[1,2,4]триазолопіридини і їх застосування як інгібіторів фосфодіестерази
Номер патенту: 111520
Опубліковано: 10.05.2016
Автори: Ларсен Йєнс Хрістіан Хойланн, Нільсен Сімон Фельдбек







Формула / Реферат
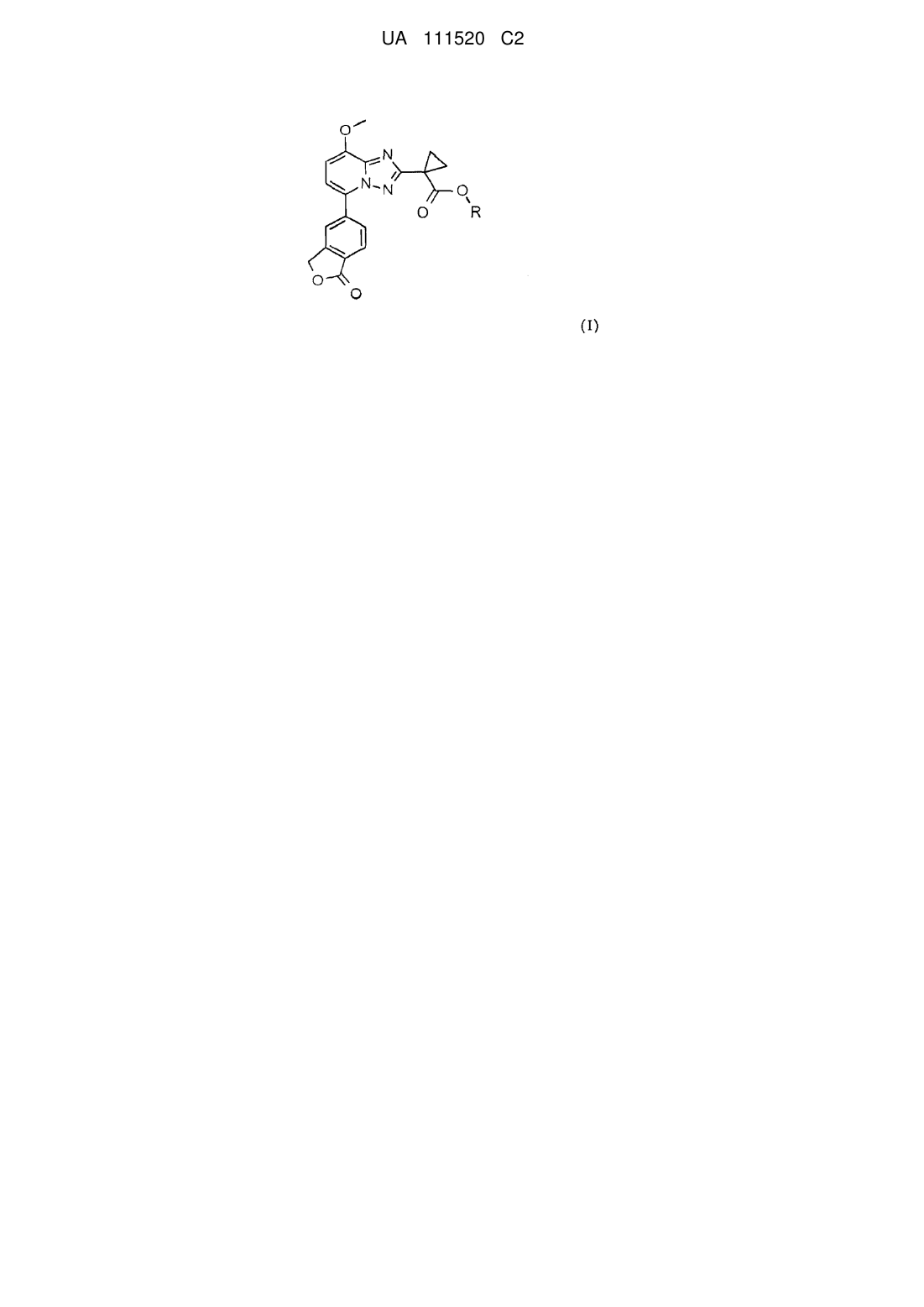
1. Сполука загальної формули (І):
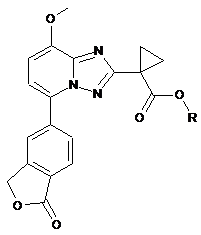 (І)
(І)
або будь-який з її стереоізомерів, або будь-яка суміш її стереоізомерів, або її фармацевтично прийнятна сіль, у яких R являє собою розгалужений бутил.
2. Сполука за п. 1 або будь-який з її стереоізомерів, або будь-яка суміш її стереоізомерів, або її фармацевтично прийнятні солі, де R являє собою 1-метилпропіл, 2-метилпропіл або трет-бутил.
3. Сполука за п. 1, яка являє собою [(1S)-1-метилпропіл]-1-[8-метоксі-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5-а]піридин-2-іл]циклопропанкарбоксилат, вільну основу.
4. Сполука за п. 1, яка являє собою [(1R)-1-метилпропіл]-1-[8-метоксі-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5-а]піридин-2-іл]циклопропанкарбоксилат, вільну основу.
5. Сполука за п. 1, яка являє собою [2-метилпропіл]-1-[8-метоксі-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5-а]піридин-2-іл]циклопропанкарбоксилат, вільну основу.
6. Сполука за п. 1, яка являє собою трет-бутил-1-[8-метоксі-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5-а]піридин-2-іл]циклопропанкарбоксилат, вільну основу.
7. Фармацевтична композиція, яка містить сполуку за будь-яким із пп. 1-6 разом із фармацевтично прийнятним наповнювачем або ексципієнтом, або фармацевтично прийнятним носієм(ями).
8. Фармацевтична композиція за п. 7, яка додатково містить одну або кілька інших фармацевтично прийнятних сполук.
9. Застосування сполуки за будь-яким із пп. 1-6 для отримання фармацевтичної композиції.
10. Застосування сполуки за п. 9 для отримання фармацевтичної композиції для лікування або зменшення інтенсивності симптомів захворювання, порушення або стану, сприйнятливого до інгібуючої активності PDE4.
11. Застосування за п. 10, де захворювання, порушення або стан є шкірним захворюванням або станом.
12. Застосування за п. 11, де захворювання, порушення або стан є проліферативним і запальним порушенням шкіри, дерматитом, атопічним дерматитом, себорейним дерматитом, контактним дерматитом, псоріазом, раком, епідермальним запаленням, алопецією, шкірною атрофією, індукованою стероїдами шкірною атрофією, старінням шкіри, фотостарінням шкіри, вугровим висипом, кропивницею, свербежем і екземою.
13. Сполука за будь-яким із пп. 1-6 для застосування як лікарського засобу.
14. Сполука за п. 13 для застосування при лікуванні або зменшенні інтенсивності симптомів захворювання, порушення або стану, сприйнятливого до інгібуючої PDE4 активності.
15. Сполука за п. 13 для застосування при лікуванні або зменшенні інтенсивності симптомів шкірних захворювань або станів.
16. Сполука за п. 13 для застосування при лікуванні проліферативних і запальних захворювань шкіри, дерматиту, атопічного дерматиту, себорейного дерматиту, контактного дерматиту, псоріазу, раку, епідермального запалення, алопеції, шкірної атрофії, індукованої стероїдами шкірної атрофії, старіння шкіри, фотостаріння шкіри, вугрового висипу, кропивниці, свербежу і екземи.
17. Спосіб лікування або зменшенні інтенсивності симптомів захворювання, порушення або стану, сприйнятливого до інгібуючої PDE4 активності, який включає стадію введення в організм живого організму терапевтично ефективної кількості сполуки за будь-яким із пп. 1-6.
18. Спосіб лікування або зменшення інтенсивності симптомів шкірних захворювань або станів, який включає введення пацієнту, який страждає щонайменше на одне із вказаних захворювань, ефективної кількості однієї або декількох сполук за будь-яким із пп. 1-6, необов'язково разом із фармацевтично прийнятним носієм або одним або декількома ексципієнтами, необов'язково в комбінації з іншими терапевтично активними сполуками.
19. Спосіб за п. 18, в якому шкірне захворювання або стан вибрані з групи, яка складається з проліферативних і запальних захворювань шкіри, дерматиту, атопічного дерматиту, себорейного дерматиту, контактного дерматиту, псоріазу, раку, епідермального запалення, алопеції, шкірної атрофії, індукованої стероїдами шкірної атрофії, старіння шкіри, фотостаріння шкіри, вугрового висипу, кропивниці, свербежу і екземи.
Текст
Реферат: Даний винахід стосується нових похідних [1,2,4]триазолопіридину з інгібуючою фосфодіестеразу активністю, а також їх застосування як терапевтичних агентів при лікуванні запальних захворювань і станів. UA 111520 C2 (12) UA 111520 C2 UA 111520 C2 5 10 15 20 25 30 35 40 45 50 55 60 Галузь техніки, до якої належить винахід Даний винахід стосується нових похідних [1,2,4]триазолопіридину з інгібуючою фосфодіестеразу активністю, а також їх застосування як терапевтичних агентів при лікуванні запальних захворювань і станів. Рівень техніки винаходу Фосфодіестерази являють собою ферменти, які каталізують гідроліз циклічного АМФ і/або циклічного ГМФ в клітинах в 5-АМФ і 5-ГМФ відповідно, і як такі, вони є такими, що визначають для клітинної регуляції рівнів цАМФ або цГМФ. З 11 фосфодіестераз, ідентифікованих досі, фосфодіестераза (РDE) 4, PDE7 і PDE8 є селективними для цАМФ. PDE4 є найбільш важливим модулятором цАМФ, який експресується в імунних і запальних клітинах, таких, як нейтрофіли, макрофаги і Т-лімфоцити (Z. Huang and J.A. Mancini, Current Med. Chem. 13, 2006, pp. 32533262). Оскільки цАМФ є ключовим вторинним месенджером при модуляції запальних реакцій, було виявлено, що PDE4 регулює запальні реакції запальних клітин шляхом модуляції прозапальних цитокінів, таких, як TNF-α, IL-2, IFN-y, GM-CSF і LTB4. Інгібування PDE4, таким чином, стає привабливою мішенню для лікування запальних захворювань, таких, як астма, хронічна обструктивна хвороба легень (COРD), ревматоїдний артрит, атопічний дерматит, запальне захворювання кишечнику, таке, як хвороба Крона, і т. д. (M.D. Houslay et al., Drug Discovery Today 10 (22), 2005, рр. 1503-1519). Коли пацієнти з атопічним дерматитом (AD) мають збільшену PDE-активність, виявляється, що інгібування РDE4 може бути також ефективним способом лікування AD (Journal of Investigative Dermatology (1986), 87 (3), 372-6). Сімейство генів PDE4 складається щонайменше з чотирьох генів, А, В, С і D, які мають високий ступінь гомології (V. Boswell Smith and D. Spina, Curr. Opinon Investig. Drugs 6(11), 2006, pp. 1136-1141). Чотири ізоформи PDE4 диференціально експресуються в різних тканинах і типах клітин. Так, PDE4B переважно експресується в моноцитах і нейтрофілах, але не експресується в корі головного мозку і епітеліальних клітинах, тоді як PDE4D експресується в легенях, корі головного мозку, мозочку і Т-клітинах (С. Kroegel and М. Foerster, Exp. Opinion Investig. Drugs 16(1), 2007, рр. 109-124). Було передбачено, що інгібування PDE4D в головному мозку пов'язане з негативними діями, виявленими при введенні інгібіторів PDE4 клінічно, головним чином нудота і блювота, в той час як інгібування PDE4B пов'язано з протизапальними діями (В. Lipworth, Lancet 365, 2005, рр. 167-175). Однак, не вважається, що інгібітори PDE, розроблені досі, є специфічними для будь-якої з чотирьох ізоформ PDE4. Вивчені численні інгібітори PDE4 відносно їх терапевтичної дії на запальні захворювання, в основному астми і COРD. Перший з них, теофілін, є слабким, неселективним інгібітором фосфодіестерази, що застосовується при лікуванні респіраторних захворювань, таких, як астма і COРD. Лікування теофіліном може, однак, викликати як слабкі, так і важкі побічні дії, наприклад, аритмію і судоми, обмежуючи тим самим клінічну застосовність теофіліну (Kroegel and Foerster, див. вище). Оскільки фосфодіестераза залишалася привабливою мішенню для протизапальної терапії, були розроблені і досліджені в клінічних умовах декілька інших, більш селективних інгібіторів PDE4. Клінічну розробку багатьох з першого покоління інгібіторів PDE4, таких, як роліпрам, припинили внаслідок обмежуючих дозу побічних дій, в основному нудоти і блювоти. Проте, рофлуміласт в 2010 р. був схвалений для важкого COРD, пов'язаного з хронічним бронхітом, після того, як обмежуючі дозу побічні дії, нудота, діарея і головний біль були зведені до мінімуму. Інгібітори PDE4 другого покоління зі зрозуміло менш вираженими побічними діями в цей час перебувають на стадії клінічних випробувань (Houslay, див. вище). Інгібітори PDE4 описані, наприклад, в ЕР 0771794 і ЕР 0943613. У WO 2008/125111, LEO Pharma А/S, описуються похідні триазолопіридину з сильною інгібуючою PDE4 активністю. Ці сполуки включають в себе лінкер, який включає в себе карбонільну групу між біциклічною гетероциклічною системою кілець і моноциклічною системою кілець. Для спорідненої сполуки, пікламіласту, було показано, що лінкер є надзвичайно важливим для положення моноциклічного кільця так, щоб він міг взаємодіяти з ферментом PDE4 (Card G.L., et al., "Структурна основа для активності лікарських засобів, які інгібують фосфодіестерази", Structure 2004 Dec; 12(12); 2233-47), тим самим забезпечуючи бажану інгібуючу дію. У WO 2010/069322, LEO Pharma А/S, описуються похідні триазолопіридину без карбонільного лінкера між біциклічною і моноциклічною системами кілець. Виявлено, що дані сполуки виявляють інгібуючу PDE4 активність. Існує постійна потреба в розробці нових інгібіторів PDE4, які мають більш сприятливе терапевтичне вікно, тобто мають більш слабкі побічні дії, зберігаючи при цьому свою терапевтичну протизапальну дію. 1 UA 111520 C2 5 10 15 20 25 30 35 Суть винаходу Задачею даного винаходу є створення нових сполук, які є сильнодіючими інгібіторами PDE4, що мають профіль стабільності в біологічній тканині, яка означає, що тільки дуже низький системний вплив сполук буде спостерігатися, наприклад, при місцевому введенні. Більш точно, сполуки даного винаходу мають високий кліренс у мікросомах печінки людини. Вони швидко гідролізуються в цільній крові людини, але в той же час виявляють стабільність відносно ферментативного гідролізу в кератиноцитах людини. В одному аспекті винаходу запропонована сполука формули (I) , в якій R має значення, вказані нижче. У іншому аспекті даний винахід стосується фармацевтичних композицій, які містять сполуки загальної формули (I), що визначається вище, разом з фармацевтично прийнятним носієм або ексципієнтом або фармацевтично прийнятним носієм(ями), необов'язково разом з одним або декількома іншими терапевтично активними сполуками. У іншому аспекті винахід стосується застосування сполуки винаходу для виготовлення фармацевтичних композицій для профілактики, лікування, запобігання або зменшення інтенсивності симптомів захворювання, порушення або стану, сприйнятливих до інгібуючої активності PDE4. У ще одному аспекті винахід стосується способу профілактики, лікування, запобігання або зменшення інтенсивності симптомів захворювань, порушень або станів, сприйнятливих до інгібуючої активності PDE4, причому спосіб містить стадію введення в організм живої тварини терапевтично ефективної кількості сполуки формула (I) даного винаходу. Інші задачі даного винаходу будуть очевидні фахівцеві в даній галузі техніки з подальшого докладного опису і прикладів. Докладний опис винаходу В одному аспекті даний винахід стосується сполуки формули (I) будь-якого з її стереоізомерів або будь-якої суміші її стереоізомерів або її фармацевтично прийнятної солі, у яких R являє собою розгалужений бутил. У одному варіанті здійснення даного винаходу R являє собою 1-метилпропіл, 2-метилпропіл або трет-бутил. У іншому варіанті здійснення R являє собою 1-метилпропіл. У іншому варіанті здійснення R являє собою 2-метилпропіл. У іншому варіанті здійснення R являє собою трет-бутил. Конкретні приклади сполук формули (I) можна вибрати з групи, яка складається з: 2 UA 111520 C2 5 10 15 20 25 30 35 40 45 50 55 60 [(1S)-1-метилпропіл]-1-[8-метокси-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5а]піридин-2-іл]циклопропанкарбоксилату; [(1R)-1-метилпропіл]-1-[8-метокси-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5а]піридин-2-іл]циклопропанкарбоксилату; [2-метилпропіл]-1-[8-метокси-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5-а]піридин2-іл]циклопропанкарбоксилату; трет-бутил-1-[8-метокси-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5-а]піридин-2іл]циклопропанкарбоксилату або їх фармацевтично прийнятних солей. Визначення Використовувані протягом всього даного опису і прикладеної формули винаходу наступні терміни мають вказані значення. Термін "лікування", що застосовується в контексті, означає спосіб лікування пацієнта і догляд за пацієнтом з метою боротьби із захворюванням, порушенням або хворобливим станом. Передбачається, що термін включає в себе затримку розвитку захворювання, порушення або стану, полегшення або ослаблення симптомів і ускладнень і/або лікування або ліквідації захворювання, порушення або стану. Пацієнт, що піддається лікуванню, переважно є ссавцем, зокрема людиною. Терміни "захворювання", "стан" і "порушення", що використовується в контексті, застосовують взаємозамінним чином для опису стану пацієнта, який є ненормальним фізіологічним станом людини. Термін "лікарський засіб", що застосовується в контексті, означає фармацевтичну композицію, придатну для введення фармацевтично активної сполуки пацієнту. Термін "фармацевтично прийнятний", що застосовується в контексті, означає, що засіб є прийнятним для звичайних фармацевтичних застосувань, тобто не викликає ніяких побічних дій у пацієнтів і т. д. Передбачається, що термін "фармацевтично прийнятна сіль" означає солі, отримані взаємодією сполуки формули I з прийнятною неорганічною або органічною кислотою, такою, як хлористоводнева, бромистоводнева, йодистоводнева, сірчана, азотна, фосфорна, мурашина, оцтова, 2,2-дихлороцтова, адипінова, аскорбінова, L-аспарагінова, L-глутамінова, галактарова, молочна, малеїнова, L-яблучна, фталева, лимонна, пропіонова, бензойна, глутарова, глюконова, D-глюкуронова, метансульфонова, саліцилова, янтарна, малонова, винна, бензолсульфонова, етан-1,2-дисульфонова, 2-гідроксіетансульфонова, толуолсульфонова, сульфамінова або фумарова кислота. Сполуки винаходу можна отримати в кристалічній формі або безпосередньо концентруванням з органічного розчинника, або кристалізацією або перекристалізацією з органічного розчинника або суміші вказаного розчинника і співрозчинника, який може бути органічним або неорганічним, таким, як вода. Кристали можна виділити в формі по суті без розчинника або у вигляді сольвату, такого, як гідрат. Винахід охоплює всі кристалічні модифікації і форми, а також їх суміші. Сполуки формули (I) можуть містити або можуть не містити асиметрично заміщені (хіральні) атоми вуглецю, які обумовлюють існування ізомерних форм, наприклад, енантіомерів. Даний винахід стосується всіх таких ізомерів або в чистому вигляді, або у вигляді їх сумішей (наприклад, рацематів). Чисті стереоізомерні форми сполук і проміжних продуктів даного винаходу можна отримати із застосуванням процедур, відомих в даній галузі. Різні ізомерні форми можна розділити фізичними методами розділення, такими, як селективна кристалізація і хроматографічні методи, наприклад, рідинна хроматографія із застосуванням хіральних стаціонарних фаз. Вказані чисті стереоізомерні форми можна також отримати з відповідних чистих стереоізомерних форм прийнятних вихідних речовин при умові, що реакція протікає стереоселективним або стереоспецифічним чином. Якщо бажаним є конкретний стереоізомер, вказану сполуку переважно буде синтезовано стереоселективними або стереоспецифічними способами отримання. У таких способах переважно будуть застосовувати хіральні чисті вихідні речовини. Терапевтичне застосування Коли сполуки винаходу мають інгібуючу PDE4 активність, сполуки можуть бути застосовними як терапевтичні агенти для запальних алергічних захворювань, таких, як бронхіальна астма, COPD, алергічний риніт і нефрит; аутоімунних захворювань, таких, як ревматоїдний артрит, розсіяний склероз, хвороба Крона і системний червоний вовчак; гострих або хронічних шкірних хвороб; захворювань центральної нервової системи, таких, як депресія, амнезія і деменція; органопатії, пов'язаної з ішемічним рефлюксом, викликаним серцевою 3 UA 111520 C2 5 10 15 20 25 30 35 40 45 50 55 60 недостатністю, шоком і цереброваскулярними захворюваннями і т. д.; інсулін-резистентного цукрового діабету; ран; СНІДу і тому подібного. У одному варіанті здійснення сполуки даного винаходу вважаються застосовними для лікування, запобігання або ослаблення симптомів шкірних захворювань або станів. У іншому варіанті здійснення сполуки даного винаходу вважаються застосовними для лікування, запобігання або ослаблення симптомів шкірних захворювань або станів, вибраних із групи, яка складається з проліферативних і запальних захворювань шкіри, дерматиту, атопічного дерматиту, себорейного дерматиту, контактного дерматиту, псоріазу, раку, епідермального запалення, алопеції, шкірної атрофії, індукованої стероїдами шкірної атрофії, старіння шкіри, фотостаріння шкіри, вугрового висипу, кропивниці, свербіжу і екземи. У іншому варіанті здійснення сполуки даного винаходу вважаються застосовними для лікування або ослаблення симптомів атопічного дерматиту. У іншому варіанті здійснення сполуки даного винаходу вважаються застосовними для лікування або ослаблення псоріазу. Сполуки винаходу, необов'язково в комбінації з іншими активними сполуками, можуть бути застосовними для лікування шкірних захворювань або станів, зокрема для лікування проліферативних і запальних захворювань шкіри, дерматиту, атопічного дерматиту, себорейного дерматиту, контактного дерматиту, псоріазу, раку, епідермального запалення, алопеції, атрофії шкіри, індукованої стероїдами атрофії шкіри, старіння шкіри, фотостаріння шкіри, вугрового висипу, кропивниці, свербіжу і екземи. Крім того, що сполуки даного винаходу є застосовними для лікування людини, вони можуть бути також застосовними у ветеринарії для лікування тварин, включаючи ссавців, таких, як коні, велика рогата худоба, вівці, свині, собаки і кішки. Для застосування в терапії сполуки даного винаходу звичайно знаходяться в формі фармацевтичної композиції. Тому винахід стосується фармацевтичної композиції, яка містить сполуку формули (I), необов'язково разом з одним або декількома іншими терапевтично активними сполуками, разом із фармацевтично прийнятним ексципієнтом або носієм. Наповнювач повинен бути "прийнятним" в значенні сумісності з іншими інгредієнтами композиції і не бути шкідливим для його реципієнта. У формі уніфікованої дози сполуку можна вводити один або декілька разів на день із відповідними інтервалами часу, залежно завжди, однак, від стану пацієнта і відповідно до рецепта, виписаного практикуючим лікарем. Уніфікована доза препарату для місцевого застосування переважно містить від 0,1 мг до 1000 мг, переважно від 1 мг до 100 мг, наприклад, 5-50 мг сполуки формули (I). Прийнятна доза сполуки даного винаходу буде залежати, крім іншого, від віку і стану пацієнта, тяжкості захворювання, що піддається лікуванню, і інших чинників, добре відомих практикуючому лікареві. Сполуки можна вводити або перорально, парентерально, або місцево згідно з різними схемами застосування, наприклад, щодня або з тижневими інтервалами. Загалом разова доза буде в діапазоні від 0,001 до 10 мг/кг маси тіла, наприклад, в діапазоні від 0,01 до 1 мг/кг маси тіла. Сполуки можна вводити у вигляді болюсу (тобто всю добову дозу вводити відразу) або у вигляді дробних доз два або більше разів на день. У контексті місцевого лікування дозу можна більш прийнятним чином називати "уніфікованою дозою", яка означає унітарну, тобто разову дозу, яку можна вводити пацієнту і яку можна легко приготувати і упакувати, причому вона залишається як фізично, так і хімічно стабільною уніфікованою дозою, що містить або активну речовину як таку, або її суміш з твердими або рідкими фармацевтичними розріджувачами або носіями. "Уніфіковану дозу" можна вводити місцево пацієнту при нанесенні на квадратний сантиметр шкіри в кількості від 0,1 мг до 50 мг, переважно від 0,2 мг до 5 мг кінцевої композиції, що розглядається. Передбачається також, що при деяких схемах лікування може бути сприятливим введення з більш тривалими інтервалами, наприклад, через день, кожний тиждень, або навіть з більш тривалими інтервалами. Якщо лікування включає в себе введення іншої терапевтично активної сполуки, рекомендується брати до уваги публікацію Goodman & Gilman's The Pharmacological Basis of th Therapeutics, 9 Ed., J.G. Hardman and L.E. Limbird (Eds.), McGraw-Hill 1995, для визначення придатних дозувань вказаних сполук. Введення сполуки даного винаходу з однією або декількома іншими активними сполуками можна провести або одночасно, або послідовно. Препарати включають в себе, наприклад, препарати в формі, прийнятній для перорального (зокрема препарати зі стійко підтримуваним або регульованим у часі вивільненням), ректального, парентерального (зокрема підшкірного, внутрішньочеревного, 4 UA 111520 C2 5 10 15 20 25 30 35 40 45 50 55 внутрішньом'язового, внутрішньосуглобного і внутрішньовенного), трансдермального, офтальмічного, місцевого, дермального, назального або трансбукального введення. Місцеве введення заявленого препарату є особливо прийнятним. Препарати можна переважно представляти в дозованій лікарській формі і їх можна отримувати будь-яким зі способів, добре відомих в галузі фармації, наприклад, як описано в публікації Remington, The Science and Practice of Pharmacy, 20th ed., 2000. Всі способи включають в себе стадію змішування активного інгредієнта з носієм, який складається з одного або декількох допоміжних інгредієнтів. Звичайно препарати отримують однорідним і тісним змішуванням активного інгредієнта з рідким носієм або тонкоподрібненим твердим носієм, або обома такими носіями і потім, якщо необхідно, формуванням продукту в потрібний препарат. Препарати даного винаходу, прийнятні для перорального введення, можуть бути в формі дискретних одиниць, таких, як капсули, саше, таблетки або пігулки, причому кожна з них містить заздалегідь визначену кількість активного інгредієнта; в формі порошку або гранул; в формі розчину або суспензії у водній рідині або неводній рідині, такій, як етанол або гліцерин; або в формі емульсії масло у воді або емульсії вода в маслі. Такими маслами можуть бути харчові олії, такі, як наприклад, бавовняна олія, кунжутна олія, кокосова олія або арахісова олія. Прийнятні диспергуючі або суспендуючі агенти для водних суспензій включають в себе синтетичні або природні смоли, такі, як трагакант, альгінат, аравійська камедь, декстран, натрієва сіль карбоксиметилцелюлози, желатин, метилцелюлоза, гідроксипропілметилцелюлоза, гідроксипропілцелюлоза, карбомери і полівінілпіролідон. Активні інгредієнти можна також вводити у вигляді болюсу, лікарської кашки або пасти. Таблетку можна приготувати пресуванням або формуванням активного інгредієнта, необов'язково з одним або декількома додатковими інгредієнтами. Пресовані таблетки можна отримувати пресуванням у придатній машині активного інгредієнта(ів) в сипкій формі, такій, як порошок або гранули, необов'язково змішаного зі зв'язувальною речовиною, такою, як, наприклад, лактоза, глюкоза, крохмаль, желатин, аравійська камедь, трагакантова камедь, альгінат натрію, карбоксиметилцелюлоза, метилцелюлоза, гідроксипропілметилцелюлоза, поліетиленгліколь, віск або тому подібне; змащувальною речовиною, такою, як, наприклад, олеат натрію, стеарат натрію, стеарат магнію, бензоат натрію, ацетат натрію, хлорид натрію або тому подібне; дезінтегруючим агентом, таким, як, наприклад, крохмаль, метилцелюлоза, агар, бентоніт, натрієва сіль кроскармелози, натрієва сіль гліколяту крохмалю, кросповідон або тому подібне, або диспергуючим агентом, таким, як полісорбат 80. Формовані таблетки можна отримати формуванням у придатній машині суміші порошкоподібного активного інгредієнта і прийнятного носія, зволоженого інертним рідким розріджувачем. Препарати для ректального введення можуть бути в формі супозиторіїв, в яких сполука даного винаходу змішана з низькоплавкими, розчинними або нерозчинними у воді твердими речовинами, такими, як масло-какао, гідрогенізовані рослинні олії, поліетиленгліколь або ефіри жирних кислот і поліетиленгліколей, тоді як еліксири можна отримати із застосуванням міристилпальмітату. Препарати, прийнятні для парентерального введення, переважно містять стерильний масляний або водний препарат активних інгредієнтів, який переважно є ізотонічним з кров'ю реципієнта, наприклад, ізотонічний сольовий розчин, ізотонічний розчин глюкози або буферний розчин. Препарат можна переважно стерилізувати, наприклад, фільтруванням через фільтр, який затримує бактерії, додаванням стерилізуючого агента до препарату, опроміненням препарату або нагріванням препарату. Ліпосомні препарати, що описуються, наприклад, в Encyclopedia of Pharmaceutical Technology, vol. 9, 1994, також є придатними для парентерального введення. Альтернативно, сполуки формули (I) можна представити у вигляді стерильного твердого препарату, наприклад ліофілізованого порошку, який легко розчиняють у стерильному розчиннику безпосередньо перед застосуванням. Трансдермальні препарати можуть бути в формі пластиру або наклейки. Препарати, придатні для офтальмічного введення, можуть бути в формі стерильного водного препарату активних інгредієнтів, які можуть бути в мікрокристалічній формі, наприклад, в формі водної мікрокристалічної суспензії. Ліпосомні склади або біорозкладані полімерні системи, наприклад, які описуються в Encyclopedia of Pharmaceutical Technology, vol. 2, 1989, можна також застосовувати для надавання активного інгредієнта для офтальмічного введення. Препарати, придатні для місцевого або офтальмологічного введення, включають в себе рідкі або напіврідкі препарати, такі, як лініменти, лосьйони, гелі, аплікації, емульсії масло у воді або вода в маслі, такі, як креми, мазі або пасти; або розчини або суспензії, такі, як краплі. 5 UA 111520 C2 5 10 15 20 25 30 35 40 45 Композиції для офтальмологічного лікування можуть переважно додатково містити циклодекстрин. Для місцевого введення сполука формули (I) звичайно може бути присутньою в кількості від 0,01 до 5 % (з розрахунку на масу композиції), наприклад, від 0,01 % до 1 % (з розрахунку на масу композиції). Препарати, прийнятні для назального або трансбукального введення, включають в себе порошок, саморухомі препарати і препарати, що розпилюються, такі, як аерозолі і розпилювачі. nd Такі препарати описані більш детально, наприклад, в Modern Pharmaceutics, 2 ed., G.S. Banker th and C.T. Rhodes (Eds.), page 427-432, Marcel Dekker, New York; Modern Pharmaceutics, 3 ed., G.S. Banker and C.Т. Rhodes (eds.), page 618-619 and 718-721, Marcel Dekker, New York, and Encyclopedia of Pharmaceutical Technology, vol. 10, J. Swarbrick and J.C. Boylan (Eds.), page 191221, Marcel Dekker, New York. Крім вищезгаданих інгредієнтів, препарати сполуки формули (I) можуть включати в себе один або декілька додаткових інгредієнтів, таких, як розріджувачі, буфери, коригенти, забарвлювальну речовину, поверхово-активні речовини, загущувачі, консерванти, наприклад метилгідроксибензоат (зокрема антиоксиданти), емульгуючі агенти і тому подібне. Фармацевтична композиція може додатково містити один або кілька інших активних компонентів, які звичайно застосовуються при лікуванні шкірних захворювань або станів, наприклад, вибраних з групи, яка складається з глюкокортикоїдів, вітаміну D і аналогів вітаміну D, антигістамінів, антагоністів фактору активації тромбоцитів (PAF), антихолінергічних агентів, метилксантинів, β-адренергічних агентів, інгібіторів СОХ-2, саліцилатів, індометацину, флуфенамату, напроксену, тимегадину, солей золота, пеніциламіну, агентів, що знижують вміст холестерину сироватки крові, ретиноїдів, солей цинку, саліцилазосульфапіридину і інгібіторів кальциневрину. Способи отримання Сполуки даного винаходу можна отримати декількома шляхами, добре відомими фахівцям в галузі синтезу. Сполуки формули (I) можна, наприклад, отримати із застосуванням реакцій і методів, описаних нижче, разом з методами, відомими в галузі синтетичної органічної хімії, або їх варіантами, відомими фахівцям в даній галузі. Переважні способи включають в себе, але не обмежуються способами, описаними нижче. Реакції проводять у розчинниках, придатних для реагентів і речовин, що використовуються, і придатних для здійснюваних перетворень. Крім того, в синтетичних способах, описаних нижче, повинно бути зрозуміло, що всі запропоновані умови реакцій, зокрема вибір розчинника, атмосфери реакції, температури реакції, тривалості експерименту і процедур обробки, вибирають так, щоб вони були умовами, стандартними для даної реакції, які повинні бути легко визнані фахівцем в даній галузі органічного синтезу. Не всі сполуки, які належать до даного класу, можуть бути сумісні з деякими з умов реакції, необхідних в деяких з описаних методів. Такі обмеження для замісників, які сумісні з умовами реакції, будуть легко очевидні для фахівців в даній галузі, можна застосовувати альтернативні методи. Вихідні речовини є або відомими, або комерційно доступними сполуками, або їх можна отримати за допомогою звичайних синтетичних способів, добре відомих фахівцям в даній галузі. Спосіб РХ-МС "XE Metode 7 CM" Контроль властивостей проводили на приладі Waters LCT Premier MS і Waters Aquity UPLC. Колонка: Waters Aquity UPLC HSS T3 1,8 мкм, 2,150 мм, при 40С. Розчинники: А=10 мМ ацетату амонію+0,1 % HCOOH, В=MeCN+0,1 % НСООН. Витрата потоку: 0,7 мл/хв. Об'єм упорскування — 2 мкл. Діапазон УФ-детектування — 240400 нм. Градієнт 50 Час 0,00 хв. 0,50 хв. 1,00 хв. 2,60 хв. 3,80 хв. 3,81 хв. 4,80 хв. %А 99 94 94 5 5 99 99 %В 1 6 6 95 95 1 1 Підтвердження молекулярної маси і чистоту отримували і перевіряли OpenLynx. 1 Спектри H ядерного магнітного резонансу (ЯМР) реєстрували при 400 або 600 МГц. Величини хімічних зсувів (δ, в мільйонних частках) отримували при вимірюванні у вказаному 6 UA 111520 C2 5 10 15 20 25 розчиннику відносно внутрішніх стандартів тетраметилсилану (δ=0,00) або хлороформу (δ=7,25). Величина мультиплету, або визначеного (дублет (д), триплет (т), квартет (к)), або невизначеного (м), вказана приблизно для середньої точки, якщо не вказаний діапазон. (bs) означає уширений синглет. Органічні розчинники, що застосовуються, звичайно були безводними. Хроматографію проводили на силікагелі Мерк 60 (0,040-0,063 мм). Вказані відношення розчинників належать до відношень об.:об., якщо не вказане інше. Протягом всього контексту застосовували наступні абревіатури: DBU 1,8-діазабіцикло[5.4.0]ундец-7-ен DCE 1,2-дихлоретан DCM дихлорметан DIAD діізопропілазодикарбоксилат DMAP N, N-диметилпіридин-4-амін ДМФА N, N-диметилформамід ДМСО диметилсульфоксид EDCI (3-диметиламінопропіл)етилкарбодіімід EtOH етанол MeOH метанол EtOAc етилацетат л літр Ме метил ЯМР ядерний магнітний резонанс КТ кімнатна температура ТГФ тетрагідрофуран Пет. петролейний ефір Загальні методи Сполуки винаходу можна отримати, наприклад, згідно з наступними необмежуючими загальними методами і прикладами. R має значення, вказані раніше для сполук формули (I). 7 UA 111520 C2 Отримання 1 трет-Бутилгідроксикарбамат 5 10 До переміщуваної суспензії солі гідроксиламін HCl (150 г, 2,17 моль) і К2CО3 (150 г, 1,09 моль) в діетиловому ефірі (940 мл) і воді (30 мл) при 0С повільно додавали розчин ди-третбутилдикарбонат (308 г, 1,41 ммоль) в діетиловому ефірі (600 мл) протягом 15 хв. Після додавання реакційну суміш перемішували при КТ протягом 2 годин. Реакційну суміш фільтрували і фільтрат сушили над безводним Na2S04 і концентрували. Отриманий сирий продукт промивали циклогексаном (50 мл3) і сушили, отримуючи при цьому вказану в 1 заголовку сполуку (150 г, 52 %, біла тверда речовина). H ЯМР (400 МГц, CDCl3): δ=7,18 (ушир., 2H), 1,47 (с, 9Н) м. ч. Отримання 2 трет-Бутил-4-нітробензоїлоксикарбамат 8 UA 111520 C2 5 10 15 20 25 30 35 40 До перемішуваного розчину трет-бутилгідроксикарбамату (150 г, 1,128 моль) в дихлорметані (2 л) при 0С додавали триетиламін (174 мл, 1,24 моль) з подальшим додаванням 4нітробензоїлхлориду (205 г, 1,105 моль) в рівних відношеннях. Після того, як додавання було завершене, реакційну суміш перемішували при КТ протягом 1 години. Реакційну суміш гасили водою (500 мл) і екстрагували. Відділений шар дихлорметану промивали насиченим розчином солі (200 мл), сушили над безводним Na2S04 і концентрували. Отриманий неочищений продукт промивали гексаном (100 мл2) і сушили, отримуючи при цьому вказану в заголовку сполуку 1 (300 г, 94 %, жовта тверда речовина). H ЯМР (400 МГц, CDCl3): δ=8,34-8,27 (м, 4H), 2,97-2,92 (м, 1Н), 1,53 (с, 9Н) м. ч. Отримання 3 О-(4-Нітробензоїл)гідроксиламін До перемішуваного розчину трет-бутил-4-нітробензоїлоксикарбамату (300 г, 1,06 моль) в дихлорметані (2 л) при 0С повільно додавали метансульфонову кислоту (69 мл, 1,06 моль). Після того, як додавання було закінчене, реакційну суміш залишали для перемішування при КТ протягом 16 годин. Реакційну суміш розбавляли дихлорметаном (1 л), промивали 10 % водним розчином NaHCO3 (300 мл), водою (200 мл), насиченим розчином солі (200 мл), сушили над безводним Na2SО4 і концентрували. Отриманий неочищений продукт промивали гексаном (100 мл2) і сушили, отримуючи при цьому вказану в заголовку сполуку (150 г, 77 %, блідо-жовта 1 тверда речовина). H ЯМР (400 МГц, CDCl3): δ=8,33-8,30 (м, 2H), 8,22-8,19 (м, 2H), 6,73 (ушир. с, 2H) м. ч. Отримання 4 Етиловий ефір 1-гідроксиметилциклопропанкарбонової кислоти До перемішуваного розчину діетилциклопропан-1,1-дикарбоксилату (2,13 г, 11,4 ммоль) в ТГФ (80 мл) при КТ повільно додавали літійалюмінійтри-трет-бутоксигідрид (38,76 мл, 38,76 ммоль, 1,0М розчин у ТГФ). Після того, як додавання було завершене, реакційну суміш перемішували при КТ протягом 18 годин. Реакційну суміш розбавляли етилацетатом (100 мл), промивали 1 н водним HCl (20 мл), водою (20 мл), 5 % водним NaHCО3 (25 мл), насиченим розчином солі (20 мл), сушили над безводним Na2SО4, фільтрували і концентрували, отримуючи 1 при цьому вказану в заголовку сполуку (1,3 г, 79 %, жовте масло). H ЯМР (400 МГц, CDCl3): δ=4,20-4,13 (м, 2Н), 3,62 (м, 2Н), 2,61 (м, 1H), 1,29-1,24 (м, 5H), 0,88-0,85 (м, 2Н) м. ч. Отримання 5 Етиловий ефір 1-формілциклопропанкарбонової кислоти До переміщуваного розчину етилового ефіру 1-гідроксиметилциклопропанкарбонової кислоти (1,1 г, 7,63 ммоль) в дихлорметані (45 мл) додавали NaHCО3 (2,5 г, 29,76 ммоль) і періодинан Дес-Мартіна (6,46 г, 15,23 ммоль). Потім суспензію перемішували при КТ протягом 30 хв. Реакційну суміш гасили сумішшю 1:1 10 % водного розчину Na2S2О3 і 10 % водного розчину NaHCО3 (20 мл), підтримуючи температуру нижче 20С, перемішували протягом 30 хв. Потім реакційну суміш розбавляли дихлорметаном (100 мл) і екстрагували. Органічний шар 9 UA 111520 C2 5 10 15 20 25 30 35 40 промивали насиченим розчином солі (30 мл), сушили над безводним Na2SО4, фільтрували і концентрували. Неочищений продукт очищували колонковою хроматографією на силікагелі (010 % EtOAc в петролейному ефірі в ролі елюенту), отримуючи при цьому вказану в заголовку 1 сполуку (800 мг, 76 %, жовте масло). H ЯМР (400 МГц, CDCl3): δ=10,40 (с, 1H), 4,25 (м, 2H), 1,68-1,65 (м, 2H), 1,62-1,59 (м, 2H), 1,33-1,26 (м, 3H) м. ч. Отримання 6 3-Метоксипіридин-2-іламін Суспензію 3-метокси-2-нітропіридину (30 г, 194,8 ммоль) і 10 % Pd/C (10 г) в етанолі (1 л) гідрували в звичайному гідрогенізаторі (Н2, тиск 275790 Па (40 фунтів на квадратний дюйм)) при КТ протягом 4 годин. Реакційну суміш фільтрували через целіт і фільтрат концентрували, 1 отримуючи при цьому вказану в заголовку сполуку (22 г, 91 % твердої коричневої речовини). H ЯМР (400 МГц, CDCl3): δ=7,66 (д, J=5,2 Гц; 1Н), 6,91 (д, J=7,6 Гц, 1Н), 6,63-6,60 (м, 1Н), 4,65 (ушир., 2H), 3,84 (с, 3H) м. ч. Отримання 7 1,2-Діаміно-3-метоксипіридинієва сіль 4-нітробензойної кислоти До перемішуваного розчину 3-метоксипіридин-2-іламіну (30 г, 164,8 ммоль) в дихлорметані (400 мл) при 10С додавали О-(4-нітробензоїл)гідроксиламін (13,2 г, 214,2 ммоль). Після додавання реакційну суміш перемішували при КТ протягом 16 годин. Отриманий осад відфільтровувати, промивали дихлорметаном (25 мл2) і сушили, отримуючи при цьому вказану в заголовку сполуку (40 г, 91 %, коричнева тверда речовина). (Попередження: сіль є термічно нестабільною). 1 H ЯМР (400 МГц, ДМСО): δ=8,53 (шир. 2Н), 8,12 (д, J=8 Гц, 2Н), 8,01 (д, J=8,8 Гц, 2Н), 7,73 (д, J=6,4 Гц, 1Н), 7,33 (д, J=7,2 Гц, 1Н), 7,17 (шир., 2Н), 6,77 (т, J=6,8 Гц, 1Н), 3,93 (с, 3H) м. ч. Отримання 8 Етиловий ефір 1-(8-метокси-[1,2,4]триазоло[1,5-а]піридин-2-іл)циклопропанкарбонової кислоти До переміщуваного розчину діаміно-3-метоксипіридинієвої солі 4-нітробензойної кислоти (1 г, 7,14 ммоль) в етанолі (10 мл) при 0С додавали DBU (2,1 мл) із подальшим додаванням етилового ефіру 1-формілциклопропанкарбонової кислоти (1,5 г, 10,71 ммоль). Після додавання реакційну суміш перемішували при КТ протягом 2 годин. Реакційну суміш концентрували, отриманий залишок розбавляли EtOAc (100 мл), промивали водою (20 мл2), насиченим розчином солі (20 мл), сушили над безводним Na2SО4, фільтрували і концентрували. Неочищений продукт очищували колонковою хроматографією на силікагелі (із застосуванням 015 % розчину EtOAc в CH2Cl2 як елюенту),отримуючи при цьому вказану в заголовку сполуку (800 мг, 53 %, біла тверда речовина). 1 H ЯМР (400 МГц, CDCl3): δ=8,18-8,16 (д, J=6,4 Гц; 1Н), 6,89 (д, J=7,2 Гц, 1Н), 6,76 (д, J=8 Гц, 1Н), 4,20-4,14 (м, 2Н), 4,03 (с, 3H), 1,73-1,70 (м, 2H), 1,59-1,56 (м, 2H), 1,20 (т, J=6,8 Гц; 3Н) м. ч. Отримання 9 Етиловий ефір 1-(5-бром-8-метокси-[1,2,4]триазоло[1,5-а]піридин-2іл)циклопропанкарбонової кислоти 10 UA 111520 C2 5 10 15 20 25 30 35 До переміщуваного розчину етилового ефіру 1-(8-метокси-[1,2,4]триазоло[1,5-а]піридин-2іл)циклопропанкарбонової кислоти (25 г, 95,7 ммоль) в ацетонітрилі (300 мл) при КТ порціями додавали N-бромсукцинімід (34 г, 191,5 ммоль). Після додавання реакційну суміш перемішували при КТ протягом 6 годин. Реакційну суміш розбавляли EtOAc (600 мл), промивали водою (100 мл2), насиченим розчином солі (50 мл), сушили над безводним Na2SО4, фільтрували і концентрували. Неочищений продукт очищували колонковою хроматографією на силікагелі (0-10 % EtOAc в дихлорметані як елюенті), отримуючи при цьому вказану в заголовку сполуку (25 г, 77 %, безбарвна тверда речовина). 1 H ЯМР (400 МГц, ДМСО): δ=7,44 (д, J=8,4 Гц; 1Н), 7,07 (д, J=8 Гц; 1Н), 4,43-4,08 (м, 2Н), 3,97 (с, 3H),, 60-1,57 3,93 (м, 2H), 1,48-1,45 (м, 2H), 1,13 (т, J=7,4 Гц; 3Н) м. ч. Отримання 10 1-(5-Бром-8-метокси-[1,2,4]триазоло[1,5-а]піридин-2-іл)циклопропанкарбонова кислота До розчину етил-1-(5-бром-8-метокси-[1,2,4]триазоло[1,5-а]піридин-2іл)циклопропанкарбоксилату (3,00 г, 8,82 ммоль) в ТГФ (25 мл) додавали водний 1 М розчин LiOH (25 мл). Суміш перемішували при 80С протягом 30 хвилин, охолоджували до кімнатної температури і розбавляли EtOAc (50 мл) і водою (50 мл). Органічну фазу екстрагували водним 0,1М розчином NaOH (25 мл) і об'єднані водні фази підкислювали конц. HCI до рН 0-1 і екстрагували чотири рази DCМ (30 мл). Випарювання досуха об'єднаних органічних фаз давало 1 вказану в заголовку сполуку (2,54 г, 94 %). H ЯМР (ДМСО, 400 МГц): δ=12,61 (с, 1H), 7,42 (д, 1H, J=8,3 Гц), 7,06 (д, 1H, J=8,3 Гц), 3,97 (с, 3H), 1,53 (д, 2Н, J=3,9 Гц), 1,40 (д, 2Н, J=3,9 Гц) м. ч. Отримання 11 1-[8-Метокси-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5-а]піридин-2іл]циклопропанкарбонова кислота 1-(5-Бром-8-метокси-[1,2,4]триазоло[1,5-а]піридин-2-іл)циклопропанкарбонову кислоту (1,00 г, 3,20 ммоль) і 5-(4,4,5,5-тетраметил-1,3,2-діоксаборолан-2-іл)-3Н-ізобензофуран-1-1-он (отримання боронату описане в WO2011/134468) (1,67 г, 6,40 ммоль) розчиняли в дегазованому діоксані (16 мл). Pd2(dba)3 (29 мг, 32 мкмоль)), PCy3 (18 мг, 64 мкмоль) і K3PО4 (2,38 г, 11,2 ммоль) змішували в дегазованій воді (10 мл). Два розчини змішували і потім нагрівали в мікрохвильовій печі до 110 °C протягом 10 хвилин, охолоджували до кімнатної температури і розбавляли EtOAc (40 мл). Органічну фазу екстрагували водою (25 мл) і водним 0,1М розчином NaOH (25 мл) і об'єднані водні фази підкислювали конц. HCl до рН 0-1 і екстрагували DCM (30 мл4). Після упарювання об'єднаних органічних фаз вказана в заголовку сполука кристалізувалася (746 мг, 64 %). Час утримування при ВЕРХ (XE Metode 7 СМ): 1,97 хвилин. Визначена "М+1"-маса: 366,11. Обчислена "М+1"-маса: 366,11. 11 UA 111520 C2 1 5 10 15 20 25 30 35 H ЯМР (ДМСО, 300 МГц): δ=8,25-8,20 (м, 1H), 8,13 (дд, 1H, J=8,2 Гц, 1,4 Гц), 8,00 (д, 1H, J=8,0 Гц), 7,40 (д, 1H, J=8,2 Гц), 7,24 (д, 1H, J=8,3 Гц), 5,51 (с, 2H), 4,04 (с, 3H), 1,58-1,48 (м, 2H), 1,48-1,39 (м, 2H) м. ч. Приклад 1 [(1S)-1-Метилпропіл]-1-[8-метокси-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5а]піридин-2-іл]циклопропанкарбоксилат (сполука 1) Суміш 1-[8-метокси-5-(1-оксо-3Н-ізобензофуран-5-іл)[1,2,4]триазоло[1,5-а]піридин-2іл]циклопропанкарбонової кислоти (10 мг, 27 мкмоль), (1S)-1- метилпропанолу (10 мкл, 108 мкмоль), DMAP (6,7 мг, 54 мкмоль) і EDCHCl (10,5 мг, 54 мкмоль) в DCМ (0,5 мл) перемішували в герметизованій ампулі при КТ протягом ночі, потім додавали ще (1S)-1-метилпропанол (10 мкл, 108 мкмоль) і DMAP (6,7 мг, 54 мкмоль) і суміш нагрівали до 50С протягом 3 годин. Випарювання досуха і очищення кислотною препаративною ВЕРХ давали вказану в заголовку сполуку. Час утримування за ВЕРХ (XE Metode 7 СМ): 2,35 хвилини. Визначена "М+1"-маса: 422,16. Обчислена "M+1"-маса: 422,17. 1 H ЯМР (ДМСО, 600 МГц): δ=8,25-8,21 (м, 1H), 8,14 (дд, 1H, J=8,0 Гц, 1,2 Гц), 8,01-7,97 (м, 1H), 7,41 (д, 1H, J=8,2 Гц), 7,23 (д, 1H, J=8,2 Гц), 5,51 (с, 2H), 4,78 (h, 1H, J=6,3 Гц), 4,04 (с, 3H), 1,61-1,40 (м, 6H), 1,13 (д, 3H, J=6,2 Гц), 0,78 (т, 3H, J=7,4 Гц) м. ч. Приклад 2 [(1R)-1-Метилпропіл]-1-[8-метокси-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5а]піридин-2-іл]циклопропанкарбоксилат (сполука 2) Суміш 1-[8-метокси-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5-а]піридин-2іл]циклопропанкарбонової кислоти (400 мг, 1,095 ммоль), (2R)-бутан-2-олу (122 мг, 1,64 ммоль), DMAP (147 мг, 1,20 ммоль) і EDClHCl (231 мг, 1,20 ммоль) в DCM (10 мл) перемішували при КТ протягом ночі перед тим, як її випарювали досуха. Колонкова хроматографія (градієнт 0-5 % МеОН в DCМ) з подальшою перекристалізацією в EtOH і ліофілізацією давали вказану в заголовку сполуку у вигляді безбарвного порошку (138 мг, 30 %). Час утримування при ВЕРХ (XE Metode 7 СМ): 2,33 хвилини. Визначена "М+1"-маса: 422,15, обчислена "M+1"-маса: 422,17. 1 H ЯМР (ДМСО, 600 МГц): δ=8,23 (шир. с, 1H), 8,14 (дд, 1H, J=8,0 Гц, 1,6 Гц), 7,99 (д, 1H, J=8,0 Гц), 7,41 (д, 1H, J=8,2 Гц), 7,24 (д, 1H, J=8,2 Гц), 5,51 (с, 2H), 4,78 (h, 1H, J=6,3 Гц), 4,04 (с, 3H), 1,62-1,38 (м, 6H), 1,13 (д, 3H, J=6,3 Гц), 0,78 (т, 3H, J=7,4 Гц) м. ч. Приклад 3 [2-Метилпропіл]-1-[8-метокси-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[,5-а]піридин2-іл]циклопропанкарбоксилат (сполука 3) 12 UA 111520 C2 5 10 15 20 25 30 35 Суміш 1-[8-метокси-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5-а]піридин-2іл]циклопропанкарбонової кислоти (250 мг, 685 мкмоль), ізобутанолу (100 мкл, 1,37 ммоль), DMAP (250 мг, 2,05 ммоль) і EDCHCl (262 мг, 1,37 ммоль) в DCМ (14 мл) перемішували при 50С протягом 2 годин в герметизованій ампулі, потім її розбавляли DCM (80 мл), промивали водним 1 М розчином HCl (40 мл) і упарювали досуха. Неочищену суміш знову розчиняли в MeCN (~2 мл) і неочищений продукт кристалізували при додаванні води (~2 мл). Колонкова хроматографія (градієнт 20-100 % розчин EtOAc в петр. ефірі) і подальша перекристалізація в MeCN і воді давали вказану в заголовку сполуку у вигляді безбарвних кристалів (178 мг, 62 %). Час утримування при ВЕРХ (XE Metode 7 СМ): 2,34 хвилини. Визначена "М+1"-маса: 422,16. Обчислена "M+1"-маса: 422,17. 1 H ЯМР (ДМСО, 600 МГц): δ=8,22 (шир. с, 1H), 8,13 (дд, 1H, J=8,1 Гц, 1,5 Гц), 8,00 (д, 1H, J=8,0 Гц), 7,40 (д, 1H, J=8,2 Гц), 7,24 (д, 1H, J=8,2 Гц), 5,51 (с, 2H), 4,04 (с, 3H), 3,84 (д, 2H, J=6,5 Гц), 1,79 (м, 1H), 1,61-1,54 (м, 2H), 1,54-1,46 (м, 2H), 0,78 (д, 6H, J=6,7 Гц) м. ч. Приклад 4 трет-Бутил-1-[8-метокси-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[,5-а]піридин-2іл]циклопропанкарбоксилат (сполука 4) Суспензію 1-[8-метокси-5-(1-оксо-3Н-ізобензофуран-5-іл)-[1,2,4]триазоло[1,5-а]піридин-2іл]циклопропанкарбонової кислоти (30 мг, 83 мкмоль) і хлориду бензилтриетиламонію (19 мг, 83 мкмоль) в ДМФА (1,0 мл) обережно нагрівали доти, поки вона не ставала розчином. Додавали трет-бутилбромід (297 мкл, 2,64 ммоль) і К2CО3 (171 мг, 1,24 ммоль) і суміш перемішували при 55С протягом трьох днів. Додавали додатковий трет-бутилбромід (139 мкл, 1,24 ммоль) і К2CО3 (171 мг, 1,24 ммоль) і суміш перемішували при 55С протягом ще одного дня. Очищення препаративною ВЕРХ давало вказану в заголовку сполуку. Час утримування за ВЕРХ (XE Metode 7 СМ): 2,29 хвилини. Визначена "М+1"-маса: 422,18. Обчислена "M+1"-маса: 422,17. 1 H ЯМР (ДМСО, 300 МГц): δ=8,27-8,22 (м, 1H), 8,16 (дд, 1H, J=8,0 Гц, 1,5 Гц), 8,00 (д, 1H, J=8,2 Гц), 7,40 (д, 1H, J=8,2 Гц), 7,22 (д, 1H, J=8,3 Гц), 5,51 (м, 2H), 4,04 (с, 3H), 1,54-1,40 (м, 4H), 1,38 (с, 9H) м. ч. Аналізи Аналіз PDE4 Рекомбінантну PDE4 людини (GenBank acession no NM_006203) інкубували протягом 1 години із тестованою сполукою при концентрації аж до 10 мкМ, з цАМФ (110-5М) і з невеликою кількістю (0,021 МВк) радіоактивно міченого цАМФ. В кінці інкубації розщеплення субстрату оцінювали зв'язуванням продукту AMФ з SPA-гранулами, які генерують хемілюмінесценцію при зв'язуванні з радіоактивною міткою. Продукт АМФ інгібував зв'язування радіоактивної мітки з гранулами, і виявляли конкуренцію з люмінесцентним сигналом. 13 UA 111520 C2 5 10 15 20 25 30 35 40 45 50 55 Результати обчислювали як молярні концентрації, що приводять до 50 % інгібування розщеплення субстрату, порівняно з контрольними зразками, і виражали в діапазоні величин IC50 (нМ). Сполуки даного винаходу випробовували в аналізі PDE4, IC50 (нМ): сполука 1, 10,6 нМ; сполука 2, 13,0 нМ; сполука 3, 12,3 нМ; сполука 4, 20,7 нМ (на основі середньої величини 2-5 випробувань для кожної сполуки). Вивільнення TNF-α Мононуклеарні клітини периферичної крові (РВМС) людини виділяли з лейкоцитарних плівок. Кров змішували з фізіологічним розчином у відношенні 1:1, і РВМС виділяли з використанням пробірок Lymphoprep TM (Nycomed, Норвегія). РВМС суспендували в RPMI1640 з 0,5 % сироватковим альбуміном людини, пеніцилін/стрептоміцин (pen/strep) і 2 мМ Lглутаміном при концентрації 5105 с/мл. Клітини заздалегідь інкубували протягом 30 хвилин із випробовуваними сполуками в 96-ямкових планшетах для культури тканин і стимулювали протягом 18 годин із ліпополісахаридом в кількості 1 мг/мл (Sigma). Концентрацію TNF-α в супернатантах вимірювали з використанням гомогенного флуоресцентного резонансу з тимчасовим розрізненням (TR-FRET). Кількісні дані аналізу отримували вимірюванням флуоресценції при 665 нм (пропорціонально до концентрації TNF-α) і 620 нм (контроль). Результати представлені у вигляді величин IC50 (нМ), обчислених із кривих інгібування із застосуванням як позитивних контролів секреції в стимульованих LPS ямках і як негативних контролів секреції в нестимульованих клітинах. Сполуки даного винаходу випробовували в аналізі вивільнення TNF-α, IC50(нМ): сполука 1 12,8 нМ; сполука 2, 15,7 нМ; сполука 3, 14,6 нМ; сполука 4, 15,3 нМ (на основі середньої величини 2-5 випробувань для кожної сполуки). Аналіз HLM (мікросом печінки людини) Інкубації випробовуваних сполук в ДМСО, розбавленому фосфатним буфером, рН 7,4, при концентрації 0,5 мкМ проводили з мікросомами печінки людини (0,5 мг/мл). Процент органічного розчинника при інкубації становив 1 %. Суспензію мікросом печінки людини в фосфатному буфері змішували з NADPH (1 мМ) і заздалегідь нагрівали до 37С перед додаванням випробовуваної сполуки. Аліквоти брали через 0, 5, 10, 20 і 30 хвилин і реакції термінували додаванням метанолу, що містить аналітичний внутрішній стандарт (IS). Результати виражали у вигляді апарентного кліренсу (CIарр) (мл/хв./кг) і індексу екстракції -1 печінки (Еh) (%), обчисленого з константи швидкості (к) (хв. ) виснаження сполуки, що тестується. Сполуки даного винаходу випробовували в аналізі HLM, Еh (%): сполука 1, >91 %; сполука 2, >91 %; сполука 3, >91 %; сполука 4, >91 % (на основі середнього значення 2-3 випробувань для кожної сполуки). Аналіз цільної крові (WB) людини Інкубацію випробовуваних сполук в ДМСО, розбавленому фосфатним буфером, рН 7,4, при концентрації 1 мкМ проводили з цільною кров'ю людини. Процент органічного розчинника при інкубаціях був 1 %. Інкубацію проводили при 37С з аліквотами, відібраними через 0, 15, 30, 60 і 120 хвилин, і реакції зупиняли додаванням метанолу, що містить аналітичний внутрішній стандарт (IS).) 1 Результати виражали як період напіврозпаду (T /2) у хвилинах, обчислений з константи -1 швидкості (к) (хв. ) виснаження випробовуваної сполуки. 1 Зразки прикладів сполук даного винаходу випробовували в аналізі WB, T /2 (хвилини): сполука 1, 10,7 хвилини; сполука 2, 12,6 хвилини; сполука 3, 16,6 хвилини; сполука 4, 720 хвилин; сполука 2, >720 хвилин; сполука 3, >720 хвилин; сполука 4, >720 хвилин (на основі середньої величини 2-4 випробувань для кожної сполуки). 60 14 UA 111520 C2 ФОРМУЛА ВИНАХОДУ 1. Сполука загальної формули (І): O N N N O O R O O 5 10 15 20 25 30 35 40 45 (І) або будь-який з її стереоізомерів, або будь-яка суміш її стереоізомерів, або її фармацевтично прийнятна сіль, у яких R являє собою розгалужений бутил. 2. Сполука за п. 1 або будь-який з її стереоізомерів, або будь-яка суміш її стереоізомерів, або її фармацевтично прийнятні солі, де R являє собою 1-метилпропіл, 2-метилпропіл або трет-бутил. 3. Сполука за п. 1, яка являє собою [(1S)-1-метилпропіл]-1-[8-метоксі-5-(1-оксо-3Нізобензофуран-5-іл)-[1,2,4]триазоло[1,5-а]піридин-2-іл]циклопропанкарбоксилат, вільну основу. 4. Сполука за п. 1, яка являє собою [(1R)-1-метилпропіл]-1-[8-метоксі-5-(1-оксо-3Нізобензофуран-5-іл)-[1,2,4]триазоло[1,5-а]піридин-2-іл]циклопропанкарбоксилат, вільну основу. 5. Сполука за п. 1, яка являє собою [2-метилпропіл]-1-[8-метоксі-5-(1-оксо-3Н-ізобензофуран-5іл)-[1,2,4]триазоло[1,5-а]піридин-2-іл]циклопропанкарбоксилат, вільну основу. 6. Сполука за п. 1, яка являє собою трет-бутил-1-[8-метоксі-5-(1-оксо-3Н-ізобензофуран-5-іл)[1,2,4]триазоло[1,5-а]піридин-2-іл]циклопропанкарбоксилат, вільну основу. 7. Фармацевтична композиція, яка містить сполуку за будь-яким із пп. 1-6 разом із фармацевтично прийнятним наповнювачем або ексципієнтом, або фармацевтично прийнятним носієм(ями). 8. Фармацевтична композиція за п. 7, яка додатково містить одну або кілька інших фармацевтично прийнятних сполук. 9. Застосування сполуки за будь-яким із пп. 1-6 для отримання фармацевтичної композиції. 10. Застосування сполуки за п. 9 для отримання фармацевтичної композиції для лікування або зменшення інтенсивності симптомів захворювання, порушення або стану, сприйнятливого до інгібуючої активності PDE4. 11. Застосування за п. 10, де захворювання, порушення або стан є шкірним захворюванням або станом. 12. Застосування за п. 11, де захворювання, порушення або стан є проліферативним і запальним порушенням шкіри, дерматитом, атопічним дерматитом, себорейним дерматитом, контактним дерматитом, псоріазом, раком, епідермальним запаленням, алопецією, шкірною атрофією, індукованою стероїдами шкірною атрофією, старінням шкіри, фотостарінням шкіри, вугровим висипом, кропивницею, свербежем і екземою. 13. Сполука за будь-яким із пп. 1-6 для застосування як лікарського засобу. 14. Сполука за п. 13 для застосування при лікуванні або зменшенні інтенсивності симптомів захворювання, порушення або стану, сприйнятливого до інгібуючої PDE4 активності. 15. Сполука за п. 13 для застосування при лікуванні або зменшенні інтенсивності симптомів шкірних захворювань або станів. 16. Сполука за п. 13 для застосування при лікуванні проліферативних і запальних захворювань шкіри, дерматиту, атопічного дерматиту, себорейного дерматиту, контактного дерматиту, псоріазу, раку, епідермального запалення, алопеції, шкірної атрофії, індукованої стероїдами шкірної атрофії, старіння шкіри, фотостаріння шкіри, вугрового висипу, кропивниці, свербежу і екземи. 17. Спосіб лікування або зменшенні інтенсивності симптомів захворювання, порушення або стану, сприйнятливого до інгібуючої PDE4 активності, який включає стадію введення в організм живого організму терапевтично ефективної кількості сполуки за будь-яким із пп. 1-6. 15 UA 111520 C2 5 10 18. Спосіб лікування або зменшення інтенсивності симптомів шкірних захворювань або станів, який включає введення пацієнту, який страждає щонайменше на одне із вказаних захворювань, ефективної кількості однієї або декількох сполук за будь-яким із пп. 1-6, необов'язково разом із фармацевтично прийнятним носієм або одним або декількома ексципієнтами, необов'язково в комбінації з іншими терапевтично активними сполуками. 19. Спосіб за п. 18, в якому шкірне захворювання або стан вибрані з групи, яка складається з проліферативних і запальних захворювань шкіри, дерматиту, атопічного дерматиту, себорейного дерматиту, контактного дерматиту, псоріазу, раку, епідермального запалення, алопеції, шкірної атрофії, індукованої стероїдами шкірної атрофії, старіння шкіри, фотостаріння шкіри, вугрового висипу, кропивниці, свербежу і екземи. Комп’ютерна верстка А. Крижанівський Державна служба інтелектуальної власності України, вул. Василя Липківського, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 16
ДивитисяДодаткова інформація
Назва патенту англійською[1,2,4]triazolopyridine compounds and used thereof as phosphodiesterase inhibitors
Автори англійськоюNielsen, Simon Feldbaek, Larsen, Jens Christian Hojland
Назва патенту російською[1,2,4]триазолопиридины и их применение как ингибиторов фосфодиестеразы
Автори російськоюНильсэн Симон Фельдбэк, Ларсэн Йенс Христиан Хойланн
МПК / Мітки
МПК: A61P 17/14, A61P 17/06, A61P 17/04, A61P 17/02, A61P 35/00, A61K 31/437, A61P 17/08, A61P 17/10, C07D 471/04, A61P 17/00, A61P 17/16
Мітки: інгібіторів, застосування, фосфодіестерази, 1,2,4]триазолопіридини
Код посилання
<a href="https://ua.patents.su/18-111520-124triazolopiridini-i-kh-zastosuvannya-yak-ingibitoriv-fosfodiesterazi.html" target="_blank" rel="follow" title="База патентів України">[1,2,4]триазолопіридини і їх застосування як інгібіторів фосфодіестерази</a>
7-азаіндоли, їх застосування як інгібіторів фосфодіестерази 4 та спосіб їх одержання

Номер патенту: 76432
Опубліковано: 15.08.2006
Автори: Кронбах Томас, Маркс Дегенхард, Зелений Штефан, Гьофген Норберт, Егерланд Уте, Полімеропулос Еммануель, Кусс Хільдегард
МПК: A61K 31/40, C07D 471/04, A61P 11/00
Мітки: спосіб, інгібіторів, одержання, фосфодіестерази, 7-азаіндоли, застосування
Формула / Реферат:
1. 7-азаіндоли формули 1, 1деn може дорівнювати 1 або 2, іR1 може означати-(С1-С10)-алкіл лінійної або розгалуженої будови, незаміщений або заміщений одним або кількома замісниками, вибраними із групи, до якої входять -OH, -SH, -NH2, -NH(С1-С6)-алкіл, -N(С1-С6-алкіл)2, -NH(С6-С14)-арил, -N(С6-С14-арил)2,...
Гідроксііндоли, їх застосування як інгібіторів фосфодіестерази-4 та спосіб їх одержання

Номер патенту: 80567
Опубліковано: 10.10.2007
Автори: Егерланд Уте, Рундфельдт Кріс, Кусс Хільдегард, Гартенгауер Хельга, Гьофген Норберт, Гаспаріч Антьє
МПК: C07D 209/22, A61P 11/00, C07D 209/42
Мітки: застосування, фосфодіестерази-4, спосіб, одержання, інгібіторів, гідроксііндоли
Формула / Реферат:
1. Сполуки загальної формули 1,деn - 1 або 2, іR1 є(і) -С1-С10-алкіл нормальної або розгалуженої будови, факультативно заміщений одним або кількома з таких замісників: моно-, бі- або трициклічні насичені або моно- або поліненасичені 3-14-членні карбоцикли, причому ці карбоциклічні замісники, у свою чергу, є заміщеними одним або кількома...
Спіротрициклічні похідні та їх використання як інгібіторів фосфодіестерази-7

Номер патенту: 74243
Опубліковано: 15.11.2005
Автори: Лортіуа Едвіг, Бернарделлі Патрік, Вернь Фабріс, Дюкро П'єр
МПК: A61P 37/08, A61K 31/5377, A61P 35/00, C07D 498/10, A61P 37/04, A61P 29/00, C07D 487/10, C07D 239/84, A61K 31/537, C07D 413/12, A61P 11/00, C07D 401/04, A61P 11/06, C07D 403/12, A61P 1/00, C07D 403/04, A61P 37/00, C07D 401/12, A61K 31/527, A61P 19/02, A61P 19/10, C07D 239/70, C07D 405/12, C07D 239/78, C07D 239/80, A61P 25/00
Мітки: похідні, використання, інгібіторів, спіротрициклічні, фосфодіестерази-7
Формула / Реферат:
1. Сполуки, що мають наступні формули (І), (II) або (III), (I), (II), (III)в якиха) Х1, Х2, Х3 та Х4 є однаковими або різними і вибрані з:- N, за умови, що не більше, ніж дві групи Х1, Х2, Х3 та Х4 одночасно означають атом азоту, або- C-R1, де R1 вибраний з:- Q1, або- нижчого алкілу, нижчого алкенілу або нижчого алкінілу, причому ці групи є незаміщеними або заміщеними однією або...
Спосіб одержання інгібіторів фосфодіестерази-4

Номер патенту: 79518
Опубліковано: 25.06.2007
Автори: Сохейлі Араш, Албанез-Уолкер Дженніфер, Меррі Джеррі Ентоні, Спрінгфілд Шон А.
МПК: C07D 471/04
Мітки: інгібіторів, спосіб, одержання, фосфодіестерази-4
Формула / Реферат:
1. Спосіб одержання сполуки формули IX або IХа: ,який відрізняється тим, що включає стадію С: взаємодію в розчиннику А сполуки формули Va, (Va)де R1 вибраний з С1-С8-алкілу і розчинник А вибраний...
Застосування інгібіторів сукцинатдегідрогенази та інгібіторів iii комплексу дихального ланцюга для покращення співвідношення шкідливих і корисних мікроорганізмів

Номер патенту: 110487
Опубліковано: 12.01.2016
Автори: Штайгер Домінік, Лабурдетт Жільбер, Рік Хайко, Фот Лоріанн, Янг Герберт, Массон Джордж
МПК: A01P 3/00, A01N 61/00, A01N 43/40, A01N 37/50
Мітки: інгібіторів, корисних, застосування, комплексу, мікроорганізмів, ланцюга, покращення, дихального, шкідливих, сукцинатдегідрогенази, співвідношення
Формула / Реферат:
1. Застосування інгібіторів сукцинатдегідрогенази та інгібіторів III комплексу для боротьби з небажаними патогенними грибами без зменшення кількості корисних мікроорганізмів на культурних рослинах, де інгібітором сукцинатдегідрогенази є флуопірам і інгібітором III комплексу є трифлоксистробін, і де небажані патогенні гриби вибирають з групи, що складається з Botrytis spp., Rhizopus spp., Penicillium spp.,...
Попередній патент: Спосіб і система для генерування синтез-газу
Наступний патент: Електротермічний рухово-накопичувальний модуль
Випадковий патент: Різальний блок для робочої машини і робоча машина, яка містить такий різальний блок