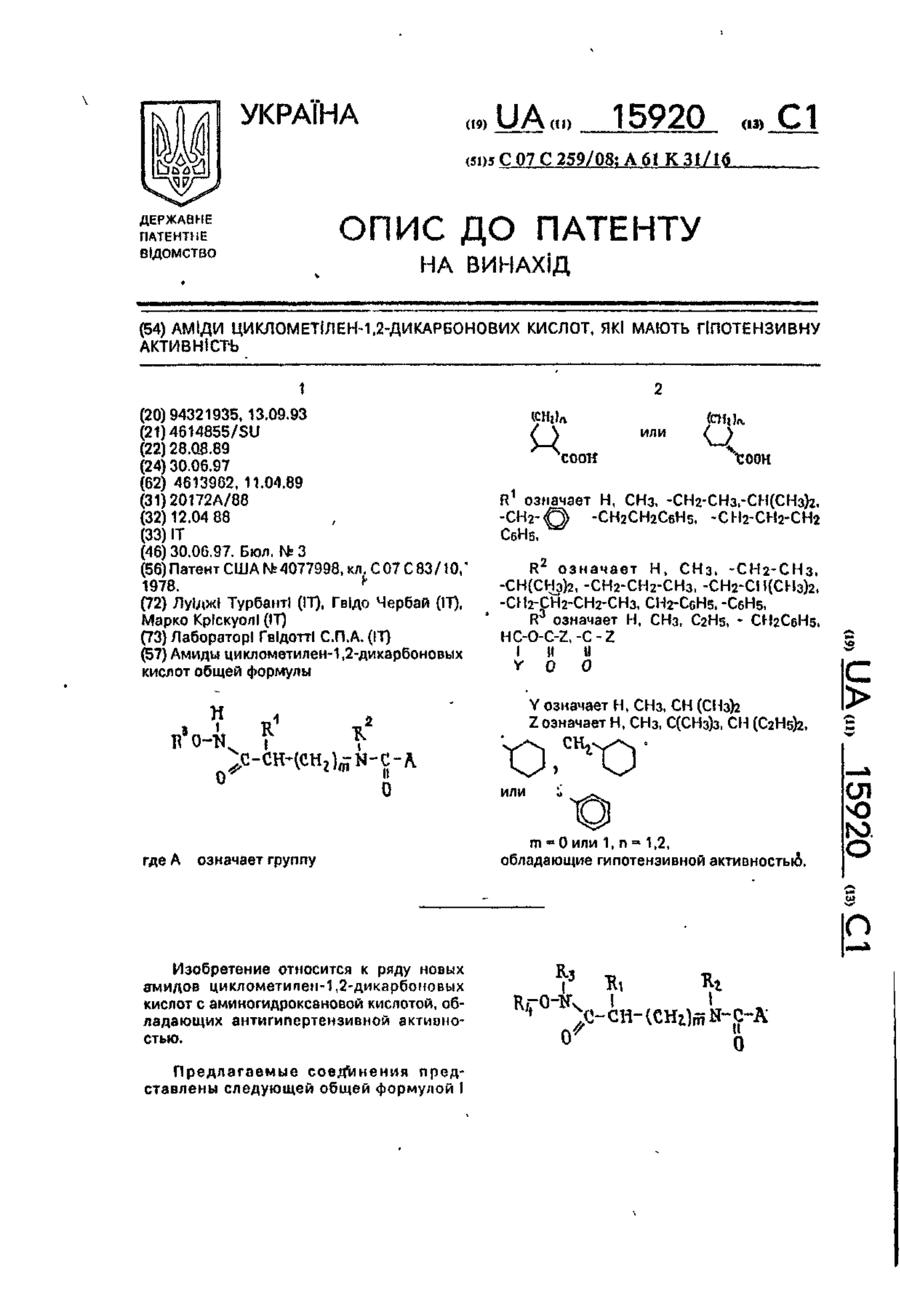
Аміди циклометілен-1, 2-дикарбонових кислот, які мають гіпотензивну активність
Номер патенту: 15920
Опубліковано: 30.06.1997
Автори: Гвідо Чербай, Марко Кріскуолі, Луіджі Турбанті










Формула / Реферат
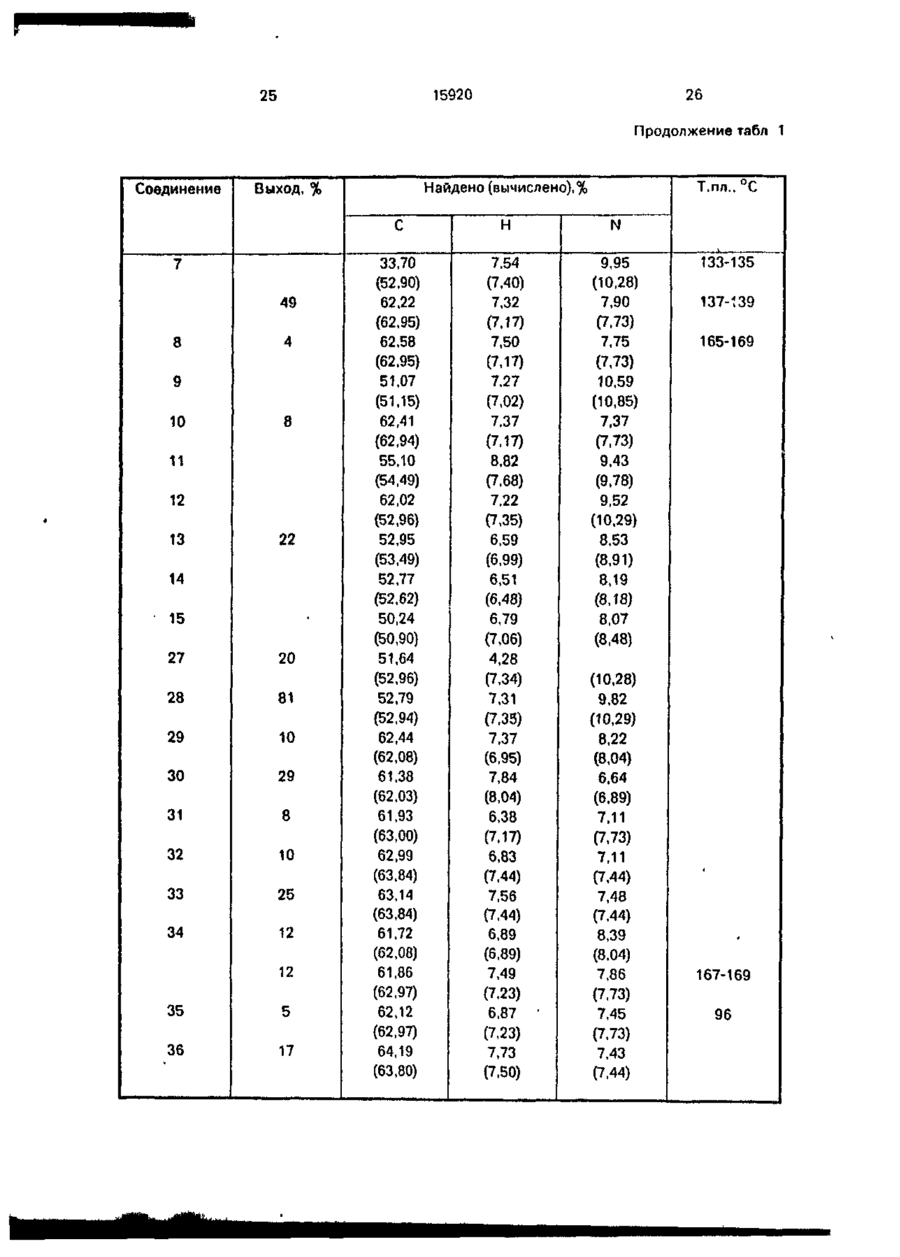
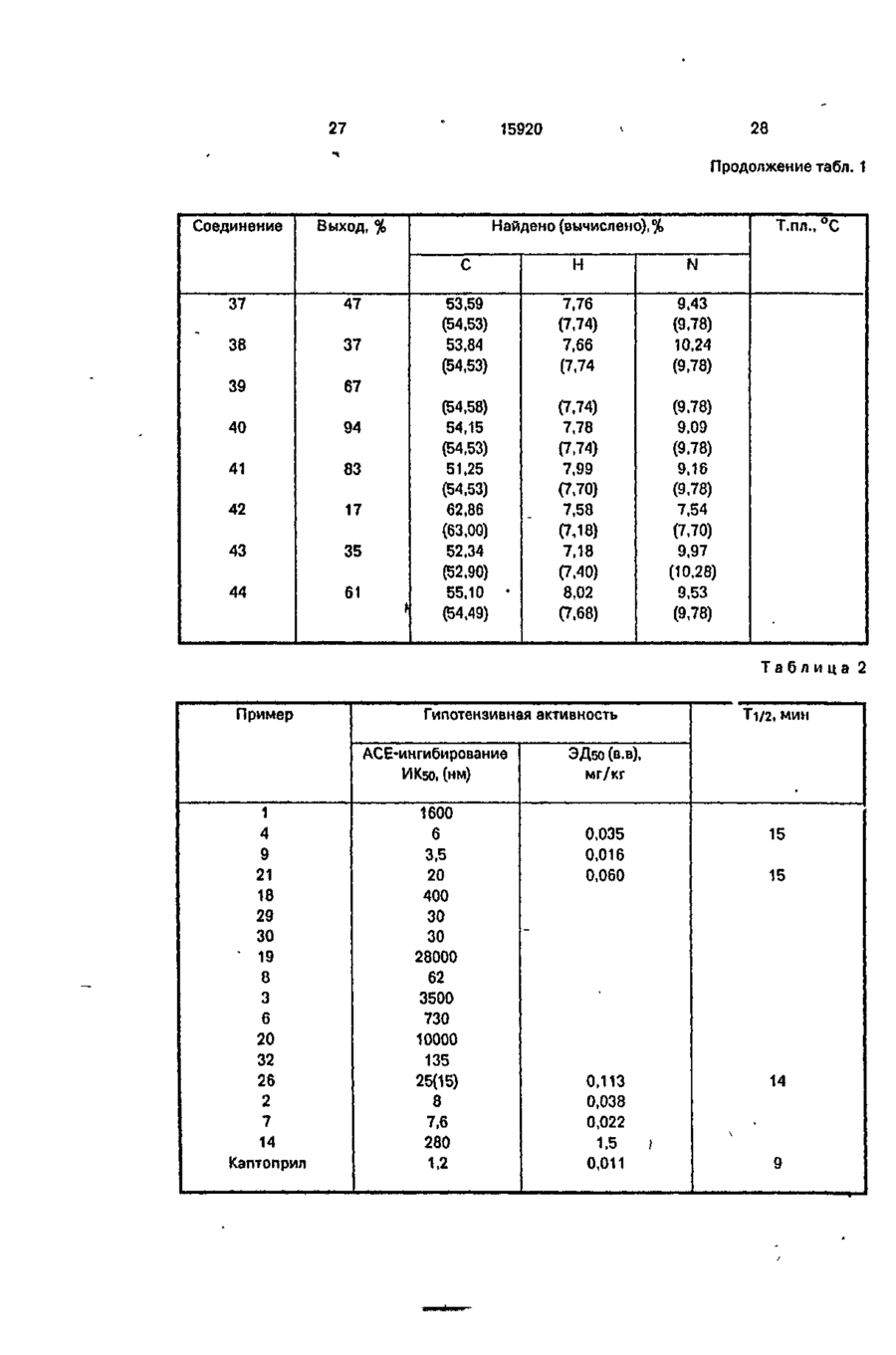
Текст
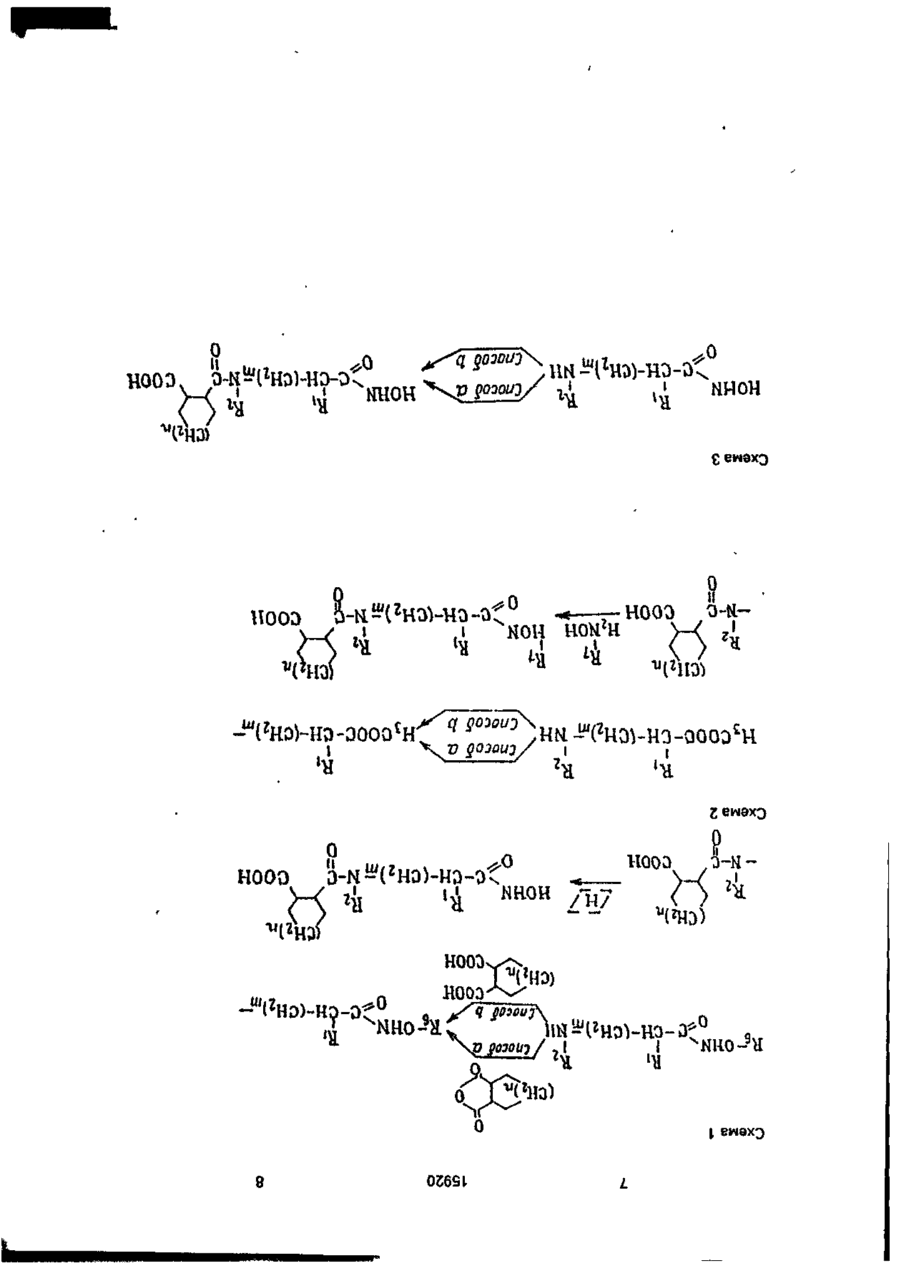
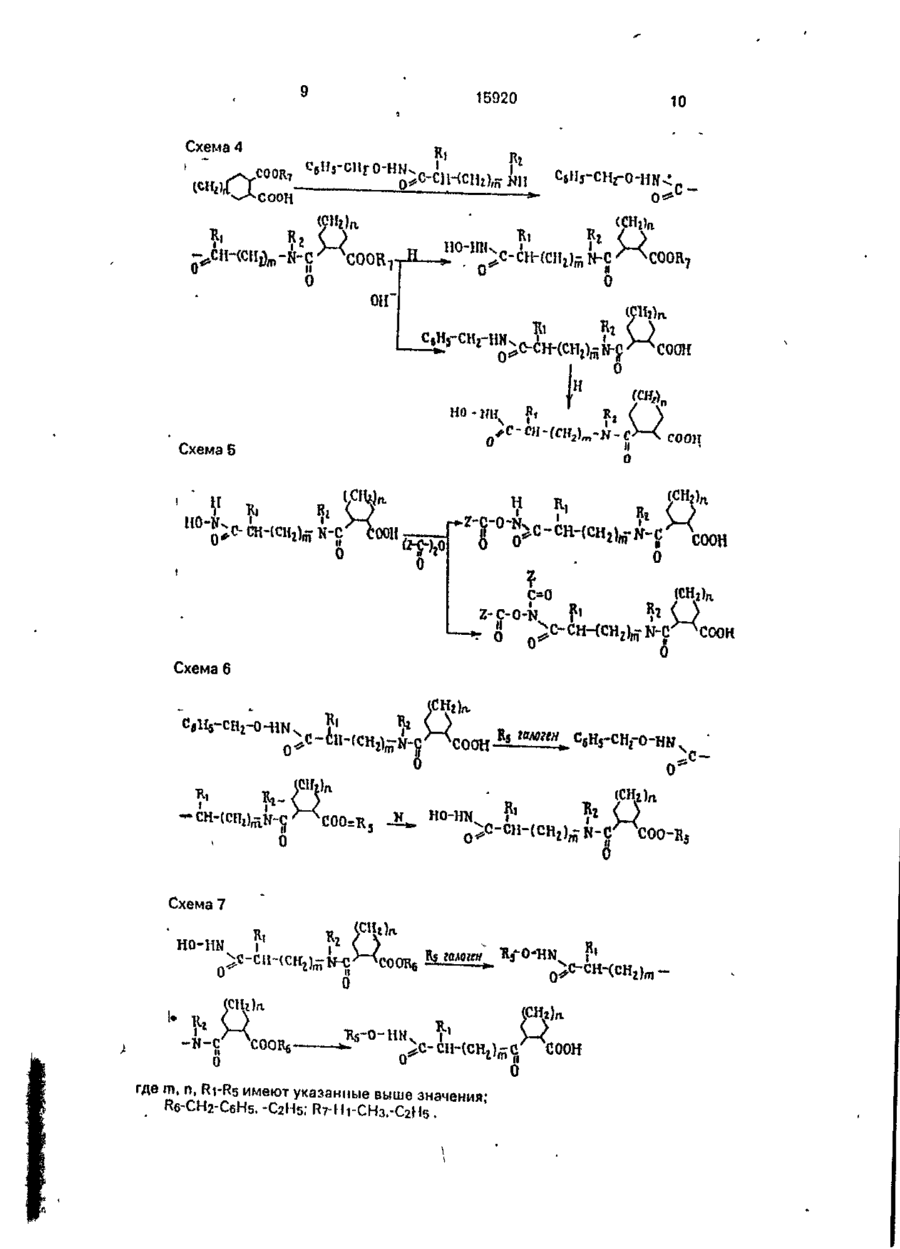
Амиды циклометилен-1,2~дикарбоновых кислот общей формулы анії соон где А 'tjOOH R1 означает Н, СНз, -СН2-СНзгСН(СНз)г, -СН2- -СН2СН2С6Н5, -CH2-CH2-CH2 R2 о з н а ч а е т Н, СНз, - С Н г - С Н з , -СН(СНз)2, -СНг-СНг-СНз, -СН2-СН(СНз)2, -СН2-СН2-СН2-СНЗ, СНг-СбНб, -СбН5, R3 означает Н, СНз, С2Н5, - СНгСбНб, HC-O-C-Z,-C-Z I И U У О О п їго-к о или Y означает Н, СНз, СН (СНзЬ Z означает Н, СНз, С(СНз)з, СН (СгНб)2, о означает группу или m -0 или 1, п = 1,2, обладающие гипотензивной активностью. С > ел о ю. о о Изобретение относится к ряду новых амидов циклометипен-1,2-дикарбоновых кислот с аминогидроксановой кислотой, обладающих антигипертензивной активностью. Предлагаемые соединения представлены следующей общей формулой I 15920 А где А ИЛИ соон 1 / \ HS, L Ri - Н, -СНз, -СНг-СНз, -СН(СНзЬ. 10 Я2 - H, СНз, -СНг-СНз, -СН(СНз)2, -СНгСНг-СНз, -СН2-СН(СНз)2, -СН2-СН2-СН2-СН3, -СНг-СеНь -Сб-Нб; R3-H; R4 - Н, СНз, С2Н5, -СН2СбН5, 15 HC-O-C-Z.-C-Z і її її Y 0 0 О- О' (И) о соон Y - H , СНз,-СН(СНз)г; Z - Н, -СНз, -С(СНз)з, -СН(СгН5)2, СООН 11 20 В настоящее время кроме каптоприла к другим АСЕ ингибирующим средствам, широко используемым в фармацевтической области, относятся эналаприл и лизиноприл. Терапевтическое действие указанных соединений, как полагают, происходит главным образом через ингибирование фермента превращения ангиотензина I как в плазматической, так и в определенных тканевых системах, сопровождаемое снижением концентраций сильного эндогенного прессорного антагониста ангиотензина II. С другой стороны, благодаря тому, что АСЕ-ингибирование приводит также к снижению метаболизма брадикинина, повышение уровней этого сосудорасширяющего и диуретического агента должно в некоторой степени разъяснить гипотензивное действие указанных лекарственных веществ. В случаях гипертензии в сочетании с низкими уровнями ангиотензина II эффект АСЕ-ингибиторов можно было бы отнести за счет косвенного действия из-за вмешательства нейрогенной конструкции сосудов (при которой облегчается нервно-симпатическая трансмиссия). Соединения изобретения отличаются от указанных соединений и других ингибиторов АСЕ, описанных в литературе, тем, что их карбоксильный концевой участок (возможно способный к взаимодействию с определенными активными центрами ферментов АСЕ) состоит из амида циклометилен-1,2-бикарбоновой кислоты (см.формулу Ilia), в то время как во всех известных АСЕ-ингибиторах указанный участок состоит из амида циклических или линейных аминокислот (формулы IIIb И НІС), m = 0 или 1; n - 1,2. 25 Более конкретно соединения изобретения включают ряд амидов циклометилен1,2-дикарбоновых кислот с цис- или транс-конфигурацией, связанных с первичной или вторичной аминогруппой аминогид- 30 роксамовой кислоты. Предлагаемые соединения, исходя из данных, полученных при испытании in vitro, обладают ингибирующим действием против АСЕ (фермента, посредством которого анги- 35 отензин I превращается в сильный эндогенный агонист давления ангиотензин II) и противогипертензиьной (гипотензивной) активностью, которая, как считают, связана с предшествующим действием, которая обна- 40 руживается в случае некоторых соединений у непроизвольно гипертензивных крыс, особенно у животных, бодрствующих или анестезированных, на которых оказывает действие гипертония, индуцированная анги- 45 отензином I. I < Известны и широко используются ле"с'тооц Р\ ? NCOOH карственные препараты, ингибирующие II С АСЕ, для лечения, некоторых форм артериальной гипертонии, которые также исполь- 50 о зуют для лечения застойной сердечной Ш b .Ща недостаточ н ости. I В качестве основного и главного АСЕ/ СН ингибирующего лекарственного препарата использовали и используют в настоящее 55 соон время (1-(3-меркапто-2-метилпропионил}-1пирролидин-2-карбоновую кислоту, известШс ную также под нехимическим названием (6Cl) как каптоприл и представленную форВследствие этой химической особенномулой сти, которая является общей для всех пред 15920 лагаемых соединений, заявленные соединения кроме новизны имеют явное преимущество в структуре по сравнению с известными соединениями, благодаря которой они обладают преимуществом также в отношении фармакологических и терапевтических свойств. С фармакологической точки зрения предлагаемые соединения, как уже указывалось, обладаются АСЕ-ингибирующему действию, установленному в испытаниях In vitro, во время которых при исследовании функциональной активности обнаружен легко возникающий и пролонгированный гипотензивный эффект. Соединения согласно изобретению могут быть получены с помощью ряда способов В соответствии с первым способом циклометилен-1,2-дикарбоновую кислоту, особенно ее кислотную часть, конденсируют с аминопроизводным, содержащим замещенную бензилом или ал килом гидроксамовую группу. Продукт конденсации, в свою очередь, если замещающей группой является бензил, подвергается каталитическому гидрированию для удаления бензильной іруппьі с получением в результате этого соединения формулы I, где R3 и Rn~ водород. В пределах данного способа стадию конденсации можно проводить двумя методами, а именно: метод а) с помощью реакции указанного аминопроизводного с ангидридом циклометилен-1,2-дикарбоновой кислоты; метод Ь) по реакции указанного аминопроизводного с дикарбоновой кислотой в присутствии конденсирующего агента. Конденсирующим агентом может быть любой из известных, но предпочтительно использование либо этил1\Г-(3-диметиламинолропил)карбодиимида (WSC) или дициклогексилкарбодиимида (DCC). В соответствии с вторым способом конденсацию осуществляют исходя из сложных аминоэфиров по методике первого способа, в результате чего получаются соответствующие амидопроизводные, а затем они превращаются $ соответствующее производное гидроксамовой кислоты взаимодействия с гидроксиламином или N-алкилгидроксиламином, где алкил - метил и этил. В данном случае также предусмотрены оба указанных метода а) и Ь) реакции конденсации. Согласно третьему способу получения соединений изобретения формулы I, где m 1, ангидрид дикарбоновой кислоты или саму циклометилен-1,2-дикгфбсновую кислоту непосредственно конденсируют с аминсодержащей гидроксамовой кислотой. 5 10 15 20 25 30 Согласно четвертому способу получения предлагаемых соединений алкиловый моноэфир циклометилен-1,2-дикарбоновой кислоты конденсируют с аминопроизводным, содержащим гидроксамовую группу, защищенную бензильной группой. Полученный в результате сложный амидоэфир альтернативно подвергают: c) каталитическому гидрированию для удаления бензильной группы с получением соединений формулы 1, где Rs - метил, этил; d) щелочному гидролизу и последующему каталитическому гидрированию с получением соединений формулы 1, где R3 - R4 = Rs-H Амидогидроксамовые кислоты, полученные в соответствии с раскрытыми вкратце указанными способами, можно использовать в качестве исходных соединений для получения дополнительного ряда производных, представленных общей формулой I при взаимодействии с ангидридом формулы (Z-C-)2O и О где Z имеет указанные значения, с получением соединений формулы I, где R3 и/или R4 означает Z-Cп О Если, наоборот, промежуточное амидосоединение, полученное до удаления защит-, 35 ной бензильной группы подвергается взаимодействию с ацилоксиметилгалогенидом, получают промежуточное соединение, которое в результате каталитического гидрирования для удаления бензильной группы 40. приводит к образованию амидопроизводного формулы I, где 45 R5-HC-O-C-Z } и Y 0 где Y и Z имеют указанные значения. Наконец, если соединения формулы I, где R5-CH3, C2H5, СбНб-СНг, взаимодействуют с ацилоксиметилгалогенидом и из полБ0 ученного промежуточного соединения удаляют защитную группу у исходной функции этерифицированной карбоновой кислоты, то получают соединения формулы I, где 55 R4-HC-O-C-Z і її Ї О В приводимых ниже схемах проиллюстрированы указанные способы синтеза предлагаемых соединений. I w ш J? V_/° I X. v goiouj/ і г НООО 0 Э-N- гНЭ)-НЭ-оС ш ,0 0 02691 15920 10 Схема А м О I f, Г соон Схема 5 И Н К» R, ОН О С»О Ь Схема 6 JU. °-™ 5 c-t ( cH 2J-cH Bj ,J-cHooR R H о Схема 7 г ?О H0-HN 0 П ) где m, n, R1-R5 имеют указанные выше значения; R6-CH2-C6H5, -С2Н5; R7-Hi-CH3,-C2H5. COOK 11 15920 При осуществлении первого способа реакцию конденсации можно проводить в растворителе, выбранном из таких растворителей, как вода, алифатические спирты, например метанол, этанол, бутанолы, и хлорированные алифатические растворители, например хлористый метилен, хлороформ, дихлорэтан, при температуре в интервале от -5 до 60°С, причем следует соблюдать осторожность, проводя процесс при низкой температуре (в интервале от -5°С до комнатной температуры), если в качестве растворителя используют воду и водную смесь. Стадию каталитического гидрирования осуществляют в алифатических спиртах, таких как метанол или этанол, с использованием водорода при нормальном давлении и комнатной температуре в присутствии стандартного катализатора, такого как палладий на угле. При проведении второго способа конденсацию осуществляют в условиях, указанных выше, однако следующая реакция с гидроксиламином предполагает использование спиртов в виде воды и растворителя и протекает при комнатной температуре. В третьем способе реакцию конденсации проводят в водно-щелочной среде при температуре не оыше 40°С, как правило при комнатной температуре. По схеме 4 в данном описании первую стадию проводят при таких же условиях, как уже указывалось для первого способа (схема 1, метод Ь). Каталитическое гидрирование проводят аналогичным методом, тогда как для альтернативного варианта включающего щелочной гидролиз, условия для егр проведения должны быть мягкими (го есть реакцию проводят при комнатной температуре в течение 2 ч, используя воду в качестве растворителя), На схемах 5-7 показан синтез производных формулы 1 с использованием в качестве исходного материала конечных и промежуточных соединений, полученных согласно способам схем 1-4,учитывая значения, указанные для замещающих групп. Более конкретно, на схеме 5 показано получение производных формулы 1,0-ацилированных у гидроксамовой группы; в этом случае реакцию с ангидридом осуществляют при низкой температуре (ниже 20°С), по крайней мере в начальной стадии, в присутствии каталитических количеств Г\1,№-диметиламинопиридина. На схемах 6 и 7 реакцию с ацетилоксиметилгалогенидом проводят при низкой температуре (ниже 20 С), по крайней мере в 12 начальной стадии, в безводных условиях в атмосфере азота. Следующие примеры иллюстрируют получение нескольких соединений изобре5 тения, следует иметь в виду, что указанные примеры не предполагают ограничение объема изобретения. Точки плавления определялись на приборе Кофлера и являются не скорректиро10 ванными. Все соединения имеют ИК- и *Н ЯМРспектры, согласующиеся с приписанными им структурами, и данные элементного анализа с отклонениями в пределах +0.4% от 15 расчетных значений, если не оговорено особо. Некоторые растворители и реагенты указаны в традиционно используемых сокращениях: ТГФ - тетрагидрофуран, ДСС 20 дициклогексилкарбодиимид, ACOEt- этилацетат, WSC - этил-N -{3-диметиламинопропил)карбодиимид. П р и м е р 1. цис-2({Ы~[2-(Гидроксиамино)-2-оксоэтил]амино}карбонил)циклогекса25 нкарбоновая кислота. К раствору, содержащему 1,2 г (4,07 ммоль) О-бензиламиноацетоксигидроксамовой кислоты трифторацетат в 30 мл НгО, прибавляют 1,0 мл 4 н. NaOH с осаждением 30 кристаллического продукта. К полученной суспензии прибавляют при перемешивании при комнатной температуре 0,628 г (4,07 ммоль) ангидрида 1,2циклогександикарбоновой кислоты и 1,02 мл 35 4 н. NaOH по каплям в течение 1 ч с поддержанием рН 1,0 реакционной смеси в течение всего периода введения. Реакционную смесь, имеющую опалесцирующую окраску, выдерживают при перемешивании при ком40 натной температуре в течение 2 ч, затем фильтруют, прозрачный раствор подкисляют до рН 1 10%-ной HCI при охлаждении льдом, в результате происходит осаждение 0-бензилированного амидопроизводного в 45 виде кристаллов цвета слоновой кости, 0,9 г, выход 66%, т.пл., 141-143°С. 0,7 г (2,09 ммоль) указанного О-бензилированного промежуточного соединения, растворенного в этаноле, гидрируют при 50 нормальном давлении и комнатной температуре в присутствии 10% палладия на угле. Примерно через 4 ч поглощается вычисленное количество водорода, 51 мл. Этанольный раствор после отфильтрования 55 катализатора выпаривается досуха, и остаток, состоящий из гигроскопичных кристаллов, очищается с помощью кристаллизации из ацетона, давая ожидаемое амидопроизводное, 0,27 г, выход53%, т.пл. 133-135°С (с разложением) 13 15920 П р и м е р 2. транс-2({Г4 [2 (Гидроксиамино)-2-оксоэтил]-1\)-этилэмино}карбонил) циклогексанкарбоновая кислота. К раствору, содержащему б г (45,7 ммоль) этилового эфира этиламиноуксусной 5 кислоты в 130 мл хлористого метилена, прибавляют при перемешивании при 5°С 7,05 г (45,7 ммоль) ангидрида транс-1,2-циклогоксандикарбоновой кислоты и полученный раствор выдерживают при комнатной тем- 10 пературе о течение 20 ч. После серии промывок 40 мл 5%-ной HCI и двумя по 40 мл порциями насыщенного водного раствора хлористого натрия реакционный раствор дегидратируют с 15 использованием безводного сульфата натрия и упаривают досуха в вакууме с получением 13 г (выход 99%) хроматографически чистого, твердого продукта белого цвета, состоящего из транс-2({Ы-[2~(гидроксиамино)- 20 2-оксоэтпл]-М-этиламиио}карбонил) циклогексанкарбоновой кислоты. Метил-Nэтил-карбамойл-1-циклогексан-карбоновая кислота. К раствору, содержащему б г (21,03 25 ммоль) указанной кислоты в 30 мл метанола, прибавляют при перемешивании при 5°С 2,77 г (69,4 ммоль) NaOH в 30 мл метанола, а затем 13 г (23,1 ммоль) хлоргидрата гидроксилэмина. 30 Полученную суспензию выдерживают при интенсивном перемешивании при 15°С в течение 4 ч, а затем реакционную смесь упаривают досуха в вакууме при комнатной температуре, получая 9,8 г смолообразного, 35 бесцветного остатка. Указанный остаток растворяют при перемешивании в 10 мл воды и полученный прозрачный раст'вор подкисляют до рН б НСІ, подсаливают до насыщения хлористым натрием и затем экс- 40 трагируют порциями по 20 и 10 мл этилацетата. Объединенные органические экстракты дегидратируют над сульфатом магния и после упаривания досуха в вакууме получают 45 5,59 г бесцветного кристаллического продукта. Полученный продукт смешивают с 50 мл хлороформа и полученную суспензию фильтруют в вакууме, а остаток обрабатывают 90 мл 1,2-дихлорэтана с образованием 50 суспензии, которую выдерживают при комнатной температуре без перемешивания. После фильтрования в вакууме получают 4,58 € (выход 80%) требуемого продукта в виде бесцветных кристаллов, т.пл. 137- 55 139°С(по Кофлеру). П р и м е р З . цис-2~({М~[2-Бензил~3 (гидроксиамино)-3-оксопропил]амино}карбонил) циклогексановая кислота. 14 К раствору, содержащему 4 г (20,0 ммоль) 2-бензил-З-аминопропионовой-гидроксамовой кислоты (полученной при взаимодействии метилового эфира 2-бензил-З-аминопропионовой кислоты с гидроксиламином) в 70 мл Н2О и 6 мл 4 н. NaOH, прибавляют при перемешивании при 20-25°С в течение 1 ч одновременно 3,1 г (20,0 ммоль) ангидрида 1,2-циклогександикарбоновой кислоты и 4 мл 4 н. NaOH при поддержании рН реакционной смеси при 11 в течение всего времени введения реактивов. После 2 ч перемешивания при 20-25°С смесь подкисляют до рН 1 10%-ной HCI и экстрагируют СНСІз, СНСІз выпаривают и после кристаллизации остатка цвега слоновой кости из ацетона получают целевое соединение в виде белых кристаллов, 1,48 г, выход 26,6%, т.пл. 171-175°С. П р и м е р 4. цис-2-({М-[2-(гидроксиамино)-2-оксоэтил]-2-Ы-метиламино}карбонил) циклогексанкарбоновая кислота. К перемешанному раствору ангидрида цис- циклогексан дика рбо но вой кислоты (1,60 г 10.4 ммоль) в дихлорметане (20 мл) в атмосфере азота прибавляют раствор, содержащий трифторацетат 0-бензилсаркозингидроксамовой кислоты (3,08 г 10,0 ммоль) и триэтиламин (3,0 мл, 21,5 ммоль) в дихлорэтане (30 мл). Полученную смесь промывают охлажденным 5%-ным раствором НСІ (10 см 3 х 2), нейтрализуют 10%-ным раствором ЫаНСОз и сушат MgSO4. Растворитель упаривают при пониженном давлении и после кристаллизации остатка из смеси ацетон/простой эфир получают промежуточный "0-бензилированный амид" (3,40 г, 95%) в виде белых кристаллов, т.пл. 130°С. Это соединение (2,35 г, 6,75 ммоль) гидрируют в метаноле (30 мл) в присутствии 2 ч. После упаривания растворителей при пониженном давлении при 5°С полученный продукт кристаллизуют в дихлорметане с получением названного соединения (1,25 г, 71%) в виде белых кристаллов: т.пл. 131133°С. П р и м е р 5. цис-2~({Ы-[2-(Гидроксиамино)-2-оксоэтил]-!М-фениламино}карбонил) циклогексанкарбоновая кислота. К раствору, содержащему 1,32 г (33 ммоль) NaOH в 32 мл метанола, прибавляют при перемешивании при 10°С 3,19 г (10 ммоль) цис-2-({Ы-[2-(метокси)-2-оксоэтил]-Ыфенламино}карбонил) циклогексанкарбоновой кислоты (полученной из ангидрида Чис-1,2-циклогександикарбоновой кислоты и метилового эфира N-фениламиноуксусной кислоты по аналогичной методике, описанной в примере 2, а затем 0,764 г (11 ммоль) 15 15920 хлоргидрата гидроксиламина. Полученную суспензию выдерживают в течение 6 ч, а затем оставляют стоять в течение ночи. После упаривания досуха в вакууме остаток растворяют в 5 мл воды, суспению обрабатывают "норитом", фильтруют и в конце подкисляют до рН 2 охлажденной 10%-ной HCI. Смолистый осадок экстрагируют двумя порциями по 60 мл хлористого метилена, объединенные органические экстракты промывают насыщенным водным раствором хлористого натрия. После дегидратации над безводным сульфатом натрия и упаривания в вакууме получают 2,7 г (выход 84%) бесцветного смолистого твердого продукта. Полученный продукт очищают при растворении его в 27 мл ацетона. После продолжительного выдерживания при 0°С для повторного осаждения получают требуемое соединение в виде бесцветных кристаллов, т.пл. 160-161°С. П р и м е р 6. транс-2({Г\І-[2-(Гидроксиамино)-2-оксоэтил]-№-метиламино}карбонил) циклопентаикарбомовая кислота. 1,16 г (7,34 ммоль) транс-1,2-циклопентандикарбоновой кислоты солюбилизируют в растворе вода/трет-бутанол. К полученному раствору прибавляют 1,42 г (7,34 ммоль) 0-бензил-Г\1-метиламиноэцетогидроксамовой кислоты и доводят рН до 4,5, используя 1 н. NaOH. Затем прибавляют порциями 1,30 г (7,34 ммоль) этил-Ы'-З-диметиламинопропилкарбодиимида при поддерживании рН 4,5. Через 22 ч при перемешивании при комнатной температуре реакционный раствор экстрагируют тремя порциями СНСІз. После выпаривания хлороформа получают 0-бензилированное амидопроизводное в виде белых кристаллов, 1,15 г (выход 4 7 % ) . Полученный таким образом продукт растворяют в 10 мл метанола и осуществляют каталитическое гидрирование при 20°С в присутствии 10% палладия на угле при обычном давлении, в результате получают ожидаемый продукт в виде белых кристаллов, т.пл. 107-111°С, 0,7 г, выход 83%. Соединения, которые перечислены далее, получают по методикам предыдущих примеров, но в целях сокращения объема описания их получение не приведено, а указаны только химические характеристики некоторых соединений. Приводимые далее примеры 7-12 осуществляют по методике примера 4. Примеры 14, 15 и 16 выполняют по аналогичным методикам примеров 1,5 и 2. . П р и м е р 7. TpaHC-(1R,2R)-2-({N-[2--1,2-4HKVIOгександикарбоновой кислоты и 1,52 г (11,6 ммоль) этилового эфира N-этиламиноуксусной кислоты в 60 мл тетрагидрофурана при 5°С, прибавляют 2,38 г(11,6 ммоль) дициклогексилкарбодиимида. Реакционный раствор перемешивают при 5°С в течение 2 ч и при комнатной температуре в течение ночи. Осажденную дициклогексилмочевину отфильтровывают, а растворитель упаривают. Полученное масло растворяют в CH2CI2, промывают водой (10 мл), а затем 15 мл 5%ного водного раствора ЫаНСОз. Водный экстракт промывают хлористым метиленом (10 мл), подкисляют 6 н. соляной кислотой, затем экстрагируют 20 мл хлористого метилена и экстракты сушат сульфатом натрия и упаривают, получая промежуточный "сложный амидоэфир": 1,9 г, 58% выход, маслянистые кристаллы. Промежуточный амидоэфир обрабатывают хлоригидратом гидроксиламина по методике, описанной в примере 2, получая 1,38 г (87%) чистого целевого соединения в виде белого твердого продукта: [ a] D ~ 10,7 (С - 1,5 этанол). П р и м е р З . цис-2-({М-[1-(2-Фенилэтил)2-{гидроксиамино)-2-оксоэтил]-!Ч-метиламино}карбонил) циклогексанкарбоновая кислота. К перемешанному раствору, содержащему 1,24 г (8,0 ммоль) ангидрида цис-циклогександикарбоновой кислоты в 50 мл хлористого метилена, медленно прибавляют при комнатной температуре раствор CH2CI2. содержащий 2,24 (8,0 ммоль) 0-бензил-2-метиламино-4-фенилбутангидроксамовой кислоты и 0,81 г (8,0 ммоль) триэтиламина. Затем реакционную смесь перемешивают в течение 5 ч при комнатной температуре. Хлористый метилен упаривают при пониженном давлении, а полученный осадок растворяют в водном 5%-ном растворе NaOH; после подкисления раствора концентрированным раствором HCI получают 2,5 г (68%) промежуточного 0-бензилированного амида, который гидрируют в 20 мл метанола с использованием 0,24 г 10% палладия на угле при комнатной температуре и нормальном давлении в течение 2 ч. После отфильтровывания катализатора и упаривания растворителя в вакууме получают осадок, который растворяют в 10 мл горячего ацетона. Затем ацетоновый раствор охлаждают, а осажденную первую порцию продукта (0,1 г) отфильтровывают. Раствор выдерживают 4 дня при 0°С, После чего начинает осаждаться вторая пор 17 15920 18 10%-ной НС! при 0°С и экстрагируют СНСІз ция кристаллов; твердый продукт отфильт(3x20 мл). Органический слой сушат над ровывают (0,27 г) и обрабатывают горячим MgSO4 и упаривают .в вакууме. После криацетоном (20 мл) при перемешивании в течесталлизации остатка из ацетона получают ние 0,5 ч. После фильтрования горячей сусбелый твердый продукт (1,42 г, 65%), т.пл. пензии получают твердый белый продукт (т.пл. 165-169°С), соответствующий одному 110-113°С,' [а]578 2 0 = + 12,7 (С = 5,0, этанол). из двух рацематов, указанных в заголовке Предшествующее соединение (0,82 г, 2,36 химическим наименованием. ммоль) гидрируют с использованием 10% Ацетоновый фильтрат упаривают с полпалладия на угле (100 мг) в метаноле (40 мл), учением осадка, который после перекри- 10 затем фильтруют, а растворитель отгоняют в сталлизации из СНСІз дает твердый продукт вакууме. После кристаллизации остатка из белого цвета, соответствующий второму расмеси метанол/простой эфир получают бецемическому соединению, указанному хилый твердый продукт (300 мг, 50%), т.пл. мическим названием целевого соединения. 20 П р и м е р 9. m«c-(1S,2R)-2-({N-[2-(rMApo- 15 127-128°С, [ а ] 5 7 8 = + 26,1°(С= 1,5, этанол). ксиамиію)-2-оксозїил}-Г\!-метилямино}карбоП р и м е р Ю . цис-2-({М-[1-Бензил-2-(менил) циклогексановая кислота. токсиамино)-2-оксоэтил]-Ы~метиламино}кар2-Метоксикарбонил-(Ш,25)-циклогексанбонил) циклогексанкарбоновая кислота. карбоповую кислоту получают по методике, К перемешанной суспенз'ии, содержаописанной в литературе (P.Mohr et al., Helv. 20 щей 1,28 г (8,32 ммоль) ангидрида цис-цикChim. Acta, 1983 г., вып. 66, с.2501). [ логександикарбоновой кислоты в 50 мл «J578 2 0 = : +4,23°, (С = 5,5, этанол), масляниСН2СІ2, прибавляют по каплям 120 мл расстый продукт, выход 63,1 %. твора хлористого метилена, содержащего 2 Образец неполного эфира (2,7 г, 14,5 г (8,3 ммоль) N-метил-О-метилфенилаланилммоль) растворяют в тетрагидрофуране (10 25 гидроксамовой кислоты (соль муравьиной мл) и охлаждают примерно до 0°С. Затем к кислоты) и 0,84 г (8,32 ммоль) триэтиламина полученному раствору последовательно с последующим перемешиванием полученприбавляют раствор, содержащий трифтоной смеси при комнатной температуре в терацетат О-бензилсаркозингидроксамовой чение 5 ч. Реакционную смесь промывают кислоты (4,47 г, 14,5 ммоль) и тризтиламин (2 30 дважды 5%-ным раствором НСІ и водой, а мл, 14,5 ммоль) в СНСІз 910 мл), 1-гидроксизатем экстоагируют 10%-ным водным расбензотриазол (16,20 % воды, 2,45 г, 14,5 твором ИаНСОз. Экстракты охлаждают и ммоль) в тетрагидрофуране (20 мл) и дицикподкисляют концентрированной соляной логексилкарбодиимид (3,29 г, 14,65 ммоль) в кислотой с получением твердого продукта, тетрагидрофуране (15 мл). Раствор переме- 35 после кристаллизации которого из метанола шивают при 0°С в течение 1 ч, а затем при получают названное соединение в виде бекомнатной температуре в течение ночи. Полых кристаллов, т.пл. 168-171 °С. сле отфильтровывания дициклогексилмочеП р и м е р 11. TpaHC-2-({N-[2-(N'-rnflpoвины и упаривания растворителей остаток 40. кси-Ы-метиламино)-2-оксоэтил]-Г\1-этиламирастворяют в етилацетате. После отфильтроно} карбонил)циклогексанкарбоновая кисвывания оставшейся дициклогексилмочевилота. ны раствор промывают последовательно К перемешанному раствору, содержаводой (2x20 мл), 10%-ным раствором лимонщему 4,28 г (15 ммоль) транс-2-({Ы-[2-(этокси) ной кислоты (3x20 мл), водой (20 мл), 5%-ным 2-оксоэтил]-1М-этиламино}карбонил) циклораствором ЫаНСОз (3x20 мл) и водой (20 мл). 45 гексанкарбоновой кислоты (промежуточный Органический слой сушат сульфатом магния амидоэфир, описанный в примере 2) в 21 мл и упаривают в вакууме. Полученное масло метанола прибавляют при 5°С 2,0 г (49,5 очищают с помощью мгновенной хроматогммоль) NaOH, растворенного в 21 мл метарафии (смесь этилацетат/петролейный нола, а затем 1,38 г (16,5 ммоль) хлоргидрата' эфир, 80/20). Маслянистый продукт (2,55 г, 50 N-метилгидроксиламина. После перемеши45%) имеет [ а ] 5 7 8 2 0 - +14,89°С (С = 6,2, вания смеси при 10°С в течение 4 ч раствоэтанол). Полученное масло (2,27 г, 6,27 ритель отгоняют в вакууме, а остаток ммоль) растворяют в 1М водном растворе растворяют в 10 мл воды и 10 мл этилацетаNaOH (50 мл) и смесь перемешивают в тече- 55 та. Перемешанную смесь медленно подкисние 2 ч при комнатной температуре. Раствор ляют 5%-ным раствором НС!, органический подкисляют 10%-ной НС1 при 0°С и экстраслой отделяют, водный раствор экстрагиругируют СНСІз (3x30 мл). Органический слой ют повторно 10 мл этилацетата. Экстракты экстрагируют 5%-ным раствором МаНСОз сушат (Na2SO4), растворитель отгоняют при (3x20 мл),основной раствор подкисляют пониженном давлении с осаждением осад 19 15920 ка, после рекристаллизации которого из простого эфира получают 3,4 г (79%) названного соединения в виде белого твердого вещества, т.пл. 132°С. П р и м е р 12. Метиловый эфир цис-({Ы- 5 [2-(гидроксиамино)-2-оксоэтил]-Ы-метиламино}карбонил] циклогексанкарбоновой кислоты. 2-Метоксикарбонилциклогексанкарбоновую кислоту (1,5 г, 8.06 ммоль) растворяют 10 в хлористом метилене (10 мл) и прибавляют раствор, содержащий трифторацетат 0-бензилсаркозингидроксамовой кислоты (2,48 г, 8,06 ммоль) и триэтиламин (1,1 мл, 8,06 ммоль). Полученный раствор охлаждают до 15 0°С, при быстром перемешивании прибавляют дициклогексилкарбодиимид (1,66 г, 8,06 ммоль), растворенный в хлористом метилене (20 мл). После выдерживания реакционного раствора при 0°С в течение 0,5 ч 20 смесь перемешивают при комнатной температуре в течение 3 ч с образованием дициклогексилмочевины, которую затем отделяют фильтрованием. После упаривания растворителя остаток растворяют в этилацетате и 25 после отфильтровывания остаточной дицик-* логексилмочевины (ДЦМ) промывают последовательно водой (30 мл), 10%-ным раствором лимонной кислоты (3x20 мл), водой, 5%-ным раствором NaHCOa (3x20 мл) и 30 водой (30 мл). Органический слой сушат над MgSO4 и упаривают в вакууме. Получают маслянистый продукт (1,85 г, 62%). Предшествующее соединение (1,57 г, 4,34 ммоль) гидрируют в метаноле (50 мл) с использова- 35 нием 10% Pd на активном угле (0,15 г). 20 ром и после его перекристаллизации и смеси простой эфир/хлористый метилен (2:1) получают названное соединение в виде белых кристаллов, т.пл. 140-141°С. П р и м е р 14. цис-2({Ы-(1-Метил-2-(Ы'ацетокси-N '-ацетил амино)-2-оксоэтил]-1Мметиламино} карбонил)циклогексанкарб6новая кислота. К раствору, содержащему соединение, полученное в примере 4 (330 мг, 1,28 ммоль), триэтиламин (0,55 мл, 3,95 ммоль) и N.N-диметиламинопиридин (10 мг) в дихлорметане (10 мл), перемешанному в атмосфере азота и охлажденному до 0°С, прибавляют уксусный ангидрид (270 мг, 2,65ммоль). Смесь нагревают до комнатной температуры, а для проверки окончательного ацилирования гидроксамового остатка некоторое количество его проверяют с использованием треххлористого железа (3). Затем смесь промывают последовательно 10%-ным раствором НСІ (2x10 мл), 5%-ным раствором ЫаНСОз (2x10 мл), водой и сушат с использованием MgSO-i. Растворитель отгоняют при пониженном давлении при комнатной температуре с выходом сырого продукта, после кристаллизации которого из диэтилового эфира получают твердое вещество белого цвета (306 мг, 70%), т.пл. 108-109°С. П р и м е р 15. Ацетоксиметиловый эфир цис-2-({Г\]-[2-(гидроксиамино-2-оксоэтил]-Мметиламино}карбонил) циклогексанкарбоновой кислоты. _ Смесь, состоящую из промежуточного 0-бензилизированного амида, полученного в примере 4 (4,5 г, 13 ммоль), и триэтиламина (i,8 мл, 13 ммоль) в тетрагидрофуране (30 После отфильтрования катализатора мл), прибавляют по каплям в атмосфере азораствор упаривают в вакууме и полученное та к раствору, содержащему модметилацемасло кристаллизуют из смеси ацетон/диэтиловый эфир. Получают белое твердое ве- 40 тат (3,0 г, 15 ммоль) в растворителе (20 мл), охлажденному до -5°С. Смесь перемешиващество (0,71 г, 60%), т.пл. 102°С. ют в течение 30 мин, а затем нагревают до П р и м е р 13. транс-2({Ы-[2-(Ацетилоккомнатной температуры. Осажденное белое сиамино)-2-оксоэтил]-1\1-этиламино}карбовещество отфильтровывают, а фильтрат упанил) циклогексанкарбоновая кислота. К перемешанной суспензии, содержа- 45 ривают в вакууме Полученный осадок растворяют в этилацетате (20 мл), промывают щей 2,0 г(7,3 ммоль) транс-2({Щ2-гидрокси5%-ным раствором ЫаСОз (2x20 мл), нодой и амино)-2-оксоэтил]-№-этиламино}карбонил) сушат (MgSO4). циклогексанкарбоновой кислоты (пример 2) в 20 мл хлористого метилена, при 5°С прибавляют 2,1 г (21,2 ммоль) уксусного ангид- 50 После отгонки растворителя при понирида, а затем по каплям 1,48 г (14,7 ммоль) женном давлении получают промежуточный триэтиламина. К полученному раствору за0-бензилированный ацетоксиметиламид тем прибавляют 0,4 г (0,36 ммоль) N.N-диме(3,38 г, 71 %) в виде желтого вязкого масла, чиламинопиридина и смесь перемешивают после гидрирования которого в течение 6 ч в течение 3 ч при комнатной температуре,. 55 в тетрагидрофуране (30 мл) в присутствии 10% палладия на угле получают названное Реакционную смесь промывают дважды соединение (количественный выход в виде 10 мл 5%-ного водного раствора HCI, затем белого стеклообразного продукта. насыщенным водным раствором NaCI (10 мл), сушат (СаСІг) и отгоняют растворитель. П р и м е р 1 6 . Метиловый эфир цис-2Остаточное масло промывают простым эфи({Ы-[2-(ацетоксиметилокси) амино-2-оксоэ 21 15920 22 бонил) циклопентанкарбоновая кислота. тил]-{\1-метиламино}кар6онил)циклогексанКристаллы цвета слоновой кости, т.пл. 143карбоновой кислоты. 146°С. К перемешанной смеси, содержащей соП р и м е р 25. транс-2-({Ы-[2-(Гидроксиединение примера 12 (1 г, 3,6 ммоль) и триэтилзмин (0,52 мл, 3,7 ммоль) в 5 амино)-2-оксоэтил}-Ы-фениламино}карбонил) циклогексанкарбоновая кислота. Белые тетрагидрофуране (25 мл), прибавляют иодкристаллы, т.пл. 151-152°С. метил ацетат (0,74 г, 3,6 ммоль) в растворитеП р и м е р 26. цис-2-({Ы-[2-(Гидроксиале (10 мл) в атмосфере азота. Смесь мино)-2-оксоэтил]-Ы-этиламино}карбонил) выдерживают при комнатной температуре в течение 98 ч. Осажденное твердое вещество 10 циклогексанкарбоновая кислота. Белые кристаллы, т.пл. 172-174°С. отфильтровывают и большу о часть раствоП р и м е р 27. цис-2-({М-[1-Метил-2рителя упаривают при пониженном давле(гидроксиамино)-2-оксоэтил]-Ыметиламино} нии. Остаток экстрагируют дихлорметаном и карбонил) циклогексанкарбоновая кислоохлажденным 5%-ным води *п раствором HCI, органический слой промывают 5%-ным 15 та. Кристаллы цвета слоновой кости, т.пл 8384°С. раствором HCI, 10%-иым раствором П р и м е р 28. транс-2-({1М-[1-Метил-2ЬІаНСОз, водой и сушат над MgSO4. (гидроксиамино)-2-оксоэтил}-М-метилзмино} Растворитель упаривают при пониженкарбонил) циклогексанкарбоновая кислоном давлении, получая названное соединение (0,9 г, 79 %) в виде масла бледно-желтого 20 та. Белые кристаллы, т.пл. 132-134°С. v цвета. П р и м е р 29. цис-2-({Ы-(1 -Бензил-2(гидроксиамино)-2-оксоэтил]-№-метиламино} В приводимых ниже примерах: соединекарбонил) циклогексанкарбоновая кислония 17-23, 27-34, получены по методике прита. Вязкое масло. мера 4, используя соответствующие П р и м е р 30. транс-2-({Ы-[1-Бензил-2исходные соединения, соединение 24 пол- 25 (гидроксиамино)-2-оксоэтил]-Ы-метиламиучено по методике примера 1, соединения но}карбонил) циклогексанкарбоновая кис25,26,37-41 получены по методике примера лота. Кристаллы цвета слоновой кости, 2, соединения 47 по методике примера 10, т.пл. 144-147°С. соединения 43-45 по методике примера 11, П р и м е р 31. транс-2-({(Ч-[1-(2-Фенилсоединение 35 по методике примера 8, 30 этил)-2-(гидроксиамино)2-оксоэтил]-ГЧ-меПример17. цис-2({Ы~[2-(Гидроксиамитиламино} карбонил)циклогексанкарбононо)-2-оксопропил]амино}карбонил)циклогевая кислота, Вязкое масло. ксанкарбоновая кислота. Белые кристаллы, т.пл. 145-148°С. П р и м е р 32. цис-2-({Ы-[1-(3-ФенилП р и м е р 18. цис-2({Ы-[1-Бензил-2-(гид- 35 пропил)-2-(гидроксиамино)-2-оксоэтил]роксиамино)-2-оксоэтилЗ амино}карбоN-метиламино) карбонил)циклогексанкарнил)циклогексанкарбоновая кислота. Белые боновая кислота. Белые кристаллы, т.пл. кристаллы, т.пл. 115-118°С. 192°С. (разложение). П р и м е р 19. цис-2({№-[1-(2-Фенилэтил)П р и м е р 33. транс-2-({1Ч-[1-(3-Фенил2-(гидроксиамино)-2-оксоэтилЗамино)карбо- 40 - пропил)-2-(гидроксиамино)-2-оксоэтил]- Nнил) циклогексанкарбоновая кислота. Беметиламино} карбонил) циклогексанлые кристаллы, т.пл. 83-85°С. карбоновая кислота. Белые кристаллы, П р и м е р 20. цие-2({Ы-[1-(3-Фенилпрот.пл. 150-155°С. пил)-2-(гидроксиамино)-2-оксоэтил] аминоП р и м е р 34. цис-2-({Ы-[1 -(2-Фенилэтил)к а р б о н и л ) ц и к л о г е к с а н к а р б о н о в а я 45 2-(гидроксиамино)-2-оксозтил]-М-метилкислота. Белые кристаллы, т.пл. 148-150°С. амино}карбонил)циклопентан карбоновая П р и м е р 21. транс-2-({Г\і-[2-(Гидроксикислота. Белые кристаллы, т.пл. 150-151 °С. амино)-2-оксоэтил)-Ы-метиламино]карбонил)П р и м е р 35. транс-2-({М-[1-Бензил-2циклогексанкарбоновая кислота. Белые (гидроксиамино)-2-оксоэтил)-Ы-этиламино}кристаллы, т.пл. 172-174°С. 50 карбонил) циклогексанкарбоновая кислота. П р и м е р 22, цис-2-({!\)-]1-(2-ФенилэДва рацемических соединения: бесцветные тил)-2-(гидроксиамино)-2-оксоэтил]амино} кристаллы, т.пл. 167-169°С, кристаллы цвета карбонил) циклопентанкарбоновая кислослоновой кости, т.пл. 96°С (разложение). та. Белые кристаллы, т.пл. 147-148°С. П р и м е р 36. транс-2-({Ы-[2-Бензил-3П р и м е р 23. цис-2-({Г\Н1-(3-Фенил 55 (гидроксиамино)-3-оксопропил]-Ы-этиламипропил)-2-(гидроксиамино)-2-оксоэтил] но)карбонил) циклогексанкарбоновая кисамино}-карбонил) циклопентанкарбоновая лота. Белое твердое вещество, т.пл. 94°С. кислота. Белые кристаллы, т пл. 122-12б°С. П р и м е р 24. цис-2-({Ы-[2-Бензил-3П р и м е р 37. цис-2-({Г\!-[3-(Гидроксиами(гидроксиамино) -3-оксопропил]амино}карно)-3-оксопропил]~г\1-этиламино}карбонил) 23 15920 циклогексанкарбоновая кислота. Бесцветные кристаллы, т.пл. 146-148°С. П р и м е р 38. транс-2-{{М-[3-{Гидроксиамино)-3-оксопропилЗ-М-этиламино}карбонил) циклогексанкарбоновая кислота. Бесцветные кристаллы, т.пл. 148-150°С. П р и м е р 39. цис-2-({Г\|-{2
ДивитисяДодаткова інформація
МПК / Мітки
МПК: A61K 31/165, A61P 9/12, C07C 259/00, A61P 43/00
Мітки: аміди, мають, кислот, гіпотензивну, циклометілен-1, 2-дикарбонових, активність
Код посилання
<a href="https://ua.patents.su/16-15920-amidi-ciklometilen-1-2-dikarbonovikh-kislot-yaki-mayut-gipotenzivnu-aktivnist.html" target="_blank" rel="follow" title="База патентів України">Аміди циклометілен-1, 2-дикарбонових кислот, які мають гіпотензивну активність</a>
Спосіб одержання аліфатичних дикарбонових кислот с -с та ефірів мнонокарбонових кислот с -с із 10 14 2 3 відходів виробництв себацинової та оцтової кислот

Номер патенту: 3386
Опубліковано: 27.12.1994
Автори: Шейко Тамара Павлівна, Ткаленко Петро Опанасович, Шейко Віктор Іванович, Кедріна Надія Миколаївна, Скубак Володимир Володимирович, Сорокін Валерій Євгенович, Постернак Світлана Михайлівна, Луб'яницький Ізраіль Якович, Терещенко Григорій Михайлович, Букаров Олексій Родіонович
МПК: C07C 51/09, C07C 55/02
Мітки: мнонокарбонових, спосіб, аліфатичних, виробництв, оцтової, дикарбонових, себацинової, кислот, ефірів, одержання, відходів
Формула / Реферат:
1. Способ получения алифатических ди-карбонових кислот С10-С14 и эфиров монокарбоиовых кислот С2-С3 из отходов производств себациновой и уксусной кислот путем взаимодействия сложных эфиров алифатических дикарбоновых кислот с монокарбоновой кислотой в присутствии воды и азотной кислоты при повышенной температуре с одновременным выводом образующегося эфира монокарбоновой кислоты из зоны реакции с последующим выделением кристаллизацией смеси...
Спосіб виділення gamma-валеролактону та суміші аліфатичних дикарбонових кислот с -с із відходу 6 10 виробництва себацинової кислоти

Номер патенту: 4572
Опубліковано: 28.12.1994
Автори: Сорокін Валерій Євгенович, Шейко Тамара Павлівна, Кедріна Надія Миколаївна, Луб'яницький Ізраіль Якович
МПК: C07D 307/33
Мітки: себацинової, суміші, аліфатичних, кислот, відходу, gamma-валеролактону, виробництва, спосіб, дикарбонових, кислоти, виділення
Текст:
...на ректификационной колонне нию степени извлечения у-валеролактона отгоняют воду и легколетучие соединения (I на стадии обработки отхода производства фракция) с содержанием у-валеролактона себациновой кислоты водно-аммиачным 30 0,4-0,9 мас,% под вакуумом, создаваемым раствором (см. пример 4). водоструйным насосом, затем при температуре 85°С и вакууме 11 мм рт.ст. отгоняют При использовании более высокого масу-валеролактон ПІ фракция) с...
2-[4-(дифенілметіл)-1-піперазиніл]-оцтові кислоти або їх аміди, або їх нетоксичні фармацевтично прийняті солі, що проявляють спазмолітичну і антигістамінну активність

Номер патенту: 8337
Опубліковано: 29.03.1996
Автори: Людовік Родрігез, Жан Де Ланноі, Ежен Бальтес
Мітки: активність, прийняті, нетоксичні, солі, спазмолітичну, антигістамінну, кислоти, аміди, проявляють, фармацевтично, 2-[4-(дифенілметіл)-1-піперазиніл]-оцтові
Формула / Реферат:
(57) 2-[4-(Дифенилметил)-1-пиперазинил]-уксусные кислоты или их амиды общей формулы ; где группа -ОН или NH2;m = 1 или 2;x - водород, хлор, фтор, метокси или трифторметилрадикал;х’ - водород или фтор, причем, х' - всегда водород, за исключением случая, когда х и х1 одновременно фтор;х - метокси или трифторметил при у - только группа NHa;m = 1 или 2 при х – водород или...
Гідрохлориди діфенілпропіламіну, що мають антиангінальну активність

Номер патенту: 3466
Опубліковано: 27.12.1994
Автори: Габор Ковач, Каталін Мармароші, Ласло Секереш, Пал Кішш, Імре Бата, Єва Удварі, Д'юла Папп, Дежьо Корбонітш, Андрео Шанта, Золтан Варгаі, Шандор Віраг, Віра Гергелі, Ласло Тардош, Петер Кьормеці
Мітки: діфенілпропіламіну, гідрохлориди, активність, мають, антиангінальну
Формула / Реферат:
Пидрохлориды дифениппропиламина формулы обладающие антиангинальной активностью.
7-алкокси-4-окса-1, 2, 6-тринітратгептантріоли, які мають кардіотонічну активність

Номер патенту: 2553
Опубліковано: 26.12.1994
Автори: Лозинський Мирон Онуфрійович, Клебанов Борис Маркович, Красавцев Іван Іванович, Шаваран Сергій Сергійович, Короткий Юрій Васильович
МПК: A61P 9/00, A61K 31/21, A61K 31/04, C07C 203/00
Мітки: 7-алкокси-4-окса-1, 6-тринітратгептантріоли, активність, кардіотонічну, мають
Формула / Реферат:
7-Алкокси-4-окса-1,2,6-тринитратгептантриолы общей формулы I:обладающие кардиотонической активностью.
Попередній патент: Спосіб гідравлічного розділення зернистих сумішей
Наступний патент: Засувка
Випадковий патент: Різцетримач