Спосіб одержання стереоселективних похідних дигідро-2,3-бензодіазепіну (варіанти) та проміжні сполуки для їх одержання
Номер патенту: 41354
Опубліковано: 17.09.2001
Автори: Змієвскі Мелтон Джозеф (мол.), Хансен Марвін Мартін, Вері Девід Лі, Андерсон Бенджамін Алан, Віченці Джеффрі Томас








Формула / Реферат
1. Способ получения стереоселективных производных дигидро-2,3-бензодиазепина, имеющих общую формулу (I):
в которой R представляет водород или (С1-С10)алкил, и
Х представляет водород, (С1-С10)алкил, ацил, арил, карбоксил или его замещенное производное или защитную группу,
или их фармацевтически приемлемых солей, отличающийся тем, что циклизуют соединение, имеющее общую формулу (VIII):
в которой Z представляет уходящий атом или группу, с получением соединения, имеющего общую формулу (I), после чего, если необходимо, превращают соединение формулы (I) в другое соединение формулы (I), и/или образуют фармацевтически приемлемую соль.
2. Способ получения соединения по п. 1, имеющего формулу (I), в которой Аr представляет п-аминофенил, отличающийся тем, что циклизуют соединение общей формулы (VIII), в которой Аr представляет п-нитрофенил, п-аминофенил или (защищенный амино)фенил, после чего, если необходимо,
(а) восстанавливают п-нитрофенильную группу с получением п-аминофенильной группы, или
(b) снимают защиту или деблокируют п-(защищенную амино)фенильную группу с получением п-аминофенильной группы.
3. Способ получения соединения по п. 2, имеющего общую формулу (I), в которой R представляет метил и Х представляет водород, формил, ацетил, пропионил или N-метилкарбамоил, или его фармацевтически приемлемой соли, отличающийся тем, что циклизуют соединение, имеющее общую формулу (VIII), в которой
R - метил, Х - водород, формил, ацетил, пропионил, N-метилкарбамоил или защитная группа, и Аr представляет п-нитрофенил, п-аминофенил или п-(защищенный амино)фенил, после чего, если необходимо:
(a) восстанавливают п-нитрофенильную группу с получением п-аминофенильной группы;
(b) деблокируют незащищённую амино)фенильную группу с получением п-аминофенильной группы;
(c) удаляют защитную группу, представленную символом Х с получением соединений формулы (I), в которой Х - водород; и/или
(d) ацилируют соединение формулы (I), в которой Х - водород, с получением соединения формулы (I), в которой Х - формил, ацетил, пропионил или N-метилкарбамоил; и, если необходимо, образуют фармацевтически приемлемые соли.
4. Способ по любому из пп. 1-3, отличающийся тем, что Z представляет атом галогена или органосульфонилоксигруппу, и циклизацию проводят в присутствии основания.
5. Способ по п. 4, отличающийся тем, что Z представляет собой (С1-С4)алкансульфонилоксигруппу, трифторметансульфонилокси или фенилсульфонилокси, в котором фенильная группа является незамещенной или замещенной одним или двумя заместителями, выбранными независимо из (С1-С4)алкила, (С1-С4)алкокси, галогена, нитро и галоид (С1-С4)алкила.
6. Способ по п. 5, отличающийся тем, что основание выбирают из гидроокисей щелочных металлов, карбонатов щелочных металлов, гидридов щелочных металлов и алкоголятов щелочных металлов.
7. Способ по любому из пп. 1-6, отличающийся тем, что циклизацию проводят при температуре в пределах от -30 до 100°С.
8. Способ по п. З, отличающийся тем, что соединение формулы (VIII), в которой Z представляет уходящий атом или группу, получают на месте из соответствующего соединения общей формулы (VIII), в которой Z представляет гидроксил, с помощью реакции с триарилфосфином в присутствии азодикарбоксилатного сложного эфира.
9. Способ по п. 8, отличающийся тем, что триарилфосфином является трифенилфосфин, а азодикарбоксилатным эфиром диэтилазодикарбоксилат.
10. Способ по любому из пп. 8 или 9, отличающийся тем, что циклизацию проводят при температуре в интервале от -30 до 100°С.
11. Соединение общей формулы (VIII):
в которой R представляет водород или (C1-C10)алкил;
Х представляет водород, (C1-C10)алкил, ацил, арил, карбоксил или его замещенное производное, или защитную группу; и
Z представляет гидроксильную группу или уходящий атом или группу;
или его соль.
12. Соединение по п. 11, в котором Аr представляет п-нитрофенил, п-аминофенил или п-(защищенный амино)фенил, или его соль.
13. Соединение по п. 12, в котором Z представляет гидрокси, Х представляет водород, формил, ацетил, пропионил или N-метилкарбамоил, и Аr представляет п-нитрофенил или п-аминофенил.
14. Соединение по п. 13, в котором Х представляет ацетил и Аr
представляет п-нитрофенил.
15. Соединение по п. 11, в котором R представляет метил, Z - атом галогена, (С1-С4)алкансульфонилокси, трифторметансульфонилокси или фенилсульфонилоксигруппу, в которой фенильная группа является незамещенной или замещенной одним или двумя заместителями, выбранными независимо из (С1-С4)алкила, (С1-С4)алкокси, галогена, нитро- и галоид (С1-С4)алкила;
Х представляет водород, формил, ацетил, пропионил или N-метилкарбамоил, и Аr представляет п-нитрофенил или п-аминофенил.
16. Соединение по п. 15, в котором Z представляет метансульфонилокси, Х представляет ацетил и Аr представляет п-нитрофенил.
17. Способ получения стереоселективных производных дигидро-2,3-бензодиазепина, имеющих общую формулу (I):
в которой R представляет водород или (C1-C10)алкил; и
Х представляет водород, (C1-C10)алкил, ацил, арил или карбоксил, или его замещенное производное, отличающийся тем, что процесс включает стадии:
(а) предоставления некоторого количества соединения, имеющего формулу (II):
(b) асимметрического восстановления соединения формулы (II) с получением соединения, имеющего формулу
(III):
(с) взаимодействия соединения формулы (III) с арилальдегидным соединением формулы Аr-СНО с получением изохроманового соединения, имеющего формулу (IV):
(d) взаимодействия соединения формулы (IV) с окисляющим агентом с получением соединения формулы (V):
(e) взаимодействия соединения формулы (V) с гидразидным производным формулы H2NNHX с получением соединения формулы (VI):
и (f) взаимодействия соединения формулы (VI) с (i) сульфонилгалогенидным реагентом и основанием для образования промежуточного сульфоната; или (ii) с помощью прямой циклизации по Мицунобу с получением соединения формулы (I).
18. Соединение, имеющее общую формулу (V):
в которой R представляет водород или (C1-C10)алкил.
19. Соединение по п. 18, в котором R представляет метил и Аr представляет 4-нитрофенил или 4-аминофенил.
20. Соединение по п.19, в котором Аr представляет 4-нитрофенил.
Текст
1. Способ получения стереоселективных производных дигидро-2,3-бензодиазепина, имеющих общую формулу (I): 2 UA 1 ДИГІДРО-2,3-БЕНЗОДІАЗЕПІНУ (19) ДЕРЖАВНИЙ ДЕПАРТАМЕНТ ІНТЕЛЕКТУАЛЬНОЇ ВЛАСНОСТІ (11) 3 41354 4 16. Соединение по п. 15, в котором Z представляили замещенной одним или двумя заместителями, ет метансульфонилокси, Х представляет ацетил и выбранными независимо из (С1-С4)алкила, (С1Аr представляет п-нитрофенил. С4)алкокси, галогена, нитро и галоид (С117. Способ получения стереоселективных произС4)алкила. водных дигидро-2,3-бензодиазепина, имеющих 6. Способ по п. 5, отличающийся тем, что оснообщую формулу (I): вание выбирают из гидроокисей щелочных металлов, карбонатов щелочных металлов, гидридов щелочных металлов и алкоголятов щелочных металлов. 7. Способ по любому из пп. 1-6, отличающийся тем, что циклизацию проводят при температуре в пределах от -30 до 100°С. 8. Способ по п. З, отличающийся тем, что соединение формулы (VIII), в которой Z представляет уходящий атом или группу, получают на месте из соответствующего соединения общей формулы в которой R представляет водород или (C1(VIII), в которой Z представляет гидроксил, с поC10)алкил; и мощью реакции с триарилфосфином в присутстХ представляет водород, (C1-C10)алкил, ацил, вии азодикарбоксилатного сложного эфира. арил или карбоксил, или его замещенное произ9. Способ по п. 8, отличающийся тем, что триаводное, отличающийся тем, что процесс включарилфосфином является трифенилфосфин, а азоет стадии: дикарбоксилатным эфиром диэтилазодикарбокси(а) предоставления некоторого количества соедилат. нения, имеющего формулу (II): 10. Способ по любому из пп. 8 или 9, отличающийся тем, что циклизацию проводят при температуре в интервале от -30 до 100°С. 11. Соединение общей формулы (VIII): (b) асимметрического восстановления соединения формулы (II) с получением соединения, имеющего формулу (III): в которой R представляет водород или (C1C10)алкил; Х представляет водород, (C1-C10)алкил, ацил, арил, карбоксил или его замещенное производное, или защитную группу; и Z представляет гидроксильную группу или уходящий атом или группу; или его соль. 12. Соединение по п. 11, в котором Аr представляет п-нитрофенил, п-аминофенил или п(защищенный амино)фенил, или его соль. 13. Соединение по п. 12, в котором Z представляет гидрокси, Х представляет водород, формил, ацетил, пропионил или N-метилкарбамоил, и Аr представляет п-нитрофенил или п-аминофенил. 14. Соединение по п. 13, в котором Х представляет ацетил и Аr представляет п-нитрофенил. 15. Соединение по п. 11, в котором R представляет метил, Z атом галогена, (С1С4)алкансульфонилокси, трифторметансульфонилокси или фенилсульфонилоксигруппу, в которой фенильная группа является незамещенной или замещенной одним или двумя заместителями, выбранными независимо из (С1-С4)алкила, (С1С4)алкокси, галогена, нитро- и галоид (С1С4)алкила; Х представляет водород, формил, ацетил, пропионил или N-метилкарбамоил, и Аr представляет п-нитрофенил или п-аминофенил. (с) взаимодействия соединения формулы (III) с арилальдегидным соединением формулы Аr-СНО с получением изохроманового соединения, имеющего формулу (IV): (d) взаимодействия соединения формулы (IV) с окисляющим агентом с получением соединения формулы (V): (e) взаимодействия соединения формулы (V) с гидразидным производным формулы H2NNHX с получением соединения формулы (VI): 2 5 41354 и (f) взаимодействия соединения формулы (VI) с (i) сульфонилгалогенидным реагентом и основанием для образования промежуточного сульфоната; или (ii) с помощью прямой циклизации по Мицунобу с получением соединения формулы (I). 18. Соединение, имеющее общую формулу (V): 6 в которой R представляет водород или (C1C10)алкил. 19. Соединение по п. 18, в котором R представляет метил и Аr представляет 4-нитрофенил или 4аминофенил. 20. Соединение по п.19, в котором Аr представляет 4-нитрофенил. R1 отсутствует, когда между N(3) и С(4) атомами существует двойная связь; R2 представляет (С1-С3)алкильную группу; или R1 и R2 вместе обозначают метиленовую группу, и между N(3) и С(4) атомами отсутствует двойная связь; R3 представляет водород или (С1С4)алифатическую ацильную группу; R4 представляет водород; (C1C6)алифатическую ацильную группу, необязательно замещенную метокси, циано, карбоксильной, амино, (С1-С4)алкиламино, ди((С1С4)алкил)амино, пирролидино, фталимидо или фенильной группой, или одним или более атомами галогена; а также бензоильную, пальмитоильную, циклопропанкарбонильную, (С1-С5)алкилкарбамоильную или фенилкарбамоильную группу; или пунктирные линии представляют валентные связи, которые необязательно присутствуют, при условии, что между N(3) и С(4) атомами двойная связь отсутствует, когда и R3 и R4 обозначает водород. В публикации международной патентной заявки №WO 92/11262 также раскрываются некоторые производные дигидро-2,3-бензодиазепина, оказывающие действие на центральную нервную систему, в особенности антидепрессивное и/или антипаркинсоновое действие. Эти соединения могут быть представлены общей формулой (А), приведенной выше, в которой R1 представляет водород, R2 представляет метил, Rа представляет водород или (С1-С4) алкильную группу, необязательно замещенную карбоксильной или (С2-С5) алкоксикарбонильной группой, R3 представляет водород и R4 представляет алифатическую (С1-С6)ацильную, бензоильную или фенилацетильную группу. В настоящее время известно, что соединения, раскрытые в патенте США №4853152, ЕР-А10492485 и WO 92/11262, являются сильными антагонистами рецепторов класса АМРА (альфаамино-3-гидрокси-5-метилизоксазол-4пропионовая кислота) центральной нервной системы млекопитающих. Они потенциально имеют широкое применение в качестве нейрозащитных агентов, особенно в качестве антиконвульсивных Данное изобретение относится к новому процессу синтеза некоторых производных дигидро2,3-бензодиазепина, и особенно применимо к процессу получения этих соединений с высокой энантимерной чистотой и выходами. Оно также относится к промежуточным соединениям, полезным в данном процессе. В патенте США №4835152 раскрывается, что соединение 1-(4-аминофенил)-4-метил-7,8метилендиокси—3,4-дигидро-5Н-2,3бензодиазепин обладает свойством оказывать действие на центральную нервную систему. Европейская патентная заявка, опубликованная под №ЕР-А1-0492485 раскрывает производные 2,3-бензодиазепина и другие дигидро-2,3бензодиазепиновые производные, оказывающие действие на центральную нервную систему, в особенности проявляющие активность расслаблять мышцы и противоконвульсивную активность. Соединения, описанные в ЕР-А1-0492485, представлены общей формулой: в которой Rа представляет (С1-С6) алифатическую ацильную группу, необязательно замещенную метокси, циано, карбоксильной, амино, (C1С4)алкиламино, ди(C1-С4)алкил)амино, пирролидино, фталимидо или фенильной группой или одним или более атомами галогена; или Rа представляет бензоильную, циклопропанкарбонильную, (С1-С5)алкилкарбамоильную или фенилкарбамоильную группу; или Rа отсутствует, когда между атомами N(3) и С(4) существует двойная связь; R1 обозначает водород; или 3 7 41354 8 агентов. Они, таким образом, могут быть, полезными при лечении эпилепсии, церебральной ишемии, травм головного и спинного мозга, эпилептических припадков, болезни Паркинсона и амиотрофического латерального склероза. Дигидро-2,3-бензодиазепины, описанные в патенте США №4835152, ЕР-А1-0492485 и WO 92/11262, имеют центр асимметрии в положении 4. В настоящее время известно, что (R) энантиомеры, например (R)-7-ацетил-5-(4-аминофенил)8,9-дигидро-8-метил-7Н-1,3-диоксоло[4,5-h[2,3]бензодиазепин, известный также как (R)-1-(4аминофенил)-3-ацетил-4-метил-7,8-метилендиокси-З,4-дигидро-5Н-2,3-бензодиазепин, являются более сильными, чем (S) энантиомеры. В патенте США №4835152, ЕР-А1-0492485 и WO 92/11262 описываются синтетические способы получения дигидро-2,3-бензодиазепинов. По этим способам дигидро-2,3-бензодиазепиновое кольцо образуется с помощью селективного восстановгде Z представляет уходящий атом или групления соответствующего 2,3-бензодиазепинового пу, такую как мезилат (метилсульфонат). Следует соединения с использованием неорганического иметь в виду, что R и фрагменты сохраняют одни или смешанного органическо-неорганического и те же значения на протяжении данного описаи/или комплекса гидрида металла, такого как борния. гидрид натрия с последующим, если необходимо, Соответственно, целью данного изобретения отделением оптически активных форм. является предоставление нового стереоселективВ настоящее время найден весьма оригиного процесса образования производных дигидрональный привлекательный способ стереоселек2,3-бензодиазепинов. тивного синтеза для получения (R) энантиомеров Еще одной целью является предоставление дигидро-2,З-бензодиазепинов, описанных в патенпроцесса получения производных дигидро-2,3те США №4835152, ЕР-А1-0492485, WO 92/11262, бензодиазепина, который является эффективным и некоторых других дигидро-2,3-бензодиазепинов. и экономичным в связи с тем, что достигаются Настоящее изобретение представляет способ высокие выхода и энантиомерная чистота с пополучения соединения, имеющего общую формумощью меньшего числа стадий и с меньшими залу: тратами, чем это было известно ранее. Еще одна цель заключается в предоставлении новых промежуточных соединений, образуемых в течение синтеза дигидро-2,3-бензодиазепинов. Другие цели станут очевидными после прочтения следующего ниже описания. Согласно одному аспекту настоящее изобретение предоставляет процесс получения соединения, имеющего общую формулу: в которой R представляет водород или (С1С10)алкил; Х представляет водород, (С1-С10)алкил, ацил, арил или карбоксил, или их замещенные производные. Во время данного процесса образуются новые и неочевидные промежуточные соединения, которые далее обеспечивают эффективный синтез целевых соединений. Некоторые из раскрытых новых промежуточных соединений включают полукетальные и гидразоновые соединения. в которой R представляет водород или (С1С10)алкил; и Х представляет водород, (С1-С10) алкил, ацил, арил, карбоксил; или их замещенные производные, или защитную группу, или их фармацевтически приемлемых солей, при этом данный процесс включает циклизацию соединения, имеющего общую формулу: 4 9 41354 10 1991. R предпочтительно представляет (С1-С3) алкильную группу, например метил. Примерами значений для Х являются водород, формил, ацетил, пропионил и метилкарбамоил. Х предпочтительно представляет (С1-С6) алифатическую ацильную группу, необязательно замещенную метокси, циано, карбоксильной, амино, в которой Z представляет уходящий атом или (С1-С4)алкиламино, ди((С1-С4)алкил)амино пиррогруппу, с получением соединения, имеющего облидино, фталимидо или фенильной группой, или щую формулу(I); после чего, если необходимо, одним или более галогенами; или R представляет превращение соединения формулы (I) в другое бензоильную, циклопропанкарбонильную, (С1-С5) соединение формулы (I) и/или образование фаралкилкарбамоильную или фенилкарбамоильную мацевтически приемлемой соли. группу. Было найдено, что соединения формулы (I) Уходящим или удаляемым атомом или групмогут получаться с высоким выходом и с высокой пой, представленными символом Z, могут быть, энантиомерной чистотой с помощью процесса например, атом галогена или органосульфонилоксогласно изобретению. сигруппа, или она может образовываться на месте Как он используется здесь, термин "(С1(ин ситу) из соответствующего соединения форС10)алкил" представляет прямую или разветвленмулы VIII, в которой Z представляет гидрокси. ную алкильную цепь, имеющую от одного до десяКонкретными значениями Z, когда он предти атомов углерода. Типичные (С1-С10) алкильные ставляет атом галогена, является хлор или бром. группы с прямой или разветвленной цепью вклюОрганосульфонилоксигруппой, представленчают метил, этил, н-пропил, изопропил, н-бутил, ной Z, может быть, например, (С1изобутил, втор-бутил, трет-бутил, н-пентил, изоС4)алкансульфонилоксигруппа, трифторметанпентил, н-гексил, 2-метил-пентил, н-октил, децил и сульфонилоксигруппа или фенилсульфонилоксианалогичные. Термин "(С1-С10)алкил" охватывает группа, в которой фенильная группа является несвоим значением и "(С1-С4)алкил" и "(С1-С6)алкил". замещенной или замещенной одним или двумя Термин "арил" обозначает ароматический заместителями, выбранными независимо из (С1фрагмент, такой как фенил, тиенил, фурил, пириС4)алкила, (С1-С4)алкокси, галогена, нитро и галодил, имидазолил, и полиядерные ароматические ид (С1-С4)алкила. Конкретными значениями Z явфрагменты, такие как нафтил, фталазинил, хиноляются метансульфонилокси, плил, флуоренил, антрацил и фенантренил. Тертолуолсульфонилокси и пмин "замещенный арил" представляет арильную нитрофенилсульфонилокси. группу, замещенную одним или большим числом Было найдено, что гораздо лучший выход пофрагментов, выбранных из группы, состоящей из лучается с помощью циклизации соединения галогена, гидрокси, циано, нитро, (С1-С6)алкила, формулы VIII, в которой Z представляет органо(С1-С4)алкокси, карбокси, ацетила, формила, карсульфонилоксигруппу, а не соединения формулы боксиметила, гидроксиметила, амино, аминометиVIII, в которой Z представляет атом галогена. ла или трифторметила. Примеры замещенных Когда Z представляет атом галогена или оргаарильных групп включают 4-метилфенил, 2носульфонилоксигруппу, циклизация предпочтиметилфенил, 4-метоксифенил, 4тельно проводится в присутствии основания, вы(изопропил)фенил, 4-циклопентилфенил, 4-(третбранного из гидроокисей щелочного металла, бутил)фенил, 4-ацетилфенил, 4например гидроокиси натрия или калия; карбонатрифторметилфенил, 4-хлорфенил, 2-бромфенил, тов щелочного металла, например карбонатов 3-иодфенил, 6-бромнафтил, 3,4-(метилнатрия или калия; гидридов щелочных металлов, ендиокси)фенил, инданил, 1,2,3,4например гидрида натрия или калия; алкоголятов тетрагидронафтил, и 1,2,4,4-тетраметил-1,2,3,4щелочных металлов, например трет-бутилата литетрагидронафтил. тия, натрия или калия. Процесс удобным образом Термин "ацил" означает водород, (С1-С6) алпроводится при температуре в интервале от кильную группу или гетероатом (например, азот, 30°C до 100°C, предпочтительно от 0° до 50°С. как в амидогруппе), присоединенные к карбонильПодходящие растворители включают алканолы, ной группе. Типичные ацильные группы включают такие как метанол или этанол, и простые эфиры, формил, ацетил, пропионил, бутанил, валерил, такие как тетрагидрофуран. гексанил карбамоил, N-метилкарбамоил и уреил. Соединение формулы VIII, в которой Z пред"Арил", используемый в формулах в данном ставляет уходящий атом или группу, может генеописании, представляет незамещенную или зарироваться на месте по реакции, например соедимещенную арильную группу. Примерами значений нения формулы VIII, в которой Z представляет для арила являются п-нитрофенил, п-аминофенил гидроксильную группу, с триарилфосфином в прии п-(замещенный амино)фенил, такой как п-((С1сутствии азидодикарбоксилатного эфира. ПредС6)алканоиламино)фенил, например ппочтительно триарилфосфином является трифеацетиламинофенил. Примеры подходящих защитнилфосфин, а азидодикарбоксилатным эфиром ных групп могут быть найдены в Mcomie, Protective диэтилазодикарбоксилат. Процесс удобно провоGroups in Organic Chemistry, Plenum Press, Н.-Й., дится при температуре в интервале от - 30° до 1973, и Greene и Wutz, Protecting Groups in Organic 100°C, предпочтительно от - 10° до 50°С. ПодхоSynthesis 2-е изд., Джон Вили энд Санз, Н.-Й., 5 11 41354 12 дящие растворители включают простые эфиры, ствии минеральной кислоты, например соляной такие как тетрагидрофуран. Следует понимать, кислоты. что в данном случае уходящей группой, представАцилирование соединения формулы (I), в коленной символом Z, является триарилфосфониторой Х представляет водород, для получения локсигруппа, такая как трифенилфосфонилокси. соединения формулы (I), в которой Способ согласно изобретению представляет Х представляет ацильную группу, такую как особый интерес для получения соединений форформил, ацетил, пропионил или Nмулы (I), в которой арил представляет пметилкарбамоил, может проводиться, как описано аминофенил. Такие соединения предпочтительно в ЕР-А1-492485. получаются с помощью циклизации соединения Соединения формулы VIII, в которой Z предобщей формулы VIII, в которой арил представляет ставляет гидроксильную группу или уходящий п-нитрофенил, п-аминофенил или п-(защищенный атом или группу, считаются новыми и предоставамино)фенил, после чего, если необходимо, осуляются в качестве дополнительного аспекта изоществляют: бретения. (a) восстановление п-нитрофенилового эфиСоединения общей формулы VIII могут полура, получая п-аминофениловый эфир, или чаться с помощью многостадийного процесса, (b) снятие защиты или деблокирование писходящего из производного метилендиоксифе(защищенный амино)-фенильной группы с полунилацетона. чением п-аминофенильной группы. Согласно еще одному аспекту, следовательВ соответствии с предпочтительным аспектом но, настоящее изобретение предоставляет пронастоящее изобретение предоставляет способ цесс получения соединения, имеющего общую получения соединения, имеющего общую формуформулу: лу (I), в которой R представляет метил и Х представляет водород, формил, ацетил, пропионил или N-метилкарбамоил или защитную группу, а арил представляет п-нитрофенил, п-аминофенил или п-(защищенный амино)фенил, после чего, если необходимо, осуществляют: (a) восстановление п-нитрофенильной группы с получением п-аминофенильной группы; (b) снятие защиты с п-(защищенный амив которой R представляет водород или (С1но)фенильной группы с получением пС10) алкил; и аминофенильной группы; Х представляет водород, (С1-С10)алкил, ацил, (c) удаление защитной группы, представленарил, карбоксил, или его замещенное производной символом X, для получения соединения форное; мулы (I), в которой Х представляет водород; и/или данный процесс включает стадии: (d) ацилирование соединения формулы (I), в а) предоставления некоторого количества сокоторой Х представляет водород, для получения единения, имеющего формулу: соединения формулы (I), в которой Х представляет формил, ацетил, пропионил, или Nметилкарбамоил; и, если необходимо, образование фармацевтически приемлемой соли. Нитрогруппа в п-нитрофенильной группе может восстанавливаться с помощью методов, известных в технике, например, как описано в ЕРА1-0492485. Так, она может восстанавливаться с помощью реакции с гидразином или гидразингидb)асимметричного восстановления соединератом в присутствии катализатора - никеля Ренея. ния формулы II, давая соединение формулы: Альтернативно, она может восстанавливаться по реакции с водородом, муравьиной кислотой, формиатом аммония, триалкиламмонийформиатом, таким как триэтиламмонийформиат, или формиатом щелочного металла, таким как формиат натрия или калия, в присутствии катализатора на основе металла группы УШ, такого как палладий с) взаимодействие соединения формулы III с на активированном угле. Подходящие раствориарилальдегидным соединением формулы Арил. тели включают спирты, такие как метанол, этанол СНО, с получением изохроманового соединения, или изопропанол, и простые эфиры, такие как тетимеющего формулу: рагидрофуран или ацетон. Восстановление может удобно проводиться при температуре в интервале от - 10° до 120°С. Защитная группа в п-(защищенный амино)фенильной группе может удаляться обычным образом. Например, (С1-С6)алканоильная группа может удаляться с помощью гидролиза в присут 6 13 41354 14 тельно энантиомерно чистым материалом. Это увеличивает как производительность, так и энантиомерную чистоту. Первая стадия процесса включает хиральное восстановление исходного материала (предпочтительно 3,4-метилендиоксифенилацетонового производного) с получением фактически энантиомерd) реакция соединения формулы IV с окисно чистого спиртового производного 1,2ляющим агентом для получения соединения форметилендиоксибензола. Предпочтительно обрамулы: зуемым энантиомером является (S) или (+) стереоизомер спирта. Наиболее предпочтительным исходным соединением является 3,4метилендиоксифенил-ацетон. Альтернативно, начальная стадия может включать сочетание галоидного производного 1,2метилендиоксибензола с энантиомерно обогащенным эпоксидом. Это также приводит в результате к получению высоко обогащенного в энане) взаимодействие соединения формулы V с тиомерном отношении спиртового производного гидразидным производным формулы Н2NNНХ для 1,2-метилендиоксибензола. получения соединения формулы: Материалом, используемым для проведения начальной стадии хирального восстановления, может быть или химический или, предпочтительно, биологический материал. В случае биологических агентов предпочтительными агентами являются восстанавливающие ферменты, наиболее предпочтительными являются дрожжи из группы Zygosaccharomyces. Другие биологические агенты, и f) взаимодействия соединения формулы VI с которые могут использоваться, включают: Pichia (і) сульфонилгалогенидным реагентом и основаfermentans, Endomycopsis fibuligera, Nematospora нием для образования промежуточного сульфонаcoryli, Saccharomyces sp, Candida famata, Sacта, с последующей реакцией полученного в реcharomyces pastorianus, Saccharomyces cerevisiae, зультате сульфоната с сильным основанием; или Saccharomyces uvarum, Candida utitis, Saccharo(іі) путем непосредственной циклизации по myces globosus, Kluyveromyces dobzhansk, KluyМицунобу для получения соединения формулы I. veromyces lactis, Candida albicans, пекарские Новый процесс данного изобретения предосдрожжи, Zydosaccharomyces rouxii, Zactobaillus тавляет синтез с помощью меньшего числа стаacidophilus, Aureobasidium pullulans, Mortierella дий, с получением более высоких выходов и стеisabellina, Rhizopus orysae, Kloeckeva javanika, Hinреоселективности, и не дает никаких тяжелых seniaspora valbyensis, Octosporomyces octospori, металлов, и приводит к очень незначительным Candida guilliermondi, Candida parapsilosis, Candida или вообще не дает почти никаких непроизводиtropicalis, Torulopsis taboadae, Torulopsis ethanoтельных затрат. Процесс предусматривает на litolerans, Torulopsis ptarmigani, Torulopsis sonorenранней стадии энантиоселективное восстановлеsis, Trigonopsis variabilis, Torulopsis enokii, Torulopние, во время которого стереохимия устанавливаsis methanothermo, быстро растворимые дрожжи ется в сторону предпочтительного изомера (в SAF, инакт. дрожжи ashland, Candida boidinii, Canданном случае (R) или (-) энантиомера конечного dida blankii и дрожжи Red star. продукта). Желательным промежуточным продуктом, обПредпочтительный процесс включает раннее разующимся на начальной стадии, является спирхиральное восстановление кетона в спирт. В мнотовое замещенное производное 1,2гостадийном процессе добавляются заместители метилендиоксибензола, причем наиболее преддля закрытия бензо-сконденсированного пиранопочтительное производное состоит из (S)-альфавого кольца перед тем, как вводится гидразиновый метил-1,3-бензодиоксол-5-этанола. реагент для открытия кольца и добавления необЖелательное промежуточное соединение, обходимых азотных компонентов. Наконец, вторичразовавшееся на начальной стадии, затем подное кольцо закрывается при добавлении сильного вергается реакции Пиктет-Спенглера, которая основания и соединение восстанавливается, обдает конвергентное слияние углеродных составразуя желаемое соединение. ных компонентов бензодиазепина. ПредпочтиНаиболее предпочтительно стадия хиральнотельным реагентом является пго восстановления является начальной стадией в нитробензальдегид, хотя могут использоваться и синтезе соединений формулы (I) из кетонов. Хидругие известные специалистам в данной области ральное восстановление может проводиться при реагенты, такие как ацетали. Предпочтительными использовании специфических химических вепромежуточными соединениями являются дигидществ, или, предпочтительно, с помощью испольробензопираны, причем наиболее предпочтительзования биологических агентов, описанных ниже. ным соединением является 7,8-дигидро-7-метилУстановление стереохимии на ранней стадии в 5-(4-нитрофенил)-5Н-1,3-диоксоло-бензо[b]пиран. процессе является благоприятным и позволяет Дигидробензопирановое производное затем более поздним стадиям осуществляться с относи 7 15 41354 16 окисляется в С-5 положении, давая полукетальное Сульфонат затем превращается в 8,9производное общей формулы: дигидро-7Н-2,3-бензодиазепиновое производное с помощью добавления сильного основания, наиболее предпочтительно - гидроокиси щелочного металла, такой как каустическая сода, алкоголята щелочного металла, такого как трет-бутилат натрия или калия, карбоната щелочного металла, такого как карбонат калия, или гидрида щелочного металла, такого как гидрид натрия. Необязательно реакция может проводиться в присутствии катализатора переноса фаз, такого как тетрабутиламмоПредпочтительные окисляющие агенты вклюнийбромид. чают перманганат калия, DDQ (2,3-дихлор-5,6Альтернативно, соединение формулы VI моциано-1,4-бензохинон) или другие, причем наибожет превращаться в соединение формулы VIII, в лее предпочтительным агентом является гидрокоторой Z представляет атом галогена, например окись натрия, диметилсульфоксид и сочетание с соединение формулы VI может подвергаться ревоздухом. акции с имидазолом, трифенилфосфином и броС-5 полукеталь затем подвергается реакции с мом, давая соединение формулы VIII, в которой Z гидразидным производным формулы H2NNНХ в представляет атом брома. Полученное соединеприсутствии кислоты для образования гидразононие формулы VIII может затем циклизоваться пового промежуточного соединения. На данной стасле той же процедуры, что используется в случае дии раскрывается бензопирановое кольцо, так что соединения формулы VIII, в которой Z представгидразоновый компонент становится связанным с ляет органосульфонилокси-группу. С-5 углеродом. Наиболее предпочтительным гидСовершенно удивительно было найдено, что разидом является уксусный гидразид и он предциклизация соединения формулы VIII, в которой Z почтительно подвергается реакции в дефлегмипредставляет органосульфонилоксигруппу, может рующем ароматическом или протонном проводиться с высоким выходом, с явно незначирастворителе, предпочтительным гидразоном явтельным, если оно и есть, элиминированием. Одляется соединение общей формулы: нако, в случае соединения формулы VIII, в которой Z представляет атом галогена, выход является значительно более низким, вследствие конкурентного элиминирования. Соответственно, предпочитается использование соединения формулы VIII, в которой Z представляет органосульфонилоксигруппу (соответствующую соединению формулы VII). Еще один метод включает циклизацию по Мицунобу, которая представляет одностадийный процесс, дающий п-нитрофенилбензодиазепиногде R представляет СН3, Х представляет ацевое промежуточное соединение. тил и арил представляет п-нитрофенил. Когда желательным является соединение Гидразоновое производное превращается в формулы I, в котором арил представляет пжелательное бензодиазепиновое кольцо через аминофенил, и получено соединение формулы IV, внутримолекулярное алкилирование. Это достив которой арил представляет п-нитрофенил, нитгается с помощью одного из нескольких возможрогруппа может восстанавливаться на любой станых методов. Первый метод включает добавление дии процесса. Предпочтительно, она восстанавсмеси сульфонилгалогенидного реагента формуливается после стадии процесса е) или f). лы YSO2Xa, в которой Xа представляет атом галоНитрогруппа может восстанавливаться с погена, такой как хлор, и в которой Y представляет мощью добавления газообразного водорода или органическую группу, такую как (С1-С4)алкил, источника водорода в присутствии катализатора. трифторметил или фенил, в которых фенильная Предпочтительным источником водорода являетгруппа является незамещенной или замещенной ся формиат калия или другая формиатная соль одним или двумя заместителями, выбранными (такая как формиат аммония), при этом предпочнезависимо из (С1-С4)алкила, (С1-С4)алкокси, галотительным катализатором является сочетание гена, нитро и галоид(С1-С4)алкила (например, меметалла палладия и активированного древесного тансульфонилхлорида) и основания, такого как угля. Стадия восстановления хорошо известна третичный амин (например, триэтиламин), с обраспециалистам в данной области техники. зованием сульфонатного промежуточного соедиПредпочтительные процессы могут быть сумнения формулы: мированы с помощью следующих ниже схем, с получением наиболее предпочтительного продукта. Схема I. 8 17 41354 18 Пример 1. Синтез (S)-альфа-метил-1,3бензодиоксол-5-этанола. 1 эквивалент 3,4-метилендиоксифенилацетона, 0,45 эквив. динатрийфосфата, 0,03 эквив. фосфорной кислоты, 12,5 объемов смолы AD-7 и 5,8 объемов воды смешивались вместе и перемешивались в течение 15-60 минут при 20°25°С. Добавлялось 2,27 эквив. глюкозы и Z rouxii ATCC 14462 добавлялся в количестве 1,5г влажной клеточной пасты на грамм кетона (это составляет 0,375г/г на сухой основе). Данная смесь разбавлялась водой до 25 объемов, а затем осторожно перемешивалась при 33°-35°C в течение 8-16 часов. Смесь фильтровалась на сите из нержавеющей стали размером 100 меш (примерно 150 микрон) и смола, которая оставалась на сите, промывалась 25 объемами воды, разделенными на 4 отдельные порции. Продукт, который адсорбировался смолой, затем десорбировался со смолы 25 объемами ацетона. Раствор ацетонпродукт затем подвергался паровой отгонке досуха под вакуумом, давая целевое промежуточное соединение в виде желтого масла средней вязкости. Выход на месте (ин ситу) был 97-100%, в то время как выход выделенного продукта составлял 85-90%. Активность составляла 80-95%, а ЕЕ 100%. Пример 2. Синтез (5RS,7S)-7,8-дигидро-7метил-6-(4-нитрофенил)-5Н-1,3-диоксоло-[4,5G][2]бензопирана. Указанное выше промежуточное соединение растворялось в 4,64 объемах толуола, фильтровалось через гифло и промывалось 1,55 объемами толуола. Добавлялось 1,05 эквив. п-нитробензальдегида и 1,05 эквив. концентрированной соляной кислоты, и смесь нагревалась до 55°C65°C и перемешивалась в течение 1 часа. Затем производился обмен растворителей, при 250мм рт.ст. толуол заменялся 12,4 объемами смеси 93% изопропанола/7% воды. Объем во время данной замены растворителя менялся от 11-14 объемов и конечный объем составлял примерно 11 объемов. Смесь охлаждалась до 0°C-10°C и перемешивалась в течение 2 часов. Продукт в виде иглообразных кристаллов отфильтровывался и промывался 2 раза 1,85 объема изопропанола и сушился под вакуумом при 50°-60°С. Выход ин ситу целевого соединения был 95%, в то время как выход выделенного продукта составлял 8793%. Активность составляла более 99%, а ЕЕ 100%. Пример 3. Альтернативный синтез (S)-альфаметил-1,З-бензодиоксол-5-этанола. 3,47г 4-бром-1,2-(метилендиокси)бензола растворялось в 100мл тетрагидрофурана при -78°С, затем добавлялось 13,9мл 1,3 М втор-бутиллития в циклогексане для потребления арилгалогенида менее, чем через 30 минут. С помощью шприца добавлялось 1,00г (S)-(-)-окиси пропилена в 2мл ТГФ, и раствор перемешивался в течение 45 минут. Раствор затем подогревался до 23°С в течение 16 часов. Реакционная смесь выливалась в 3 М раствор хлорида аммония и продукт выделялся с помощью экстракции этилацетатом. Объединенные экстракты сушились над сульфатом магния, фильтровались через флорисил и концентрирова На схеме I начальная стадия процесса включает добавление биологических агентов, наиболее предпочтительно Zygosaccharomyces rouxii, для восстановления кетона в желаемый спирт. Для предотвращения гибели организма или для адбсорбирования спирта, как только он образуется, к реакционной смеси может добавляться подходящее количество адсорбирующей смолы, такой как AD-7, XAD-7, HP2MGa (поперечно-сшитые полиметакрилаты фирмы Ром энд Хаас), НР20 (полистирольная) или SP207 (бромированный полистирол фирмы Мицунобу). Могут также использоваться другие сходные смолы. Схема П. На схеме (П) начальная стадия процесса включает взаимодействие арилгалогенидного производного, такого как 4-бром-1,2(метилендиокси)бензол, с углеводородным производным щелочного металла (предпочтительным является втор-бутиллитий) и энантиомерно чистым эпоксидом. Предпочтительным является (S)-()-пропиленоксид. Альтернативно, арилгалогенид может сначала превращаться в реагент Гриньяра по реакции с магнием, а затем вводиться в реакцию с энантиомерно чистым эпоксидом в присутствии иодида меди (1) в качестве катализатора. На обоих схемах (1) и (П), целью является как можно раньше установить стереохимию С-8 атома бензодиазепинового кольца. Было замечено, что обе эти схемы достигают данной цели, и образуются энантиомерно обогащенные (ее) спирты с 98% чистотой. Следующие примеры являются показательными примерами осуществления процесса данного изобретения. 9 19 41354 20 1 H ЯМР (СD Cl3, 300МГц): (главный изомер) лись с помощью роторного испарения. Остаточное 8,16(д, 2Н, J=6,9Гц), 7,73(д, 2Н, J=6,9Гц), 6,55(с, масло очищалось с помощью хроматографии на 1Н), 6,38(с, 1Н), 5,86(с, 1Н), 5,83 (с, 1Н), 4,38(м, силикагеле и элюировались смесью 50:50 гексана 1Н), 2,70(м, 2Н), 1,39(д, 3Н, J=6,3Гц), d (второстеи диэтилового эфира, давая 1,40г (45%) указаннопенный изомер) 8,27(д, 2Н, J=8,9Гц), 7,90(д, 2Н, го в заголовке промежуточного соединения. J=8,6Гц), 6,87(с, 1Н), 6,73(с, 1Н), 6,03(с, 1Н), Р chem.:(α)365+117,2°(c 1,0 СНСl3); 6,02(с, 1Н), 3,95(м, 1Н), 2,7(смазанный, м, 2Н), ТСХ Rf=0,26(50:50 гексан:эфир); 1,24(д, 3Н, J=6,1Гц); ИК (СНСl3):3598, 3012, 2973, 2887, 1490, 1249, масс. спектр, m/z (FD, M+) 329; 1041 см-1; 13 Анализ для C17H15NO6: С ЯМР (CD Cl3): д. 147,75, 146,19, 132,26, 122,27, 109,68, 108,30; Вычислено C 62,01 H 4,59 N 4,25 масс. спектр, m/z (FD, M+) 180. Найдено C 62,22 H 4,79 N 4,29 Анализ для С10Н12О3: Пример 6. Синтез (S)-уксусная кислота-[[6-(2Вычислено С 66,65 Н 6,71 гидроксипропил)-1,З-бензодиоксол-5-ил](4Найдено С 66,42 Н 6,66 нитрофенил)метилен]гидразида. К 350г влажного осадка с фильтра из примера Пример 4. Альтернативный синтез (5RS, 7S)5 в 2300мл этанола добавлялось 94,5г уксусного 7,8-дигидро-7-метил-5-(4-нитрофенил)-5Н-1,3гидразида и 1мл концентрированной соляной кидиоксоло[4,5-G][2]бензопирана. слоты. Получающийся раствор нагревался до 244г п-нитробензальдегида добавлялось к температуры дефлегмации в течение 2,5 часов. раствору 300г промежуточного соединения, обраСмесь охлаждалась до комнатной температуры и зовавшегося на стадии биокатализируемого восконцентрировалась до желтой пены с помощью становления примера 1, в 4,45л толуола. На пророторного испарения. Концентрат растворялся в тяжении 15-20 минут по каплям добавлялось 4,9л этилацетата и промывался 1,5л насыщенного 166,5мл концентрированной соляной кислоты и бикарбоната натрия, а затем 1,5л солевого располучающаяся в результате смесь нагревалась до твора. Органическая фаза сушилась над сульфа60°С в течение 2,5 часов. Смесь охлаждалась до том натрия, фильтровалась и концентрировалась, комнатной температуры и концентрировалась с давая 373г желтой пены (91%). Материал идентипомощью роторного испарения. Добавлялось 3л фицировался в виде 1:1 неразделимой смеси этанола и смесь концентрировалась до твердого изомеров указанного в заголовке соединения (97% вещества. Добавлялась еще 3л порция этанола и чистая по данным ВЭЖХ). Р chem: данные регистсмесь перемешивалась в течение 1 часа. Суспенрировались по 1:1 изомерной смеси; зия охлаждалась на протяжении ночи, и кристалТ.пл. 167,8°-169,7°С; лический продукт отделялся с помощью вакуумТСХ Rf=0,55(этилацетат); ной фильтрации. Осадок на фильтре промывался ИК (СНСl3): 3590, 3485, 3310, 1694, 1673, 1520, этанолом, а затем сушился в вакуумной печи при 1485, 1346 см-1; 40°-60°С, давая 450г (86%) не совсем белого 1 H ЯМР (СDСІ3, 300МГц): 8,64, 8,50(с, 1Н, NН), твердого вещества, которое по данным определе8,18(д, 2Н, Аr-Н), 7,74, 7,71(д, 2Н, J=8Гц, Ar-Н), ния представляло изомерную смесь названного 6,99, 6,95(с, 1Н, Ar-H), 6,52, 6,50(с, 1Н, Аr-Н), 6,06, выше в заголовке оптически активного промежу6,05(д, 2Н, J=5Гц, О2СH2), 2,44(с, 1Н, СН3), 3,87(м, точного соединения. 1Н, СН), 2,4-2,2(м, 2Н, СН2), 1,12, 1,10(д, 3Н, СН3); Р chem:(α)365+55° (c. 0,4, СНСl3). 13 С ЯМР (СDCl3, 75МГц): d 209,94(С), 173,38, Пример 5. Синтез (5RS, 7S)-7,8-дигидро-7173,43(С), 149,38, 149,62(С), 148,31, 148,58(С), метил-5-(4-нитрофенил)-5Н-1,3-диоксоло-[4,5147,90, 148,18(С),147,54(C),142,5, 143,04(C), G][2]бензопиран-5-ола. 132,64(C), 127,53, 127,61(CH), 123,75, 123,77(CH), 350г изомерного промежуточного соединения 122,86, 123,27(C), 112,13(CH), 110,55 (CH), 108,03, из примера 4 добавлялось к раствору 731мл ди108,10(CH), 108,03, 108,10(CH), 101,83(СН2), метилсульфоксида и 2923мл диметилформамида. 67,51, 68,08(CH), 42,37, 42,97(СН2), 23,48, Смесь охлаждалась до 8°-12°С и через смесь про23,83(СН3), 23,48, 23,83(CH3), 20,47, 20,55(СН3); пускался сжатый воздух. Добавлялось 117,5мл (α)365+103,8° (с 1, СНСІ3); 50% водной гидроокиси натрия в виде одной пормасс.спектр. m/z (FD, M+), 385; ции, и получающаяся в результате смесь перемеАнализ для C19H19C3O6: шивалась в течение 4,5 часов. Реакционная смесь Вычислено C 59,22 H 4,97 N 10,90 добавлялась с помощью трубочки на протяжении N 10,68 30-60 минут к 8,25л перемешиваемого раствора 1 Найдено C 58,99 H 5,04 Н соляной кислоты при 10°-15°C. Получающийся Пример 7. Синтез (S)-уксусной кислоты [[6-2осадок отфильтровывался и промывался 3л воды, [(метилсульфонил)окси]пропил]-1,3-бензодиоксолзатем подвергался воздушной сушке до постоян5-ил](4-нитрофенил)метилен]гидразида. ного веса (384г). Влажный осадок на фильтре пе340г промежуточного соединения из примера реносился на стадию примера 6 без дальнейшей 6 растворялось в 2380мл метиленхлорида. Рассушки. твор охлаждался до 0° - -10°С и добавлялось Р chem: данные регистрировались по 3:1 изо187мл триэтиламина. Затем добавлялось 78,2мл мерной смеси; метансульфонилхлорида, и получающаяся в реТСХ Rf=0,19 (75:25 гексан:этилацетат); зультате смесь перемешивалась в течение 15-30 ИК (СНСl3): 3605, 3590, 3015, 3000, 2960, 1608, минут. Добавлялось 510мл воды. Отделенная ор1522, 1484, 1352, 1240, 1042 см-1; ганическая фаза промывалась 460мл 1 Н раство 10 21 41354 22 медленно охлаждалась до комнатной температура соляной кислоты, а затем 500мл солевого расры. Продукт отделялся с помощью вакуумного твора. Метиленхлоридный раствор подогревался фильтрования и промывался 10-20мл воды. Отдо 35°-45°С и на протяжении 90 минут добавляделенное твердое вещество сушилось в вакууме лось 4760мл гексана. Смесь медленно охлаждадо 50°С, давая 4,17г (93%) названного в заголовке лась до комнатной температуры, а затем дополконечного соединения, которое было на 100% чиснительно охлаждалась до 0°-5°С. Продукт тым по данным анализа на содержание активного отделялся с помощью вакуумного фильтрования и вещества ВЭЖХ. сушился в вакуумной печи при 40°-50°С, давая (α)365=-303,7 (с=1, метанол). 356,2г (87%) изомерной смеси названного в загоПример 10. Синтез (5RS, 7)-7,8-дигидро-7ловке соединения в виде желтого твердого вещеметил-5-(4-нитрофенил-5Н-1,3-диоксоло[4,5ства. G][2]бензопиран-5-ола. Р chem: данные регистрировались по 3:1 изо15г промежуточного соединения из примера 4 мерной смеси; (получаемого по методу восстановления кетона с Т.пл. 150,5°-152,5°С; помощью Z rouxii) растворялось в растворе 75мл ТСХ Rf=0,80 и 0,73(этилацетат); диметилсульфоксида и 75мл диметилформамида. ИК (СНСl3): 1696, 1520, 1486, 1346, 1175, 1041, Раствор охлаждался до 7°-9°С, а затем аэриро923 см-1; 1 вался 40% кислородом в азоте. Добавлялось 7,62г H ЯМР (СDСІ3, 300МГц) δ:8,44(C, 1H, NH), 50% гидроокиси натрия в воде и получающаяся в 8,20(д, 2Н, J=8,8Гц, Ar-H), 7,73(д, 2Н, J=8,6Гц), результате смесь перемешивалась в течение 3-4 6,94(д, 1Н, J=2,7Гц, Аr-Н), 6,57(д, 1Н, =2,6Гц, Аr-Н), часов. Реакция заканчивалась и при поддержании 6,08(д, 2Н, J=5,4Гц), 4,77(м, 1Н, СН), 2,90(с, ЗН, температуры равной 12°С добавлялось 120мл СН3 основной или главный), 2,83(с, ЗН, СН3 втотолуола, а затем смесь 45мл воды и 10мл соляной ростепенный), 2,66-2,57(м, 2Н, СН2), 1,30(д, СН3 кислоты. Фазы разделялись и органический слой второстепенный), 1,26(д, ЗН, СН3 главный); промывался 75мл 10% водного раствора тиомасс.спектр, m/z (FD, M+) 385; сульфата натрия. Органический слой, содержаАнализ для С20Н21N3O8S: щий названное в заголовке промежуточное соедиВычислено C 51,83 H 4,57 N 9,07 S 6,92 нение, переносился на следующую стадию. Найдено C 52,05 H 4,53 N 8,84 S 6,96 Пример 11. Синтез (S)-уксусная кислота-[[6-(2Пример 8. Синтез (R)-7-ацетил-8,9-дигидро-8гидроксипропил)-1,3-бензодиоксол-5-ил](4метил-5-(4-нитрофенил)-7Н-1,3-диоксоло-[4,5нитрофенил)метилен гидразида. h][2,3]бензодиазепина. К толуольному раствору промежуточного со325г промежуточного соединения примера 7 единения из примера 10 добавлялось 4,26г уксусрастворялось в 3174мл метанола. К перемешиного гидразида и (0,01 объема) соляная кислота. ваемому раствору добавлялось 38,1мл 50% расПолучающаяся в результате смесь нагревалась твора каустической соды. К смеси добавлялось до кипения в течение 3,5 часов с удалением воды 6348мл воды и содержимое перемешивалось в с помощью ловушки Дина-Старка. Реакционная течение 3 часов, после чего получающийся в ресмесь концентрировалась с помощью вакуумной зультате осадок отделялся с помощью вакуумного перегонки до 1 объема. Концентрат разбавлялся фильтрования. Вещество сушилось в вакуумной 105мл метиленхлорида и промывался по 50-55мл печи при 45°-55°С, давая 255г (97%) названного в каждым из насыщенного раствора бикарбоната заголовке соединения, которое было 97,6% чистонатрия и солевого раствора. Органический расты по данным процента площади ВЭЖХ. 221г вытвор сушился над сульфатом магния (0,25вес.%) и сушенного материала дополнительно очищалось фильтровался через слой гифло. Фильтр пропос помощью ресуспендирования в 110,5мл этаноласкивался 1 объемом метиленхлорида. Объедила, который нагревался до температуры дефлегненная органическая фаза, содержащая названмации. Получающаяся в результате смесь охлажное в заголовке промежуточное соединение, далась до комнатной температуры и осадок переносилась на следующую стадию. отделялся, с помощью вакуумного фильтрования. Пример 12. Синтез (S)-уксусная кислота[[6[2Изолят сушился в вакуумной печи при 45°-55°С, [(метилсульфонил)окси]пропил]-1,3-бензодиоксолдавая 199г (90%) указанного в заголовке соедине5-ил](4-нитрофенил)метилен]гидразида. ния, которое имело 100% чистоту по данным анаМетиленхлоридный раствор, содержащий лиза ВЭЖХ. промежуточное соединение примера 11, охлажПример 9. Синтез (R)-7-ацетил-5-(4дался до 0°- -5°С и добавлялось 10мл триэтилааминофенил)-8,9-дигидро-8-метил-7Н-1,3мина. диоксоло-[4,5-h][2,3]бензодиазепина. Медленно добавлялось 4,1мл метансульфоК 5г промежуточного соединения из примера 8 нилхлорида для поддержания температуры реакв 50мл этанола добавлялось 0,5г 10% Рd/c, увции равной 0°С. К получающемуся раствору долажненного водой. Перемешиваемая взбалтывабавлялось 1,5 объема воды. Органическая фаза нием суспензия обрабатывалась раствором 4г отделялась и промывалась 2,5 объемами 1 Н расформиата калия в 4мл воды. Получающаяся в твора соляной кислоты. Органическая фаза отдерезультате смесь перемешивалась в течение 2,5 лялась и концентрировалась до половины первочасов, а затем фильтровалась через Гифло. начального объема с помощью перегонки при Фильтрат концентрировался до 10-20мл с помоатмосферном давлении. Продукт осаждался дощью перегонки и к теплому (78°С) раствору медбавлением по каплям гептана (2:1 объема гептана ленно добавлялось 22мл воды. Получающаяся в к органическому концентрату) к раствору при 45°С. результате смесь нагревалась до 90°С и затем 11 23 41354 24 Перемешиваемая смесь охлаждалась до 20°-25°С 98,3% чистым. в течение 1 часа, затем охлаждалась до 0°- -5°С в Примеры 16-18 течение 1-2 часов. Осадок отделялся с помощью 0,5мл суспензии замороженных дрожжей, совакуумной фильтрации и промывался 3 объемами держащей микроорганизм, указанный в таблице 1, смеси 4:1 гептан:метиленхлорид; затем сушился в добавлялось к 50мл солодово-дрожжевой среды в вакуумной печи при 45°-55°С. Получалось 17,43г 250мл колбе. После 48 часового встряхивания указанного в заголовке промежуточного соедине1,0мл культуры добавлялся к дополнительным ния (78%) в виде оптически активной смеси изо50мл среды и смесь встряхивалась в течение еще меров гидразона, которая имела 97,7% чистоту по 48 часов. Добавляется 3,4данным анализа содержания активного вещества метилендиоксифенилацетон до тех пор, пока кос помощью ВЭЖХ. нечная концентрация не будет 10г/л наряду с 1мл Пример 13. Синтез (R)-7-ацетил-8,9-дигидро10% глюкозы. Культуры инкубируются и встряхи8-метил-5-(4-нитрофенил)-7Н-1,3-диоксоло-[4,5ваются в течение 24 часов, затем анализируются H][2,3]бензодиазепина. с помощью ВЭЖХ на присутствие хирального 17,5г промежуточного соединения из примера спиртового промежуточного продукта примера 1. 12 суспендировалось в 175мл этилового спирта. К перемешиваемой смеси добавлялось 1,7г порошкообразной гидроокиси натрия. Получающаяся смесь перемешивалась в течение 1 часа. К смеси добавлялось 88мл воды, и содержимое перемешивалось в течение 1 часа, после чего получающийся в результате осадок отделялся с помощью вакуумного фильтрования и промывался 175мл воды. Вещество сушилось в вакуумной печи при 70°С, давая 12,2г (86%) названного в заголовке соединения, которое имело 99,9% чистоту по данПример 19. Синтез (S)уксусная кислота[[6-[2ным анализа активности с помощью ВЭЖХ. (метилсульфонил)окси]пропил-1,3-бензодиоксолПример 14. Синтез (R)-7-ацетил-5-(45-ил](4-аминофенил)-метилен]гидразина. аминофенил)-8,9-дигидро-8-метил-7Н-1,3К суспензии промежуточного соединения из диоксоло-[4,5-h][2,3]бензодиазепина. примера 7 (5,00г) в 100мл изопропилового спирта При использовании продукта примера 13 цедобавлялся 10% Рd/c (1г), а затем формиат калия левое соединение получалось с помощью той же (3,7г), растворенный в 8мл воды. Спустя 1/5 часа, самой прцедуры эксперимента, что описана в добавлялась еще одна порция формиата калия примере 9. (3,7г) с последующим добавлением 10% Pd/c(1г). Пример 15. (R)-7-ацетил-8,9-дигидро-8-метилИсходное вещество потреблялось через 30 минут. 5-(4-нитрофенил)-7Н-1,3-диоксоло-[4,5Смесь фильтровалась через слой диатомовой h][2,3]бeнзoдиaзeпин. земли и концентрировалась, остаток растворялся 1,05г (S)-уксусная кислота [[6-[2-[гидрокси пров метиленхлориде и промывался водой и солевым пил]-1,3-бензодиоксол-5-ил](4раствором. Органический раствор сушился над нитрофенил)метилен]гидразида и 0,78г трифесульфатом натрия, фильтровался и концентриронилфосфина в 70мл тетрагидрофурана охлаждавался. Целевое соединение (4,52г) отделялось в лись до 0°С. По каплям на протяжении 15 минут виде светло-желтого твердого вещества с выходобавлялось 0,57г диэтилазодикарбоксилата в дом 97% в виде 1:1,3 изомерной смеси. 5мл тетрагидрофурана. Получающаяся смесь пеДанные, зарегистрированные по 1:1,3 изомерремешивалась в течение 2 часов, затем подогреной смеси. валась до комнатной температуры в течение 2 ТСХ Rf = 0,83(ацетонитрил); часов. Смесь переносилась в делительную воронИК (СНСl3): 3010, 1670, 1628, 1332, 1174, 1041, ку и раствор промывался 1 Н НСl, водой и соле922 см-1; вым раствором. Органическая фаза сушилась над 1 Н ЯМР (СDСІ3, 300МГц) d 8,18(д, 1Н, J=9,2), сульфатом магния, фильтровалась и концентри7,39(д, 2Н, J=11,4), 7,38(д, 2Н, J=10), 6,91(с, 1Н), ровалась с помощью роторного испарения. Оста6,89(с, 1Н), 6,62(д, 2Н, J=8,5), 6,61(д, 2Н, J=8,1), ток элюировался на силикагельной колонке (1:1 6,57(с, 1Н), 6,56(с, 1Н), 6,06(м, 4Н), 4,71(секст., 2Н, этилацетат:гексан). Фракции, содержащие желаеJ=7), 3,9(шир.с, 4Н), 2,86(с, 3Н), 2,78(с, 3Н), 2,74мое соединение, концентрировались до желтого 2,49(м, 4Н), 1,29(д, 3Н, J=10,8), 1,25(д, 3Н, J=10,8); масла, которое затвердевало при стоянии. Желмасс.спектр, m/z (FD, M+) 433; тое кристаллическое вещество суспендировалось УФ макс. (этанол) 326нм (E 20767), в 30мл метиленхлорида и гексана (3:7) при 0°С. 231(17587), 205(42765). Осадок удалялся с помощью фильтрования и Пример 20. Синтез (R)-7-ацетил-5-(4фильтрат концентрировался до желтой пены. Осаминофенил)-8,9-дигидро-8-метил-7Н-1,3таток суспендировался в 10мл этанола, который диоксоло-[4,5-h][2,3]бензодиазепина. подогревался до дефлегмации, затем медленно К раствору промежуточного соединения из охлаждался до комнатной температуры. Осадок примера 19 (0,51г) в 6мл ТГФ добавлялась одна собирался с помощью фильтрования и сушился в порция трет-бутилата лития (0,17г). Раствор певакуумной печи при 60°С, давая 0,51г (50%) укаремешивался при температуре окружающей срезанного в заголовке продукта (100% ее), который ды в течение 2 часов, затем подогревался до 40°был по данным ВЭЖХ анализа активности на 12 25 41354 26 мера, 46,2%+53,1%. МС (FВ+)=400; 50°С в течение 4 часов. Реакционная смесь гасиИК: 1692, 1345 см-1; лась добавлением 10мл 3 М хлорида аммония. 1 H ЯМР (DМСО) δ: 0,90 (т, 3, J=6), 2,22(м, 2), Получающаяся в результате смесь разбавлялась 2,72(д, 3, J=5), 3,63(м, 1), 4,42(д, 1/2, J=6), 4,58(д, 15мл эфира и промывалась 15мл воды и 15мл 1/2, J=6), 6,10(с, 2), 6,69(д, 1, J=8), 7,05(д, 1, J=10), солевого раствора. Органический раствор сушил7,32(шир.т, 1, J=4), 7,85(д, 1, J=9), 8,18(д, 2, J=9), ся над сульфатом натрия, фильтровался и кон8,62(д, 1, J=8); центрировался. Остаток растворялся в 5-10мл 13 С ЯМР (DМСО) δ: 24,27, 24,51, 27,18, 43,30, теплого метиленхлорида и продукт осаждался при 43,73, 67,09, 67,47, 102,45, 108,95, 109,19, 111,50, добавлении 10-20мл эфира. Продукт отделялся с 111,54, 112,41, 112,45, 124,40, 124,48, 124,69, помощью фильтрования, повторно растворялся в 128,35, 133,47, 133,69, 144,55, 144,61, 144,68, этиловом спирте и концентрировался. Целевое 144,79, 147,71, 147,76, 148,06, 149,39, 149,54, соединение (0,23г) отделялось с выходом 58% и 156,11, 156,23; 100% ее (по данным ВЭЖХ хирального анализа). Анализ для C19H20N4O6: Пример 21. Метилсемикарбазид. Вычислено С 57,00 Н 5,03 N 13,99 В 200мл этанола, охлажденного до 2,5°С, а атмосфере азота растворялось 17мл Найдено С 57,72 Н 5,01 N 13,99 (350,4ммоля) моногидрата гидразина. К данному Пример 23. Синтез (S)-N-метил-2-[[6-[2холодному перемешивающемуся раствору добав[(метилсульфонил)окси]пропил]-1,3-бензодиоксоллялся по каплям на протяжении 2 часов раствор 5-ил](4-нитрофенил)10,3мл (174,6ммоля) метилизоцианата в 150мл метилен]гидразинкарбоксамида. толуола при поддержании внутренней температуВ колбе в атмосфере азота в 120мл ТГФ сусры менее 6°С. Прозрачный бесцветный раствор пендировалось 2,00г (5,00ммолей) промежуточноперемешивался в течение 30 минут при 5°-10°С, го соединения примера 22. Смесь нагревалась затем выпаривался и сушился под вакуумом, даслегка для растворения твердых веществ, затем вая 15,19г белого твердого вещества. Твердые медленно охлаждалась до температуры окрувещества перемешивались в 40мл толуола и нажающей среды без осаждения. К желтому раствогревались до кипения, давая мутный раствор. Пору добавлялось 1,1мл (7,89ммоля) триэтиламина. сле медленного охлаждения до комнатной темпеРаствор затем охлаждался на бане из смеси лературы и перемешивания в течение 2 часов дяной воды и NаСl и добавлялось 500мкл суспензия фильтровалась через грубого помола (6,34ммоля) метансульфонилхлорида. Спустя 30 стекло. Твердые вещества промывались гексаном минут ВЭЖХ показал завершение реакции, 99,0% и сушились под вакуумом при 50°С, давая 13,39г мезилата. Реакционная смесь гасилась 50мл воды (86,4%) целевого соединения в виде белого крии переносилась в делительную воронку с 100мл сталлического твердого вещества. этилацетата. Органический слой промывался 1 Н Т.пл. 116°С; НСl (50мл) и солевым раствором (50мл), затем МС (FD+)=89; сушился над сульфатом натрия. Растворитель ИК: 3362, 3303, 1630, 1561 см-1; выпаривался, давая 2,56г сырого целевого соеди1 H ЯМР (DМСО) δ: 2,57(д, 3, J=4), 4,06(шир.с, нения в виде смеси желтого твердого вещест2), 6,27(шир.с, 1), 6,94(шир.с, 1); ва/пены. Сырое или неочищенное целевое соеди13 С ЯМР (DМСО) δ: 160,87, 25,89. нение растворялось в 12мл метиленхлорида и Пример 22. Синтез (S)-2-[[6-(2раствор нагревался до кипения с обратным хологидроксипропил)-1,3-бензодиоксол-5-ил](4дильником. К раствору добавлялось 6мл гексанов нитрофенил)-метилен]-Nпо каплям, что вызывало осаждение желтых тверметилгидразинкарбоксамида. дых веществ. Смесь медленно охлаждалась до В колбе в атмосфере азота растворялось температуры окружающей среды при перемеши5,17г (15,70ммоля) промежуточного продукта привании. После нахождения в течение 1 часа при мера 5 в 30мл этанола. К данному перемешиваетемпературе окружающей среды, смесь фильтромому раствору добавлялось 1,75г (19,7ммоля) валась через грубый стеклянный фильтр и тверметилсемикарбазида. Смесь нагревалась до темдые вещества промывались гексанами. После пературы дефлегмации для растворения твердых сушки при 50°С и 30" Нg, собиралось целевое совеществ, а затем добавлялось 5 капель конценединение в виде 2,19г (91,6%) желтых кристаллов, трированной НСl. После 1 часа нагревания с обт.пл. 164°С. ратным холодильником желтое твердое вещество МС (FD)=478; выпадало в осадок из раствора. Спустя 1 час анаИК: 1696, 1346см-1; 1 лиз ВЭЖХ показал завершение реакции, 38,2% и H ЯМР (DМСО) δ: 1,12 и 1,19(д, 3, J=6), 55,4% двух изомеров продукта и полное отсутст2,52(м, 2), 2,73(д, 3, J=3), 2,98 т 3,03(с, 3), 4,76 и вие какого-либо остатка исходного материала. 4,84(кв., 1,J=6, 12), 6,13(c, 2), 6,74 и 6,78(с, 1), 7,16 Желтая суспензия медленно охлаждалась до теми 7,20(с, 1), 7,33(шир.т, 1, J=5), 7,86(д, 2, J=9), 8,18 пературы окружающей среды и перемешивалась в и 8,22 (д, 2, J=9), 8,76(с, 1); 13 течение 1 часа, а затем перемешивалась в течеС ЯМР (DMCO) δ: 21,35, 21,47, 27,09, 38,55, ние 30 минут на бане из ледяной воды. Смесь 79,50, 79,91, 102,56, 109,34, 109,46, 111,34, 111,37, фильтровали через грубое стекло. Твердые веще111,72, 111,74, 124,37, 124,42, 124,86,128,26, ства промывались этанолом и сушились под ва128,36, 130,01, 130,14, 143,74, 143,81, 144,22, куумом при 50°С, давая 5,08г (81,2%) целевого 144,32, 147,93, 147,98, 148,19, 148,25, 149,65, соединения в виде желтого твердого вещества. 155,97; Т.пл. 238°С. Анализ ВЭЖХ показал два изоАнализ для C20H22N4O8S: 13 27 41354 28 торый охлаждался до 0°С на бане из ледяной воВычислено С 50,21 Н 4,63 N 11,71 ды. После охлаждения до 0OC добавлялось 0,414г Найдено С 50,46 Н 4,71 N 11,65 (2,59ммоля) брома. Получающийся в результате Пример 24. Синтез (R)-7-N-метилкарбамоилраствор перемешивался в течение 90 минут при 8,9-дигидро-8-метил-5-(4-нитрофенил)-7Н-1,30°С, в течение которых наблюдалось, что он стадиоксоло[4,5-h][2,3]бензодиазепина. новился слегка мутным. Реакционная смесь затем В колбе в атмосфере азота в 40мл ТГФ сусгасилась добавлением 6 мл 1 Н НСl. Смесь перепендировалось 1,50г промежуточного соединения носилась в делительную воронку, где она промыиз примера 23. После перемешивания желтая валась дважды 20мл 1 Н НСl, а затем промывасмесь охлаждалась на бане из смеси ацетона и лась 10мл насыщенного солевого раствора. льда и добавлялось 276мг (3,45ммоля) третОрганический раствор сушился над сульфатом бутилата лития. После перемешивания в течение натрия и выпаривался до красно-коричневого ка1 часа анализ ВЭЖХ мутной оранжево-красной менеобразного твердого вещества. Хроматограсмеси показал 96,5% желаемого соединения и фия на силикагеле с использованием 2:1 смеси лишь 3,1% оставшегося исходного материала. этилацетата и гексанов давала 0,71г (78%) целеСпустя 90 минут реакционная смесь гасилась 5мл вого соединения. В качестве загрязняющей принасыщенного водного хлористого аммония и меси от реакции элиминирования брома присутстсмесь переносилась в делительную воронку с 5мл вовало около 5% стирола. Наблюдалась смесь воды и 60мл метиленхлорида. Желтый органичегидразоновых изомеров с двойной связью и амидский слой промывался 20мл 1 Н НСl, насыщенным ных ротомеров, что осложняло 1Н ЯМР спектр. водным бикарбонатом натрия и солевым раство1 Н ЯМР (СDСІ3): 1,39 и 1,44, и 1,58 и 1,65(д, 3, ром, затем сушился над сульфатом натрия. РасJ=6), 2,48(с 3), 2,50-2,85(м, 2), 3,85-4,15(м, 1), творитель удаляли путем выпаривания, давая 6,09(с, 1), 6,11(с, 1), 6,55(с, 1), 6,94 (c, 1), 7,73 и 1,29г сырого целевого соединения в виде смеси из 7,80(д, 2, J=9), 8,19 и 8,20(д, 2, J=9), 8,37 и 8,45(с, желтого твердого вещества и пены. 1); МС (FD+)=382,2; MC (FD+) М+=450, наблюдаемый для Анализ для C19H18N4O5: C19H18N3O5Br. Вычислено С 59,68 Н 4,74 N 14,65 Пример 27. Синтез 7-ацетил-8,9-дигидро-8Найдено С 60,00 Н 5,13 N 13,75 метил-5-(4-нитрофенил)-7Н-1,3-диоксоло[4,5Пример 25. Синтез (R)-5-(4-аминофенил)-8,9H][2,3]бензодиазепина. дигидро-8-метил-7-N-метилкарбамоил-7Н-1,30,100г (0,22ммоля) промежуточного соединедиоксоло[4,5-h][2,3]-бензодиазепина. ния из примера 26 растворялось в 2мл сухого ТГФ В колбе в атмосфере азота в 9мл этанола и охлаждалось до 0°С на бане из ледяной воды в растворялось 902мг (2,36ммоля) промежуточного атмосфере азота. Затем добавлялось 0,018г соединения из примера 24. К данному раствору (0,22ммоля) трет-бутилата лития. Смесь перемедобавлялось 90мг 10% Рd/c с последующим дошивалась при 0°С в течение 2 часов, в течение бавлением раствора 690мг (8,20ммолей) формиакоторых за ходом реакции следили с помощью та калия в 0,7мл воды. Формиатный раствор доВЭЖХ. Поскольку ВЭЖХ обнаруживал, что обрабавлялся с помощью пипетки на протяжении зовалось приблизительно 10% продукта и все еще примерно 30 секунд, и инициировалась изотероставалось значительное количество исходного мия, которая достигала 53°С через 2-3 минуты. материала, колба закрывалась стеклянной крышСпустя 15 минут ВЭЖХ анализ аликвоты реакцикой и помещалась в морозильник при -35°С на 3 онной смеси показал только желаемый продукт. дня. После дополнительного нахождения в течеЧерная реакционная смесь фильтровалась через ние 8 часов при комнатной температуре ВЭЖХ смоченный этанолом фильтр из целита на микропоказал наличие 16% желаемого продукта. Реакволокнистой бумаге, и фильтр промывался этаноционная смесь гасилась 1мл 50% насыщенного лом. Фильтрат выпаривался, давая 953мг сырого водного раствора хлористого аммония и переноцелевого соединения в виде светло-желтых тверсилась в делительную воронку с 10мл метиленхдых веществ. После попытки перекристаллизации лорида. Смесь промывалась дважды 10мл 1 Н из водного этанола материал распределялся меНСІ, а затем 10мл насыщенного солевого раствожду водой и этилацетатом. Органический слой ра. Органический раствор сушился над сульфатом промывался солевым раствором и сушился над натрия и выпаривался до желто-коричневого смосульфатом натрия. Растворитель выпаривался и лообразного твердого вещества. Выход 0,05г. Жеэтанол отгонялся с паром из продукта несколько лаемый продукт образовывался с выходом около раз, давая 647мг (97%) целевого соединения в 15%, о чем судили по сравнению 1Н ЯМР спектра виде от светло-рыжеватокоричневого до желтого и следов ВЭЖХ с данными аутентичного продукта. твердого вещества, т.пл.=142,4°С. По данным Основной продукт получался в результате элимианализа хиральной ВЭЖХ определено 99,5% ее. нирования в соответствующее стирольное произПример 26. Синтез (S)-уксусная кислота[[6-(2водное. бромпропил)-1,3-бензодиоксол-5-ил]-(4Пример 28. Альтернативный синтез (S)нитрофенил)метилен]гидразида. альфа-метил-1,3-бензодиоксол-5-этанола. 1,0г (2,59ммоля) промежуточного соединения К суспензии магниевой стружки (17г) в 50мл примера 6, 0,265г (3,89ммоля) имидазола и 0,849г тетрагидрофурана добавлялся по каплям раствор (3,24ммоля) трифенилфосфина объединялись в 5-бром-1,3-бензодиоксола (93,6г). После полного 10мл метиленхлорида при температуре окружаюдобавления смесь разбавлялась 250мл тетрагидщей среды, давая золотисто-желтый раствор, корофурана и получающаяся смесь перемешива 14 29 41354 30 лась на протяжении ночи. 13мл раствора (0,78М) Пример 29. Синтез (R)-7-ацетил-8,9-дигидропереносилось в круглодонную колбу, содержащую 8-метил-5-(4-нитрофенил)-7Н-1,3-диоксоло[4,5иодистую медь(1) (0,12г). Получающаяся в реH][2,3]бензодиазепина. зультате смесь охлаждалась до -50°С и медленно К суспензии промежуточного соединения из добавлялся раствор (S)-(-)-окиси пропилена в 3мл примера 7 (1,53) в 60мл толуола добавлялись тетрагидрофурана, а затем смесь перемешива10мл 1 Н гидроокиси натрия и тетрабутиламмолась 10 минут. Смесь разбавлялась эфиром. Отнийбромида (0,053г). Получающаяся смесь переделенная органическая фаза промывалась водой мешивалась энергично в течение 72 часов. Смесь и солевым раствором. Водные смывки экстрагипромывалась солевым раствором и органическая ровались эфиром (2х) и объединенные органичефаза сушилась над сульфатом магния, фильтроские растворы сушились над сульфатом магния, валась и концентрировалась с помощью роторнофильтровались и концентрировались. Остаток го испарения. Остаток растворялся в этаноле и очищался с помощью хроматографии на силикаконцентрировался досуха, давая 1,05г целевого геле (50% эфира в пентане), давая 1,66г желаемосоединения (86%). Анализ ВЭЖХ показал, что го продукта (91%). Анализ хиральной ВЭЖХ покаобразовалось менее 0,5% соответствующего прозал, что оптическая чистота материала была дукта элиминирования. 98,3%. ____________________________________________________ ДП "Український інститут промислової власності" (Укрпатент) Україна, 01133, Київ-133, бульв. Лесі Українки, 26 (044) 295-81-42, 295-61-97 __________________________________________________________ Підписано до друку ________ 2001 р. Формат 60х84 1/8. Обсяг ______ обл.-вид. арк. Тираж 50 прим. Зам._______ __________________________________________________________ УкрІНТЕІ, 03680, Київ-39 МСП, вул. Горького, 180. (044) 268-25-22 __________________________________________________________ 15
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for producing stereoselective derivatives of dihydro-2,3-benzodiazepine (variants) and intermediary compounds for producing thereof
Автори англійськоюAnderson Benjamin Alan, Hansen Marvin Martin, Vicenzi Jeffry Thomas, Very David Li, Zmievski Melton Foseph
Назва патенту російськоюСпособ получения стереоселективных производных дигидро-2,3-бензодиазепина (варианты) и промежуточные соединения для их получения
Автори російськоюАндерсон Бенджамин Алан, Хансен Марвин Мартин, Виченци Джеффри Томас, Вери Дэвид Ли, Змиевски Мелтон Джозеф (мл.)
МПК / Мітки
МПК: C07D 491/056, A61P 25/00, C07D 317/58, A61K 31/55, C07D 493/04, C07D 491/04
Мітки: варіанти, одержання, стереоселективних, дигідро-2,3-бензодіазепіну, сполуки, проміжні, похідних, спосіб
Код посилання
<a href="https://ua.patents.su/15-41354-sposib-oderzhannya-stereoselektivnikh-pokhidnikh-digidro-23-benzodiazepinu-varianti-ta-promizhni-spoluki-dlya-kh-oderzhannya.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання стереоселективних похідних дигідро-2,3-бензодіазепіну (варіанти) та проміжні сполуки для їх одержання</a>
Спосіб одержання похідних хінолінкарбонової кислоти або її фармацевтично придатних солей та проміжні сполуки для їх одержання
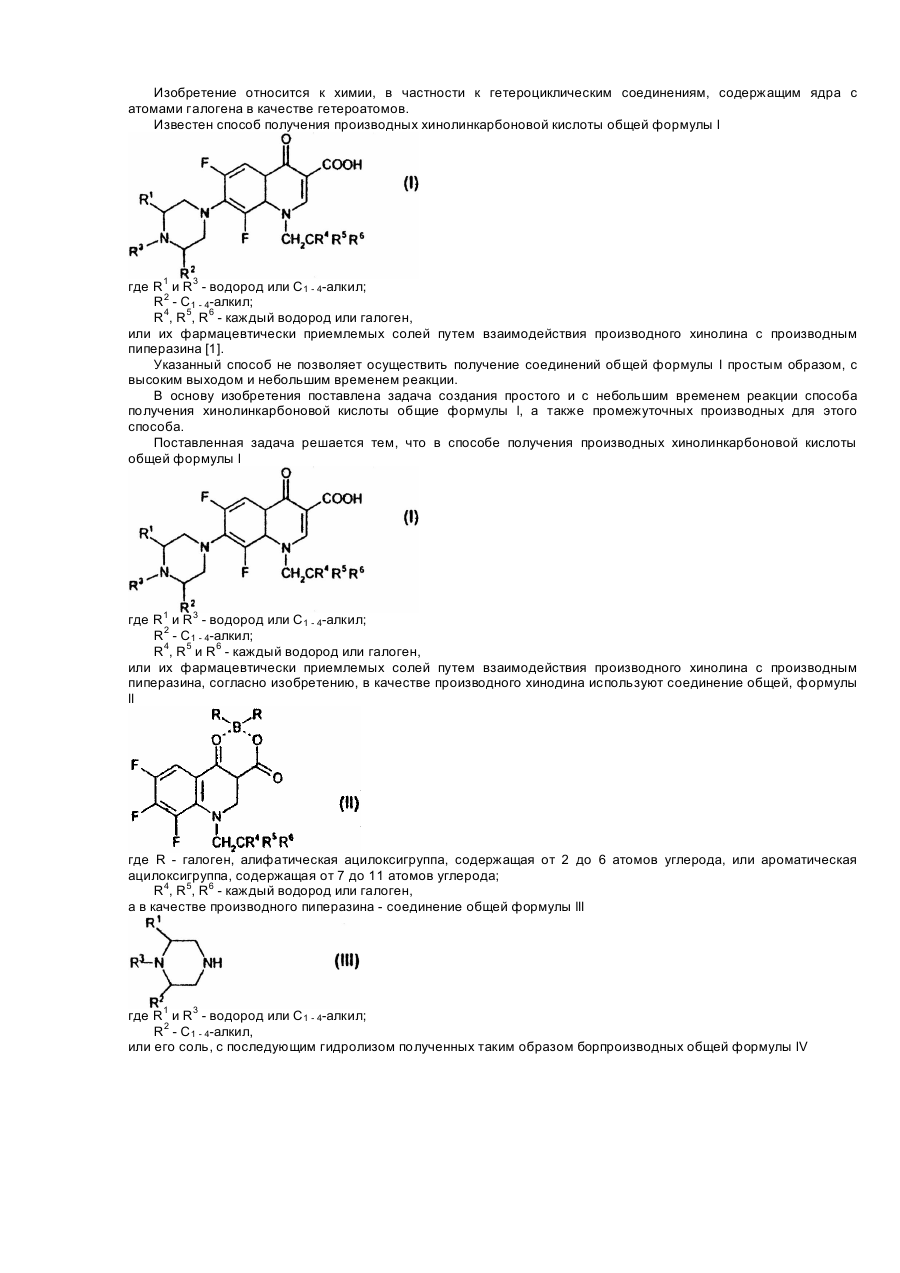
Номер патенту: 26568
Опубліковано: 11.10.1999
Автори: Керестурі Геза, Шіпош Юдіт, Хорват Агнеш, Рітлі Петер, Балог Марія, Вашварі Лелле, Пайор Аніко, Хермец Іштван
МПК: A61P 31/04, C07D 215/56, C07F 5/00, A61K 31/495, C07D 401/04
Мітки: проміжні, похідних, солей, одержання, спосіб, кислоти, хінолінкарбонової, сполуки, придатних, фармацевтично
Формула / Реферат:
1. Способ получения производных хинолинкарбоновой кислоты общей формулы lгде R1 и R3 - водород или C1 - 4-алкил;R2 - C1 - 4-алкил;R4, R5 и R6 - каждый - водород или галоген,или ее фармацевтически приемлемых солей, взаимодействием производного хинолина с производным пиперазина, отличающийся тем, что в качестве производного хинолина используют соединение общей формулы llгде R - галоген,...
Сполуки,які містять біциклічне конденсоване ядро, спосіб їх одержання, проміжні сполуки, спосіб їх одержання (варіанти) та фармацевтична композиція для інгібування перетворення ферменту ангіотензину та нейтраль

Номер патенту: 39924
Опубліковано: 16.07.2001
Автори: Робл Джефрі А., Годфрей Джоллі Д., Кронентал Девід Р.
МПК: C07D 513/04, A61K 31/542, A61K 31/535, C07D 498/04, C07C 317/50, A61P 43/00, C07K 5/078, A61K 31/54, A61P 9/04, C07C 327/00, A61K 31/553, A61K 38/00, A61K 31/554, C07C 319/00, A61P 9/12, C07C 323/59, A61K 31/55
Мітки: конденсоване, ядро, біциклічне, інгібування, перетворення, спосіб, містять, ангіотензину, сполуки,які, проміжні, нейтраль, композиція, сполуки, одержання, варіанти, фармацевтична, ферменту
Формула / Реферат:
1. Соединения, содержащие конденсированное бициклическое ядро формулы,или их фармацевтически приемлемая соль, где А представляетили,Χ представляет О или S,r1 и R12 независимо...
Похідні 17,20-епоксипрегнану, спосіб їх одержання, спосіб одержання похідних кортизону і проміжні сполуки
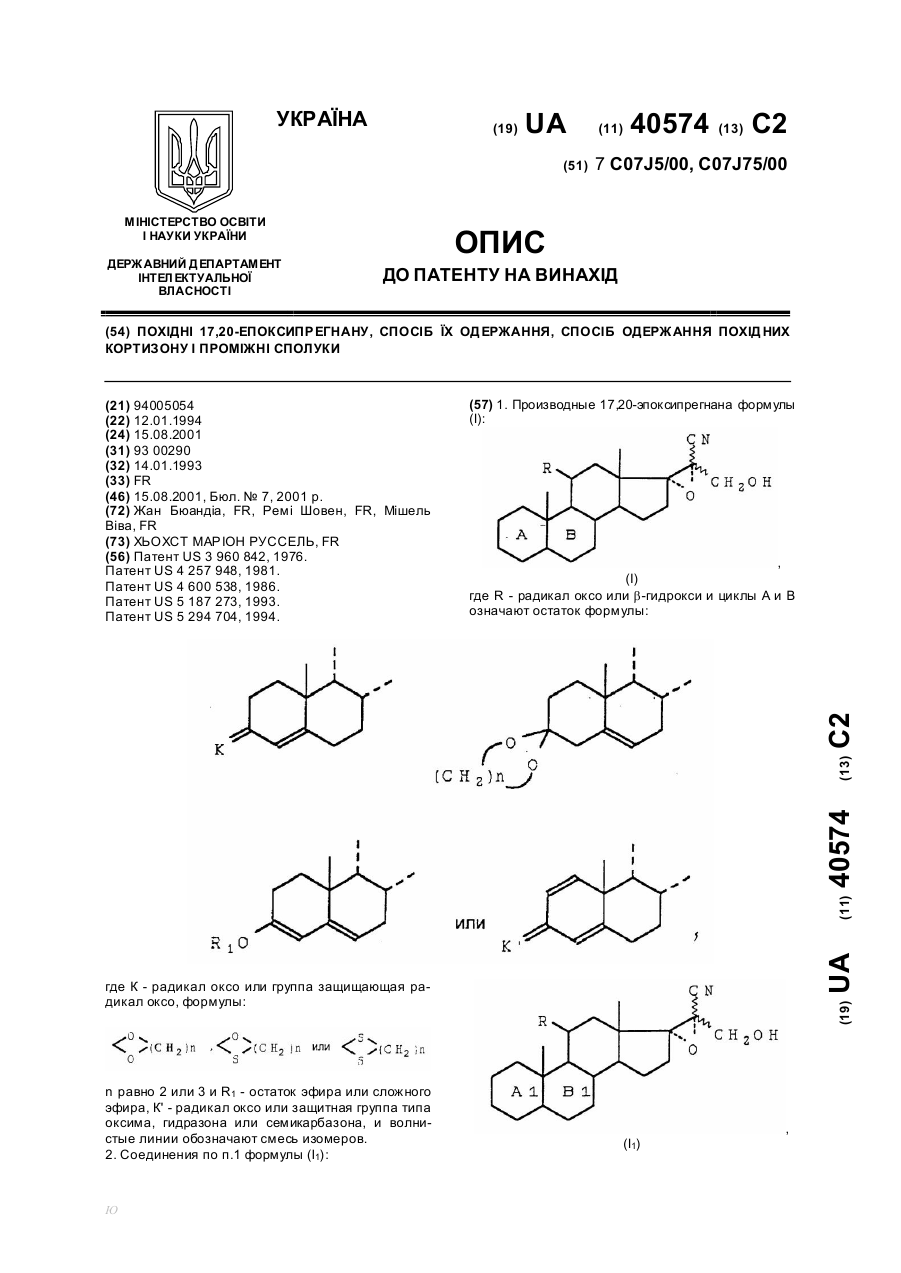
Номер патенту: 40574
Опубліковано: 15.08.2001
Автори: Мішель Віва, Ремі Шовен, Жан Бюандіа
МПК: C07J 7/00, C07J 41/00, C07J 5/00, C07J 21/00, C07J 75/00
Мітки: 17,20-епоксипрегнану, одержання, спосіб, похідні, похідних, проміжні, сполуки, кортизону
Формула / Реферат:
1, Производные 17,20-эпоксипрегнана формулы (I): где R - радикал оксо или ß-гидрокси и циклы А и В означают остаток формулы: где К - радикал оксо или группа защищающая радикал оксо, формулы:n равно 2 или 3 и R1 - остаток эфира или сложного эфира, К' - радикал оксо или защитная группа типа оксима, гидразона или семикарбазона, и волнистые линии обозначают смесь изомеров.2....
Комплексна сполука платини (іі), спосіб її одержання, проміжні сполуки та спосіб їх одержання

Номер патенту: 26686
Опубліковано: 12.11.1999
Автори: Кім Дае-Кі, Кім Гангхіок, Кім Кей Хіуп, Чо Енг Байк, Тай Йо Хо, Кім Хан Таєк, Гем Енгсік
МПК: C07F 15/00, C07D 317/28, C07D 317/72
Мітки: спосіб, комплексна, проміжні, сполука, іі, одержання, платини, сполуки
Формула / Реферат:
1. Комплексные соединения платины (II), представленные формулой (I)где R1 и R2, которые могут быть одинаковыми или разными, представляют собой атом водорода или C1-C4 алкильную группу, или совместно образуют C3-C9 циклоалкил группу вместе с присоединенным атомом углерода; два X совместно образуют группу, представленную формулойгде R3 представляет собой атом водорода или метильную группу; R4 и R5, которые могут быть...
Спосіб одержання похідних фенілгуанідину та похідні сульфокислоти як проміжні продукти в синтезі похідних фенілгуанідину

Номер патенту: 13323
Опубліковано: 28.02.1997
Автори: Єва Шомфаї, Чаба Хусар, Ференц Шпербет, Лайошне Палі, Петер Шаркезі, Атілла Немет
МПК: C07C 317/42, C07C 277/00, C07C 319/00, C07C 323/43, C07C 279/00, C07C 309/00
Мітки: сульфокислоти, проміжні, похідні, спосіб, синтезі, фенілгуанідину, похідних, продукти, одержання
Формула / Реферат:
(57) 1. Способ получения производных фенилгуанидина общей формулы I:где R - алкил с 1-4 атомами углерода; М - водород, натрий, калий или кальций; А - группа -S-, -SO- или -SO2-,отличающийся тем, что производное тиокарбамида общей формулы II:где R и М имеют вышеуказанные значения, подвергают окислению с последующей обработкой полученного при этом производного сульфокислоты общей формулы...
Попередній патент: Похідні алкілбензоїлгуанідинів, спосіб їх одержання, фармацевтична композиція, що має аритмічну активність
Наступний патент: Портал вантажопідйомного крану
Випадковий патент: Цех для приготування комбікормів