Сполука 3-амінокарбазолу, фармацевтична композиція, яка її містить, та спосіб її одержання
Номер патенту: 102541
Опубліковано: 25.07.2013
Автори: Гарофало Барбара, Каццолла Нікола, Гугліелмотті Анджело, Колетта Ізабелла, Фурлотті Гвідо, Мауджері Катеріна, Алісі Марія Алессандра, Мангано Джорджина, Драгоне Патріція








Формула / Реферат
1. Сполука 3-амінокарбазолу, яка характеризується тим, що має загальну формулу (І):
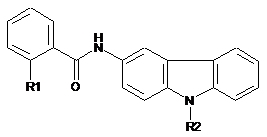 , (I)
, (I)
де
R1 є атомом галогену, метильною групою або тригалометильною групою, нітрогрупою, ціаногрупою або трифлатною групою, і
R2 є лінійною або розгалуженою гідроксіалкільною групою, яка включає від 1 до 8 атомів вуглецю, або лінійною або розгалуженою карбонілалкільною групою, яка включає від 1 до 8 атомів вуглецю, або
її фармацевтично прийнятна сіль, її поліморфний кристал, її стереоізомер, її енантіомер або її проліки.
2. Сполука 3-амінокарбазолу за п. 1, яка відрізняється тим, що R1 є атомом фтору або хлору або трифторометильною або трихлорометильною групою, і R2 є лінійною або розгалуженою гідроксіалкільною групою, яка включає від 1 до 6 атомів вуглецю, або лінійною або розгалуженою карбонілалкільною групою, яка включає від 1 до 4 атомів вуглецю.
3. Сполука 3-амінокарбазолу за п. 1, яка відрізняється тим, що R1 та R2 мають значення, вказані нижче у Таблиці 1
Таблиця 1
Сполука
R1
R2
1
CF3
СН2СН2ОН
2
CF3
CH2C(CH3)2OH
3
CF3
СН2СН2С(СН3)2ОН
4
CF3
CH2COCH3
5
Cl
СН2СН2ОН
6
Cl
СН2СН2С(СН3)2ОН
4. Фармацевтична композиція, яка відрізняється тим, що містить терапевтично ефективну дозу сполуки 3-амінокарбазолу за будь-яким з пунктів з 1 по 3 або її фармацевтично прийнятної солі, її поліморфного кристала, її стереоізомера, її енантіомера або її проліків разом з принаймні одним фармацевтично прийнятним інертним носієм.
5. Спосіб одержання сполуки 3-амінокарбазолу за будь-яким з пунктів з 1 по 3, який відрізняється тим, що здійснюють такі етапи:
а) реакцію аміну формули (II)
 , (II)
, (II)
де
R2 має значення за будь-яким з пунктів з 1 по 3,
зі сполукою формули (III)
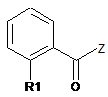 , (III)
, (III)
де
R1 має значення за будь-яким з пунктів з 1 по 3, і
Z є вибраним з групи, яка включає Сl, Br, OH, OR та OC(O)R, де R є лінійним або розгалуженим алкілом, який включає від 1 до 6 атомів вуглецю,
з одержанням сполуки 3-амінокарбазолу формули (І)
, (I)
де R1 та R2 мають вищевказані значення, та
b) необов'язкове утворення фармацевтично прийнятної солі, поліморфної кристалічної форми, стереоізомера або енантіомера одержаної таким чином сполуки формули (І).
6. Застосування сполуки 3-амінокарбазолу за будь-яким з пунктів з 1 по 3 або її фармацевтично прийнятної солі, її поліморфного кристала, її стереоізомера, її енантіомера або її проліків для виробництва медикаменту для профілактичного або терапевтичного лікування порушення, вибраного з групи, яка включає запальні процеси, біль, лихоманку, пухлини, хворобу Альцгеймера та атеросклероз.
7. Застосування сполуки 3-амінокарбазолу за п. 6, яке відрізняється тим, що вищезгадані запальні процеси є вибраними з групи, яка включає набряк, еритему, суглобне запалення, ревматоїдний артрит та артроз.
8. Застосування сполуки 3-амінокарбазолу за п. 6, яке відрізняється тим, що вищезгадані пухлини є вибраними з групи, яка включає колоректальну та легеневу карциному та аденокарциному.
9. Спосіб лікування або профілактики запальних процесів, болю, лихоманки, пухлин, хвороби Альцгеймера та атеросклерозу у ссавців, який включає введення терапевтично ефективної кількості сполуки 3-амінокарбазолу за будь-яким з пунктів з 1 по 3 або її фармацевтично прийнятної солі, її поліморфного кристала, її стереоізомера, її енантіомера або її проліків особі, яка цього потребує.
Текст
Реферат: Даний винахід стосується нових бензоїльних похідних 3-амінокарбазолу, фармацевтичної композиції, яка їх містить, способу одержання та застосування таких сполук для виготовлення медикаменту, який є корисним для лікування або профілактики порушень, пов'язаних з продукуванням простагландину Е2 (PGE2), наприклад, запальних процесів, болю, лихоманки, пухлин, хвороби Альцгеймера та атеросклерозу. UA 102541 C2 (12) UA 102541 C2 UA 102541 C2 5 10 15 20 25 30 35 40 45 50 55 60 МПК: C07D 209/88 (2006.01) A61P 29/00 (2006.01) A61K 31/403 (2006.01) сполука 3-амінокарбазолу, фармацевтична композиція, яка її містить, та спосіб її одержання ГАЛУЗЬ ВИНАХОДУ Даний винахід стосується нових сполук 3-амінокарбазолу, фармацевтичної композиції, яка їх містить, способу їх одержання та застосування таких сполук для виготовлення медикаментів, які застосовують для лікування порушень, пов'язаних з продукуванням простагландину E 2 (PGE2), наприклад, запальних процесів, болю, лихоманки, пухлин, хвороби Альцгеймера та атеросклерозу. Більш конкретно даний винахід стосується нових бензоїльних похідних 3-амінокарбазолу, які застосовують для лікування або профілактики порушень, пов'язаних з продукуванням простагландину E2 (PGE2), наприклад, запальних процесів, болю, лихоманки, пухлин, хвороби Альцгеймера та атеросклерозу. РІВЕНЬ ТЕХНІКИ Цінність простагландину E2 (PGE2) полягає у ролі, яку він відіграє як біорегулятор, разом з іншими простаноїдами, які продукуються у процесі обміну речовин з арахідонової кислоти, та як медіатор запалення. Простаноїди являють собою клас сполук, до якого належать простагландини, тромбоксани та простацикліни. Простаноїди є ліпідними медіаторами, що діють як місцеві гормони на клітинах, які межують з місцем їх вивільнення. Простаноїди здебільшого продукуються з арахідонової кислоти шляхом активованого циклооксигеназою ферментативного окиснення. Циклооксигенази (простагландин G/H синтази) каталізують послідовне утворення PGG 2 та PGH2 з арахідонової кислоти. PGH2 після цього перетворюється за допомогою специфічних ферментів на різні простаноїди. Простагландин D2 (PGD2), простагландин E2 (PGE2), простагландин F2α (PGF2α), простагландин I2 (PGI2) та тромбоксан A2 (TXA2) утворюються у такий спосіб. За винятком сім'яної рідини, простаноїди не накопичуються. Після різних подразників (запальних, імунологічних, гормональних, ультрафіолетових променів, пухлинних агентів, а також механічного збудження) вони синтезуються й вивільнюються у позаклітинний простір, звідки вони проходять у плазму з сечею та іншими біологічними рідинами. Простаноїди відіграють важливу роль у механізмах захисту функціонування органів і у цілісності організму. Це демонструється через цитозахисну функцію у шлунково-кишковому тракті шляхом регулювання ниркової функції та капілярного кровообігу, шляхом регулювання агрегації тромбоцитів та згортання крові, участь у диференціації імунних клітин і у загоєнні ран, метаболізмі клітин та овуляції. Зокрема, слід відзначити судинозахисну дію PGI 2, яка є суттєвою для підтримання судинного тонусу та для профілактики тромбоемболії та атеросклерозу на ендотеліальному рівні, і протизапальну та антипроліферативну дію PGD 2, метаболіт якого, 15d-PGJ2, здатен забезпечувати протизапальний ефект через активацію PPARγ (активованого проліфератором пероксисом гамма-рецептора) ядерних рецепторів (Inoue et al., 2000, "Feedback control of cyclooxygenase-2 expression through PPARgamma" J. Biol. Chem. 2000, 275(36): 28028-28032). Таким чином, простаноїди є біорегуляторами, а також важливими медіаторами запалення та інших патологій. Зокрема, PGE2 у великій кількості міститься у місцях запалення і відповідає за багато патологічних аспектів гострого та хронічного запалення, наприклад, набряку, утворення еритем, запального болю, суглобного запалення та лихоманки. Фактично PGE 2 являють собою сильні прозапальні та альгогенні агенти. Антитіла проти PGE2 мають протизапальну дію, і тварини, у яких відсутні рецептори PGE2, демонструють знижену реакцію на запальні подразники (Portanova et al., "Selective neutralization of prostaglandin E2 blocks inflammation, hyperalgesia, and interleukin 6 production in vivo", J. Exp. Med. 1996, 184(3): 883; Ueno et al., "Major roles of prostanoid receptors IP and EP(3) in endotoxin-induced enhancement of pain perception" Biochem. Pharmacol. 2001, 62(2): 157-160) і відсутність фебрильної реакції на пірогенні подразники (Ushikubi et al., "Impaired febrile response in mice lacking the prostaglandin E receptor subtype EP3" Nature 1998,395:281-284). Нестероїдні протизапальні медикаменти (NSAID) та селективні медикаменти проти COX-2, які нині застосовуються, зменшують пов'язані з запаленням симптоми через неселективне інгібування продукування ейкозаноїдів (PGE2, PGD2, PGF2α, PGI2 та TXA2) за рахунок їх інгібіторної дії на циклооксигенази 1 та 2 (Fitzgerald and Patrono, 2001). Зокрема, селективні медикаменти проти COX-2, які нині реалізуються на ринку, мають знижену шлунково-кишкову токсичність порівняно з традиційними нестероїдними 1 UA 102541 C2 5 10 15 20 25 30 35 40 45 50 протизапальними медикаментами (NSAID). Однак вищезгадані селективні медикаменти проти COX-2 знижують продукування судинного простацикліну (PGI2, який продукується переважно з COX-2), змінюючи нормальну рівновагу між протромботичними та антитромботичними ейкозаноїдами на користь протромботичних (TXA2, який продукується переважно з COX-1) і створюють підвищений ризик тромботичних серцево-судинних патологій (S. Malhotra, MD, DM; N. Shafiq, MD; P. Pandhi, MD Medscape General Medicine 6(1), 2004; D. Mukherjee and E.J. Topol Cardiovascular risk and COX-2 inhibitors, Arthritis Res. Ther. 2003,5:8-11-2002). Різні сполуки 3-амінокарбазолу було досліджено на їхню здатність до селективного зв'язування з людським рецептором Y5 та модулювання його активності. Ця здатність робить їх корисними для лікування порушень апетиту та обміну речовин, наприклад, ожиріння, нейрогенної булімії, нейрогенної анорексії, порушень сну, залежності від морфію та епілептичних нападів (WO 01/07409 A1, WO 02/051806, WO 02/096902 та US 6 399 631). У патентній заявці WO 2006/122 680 описується застосування багатьох сполук 3амінокарбазолу для лікування порушень, пов'язаних з продукуванням простагландину E2 (PGE2). Крім того, у патентній заявці WO 2007/014 687 описується багать нових сполук 3амінокарбазолу та їх застосування для лікування порушень, пов'язаних з продукуванням простагландину E2 (PGE2). КОРОТКИЙ ОПИС ВИНАХОДУ Несподівано було виявлено, що деякі нові сполуки 3-амінокарбазолу, крім здатності до селективного інгібування продукування простагландину E2 (PGE2), несподівано продемонстрували поліпшену біодоступність та фармакокінетичні властивості. Ці сполуки здатні знижувати продукування PGE2 і, таким чином, є активними при всіх патологічних станах, у яких PGE2 діє як медіатор, наприклад, при запальних процесах, болі, лихоманці, пухлинах, хворобі Альцгеймера та атеросклерозі. Крім того, ці сполуки несподівано продемонстрували високу метаболічну стійкість, високу абсорбцію in vitro та високу біодоступність. Типовими прикладами таких запальних процесів є набряк, еритема, суглобне запалення, ревматоїдний артрит та артроз. Типовими прикладами таких пухлин є колоректальна та легенева карцинома та аденокарцинома. Сполуки згідно з даним винаходом селективно інгібують синтез PGE 2. Ця селективність має перевагу інгібування сильного медіатора запалення, болю та лихоманки, водночас залишаючи незмінним продукування інших простаноїдів, які продукуються одночасно у каскаді арахідонової кислоти, таких, як PGF2α, TXA2, PGI2 та PGD2. Усі механізми захисту функціонування органів та цілісності організму, які є типовими для активності інших простаноїдів, таким чином, залишаються незмінними. Так само, як і традиційні нестероїдні протизапальні медикаменти, сполуки згідно з даним винаходом мають протизапальні, антипіретичні та анальгетичні властивості і, таким чином, є активними при таких патологіях, як запалення, біль, лихоманка, ревматоїдний артрит та артроз. Крім того, оскільки участь PGE2 при пухлинах, хворобі Альцгеймера та атеросклерозі є відомою з літератури, сполуки згідно з даним винаходом також застосовують у профілактиці та лікуванні цих патологій. Однак вигідним є те, що ці сполуки мають менше побічних ефектів порівняно з NSAID та селективними медикаментами проти COX-2, які при інгібуванні циклооксигеназ не розрізняють простаноїди. Зокрема, ці сполуки є корисними як для лікування, так і для профілактики запальних процесів. Зокрема, сполуки згідно з даним винаходом демонструють знижену шлунково-кишкову, ниркову та судинну токсичність. ДЕТАЛЬНИЙ ОПИС ВИНАХОДУ У першому аспекті даний винахід стосується сполуки 3-амінокарбазолу, яка має таку загальну формулу (I): 2 UA 102541 C2 5 10 15 20 (I) де R1 є атомом галогену, метильною групою або тригалометильною групою, нітрогрупою, ціаногрупою або трифлатною групою, і R2 є лінійною або розгалуженою гідроксіалкільною групою, яка включає від 1 до 8 атомів вуглецю, або лінійною або розгалуженою карбонілалкільною групою, яка включає від 1 до 8 атомів вуглецю, або її фармацевтично прийнятної солі, її поліморфного кристала, її стереоізомеру або її енантіомеру. Зокрема, даний винахід стосується сполуки 3-амінокарбазолу загальної формули (I) де R1 є атомом фтору або хлору або трифторометильною або трихлорометильною групою, і R2 є лінійною або розгалуженою гідроксіалкільною групою, яка включає від 1 до 6 атомів вуглецю, або лінійною або розгалуженою карбонілалкільною групою, яка включає від 1 до 4 атомів вуглецю. З точки зору даного винаходу термін "гідроксіалкіл" означає алкільну групу, яка включає від 1 до 3 гідроксильних груп (-OH), зв'язаних з одним або кількома атомами вуглецю, і термін "карбонілалкіл" означає алкільну групу, яка включає від 1 до 3 оксигруп (=O), зв'язаних з одним або кількома атомами вуглецю. Згідно з оптимальним аспектом, даний винахід стосується сполуки 3-амінокарбазолу загальної формули (I), де R1 та R2 мають значення, вказані нижче у Таблиці 1. Таблиця 1 Сполука 1 2 3 4 5 6 25 30 35 40 R1 CF3 CF3 CF3 CF3 Cl Cl R2 CH2CH2OH CH2C(CH3)2OH CH2CH2C(CH3)2OH CH2COCH3 CH2CH2OH CH2CH2C(CH3)2OH Описана вище Формула (I) включає сполуки, у яких фенільна група має, крім R1, один або кілька замісників, таких, як, наприклад, атом галогену, алкільна група, яка включає від 1 до 3 атомів вуглецю, трифторометильна група, нітрогрупа, трифлатна група (CF 3SO3-), алкілкарбоксильна група, яка включає від 1 до 3 атомів вуглецю (-(CH2)nCOOH), амідна група (CONH2), метилсульфокси-група (-SO2CH3), N-метилсульфонамідна група -SO2NHCH3 або метансульфонамідна група NHSO2CH3. Як відомо спеціалістам у даній галузі, фармацевтично прийнятні солі сполук загальної формули (I) можуть бути основними адиційними солями. Прикладами фармацевтично + + ++ ++ прийнятних основ є лужні метали та лужноземельні метали, такі, як Na , K , Mg , Ca , та органічні основи, такі, як трометамін, холін та лізин. Сполуки загальної формули (I) згідно з даним винаходом можуть мати більш, ніж одну кристалічну структуру або форму, або можуть існувати в аморфній формі. Сполуки, які мають цю характеристику, зазвичай називають поліморфними. Різні поліморфи цієї сполуки можуть демонструвати різні хімічні, фізичні та спектроскопічні властивості. Крім того, у разі певних замісників сполуки загальної формули (I) згідно з даним винаходом можуть мати один або кілька асиметричних атомів вуглецю і, таким чином, можуть існувати у формі стереоізомерів та енантіомерів. Таким чином, сполуки згідно з даним винаходом також включають фармацевтично прийнятні солі, поліморфні кристалічні форми, стереоізомери та енантіомери сполуки, представленої формулою (I), описаною у формулі винаходу. 3 UA 102541 C2 5 10 15 20 25 30 35 40 45 50 55 У другому аспекті даний винахід стосується фармацевтичної композиції, яка характеризується тим, що включає терапевтично ефективну дозу сполуки 3-амінокарбазолу, що має вищезгадану загальну формулу (I), або її фармацевтично прийнятну сіль разом з принаймні одним фармацевтично прийнятним інертним носієм. В оптимальному варіанті фармацевтичні композиції згідно з даним винаходом приготовляють у прийнятних дозованих формах. Прикладами прийнятних дозованих форм є таблетки, капсули, вкриті таблетки, гранули і розчини та сиропи для перорального введення; креми, мазі та антисептичні пластирі для місцевого нанесення; супозиторії для ректального введення та стерильні розчини для введення шляхом ін'єкції або аерозольного або очного введення. В оптимальному варіанті ці дозовані форми рецептують з метою забезпечення контрольованого вивільнення з часом сполуки вищезгаданої загальної формули (I) або її фармацевтично прийнятної солі. Зокрема, залежно від типу терапії, потрібний час вивільнення може бути дуже коротким, нормальним або довгим. Дозовані форми також можуть містити інші традиційні інгредієнти, наприклад: консерванти, стабілізатори, поверхнево-активні речовини, буфери, солі для регулювання осмотичного тиску, емульгатори, підсолоджувачі, барвники, ароматизатори і т. ін. Крім того, якщо вимагається для конкретних типів терапії, фармацевтична композиція згідно з даним винаходом також може містити інші фармакологічно активні інгредієнти, які зазвичай вводять одночасно. В оптимальному варіанті фармацевтична композиція згідно з даним винаходом може містити проліки сполуки формули (I). Проліки сполуки формули (I) є речовиною у практично неактивній формі, яка при введенні у живий організм метаболізується у сполуку формули (I). Як відомо спеціалістам у даній галузі, проліки сполук загальної формули (I) можуть бути естерними похідними, одержаними шляхом реакції гідроксигрупи R2 з кислотою, такою, як монокарбонова кислота, бікарбонова кислота, жирна кислота, амінокислота, (алкіл)фосфорна кислота або (алкіл)тіофосфорна кислота. Прикладами прийнятних проліків є ацетиловий естер, пропіоніловий естер, сукциніловий естер, стеаратний естер, пальмітатний естер, гліциновий естер, лейциновий естер, лізиновий естер, фосфатний естер, метилфосфатний естер, метилтіофосфатний естер та фосфонатний естер. Приклади прийнятних проліків описуються, наприклад, у публікації Stella V.J. et al, "Prodrug strategies to overcome poor water solubility", Advance Drug Delivery Reviews 59 (2007) 677-694. Композиція згідно з даним винаходом також може включати фармацевтично прийнятні солі, поліморфні кристалічні форми, стереоізомери та енантіомери проліків сполуки, представленої формулою (I), описаною у формулі винаходу. Кількість сполуки згідно з даним винаходом у фармацевтичній композиції може бути різною у широких межах, залежно від відомих чинників, наприклад, типу хвороби, яка підлягає лікуванню, тяжкості хвороби, маси тіла пацієнта, дозованої форми, вибраного шляху введення, кількості введень на день та ефективності вибраної сполуки. Однак оптимальна кількість може бути легко й звично визначена спеціалістом у даній галузі. Як правило, кількість сполуки згідно з даним винаходом у фармацевтичній композиції є такою, що забезпечує рівень введення від 0,0001 до 100 мг/кг/день, у ще кращому варіанті – від 0,01 до 10 мг/кг/день. Дозовані форми фармацевтичної композиції згідно з даним винаходом приготовляють згідно зі способами, які є добре відомими спеціалістам у галузі фармацевтичної хімії, включаючи змішування, гранулювання, таблетування, розчинення, стерилізацію і т. ін. У третьому аспекті даний винахід стосується способу лікування або профілактики запальних процесів, болю, лихоманки, пухлин, хвороби Альцгеймера та атеросклерозу у ссавців, який включає введення терапевтично ефективної кількості сполуки 3-амінокарбазолу, яка має вищезгадану загальну формулу (I), її фармацевтично прийнятної солі, її поліморфного кристала, її стереоізомеру або її енантіомеру, особі, яка цього потребує. Як правило, запальний процес є вибраним з групи, яка включає набряк, еритему, суглобне запалення, ревматоїдний артрит та артроз, і пухлина є вибраною з групи, яка включає колоректальну або легеневу карциному та аденокарциному. 3-амінокарбазоли, які мають вищезгадану загальну формулу (I), можуть бути одержані згідно з відомими способами, наприклад, шляхом реакції кислоти або її реакційноздатної похідної з вибраним аміном (патентна заявка WO 02/096902 A1, WO 02/051806, J. Med. Chem. 2002, т. 45, стор. 3509-3523). Типовими прикладами реакційноздатних похідних є ацилгаліди, ангідриди або естери. 4 UA 102541 C2 У четвертому аспекті даний винахід, таким чином, стосується способу одержання 3амінокарбазолу, який має вищезгадану загальну формулу (I), який характеризується тим, що включає такі етапи: a) реакцію аміну формули (II) (II) 5 де R2 має вищевказане значення, зі сполукою формули (III) 10 15 20 25 30 35 40 (III) де R1 має вищевказане значення, і Z є вибраним з групи, яка включає Cl, Br, OH, OR та OC(O)R, де R є лінійним або розгалуженим алкілом, який має від 1 до 6 атомів вуглецю, для одержання сполуки 3амінокарбазолу формули (I) (I) де R1 та R2 мають вищевказані значення, і b) необов'язкове утворення фармацевтично прийнятної солі, поліморфної кристалічної форми, стереоізомеру або енантіомеру одержаної таким чином сполуки формули (I). Як правило, етап (a) здійснюють у присутності прийнятного розріджувача при температурі у межах від 0 до 140 °C, протягом часу у межах від 0,5 до 24 годин. В оптимальному варіанті температура реакції становить у межах від 15 до 40 °C. В оптимальному варіанті час реакції становить від 2 до 18 годин. В оптимальному варіанті розріджувач є апротонним, полярним або аполярним. У ще кращому варіанті він є полярним апротонним розріджувачем. Прикладами прийнятних полярних апротонних розріджувачів є диметилформамід та дихлорометан. У варіанті втілення у якому Z є Cl або Br, реакцію бажано здійснювати у присутності прийнятного органічного або неорганічного кислотного акцептора. Прикладами прийнятних органічних акцепторів є діізопропілетилендіамін, триетиленамін, піридин та диметиламінопіридин. Прикладами прийнятних неорганічних акцепторів є карбонати або бікарбонати лужних металів. У варіанті втілення, де Z є OH, реакцію бажано здійснювати у присутності прийнятного зв'язувального агента, наприклад, дициклогексилкарбодііміду (також на основі з полістиролової смоли) або карбонілдіімідазолу. Представлені нижче приклади подано лише для більш детального пояснення винаходу, однак жодним чином його не обмежують. Приклад 1 Одержання Сполуки 1 R1=CF3, R2=CH2CH2OH a) 2-(3-нітро-9H-карбазол-9-іл)етанол До розчину 2-(9H-карбазол-9-іл)етанолу (25 г; 0,12 моль) у крижаній оцтовій кислоті (374 мл) протягом 30 хвилин по краплях додавали розчин, який містив азотну кислоту (6,8 мл; 0,17 моль) у крижаній оцтовій кислоті (20 мл) з інтенсивним перемішуванням. Через п'ять хвилин після завершення додавання відокремлювався зелений осад. Реакційну суміш повільно виливали у 5 UA 102541 C2 5 10 15 20 25 30 35 H2O з льодом (1 л), перемішували протягом 1 години, фільтрували і наприкінці промивали H 2O. Відокремлену тверду речовину спочатку захоплювали у H 2O (500 мл), а потім 10 % розчин карбонату натрію для досягнення pH 7 і зрештою відфільтровували. Утворену тверду речовину кристалізували з розчину ацетону / абсолютного етанолу (1:1) для одержання 20 г 2-(3-нітро-9Hкарбазол-9-іл)етанолу 1 H ЯМР (300 MГц, DMSO-d6) δ 9,16 (d, J=2,34 Гц, 1H), 8,40 (d, J=7,75 Гц, 1H), 8,33 (dd, J=2,34, 9,21 Гц, 1H), 7,79 (d, J=9,06 Гц, 1H), 7,73 (d, J=8,33 Гц, 1H), 7,57 (ddd, J=1,24, 7,13, 8,29 Гц, 1H), 7,33 (ddd, J=0,95, 7,13, 7,86 Гц, 1H), 4,89 (t, J=5,90 Гц, 1H), 4,54 (t, J=5,41 Гц, 2H), 3,82 (q, J=5,41 Гц, 2H). b) гідрохлорид 2-(3-аміно-9H-карбазол-9-іл)етанолу Продукт, одержаний, як описано на попередньому етапі a) (10 г; 0,04 моль), розчиняли у тетрагідрофурані (550 мл). Після цього додавали дигідрат хлориду олова (87 г; 0,4 моль). Одержану таким чином суміш піддавали дефлегмації протягом 16 годин. Реакційній суміші давали охолонути до кімнатної температури, а потім видаляли розчинник при зниженому тиску. Залишок захоплювали у H2O та дихлорометан і інтенсивно перемішували. Рівень pH доводили до 7,5 шляхом додавання насиченого розчину бікарбонату натрію, суміш фільтрували крізь Celite і фільтрат переносили до ділильної лійки. Органічну фазу відокремлювали і висушували над Na2SO4. Розчинник видаляли шляхом випарювання при зниженому тиску і одержаний таким чином залишок (9 г) розчиняли в етанолі й перетворювали на відповідний гідрохлорид шляхом додавання етанольного розчину 5 M хлористого водню. Осаджену тверду речовину відфільтровували для одержання гідрохлориду 2-(3-аміно-9Hкарбазол-9-іл)етанолу (9 г). 1 H ЯМР (300 MГц, DMSO-d6) δ 10,41 (broad s, 3H), 8,17 (d, J=7,76 Гц, 1H), 8,12 (d, J=1,98 Гц, 1H), 7,73 (d, J=8,75 Гц, 1H), 7,66 (d, J=8,26 Гц, 1H), 7,38-7,56 (m, 2H), 7,23 (t, J=7,27 Гц, 1H), 4,70 (broad s, 1H), 4,47 (t, J=5,53 Гц, 2H), 3,78 (t, J=5,45 Гц, 2H). c) N-[9-(2-гідроксіетил)-9H-карбазол-3-іл]-2-(трифторометил)бензамід Продукт, одержаний, як описано на попередньому етапі b) (26 г; 0,1 моль), суспендували у дихлорометані (300 мл). Після цього до розчину додавали триетиламін (28 мл; 0,2 моль) та 2трифторометилбензоїлхлорид (15.6 мл; 0,11 моль). Одержану таким чином суміш перемішували при кімнатній температурі протягом 16 годин. Розчинник випарювали при зниженому тиску, залишок захоплювали у розчин 2N NaOH (200 мл) і утворений в результаті розчин піддавали дефлегмації протягом 2 годин. Одержану таким чином суспензію виливали у воду і продукт відфільтровували, висушували й кристалізували з суміші ізопропілового етеру / ізопропанолу (1:1). Таким чином, одержували N-[9-(2-гідроксіетил)-9H-карбазол-3-іл]-2(трифторометил)бензамід (24 г). т. пл.: 176-177 °C Елементний аналіз для C22H17F3N2O2 C 66,14 66,33 Виявлено % Розраховано % 40 45 50 55 1 H 4,06 4,30 N 6,85 7,03 H ЯМР (300 MГц, DMSO-d6) δ 10,52 (s, 1H), 8,50 (d, J=1,75 Гц, 1H), 8,08 (d, J = 7,31 Гц, 1H), 7,54-7,93 (m, 7H), 7,44 (t, J=7,02 Гц, 1H), 7,18 (t, J=7,45 Гц, 1H), 4,85 (t, J = 5,45 Гц, 1H), 4,43 (t, J=5,70 Гц, 2H), 3,79 (q, J=5,75 Гц, 2H). Приклад 2 Одержання Сполуки 2 R1=CF3, R2=CH2C(CH3)2OH a) 1 -(9H-карбазол-9-іл)-2-метилпропан-2-ол До розчину, який містив карбазол (20 г; 0,12 моль) у DMSO (300 мл), додавали 50 % розчин гідроксиду натрію (300 мл), бензилтриметиламонійхлорид (5,5 г; 0,024 моль) та по краплях 2хлоро-2-метилпропан-2-ол (39,1 г; 0,36 моль). Одержану таким чином суміш перемішували при кімнатній температурі протягом 16 годин. Суміш виливали у H2O з льодом (3 L), перемішували протягом 1 години, фільтрували і утворену тверду речовину кристалізували з суміші гексану/етилацетату (9:1) для одержання 1(9H-карбазол-9-іл)-2-метилпропан-2-олу (15 г). 1 H ЯМР (300 MГц, DMSO-d6) δ 8,07-8,15 (m, 2H), 7,68 (d, J=8,33 Гц, 2H), 7,40 (ddd, J=1,24, 7,13, 8,29 Гц, 2H), 7,13-7,20 (m, 2H), 4,64 (s, 1H), 4,26 (s, 2H), 1,21 (s, 6H). b) 1-(3-нітро-9H-карбазол-9-іл)-2-метилпропан-2-ол 6 UA 102541 C2 5 10 15 20 25 30 35 40 До розчину продукту, одержаного, як описано на попередньому етапі a) (21 г; 0,088 моль) у крижаній оцтовій кислоті (400 мл), протягом 30 хвилин по краплях додавали розчин, який містив азотну кислоту (5 мл; 0,123 моль) у крижаній оцтовій кислоті (15 мл; 0,263 моль) з інтенсивним перемішуванням. Через 5 хвилин після завершення додавання відокремлювався зелений осад. Реакційну суміш повільно виливали у H2O з льодом (1 л), перемішували протягом 1 години, фільтрували і наприкінці промивали H2O. Відокремлену тверду речовину спочатку захоплювали у H2O (500 мл), а потім у 10 % розчин карбонату натрію до досягнення pH 7 і зрештою відфільтровували. Утворену тверду речовину кристалізували з суміші етилацетату/етанолу (8:2) для одержання 1-(3-нітро-9H-карбазол-9-іл)-2-метилпропан-2-олу (19 г). 1 H ЯМР (300 MГц, DMSO-d6) δ 9,15 (d, J=2,05 Гц, 1H), 8,38 (d, J=7,31 Гц, 1H), 8,30 (dd, J=2,48, 9,21 Гц, 1H), 7,87 (d, J=9,06 Гц, 1H), 7,81 (d, J=8,48 Гц, 1H), 7,54 (ddd, J=1,17, 7,16, 8,33 Гц, 1H), 7,31 (td, J=0,88, 7,60 Гц, 1H), 4,73 (s, 1H), 4,37 (s, 2H), 1,21 (s, 6H). c) 1 -(3-аміно-9H-карбазол-9-іл)-2-метилпропан-2-ол гідрохлорид Продукт, одержаний, як описано на попередньому етапі b) (7,9 г; 0,028 моль) розчиняли у тетрагідрофурані (350 мл). Після цього додавали дигідрат хлориду олова (62.8 г; 0,28 моль). Одержану таким чином суміш піддавали дефлегмації протягом 16 годин. Реакційній суміші давали охолонути до кімнатної температури, а потім видаляли розчинник при зниженому тиску. Залишок захоплювали у H2O та дихлорометан і піддавали інтенсивному перемішуванню. Рівень pH доводили до 7,5 шляхом додавання насиченого розчину бікарбонату натрію, суміш фільтрували крізь Celite і фільтрат переносили до ділильної лійки. Органічну фазу відокремлювали і висушували над Na2SO4. Розчинник видаляли шляхом випарювання при зниженому тиску і одержаний таким чином залишок (9 г) розчиняли в етанолі й перетворювали на відповідний гідрохлорид шляхом додавання етанольного розчину 5 M хлористого водню. Утворену тверду речовину кристалізували з суміші ізопропанолу / води (8:2) для одержання 1(3-аміно-9H-карбазол-9-іл)-2-метилпропан-2-ол гідрохлориду (6 г). 1 H ЯМР (300 MГц, DMSO-d6) δ 10,32 (broad s, 3H), 8,16 (d, J=7,60 Гц, 1H), 8,10 (d, J=2,31 Гц, 1H), 7,81 (d, J=8,92 Гц, 1H), 7,74 (d, J=8,26 Гц, 1H), 7,47 (ddd, J=0,99, 7,10, 8,42 Гц, 1H), 7,42 (dd, J=2,15, 8,75 Гц, 1H), 7,22 (t, J=7,43 Гц, 1H), 4,70 (broad s, 1H), 4,30 (s, 2H), 1,20 (s, 6H). d) N-[9-(2-гідрокси-2-метилпропіл)-9H-карбазол-3-іл]-2-(трифторометил)бензамід Продукт, одержаний, як описано на попередньому етапі c) (3,3 г; 0,011 моль), суспендували у дихлорометані (30 мл). Після цього до розчину додавали триетиламін (3 мл; 0,022 моль) та 2трифторометилбензоїлхлорид (1,7 мл; 0,012 моль). Одержану таким чином суміш перемішували при кімнатній температурі протягом 16 годин. Розчинник випарювали при зниженому тиску, залишок захоплювали у розчин 2N NaOH (20 мл) і утворений в результаті розчин піддавали дефлегмації протягом 2 годин. Одержану таким чином суспензію виливали у воду і продукт відфільтровували, висушували й кристалізували з суміші ізопропілового етеру / ізопропанолу (1:1). Таким чином, одержували N-[9-Гідрокси-2-метилпропіл)-9H-карбазол-3-іл]-2(трифторометил)-бензамід (2,7 г). т. пл.: 179-181 °C Елементний аналіз для C24H21F3N2O2 C 67,51 67,60 Виявлено % Розраховано % 45 50 55 1 H 4,82 4,96 N 6,52 6,57 H ЯМР (300 MГц, DMSO-d6) δ 10,50 (s, 1H), 8,49 (s, 1H), 8,06 (d, J=7,93 Гц, 1H), 7,56-7,92 (m, 7H), 7,42 (t, J=7,76 Гц, 1H), 7,17 (t, J=7,43 Гц, 1H), 4,65 (s, 1H), 4,26 (s, 2H), 1,21 (s, 6H). Приклад 3 Одержання Сполуки 3 R1=CF3, R2=CH2CH2C(CH3)2OH a) Етил 3-(9H-карбазол-9-іл)пропаноат До розчину, який містив карбазол (20 г; 0,12 моль) у DMF (130 мл), порціями додавали гідрид натрію (50 % суспензія) (6,7 г; 0,14 моль); одержану таким чином суспензію перемішували при кімнатній температурі протягом 30 хвилин, а потім нагрівали до 60 °C. По краплях додавали розчин, який містив етил 3-бромопропаноат (17,9 мл; 0,14 моль), у DMF (20 мл) і суміш перемішували протягом 16 годин. Суміш виливали у H2O (0,5 л) і фільтрували. Утворену тверду речовину очищали шляхом флеш-хроматографії на силікагелі, застосовуючи як елюент суміш гексану/етилацетату (8:2), 7 UA 102541 C2 5 10 15 20 25 30 35 для одержання 16 г етил 3-(9H-карбазол-9-іл)пропаноату, і цей продукт використовували у наступній реакції без подальшого очищення. b) 4-(9H-карбазол-9-іл)-2-метилбутан-2-ол До розчину продукту, одержаного на попередньому етапі a) (15,2 г; 0,057 моль), у тетрагідрофурані (200 мл) додавали 3M розчин метилмагніййодиду у діетиловому етері (57 мл; 0,171 моль). Одержану таким чином суміш перемішували при кімнатній температурі протягом 16 годин. Після цього до суміші додавали розчин 1M NH 4Cl (500 мл). Утворену в результаті суміш переносили до ділильної лійки і екстрагували етилацетатом. Органічну фазу висушували над Na2SO4 і розчинник випарювали при зниженому тиску. Одержаний залишок кристалізували з суміші гексану/етилацетату (8:2) для одержання 4-(9H-карбазол-9-іл)-2-метилбутан-2-олу (9 г) і цей продукт використовували у наступній реакції без подальшого очищення. c) 2-метил-4-(3-нітро-9H-карбазол-9-іл)бутан-2-ол Продукт, одержаний на попередньому етапі b) (7,2 г; 0,028 моль), піддавали реакції у спосіб, подібний до описаного у Прикладі 1a). Одержаний продукт кристалізували з етилацетату для одержання 2-метил-4-(3-нітро-9H-карбазол-9-іл)бутан-2-олу (6 г). 1 H ЯМР (300 MГц, DMSO-d6) δ 9,17 (d, J=2,34 Гц, 1H), 8,41 (d, J=7,60 Гц, 1H), 8,36 (dd, J=2,34, 9,35 Гц, 1H), 7,73 (d, J=9,35 Гц, 1H), 7,66-7,71 (m, 1H), 7,56-7,64 (m, 1H), 7,31-7,38 (m, 1H), 4,62 (s, 1H), 4,48-4,58 (m, 2H), 1,79-1,89 (m, 2H), 1,24 (s, 6H). d) 4-(3-аміно-9H-карбазол-9-іл)-2-метилбутан-2-ол гідрохлорид До суспензії продукту, одержаного на попередньому етапі c) (5,9 г; 0,020 моль) у 95° етанолі (80 мл) додавали 10 % Pd/C (0,5 г; 0,0005 моль) і суміш піддавали гідрогенізації у гідрогенізаторі Парра (30 psi) протягом 4 годин. Реакційну суміш фільтрували, розчин випарювали при зниженому тиску і одержаний продукт розчиняли в етилацетаті і перетворювали на відповідний гідрохлорид шляхом додавання етанольного розчину 5M хлористого водню. Утворену таким чином тверду речовину кристалізували з суміші ізопропілового етеру / ізопропанолу (1:1) для одержання 4-(3-аміно-9H-карбазол-9-іл)-2-метилбутан-2-ол гідрохлориду (5,5 г). 1 H ЯМР (300 MГц, DMSO-d6) δ 10,34 (broad s, 3H), 8,19 (d, J=7,60 Гц, 1H), 8,13 (d, J=1,98 Гц, 1H), 7,68 (d, J=8,92 Гц, 1H), 7,62 (d, J=8,20 Гц, 1H), 7,44-7,58 (m, 2H), 7,25 (t, J=6,94 Гц, 1H), 4,084,83 (m, 3H), 1,73-1,88 (m, 2H), 1,23 (s, 6H). e) N-[9-(3-гідрокси-3-метилбутил)-9H-карбазол-3-іл]-2-трифторометил-бензамід Продукт, одержаний, як описано на попередньому етапі d) (3,9 г; 0,013 моль), піддавали реакції у спосіб, подібний до описаного у Прикладі 1c). Утворену тверду речовину кристалізували з етанолу для одержання N-[9-(3-гідрокси-3метилбутил)-9H-карбазол-3-іл]-2-(трифторометил)бензаміду (2,3 г). т. пл.: 188-189 °C Елементний аналіз для C25H23F3N2O2 C 67,75 68,17 Виявлено % Розраховано % 1 40 45 50 55 H 5,31 5,26 N 6,23 6,36 H ЯМР (300 MГц, DMSO-d6) δ 10,53 (s, 1H), 8,52 (d, J=1,98 Гц, 1H), 8,10 (d, J = 7,60 Гц, 1H), 7,68-7,90 (m, 4H), 7,66 (dd, J=2,00, 8,70 Гц, 1H), 7,52-7,59 (m, 2H), 7,47 (t, J = 7,10 Гц, 1H), 7,19 (t, J=7,43 Гц, 1H), 4,55 (s, 1H), 4,38-4,51 (m, 2H), 1,71-1,91 (m, 2H), 1,23 (s, 6H). Приклад 4 Одержання Сполуки 4 R1=CF3, R2=CH2COCH3 a) Етил 9H-карбазол-9-ілацетат До розчину, який містив карбазол (20 г; 0,12 моль), у DMF (130 мл) порціями додавали гідрид натрію (50 % суспензія) (6,9 г; 0,14 моль); одержану таким чином суспензію перемішували при кімнатній температурі протягом 30 хвилин, а потім нагрівали до 60 °C. По краплях додавали розчин, який містив етил 2-бромоацетат (24 г; 0,14 моль), у DMF (20 мл) і утворену в результаті суміш перемішували протягом 16 годин. Суміш виливали у H 2O (0.5 L) і фільтрували і утворену тверду речовину кристалізували з гексану для одержання етил 9Hкарбазол-9-ілацетату (20 г). 1 H ЯМР (300 MГц, DMSO-d6) δ 8,15 (d, J=7,60 Гц, 2H), 7,54 (d, J=8,20 Гц, 2H), 7,43 (td, J=1,02, 7,67 Гц, 2H), 7,17-7,27 (m, 2H), 5,33 (s, 2H), 4,14 (q, J=7,02 Гц, 2H), 1,20 (t, J=7,16 Гц, 3H). b) 1-(9H-карбазол-9-іл)ацетон До розчину продукту, одержаного на попередньому етапі a) (14,1 г; 0,056 моль), у тетрагідрофурані (130 мл) додавали 3M розчин метилмагніййодиду у діетиловому етері (28 мл; 8 UA 102541 C2 5 10 15 20 25 30 35 0,084 моль). Одержану таким чином суміш перемішували при кімнатній температурі протягом 16 годин. Після цього до суміші додавали розчин 1M NH 4Cl (100 мл). Утворену в результаті суміш переносили до ділильної лійки і екстрагували етилацетатом. Органічну фазу висушували над Na2SO4 і розчинник випарювали при зниженому тиску. Одержаний залишок очищали шляхом флеш-хроматографії на силікагелі, застосовуючи як елюент суміш гексану/етилацетату (95:5) для одержання 1-(9H-карбазол-9-іл)ацетону (8 г), і цей продукт використовували без подальшого очищення у наступній реакції. 1 H ЯМР (300 MГц, DMSO-d6) δ 8,15 (d, J=7,89 Гц, 2H), 7,49 (d, J=8,20 Гц, 2H), 7,41 (ddd, J=1,10, 7,00, 8,20 Гц, 2H), 7,21 (ddd, J=1,10, 7,00, 7,89 Гц, 2H), 5,39 (s, 2H), 2,24 (s, 3H). c) 1-(3-нітро-9H-карбазол-9-іл)ацетон Продукт, одержаний на попередньому етапі b) (5 г; 0,022 моль), піддавали реакції у спосіб, подібний до описаного у Прикладі 1a). Одержаний залишок очищали шляхом флешхроматографії на силікагелі, застосовуючи як елюент 8:2 суміш гексану/етилацетату для одержання 1-(3-нітро-9H-карбазол-9-іл)ацетону (4,5 г), і цей продукт використовували без подальшого очищення для наступної реакції. 1 H ЯМР (300 MГц, DMSO-d6) δ 9,20 (d, J=2,34 Гц, 1H), 8,42 (d, J=7,89 Гц, 1H), 8,33 (dd, J=2,34, 9,06 Гц, 1H), 7,70 (d, J=9,35 Гц, 1H), 7,60-7,67 (m, 1H), 7,54 (td, J=1,17, 7,75 Гц, 1H), 7,30-7,39 (m, 1H), 5,57 (s, 2H), 2,32 (s, 3H). d) гідрохлорид 1-(3-аміно-9H-карбазол-9-іл)ацетону До суспензії продукту, одержаного на попередньому етапі c) (1,3 г; 0,005 моль), у 95° етанолі (80 мл) додавали 10 % Pd/C (0,5 г; 0,0005 моль) і суміш піддавали гідрогенізації у гідрогенізаторі Парра (30 psi) протягом 4 годин. Реакційну суміш фільтрували і розчин випарювали при зниженому тиску. Одержаний продукт розчиняли в етилацетаті і перетворювали на відповідний гідрохлорид шляхом додавання етанольного розчину 5M хлористого водню. Осаджену тверду речовину відфільтровували для одержання гідрохлориду 1-(3-аміно-9H-карбазол-9-іл)ацетону (1,1 г). 1 H ЯМР (300 MГц, DMSO-d6) δ 10,39 (broad s, 3H), 8,19 (d, J=7,60 Гц, 1H), 8,14 (d, J=1,98 Гц, 1H), 7,62 (d, J=8,59 Гц, 1H), 7,52-7,58 (m, 1H), 7,40-7,52 (m, 2H), 7,25 (ddd, J = 0,99, 6,94, 7,93 Гц, 1H), 5,46 (s, 2H), 2,27 (s, 3H). e) N-[9-(2-оксопропіл)-9H-карбазол-3-іл]-2-(трифторометил)бензамід Продукт, одержаний на попередньому етапі d) (1,1 г; 0,004 моль), піддавали реакції у спосіб, подібний до описаного у Прикладі 1c). Утворену тверду речовину кристалізували з суміші ізопропілового етеру / ізопропанолу (1:1) для одержання N-[9-(2-оксопропіл)-9H-карбазол-3-іл]-2-(трифторометил)бензаміду (1,2 г). т. пл.: 223-226 °C Елементний аналіз для C23H17F3N2O2 C 67,02 67,31 Виявлено % Розраховано % 1 40 45 50 55 H 3,91 4,18 N 6,78 6,83 H ЯМР (300 MГц, DMSO-d6) δ 10,52 (s, 1H), 8,50 (d, J=1,75 Гц, 1H), 8,10 (d, J = 7,60 Гц, 1H), 7,66-7,91 (m, 4H), 7,61 (dd, J=2,05, 8,77 Гц, 1H), 7,36-7,53 (m, 3H), 7,20 (t, J = 6,87 Гц, 1H), 5,38 (s, 2H), 2,24 (s, 3H). Приклад 5 Одержання Сполуки 5 R1=Cl, R2=CH2CH2OH a) 2-хлоро-N-[9-(2-гідроксіетил)-9H-карбазол-3-іл]бензамід Одержаний продукт, як описано Прикладі 1b) (6,4 г; 0,028 моль), суспендували у дихлорометані (70 мл). Після цього до розчину додавали триетиламін (7,9 мл; 0,2 моль) та 2хлоробензоїлхлорид (3,95 мл; 0,031 моль). Одержану таким чином суміш перемішували при кімнатній температурі протягом 16 годин. Розчинник випарювали при зниженому тиску, залишок захоплювали у розчин 2N NaOH (80 мл) і утворений в результаті розчин піддавали дефлегмації протягом 2 годин. Одержану таким чином суспензію виливали у воду і продукт відфільтровували, висушували й кристалізували з 95° етанолу. Таким чином, одержували 2-хлоро-N-[9-(2-гідроксіетил)-9H-карбазол-3-іл]бензамід (5,5 г). т. пл.: 168-169 °C Елементний аналіз для C21H17CIN2O2 9 UA 102541 C2 C 68,83 69,14 Виявлено % Розраховано % H 4,63 4,70 N 7,58 7,68 1 5 10 15 H ЯМР (300 MГц, DMSO-d6) δ 10,47 (s, 1H), 8,55 (d, J=1,98 Гц, 1H), 8,08 (d, J = 7,27 Гц, 1H), 7,39-7,71 (m, 8H), 7,18 (t, J=7,43 Гц, 1H), 4,86 (t, J=5,45 Гц, 1H), 4,43 (t, J = 5,78 Гц, 2H), 3,78 (q, J=5,83 Гц, 2H). Приклад 6 Одержання Сполуки 6 R1=Cl, R2=CH2CH2C(CH3)2OH a) 2-хлоро-N-[9-(3-гідрокси-3-метилбутил)-9H-карбазол-3-іл]бензамід Одержаний продукт, як описано Прикладі 3d) (1,1 г; 0,0037 моль) піддавали реакції з 2хлоробензоїлхлоридом (0,52 мл; 0,0041 моль) у спосіб, подібний до описаного у Прикладі Приклад 1c). Утворену тверду речовину кристалізували з етилацетату для одержання 2-хлоро-N-[9-(3гідрокси-3-метилбутил)-9H-карбазол-3-іл]бензамід (0,63 г). т. пл.: 120-124 °C Елементний аналіз для C24H23CIN2O2 Виявлено % Розраховано % C 70,52 70,84 1 20 25 30 35 40 45 50 H 5,62 5,70 N 6,71 6,88 H ЯМР (300 MГц, DMSO-d6) δ 10,48 (s, 1H), 8,57 (d, J=1,98 Гц, 1H), 8,09 (d, J = 7,60 Гц, 1H), 7,41-7,73 (m, 8H), 7,19 (t, J=7,43 Гц, 1H), 4,55 (s, 1H), 4,37-4,51 (m, 2H), 1,73-1,88 (m, 2H), 1,23 (s, 6H). Приклад 7 Приготування порівняльної сполуки A Порівняльна сполука A відповідає сполуці 1 з патентної заявки WO 2006/122 680, і її приготовляли, як описано у вищезгаданій патентній заявці. Приклад 8 Приготування порівняльної сполуки B Порівняльна сполука B відповідає сполуці 6 з патентної заявки WO 2007/014 687, і її приготовляли, як описано у вищезгаданій патентній заявці. Приклад 9 Приготування порівняльної сполуки C Порівняльна сполука C відповідає сполуці 13 з патентної заявки WO 2007/014 687, і її приготовляли, як описано у вищезгаданій патентній заявці. Приклад 10 Випробування in vitro активності Це випробування дозволяє визначити здатність до інгібування продукування PGE 2 та селективність стосовно продукування PGF2α. Використовували лінію клітин A549 людської легеневої аденокарциноми, яка є особливо чутливою до стимуляції прозапальними цитокінами, наприклад, IL-1β, і у відповідь на цю стимуляцію є особливо активною при продукуванні та вивільненні двох простаноїдів: PGE2 та PGF2α (Thoren S. Jakobsson P-J, 2000). Клітини стимулювали IL-1β (1 нг/мл) і одночасно обробляли випробуваною сполукою протягом 22 годин у відповідному культуральному середовищі (DMEM - модифікованому Дульбекко середовищі Ігла) з додаванням 5 % ембріональної телячої сироватки та L-глутаміну (остаточна кількість – 4 мM) в інкубаторі при 37 °C і при концентрації CO2 5 %. Після інкубації кількість PGE2 та PGF2α, продукованих і вивільнених у супернатант, аналізували за допомогою комплекту EIA (який виробляється й реалізується Cayman Chemicals, Ann Arbor, MІ, USA). Застосовуваною порівняльною сполукою був індометацин у концентрації 10 нM (SigmaAldrich), нестероїдний протизапальний медикамент, якій однаковою мірою інгібує PGE 2 та PGF2α. Результати, виражені як значення IC50, тобто, як концентрація сполуки, яка інгібує 50 % продукування PGE2 та PGF2α відносно клітин, які піддавали стимуляції, але не обробляли тією ж сполукою, представлено у Таблиці 2. Відсутність активності або знижена активність сполуки у 10 UA 102541 C2 біосинтезі PGF2α є показником селективності щодо продукування PGE2, а отже, селективного інгібування mPGES-1. Таблиця 2 Сполука IC50 PGE2 2,9 2,3 1,4 5,6 0,6 0,005 1 2 3 4 6 Індометацин 5 10 15 20 [мкM] PGF2α >100 >100 >100 >100 >100 0,003 Приклад 11 Випробування in vivo активності Випробувану сполуку оцінювали у моделі викликаних оцтовою кислотою судом у мишей (Stock J.L. et al., J. Clin. Inv. 2001, 107: 325-331). Це випробування дозволяє визначати антиноцицептивну активність сполук згідно з винаходом у моделі запального болю. Для випробування використовували самиць мишей CD-1 масою 25-30 г. Тваринам внутрішньочеревинно вводили випробувану сполуку (0,1-10 мг/кг), суспендовану у метилцелюлозі (MTC). Контрольним тваринам вводили лише індиферентний носій (MTC) таким самим шляхом. Через півгодини після введення тваринам вводили внутрішньочеревинну ін'єкцію оцтової кислоти (0,7 % (об'єм/об'єм) у фізіологічному розчині, 16 мкл/г маси тіла) для викликання запального болю і для перевірки впливу випробуваної сполуки на ноцицептивну реакцію. Відразу після введення оцтової кислоти і протягом наступних 20 хвилин вимірювали кількість судом, що є параметром оцінки ноцицептивної реакції. Як показано у Таблиці 3, сполука згідно з винаходом викликала залежне від дози зниження кількості судом за 20 хвилин після введення оцтової кислоти, порівняно з тваринами, які отримували лише MTC. Таблиця 3 Введення Носій Сполука 1 25 30 35 40 Доза (мг/кг) 0,1 1 10 Кількість судом 48,4±3,66 38,4±3,99 31,5±5,72 12,8±2,46 % інгібування 21 35 74 Приклад 12 Випробування метаболічної стійкості у мікросомах печінки людини та щура. Це випробування дозволяє визначити метаболічну стійкість сполук згідно з винаходом та порівняльних сполук у щурів та людини. Випробувані сполуки інкубували у печінкових мікросомах людини (група донорів, Xenotech) та печінкових мікросомах щурів Sprague-Dawley (група донорів, Xenotech) і порівняння випробуваної сполуки вимірювали таким чином, щоб отримати попередній показник метаболічної стійкості у різних видів, застосовуючи HPLC/MS/MS з мас-спектрометром Applied Biosystems 4000 QTrap. Сполуки, які піддавали аналізові, у кінцевій концентрації 0 та 1 мкM, поміщали у суспензію, яка містила пул мікросом у кінцевій концентрації 0,5 мг/мкл у кінцевому об'ємі 200 мкл, у 96лункових планшетах. Випробування стандартизували з застосуванням фосфатного буфера (75 мM, pH 7,4) та регенеруючої системи NADPH (MgCl2: 3,3 мM; G6P: 3,3 мM; G6PD: 0,4 U/мл; NADP+: 1,3 мM). Контрольні сполуки варфарин, пропанолол та тестостерон (Sigma) інкубували як коктейль і піддавали обробці як для випробуваних сполук. Зразки інкубували при 37 °C у зволоженому інкубаторі. На початку відліку часу і через 60 хвилин додавали 100 мкл ацетонітрилу, що містив внутрішній стандарт (0,2 мкM метопрололу та 0,4 мкM диклофенаку) для припинення реакції. Перед аналізом зразки центрифугували. Аналіз HPLC/MS/MS здійснювали, застосовуючи джерело електророзпилювальної позитивної іонізації та SRM (Single Reaction Mode). 11 UA 102541 C2 5 10 Хроматографічні умови передбачають застосування колонки XDB-C18 (2,1 × 50 мм, Agilent) та градієнта від 5 % до 91 % ацетонітрилу у воді, що містить 0,1 % мурашиної кислоти (загальний час прогону дорівнює 6 хвилинам); швидкість потоку становила 0,5 мл/хв. Площі під піками для випробуваних сполук об'єднували і результати виражали як співвідношення площ аналізованого зразка / внутрішнього стандарту (PAR). Щоразу аналізували два зразки і розраховували середнє значення. Відсоток показника сполуки, що залишилася, розраховували таким чином: % неметаболізованої сполуки = 100 * (середн. PAR Tfinal/середн. PART0). Результати для сполук з 1 по 6 представлено у Таблиці 4 разом з результатами для порівняльних сполук A, B та C і для контрольних сполук. Сполуки згідно з даним винаходом демонстрували поліпшену метаболічну стійкість відносно порівняльних сполук. Таблиця 4 Сполука 1 2 3 4 5 6 A B C Варфарин Пропанолол Тестостерон 15 20 25 30 35 40 45 Щур 58 % 68 % 65 % 58 % 63 % 74 % 0,2 % 0,4 % 4,0 % 97 % 0,4 % 0% Людина 81 % 82 % 77 % 7% 76 % 65 % 51 % 55 % 22 % 103 % 66 % 27 % Приклад 13 Випробування in vitro абсорбції Це випробування дозволяє визначити абсорбовану кишковим бар'єром кількість сполук згідно з винаходом та порівняти сполуки за допомогою лінії клітин Caco-2 як в in vitro моделі кишкового бар'єру. Випробування проникності на клітинах Caco-2 являє собою in vitro систему, затверджену для прогнозування та оцінки in vivo кишкової абсорбції медикаменту. Коли клітини Caco-2 культивують на пористому фільтрі протягом приблизно 21 дня, вони мають здатність диференціюватися до ентероцитів. На практиці протягом цього періоду клітини Caco-2 зазнають спонтанних морфологічних та біохімічних змін, які в результаті ведуть до утворення поляризованого клітинного моношару, який на апікальній поверхні має чітко визначену "щіткову облямівку", і утворюють "щільні з'єднання" між клітинами, таким чином, являючи собою придатну модель для аналізу кишкової проникності медикаментів. Для здійснення випробування застосовували такі матеріали: Lucifer yellow (Sigma) Збалансований сольовий розчин Хенка (HBSS) (Invitrogen) радіоактивний стандартний зразок (Perkin Elmer) Клітини Caco-2 (ATCC) Планшети Caco-2 Multiscreen™ (Millipore) HPLC/MS/MS за допомогою мас-спектрометра Applied Biosystems 4000 QTrap Ацетонітрил, який містить 0,2 мкM метопрололу як внутрішній стандарт. Сполуки, які піддавали випробуванню, розводили від 10 мM вихідного розчину у HBSS до кінцевої концентрації 10 мкM. Система складалася з конфлюентного клітинного моношару у культурі протягом 21-28 днів. Контрольні сполуки (Lucifer yellow, атенолол, пропанолол та дигоксин) включали до кожного випробування як контроль якості і для порівняння зі сполуками, які піддаються випробуванню. Кожну сполуку випробували у трьох примірниках, двоспрямовано, при pH 7,4, від апікального до базолатерального відділу (A→B) і від базолатерального до апікального відділу (B→A). Зразки, зібрані у певний час, аналізували шляхом HPLC-MS/MS, застосовуючи джерело електророзпилювальної позитивної іонізації та SRM (Single Reaction Mode). Хроматографічні умови передбачають застосування колонки XDB-C18 (2,1 × 50 мм, Agilent) з градієнтом від 5 % до 91 % ацетонітрилу у воді, що містить 0,1 % мурашиної кислоти (загальний час прогону 12 UA 102541 C2 5 10 15 20 дорівнює 6,5 хвилини) та швидкістю потоку 0,5 мл/хв. Метопролол використовували як внутрішній стандарт. Дані концентрації використовували для розрахунку значень ефективної проникності (P app) і розраховували середнє та стандартне відхилення Papp. Співвідношення потоків розраховували як Papp(B→A)/Papp(A→B). Відсоток видобутку розраховували таким чином: (кількість у приймаючому відділі + кількість у донорному відділі) / нормальна кількість Площі під піками випробуваних сполук об'єднували і результати виражали як співвідношення площ аналізованого зразка / внутрішнього стандарту і коректували з врахуванням коефіцієнта розведення, який застосовували під час приготування зразка. Коефіцієнти ефективної проникності розраховували за допомогою такого рівняння: де: VA об'єм у приймальному резервуарі (0,25 мл для випробування від A→B, 0,075 мл для випробування від B→A) 2 Area площа поверхні мембрани (0,11 см ) Time загальний час перенесення (3600 секунд) Отримані значення класифікували на основі такого критерію оцінки. 6 Низьке Papp < 2 × 10- см/сек 6 6 Середнє 2 × 10- см/сек < Papp 20 × 10- см/сек Результати для сполук з 1 по 6 представлено у Таблиці 5 разом з результатами для порівняльних сполук A, B та C і для контрольних сполук. Сполуки згідно з даним винаходом демонстрували поліпшені очікувані показники абсорбції відносно порівняльних сполук. 25 Таблиця 5 Сполука 1 2 3 4 5 6 A B C Lucifer yellow Атенолол Пропанолол Дигоксин 30 35 40 Абсорбція Висока Висока Висока Висока Висока Висока Низька Низька Середня Низька Низька Висока Низька Приклад 14 Випробування in vivo біодоступності Це випробування дозволяє визначити in vivo біодоступність сполук згідно з винаходом, таким чином, дозволяючи оцінити й порівняти фармакокінетичний профіль випробуваних сполук. Випробування здійснювали, застосовуючи касетний спосіб, тобто, пероральне введення кількох продуктів одночасно одній тварині у дозі 5 мг/кг. Продукти суспендували у метилцелюлозі (MTC). Підданих лікуванню тварин катетеризували для серійного відбирання проб крові, яке здійснювали за допомогою автоматичної системи відбирання проб. Плазматичну концентрацію продуктів вимірювали шляхом HPLC/MS/MS. Профілі плазматичної концентрації у часі дозволяли визначити відносну біодоступність випробуваних продуктів з точки зору швидкості (tmax та Cmax) та виду (AUC). Нахил кривої у кінцевій частині також дозволяв здійснити порівняльну оцінку швидкості видалення сполук з плазми – чим повільніша швидкість, тим нижче нахил. Для кожної комбінації сполук випробували по три тварини. Відбирали сполуку, яка 13 UA 102541 C2 5 10 мала вищі показники Cmax та AUC і очікуваний показник tmax відносно інших, оскільки вона демонструвала добру швидкість in vivo абсорбції. Застосовуваним порівняльним продуктом була сполука C, яка демонструвала обмежену абсорбцію, тоді, як сполуки 1, 2 та 3 згідно з даним винаходом демонстрували добрі характеристики біодоступності. Результати, виражені як Cmax, тобто, максимальна концентрація медикаменту, яка досягається у плазмі, Tmax, тобто, час, який вимагається для досягнення максимальної концентрації медикаменту у плазмі, та AUC0-7, тобто, площа під кривою плазматичної концентрації медикаменту у часі, виміряна за перші сім годин після введення, представлено у Таблиці 6. Таблиця 6 Cmax нг/мл 1200 984 457 165 Сполука 1 2 3 C 15 Tmax год. 1,5 4,3 1,8 1,7 AUC0-7 нг/мл*год. 5754 5403 2668 751 сполука 3-амінокарбазолу, фармацевтична композиція, яка її містить, та спосіб її одержання Реферат Даний винахід стосується нових бензоїльних похідних 3-амінокарбазолу, фармацевтичної композиції, яка їх містить, способу одержання та застосування таких сполук для виготовлення медикаменту, який є корисним для лікування або профілактики порушень, пов'язаних з продукуванням простагландину E2 (PGE2), наприклад, запальних процесів, болю, лихоманки, пухлин, хвороби Альцгеймера та атеросклерозу. 20 ФОРМУЛА ВИНАХОДУ 1. Сполука 3-амінокарбазолу, яка характеризується тим, що має загальну формулу (І): H N R1 O N R2 , (I) 25 30 35 де R1 є атомом галогену, метильною групою або тригалометильною групою, нітрогрупою, ціаногрупою або трифлатною групою, і R2 є лінійною або розгалуженою гідроксіалкільною групою, яка включає від 1 до 8 атомів вуглецю, або лінійною або розгалуженою карбонілалкільною групою, яка включає від 1 до 8 атомів вуглецю, або її фармацевтично прийнятна сіль, її поліморфний кристал, її стереоізомер, її енантіомер або її проліки. 2. Сполука 3-амінокарбазолу за п. 1, яка відрізняється тим, що R1 є атомом фтору або хлору або трифторометильною або трихлорометильною групою, і R2 є лінійною або розгалуженою гідроксіалкільною групою, яка включає від 1 до 6 атомів вуглецю, або лінійною або розгалуженою карбонілалкільною групою, яка включає від 1 до 4 атомів вуглецю. 3. Сполука 3-амінокарбазолу за п. 1, яка відрізняється тим, що R1 та R2 мають значення, вказані нижче у Таблиці 1 40 14 UA 102541 C2 Таблиця 1 5 Сполука R1 R2 1 CF3 СН2СН2ОН 2 CF3 CH2C(CH3)2OH 3 CF3 СН2СН2С(СН3)2ОН 4 CF3 CH2COCH3 5 Cl СН2СН2ОН 6 Cl СН2СН2С(СН3)2ОН 4. Фармацевтична композиція, яка відрізняється тим, що містить терапевтично ефективну дозу сполуки 3-амінокарбазолу за будь-яким з пунктів з 1 по 3 або її фармацевтично прийнятної солі, її поліморфного кристала, її стереоізомера, її енантіомера або її проліків разом з принаймні одним фармацевтично прийнятним інертним носієм. 5. Спосіб одержання сполуки 3-амінокарбазолу за будь-яким з пунктів з 1 по 3, який відрізняється тим, що здійснюють такі етапи: а) реакцію аміну формули (II) H2N N R2 10 , (II) де R2 має значення за будь-яким з пунктів з 1 по 3, зі сполукою формули (III) Z R1 15 O , (III) де R1 має значення за будь-яким з пунктів з 1 по 3, і Z є вибраним з групи, яка включає Сl, Br, OH, OR та OC(O)R, де R є лінійним або розгалуженим алкілом, який включає від 1 до 6 атомів вуглецю, з одержанням сполуки 3-амінокарбазолу формули (І) H N R1 O N R2 20 25 30 , (I) де R1 та R2 мають вищевказані значення, та b) необов'язкове утворення фармацевтично прийнятної солі, поліморфної кристалічної форми, стереоізомера або енантіомера одержаної таким чином сполуки формули (І). 6. Застосування сполуки 3-амінокарбазолу за будь-яким з пунктів з 1 по 3 або її фармацевтично прийнятної солі, її поліморфного кристала, її стереоізомера, її енантіомера або її проліків для виробництва медикаменту для профілактичного або терапевтичного лікування порушення, вибраного з групи, яка включає запальні процеси, біль, лихоманку, пухлини, хворобу Альцгеймера та атеросклероз. 7. Застосування сполуки 3-амінокарбазолу за п. 6, яке відрізняється тим, що вищезгадані запальні процеси є вибраними з групи, яка включає набряк, еритему, суглобне запалення, ревматоїдний артрит та артроз. 15 UA 102541 C2 5 8. Застосування сполуки 3-амінокарбазолу за п. 6, яке відрізняється тим, що вищезгадані пухлини є вибраними з групи, яка включає колоректальну та легеневу карциному та аденокарциному. 9. Спосіб лікування або профілактики запальних процесів, болю, лихоманки, пухлин, хвороби Альцгеймера та атеросклерозу у ссавців, який включає введення терапевтично ефективної кількості сполуки 3-амінокарбазолу за будь-яким з пунктів з 1 по 3 або її фармацевтично прийнятної солі, її поліморфного кристала, її стереоізомера, її енантіомера або її проліків особі, яка цього потребує. Комп’ютерна верстка Д. Шеверун Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 16
ДивитисяДодаткова інформація
Назва патенту англійською3-aminocarbazole compound, pharmaceutical composition containing it and preparation method therefor
Автори англійськоюAlisi, Maria Alessandra, Cazzolla, Nicola, Coletta, Isabella, Dragone Patrizia, Furlotti, Guido, Garofalo, Barbara, Guglielmotti, Angelo, Mangano, Giorgina, Maugeri, Caterina
Назва патенту російськоюСоединение 3-аминокарбазола, фармацевтическая композиция, которая его содержит, и способ его получения
Автори російськоюАлиси Мария Алессандра, Каццолла Никола, Колетта Изабелла, Драгоне Патриция, Фурлотти Гвидо, Гарофало Барбара, Гуглиелмотти Анджело, Гуглиэлмотти Анджело, Мангано Джорджина, Мауджери Катерина
МПК / Мітки
МПК: A61P 29/00, C07D 209/88, A61K 31/403
Мітки: композиція, фармацевтична, одержання, 3-амінокарбазолу, яка, сполука, містить, спосіб
Код посилання
<a href="https://ua.patents.su/18-102541-spoluka-3-aminokarbazolu-farmacevtichna-kompoziciya-yaka-mistit-ta-sposib-oderzhannya.html" target="_blank" rel="follow" title="База патентів України">Сполука 3-амінокарбазолу, фармацевтична композиція, яка її містить, та спосіб її одержання</a>
3-амінокарбазольна сполука, фармацевтична композиція, яка її містить, та спосіб її одержання

Номер патенту: 93378
Опубліковано: 10.02.2011
Автори: Фурлотті Гвідо, Алісі Марія Алессандра, Колетта Ізабелла, Мангано Джорджина, Поленцані Лоренцо, Каццолла Нікола, Руссо Вінченцо, Драгоне Патріція
МПК: C07D 209/88, A61P 35/00, A61K 31/403
Мітки: композиція, фармацевтична, одержання, спосіб, яка, 3-амінокарбазольна, містить, сполука
Формула / Реферат:
1. 3-Амінокарбазольна сполука, яка характеризується тим, що вибрана із групи, яка складається зі сполук таблиці 1 нижче:Таблиця 1 (I) № сполуки R1 R2 R3 R4 R5 R6 X Y 1 ...
Сполука, що є похідним 2-амінобіцикло[3.1.0]гексан-2,6-дикарбонової кислоти, спосіб одержання цієї сполуки, фармацевтична композиція, що містить цю сполуку, проміжна сполука

Номер патенту: 43332
Опубліковано: 17.12.2001
Автори: Тіззано Джозеф Патрік, Монн Джеймс Аллен, Хелтон Девід Рід, ШЕПП Дарріл Дарвін, Роубі Роджер Льюіс
МПК: A61K 31/198, C07D 233/78, C07D 235/02, A61P 25/18, A61K 31/195, C07C 69/757, A61K 31/215, C07C 62/00, A61K 31/00, C07C 229/50
Мітки: кислоти, проміжна, сполуки, сполуку, цієї, фармацевтична, сполука, похідним, 2-амінобіцикло[3.1.0]гексан-2,6-дикарбонової, композиція, містить, одержання, спосіб
Формула / Реферат:
1. Соединение, являющееся производным 2-аминобицикло[3.1.0]гексан-2,6-дикарбоновой кислоты, формулы (1)гдеХ-(СН2)n,R2- CO2R4 и R3 - водород или R2 - водород и R3 - CO2R4,R1 и R4, независимо друг от друга - водород, алкил С1-С10, алкенил С2-С10, арил или арилалкил, n -1,или его фармацевтически приемлемая соль,предназначенное для использования в качестве фармацевтического препарата....
Похідні гетероциклічних оксимів, спосіб їх одержання (варіанти), фармацевтична композиція, яка їх містить, проміжна сполука

Номер патенту: 80814
Опубліковано: 12.11.2007
Автори: Кеньяр Даніель-Енрі, Ренар Пьєр, Лебег Ніколя, Інтровінь Карін, Бутен Жан, Альбер, Даке Катрін, Карато Паскаль, Леклерк Веронік, Бертело Паскаль, Пайу Сільві
МПК: A61P 25/28, A61P 17/06, A61P 15/00, A61P 3/10, A61P 13/12, C07D 417/10, A61P 1/04, A61P 21/04, A61P 19/10, C07D 277/68, A61K 31/427, A61P 27/02, A61P 9/10, A61P 9/00, A61P 1/18, A61P 35/00, A61K 31/426, A61P 3/06, C07D 417/12, A61P 13/04
Мітки: варіанти, похідні, гетероциклічних, оксимів, містить, сполука, проміжна, композиція, фармацевтична, одержання, спосіб, яка
Формула / Реферат:
1. Сполуки формули (І):, (I)де:Х являє собою атом кисню або сірки або групу СН2 або (де R'2 разом з R2 утворює додатковий зв'язок),R1 і R2, які можуть бути однаковими або різними, кожний являє собою атом водню, лінійну або розгалужену (С1-С6)алкільну групу, арильну групу,...
Похідні 1,2-анельованого хіноліну, спосіб їх одержання (варіанти), фармацевтична композиція, що їх містить, проміжна сполука та спосіб її одержання

Номер патенту: 71592
Опубліковано: 15.12.2004
Автори: Бурдрез Ксав'єр Марк, Венет Марк Гастон, Анжибо Патрік Рене
МПК: A61K 31/519, A61K 31/4709, C07D 471/04, C07D 215/18, A61P 35/00, C07D 487/04, C07D 401/06, C07D 403/06, C07D 215/06
Мітки: сполука, спосіб, 1,2-анельованого, варіанти, проміжна, одержання, хіноліну, містить, похідні, композиція, фармацевтична
Формула / Реферат:
1. Похідні 1,2-анельованого хіноліну формули (І)(І)або їх фармацевтично прийнятна кислотно-адитивна сіль чи стереохімічно ізомерна форма,у якій=Х1-Х2-Х3- означає тривалентний радикал формули =N-СR6=СR7- (х-1), =СR6-СR7=СR8- (х-6), =N-N=СR6- (х-2), =СR6-N=СR7- (х-7), ...
Тієно[2,3-d]піримідинова сполука, спосіб її одержання, фармацевтична композиція, що її містить, і спосіб її одержання, спосіб здійснення імуносупресії і спосіб лікування

Номер патенту: 61111
Опубліковано: 17.11.2003
Автори: Кук Ендрю, Купер Мартін, Перрі Меттью, Тоурн Філіп, ФЕРБЕР Марк, Дональд Девід, Чешір Девід
МПК: A61P 37/06, A61P 1/00, A61P 1/04, A61P 11/06, A61P 37/02, A61P 19/02, A61P 11/00, A61P 9/10, A61P 43/00, A61P 17/00, A61P 17/06, A61P 31/18, A61P 37/04, A61P 29/00, A61K 31/519, C07D 495/04
Мітки: фармацевтична, здійснення, спосіб, сполука, імуносупресії, тієно[2,3-d]піримідинова, лікування, одержання, композиція, містить
Формула / Реферат:
1. Тієно[2,3-d]піримідинова сполука формули (І):, (I)деR являє собою -C(O)Ar1, -C(R4)(R5)Ar1 або Аr2;Аr1 являє собою нафтил, хіноліл, ізохіноліл, індоліл, бензофураніл або бензотієніл, кожен з яких може бути необов'язково заміщеним одним чи кількома замісниками, вибраними з С1-4-алкілу, С1-4-алкокси, галогену або трифторметилу, або Аr1 являє собою феніл, необов'язково заміщений одним чи кількома замісниками,...
Попередній патент: П’ятичленні гетероцикли, їх конденсовані похідні та їх застосування в лікуванні діабету
Наступний патент: Жорсткий пакет для дрібних предметів
Випадковий патент: Зносостійка полімерна композиція