Піридо[2,3-d]піримідинові та піримідо[4,5-d]піримідинові нуклеозиди, проліки та спосіб інгібування ракових клітин
Номер патенту: 72612
Опубліковано: 15.03.2005
Автори: Пєтшковский Збігнєв, Гунік Есмір, Гонг Жі, Ванг Гуанжі, Лау Джонсон, Жирарде Жан-Лу








Формула / Реферат
1. Нуклеозидний аналог згідно з формулою (І)
(І),
де
А - це О, S, CH2,
R2, R2', R3 та R3' є незалежно вибраними з Н, F, OH, NH2, CN, N3, CONH2 та R, де R - це нижчий алкіл, нижчий алкеніл, нижчий алкініл або нижчий ацил, та він необов'язково містить принаймні один гетероатом та функціональну групу,
або R2 та R2' разом, або R3 та R3' разом є вибраними з =СН2, =CHR", =CR"2, =NR", де R" - це Η, F, ОН, CN, N3, CONH2, нижчий алкіл, нижчий алкеніл, нижчий алкініл або нижчий ацил,
R4 та R5' є незалежно вибраними з Н, нижчого алкілу, нижчого алкенілу, нижчого алкінілу або аралкілу та необов'язково містять принаймні один гетероатом та функціональну групу,
R5 - це Н, ОН, ОР(О)(ОН)2, Р(О)(ОН)2, OP(О)(OR'")2 або P(О)(OR'")2, де R'" - це маскувальна група, та
В є вибраним з групи гетероциклічних радикалів, що складається з формул (II), (III), (IV) та (V)
(ІІ),
(ІІІ),
(ІV),
(V),
де Χ - це Η, ΝΗ2 або ОН,
Υ - це Η, ΝΗ2 або галоген,
Ζ1 у формулах (II) і (IV) та Z3 у формулах (III) і (V) - це О, S, NR", СНМ або СМ2,
Ζ1 у формулах (III) і (V) та Z3 у формулах (II) і (IV) - це N, СН або CM,
Z2 - це Ν, СН або CM,
де Μ - це F, Сl, Br, OH, SH, NH2, CN, COOR", C(=NH)NH2, нижчий алкіл, нижчий алкеніл, нижчий алкініл, аралкіл або арил, та
де цукор у нуклеозиді може мати D- або L-конфігурацію.
2. Нуклеозидний аналог за п. 1, де А - це О та В - це гетероциклічний радикал за формулою (II).
3. Нуклеозидний аналог за п. 2, де Χ - це ΝΗ2, Ζ1 - це О та Z2 i Z3- це СН.
4. Нуклеозидний аналог за п. 3, де R4 і R5' - це водень та R5 - це ОН.
5. Нуклеозидний аналог за п. 1, який має структуру за формулою (VI)
(VI).
6. Проліки, що містять нуклеозидний аналог за п. 5.
7. Проліки за п. 6, де проліки містять фосфат або фосфонат, ковалентно зв'язаний з атомом С5 рибози.
8. Проліки за п. 6, де проліки містять складову, яка є ковалентно з'єднаною з принаймні однією гідроксильною групою рибози та яка відщеплюється від принаймні однієї гідроксильної групи у межах клітини-мішені.
9. Проліки за п. 6, де проліки містять складову, яка є ковалентно з'єднаною з аміногрупою основи та яка відщеплюється від аміногрупи у межах клітини-мішені.
10. Спосіб інгібування росту ракових клітин, який включає:
забезпечення сполукою за п. 1 та
введення сполуки у клітину в дозі, яка є ефективною для інгібування росту клітини.
11. Спосіб за п. 10, де А - це О та В - це гетероциклічний радикал за формулою (II).
12. Спосіб за п. 11, де Χ - це ΝΗ2, Ζ1 - це О та Z2 і Z3 - це СН.
13. Спосіб за п. 12, де R4 та R5' - це водень та R5 - це ОН.
14. Спосіб за п. 10, де сполука має структуру згідно з формулою (VI)
(VI).
15. Спосіб за п. 14, де сполука містить фосфат або фосфонат, ковалентно зв'язаний з атомом С5 рибози.
16. Спосіб за п. 14, де сполука містить складову, яка є ковалентно з'єднаною з принаймні однією гідроксильною групою рибози та яка відщеплюється від принаймні однієї гідроксильної групи у межах клітини-мішені.
17. Спосіб за п. 14, де сполука містить складову, яка є ковалентно з'єднаною з аміногрупою основи та яка відщеплюється від аміногрупи у межах клітини-мішені.
18. Спосіб за п. 14, де ракова клітина - це клітина, яку вибирають з групи, що складається з клітини раку товстої кишки, клітини раку молочної залози, клітини меланоми, клітини гліоми, клітини раку простати, клітини раку легенів, клітини раку печінки, клітини раку підшлункової залози та клітини раку яєчника.
19. Спосіб за п. 14, де інгібування росту клітини включає апоптоз.
20. Спосіб за п. 19, де апоптоз розпочинається принаймні частково завдяки МЕК-фосфорилюванню.
21. Спосіб за п. 14, де інгібування росту клітини включає інгібування принаймні однієї з РНК-полімерази І, РНК-полімерази II та РНК-полімерази III.
Текст
Для цієї заявки заявляється право пріоритету попередньої патентної заявки США за номером 60/216,418, поданої 6 липня 2000 року, яку в повному її обсязі включено до цього опису шляхом посилання. Галузь винаходу - це н уклеозидні аналоги. Нуклеозидні аналоги протягом тривалого часу застосовуються як антиметаболіти для лікування раку та вірусних інфекцій. Після входження у клітину багато нуклеозидних аналогів фосфорилюються нуклеозидними реутилізаційними шляхами перетворення до відповідного монофосфату нуклеозидкіназами, та монофосфати послідовно фосфорилюються кіназою до ди- та трифосфатів. Якщо нуклеозидний аналог перетворюється у його трифоефат усередині клітини, то він може служити як субстрат ДНК- або РНК-полімераз, і він може включатися у ДНК або РНК. Включення певних неприродних нуклеозидних аналогів у репл ікати або транскрипти нуклеїнових кислот може припиняти генну експресію шляхом ранньої термінації ланцюга модифікованих нуклеїнових кислот або втрати ними функції. Крім того, певні нуклеозидні аналоги є дуже ефективними інгібіторами ДНК- та РНК-полімераз, що може значно знижувати швидкість, з якою може приєднуватися природний нуклеозид. Крім того, нуклеозидні аналоги можуть також завдавати клітині шкоди за способом, відмінним від синтезу ДНК та/або РНК. Наприклад, деякі нуклеозидні аналоги можуть викликати апоптоз ракових клітин або інгібувати певні ферменти, відмінні від полімераз. Стосовно подальших альтернативних біологічних ефектів, відомо, що деякі нуклеозидні аналоги модулюють імунну систему. Звичайні приклади біологічних ефектів нуклеозидних аналогів включають інгібування тимідилатсинтази 5-фторуридином або інгібування аденозиндезамінази 2-хлораденозином. Подальші приклади включають інгібування S-аденозилгомоцистеїнгідролазену планоцином А. На жаль, проте, більшість з відомих нуклеозидних аналогів, які інгібують ріст пухлини або вірусні інфекції, також представляють загрозу нормальним клітинам ссавців, здебільшого через те, що таким аналогам не вистачає адекватної селективності стосовно нормальних та вірусних або пухлинних клітин. Отже, все ще існує необхідність у забезпеченні способами та композиціями щодо нуклеозидних аналогів з удосконаленою специфічністю та зниженою токсичністю. Цей винахід забезпечує новими нуклеозидами, що мають модифікації на нуклеозидній основі та/або цукровій складовій, які можуть суттєво підвищити селективність стосовно цитотоксичності до ракових клітин та/або знизити токсичність нуклеозидних аналогів до нормальних клітин. Особливо передбачені нуклеозиди включають піридо[2,3-d]піримідин, піримідо[4,5-d]піримідин та їхні похідні. Цукрові складові передбачених сполук можуть далі містити модифікації у положенні С 2, С3, С4 та/або C5. Більш специфічно, цим винаходом пропонуються нуклеозиди, які мають структуру за формулою (І): де А - це О, S, CH2; R2, R21, R3 та R3' є незалежно обраними з Н, F, OH, NH2 CN, N 3, CONH2 та R, де R - це нижчий алкіл, нижчий алкеніл, нижчий алкініл або нижчий ацил, та він необов'язково містить, принаймні, один гетероатом та функціональну груп у; або R2 та R2' разом, або R3 та R3' разом є обраними з =СН2, =CHR", =CR"2, =NR", де R" - це Η, F, OH, CN, N3, CONH2, нижчий алкіл, нижчий алкеніл, нижчий алкініл або нижчий ацил; R4 та R5' є незалежно обираними з Н, нижчого алкілу, нижчого алкенілу, нижчого алкінілу або аралкілу та необов'язково містять, принаймні, один гетероатом та функціональну групу; R5 - це Н, ОН, ОР(О)(ОН)2, Р(О)(ОН)2, OP(O)(OR'")2 або P(O)(OR'")2, де R"1 - це маскувальна група, та В є обраним з групи гетероциклічних радикалів, що складається з формул (II), (III), (IV) та(У) де Χ - це Η, ΝΗ2 або ОН; Υ - це Η, ΝΗ2 або галоген; Ζ1 у формула х (II) і (IV) та Z3 у формула х (III) і (V) - це О, S, NR", СН М або СМ2 ; Ζι у формулах (III) і (V) та Ζ3 у формулах (II) і (IV) - це Ν, СН або CM; Z 2 - це Ν, СН або CM, де Μ - це F, СІ, Вr, ОН, SH, ΝΗ2, CN, COOR", C(=NH)NH2, нижчий алкіл, нижчий алкеніл, нижчий алкініл, аралкіл або арил. В особливо переважному аспекті нуклеозидний аналог має структур у згідно з формулою (VI): У подальших передбачених аспектах предмету винаходу нуклеозидний аналог може бути модифікований для утворення відповідних проліків, та особливо передбачені модифікації включають фосфорилювання або додавання фосфонатної гр упи у положенні С 5 цукрової складової, модифікації на гідроксильних гр упах у цукровій складовій та модифікації на аміногрупі нуклеїнової основи. Особливо переважним є те, щоб такі модифікації можна було б відщеплювати від передбачених сполук у компартменті-мішені, клітині-мішені або органі-мішені. У іншому аспекті предмету винаходу спосіб інгібування росту пухлинних клітин містить етап, на якому передбачені сполуки згідно з формулою (І) вводять у систему, переважно ссавцю, та більш переважно людині. Особливо передбачені пухлинні клітини включають клітини раку товстої кишки, клітини раку молочної залози, клітини меланоми, клітини гліоми та клітини раку простати. Далі передбачається, що інгібування росту включає інгібування РНК-полімерази І, РНК-полімерази II та/або РНК-полімерази III та/або індукування апоптозу, який може викликатися, принаймні частково, фосфорилюванням мітоген активоаною кіназою керованою ззовні кіназою (МЕК). Стислий опис ілюстративного матеріалу Фігура 1 - графік, що зображує порівняльну цитотоксичність прикладу передбаченої сполуки стосовно різних ракових клітин. Фігура 2 - графік, що зображує порівняльне інгібування проліферації різних ракових клітин прикладами передбачених сполук. Фігура 3 - графік, що зображує порівняльну антиклоногенну активність прикладів передбачених сполук стосовно різних ракових клітинах. Фігури 4 А та 4В - графіки, що зображують індукування апоптозу різних ракових клітин прикладами передбачених сполук. Фігура 5 - графік, що зображує інгібування синтезу РНК у клітинах К562 прикладами передбачених сполук. Фігура 6 - графік, що зображує інгібування РНК-полімерази І та III прикладами передбачених сполук. Фігура 7 - графік, що зображує інгібування РНК-полімерази II прикладами передбачених сполук. Фігура 8 - авторадіограма, що зображує вплив прикладів передбачених сполук на MEK-ERKфосфорилювання. Докладний опис винаходу Передбачені сполуки Взагалі передбачається, що сполуки згідно з предметом винаходу мають структуру за формулою (І): де А - це О, S, CH2; R2, R2', R3 та R3' є незалежно обраними з Н, F, OH, NH 2, CN, N3, CONH2 та R, де R - це нижчий алкіл, нижчий алкеніл, нижчий алкініл або нижчий ацил, та він необов'язково містить, принаймні, один гетероатом та функціональну груп у; або R2 та R21 разом, або R3 та R31 разом є обраними з =СН2, =CHR", =CR''2, =NR", де R" - це Η, F, OH, CN, N3, CONH2, нижчий алкіл, нижчий алкеніл, нижчий алкініл або нижчий ацил; R4 та R5' є незалежно обраними з Н, нижчого алкілу, нижчого алкенілу, нижчого алкінілу або аралкілу та необов'язково містять, принаймні, один гетероатом та функціональну групу; R5 - це Н, ОН, ОР(О)(ОН)2, Р(О)(ОН)2, OP(0)(OR'") 2 або P(O)(OR''')2, де R'" - це маскувальна група, та В є обраним з групи гетероциклічних радикалів, що складається з формул (II), (III), (IV) та (V) де Χ - це Η, NHb або ОН; Υ - це Η, ΝΗ2 або галоген; Ζ1 у формулах (II) і (IV) та Ζ3 у формулах (ІII) і (V) - це О, S, NR", СНМ або СМ2; Ζ1 у формулах (III) і (V) та Ζ3 у формула х (II) і (IV) - це Ν, СН або CM; Z2 - це Ν, СН або CM, де Μ - це F, СІ, Вr, ОН, SH, NH2, CN, COOR", C(=NH)NH 2, нижчий алкіл, нижчий алкеніл, нижчий алкініл, аралкіл або арил. У переважному аспекті предмету винаходу передбачені сполуки мають гетероциклічний радикал за формулою (II), А - це кисень у цукровій складовій нуклеозидного аналога, та навіть більш переважно, що у таких нуклеозидних аналогах X - це ΝΗ2, Ζ 1 - це О, та Ζ2 і Z 3 - це СН. Не обмежуючи предмету винаходу, іще далі переважно, що R4 та R5 ' - це водень, та R5 - це ОН у цукровій складовій. Далі в особливо переважних аспектах предмету винаходу передбачені нуклеозидні аналоги мають структур у за формулою (VI): Стосовно стереохімічної конфігурації передбачених сполук слід відзначити, що цукор не слід обов'язково обмежувати D-конфігурацією, він може також мати L-конфігурацію. Подібно до цього, передбачається, що придатні молекули можуть включати один або більше хіральних центрів, що вони можуть бути енантіомерно чистими (тобто, у R- або S-конфігурації) або у рацемічній суміші (тобто, у R- та S-конфігурації). Подібно до цього, там, де замісники (наприклад, основа або група ОН) можуть демонструвати орієнтацію у a- або βположенні, передбачаються обидва положення. Крім того, слід відзначити, що передбачені сполуки можна модифікувати до їх відповідних проліків. Термін "проліки", що застосовується тут, позначає будь-яку модифікацію передбачених сполук, яка (а) змінює молекулярну вагу передбачених сполук та/або (б) змінює біологічну доступність передбачених сполук стосовно клітини-мішені та клітини, яка не ε мішенню. Наприклад, проліки можна приготувати шляхом естерифікації гідроксильної групи передбачених сполук органічною кислотою (тим самим змінюючи молекулярну вагу, при цьому не обов'язково змінюючи біологічну доступність стосовно клітини-мішені). З іншого боку, передбачені сполуки можна перетворити у такі проліки, які б включали циклічний фосфонатний естер (при цьому підвищуючи біологічну доступність стосовно печінкових клітин). Крім того, слід особливо відзначити, що пролікарські форми передбачених сполук можуть повністю або частково знов перетворюватися у передбачені сполуки в органі-мішені, клітині-мішені або компартменті-мішені (або у будь-якому середовищі, яке не є мішенню). Зворотне перетворення може включати різні механізми, та особливо передбачені механізми - це ферментативна конверсія, окиснення та/або відновлення. Проліки можна також застосовувати для підвищення специфічності передбачених сполук стосовно органамішені, клітини-мішені або компартмента-мішені. Наприклад, передбачені сполуки можна сполучити з холестерином (або похідним холестерину) для підвищення концентрації передбачених сполук у межах гепатобіліарної циркуляції. Альтернативно, передбачені сполуки можна з'єднати зі сполукою, що має відповідний рецептор на клітині-мішені, тим самим підвищуючи концентрацію передбачених сполук на або у клітині-мішені. У іще іншому прикладі передбачені сполуки можна з'єднати з сигналом ядерної транслокації для підвищення концентрації передбачених сполук у межах ядра клітини. Крім того, проліки можна застосовувати для зменшення акумуляції передбачених сполук в органах, що не є мішенями, клітинах, що не є мішенями, або у компартментах, що не є мішенями. Наприклад, передбачені сполуки можна модифікувати так, щоб клітина, яка не є мішенню, мала б значно знижену швидкість поглинання передбачених сполук, коли такі сполуки модифіковані у проліки. Отже, та особливо там, де передбачені сполуки демонструють цитотоксичність до клітини, що не є мішенню, проліки можна застосовувати для зниження цитотоксичності передбачених сполук у клітинах та органах, відмінних від клітинмішеней або органів-мішеней. У подальших передбачених аспектах предмету винаходу придатні сполуки можуть бути ковалентно з'єднаними з фармакологічно активними або неактивними складовими. Наприклад, фармакологічно активні складові включають протипухлинні ліки, такі як антиметаболіти (наприклад, Pentostatin™), інгібітори ДНКполімерази (наприклад, Gemzar™), інгібітори РНК-полімерази (наприклад, ECyd™), платинові похідні (наприклад, Paraplatin™), антиестрогени (наприклад, Nolvadex™), таксани (наприклад, Taxotere™), аналоги GnRH (наприклад, Lupron™), інгібітори ДНК-полімерази (наприклад, Gemzar™), інгібітори топоізомерази (наприклад, Hycamptin™), біфосфонати (наприклад, Aredia™), соматостатини (наприклад, Sandostatin™), інтерферони (наприклад, IntronA™), н уклеозидні аналоги (наприклад, Ribavirin™) та інгібітори IMPDH (наприклад, Tiazofutin™). Фармакологічно неактивні складові включають біологічні та небіологічні складові. Наприклад, там, де специфічність до мішені є особливо бажаною, передбачені сполуки можна з'єднати з антитілом, фрагментом антитіла або синтетичним антитілом (наприклад, scFv). У подальшому прикладі передбачені сполуки можуть бути з'єднані з хелатом (наприклад, з хелатом, який зв'язує радіоізотоп). Альтернативно, там, де особливо переважним є подовження періоду напівжиття у кров'яному руслі або знижена імуногенність, передбачені сполуки можуть бути з'єднаними з інертними полімерами або полімерами, здатними до біологічного розкладу (наприклад, декстрином, поліетиленгліколем тощо). Стосовно способу з'єднання передбачених сполук з іншими складовими вважають, що усі відомі способи з'єднання є доречними, та вони особливо включають ковалентний зв'язок (з або без окремої лінкерної молекули), водневий зв'язок та гідрофобні/гідрофільні взаємодії. У подальших аспектах предмету винаходу передбачені сполуки можуть також бути у виді їх відповідних солей, де сіль може бути сіллю органічної або неорганічної кислоти, або основи (наприклад, ацетатна сіль або морфоліно-сіль, сіль НСl). У галузі відомо багато фармакологічно прийнятних солей, та вважають, що усі відомі солі є прийнятними для застосування у зв'язку з концепцією, запропонованою тут. Синтез передбачених сполук Синтез модифікованих рибофураноз Приклади схем, які наведено нижче, демонструють способи синтезу деяких передбачених сполук. Сполуку 1, яку приготували згідно з опублікованим способом (Jones et al. Methods in Carbohydrate Chemistry (edited by Whistier and Moffat), vol. VI, pp 315-322, Academic Press, New York, (1972)), обробляли рядом нуклеофілів, такими як реактиви Гріньяра, внаслідок чого одержали сполуку 2, яку бензоїлували або ацетилювали та далі обробили трифторооцтовою кислотою, внаслідок чого одержали сполуку 4. Внаслідок бензоїлування та наступної обробки ацетангідридом/оцтовою кислотою у присутності сірчаної кислоти одержали сполуку 6, яку застосовували для конденсації з піридо[2,3-d]піримідиновими або піримідо[4,5-d]піримідиновими основами. 6а R = Me 6b R = етиніл 6с R = вініл 6d R = аліл Сполуку 2а (5(R)-С-метильне похідне) перетворили у еульфонат 7, який піддали нуклеофільному заміщенню, внаслідок чого одержали сполуку 8 зі зворотною конфігурацією. Внаслідок усування захисту ізопропілідену та наступного ацетилування одержали тетраацетат 9. Сполуку 1 обробили формальдегідом у водному гідроксиді натрію, внаслідок чого одержали 4'гідроксиметильне похідне 10, яке була селективно захищено, внаслідок чого одержали сполуку 11. Внаслідок наступного захищення диметилтерефталатом (DMT) та видалення трет-бутилстиролу (TBS) одержали сполуку 13, яку можна перетворити у ряд замісників. 4-С-заміщені похідні, які зазнали схожих трансформацій, такі як 5-С-заміщені рибофуранози, можна перетворити у сполуку 17, яка застосовується для конденсації з нуклеозидними основами. Сполуку 13 перетворили у сполуку 18, яку піддали реакції Віттига, внаслідок чого одержали сполуку 19. Внаслідок гідрування сполуки 19 над паладієм одержали сполуку 20, Сполуку 13 перетворили у 4-Сфенокситіокарбонілоксиметильне похідне 21, яке піддали реакції з трис(триметилсиліл)силаном (9,0 мл, 29 ммоль), а потім з 1,1-азобіс(циклогексанкарбонітрилом), внаслідок чого одержали сполуку 22. Там, де передбачені сполуки містять цукрову складову, яка не модифікована у положенні С 4' або C5' (наприклад, L-рибофуранозу, 2'-гідрокси-2'-етиніл-L-рибофуранозу), конденсацію відповідних захищених цукрових складових (у D- або L-конфігурації) здійснюють за способами, добре відомими у галузі. Синтез модифікованих піридопіримідинових нуклеозидів 5'-заміщені нуклеозидні аналоги одержали конденсацією силілованих піридо[2,3-d]піримідинових основ та належним чином захищених модифікованих рибофураноз. Наступна схема демонструє синтез 4-аміно-5-оксопіридо[2,3-d]піримідину 25. Сполуку 23, одержану згідно з опублікованим способом (Archive der Pharmazie 1985, 318,481 -486), кип'ятили зі зворотним холодильником з хлортриметилсиланом та йодидом натрію в ацетонітрилі, внаслідок чого одержали сполуку 24. Внаслідок реакції сполуки 24 з формамідинацетатом в умовах кип'ятіння зі зворотним холодильником одержали сполуку 25. Наступні схеми демонструють конденсацію піридо[2,3-сі]піримідину 25 та модифікованих рибофураноз 6 та 17. Сполуку 25 обробили (біс(триметилсилі)ацетамідом, внаслідок чого одержали силілований піридопіримідин, який реагував зі сполукою 6 або 17, внаслідок чого утворилася сполука 26 або 28, відповідно. Видалення бензоїлу дозволило одержати піридо[2,3 d]піримідинові нуклеозиди 27 та 29, відповідно. R = Me, етил, вініл, пропіл, аліл, гідроксиметил Інші піридопіримідинові або піримідопіримідинові нуклеозиди з модифікованим цукром можна одержати як шляхом конденсації нуклеозидних основ та модифікованих цукрів, так і шля хом модифікацій нуклеозидів. Наприклад, 4-аміно-5-оксо-піридо[2,3-d]піримідинрибозу (27 або 29 R = Н) перетворили у 2'-дезокси-похідне 30 за процедурою, подібною до процедури, описаної для 2'-дезоксиаденозину, та 4-аміно-5-оксо-піридо[2,3d]піримідинксилозид 34 приготували шляхом конденсації. 4-Аміно-5-оксопіридо[2,3-d]піримідин конденсували з рядом 1--ацетильованих пентозних цукрів шля хом реакцій за Vorbruggen'oM. Внаслідок наступного утворення нуклеозидних похідних одержали додаткову гр упу піридо[2,3-а]піримідинових нуклеозидів. Синтез модифікованих піримідопіримідинових нуклеозидів 5'-Заміщені нуклеозидні аналоги одержали конденсацією силілованих піримідо[4,5-d]піримідинових основ та належним чином захищених модифікованих рибофураноз. Синтез піримідо[4,5-d]піримідинових нуклеозидів проводиться за процедурою, яка є суттєво подібною до процедури синтезу, описаної вище. Залежно від умов реакцій конденсації, які застосовуються для приєднання цукрової складової до піридо[2,3-d]піримідинової або піримідо[4,5-d]піримідинової основи, відповідні нуклеозидні аналоги можна приєднати на атомі Νi або Ns основи. У будь-якому випадку місце глікозилування основи встановили за рентгенівською кристалічною структурою. Застосування передбачених сполук Слід взагалі признати, що передбачені сполуки можна застосовувати у будь-якому лікуванні або терапії системи, яка позитивно реагує на введення передбачених сполук. Проте, особливо переважно, щоб передбачені сполуки можна було б застосовувати у протипухлинному лікуванні, та у противірусному лікуванні (як пряму противірусну сполуку та/або як непряму противірусну сполуку), та у лікуванні, спрямованому на модулювання імунної системи. Протипухлинне лікування Взагалі передбачається, що сполуки згідно з предметом винаходу можна застосовувати як протипухлинні агенти, які безпосередньо або опосередковано інгібують ріст, інвазивність та/або розповсюдження пухлинної клітини або популяції пухлинних клітин. Особливо передбачається, що спосіб лікування пухлинних хвороб у пацієнта містить етап, на якому передбачені сполуки вводять пацієнтові у дозі, яка є ефективною у інгібуванні росту п ухлинної клітини, та особливо переважною сполукою є сполука за формулою (VI, вище). Передбачені дози становлять діапазон між 0,01 - 100 мг/кг, та більш переважні дози становлять діапазон між 5 - 50 мг/кг. Однак також передбачаються альтернативні дози, способи та режими введення та фармацевтичні склади. Придатні альтернативні варіанти введення описано нижче. Незважаючи на те, що застосування передбачених сполук не обмежується певною пухлинною клітиною або пухлинною хворобою, особливо передбачені пухлинні клітини включають клітини раку товстої кишки, клітини раку молочної залози, клітини меланоми, клітини гліоми та клітини раку простати. Противірусне лікування Взагалі передбачається, що сполуки згідно з предметом винаходу можна застосовувати як прямий та/або непрямий противірусний агент у лікуванні вірусної інфекції. Особливо передбачається, що спосіб лікування вірусної інфекції у пацієнта містить етап, на якому передбачені сполуки вводять пацієнтові у дозі, ефективній інгібувати репродукцію вірусу (тобто, процес, який залучає клітину хазяїна, у якій один або більш ніж один вірус примушує клітину-хазяїна виробляти одну або більше копій вірусу, де термін "виробляти" позначає синтез нуклеотиду, процесінг білка та складання білкової структури), та де композиція містить, принаймні, одну з передбачених сполук. Передбачені дози становлять діапазон між 0,1-100 мг/кг, та більш переважні дози становлять діапазон між 5 - 50 мг/кг. Проте також передбачаються альтернативні дози, способи, режими введення та фармацевтичні склади, та придатні альтернативні варіанти введення описано нижче. Незважаючи на те, що застосування передбачених сполук не обмежується певним вірусом у певній вірусній інфекції, особливо передбаченими вірусними інфекціями є інфекція ВІЛ, інфекція вірусу гепатиту С, інфекція вірусу гепатиту В, інфекція респіраторного синцитіального вірусу, інфекція вірусу грипу та інфекція вірусу парагрипу. Імуномодуляція Взагалі передбачається, що сполуки згідно з предметом винаходу можна застосовувати як імуномодуляторні сполуки, та особливо передбачається, що такі сполуки можна застосовувати для модуляції балансу між реакцією Типу 1 та реакцією Типу 2 імунокомпетентної клітини (наприклад, Т-клітини) у напрямку стимуляції. Більш специфічно, передбачається, що сполуки згідно з предметом винаходу можуть підвищувати реакцію Типу 1 відносно реакції Типу 2 (як шляхом підвищення реакції Типу 1, так і шля хом пригнічення реакції Типу 2). Проте також передбачається, що сполуки згідно з предметом винаходу можуть підвищувати реакцію Типу 2 відносно реакції Типу 1 (як шляхом підвищення реакції Типу 2, так і шля хом пригнічення реакції Типу 1). Щодо подальших передбачених варіантів застосування сполук згідно з предметом винаходу слід відзначити, що передбачені сполуки можна також застосовувати як імуносупресори за концентрацією, ефективною для пригнічування як реакції Типу 1, так і реакції Типу 2. Введення передбачених сполук Стосовно введення передбачених сполук слід відзначити, що сполуки можна вводити за будь-яким відповідним протоколом у будь-якому відповідному фармацевтичному складі. Взагалі краще за все, проте, щоб передбачені сполуки вводилися перорально. У подальших аспектах предмету винаходу слід відзначити, що різні альтернативні варіанти введення є також доречними, та також слід іще далі признати, що певний варіант введення буде взагалі залежати від хімічної стійкості, біологічної доступності, дози, фармацевтичного складу та/або бажаних фармакокінетичних/фармакодинамічних властивостей передбачених сполук. Отже, відповідні варіанти введення будуть включати пероральне введення (наприклад, таблетки, сиропи тощо), місцеве введення (наприклад, мазь, аерозоль, крем тощо), парентеральне системне введення (наприклад, інгаляція) та прямий або непрямий спосіб введення у кровотік (наприклад, внутрішньовенна або внутрішньом'язова ін'єкція тощо). Отже, фармацевтичний склад передбачених сполук може різнитися суттєво. Наприклад, там, де ліки або склад ліків демонструють суттєву стійкість під час проходження крізь шлунково-кишкову систему без небажаних хімічних або ферментативних модифікацій, фармацевтичні склади для перорального введення можуть включати сироп, таблетки, гелеві капсули, порошки тощо. З іншого боку, там, де абсорбція або проникнення передбачених сполук крізь шлунково-кишковий тракт у кровотік є проблемним, придатні фармацевтичні склади особливо включають розчини або суспензії для ін'єкцій (наприклад, фізіологічний соляний розчин, забуферений до рН приблизно 7,2 - 7,5). Стосовно дози передбачених сполук слід відзначити, що різні дози є доречними, та передбачені дози звичайно становлять діапазон від 0,1 мг/кг до декількох 100 мг/кг, та навіть більше. Наприклад, там, де передбачені сполуки виділяються або метаболізуються за відносно низькою швидкістю, або там, де бажаним є довготермінове лікування, дози будуть звичайно становити діапазон між 0,5 мг -10 мг/кг. З іншого боку, там, де біологічна доступність передбачених ліків є відносно низькою, або там, де метаболічне перетворення є відносно швидким, дози звичайно становитимуть діапазон між 10 мг/кг -100 мг/кг. Стосовно дози передбачених сполук, слід далі відзначити, що, принаймні, деякі сполуки згідно з предметом винаходу можуть фосфорилюватися in vivo. Отже, та особливо там, де бажаною є негайна біологічна доступність, дози можна знизити, коли передбачені сполуки вводяться у фосфорилованій формі. Режим введення може різнитися суттєво, та передбачені режими включають введення разової дози протягом усього курсу лікування, введення декількох разових щоденних доз протягом усього курсу лікування, введення декількох щоденних доз та постійне введення (наприклад, перманентна інфузія, імплантована осмотична помпа тощо) протягом, принаймні, частини курсу лікування. Незважаючи на те, що взагалі переважно, щоб доречні режими підтримували постійне введення передбачених сполук, імпульсне введення (тобто, принаймні одне введення за першою дозою з наступним, принаймні, іще одним введенням за дозою, нижчою ніж перша доза) є також доречним. Стосовно тривалості лікування, передбачається, що відповідні терміни можуть різнитися від разового введення до декількох днів, декількох тижнів, декількох років та навіть триваліше. Наприклад, там, де передбачені сполуки застосовуються у клітинній культурі, може вистачити разового введення або відносно короткого терміну введення. З іншого боку, там, де передбачені сполуки вводяться для лікування гострої фази хвороби, відповідний термін лікування може становити діапазон від декількох днів до декількох тижнів. Подібно до цього, там, де хронічні хвороби лікуються шляхом введення передбачених сполук, триваліше введення протягом одного або більше років може бути доречним. У подальших альтернативних аспектах предмету винаходу передбачені сполуки можуть поєднуватися з додатковими фармацевтично активними речовинами для сприяння лікуванню різних хвороб та особливо пухлинних хвороб. Додаткові фармацевтично активні речовини можуть вводитися окремо або разом, та коли вони вводяться окремо, введення можна здійснювати одночасно або окремо у будь-якій черзі. Особливо передбачені додаткові фармацевтично активні речовини включають ліки, що звичайно застосовуються як хіміотерапевтичні агенти для лікування раку та як імуномодулятори. Наприклад, хіміотерапевтичні агенти включають антиметаболіти (наприклад, Pentostatin™), інгібітори ДНК-полімерази (наприклад, Gemzar™), інгібітори РНК-полімерази (наприклад, ECyd™), платинові похідні (наприклад, Paraplatin™), антиестрогени (наприклад, Nolvadex™), таксани (наприклад, Taxotere™), аналоги GnRH (наприклад, Lupron™), інгібітори ДНК-полімерази (наприклад, Gemzar™), інгібітори топоізомерази (наприклад, Hycamptin™), біфосфонати (наприклад, Aredia™), соматостатини (наприклад, Sandostatin™), нуклеозидні аналоги (наприклад, Ribavirin™) та інгібітори IMPDH (наприклад, Tiazofutin™). Передбачені імуномодулятори включають цитокіни (наприклад, α та γ, IL2, IL4, IL6, IL8, IL10 та IL12), цитокініни (наприклад, кінетин) та хемокіни (наприклад, МІР-1). Приклади Наступні приклади передбачають приклади синтезу експериментів in vitrolin vi vo, та призначені для ілюстрації винаходу без його обмеження. Синтез Одержання 2,3-О-ізопропіліден-5 (R,S)-С-етиніл-1-О-метил-b-D-рибофуранози До перемішаного розчину метил-4-С,5-O-дидегідро-2,3-0-ізопропіліден-b-D-рибофуранозиду (Jones et al. Methods in Carbohydrate Chemistry Vol. 1, pp 315-322 (1972), 4,00 r, 19,78 ммоль) у безводному тетрагідрофурані (THF) (20 мл) при 42°С в умовах аргону додавали по краплях етинілмагнійбромід (0,5 Μ у те трагідрофурані, 80 мл, 40 ммоль). Після додавання одержану суміш повільно нагрівали до 0°С (приблизно 90 хвилин). Реакцію загасили шляхом додавання льоду (50 г)/води (50 мл), та суміш перемішували протягом 30 хвилин. Після нейтралізації 10% водною оцтовою кислотою суміш двічі екстрагували етилацетатом. Комбінований органічний шар висушили (Na2SO4) та концентрували. Внаслідок хроматографії на діоксиді кремнію (етилацетат-гексани, 1:4) одержали 3,48 г сполуки, зазначеної в заголовку, (співвідношення R/S 1:1) у вигляді білої твердої речовини. Подібно до цього приготували наступні сполуки: 1-0,5(R)-С-Диметил-2,3-0-ізопропіліден-b-D-рибофуранозу з 4-С,5-Oдидегідро-2,3-O-ізопропіліден-1-О-метил-Р-О-рибофуранози та метилмагнійброміду, 2,3-0-ізопропіліден-1-0метил-5(ІІ)-С-вініл-р-О-рибофуранозуз4-С,5-0-дидегідро-2,3-0-ізопропіліден-1-0-метил-р-0-рибофуранози та вінілмагнійброміду, 5(R)-С-Аліл-2,3-0-ізопропіліден-1-0-метил-b-D-рибофуранозу з 4-С,5-О-дидегідро-2,3-Оізопропіліден-1 -0-метил--b-D-рибофуранози та алілмагнійброміду. Одержання 5-0-ацетил-і-0,5 (S)-C-диметіл-2,3-О-ізопропіліден-b-D-рибофуранози До перемішаного розчину 1-0,5(R)-С-диметил-2,3-0-ізопропіліден-b-D-рибофуранози (7,24 г, 33,17 ммоль) у безводному піридині (50 мл) при 0°С додали метансульфонілхлорид (3,1 мл, 39,92 ммоль). Одержану суміш перемішували при кімнатній температурі протягом 1 години, охолодили до 0°С, загасили шляхом додавання води (1,0 мл) та перемішували при кімнатній температурі протягом 30 хвилин. Розчинник випарили та залишок розчинили в етилацетаті, тричі промили соляним розчином, висушили (Na2SO 4) та концентрували. Внаслідок хроматографії на діоксиді кремнію (30% ЕtOАс у гексанах) одержали 8,62 г метилату у вигляді безбарвного сиропу. Перемішану суспензію 1-0,5(R)-С-диметил-2,3-0-ізопропіліден-5-0-метансульфоніл-b-D-рибофуранози (8,62 г, 29,1 ммоль) та NaOAc (безводний, 3,5 г, 42,5 ммоль) у безводному Ν,Ν-диметилформаміді (DMF) (350 мл) нагрівали при 125°С в умовах аргону протягом 4 днів. Розчинник випарили та внаслідок хроматографії залишку на діоксиді кремнію (25% ЕtOАс у гексанах) одержали 4,0 г сполуки, зазначеної в заголовку, у вигляді білої твердої речовини. Одержання 5-дезокси-2,3-0-ізопропіліден-1-0-метил--b-D-рибофуранози До перемішаного розчину 2,3-0-ізопропіліден-1-0-метил-b-D-рибофуранози (14,2 г, 70,0 ммоль) у безводному піридині (250 мл) при 10°С додавали частинами (протягом 30 хвилин) п-толуолсульфонілхлорид (19,1 г, 100 ммоль). Одержану суміш перемішували при кімнатній температурі протягом 18 годин, охолодили до 0°С, загасили шляхом додавання води (5,0 мл) та перемішували при кімнатній температурі протягом 30 хвилин. Розчинник випарили. Залишок розчинили в етилацетаті, промили тричі соляним розчином, висушили (Na2SO4) та концентрували до сухого стан у. Внаслідок хроматографії на діоксиді кремнію (етилацетат-гексани, 1:3) одержали 24,1 г тозилату у вигляді білої твердої речовини. До перемішаної суспензії LiAlH4 (4,58 г, 120,5 ммоль) у безводному діетиловому ефірі (120 мл) додали тозилат (13,1 г, 36,55 ммоль) у діетиловому ефірі-толуолі (2,5:1, 140 мл). Одержану суміш кип'ятили зі зворотним холодильником протягом 22 годин, охолодили до кімнатної температури, розвели етилацетатом (25 мл), загасили шляхом додавання води (5,0 мл). Розчинник випарили. Залишок розчинили в етилацетаті, тричі промили соляним розчином, висушили (Na 2SO4) та концентрували до сухого стан у. Внаслідок хроматографії на діоксиді кремнію (етилацетат-гексани, 1:3) одержали 3,58 г сполуки, зазначеної в заголовку, у вигляді безбарвної рідини. Одержання 5 (R)-С-аліл-5-О-бензоїл-1-О-метил-b-D-рибофуранози До перемішаного розчину 5(Е)-С-аліл-2,3-0-ізопропіліден-1-0-метил-р-О-рибофуранози (4,49 г, 18,38 ммоль) у безводному піридині (40 мл) при 0°С додали бензоїлхлорид (2,7 мл, 23,0 ммоль). Одержану суміш перемішували при кімнатній температурі протягом 18 годин, охолодили льодом, загасили шляхом додавання води (1 мл) та перемішували при кімнатній температурі протягом 30 хвилин. Розчинник випарили та залишок розчинили в етилацетаті, промили тричі соляним розчином, висушили (Na2SO 4) та концентрували. Внаслідок хроматографії на діоксиді кремнію (12% етилацетат у гексанах) одержали 6,26 г 5(ІІ)-С-аліл-5-0-бензоїл-2,3-0ізопропіліден-і-О-метил-b-D-рибофуранози у вигляді безбарвного сиропу. Розчин 5(R)-С-аліл-5-0-бензоїл-2,3-0-ізопропіліден-1-Ο-метил-b-D-рибофуранози (6,2 г, 17,8 ммоль) у суміші тетрагідрофуран-вода (9:1) перемішували при 0°С протягом 90 хвилин та концентрували до сухого стану при 0°С. Залишок розчинили у суміші метанолу-толуолу (20 мл, 1:1) та концентрували до сухого стану. Внаслідок хроматографії на діоксиді кремнію (етилацетат-гексани, 1:1) одержали 3,70 г сполуки, зазначеної в заголовку, у вигляді білої твердої речовини. Подібно до цього приготували наступні сполуки: 5-0-Бензош5(R,S)-С-етиніл-1-0-метил-b-D-рибофуранозу (співвідношення R/S 1:1) з 5-0-бензоїл-5(R,S)-C-етеніл-2,3-0ізопропіліден-1-О-метил-b-D-рибофуранози, 5-О-Бензоїл-4-С-бензоїлоксиметил-1-О-метил-b-D-рибофуранозу з 5-О-бензоіл-4-С-бензоїлоксиметил-2,3-(9-ізопропіліден-1-О-метил-b-D-рибофуранози, 5-0-Бензоїл-1-0-метил5(R)-С-вініл-Р-О-рибофуранозу з 2,3-0-ізопропіліден-1-O-метил-5(R)-С-вініл-Р-В-рибофуранози. Одержання 1-0-ацетил-5(R)-С-аліл-2,3,5-три-O-бензоїл-В-рибофуранози До перемішаного розчину 5(R)-С-аліл-5-O-бензоїл-1-O-метил-b-D-рибофуранози (3,60 мг, 11,68 ммоль) у безводному піридині (80мл) при 0°С додали бензоїлхлорид (3,0 мл, 25,84 ммоль). Одержану суміш перемішували при кімнатній температурі протягом 18 годин, охолодили льодом, загасили шляхом додавання води (1 мл), потім перемішували при кімнатній температурі протягом 30 хвилин. Суміш концентрували, розвели етилацетатом, промили тричі соляним розчином, висушили (Na 2SO4) та концентрували до сухого стану. Внаслідок хроматографії на діоксиді кремнію (15% етилацетат у гексанах) одержали 5,3 г сполуки, зазначеної в заголовку, у вигляді безбарвного сиропу. До перемішаного розчину 5(R)-С-аліл-2,3,5-три-O-бензоліл-1-O-метил-b-D-рибофуранози (4,0 г, 7,74 ммоль) в оцтовій кислоті (14 мл) та ацетангідриді (1,75 мл, 18,36 ммоль) при 0°С додали концентровану сірчану кислоту (200 мкл, 3,79 ммоль у 4,0 мл оцтової кислоти). Одержану суміш перемішували при кімнатній температурі протягом 20 годин, охолодили до 0°С, розвели холодним етилацетатом, промили водою, розвели NаНСО3 та потім соляним розчином, висушили (Na2SO4) та концентрували. Внаслідок хроматографії на діоксиді кремнію (етилацетат-гексани, 1:4) одержали 2,82 г сполуки, зазначеної в заголовку, (співвідношення α/β 1:2) у ви гляді безбарвної піни. Подібно до цього одержали наступні сполуки: 1-О-Ацетил-5(R,S)-С-етиніл2,3,5-три-О-бензоліл-b-D-рибофуранозу (співвідношення R/S 1:1 та співвідношення α/β 1:2) з метил 5(R,S)-Сетиніл-2,3,5-три-O-бензоліл-b-D-рибофуранозиду, 1-O-Ацетил-4-С-бензоїлоксиметил-2,3,5-три-O-бензоїл-Врибофуранозу (співвідношення α/β 1:3) з метил 4-С-бензоїлоксиметил-2,3,5-три-O-бензоїл-b-Dрибофуранозиду, 5(R)-C-метил-1,2,3,5-тетра-О-ацетил-b-D-рибофуранозу з 1-(9,5(R)-С-диметил-2,3-Oізопропіліден-b-D-рибофуранози, 5(S)-C-Метил-1,2,3,5-тетра-O-ацетил-b-D-рибофуранозу з 5-O-ацетил-1О,5(R)-С-диметил-2,3-О-ізопропіліден-b-D-рибофуранози, 5-Дезокси-1,2,3-три-O-ацетил-b-D-рибофуранозу з 5-O-ацетил-2,3-O-ізопропіліден-1-O-метил-b-D-рибофуранози, 1-O-Ацетил-2,3,5-три-O-бензоїл-5(R)-С-вініл-bD-рибофуранозу з 1-O-метил-2,3,5-три-O-бензоїл-5(R)-С-вініл-b-D-рибофуранози. Одержання 1-O-метил-5-O-бензоїл-4-С-бензоїлоксиметил-b-D-рибофуранози До перемішаного розчину 4-С,5-O-дидегідро-2,3-O-ізопропіліден-1-O-метил-b-D-рибофуранози 1 (20,22 г, 100 ммоль) у діоксані (380 мл) при 0°С додавали по краплях формальдегід (37% розчин, 76 мл), а потім 2 Μ NaOH (188 мл). Одержану реакційну суміш перемішували при кімнатній температурі протягом 20 годин, охолодили до 0°С, нейтралізували (10% оцтовою кислотою), концентрували (приблизно 50%) та двічі екстрагували метиленхлоридом. Комбінований органічний шар висушували над Na2SO4 та концентрували до сухого стан у. Внаслідок хроматографії на діоксиді кремнію (4% метанол у хлороформі) одержали 20,2г 1-0метил-5-О-бензоїл-2,3-О-ізопропіліден-4-С-бензоїлоксиметил-b-D-рибофуранозиу вигляді білої твердої речовини. Розчин 1-0-метил-5-O-бензоїл-2,3-O-ізопропіліден-4-С-бензоїлоксиметил-b-D-рибофуранози (2,0 г, 4,5 ммоль) у суміші 9:1 (об'єм/об'єм) трифторооцтової кислоти та води (11 мл) перемішували при 0°С протягом 2 годин та випарили до сухого стану. Залишок розчинили у метанолі та випаровували (тричі), потім розчинили у піридині та випарили. Залишок піддали хроматографії на силікагелі (метанол (0 - 0,5%) у дихлорометані), внаслідок чого одержали 1,7г сполуки, зазначеної в заголовку, у вигляді олії. Одержання 1-О-ацетіл-2,3,5-три-О-бензоїл-4-С-бензоїлоксиметіл-b-D-рибофуранози До розчину 1 -О-метил-5-О-бензоїл-4-С-бензоїлоксиметил-b-D-рибофуранози (1,7 г, 4,2 ммоль) у піридині (14 мл) додали бензоїлхлорид (1,2 мл, 10 ммоль). Реакційну суміш перемішували при 25°С протягом 16 годин та додали метанол (5 мл). Розчинники випарили та залишок розчинили етилацетатом (20 мл) та водою (10 мл). Органічний шар висушили над сульфатом натрію, профільтрували та фільтрат випарили до сухого стану. Залишок піддали хроматографії на силікагелі (метанол (0 -0,5%) у дихлорометані), внаслідок чого одержали 2,4 г препарату 1-О-метил-2,3,5-три-O-бензоїл-4-С-бензоїлоксиметил-b-D-рибофуранози у вигляді білої твердої речовини. Сірчану кислоту (97%, 75 мл) додали до розчину 1-О-метил-2,3,5-три-О-бензоїл-4-С-(бензоїлоксиметил)-bD-рибофуранози (1,7 г, 2,8 ммоль) у суміші оцтової кислоти (6,7 мл) та ацетангідриду (0,67 мл) при 0°С. Реакційну суміш перемішували при 25°С протягом 15 годин та розвели етилацетатом (50 мл) та водою (10 мл). Цей розчин промили соляним розчином (тричі), насиченим водним розчином бікарбонату натрію, висушили над сульфатом натрію, профільтрували та фільтрат випарили до сухого стан у. Залишок піддали хроматографії на силікагелі (етанол (0 - 2%) у дихлорометані), внаслідок чого одержали 1,4 г сполуки, зазначеної в заголовку, у вигляді білої твердої речовини. Одержання 4-С-(4,4'-диметокситритилоксиметил)-2,3-О-ізопропіліден-1-O-метіл-b-D-рибофуранози Розчин 4,4'-диметокситритилхлориду (6,0 г, 18 ммоль) у піридині (18 мл) додали до розчину 4-Сгідроксиметил-2,3-0-ізопропіліден-1-0-метил-р-0-рибофуранози (3,5 г, 15 ммоль) у піридині (60 мл), перемішаний при 0°С. Реакційну суміш перемішували при кімнатній температурі протягом 18 годин, потім охолодили до 0°С. Додали метанол (6 мл), розчинники випарили в умовах зниженого тиску. Додали етилацетат та соляний розчин, та органічний екстракт промили у соляному розчині, висушили над сульфатом натрію, профільтрували та випарили до сухого стану. Залишок піддали хроматографії на силікагелі (етилацетат (20 - 25%) у гексанах), внаслідок чого одержали 6,2 г сполуки, зазначеної в заголовку, у вигляді білої твердої речовини. Одержання 2,3,5-три-O-бензоїл-1-O-метіл-4-С-метил-b-D-рибофуранози Розчин бензоїлхлориду (1,5 мл, 13 ммоль) у піридині (6 мл) додали до розчину 4-С-(4,4'-диметокситритилоксиметил)2,3-О-ізопропіліден-1-О-метил-b-D-рибофуранози (6,2 г, 12 ммоль) у піридині (52 мл), перемішаному при 0°С. Реакційну суміш перемішували при кімнатній температурі протягом 15 годин, потім охолодили до 0°С. Додали метанол (5 мл) та розчинники випарили в умовах зниженого тиску. Додали етилацетат та соляний розчин, органічний екстракт промили соляним розчином, висушили над сульфатом натрію, профільтрували та випарили до сухого стану. Залишок випаровували разом з толуолом та розчинили у розчині 80% оцтової кислоти у воді (174 мл). Реакційну суміш перемішували при кімнатній температурі протягом 2 годин, потім розчинник випарили в умовах зниженого тиску. Залишок піддали хроматографії на силікагелі (метанол (1 - 2%) у ди хлорометані) з метою видалення більшості домішок, та білу піну, яку одержали, розчинили в ацетонітрилі (174 мл). Цей розчинник перемішували при 0°С, а потім додали Ν,Ν-диметиламінопіридин (4,3 г, 35 ммоль) та фенокситіокарбонілхлорид (2,4 мл, 17 ммоль). Реакційну суміш знов перемішували при кімнатній температурі протягом 2 годин та розчинник випарили. Залишок розчинили дихлорометаном та водою та одержаний органічний екстракт промили 0,5 N розчином соляної кислоти, потім водою та зрештою соляним розчином. Його висушили над сульфатом натрію, профільтрували та фільтрат випарили в умовах зниженого тиску. Залишок розчинили толуолом та додали трис(триметилсиліл)силан (9,0 мл, 29 ммоль) та 1,1іазобіс(циклогексанкарбонітрил) (0,71 г, 2,9 ммоль). Реакційну суміш перемішували при 100°С протягом 15 годин, потім охолодили до кімнатної температури та розчинник випарили в умовах зниженого тиску. Залишок піддали хроматографії на силікагелі (метанол (1 - 2%) у дихлорометані) з метою видалення більшості домішок, та олію, яку одержали, розчинили у розчині трифторооцтової кислоти у воді (90% об'єм/об'єм, 21 мл) з температурою -15°С. Реакційну суміш перемішували при -10°С протягом 1 години, потім розчинник випарили в умовах високого вакууму та низької температури. Залишок випаровували разом з метанолом, та його піддали хроматографії на силікагелі (метанол (0 - 3%) у дихлорометані) з метою видалення більшості домішок. Олію, яку одержали, розчинили у піридині (18 мл) та розчин перемішували при 0°С. Додали бензоїлхлорид (1,4 мл, 12 ммоль) та реакційну суміш перемішували при кімнатній температурі протягом 16 годин, потім охолодили до 0°С. Додали метанол та розчинники випарили в умовах зниженого тиску. До залишку додали етилацетат, гексани та соляний розчин та одержаний органічний екстракт висушили над сульфатом натрію, профільтрували та фільтрат випарили до сухого стану. Залишок піддали хроматографії на силікагелі (етилацетат (25%) у гексанах), внаслідок чого одержали 1,8 г (32%, шість етапів) сполуки, зазначеної в заголовку, у вигляді сиропу. Одержання 1-О-ацетил-2,3,5-три-О-бензоїп-4-С-метил-b-D-рибофуранози Концентровану сірчану кислоту (97%, 99 мл) додали до розчину 2,3,5-три-О-бензоїл-1-О-метил-4-С-метилb-D-рибофуранози (1,8 г, 3,6 ммоль) у суміші оцтової кислоти (9,0 мл) та ацетангідриду (0,90 мл) при 0°С. Реакційну суміш перемішували при 25°С протягом 16 годин та розвели етилацетатом (50 мл) та соляним розчином (10 мл). Органічний екстракт промили соляним розчином (тричі), насиченим водним розчином бікарбонату натрію, висушили над сульфатом натрію, профільтрували та фільтрат випарили до сухого стану. Залишок розчинили сумішшю етилацетату та гексанів (у відношенні об'ємів 1:4), та b-аномер сполуки, зазначеної в заголовку, кристалізувався миттєво. Білі кристали відфільтрували, внаслідок чого одержали 1,1 г (56%) сполуки, зазначеної в заголовку, у формі її b-аномеру. Фільтрат піддали хроматографії на силікагелі (етилацетат (20%) у гексанах), внаслідок чого одержали 0,6 г (32%) сполуки, зазначеної в заголовку, у вигляді олії (суміш а/b-аномерів 3:1). Одержання 5-О(4,4'-диметокситритил) -4-С-гідроксиметж-2,3-О-ізопропіліден-1-O-метил-b-Dрибофуранози Розчин трет-бутилдиметилсилілхлориду (3,4 г, 23 ммоль) у піридині (16 мл) додали до розчину 4-Сгідроксиметил-2,3-О-ізопропіліден-1-О-метил-b-D-рибофуранози (4,5 г, 19 ммоль) у піридині (80 мл) та перемішували при 0°С. Реакційну суміш потім перемішували при кімнатній температурі протягом 24 годин, потім охолодили до 0°С. Додали воду (5 мл) та розчинник випарили в умовах зниженого тиску. Додали етилацетат та соляний розчин та органічний екстракт промили соляним розчином, висушили над сульфатом натрію, профільтрували та випарили до сухого стан у. Залишок розчинили у піридині та розчин перемішували при 0°С. Додали 4,4'-диметокситритилхлорид (8,4 г, 25 ммоль) та реакційну суміш перемішували при кімнатній температурі протягом 16 годин, потім охолодили до 0°С. Додали метанол (10 мл) та розчинники випарили в умовах зниженого тиску. Додали етилацетат, гексани та соляний розчин та органічний екстракт промили 0,5 N розчином соляної кислоти, потім соляним розчином, висушили над сульфатом натрію, профільтрували та випарили до сухого стан у. Залишок розчинили у тетрагідрофурані (57 мл) та додали розчин тетрабутиламонійфториду (TBAF, 1 Μ у тетрагідрофурані, 23 мл). Після перебування протягом 24 годин при кімнатній температурі додали 0,2 еквівалента TBAF та суміш перемішували протягом додаткових 36 годин. Розчинник випарили та залишок піддали хроматографії на силікагелі (етилацетат (50%) у гексанах), внаслідок чого одержали 6,6 г (65%, 3 етапи) сполуки, зазначеної в заголовку, у вигляді білої твердої речовини. Одержання 5-О-(4,4'-диметокситритил)-2,3-0-ізопропіліден-1-0-метил-4-Свініл-0-В-рибофуранози Розчин трифторооцтової кислоти (0,49 мл, 6,4 ммоль) та піридину (1,6 мл, 19 ммоль) у диметилсульфоксиді (11 мл) додали до розчину 5-0-(4,4'-диметокситритил)-4-С-гідроксиметил-2,3-Оізопропіліден-1-О-метил-b-D-рибофуранози (6,9 г, 13 ммоль) та Ν,Ν'-дициклогексилкарбодііміду (6,6 г, 32 ммоль) у суміші толуолу (26 мл) та диметилсульфоксиду (66 мл), перемішаній при 5°С. Реакційну суміш потім перемішували при кімнатній температурі протягом 8 годин, потім охолодили до 0°С. Додали етилацетат (80 мл) та розчин щавлевої кислоти (1,8 г, 19 ммоль) у метанолі (10 мл) та суміш перемішували при кімнатній температурі протягом 15 годин. Осад відфільтрували та промили сумішшю у співвідношенні 1:1 гексанів та етилацетату. Фільтрат промили соляним розчином, насиченим водним розчином бікарбонату натрію, знов промили соляним розчином, висушили над сульфатом натрію, профільтрували та фільтрат випарили до сухого стан у. Залишок піддали хроматографії на силікагелі (етилацетат (25%) у гексанах), внаслідок чого одержали 6,1 г (89%) 5-O-(4,4'-димeтoкcитpитил)-4-C-фopмiл-2,3-O-iзoпpoпІлiдeн-1-O-мeтил-β-Dрибофуранози у вигляді білої твердої речовини. Розчин пентоксиду натрію (2,5 г, 22 ммоль) у бензолі (34 мл) додали до суспензії метилфосфонійброміду (8,8 г, 25 ммоль) у етері, перемішаної при кімнатній температурі. Суміш перемішували при кімнатній температурі протягом 6 годин та додали розчин 5-0-(4,4'-диметокситритил)-4-С-форміл-2,3-(9-ізопропіліден-1 О-метил-b-D-рибофуранози (6,0 г, 11 ммоль) у е тері (30 мл). Одержану суміш перемішували при кімнатній температурі протягом 18 годин, потім охолодили до 0°С. Додали соляний розчин (100 мл), а потім етилацетат (300 мл). Органічний екстракт промили соляним розчином, висушили над сульфатом натрію, профільтрували та фільтрат випарили до сухого стану. Залишок піддали хроматографії на силікагелі (етилацетат (25%) у гексанах), внаслідок чого одержали 5,9г (99%) сполуки, зазначеної в заголовку, у вигляді твердої білої речовини. Одержання 5-О-бензоїл-4-С-етил-1-О-метил-b-D-рибофуранози Паладій на активованому вуглеці (10% Pd, 50% води, 508 мг) додали до розчину 5-О-(4,4'-диметокситритил)-2,3-О-ізопропіліден-1 -0-метил-4-С-вініл-b-Dрибофуранози (5,0 г, 9,4 ммоль) у метанолі (254 мл). Колбу струшували в умовах тиску 34,47 кПа (5 фунтів на квадратний дюйм) водню протягом 6 годин та каталізатор відфільтрували та промили метанолом. Розчинник випарили, а залишок випаровуваили разом з піридином. Потім його розчинили у піридині (75 мл) та перемішували при 0°С. Додали бензоїлхлорид (1,2 мл, 10 ммоль), реакційну суміш перемішували при кімнатній температурі протягом 15 годин, потім охолодили до 0°С. Додали метанол (5 мл), розчинники випарили в умовах зниженого тиску. Додали етилацетат, гексан та соляний розчин та органічний екстракт промили соляним розчином, висушили над сульфатом натрію, профільтрували та випарили до сухого стану. Залишок випаровували разом з толуолом та розчинили у розчині трифторооцтової кислоти у воді (90% об'єму, 57мл) з температурою - 15°С. Реакційну суміш перемішували при - 10°С протягом 2 годин, потім розчинник випарили в умовах високого вакууму та низької температури. Залишок випаровуваили разом з метанолом та одержали білий осад. Його видалили шляхом фільтрування та фільтрат, який містив сполуку, зазначену в заголовку, випарили до сухого стану та розчинили в етилацетаті, гексані та соляному розчині. Органічний екстракт промили насиченим водним розчином бікарбонату натрію, промили соляним розчином, висушили над сульфатом натрію, профільтрували та фільтрат випарили до сухого стан у. Залишок піддали хроматографії на силікагелі (етилацетат (33%) у гексанах), внаслідок чого одержали 1,8г (63%, 3 етапи) сполуки, зазначеної в заголовку, у вигляді олії. Одержання 1-О-ацетіл-2,3,5-три-О-бензоїл-4-С-етил-b-D-рибофуранози Бензоїлхлорид (1,5 мл, 13 ммоль) додали до розчину 5-0-бензоїл-4-С-етил-1-0-метил-b-D-рибофуранози у піридині (41 мл), перемішаному при 0°С, та реакційну суміш перемішували при кімнатній температурі протягом 18 годин, потім охолодили до 0°С. Додали метанол (5мл) та розчинники випарили в умовах зниженого тиску. Додали етилацетат, гексан та соляний розчин та органічний екстракт промили 0,5N розчином соляної кислоти, потім соляним розчином, висушили над сульфатом натрію, профільтрували та випарили до сухого стану. Залишок випаровували разом з толуолом та розчинили у суміші оцтової кислоти (14 мл) та ацетангідриду (1,5 мл). Сірчану кислоту (97%, 165 мл), розчинену в оцтовій кислоті (1 мл), додали при 5°С. Реакційну суміш перемішували при 25°С протягом 4 годин та розвели етилацетатом (50 мл) та соляним розчином (10 мл). Органічний екстракт промили соляним розчином (тричі), насиченим водним розчином бікарбонату натрію, промили соляним розчином, висушили над сульфатом натрію, профільтрували та фільтрат випарили до сухого стан у. Залишок піддали хроматографії на силікагелі (етилацетат (0 - 3%) у дихлорометані), внаслідок чого одержали 2,9 г (92%) сполуки, зазначеної в заголовку, у вигляді олії (суміш b/а-аномерів 2:1). Одержання 4-аміно-5-оксо-піридо[2,3-а]піримідину Триметилсилілхлорид (7,6 мл, 60 ммоль) додали до перемішаної суспензії 2-аміно-3-ціано-4-метоксипіридину (Archiv der Pharmazie 1985, 318,481-486,7,5 г, 50 ммоль) та йодиду натрію (7,50 г, 50 ммоль) у ацетонітрилі (225 мл). Одержану суміш нагрівали при температурі дефлегмації протягом 24 годин. Осад відфільтрували та промили етилацетатом, внаслідок чого одержали 10,4 г коричневого порошку (2-аміно-3ціано-4-оксо-піридину). Цей порошок висушили в умовах зниженого тиску та суспендували у 2-етоксиетанолі (300 мл). Додали формамідинацетат (31,2 г, 300 ммоль) та суспензію нагрівали при температурі дефлегмації протягом 2 днів, а потім профільтрували. Сірий залишок, який одержали, розчинили у киплячій суміші 2:1 оцтової кислоти та води та додали вугілля. Чорну суспензію профільтрували та фільтрат випарили до сухого стану, внаслідок чого одержали білу тверду речовину, яку суспендували у гарячому насиченому розчині гідрокарбонату натрію у воді. Суспензію профільтрували та одержали 4-аміно-5-оксо-піридо[2,3-d]піримідин (2,0 г) у ви гляді білої твердої речовини. Одержання 4-аміно-5-оксо-8-(2,3,5-три-O-бензоїл-4-C-метил-b-D-рибофуранозил) піридо[2,3-d]піримідину 4-Аміно-5-оксо-піридо[2,3-d]-піримідин (0,23 г, 1,4 ммоль) суспендували у 1,2-дихлороетані (20 мл) та суміш перемішували при 55°С. Додали BSA (0,87 мл, 3,5 ммоль), реакційну суміш перемішували при температурі дефлегмації протягом 90 хвилин, потім охолодили до 40°С. 1-О-Ацетил-2,3,5-три-О-бензоїл-4-Сметил-b-D-рибофуранозу (0,60 г, 1,2 ммоль) у 1,2-дихлороетані (3 мл) та TMSOTf (0,42 мл, 2,3 ммоль) додали у прозорий розчин та суміш перемішували при 100°С протягом 48 годин. Суміш охолодили до кімнатної температури та додали насичений розчин гідрокарбонату натрію у воді. С уміш розвели етилацетатом (100 мл) та органічний екстракт промили соляним розчином, висушили над суль фатом натрію, профільтрували та фільтрат випарили до сухого стан у. Залишок піддали хроматографії на силікагелі (ацетон (15 - 25%) у дихлорометані), внаслідок чого одержали 0,30 г (41%) сполуки, зазначеної в заголовку, у вигляді білої твердої речовини та 0,14 г (19%) 4-аміно-5-оксо-1-(2,3,5-три-О-бензоїл-4-С-метил-b-D-рибофуранозил)піридо[2,3d]піримідину у вигляді білої твердої речовини. Подібно до цього приготували наступні сполуки: 4-Аміно-5-оксо8-(2,3,5-три-O-бензоїл-4-С-бензоїлоксиметил-b-D-рибофуранозил)піридо[2,3-d]піримідин у вигляді білої твердої речовини з 4-аміно-5-оксо-піридо[2,3-d]піримідину та 1-О-ацетил-2,3,5-три-О-бензоїл-4-Сбензоїлоксиметил-b-D-рибофуранози;4-Аміно-5-оксо-8-(2,3,5-три-O-бензоїл-5(R,S)-С-етиніл-b-Dрибофуранозил)піридо[2,3-d]піримідин у вигляді білої твердої речовини з 4-аміно-5-оксо-піридо[2,3d]піримідину та 1-O-ацетил-2,3,5-три-O-бензоїл-5(R,S)-С-етиніл-b-D-рибофуранози;4-Аміно-5-оксо-8-(2,3,5-триO-бензоїл-b-D-рибофуранозил)піридо[2,3-d]піримідин у вигляді білої твердої речовини з 4-аміно-5-оксопіридо[2,3-d]піримідину та 1-O-ацетил-2,3,5-три-O-бенензоїл-b-D-рибофуранози; 4-Аміно-5-оксо-8-(2,3,5-три-Обензоїл-b-D-рибофуранозил)піридо[2,3-d]піримідину вигляді білої твердої речовини з 4-аміно-5-оксопіридо[2,3-d]піримідину та 1-O-ацетил-2,3,5-три-(9-бензоїл-b-D-рибофуранози;4-Аміно-5-оксо-8-(2,3,5-три-Oбензоїл-5(R)-С-аліл-b-D-рибофуранозил)піридо[2,3-d]піримідин у вигляді білої твердої речовини з 4-аміно-5 оксо-піридо[2,3-d]піримідину та 1-О-ацетил-2,3,5-три-О-бензоїл-5(R)-С-аліл-b-D-рибофуранози;4-Аміно-5-оксо8-(2,3,5-три-O-бензоїл-5(R)-С-метил-b-D-рибофуранозил)піридо[2,3-d]піримідин у вигляді білої твердої речовини з 4-аміно-5-оксо-піридо[2,3-d]піримідину та 1-О-ацетил-2,3,5-три-О-бензоїл-5(R)-С-метил-b-Dрибофуранози;4-Аміно-5-оксо-8-(2,3,5-три-0-бензоїл-4-С-етил-b-D-рибофуранозил)піридо[2,3-d]-піримідин у вигляді білої твердої речовини з 4-аміно-5-оксо-піридо[2,3-d]піримідину та 1-O-ацетил-2,3,5-три-O-бензоїл-4-Сетил-b-D-рибофуранози;4-Аміно-5-оксо-8-(2,3,5-три-О-бензоїл-5(R,S)-С-вініл-b-D-рибофуранозил)піридо[2,3d]піримідин у вигляді білої твердої речовини з 4-аміно-5-оксо-піридо[2,3-d]піримідину та 1-O-ацетил-2,3,5-триO-бензоїл-5(R,S)-С-вініл-b-D-рибофуранози. Одержання 4-аміно-5-оксо-8-(4-С-гідроксиметіл-b-D-рибофуранозил)піридо[2,3-d]піримідину Розчин 4-Аміно-5-оксо-8-(2,3,5-три-О-бензоїл-4-С-бензоїлоксиметил-b-D-рибофуранозил)піридо[2,3d]піримідину (0,82 г, 1,1 ммоль) у насиченому аміаком метанолі (насичений при 0°С) перемішували у герметичній колбі при 25°С протягом 15 хвилин. Розчинники випарили та залишок піддали хроматографії на силікагелі (метанол (30%) у дихлорометані), внаслідок чого одержали 0,36 г сполуки, зазначеної в заголовку, у вигляді білої твердої речовини. Подібно до цього приготували наступні сполуки: 4-Аміно-5-оксо-8-(5(R)-Сметил-b-D-рибофуранозил)піридо[2,3-d]піримідина-4-аміно-5-оксо-8-(2,3,5-три-О-бензоїл-5(R)-С-метил-b-Dрибофуранозил)піридо[2,3-d]піримідину;4-Аміно-5-оксо-8-(5(R)-С-аліл-b-D-рибофуранозил)піридо[2,3d]піримідина-4-аміно-5-оксо-8-(2,3,5-три-О-бензоїл-5(R)-С-аліл-b-D-рибофуранозил)піридо[2,3-піримідину;4Аміно-5-оксо-8-(5(R,S)-С-етиніл-b-D-рибофуранозил)піридо[2,3-d]піримідинa4-аміно-5-оксо-8-(2,3,5-три-Oбензоїл-5(R,S)-С-етиніл-b-D-рибофуранозил)піридо[2,3-d]піримідину;4-Аміно-5-оксо-8-(5(R,S)-С-вініл-b-Dрибофуранозил)піридо[2,3-d]піримідинз4-аміно-5-оксо-8-(2,3,5-три-O-бензоїл-5(R,S)-С-вініл-b-Dрибофуранозил)піридо[2,3-d]піримідину;4-Аміно-5-оксо-8-b-D-рибофуранозил)піридо[2,3-d]піримідин з 4-аміно5-оксо-8-(2,3,5-три-О-бензоїл-b-D-рибофуранозил)піридо[2,3-d]піримідину;4-Аміно-5-оксо-8-(-b-Dрибофуранозил)піридо[2,3-d]піримідин з 4-аміно-5-оксо-8-(2,3,5-три-0-бензоїл-b-D-рибофуранозил)піридо[2,3d]піримідину;4-аміно-5-оксо-8-(4-С-метил-b-D-рибофуранозил)піридо[2,3-d]пІримідин з 4-аміно-5-оксо-8-(2,3,5три-О-бензоїл-4-С-метил-b-D-рибофуранозил)піридо[2,3-d]піримідину;4-Аміно-5-оксо-8-(4-С-етил-b-Dрибофуранозил)піридо[2,3-d]піримідин з 4-аміно-5-оксо-8-(2,3,5-три-0-бензоіл-4-С-етил-b-Dрибофуранозил)піридо[2,3-d]піримідину. Одержання 4-аміно-5-оксо-8-(5(R,S)-С-етіл-b-D-рибофуранозил) піридо[2,3-d]піримідину Паладій на активованому вуглеці (10% Pd, 200 мг) додали до розчину 4-аміно-5-оксо-8-(5-С-етиніл-b-Dрибофуранозил)піридо[2,3-d]піримідину (0,14 г, 0,45 ммоль) у метанолі (50 мл). Колбу струшували в умовах тиску 20,68 кПа (3 фунти на кв. дюйм) водню протягом 2 годин та каталізатор відфільтрували та промили метанолом. Розчинники випарили та залишок піддали хроматографії на силікагелі (метанол (10%) у дихлорометані), внаслідок чого одержали 0,12 г сполуки, зазначеної в заголовку, у вигляді білої твердої речовини. Подібно до цього приготували наступні сполуки: 4-Аміно-5-оксо-8-(5(R)-С-пропіл-b-Dрибофуранозил)піридо[2,3-d]піримідин з 4-аміно-5-оксо-8-(5(R)-С-аліл-b-D-рибофуранозил)піридо[2,3d]піримідину. Одержання 4-аміно-5-оксо-8-(2-дезокси-b-D-рибофуранозил)піридо[2,3-d] піримідину Реакційну суміш 4-аміно-5-оксо-8-(-b-D-рибофуранозил)піридо[2,3-d]піримідину (1,8 г, 6,20 ммоль) та 1,3дихлор-1,1,3,3-тетраізопропілдисилоксану (2,15 мл, 6,73 ммоль) у безводному піридині (25 мл) перемішували при кімнатній температурі протягом 20 годин та охолодили льодом. Додали воду (0,5 мл) та суміш перемішували при кімнатній температурі протягом 30 хвилин та концентрували. Залишок розчинили в етилацетаті, промили розведеним бікарбонатом натрію, висушили над сульфатом натрію та концентрували. Внаслідок хроматографії на діоксиді кремнію (ЕtOАс-гексани, 3:2) одержали 2,0 г 4-аміно-5-оксо-8-[3,5-0(1,1,3,3-тетраізопропілдисилокси)-b-D-рибофуранозил)]піридо[2,3-d]піримідину. До розчину 4-аміно-5-оксо-8-[3,5-0-(1,1,3,3-тетраізопропілдисилокси)-b-D-рибофуранозил)]піридо[2,3d]піримідину (650 мг, 1,21 ммоль) та DMAP (295 мг, 2,42 ммоль) в ацетонітрилі (10 мл) додали фенілхлоротіоформат (185 мл, 1,33 ммоль). Суміш перемішували при кімнатній температурі протягом 2 годин та концентрували до сухого стану. Залишок розчинили у хлороформі, промили водою, висушили (сульфат натрію) та концентрували. Залишок висушували в умовах вакууму протягом 30 хвилин, а потім розчинили у толуолі (10 мл). Додали 1,Г-азобіс(циклогексанкарбо-нітрил) (74 мг, 0,30 ммоль) та одержаний розчин барботували аргоном протягом 30 хвилин. Додали трис(триметилсиліл)силан (0,56мл, 1,82ммоль) та одержану суміш перемішували протягом 2 годин при температурі 80°С, а потім протягом ночі при температурі 105°С. Розчинник випарили та залишок розчинили у тетрагідрофурані (5 мл). Додали TBAF (1,0 Μ у тетрагідрофурані, 2,5 мл) та одержаний розчин залишили при кімнатній температурі на 2 години, а потім концентрували. Внаслідок хроматографії на діоксиді кремнію (10% МеОН у СН 2Сl2) одержали 240 мг сполуки, зазначеної в заголовку. Експерименти in vitro/in vi vo Якщо не підкреслюється інше, наступні експерименти здійснювали з 4-аміно-5-оксо-8-(-b-Dрибофуранозил)піридо[2,3-d]піримідином. Визначення ЕС 50 Ракові клітини інкубували з 4-аміно-5-оксо-8-(-b-D-рибофуранозил)піридо[2,3-d]піримідином за різними концентраціями протягом, принаймні, 24 годин та визначили ЕС 50. Таблиця 1 демонструє активність in vitro 4аміно-5-оксо-8-(b-D-рибофуранозил)піридо[2,3-d]піримідину в концентраціях у н М. Таблиця 1 Тип пухлини Клітинні лінії EC50[н M] Молочна MCF 7, NCI/ADR-RES, MD A18 залоза Простата Нирка Яєчник Меланома Центральна нервова система Товста кишка Легені Лейкоз Печінка Підшлункова залоза MB-435, ВТ-549, T-47D PC-3,DU-145 786-0, А498, ACHN, САКІ-1, RXF 393, UO-31 IGROV1, OVC AR-3, OVC AR4, OVCAR-5, SK-OV-3 LOX IМ VI, MAL ME-3M, Μ14, SK-MEL-2, SK-MEL-28, АСС-257, UACC-62 SF-268, SF-295, SF-539, SNB-19, SNB-75, U251 19 44 21 30 125 SW-620, KM 12, НТ29, НСТ15, СНСТ-116, НСС-2998, COLO-205 NCI-H522, NCI-H460, NCIH322M, NCI-H23, НОР-92, НОР-62, EKVX, A549 SR, RPMI 8226, MOLT-4, К562, HL-60, CCRF-CEM PLC/PRF5, НерЗВ, Huh7 288 PaCa-2,PANC-l 188 16 68 34 Відносно висока ефективність передбачених сполук та особливо 4-аміно-5-оксо-8-(-b-Dрибофуранозил)піридо[2,3-d]піримідину відображена далі у ряді експериментів, у яких цитотоксичність та інгібування проліферації різних ракових клітинних ліній порівнюється з комерційно доступними цитостатичними агентами. Результати експериментів представлено на Фігурах 1 та 2. Антиклоногенна активність передбачених сполук Здійснили визначення антиклоногенної активності передбачених сполук в експериментах з різними раковими клітинами. Результати цих експериментів представлені на Фігурі 3. Вони ясно вказують на те, що передбачені сполуки демонструють суттєву антиклоногенну активність, особливо у клітинах меланоми В16 та 140, клітинах лейкозу (К562, М) та у клітинах раку товстої кишки НТ-29. У цих експериментах знов застосовували 4-аміно-5-оксо-8-(-b-D-рибофуранозил)піридо[2,3-d]піримідин як сполуку-представника передбачених сполук та порівнювали цю сполуку з ECyd та Gemzar. Індукція апоптозу З метою визначення, як передбачені сполуки взаємодіють з раковими клітинами, клітини NB4 (лейкоз) та клітини Prostate 81 (клітини раку простати) інкубували з 4-аміно-5-оксо-8-(-b-D-рибофуранозил)піридо[2,3d]піримідином, ECyd та Gemzar та здійснювали моніторинг апоптозу шляхом твердофазного імуноферментного аналізу гістону-ДНК. Результати наведено на Фігурах 4А та 4В, які дозволяють припустити, що передбачені сполуки індукують апоптоз залежно від дози. Інгібування синтезу РНК Здійснили моніторинг синтезу РНК у клітинах К562 із застосуванням включення Н 3-уридину згідно із загальним протоколом, описаним нижче. Фігура 5 демонструє інгібування синтезу РНК прикладом передбачених сполук (тут 4-аміно-5-оксо-8-(-b-D-рибофуранозил)піридо[2,3-d]піримідином) протягом різних періодів часу та за різними концентраціями. Інгібування РНК-полімерази І, II та НІ Здійснили експерименти з метою подальшого дослідження впливу передбачених сполук (тут знов 4-аміно5-оксо-8-(-b-D-рибофуранозил)піридо[2,3-d]піримідину) на синтез РНК, який залежить від РНК-полімерази І, II та III. Результати цих експериментів представлено на Фігурах 6 та 7, та вони дозволяють припустити, що передбачені сполуки можуть інгібувати процесинг рРНК 28S та 18S, проте не можуть суттєво інгібува ти РНКполімеразу III. Експерименти далі вказують на те, що передбачені сполуки інгібують синтез РНК, що залежить від РНК-полімерази II, залежно від концентрації. Фосфорилювання мітоген активоаною кіназою - керованою ззовні кіназою (MEK-ERK) Нещодавно продемонстрували (Wang, et al., JBC 275:39435-43 (2000), Pavlovic et al., Eur. Cytokine Netw 2:267-74 (2000)), що апоптоз можна індукувати через шлях трансдукції сигналу MEK-ERK. Здійснили експерименти з метою дослідження можливості активації шляху трансдукції сигналу MEK-ERK передбаченими сполуками. Фігура 8 - це авторадіограма, що зображує вплив передбачених сполук (представлених 4-аміно-5оксо-8-ф-0-рибофуранозил)піридо[2,3-сі]піримідином) на МЕК- та ERK-фосфорилювання у порівнянні з впливом на фосфорилювання онкогеном з функцією кінази (Raf) у лейкозних клітинах NB4. Цей та інші експерименти дозволяють припустити, що передбачені сполуки підсилюють МЕК-фосфорилювання (на відміну від ECyd та Gemzar), проте, вони не підсилюють Raf-фосфорилювання. Отже, передбачається, що сполуки згідно з предметом винаходу можуть індукувати апоптоз через активацію МЕК (який можна припинити інгібіторами шляху MEK-ERK). Метаболіти передбачених сполук Численні експерименти (дані не представлено) дозволяють припустити, що передбачені сполуки фосфорилюються у межах (пухлинної) клітини, та що продукти включають моно-, ди- та трифосфорильовані форми (наприклад, які можна визначити рідинною хроматографією та мас-спектрометрією). Далі передбачається, що метаболіти можуть мати суттєву (або навіть підвищену) біологічну активність. Отже, були описані специфічні варіанти здійснення та застосування піридо[2,3-d]піримідинових та піримідо[4,5-d]піримідинових нуклеозидів. Фахівцям у галузі слід розуміти, однак, що можливими є набагато більше модифікацій окрім вже описаних, які не відходять від концепцій винаходу, описаних тут. Предмет винаходу, отже, не слід обмежувати, але слід дотримуватися духу формули винаходу, що додається. Крім того, під час інтерпретації як опису, так і формули винаходу, усі терміни слід інтерпретувати у як можливо більш широкому сенсі, який узгоджується з контекстом. Зокрема, терміни "включає" ("містить") та "що включає" ("що містить") слід інтерпретува ти з посиланням на елементи, компоненти або етапи у невиключній манері, вказуючи на те, що елементи, компоненти або етапи, на які посилаються, можуть існувати, або застосовуватися, або комбінуватися з іншими елементами, компонентами або етапами, на які не посилаються спеціально.
ДивитисяДодаткова інформація
Назва патенту англійськоюPyrido[2.3-d]pyrimidine and pyrimido[4.5-d]pyrimidine nucleoside analogues, prodrugs and method for inhibiting growth of neoplastic cells
Назва патенту російськоюПиридо[2,3-d]пиримидиновые и пиримидо[4,5-d]пиримидиновые нуклеозиды, пролекарственные формы и способ ингибирования роста раковых клеток
МПК / Мітки
МПК: A61P 35/00, A61K 31/70
Мітки: спосіб, ракових, піримідо[4,5-d]піримідинові, інгібування, нуклеозиди, проліки, клітин, піридо[2,3-d]піримідинові
Код посилання
<a href="https://ua.patents.su/16-72612-pirido23-dpirimidinovi-ta-pirimido45-dpirimidinovi-nukleozidi-proliki-ta-sposib-ingibuvannya-rakovikh-klitin.html" target="_blank" rel="follow" title="База патентів України">Піридо[2,3-d]піримідинові та піримідо[4,5-d]піримідинові нуклеозиди, проліки та спосіб інгібування ракових клітин</a>
Спосіб визначення ракових клітин в ексудативній рідині серозних порожнин

Номер патенту: 5662
Опубліковано: 28.12.1994
Автори: Абраменко Ірина Вікторівна, Глузман Данііл Фішелевич, Скляренко Лілія Михайлівна
МПК: G01N 21/00, G01N 33/535, G01N 1/28
Мітки: клітин, рідини, серозних, ексудативній, визначення, ракових, порожнин, спосіб
Формула / Реферат:
Способ определения раковых клеток в экссудативной жидкости серозных полостей, включающий осаждение из нее клеточных элементов, отмывание их забуференным физиологическим раствором рН 7,2-7,4, разведение суспензии клеток до концентрации 2x10б/мл, приготовление цитологических препаратов, их фиксацию раствором формол-ацетона, инкубацию в 0,1 М растворе МН4СІ с последующей инкубацией с пектинами, конъюгированными с пероксидазой хрена, причем все...
Засіб для інгібування проліферації клітин гладкої мускулатури та для інгібування рестенозу
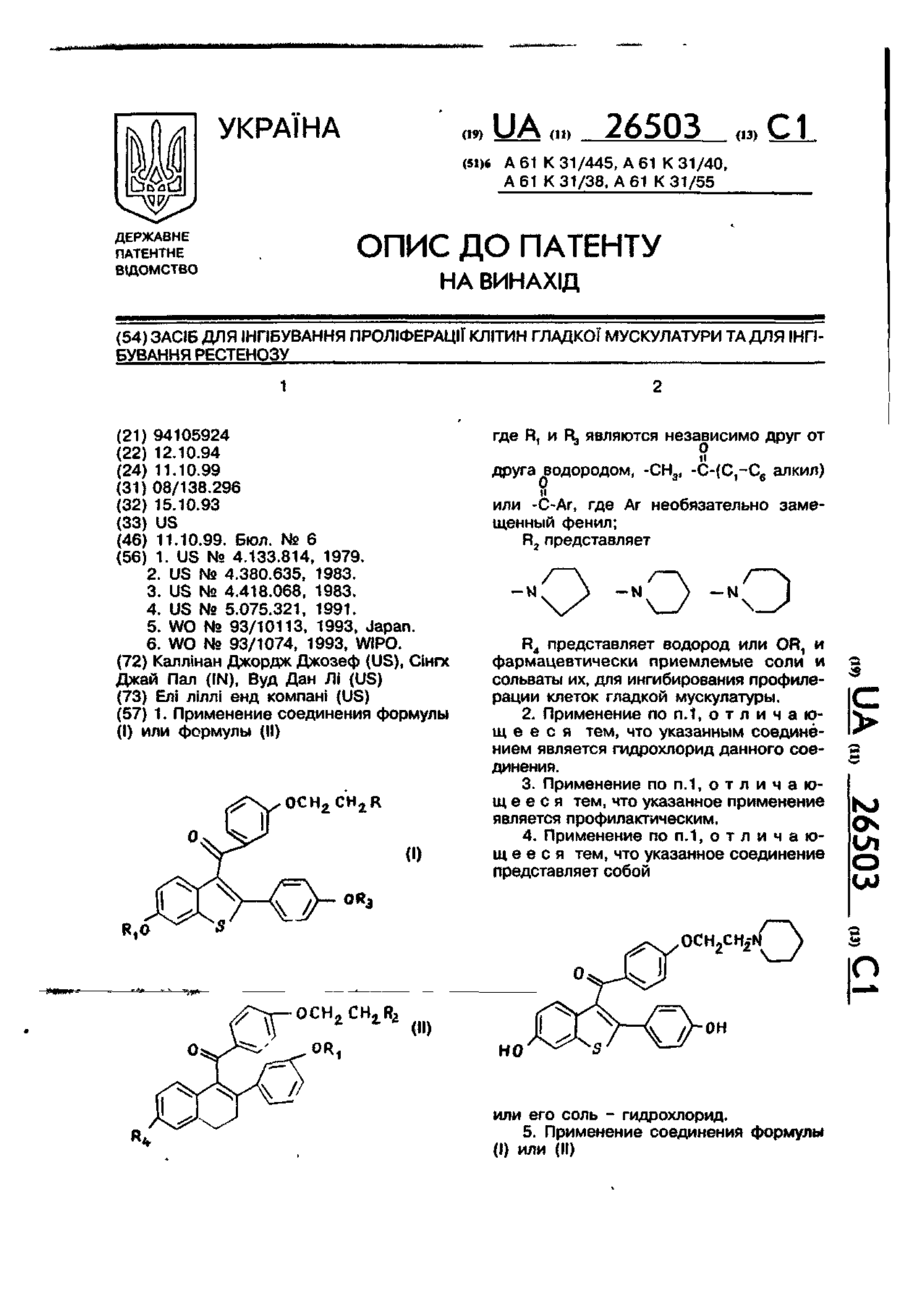
Номер патенту: 26503
Опубліковано: 11.10.1999
Автори: Вуд Дан Лі, СІНГХ Джай Пал, КАЛЛІНАН Джордж Джозеф
МПК: A61K 31/40, A61K 31/55, A61K 31/445, A61K 31/38
Мітки: інгібування, клітин, мускулатури, засіб, проліферації, рестенозу, гладкої
Формула / Реферат:
1. Применение соединения формулы (l) или (ll)где R1 и R3 являются независимо друг от друга водородом, или , где Ar необязательно замещенный фенил;R2 представляетR4 представляет водород или OR1 и фармацевтически приемлемые соли и сольваты их, для ингибирования профилерации клеток гладкой мускулатуры.2. Применение по п.1, отличающееся тем, что указанным соединением является гидрохлорид данного...
Інгібітори адгезії клітин, фармацевтична композиція, що їх містить (варіанти), та спосіб інгібування або пригнічення адгезії клітин у ссавця (варіанти)

Номер патенту: 71888
Опубліковано: 17.01.2005
Автори: Кастро Альфредо К., Хаммонд Чарльз І., Енсінгер Керол Лі, Лі Вен-Чернг, Адамс Стівен П., Зіммерман Крейг Н., Олмквіст Рональд Джі., Лін Ко Чунг, Куерво Хуліо Хернан, Картер Мері Бет
МПК: C07K 5/103, A61K 31/00, C07K 5/083, A61P 3/10, A61P 29/00, C07K 5/09, C07K 7/06, C07K 5/097, A61P 17/06, C07K 5/093, A61P 3/00, A61P 17/00, A61P 19/00, A61P 11/06, C07K 5/00, A61K 38/00, A61P 19/02, C07K 5/06, A61P 37/06, A61P 43/00, A61P 1/04, C07K 5/02, C07K 5/072, C07K 5/107, C07K 5/117, A61P 11/00, A61P 1/00, C07K 14/78, A61P 37/00
Мітки: інгібування, адгезії, клітин, пригнічення, ссавця, інгібітори, містить, композиція, спосіб, фармацевтична, варіанти
Формула / Реферат:
1. Сполука, що інгібує адгезію клітин, яка має формулу (І):Z-(Y1)-(Y2)-(Y3)n-X ,(I)в якій:Y1 являє собою -N(R1)-C(R2)(А1)-C(O)-;Y2 являє собою -N(R1)-С(R2)(А2)-C(O)-;кожен Y3, незалежно, представлено формулою -N(R1)-C(R2)(А3)-C(O)-; іn є цілим числом від 0 до 8; декожен R1, незалежно, являє собою водень, алкіл або аралкіл; ікожен R2, незалежно, являє собою водень або алкіл;і де...
Фармацевтична композиція для лікування раку або доброякісних проліферативних хвороб у ссавців, спосіб лікування, фармацевтична композиція для інгібування ненормального росту клітин у ссавців та спосіб інгібування

Номер патенту: 57081
Опубліковано: 16.06.2003
Автор: Каджиджи Шама Мохаммед
МПК: A61K 31/435, A61K 31/405, A61K 31/40
Мітки: ссавців, клітин, раку, спосіб, композиція, доброякісних, лікування, проліферативних, хвороб, фармацевтична, інгібування, ненормального, росту
Формула / Реферат:
1. Фармацевтична композиція для лікування раку або доброякісних проліферативних хвороб у ссавців, що включає інгібітор ФTази, інгібітор HMG CoA редуктази і фармацевтично прийнятний носій, в якій інгібітор ФTази і інгібітор HMG CoA редуктази присутні в кількостях, що роблять композицію ефективною при лікуванні раку або доброякісних проліферативних хвороб, і в якій інгібітор ФTази вибирають з:(a) сполуки формули I
Спосіб інгібування міграції клітин гладеньких м’язів судин

Номер патенту: 42835
Опубліковано: 15.11.2001
Автори: Сінгх Джай Пол, Верніцкі Тодд Р.
МПК: A61K 31/445
Мітки: м'язів, гладенькіх, клітин, міграції, інгібування, судин, спосіб
Формула / Реферат:
1.Спосіб інгібування міграції клітин гладеньких м'язів судин, що полягає у введенні людині або іншому ссавцеві, що потребує лікування, ефективної дози сполуки за формулою,у якій R1 та R3, незалежно один від одного, - водень, -СНз, (С1-С6 алкіл), або , де Аr — довільно заміщений феніл;R3 — це піролідино, гексаметиленіміно або піперидино; та її фармацевтично прийнятні солі та сольвати.2. Спосіб за п. 1, де...
Попередній патент: Карбамати, похідні від арилалкіламінів
Наступний патент: Композиція інгредієнтів для горілки особливої “кльова”
Випадковий патент: Спосіб імунокорекції у хворих із переломами проксимального відділу стегна на фоні абдомінального ожиріння