие эрготалкалоиды, как а-эргокриптинин, эргоин, эргозинин или их...
Спосіб одержання макролідних сполучень

Номер патенту: 6045
Опубліковано: 29.12.1994
Автори: Мануель Дебоно, Герберт Ендрю Кірст
МПК: A61K 31/70, A23K 1/17, A61K 31/7042, A61P 31/04, A61K 31/7048, C07H 17/08
Мітки: одержання, спосіб, макролідних, сполучень
Формула / Реферат:
Формула изобретенияСпособ получения макролидных соединений общей формулыACH2CH2R,где А -R - октагидроазоцин-1-ил, 4-фенил-пиперидин-1-ил, гексагидроазепин-1-ил, 1-азаспиро(4,5)дец-1-ил, 1,2,3,4-тетрагидрохинолин-1-ил, 1,2,3,4-тетрагидроизохинолин-2-ил, 1,2,3,6-тетрагидропиридин-1-ил, пирролидин-1-ил, азациклотридецин-1-ил, 3-азабицикло(3,2,2)-нон-3-ил, 1,3,3-триметил-6-азабицикло [3,2,1]окт-6-ил; цис- и...
Попередній патент: Набір для самостійного виготовлення сигарет з фільтром
Наступний патент: Вікно або двері з коробом для жалюзів, які згортаються
Випадковий патент: Спосіб запису двоїчної інформації на магнітний носій