Спосіб синтезу b-l-5-фтор-2′,3′-дидезокси-2′,3′-дидегідроцитидину (b-l-fd4c)
Номер патенту: 77291
Опубліковано: 15.11.2006
Автори: Чжао Лей, Ковальчик Бо, Шуре Ральф М., Данкл Ліза






Формула / Реферат
1. Спосіб синтезу ![]() -фтор-2',3'-дидезокси-2',3'-дидегідроцитидину (
-фтор-2',3'-дидезокси-2',3'-дидегідроцитидину (![]() ), при якому здійснюють:
), при якому здійснюють:
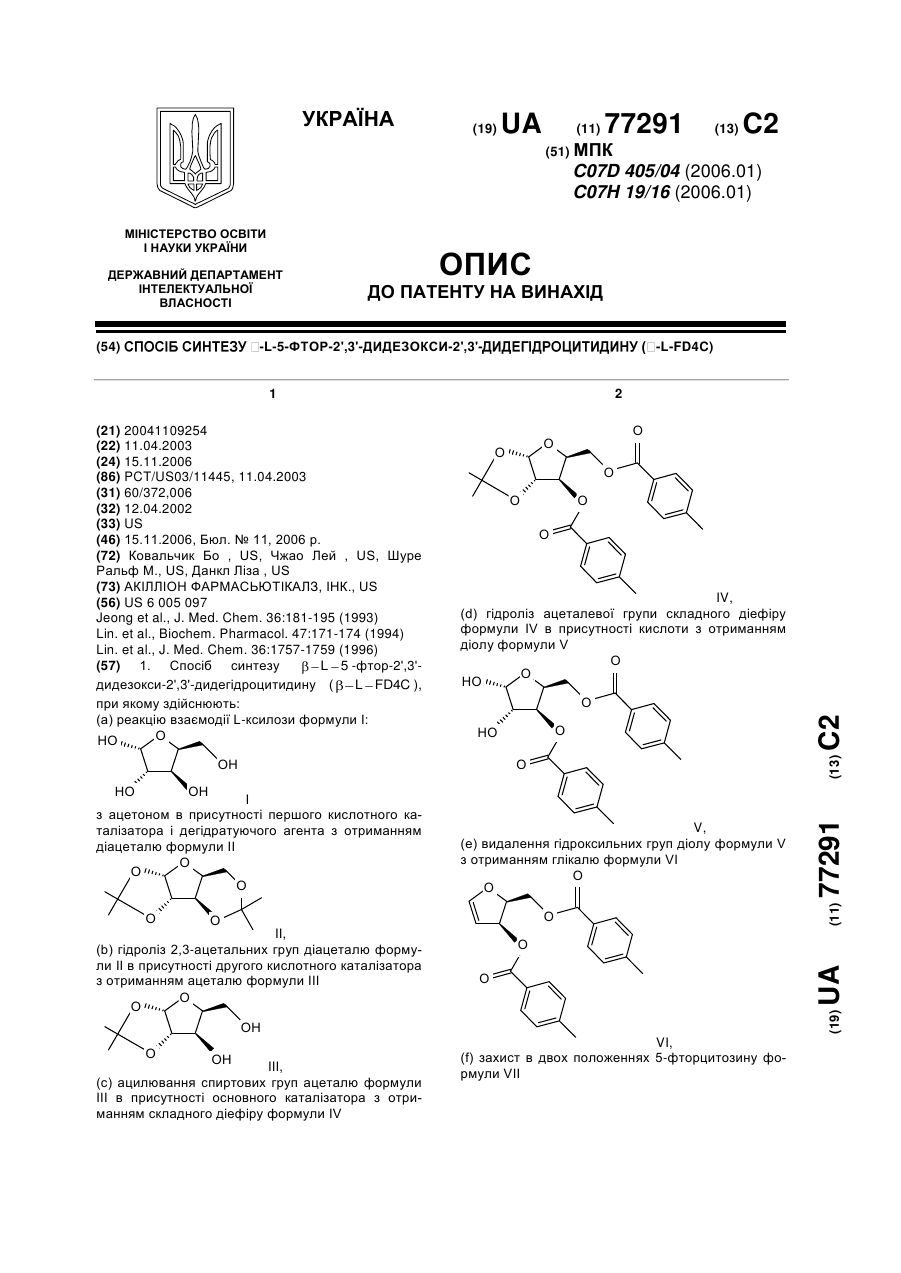
(a) реакцію взаємодії L-ксилози формули І:
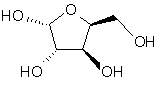 I
I
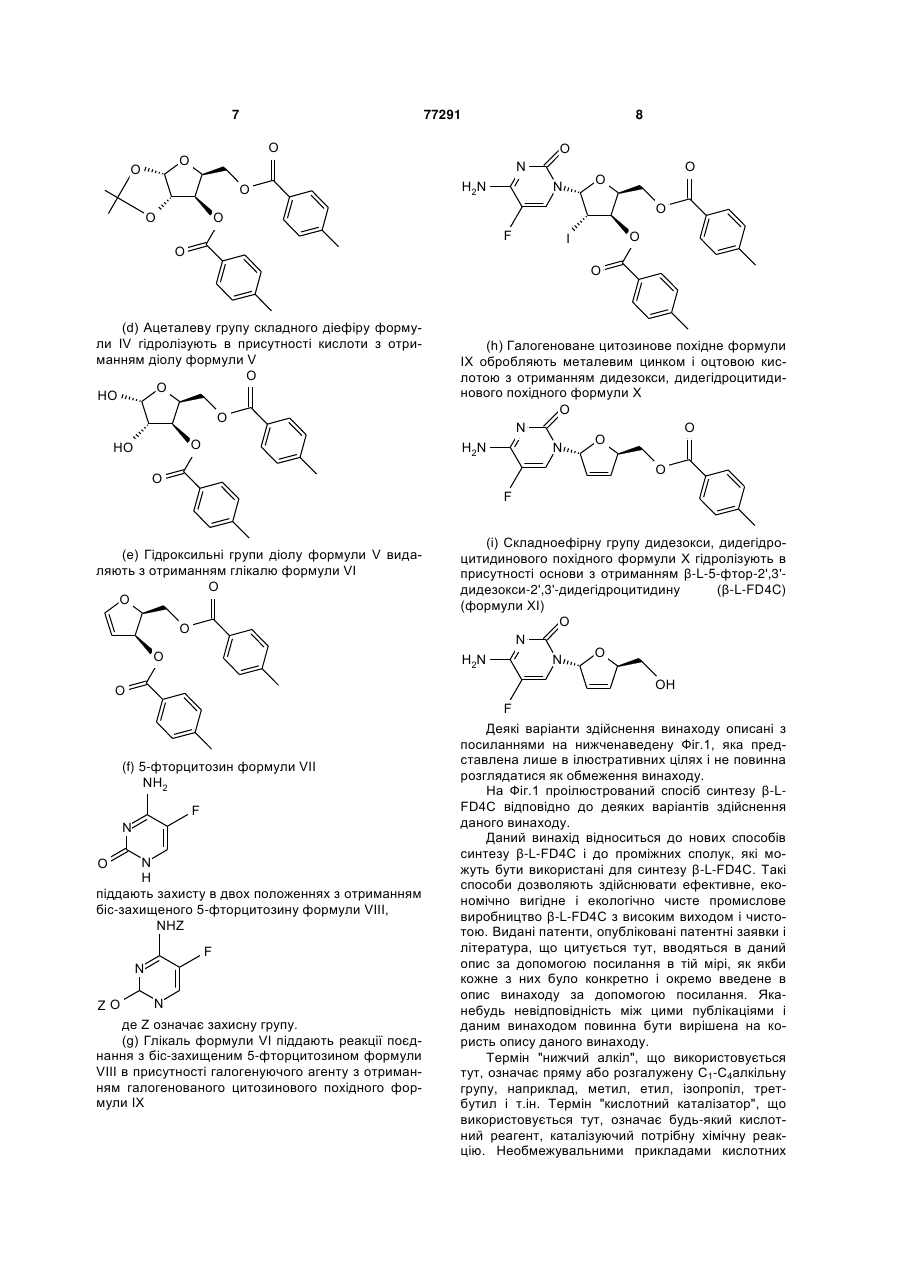
з ацетоном в присутності першого кислотного каталізатора і дегідратуючого агента з отриманням діацеталю формули II
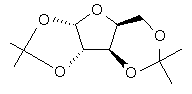 II,
II,
(b) гідроліз 2,3-ацетальних груп діацеталю формули II в присутності другого кислотного каталізатора з отриманням ацеталю формули III
 III,
III,
(c) ацилювання спиртових груп ацеталю формули III в присутності основного каталізатора з отриманням складного діефіру формули IV
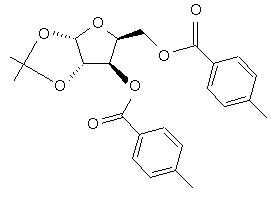 IV,
IV,
(d) гідроліз ацеталевої групи складного діефіру формули IV в присутності кислоти з отриманням діолу формули V
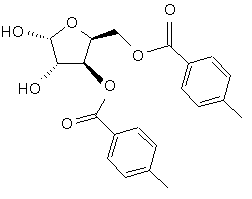 V,
V,
(e) видалення гідроксильних груп діолу формули V з отриманням глікалю формули VI
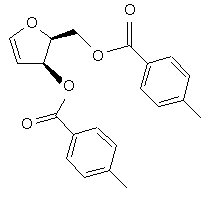 VI,
VI,
(f) захист в двох положеннях 5-фторцитозину формули VII
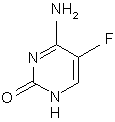 VII
VII
з отриманням біс-захищеного 5-фторцитозину формули VIII,
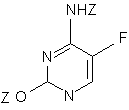 VIII,
VIII,
де Z означає захисну групу,
(g) реакцію поєднання глікалю формули VI з біс-захищеним 5-фторцитозином формули VIII в присутності галогенуючого агента з отриманням галогенованого цитозинового похідного формули IX
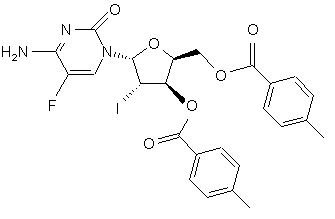 IX,
IX,
(h) обробку галогенованого цитозинового похідного формули IX металевим цинком і оцтовою кислотою з отриманням дидезокси, дидегідроцитидинового похідного формули Х
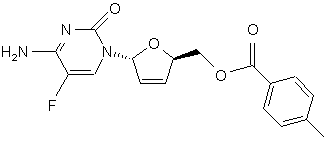 X,
X,
(і) гідроліз складноефірної групи дидезокси, дидегідроцитидинового похідного формули Х в присутності основи з отриманням ![]() -фтор-2',3'-дидезокси-2',3'-дидегідроцитидину (
-фтор-2',3'-дидезокси-2',3'-дидегідроцитидину (![]() ) формули XI
) формули XI
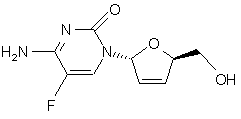 XI.
XI.
2. Спосіб за п. 1, що додатково включає виділення продукту ![]() -фтор-2',3'-дидезокси-2',3'-дидегідроцитидину (
-фтор-2',3'-дидезокси-2',3'-дидегідроцитидину (![]() ).
).
3. Спосіб за п. 2, де вказане виділення передбачає обробку розчинником, який викликає осадження ![]() -фтор-2',3'-дидезокси-2',3'-дидегідроцитидину (
-фтор-2',3'-дидезокси-2',3'-дидегідроцитидину (![]() ).
).
4. Спосіб за п. 3, де вказаним розчинником є етилацетат або ізопропанол.
5. Спосіб за п. 1, де видалення гідроксильних груп на стадії (е) передбачає обробку йодом, трифенілфосфіном і імідазолом.
6. Спосіб за п. 1, де реакція захисту вказаної сполуки в двох положеннях на стадії (f) передбачає обробку 1,1,1,3,3,3-гексаметилдисилізаном в присутності каталізатора.
7. Спосіб за п. 6, де вказаним каталізатором є сульфат амонію.
8. Спосіб за п. 1, де вказаною захисною групою Z в кожному положенні сполуки формули VIII є триметилсиліл.
9. Спосіб за п. 1, де вказані перший і другий кислотні каталізатори на стадіях (а) і (b) незалежно вибрані з групи, що складається з сірчаної кислоти, соляної кислоти і катіонообмінних смол.
10. Спосіб за п. 1, де вказаний дегідратуючий агент на стадії (а) вибраний з групи, що складається з сульфату міді, сульфату магнію і сульфату натрію.
11. Спосіб за п. 1, де основний каталізатор на стадії (с) вибраний з групи, що складається з піридину, триетиламіну, диметиламінопіридину і їх комбінацій.
12. Спосіб за п. 1, де вказаною кислотою на стадії (d) є мурашина кислота або трифтороцтова кислота.
13. Спосіб за п. 1, де вказана основа на стадії (і) вибрана з групи, що складається з аміаку, метоксиду натрію, карбонату калію, 1,8-діазабіцикло[5.4.0]-7-ундецену (DBU) і ізопропіламіну.
14. Спосіб за п. 1, де вказаним галогенуючим агентом на стадії (g) є N-йодосукцинімід (NIS).
15. Спосіб за п. 1, де стадію (b) здійснюють в такому розчиннику, як нижчий алкіловий спирт.
16. Спосіб за п. 1, де стадію (с) здійснюють в розчиннику, вибраному з групи, що складається з дихлорметану, толуолу, трет-бутилметилового ефіру і їх комбінацій.
17. Спосіб за п. 1, де стадію (d) здійснюють в розчиннику, вибраному з групи, що складається з ацетонітрилу, толуолу і їх комбінацій.
18. Спосіб за п. 1, де стадію (е) здійснюють в дихлорметані.
19. Спосіб за п. 1, де стадію (f) здійснюють в розчиннику, вибраному з групи, що складається з толуолу, хлорбензолу, хлороформу, дихлоретану, дихлорметану, ізопропілового ефіру і їх комбінацій.
20. Спосіб за п. 1, де стадію (g) здійснюють в розчиннику, вибраному з групи, що складається з дихлоретану, дихлорметану, хлорбензолу і їх комбінацій.
21. Спосіб за п. 1, де стадію (h) здійснюють в присутності нижчого алкілового спирту і нижчого алкілацетату.
22. Спосіб за п. 1, де стадію (і) здійснюють в метанолі.
23. Спосіб за п. 1, де ![]() -фтор-2',3'-дидезокси-2',3'-дидегідроцитидин (
-фтор-2',3'-дидезокси-2',3'-дидегідроцитидин (![]() ) синтезують з L-ксилози без проведення стадії випаровування досуха будь-якої з проміжних сполук формул ІІ-Х.
) синтезують з L-ксилози без проведення стадії випаровування досуха будь-якої з проміжних сполук формул ІІ-Х.
Текст
1. Спосіб синтезу L 5 -фтор-2',3'дидезокси-2',3'-дидегідроцитидину ( L FD4C ), при якому здійснюють: (a) реакцію взаємодії L-ксилози формули І: O HO 2 C2 ДЕРЖАВНИЙ ДЕПАРТАМЕНТ ІНТЕЛЕКТУАЛЬНОЇ ВЛАСНОСТІ II, (b) гідроліз 2,3-ацетальних груп діацеталю формули II в присутності другого кислотного каталізатора з отриманням ацеталю формули III O O O O OH O OH III, (c) ацилювання спиртових груп ацеталю формули III в присутності основного каталізатора з отриманням складного діефіру формули IV (11) O O (19) O V, (e) видалення гідроксильних груп діолу формули V з отриманням глікалю формули VI O O UA I з ацетоном в присутності першого кислотного каталізатора і дегідратуючого агента з отриманням діацеталю формули II O O O 77291 OH VI, (f) захист в двох положеннях 5-фторцитозину формули VII 3 77291 NH2 F N N H VII з отриманням біс-захищеного 5-фторцитозину формули VIII, NHZ O F N N ZO VIII, де Z означає захисну групу, (g) реакцію поєднання глікалю формули VI з бісзахищеним 5-фторцитозином формули VIII в присутності галогенуючого агента з отриманням галогенованого цитозинового похідного формули IX O N O O H2N N O F O I O IX, (h) обробку галогенованого цитозинового похідного формули IX металевим цинком і оцтовою кислотою з отриманням дидезокси, дидегідроцитидинового похідного формули Х O N O O H2N N O F X, (і) гідроліз складноефірної групи дидезокси, дидегідроцитидинового похідного формули Х в присутності основи з отриманням L 5 -фтор-2',3'дидезокси-2',3'-дидегідроцитидину ( L FD4C ) формули XI O N O H2N N OH F XI. 2. Спосіб за п. 1, що додатково включає виділення продукту L 5 -фтор-2',3'-дидезокси-2',3'дидегідроцитидину ( L FD4C ). 3. Спосіб за п. 2, де вказане виділення передбачає обробку розчинником, який викликає осадження 4 L 5 -фтор-2',3'-дидезокси-2',3'дидегідроцитидину ( L FD4C ). 4. Спосіб за п. 3, де вказаним розчинником є етилацетат або ізопропанол. 5. Спосіб за п. 1, де видалення гідроксильних груп на стадії (е) передбачає обробку йодом, трифенілфосфіном і імідазолом. 6. Спосіб за п. 1, де реакція захисту вказаної сполуки в двох положеннях на стадії (f) передбачає обробку 1,1,1,3,3,3-гексаметилдисилізаном в присутності каталізатора. 7. Спосіб за п. 6, де вказаним каталізатором є сульфат амонію. 8. Спосіб за п. 1, де вказаною захисною групою Z в кожному положенні сполуки формули VIII є триметилсиліл. 9. Спосіб за п. 1, де вказані перший і другий кислотні каталізатори на стадіях (а) і (b) незалежно вибрані з групи, що складається з сірчаної кислоти, соляної кислоти і катіонообмінних смол. 10. Спосіб за п. 1, де вказаний дегідратуючий агент на стадії (а) вибраний з групи, що складається з сульфату міді, сульфату магнію і сульфату натрію. 11. Спосіб за п. 1, де основний каталізатор на стадії (с) вибраний з групи, що складається з піридину, триетиламіну, диметиламінопіридину і їх комбінацій. 12. Спосіб за п. 1, де вказаною кислотою на стадії (d) є мурашина кислота або трифтороцтова кислота. 13. Спосіб за п. 1, де вказана основа на стадії (і) вибрана з групи, що складається з аміаку, метоксиду натрію, карбонату калію, 1,8діазабіцикло[5.4.0]-7-ундецену (DBU) і ізопропіламіну. 14. Спосіб за п. 1, де вказаним галогенуючим агентом на стадії (g) є N-йодосукцинімід (NIS). 15. Спосіб за п. 1, де стадію (b) здійснюють в такому розчиннику, як нижчий алкіловий спирт. 16. Спосіб за п. 1, де стадію (с) здійснюють в розчиннику, вибраному з групи, що складається з дихлорметану, толуолу, трет-бутилметилового ефіру і їх комбінацій. 17. Спосіб за п. 1, де стадію (d) здійснюють в розчиннику, вибраному з групи, що складається з ацетонітрилу, толуолу і їх комбінацій. 18. Спосіб за п. 1, де стадію (е) здійснюють в дихлорметані. 19. Спосіб за п. 1, де стадію (f) здійснюють в розчиннику, вибраному з групи, що складається з толуолу, хлорбензолу, хлороформу, дихлоретану, дихлорметану, ізопропілового ефіру і їх комбінацій. 20. Спосіб за п. 1, де стадію (g) здійснюють в розчиннику, вибраному з групи, що складається з дихлоретану, дихлорметану, хлорбензолу і їх комбінацій. 21. Спосіб за п. 1, де стадію (h) здійснюють в присутності нижчого алкілового спирту і нижчого алкілацетату. 22. Спосіб за п. 1, де стадію (і) здійснюють в метанолі. 5 77291 6 23. Спосіб за п. 1, де L 5 -фтор-2',3'дидезокси-2',3'-дидегідроцитидин ( L FD4C ) синтезують з L-ксилози без проведення стадії ви паровування досуха будь-якої з проміжних сполук формул ІІ-Х. Даний винахід відноситься до отримання нуклеозидних аналогів для їх використання як противірусних агентів. Зокрема, даний винахід відноситься до синтезу β-L-5-фтор-2',3'-дидезокси-2',3'дидегідроцитидину (β-L-FD4C). Синдром набутого імунодефіциту (СНІД), який називається вірусом імунодефіциту людини (ВІЛ), являє собою серйозну загрозу здоров'ю населення всього світу. За оцінками Всесвітньої організації охорони здоров'я, у всьому світі до кінця 2001 року було зареєстровано 40 мільйонів чоловік, ВІЛінфікованих або хворих на СНІД. З них, приблизно 5 мільйонів чоловік стали ВІЛ-інфікованими тільки в 2001 році. ВІЛ/СНІД займають четверте місце по причинах смертності у всьому світі, і тільки в 2001 році від цього захворювання померло 3 мільйони чоловік [Weekly Epidemiological Record 76:381-388 (2001)]. Іншим вірусом, що являє собою серйозну загрозу здоров'ю людини є вірус гепатиту В (HBV). Крім гострого гепатиту, HBV може викликати хронічні інфекції, що часто приводять до цирозу печінки і раку печінки з летальним кінцем. За даними, що є на 2000 рік, вірусом HBV було інфіковано 2 мільярди чоловік [Fact Sheet WHO/204, World Health Organization (October 2000)]. Різні синтетичні нуклеозиди були ідентифіковані як потенційні противірусні засоби для лікування ВІЛ і HBV. Після розробки 3'-азидо-3'дезокситимідину (АЗТ), що використовується в анти-ВІЛ-терапії [Mitsuya et al., Proc. Natl. Acad. Sci. USA 82:7096-7100)(1985)] як потенційні засоби проти ВІЛ- і HBV-інфекції були ідентифіковані деякі 2',3'-дидезокси(dd)і 2',3'-дидегідро-2',3'дидезокси(D4)-нуклеозиди. Так, наприклад, нуклеозидними аналогами, дозволеними для клінічного застосування як противірусні засоби, є 2',3'дидезоксиинозин (ddl), 2',3'-дидезоксицитидин (ddC) [Mitsuya et al., Proc. Natl. Acad. Sci. USA 83:1911-1915 (1986)] і 2',3'-дидегідро-3'дезокситимідин (D4T) [Mansuri et al., J. Med. Chem. 32:461-466 (1989)]. Хоча ці нуклеозидні аналоги використовуються в формі природного "D''енантіомеру, однак, недавні дослідження, що проводяться в цій галузі, були спрямовані також на оцінку деяких нуклеозидних аналогів, що мають неприродну "L"-конфігурацію. Так, наприклад, як потенційні засоби для ВІЛ- і HBV-терапія були ідентифіковані, наприклад, β-L-5-фтор-2',3'дидезокси-3'-тіацитидин (FTC) [Jeong et al., J. Med. Chem. 36:181-195 (1993)], β-L-5-фтор-2',3'дидезоксицитидин (β-L-FddC) [Lin et al., Biochem. Pharmacol. 47:171-174 (1994)] і β-L-5-фтор-2',3'дидезокси-2',3'-дидегідроцитидин (β-L-FD4C) [Lin et al., J. Med. Chem. 39:1757-1759 (1996)]. Було підтверджено, що β-L-FD4C є особливо цінним противірусним засобом для лікування ВІЛ- і HBV-інфекцій [Lin et al., J. Med. Chem. 39:17571759 (1996)]. Методи синтезу β-L-FD4C, що засто совуються в цей час [Lin et al., J.Med. Chem. 39:1757-1759 (1996)], дають низький вихід продукту, а тому, вони є невідповідними для великомасштабного виробництва. Таким чином, були запропоновані альтернативні методи синтезу β-L-FD4C [патент США №6005097]. Однак, потреба в розробці нових способів синтезу, що дозволяють здійснювати ефективне, економічно вигідне і екологічно чисте промислове виробництво сполуки β-L-FD4C, яка може бути використана для запобігання епідеміям ВІЛ- і HBV-інфекцій у всьому світі, досі залишається актуальною. Даний винахід був направлений на розв'язання вищезгаданої проблеми, пов'язаної з необхідністю розробки нових синтетичних способів, прийнятних для великомасштабного виробництва β-LFD4C. Такі способи дають більш високий вихід і більш високу ефективність, і при цьому, є більш економічно вигідними і не надають несприятливого впливу на навколишнє середовище. В одному з своїх аспектів, даний винахід відноситься до способів синтезу β-L-5-фтop-2',3'дидeзoкcи-2',3'-дидeгiдpoцитидинy (β-L-FD4C). Відповідно до цих способів: (а) L-ксилозу формули І: O HO OH HO OH піддають взаємодії з ацетоном в присутності першого кислотного каталізатора і дегідратуючого агенту з отриманням діацеталю формули II O O O O O (b) 2,3-ацеталь діацеталю формули II гідролізують в присутності другого кислотного каталізатора з отриманням ацеталю формули III O O OH O OH (c) Спиртові групи ацеталю формули III ацилюють в присутності основного каталізатора з отриманням складного діефіру формули IV 7 8 O O O 77291 O N O O H2N O O N O O F O O I O (d) Ацеталеву групу складного діефіру формули IV гідролізують в присутності кислоти з отриманням діолу формули V O O HO O O HO (h) Галогеноване цитозинове похідне формули IX обробляють металевим цинком і оцтовою кислотою з отриманням дидезокси, дидегідроцитидинового похідного формули X O N O O H2N N O O F (е) Гідроксильні групи діолу формули V видаляють з отриманням глікалю формули VI O O O O OH O (f) 5-фторцитозин формули VII NH2 F N N H піддають захисту в двох положеннях з отриманням біс-захищеного 5-фторцитозину формули VIII, NHZ O F N ZO (і) Складноефірну групу дидезокси, дидегідроцитидинового похідного формули X гідролізують в присутності основи з отриманням β-L-5-фтор-2',3'дидезокси-2',3'-дидегідроцитидину (β-L-FD4C) (формули XI) O N O H2N N N де Ζ означає захисну групу. (g) Глікаль формули VI піддають реакції поєднання з біс-захищеним 5-фторцитозином формули VIII в присутності галогенуючого агенту з отриманням галогенованого цитозинового похідного формули IX F Деякі варіанти здійснення винаходу описані з посиланнями на нижченаведену Фіг.1, яка представлена лише в ілюстративних цілях і не повинна розглядатися як обмеження винаходу. На Фіг.1 проілюстрований спосіб синтезу β-LFD4C відповідно до деяких варіантів здійснення даного винаходу. Даний винахід відноситься до нових способів синтезу β-L-FD4C і до проміжних сполук, які можуть бути використані для синтезу β-L-FD4C. Такі способи дозволяють здійснювати ефективне, економічно вигідне і екологічно чисте промислове виробництво β-L-FD4C з високим виходом і чистотою. Видані патенти, опубліковані патентні заявки і література, що цитується тут, вводяться в даний опис за допомогою посилання в тій мірі, як якби кожне з них було конкретно і окремо введене в опис винаходу за допомогою посилання. Яканебудь невідповідність між цими публікаціями і даним винаходом повинна бути вирішена на користь опису даного винаходу. Термін "нижчий алкіл", що використовується тут, означає пряму або розгалужену С1-С4алкільну групу, наприклад, метил, етил, ізопропіл, третбутил і т.ін. Термін "кислотний каталізатор", що використовується тут, означає будь-який кислотний реагент, каталізуючий потрібну хімічну реакцію. Необмежувальними прикладами кислотних 9 каталізаторів, що використовуються в описаних тут способах синтезу, є неорганічні кислоти, такі як сірчана кислота або соляна кислота, і катіонообмінні смоли. Катіонообмінними смолами є нерозчинні кислотні смоли, включаючи, але не обмежуючись ними, сульфовані полістиролові смоли, сульфовані поліфторвуглецеві смоли і інші катіонообмінні смоли на основі полістиролу, декстрану, агарози і т.п. Термін "основний каталізатор" означає будь-який основний реагент, що каталізує потрібну хімічну реакцію. Необмежувальними прикладами основних каталізаторів, що використовуються в описаних тут способах синтезу, є піридин, триетиламін і диметиламінопіридин (ОМАР). Термін "галогенуючий агент" означає будь-який агент, здатний здійснювати галогенування, тобто, введення атома галогену в сполуку. Термін "дегідратуючий агент" означає будь-який агент, що сприяє видаленню води. Термін "захисна група" означає будь-яку групу, яка зв'язується в одному або декількох реакційноспроможних положеннях сполуки, запобігаючи тим самим, проходженню реакцій в цих положеннях, і яка може бути видалена з вказаних положень стандартними хімічними методами. Термін "похідне" або "аналог" першої сполуки означає другу сполуку, що має хімічну структуру, аналогічну хімічній структурі першої сполуки, але, при цьому, або не містить однієї або декількох функціональних груп або одного або декількох замісників, присутніх в першій сполуці, або містить одну або декілька додаткових функціональних груп, або один або декілька додаткових замісників, відсутніх в першій сполуці. Термін "дидезокси", що використовується тут, означає нуклеозидну частину, яка має цукрову групу, в якій у кожного атома вуглецю в двох положеннях замість гідроксильної групи присутній водень. Термін "дидегідро", що використовується тут, означає нуклеозидну частину, що має цукрову групу, яка містить подвійний зв'язок. Так, наприклад, Р-Ь-5фтор-2',3'-дидезокси-2',3'-дидегідроцитидин (β-LFD4C) в 2'- і 3'-положеннях атомів вуглецю цукрової частини замість гідроксильних груп містить атоми водню, а між атомами вуглецю - подвійний зв'язок. На Фіг.1 проілюстрований синтез P-L-FD4C відповідно до деяких варіантів здійснення винаходу. β-L-FD4C 11 синтезують шляхом проведення 7стадійної процедури з використанням L-ксилози 1 як вихідного матеріалу. Принаймні, в деяких варіантах здійснення винаходу, β-L-FD4C синтезують з L-ксилози 1 без проведення процедури випаровування досуха будь-якої з проміжних сполук 2-10. Як показано на Фіг.1, процедуру синтезу починають з перетворення L-ксилози 1 в 1,2захищений ацеталь 3. Таке перетворення здійснюють шляхом отримання діацеталю 2 з подальшим гідролізом, в м'яких умовах, 3,4-ацеталевої частини в сполуці 2 з утворенням ацеталю 3. Для отримання діацеталю 2, L-ксилозу 1 об'єднують з ацетоном, з дегідратуючим агентом і з кислотним каталізатором. Дегідратуючим агентом, що використовується в процедурі синтезу, проілюстрованій на Фіг.1, є сульфат міді, який, як було виявлено, дає чудові виходи. Необмежувальними 77291 10 прикладами альтернативних дегідратуючих агентів є MgSO4 і Na2SО4. Кислотним каталізатором, що використовується в процедурі, проілюстрованій на Фіг.1, є смола Amberlyst® 15 (макросітчата катіонообмінна смола на основі сильної кислоти з функціональною групою сульфонової кислоти, Rohm & Haas, Philadelphia, PA). Така смола, що використовується як каталізатор, є найбільш відповідною, оскільки вона дає ефективну реакцію з отриманням продукту достатньої чистоти, зручна в поводженні, не вимагає значних економічних витрат і може бути легко видалена шляхом фільтрації. Необмежувальними прикладами альтернативних смол, що використовуються як каталізатори, є інші катіонообмінні смоли, такі як сульфовані полістиролові смоли, сульфовані поліфторвуглецеві смоли і інші катіонообмінні смоли на основі полістиролу, декстрану, агарози і т.п. Альтернативними кислотними каталізаторами, що не відносяться до смол і використовуються в такій реакції, є, але не обмежуються ними, сірчана кислота і соляна кислота. Гідроліз дизахищеної сполуки 2 з утворенням ацеталю 3 здійснюють шляхом додання води в органічному розчиннику, такому як, наприклад, нижчий алкіловий спирт, з подальшим доданням кислотного каталізатора. Принаймні, в деяких варіантах здійснення винаходу, перед проведенням такої процедури, розчин сполуки 2, отриманий з Lксилози 1, нейтралізують для гарантії того, що подальша заміна розчинника, тобто, ацетону на нижчий алкіловий спирт або інший органічний розчинник, не буде приводити до розкладання сполуки 2 внаслідок попереднього гідролізу з утворенням сполуки 3 або навіть із зворотним перетворенням в ксилозу. Нейтралізацію здійснюють шляхом заміни розчинника в присутності основи, такої як, наприклад, твердий карбонат калію. Заміна розчинника дозволяє безпосередньо використати діацеталь 2 в подальшій реакції гідролізу з утворенням ацеталю 3, що приводить до збільшення ефективності, оскільки, в цьому випадку, немає необхідності в проведенні стадії випаровування досуха між реакційними стадіями. У деяких варіантах здійснення винаходу, розчинником для стадії гідролізу є нижчий спирт, такий як, наприклад, метанол, як показано на Фіг.1; етанол, який є менш токсичним; технічний метильований спирт (IMS), який являє собою економічно вигідну альтернативу абсолютному етанолу при промисловому виробництві; або їх комбінацію. Необмежувальним прикладом альтернативного розчинника є толуол. Кислотним каталізатором, що використовується на стадії гідролізу, як показано на Фіг.1, є смола Amberlyst® 15. Альтернативними кислотними каталізаторами є, але не обмежуються ними, інші катіонообмінні смоли і неорганічні кислоти, такі як сірчана кислота і соляна кислота. Використання інших каталізаторів, що не є смолами, "приводить до зменшення їх впливу на навколишнє середовище завдяки тому, що ця процедура дозволяє знизити кількість отримуваних твердих відходів. Небажану ксилозу, присутню в отриманому розчині ацеталю 3, видаляють шляхом розтирання. Необмежувальними прикладами розчинників, прийн 11 ятних для розтирання, є трет-бутилметиловий ефір (ТВМЕ), толуол/ТВМЕ, толуол/етилацетат і дихлорметан (ДХМ)/етилацетат. Подальшу заміну на розчинник для розтирання здійснюють без розкладання ацеталю 3 з утворенням ксилози шляхом заміни розчинника в присутності стабілізуючої основи, такої як, наприклад, бікарбонат натрію. Використання розчинника для розтирання, такого як толуол/ТВМЕ, який також є відповідним для використання в наступній стадії реакції, приводить до збільшення ефективності реакції, оскільки, в цьому випадку, розчин ацеталю 3 може бути використаний безпосередньо в наступній стадії реакції після розтирання. Спиртові групи в сполуці 3 ацилюють з утворенням відповідних складноефірних груп в сполуці 4 шляхом обробки хлорангідридом, таким як, наприклад, п-толуоїлхлорид, і основним каталізатором. У синтезі, проілюстрованому на Фіг.1, як основний каталізатор використовують піридин, а як розчинник для реакції використовують ДХМ. Необмежувальними прикладами відповідних основних каталізаторів є піридин, триетиламін, диметиламінопіридин (DMAP) і їх комбінації. Розчинниками, придатними для використання в реакції ацилювання, є ДХМ, толуол, ТВМЕ і їх комбінації. Використання розчинника приводить до підвищення ефективності реакції; так, наприклад, використання толуолу, який є відповідним для проведення подальшої стадії реакції, дозволяє уникнути необхідності в проведенні стадії випаровування досуха між реакційними стадіями. У деяких варіантах здійснення винаходу, а зокрема, для крупномасштабного синтезу, використання більш токсичних сполук, таких як піридин і дихлорметан, обмежене через їх негативний вплив на навколишнє середовище. 1,2-ацеталеву групу сполук 4 гідролізують з отриманням відповідних спиртових груп в сполуці 5. Гідроліз здійснюють шляхом введення кислоти, такий як, наприклад, мурашина кислота або трифтороцтова кислота у воді. На Фіг.1 проілюстрований гідроліз з використанням мурашиної кислоти у воді. Розчинниками, прийнятними для проведення стадії гідролізу, є, але не обмежуються ними, ацетонітрил, толуол і їх комбінації. У конкретних варіантах здійснення винаходу, мурашину кислоту у воді використовують разом з сумішшю толуол/ацетонітрил для досягнення регульованої гомогенної реакції і отримання діолового продукту 5 хорошої міри чистоти. У деяких варіантах здійснення винаходу, продукт 5 очищають шляхом розтирання, наприклад, в гексані/ТВМЕ або в толуолі/ТБМЕ/гептані. У деяких альтернативних варіантах здійснення винаходу, заміну розчинника і осадження з розчинника, такого як, наприклад, ізопропіловий ефір, проводять для виділення діолового продукту 5, що має більш високу міру чистоти. Діол 5 перетворюють в галогеноване 5фторцитозинове похідне 9 шляхом проведення двостадійної реакції поєднання. Спочатку діол 5 перетворюють в глікаль 6 шляхом реакції взаємодії з йодом, імідазолом і трифенілфосфіном, як показано на Фіг.1. Необмежувальним прикладом 77291 12 відповідного розчинника для цієї реакції є дихлорметан. Отриманий глікаль 6 зберігають в умовах, що запобігають розкладанню. Необмежувальними прикладами таких умов є: зберігання при температурі приблизно нижче 0°С у вигляді концентрованої олії і зберігання при температурі приблизно від 5°С до 6°С в ДХМ або ТВМЕ протягом періоду часу приблизно до 3 днів. У деяких варіантах здійснення винаходу, отриманий розчин сполуки 6 використовують безпосередньо на стадії реакції поєднання, описаній нижче, що приводить до підвищення ефективності процесу. Для проведення стадії реакції поєднання, де глікаль 6 перетворюють в галогеноване 5фторцитозинове похідне 9, також необхідно використати біс-захищений 5-фторцитозин 8. На Фіг.1 проілюстрована реакція захисту 5-фторцитозину 7 двома триметилсилільними (ТМС) групами з отриманням біс-захищеної сполуки 8. Захист здійснюють шляхом контактування сполуки 7 з 1,1,1,3,3,3гексаметилдисилізаном і каталізатором. У деяких варіантах здійснення винаходу, вказаним каталізатором є сульфат амонію. Альтернативні захисні групи добре відомі фахівцям, і такими групами є, але не обмежуються ними, диметилгексилсиліл, трет-бутилдиметилсиліл, трет-бутилдифенілсиліл, трифенілметил. Необмежувальними прикладами розчинників, придатних для використання у вказаній реакції зашиті, є толуол, хлорбензол, хлороформ, дихлоретан, дихлорметан, ізопропіловий ефір і їх комбінації. У деяких варіантах здійснення винаходу використовують розчинник, який є прийнятним для проведення подальшої реакції поєднання, описаної нижче, і розчин біс-захищеного 5фторцитозину 8 вводять безпосередньо в реакцію поєднання. Проведення стадії захисту в розчиннику, прийнятному для використання в подальшій стадії поєднання, наприклад, в хлорбензолі, дихлоретані (ДХЕ) або в дихлорметані (ДХМ), підвищує ефективність процесу, а також вихід і якість продукту, оскільки таке використання дозволяє уникнути виділення або випаровування досуха нестабільного продукту 8. В деяких варіантах здійснення винаходу, а зокрема при крупномасштабному виробництві, використання більш токсичних сполук, таких як ДХЕ і ДХМ, обмежене через їх негативний вплив на навколишнє середовище. Глікаль 6 і біс-захищений 5-фторцитозин 8 піддають реакції поєднання в присутності галогенуючого агента з отриманням галогенованого 5фторцитозинового похідного 9. В деяких варіантах здійснення винаходу, вказаним галогенуючим агентом є N-йодсукцинімід (NIC), як проілюстровано на Фіг.1. Принаймні, в деяких варіантах, реакцію поєднання проводять в хлорованому розчиннику, такому як, наприклад, ДХМ, ДХЕ, хлорбензол і їх комбінації. У деяких варіантах, використання ДХЕ дозволяє зменшити негативний вплив цієї процедури на навколишнє середовище. Продукт 9 виділяють з хлорованого розчинника доданням нижчого алкілового спирту, такого як етанол, що приводить до осадження сполуки 9. Виділення продукту 9 без проведення стадії випаровування досуха, дозволяє знизити час на його отримання і дозволяє уникнути проведення тривалої стадії 13 нагрівання сполуки 9, яке приводить до певної міри розкладання. Альтернативно, продукт 9 виділяють шляхом розтирання з етанолом. У деяких варіантах здійснення винаходу, продукт 9 розчиняють, наприклад, в нижчому алкілацетаті, такому як, наприклад, метилацетат або етилацетат, і використовують в подальшій стадії реакції. Додання розчину сполуки 9 до реагентів, що залишилися, які використовуються в подальшій стадії синтезу, дозволяє уникнути необхідності в доданні твердої речовини в реакційну посудину. Як показано на Фіг.1, галогеноване 5фторцитозинове похідне 9 обробляють металевим цинком і оцтовою кислотою з отриманням дидезокси, дидегідро-5-фторцитидинового похідного 10 за допомогою дегалогенування і видалення толуолової кислоти. Таку реакцію проводять в спирті і в алкілацетаті. Так, наприклад, використовують комбінацію нижчого алкілового спирту і нижчого алкілацетату, таку як метанол і етилацетат, як показано на Фіг.1. У деяких варіантах здійснення винаходу, проведення складної реакції переетерифікації між спиртом і алкілацетатом можна уникнути завдяки використанню спирту і алкілацетату, що має ту ж саму алкільну групу, наприклад, метанолу і метилацетату, або етанолу і етилацетату. У деяких варіантах здійснення винаходу, продукт 10 виділяють шляхом розтирання з розчином гексану/етанолу. Альтернативно, до розчину продукту 10 додають ацетон. Використання ацетону дозволяє видаляти слідові кількості вихідної сполуки 9 і асоційованих з толуоїлом побічних продуктів, а також приводить до осадження продукту 10, що дає можливість провести виділення сполуки 10 без проведення стадії випаровування досуха. Щоб уникнути втрат водорозчинного продукту 10, після промивання водою, яке здійснюють шляхом зворотної екстракції, додають ацетон. Складноефірну групу сполуки 10 гідролізують і отримують кінцевий продукт β-L-FD4C 11. Відповідними розчинниками для реакції гідролізу є, але не обмежуються ними, полярні спирти, такі як метанол. Принаймні, в деяких варіантах здійснення винаходу, гідроліз здійснюють з використанням основи. Ця основа присутня в стехіометричній або в каталітичній кількості. Необмежувальними прикладами відповідних основ є аміак, метоксид натрію, карбонат калію, 1,8-діазабіцикло[5.4.0]-7ундецен (DBU) і ізопропіламін. У синтезі, проілюстрованому на Фіг.1, гідроліз здійснюють з використанням аміаку в метанолі. У деяких варіантах здійснення винаходу, використання газоподібного аміаку дозволяє знизити ризик, який існує при роботі з токсичними газами. Комерційно доступні розчини аміаку в метанолі, зокрема використовуються в подальшому виділенні продукту β-L-FD4C 11 у вигляді твердої речовини. Принаймні, в деяких варіантах винаходу виділяють продукт β-L-FD4C. Так, наприклад, неочищений продукт 11 очищають стандартними методами, відомими з рівня техніки, такими як розтирання, кристалізація і/або фільтрація через шар двоокису кремнію. Необмежувальним прикладом відповідної процедури очищення є розтирання в етилацетаті або в суміші етилацетат/метанол з 77291 14 подальшим проведенням колонкової хроматографії. Іноді, а саме, коли чистота неочищеного β-LFD4C 11 складає нижче 95%, для "отримання продукту з потрібною чистотою проводять кристалізацію або фільтрацію через силікагель, незважаючи на втрату певної кількості речовини при здійсненні такої процедури. З використанням більш чистої вихідної сполуки 10 також поліпшується якість продукту β-L-FD4C 11. Сполука 10 володіє більш нестійкою розчинністю, ніж β-L-FD4C 11. Так, наприклад, для підвищення якості вихідного матеріалу 10 можуть бути здійснені різні процедури очищення більш широкого ряду, включаючи кристалізацію, розтирання і/або фільтрацію через шар двоокису кремнію, і таким чином, може бути опосередковано підвищена чистота кінцевої сполуки β-L-FD4C 11. У деяких альтернативних варіантах здійснення винаходу, β-L-FD4C 11 виділяють шляхом додання розчинника, який викликає його осадження. Прикладами розчинників, прийнятних для ініціації осадження чистого β-L-FD4C 11, є етилацетат і ізопропанол. Осадження кінцевого продукту β-L-FD4C 11 дозволяє уникнути необхідності в проведенні стадії випаровування досуха і подальших процедур очищення. Відсутність необхідності в проведенні хроматографії на силікагелі є особливо привабливою з точки зору охорони навколишнього середовища, оскільки вона дозволяє зменшити об'єм розчинників, що використовуються, і кількість відходів, що утворюються. При цьому, потрібно зазначити, що способи даного винаходу можуть бути також застосовані для отримання сполук, споріднених з β-L-FD4C. Такими спорідненими сполуками є нуклеозидні аналоги, наприклад, 2',3'-дидезоксинуклеозиди або 2',3'-дидезокси-2',3'-дидегідронуклеозиди, що мають пуринову або піримідинову основу, пов'язану з рибозною частиною. Піримідиновою основою є гетероциклічна сполука спільного класу, що містить такі сполуки, як урацил, тимін, цитозин і споріднені аналоги. Пуриновою основою є гетероциклічна сполука спільного класу, що містить такі сполуки, як гіпоксантин, ксантин, аденін, гуанін і їх аналоги. Необмежувальними прикладами аналогів пурину або піримідину є основи, в яких СН-група заміна атомом азоту, і основи, що мають один або декілька замісників на кільці, які можуть бути введені або видалені, або вони можуть бути модифіковані звичайними замісниками, відомими фахівцям, наприклад, галогеном, гідроксилом, аміно або С1-С6алкілом. Способи даного винаходу можуть бути також використані для синтезу різних синтетичних проміжних сполук, описаних в даній заявці, або їх аналогів. Для докладної ілюстрації деяких варіантів здійснення винаходу приводяться нижченаведені необмежувальні приклади. Приклад 1 β-L-FD4C отримували відповідно до процедури, проілюстрованої на Фіг.1 Отримання ацеталю 3 L-ксилозу 1 (1000г, 6,66моль, 1екв.), ацетон (10л), сульфат міді (1,33кг, 8,3моль, 1,25екв.) і смолу Amberlyst® 15 (1000г) об'єднували в 22 15 літровій круглодонній колбі, забезпеченій механічною мішалкою, датчиком температури і пристроєм для впуску/випуску азоту. Реакційну суміш перемішували в атмосфері азоту при кімнатній температурі протягом 16 годин. ТШХ (100% етилацетат, візуалізація фосфомолібденовою кислотою (РМА)) вказувала на відсутність вихідної сполуки (Rf~0,05). Потім додавали 750г твердого карбонату натрію і суміш перемішували протягом 30 хвилин. Розчин фільтрували через Celite® (діатоміт, World Minerals Inc., Santa Barbara, CA) для видалення твердих речовин. Фільтрат концентрували у вакуумі і отримували 1,4кг прозорої олії. 1H-ЯМР підтвердив відсутність L-ксилози 1. Потім олію розчиняли в 7л розчину метанол/вода, 4:1. Потім, при кімнатній температурі, перемішуючи, додавали 1,4кг смоли Amberlyst® 15. Розчин перемішували при кімнатній температурі доти, поки ТШХ (100% етилацетат, візуалізація РМА) не показала на відсутність дизахищеної ксилози 2 (Rf~0,75). Отриманий розчин фільтрували і фільтрат доводили до рН 8 шляхом додання твердого бікарбонату натрію (приблизно 20г). Розчинник видаляли у вакуумі на 50% і отримували 1160г легкої олії. Отриману олію розтирали з 10л розчину дихлорметан/етилацетат, 3:2, і сушили над сульфатом натрію. Потім осушений розчин фільтрували через Celite® і концентрували у вакуумі з отриманням 1055г (83%) сполуки 3. 1H-ЯМР і ТШХ підтвердили дану структуру. Отримання складного діефіру 4 Ацеталь 3 (1050г, 5,52моль, 1екв.), піридин (1800мл, 23,18моль, 4,2екв.) і дихлорметан (5,65л) об'єднували в 22-літровій круглодонній колбі, забезпеченій краплинною лійкою, пристроєм для впуску/випуску азоту, датчиком температури, механічною мішалкою і крижаною банею. Розчин охолоджували до 5°С в крижаній бані в атмосфері азоту. Потім через краплинну лійку додавали птолуоїлхлорид (1,76кг, 11,48моль, 2,08екв.), підтримуючи при цьому температуру нижче 25°С. Розчин залишали для перемішування на 16 годин в атмосфері азоту. ТШХ (етилацетат/гексан, 1:1, РМА-візуалізація) вказувала на завершення реакції. Потім розчин промивали 1x5л води, 1x5л 3н розчину НСІ і 1x5л води, і сушили над сульфатом магнію протягом трьох годин. Після відфільтровування осушувача, фільтрат концентрували у вакуумі з отриманням 2460г (кількісний вихід) сполуки 4 у вигляді світлої олії. ТШХ і 1Н-ЯМР підтвердили структуру даного продукту. Отримання діолу 5 9,3л мурашиної кислоти і 2л води об'єднували в 22-літровій круглодонній колбі, забезпеченій датчиком температури, механічною мішалкою і нагрівальним кожухом. Складний діефір 4 (2320г, 5,44моль, 1екв.) розчиняли в 2,3л ацетонітрилу і однією порцією додавали до розчину мурашиної кислоти/води. Об'єднаний розчин доводили до 50°С і перемішували протягом 2,5 години. ТШХ (етилацетат/гексан, 1:1, РМА-візуалізація) вказувала на відсутність вихідного матеріалу. Потім розчин розбавляли 6л насиченого розчину солі і екстрагували 2x8л дихлорметану (або 1,2дихлоретану або хлороформу). Об'єднані дихлор 77291 16 метанові шари промивали 2x6л води, 2x4л насичених бікарбонати натрію (до доведення рН до значення 7-8, на що вказував індикаторний папір для визначення рН), 1x6л води і 1x10л насиченого розчину солі, а потім сушили над сульфатом натрію. Після видалення осушувача, розчинник видаляли у вакуумі з отриманням 1,88кг (89%) світлої твердої речовини. Потім тверду речовину розтирали з розчином гексану/третинного метилбутилового ефіру, 4:0,5 (МТВЕ) протягом 16 годин. Отримані білі тверді речовини виділяли фільтрацією, промивали 2л гексану і сушили у вакуумній печі при 35°С з отриманням 1630г (78%) сполуки 5 у вигляді ясно-коричнюватої твердої речовини. ВЕРХ показала чистоту 85%, а 1Н-ЯМР підтвердив дану структуру. Отримання галогенованого 5фторцитозинового похідного 9 Біс-захищений 5-фторцитозин 8: Сполуку 8 отримували таким чином. 5-Фторцитозин 7 (605г, 4,69моль, 1,0екв.), 1,1,1,3,3,3-гексаметилдисилізан (5л, 23,7моль, 5екв.) і сульфат амонію (24г, каталізатор) об'єднували в атмосфері азоту в чистій сухій 12-літровій круглодонній колбі, забезпеченій механічною мішалкою, нагрівальним кожухом, датчиком температури, холодильником і пристроєм для впуску/випуску азоту. Після кип'ятіння із зворотним холодильником протягом 30 хвилин, тверді речовини розчинялися, і розчин кип'ятили із зворотним холодильником протягом ще 2 годин. Отриманий розчин залишали для охолоджування приблизно до 70°С і переносили в атмосфері азоту на роторний випарник. Розчинник видаляли у вакуумі при 85°С і піддавали азеотропній перегонці з 2x2л безводного ксилолу з отриманням сполуки 8 у вигляді білої кристалічної твердої речовини. Цю кристалічну тверду речовину розчиняли в 6л дихлорметану і отримували розчин А, який зберігали до проведення стадії реакції поєднання. Глікаль 6: Сполуку 6 отримували таким чином. Дихлорметан (25л), йод (1985г, 7,82моль, 2екв.), трифенілфосфін (2051г, 7,82моль, 2екв.) і імідазол (1170г, 17,18моль, 1,4екв.) об'єднували в 100літровому реакторі, забезпеченому охолоджуючим змійовиком, пристроєм для впуску/випуску азоту, датчиком температури, механічною мішалкою і краплинною лійкою. У міру додання імідазолу, колір розчину мінявся з пурпурного на жовтий, і температура підвищувалася приблизно до 30°С. Розчин охолоджували до 15°С в атмосфері азоту. Сполуку 5 (1510г, 3,91моль, 1екв.) розчиняли в 10л дихлорметану і порціями додавали в 100-літровий реактор, підтримуючи при цьому температуру нижче 20°С. Після додання всієї кількості сполуки 5, розчин перемішували при кімнатній температурі протягом, принаймні, 2 годин. Через 2,5 години, ТШХ (етилацетат/гексан, 1:1, РМА-візуалізація) вказувала на відсутність вихідної сполуки (Rf~0,5), на слабку пляму при Rf~0,8 (проміжна сполука) і на продукт глікаль 6 (Rf~0,9). Розчин гасили 20л 20%-ного розчину тіосульфату натрію і перемішували приблизно 20 хвилин. Шари відділяли, і органічний шар промивали 1x20л води, 1x20л насиченого розчину солі і сушили над сульфатом магнію, принаймні, протягом 1,5 годин. Після видалення 17 осушувача, розчин концентрували у вакуумі з отриманням олії, і цю олію розтирали з 4л третинного метил бутилового ефіру (МТВЕ). Тверді речовини (оксид трифенілфосфіну) видаляли фільтрацією і промивали 3л МТВЕ. Фільтрат концентрували у вакуумі з отриманням олії і зберігали в атмосфері аргону при -10°С до проведення стадії реакції поєднання. Поєднання: Стадію поєднання проводили таким чином. Розчин А, отриманий внаслідок проведення реакції захисту 5-фторцитозину, додавали в атмосфері азоту в чисту суху 22-літрову круглодонну колбу, забезпечену механічною мішалкою, датчиком температури, крижаною банею і пристроєм для впуску/випуску азоту. Глікаль 6 розчиняли в 7л дихлорметану і додавали в 22-літрову круглодонну колбу. Об'єднаний розчин перемішували в атмосфері азоту при доданні порціями Nйодсукциніміду (NIS, 1100г, 4,88моль, 1,25екв.). Температуру підтримували нижче 15°С з використанням крижаної бані. Після додання NIS, розчин перемішували при кімнатній температурі, принаймні, протягом 2 годин. Через 4 години, ТШХ (етилацетат/гексан, 1:1, РМА-візуалізація) вказувала на відсутність глікалю 6 (Rf~0,9), після чого реакцію гасили 1х20л 20%-ного тіосульфату натрію. Розчин залишали для перемішування приблизно на 20 хвилин. У міру утворення значної кількості твердих речовин, їх залишав осаджуватися на 24 години для полегшення фільтрації. Тверді речовини видаляли шляхом фільтрації через фільтрувальний мішок в 1 мікрон, що знаходиться на центрифузі. Фільтрат знов вміщували в 100літровий реактор і шари розділяли. Органічний шар промивали 1x20л води і 1x20л насиченого розчину солі, а потім сушили над сульфатом натрію. Після видалення осушувача, дихлорметан видаляли у вакуумі з отриманням темної олії. Отриману олію розтирали з 4л етанолу при 20°С і перемішували 16 годин. Тверді речовини виділяли фільтрацією, промивали 4л етанолу і сушили у вакуумній печі протягом 16 годин при 35°С з отриманням 1259г (53%) сполуки 9 у вигляді не зовсім білої твердої речовини. Чистота продукту становила 98%, на що вказувала ВЕРХ, а його структура була підтверджена 1Н-ЯМР. Отримання дидезокси, дидегідроцитидинового похідного 10 Дві окремі серії реакцій проводили таким чином: Серія 1: Сполуку 9 (935г, 1,544моль, 1екв.), етилацетат (8,4л), метанол (1л) і оцтову кислоту (93мл, 1,544моль, 1екв.) додавали в чисту суху 22літрову круглодонну колбу. Розчин перемішували протягом 10 хвилин і однією порцією додавали цинк (200г, 3,08моль, 2,0екв.). Температура підвищувалася від 15°С до 23°С протягом 15 хвилин. ТШХ (етилацетат/метанол, 9:1, РМА- і УФвізуалізація) не виявляла якої-небудь реакції. Потім однією порцією додавали ще один еквівалент цинку (100г), після чого температура підвищувалася до 41°С протягом 15 хвилин. ТШХ вказувала на завершення реакції через 30 хвилин. Реакційну суміш залишали на 16 годин (протягом ночі) при кімнатній температурі для перемішування. Цинк 77291 18 видаляли фільтрацією, і фільтрат промивали 1x10л води і 1x10л 10% розчину хлориду амонію. Потім етилацетат концентрували у вакуумі до 1,5л, і отриману суспензію залишали на ніч при кімнатній температурі для перемішування. Тверді речовини виділяли фільтрацією, промивали 1л етилацетату і сушили у вакуумній печі протягом 16 годин при 35°С з отриманням 275г (52%) сполуки 10 у вигляді білої твердої речовини. Чистота становила 98%, на що вказувала ВЕРХ, а структура отриманого продукту була підтверджена 1Н-ЯМР. Спроби отримати додаткову кількість сполуки з маточного розчину не увінчалися успіхом. Серія 2: Вихідну сполуку 9 для цієї серії зберігали при -10°С протягом приблизно 3 місяців. ТШХ не вказувала на яке-небудь розкладання. У чисту суху 22-літрову круглодонну колбу додавали етилацетат (5,5л), метанол (600мл), оцтову кислоту (61мл, 1,01моль, 1екв.) і цинк (195г, 3,01моль, 3екв.). Розчин перемішували протягом 20 хвилин. Потім однією порцією додавали сполуку 9 (615г, 101моль, 1екв.), і температуру підтримували нижче 30°С з використанням крижаної бані. ТШХ (етилацетат/метанол, 9:1, УФ- і РМА-візуалізація) вказувала на завершення реакції через 3 години. Розчин залишали на 16 годин (протягом ночі) при кімнатній температурі для перемішування. Тверді речовини відфільтровували і фільтрат промивали 1x4л води, 1x6л 10% хлориду амонію і 1x6л 10% розчину карбонату калію, насиченого хлоридом натрію, а потім сушили над сульфатом натрію. Після видалення осушувача, розчинник видаляли у вакуумі з отриманням 382г (кількісний вихід) коричнюватої твердої речовини. Цю тверду речовину розтирали з розчином гексану/етанолу, 9:1, протягом 16 годин. Тверді речовини виділяли фільтрацією, промивали 500мл вищезгаданого розчину і сушили у вакуумній печі протягом 16 годин при 35°С з отриманням 226г (65%) сполуки 10 у вигляді коричнюватого порошку. Чистота становила 1 95%, на що вказувала ВЕРХ, а Н-ЯМР вказував на присутність в даній структурі невеликої кількості домішок. Елементний аналіз вказував на 8%-ий вміст золи, яку видаляли шляхом фільтрації через Celite® в наступній стадії. Отримання β-L-FD4C 11 Дві окремі серії реакцій здійснювали таким чином. Серія 1: Сполуку 10, отриману як описано вище в серії 1 (273г, 0,79моль, 1екв.) і безводний метанол (3л) об'єднували в чистій сухій 22-літровій круглодонній колбі, забезпеченій механічною мішалкою, крижаною банею, датчиком температури і пробіркою з газовим дисперсійним середовищем. При перемішуванні і підтриманні температури при 25°С, в колбу протягом 1 години барботували безводний газоподібний аміак. Потім колбу герметично закривали і залишали на 24 години (на ніч) при кімнатній температурі для перемішування. ТШХ (етилацетат/метанол, 9:1, РМА-візуалізація) вказувала на завершення реакції. Розчин відфільтровували через Celite® і фільтрат концентрували у вакуумі з отриманням 180г світлої твердої речовини. Тверду речовину розтирали з 2л етилацетату протягом 16 годин, виділяли фільтрацією і сушили 19 у вакуумній печі протягом 16 годин при 35°С з отриманням 151г (84%) β-L-FD4C 11 у вигляді білої твердої речовини. Чистота становила 99,7%, на що вказувала ВЕРХ. 1H-ЯМР, 13С-ЯМР, МС, елементний аналіз і оптичне обертання підтверджували структуру і чистоту отриманої сполуки. Серія 2: Сполуку 10 (226г, 0,665моль, 1екв.) і безводний метанол (3л) об'єднували в чистій сухій 22-літровій круглодонній колбі. При перемішуванні і підтриманні температури нижче 25°С, в колбу протягом 1 години барботували безводний газоподібний аміак. Потім колбу герметично закривали і залишали на 24 години (на ніч) для перемішування. ТШХ вказувала на завершення реакції, і розчин фільтрували через Celite® для видалення суспендованої речовини, яка не розчинилася. Фільтрат концентрували у вакуумі з отриманням коричнювато-жовтої твердої речовини. Тверді речовини подрібнювали за допомогою ступки і товкачика, і розтирали з 3л етилацетату протягом 3 днів (під час вихідних днів). Тверді речовини виділяли фільтрацією, промивали 1л етилацетату і сушили у вакуумній печі протягом 16 годин при 35°С з отриманням 147г (98%) β-L-FD4C 11 у вигляді коричнюватої твердої речовини. Чистота становила 96,5%, на що вказувала ВЕРХ. Після проведення певного дослідження, 147г неочищеного β-L-FD4C 11 розтирали з 10мл/г розчину етилацетату/етанолу, 1:1, протягом 16 годин. Тверді речовини виділяли і сушили з отриманням 105г (70,6%) βL-FD4C 11 у вигляді коричнюватої твердої речови1 ни. ВЕРХ вказувала на 98%-ну чистоту. Однак, НЯМР показав на деяку кількість домішок, а елементний аналіз виявив 8%-ний вміст золи. Приклад 2 β-L-FD4C отримували на експериментальному обладнанні відповідно до процедури, проілюстрованої на Фіг.1, але з використанням декількох альтернативних розчинників і реагентів, описаних нижче. Всі маси і об'єми є номінальними (умовними), якщо це не обумовлено особливо. Кількості вихідного і кінцевого продукту, чистота і виходи представлені нижче в Таблиці 1. Отримання ацеталю 3 У реакційну посудину завантажували Lксилозу (1,0мас, 1,0мол.екв.) і ацетон (7,9мас, 10,0об.). Отриману суспензію інтенсивно перемішували і додавали безводний сульфат міді (1,33мас), підтримуючи температуру реакції нижче 25°С. Потім додавали смолу Amberlyst® 15 (1,00мас), підтримуючи температуру реакції нижче 25°С. Отриману суміш інтенсивно перемішували при 20-25°С доти, поки реакція з утворенням сполуки 2 не була приблизно завершена, на що вказував 1Н-ЯМР (3мас. %, то додавали толуол (3,46мас, 4,0об.), і воду видаляли шляхом вакуумної перегонки при температурі до 35°С. Потім знову визначали вміст води, і якщо це необхідно, то азеотропну перегонку з толуолом повторювали. Після цього розчин освітлювали, фільтр промивали толуолом (1,73мас, 2,0об.) і розчин концентрували приблизно до 2об. шляхом вакуумної перегонки при температурі до 35°С. Якщо необхідно, розчин доводили до 30-35°С, і повільно додавали ізопропіловий ефір (ІРЕ, 4,35мас, 6,0об.), підтримуючи температуру 30-35°С. Отриманий розчин охолоджували до 0-5°С і витримували протягом 3-4 годин, після чого тверду речовину виділяли шляхом центрифугування. Твердий осад промивали ІРЕ (2x1,45мас, 2x2,0об.) і отриману тверду речовину сушили у вакуумі при температурі до 35°С. Отримання галогенованого 5фторцитозинового похідного 9 Глікаль 6: Сполуку 6 отримували таким чином. У реакційну посудину завантажували йод (1,447мас.) і дихлорметан (ДХМ, 7,30мас, 5,5об.), а потім додавали трифенілфосфін (1,50мас.) в ДХМ (5,84мас, 4,4об.), підтримуючи при цьому температуру 20-30°С. Потім проводили промивання дихлорметаном (1,46мас, 1,1об.). В суспензію завантажували імідазол (0,85мас.) в ДХМ (5,84мас, 4,4об.), підтримуючи температуру 2030°С. Потім проводили промивання ДХМ (1,46мас, 1,1об.) і суспензію охолоджували до 0-10°С. Після 77291 22 цього повільно додавали сполуку 5 (1,00мас, 1,0мол.екв.) в ДХМ (5,84мас, 4,4об.), підтримуючи температуру 10 10->11 2 Ε F G+H G Η I 3 50,58 58,6 80 36,76 43,24 20,94 4 96,2 98,17 99,58 99,21 98,93 5 G Η І 6 57,7 48,42 20,94 7 99,58 99,21 98,93 8 72,3 52,8 46 9 114,1 82,6 26,2 J 11,4 99,76 82,7 54,4 * Означає перетворення сполук з використанням відомих сполук, позначених цифрами на Фіг.1 ** Вихід сполуки 4 був теоретично прийнятий за 100%, тому вказаний загальний вихід сполуки 5 із сполуки 3 Для кращого розуміння даного винаходу вище представлений його докладний опис, однак, для кожного фахівця очевидно, що в нього можуть бу Комп’ютерна верстка А. Рябко ти внесені різні зміни, що не виходять за рамки об'єму, визначеного в нижченаведеній формулі винаходу. Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюA method of synthesizing -fluoro-2',3'-dideoxy-2",3"-didehydrocytidine ()
Назва патенту російськоюСпособ синтеза-фтор-2',3'-дидезокси-2',3'-дидегидроцитидина (в-l-fd4c)
МПК / Мітки
МПК: C07H 19/16, C07D 405/04
Мітки: синтезу, b-l-5-фтор-2',3'-дидезокси-2',3'-дидегідроцитидину, b-l-fd4c, спосіб
Код посилання
<a href="https://ua.patents.su/13-77291-sposib-sintezu-b-l-5-ftor-23-didezoksi-23-didegidrocitidinu-b-l-fd4c.html" target="_blank" rel="follow" title="База патентів України">Спосіб синтезу b-l-5-фтор-2′,3′-дидезокси-2′,3′-дидегідроцитидину (b-l-fd4c)</a>
Похідні 1,4-діарил-2-фтор-1-бутен-3-олу, спосіб їх одержання та способи одержання похідних 1,4-діарил-2-фтор-1,3-бутадієну і 1,4-діарил-2-фтор-2-бутену

Номер патенту: 70975
Опубліковано: 15.11.2004
Автори: Ху Юлін, Хант Девід Аллен
МПК: C07C 41/00, C07C 33/00, C07C 43/29, C07C 43/295
Мітки: 1,4-діарил-2-фтор-1,3-бутадієну, одержання, похідні, спосіб, 1,4-діарил-2-фтор-2-бутену, 1,4-діарил-2-фтор-1-бутен-3-олу, похідних, способи
Формула / Реферат:
1. Сполука структурної формули (І)(I),деR означає водень, С1-С4алкіл, С1-С4галоалкіл, С3-С6циклоалкіл або С3-С6галоциклоалкіл;Аr означає феніл, необов'язково заміщений будь-якою комбінацією замісників, вибраних із ряду, який містить:один(ну)-три атом(и) галогену, С1-С4алкільну(і), С1-С4галоалкільну(і), С1-С4алкокси- або С1-С4галоалкоксигрупу(и);1- або 2-нафтил, необов'язково заміщений будь-якою...
Спосіб модернізації in situ реактора гетерогенного екзотермічного синтезу, реактор гетерогенного екзотермічного синтезу та спосіб здійснення гетерогенних екзотермічних реакцій синтезу з високою продуктивністю

Номер патенту: 73466
Опубліковано: 15.08.2005
Автори: Філліппі Ерманно, ПАГАНІ Джорджіо
МПК: B01J 8/04, B01J 35/00, C01C 1/04, B01J 8/00
Мітки: високою, екзотермічного, гетерогенних, синтезу, реактор, екзотермічних, здійснення, модернізації, реактора, продуктивністю, спосіб, реакцій, гетерогенного
Формула / Реферат:
1. Спосіб модернізації in situ реактора гетерогенного екзотермічного синтезу, що включає зовнішній кожух, в якому розміщені один на одному в просторовому взаємозв'язку каталітичні шари, за яким попередньо встановлюють принаймні перший каталітичний шар у верхній частині згаданого кожуха та принаймні другий шар каталізатора в нижній частині цього кожуха, потім перший та другий шари завантажують першим каталізатором із завчасно...
Спосіб синтезу n-[(s)-1-карбоксибутил]-(s)-аланінових ефірів та їх застосування у синтезі периндоприлу

Номер патенту: 72040
Опубліковано: 17.01.2005
Автори: Сувьє Жан-Клод, Рено Ален
МПК: A61K 38/55, C07C 227/00, A61P 43/00, C07B 61/00, C07C 229/16, A61P 9/04, A61P 9/00, A61P 9/12
Мітки: периндоприлу, спосіб, n-[(s)-1-карбоксибутил]-(s)-аланінових, синтезі, ефірів, синтезу, застосування
Формула / Реферат:
1. Спосіб промислового синтезу сполук формули (І) (І),де R являє лінійну або розгалужену (C1-C6)-алкільну групу, який відрізняється тим, що аланін формули (III): (ІІІ)конденсують зі сполукою формули (ІV):
Спосіб електрохімічного синтезу алкоголятів лужних металів та регенерації каталізатора синтезу метилформіату

Номер патенту: 25079
Опубліковано: 25.12.1998
Автори: Паздерський Юрій Антонович, Заботта Георг, Тагаєв Олег Олексійович, Скачко Володимир Петрович, Лецюк Василь Володимирович
МПК: B01J 31/40, C25B 11/00, C07C 29/70, C25B 13/00, C25B 3/00, B01D 61/42
Мітки: регенерації, каталізатора, металів, лужних, синтезу, алкоголятів, метилформіату, електрохімічного, спосіб
Формула / Реферат:
1. Спосіб електрохімічного синтезу алкоголятів лужних металів та регенерації каталізатора синтезу метилформіату із спиртових розчинів солей лужних металів в апараті електролізу з розділеними іонселективною мембраною катодним та анодним просторами, який відрізняється тим, що електроліз спиртових розчинів сполук лужних металів проводять в присутності 1 - 10г/л літієвої, калієвої або натрієвої солей пероксафторалкансульфонових кислот формулою...
Спосіб синтезу n-[(s)-1-карбоксибутил]-(s)-аланінових ефірів і їх застосування у синтезі периндоприлу

Номер патенту: 73562
Опубліковано: 15.08.2005
Автор: Сувьє Жан-Клод
МПК: A61P 9/00, C07C 229/16, C07B 61/00, C07D 209/34, C07C 227/00
Мітки: ефірів, периндоприлу, синтезу, n-[(s)-1-карбоксибутил]-(s)-аланінових, спосіб, синтезі, застосування
Формула / Реферат:
1. Спосіб для промислового синтезу сполук формули (І) (I),де R представляє лінійну або розгалужену (С1-С6)алкільну групу, який характеризується тим, що піруват натрію формули (III): (III)конденсують із сполукою формули (IV):
Попередній патент: Пристрій для підйому й опускання великовагових вантажів у шахтних вертикальних стовбурах
Наступний патент: Спосіб одержання залеплону
Випадковий патент: Пристрій для вимірювання тиску у бетонній суміші при її віброущільненні