Макроциклічні хіноксалінові сполуки як інгібітори протеази вгс ns3
Номер патенту: 100436
Опубліковано: 25.12.2012
Автори: Лівертон Найджел Дж., Сумма Вінченцо, Харпер Стівен, Макколі Джон А.








Формула / Реферат
1. Сполука формули (І) або її фармацевтично прийнятна сіль:
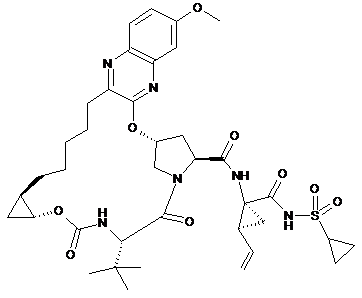 . (І)
. (І)
2. Фармацевтична композиція, яка містить ефективну кількість сполуки за п. 1 і фармацевтично прийнятний носій.
3. Фармацевтична композиція за п. 2, яка додатково містить другий терапевтичний агент, вибраний із групи, яка складається з інгібітора протеази ВГС і інгібіторів полімерази ВГС NS5B.
4. Застосування сполуки за п. 1 у медицині.
5. Застосування сполуки за п. 1 для профілактики або лікування інфекції ВГС.
6. Застосування сполуки за п. 1 для одержання лікарського засобу для інгібування активності протеази ВГС NS3 у суб'єкта, який цього потребує.
7. Застосування композиції за будь-яким з пп. 2-3 для одержання лікарського засобу для інгібування активності протеази ВГС NS3 у суб'єкта, який цього потребує.
8. Застосування сполуки за п. 1 для одержання лікарського засобу для профілактики або лікування інфекції ВГС у суб'єкта, який цього потребує.
9. Застосування композиції за будь-яким з пп. 2-3 для одержання лікарського засобу для профілактики або лікування інфекції ВГС у суб'єкта, який цього потребує.
10. Спосіб лікування пацієнта, інфікованого ВГС, що включає стадію введення вказаному пацієнту ефективної кількості сполуки за п. 1 або композиції за будь-яким з пп. 2-3.
11. Сполука за п. 1, що має структуру:
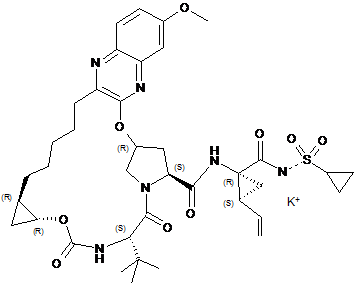 .
.
12. Фармацевтична композиція, що містить ефективну кількість сполуки за п. 11 і фармацевтично прийнятний носій.
13. Фармацевтична композиція за п. 12, що додатково містить другий терапевтичний агент, вибраний із групи, яка складається з інгібітора протеази ВГС і інгібіторів полімерази ВГС NS5B.
14. Спосіб лікування пацієнта, інфікованого ВГС, що включає стадію введення вказаному пацієнту ефективної кількості сполуки за п. 11.
15. Спосіб лікування пацієнта, інфікованого ВГС, що включає стадію введення вказаному пацієнту ефективної кількості композиції за п. 12.
16. Спосіб лікування пацієнта, інфікованого ВГС, що включає стадію введення вказаному пацієнту ефективної кількості композиції за п. 13.
Текст
Реферат: Даний винахід належить до макроциклічної сполуки формули (І): UA 100436 C2 (12) UA 100436 C2 O N N O O NH N O H N O O O N H O S O і її застосування як інгібітора протеази вірусу гепатиту С (ВГС) NS3, а також для лікування або профілактики інфекції ВГС. UA 100436 C2 5 10 15 20 25 30 35 40 45 Галузь техніки, до якої належить винахід Даний винахід належить до макроциклічних сполук, які можна використовувати як інгібітори NS3 протеази вірусу гепатиту С (ВГС), синтезу таких сполук і їх застосування для лікування інфекції ВГС і/або зменшення імовірності виникнення або тяжкості інфекції ВГС. Рівень техніки Інфекція, викликувана вірусом гепатиту С (ВГС), є основною проблемою здоров'я, оскільки в результаті цієї інфекції значне число заражених людей страждає на хронічне захворювання печінки, таке як цироз печінки і гепатоцелюлярна карцинома. Існуюча профілактика інфекції ВГС включає імунотерапію тільки рекомбінантним інтерфероном- або в комбінації з нуклеозидним аналогом рибавірином. Декілька кодованих вірусом ферментів є передбачуваними мішенями для терапевтичного втручання, що включають металопротеазу (NS2-3), серин-протеазу (NS3), геліказу (NS3) і РНКзалежну РНК-полімеразу (NS5B). Протеаза NS3 знаходиться в N-термінальній ділянці білка NS3. NS4A забезпечує кофактор для активності NS3. Потенційні способи лікування інфекції ВГС обговорюються в різних джерелах, включаючи Balsano, Mini-Rev. Med. Chem. 8(4):307-318, 2008, Ronn et al., Current Topics in Medicinal Chemistry 5:533-562, 2008, Sheldon et al., Expert Opin. Investig. Drugs 16(8):1171-1181, 2007, і De Francesco et al., Antiviral Research 58:1-16, 2003. СУТЬ ВИНАХОДУ Даний винахід належить до макроциклічної сполуки формули (I) і її фармацевтично прийнятних солей. Дана сполука і її солі являють собою інгібітори протеази ВГС NS3. Ця сполука і її солі застосовуються як у терапії, так і в наукових дослідженнях. Таким чином, перший аспект даного винаходу описує сполуку формули (I), або її фармацевтично прийнятну сіль: Даний винахід також включає фармацевтичні композиції, що містять сполуку за даним винаходом, і способи одержання таких фармацевтичних композицій. Даний винахід також включає способи лікування або зменшення імовірності виникнення або тяжкості інфекції ВГС. Інші варіанти здійснення, аспекти й ознаки даного винаходу або описані в даному описі нижче, або є очевидними з приведеного нижче опису, прикладів і прикладеної формули винаходу. ДОКЛАДНИЙ ОПИС ВИНАХОДУ Даний винахід включає сполуку формули (I) і її фармацевтично прийнятні солі. Дана сполука і її фармацевтично прийнятні солі можуть використовуватися для інгібування протеази ВГС NS3, лікування інфекції ВГС і/або зменшення імовірності виникнення або тяжкості інфекції ВГС. Профілактичне застосування включає, наприклад, лікування після появи підозри на наявність ВГС у результаті, наприклад, переливання крові, заміни рідини тіла, укусу, випадкового уколу голкою або зараження пацієнта через кров під час хірургічного втручання. Як компоненти фармацевтичної композиції, дані сполуки і солі можуть являти собою основний активний терапевтичний агент. Дана сполука також може бути об'єднана з іншими терапевтичними агентами, включаючи, без обмежень, інші анти-ВГС агенти, протиінфекційні агенти, імуномодулятори, антибіотики або вакцини. Інгібітори NS3 також придатні для підготовки і здійснення скринінгових тестів антивірусних сполук. Наприклад, такі сполуки можуть використовуватися для виділення ферментів-мутантів, які є відмінними інструментами для скринінгу більш сильних антивірусних сполук. Крім того, ці сполуки можна використовувати для виявлення або визначення сайту зв'язування іншого антивірусного агента з протеазою ВГС, наприклад, шляхом конкурентого інгібування. 1 UA 100436 C2 5 10 15 20 25 30 35 40 45 50 55 60 Як описано нижче в Прикладі 2, при проведенні порівняння сполуки формули (I) зі сполукою, описаною у прикладах 110 і 118 WO 2008/057209, показано, що заявлена сполука має декілька переваг. WO 2008/057209 не може розглядатися як найближчий аналог заявленого винаходу. I. Композиції і способи Різні варіанти здійснення включають наступне: (а) Фармацевтичну композицію, що містить ефективну кількість сполуки формули (I) і фармацевтично прийнятний носій. (b) Фармацевтичну композицію (a), що додатково містить другий терапевтичний агент, вибраний із групи, яка складається з анти-ВГС агентів, імуномодуляторів і протиінфекційних агентів. (с) Фармацевтичну композицію (b), у якій анти-ВГС агент являє собою антивірусний агент, вибраний із групи, яка складається з інгібіторів протеази ВГС і інгібіторів полімерази ВГС NS5B. (d) Фармацевтичну комбінацію зі (і)сполуки формули (I) і (ii) другого терапевтичного агента, вибраного з групи, яка складається з анти-ВГС агентів, імуномодуляторів і протиінфекційних агентів; причому і сполука формули (I), і другий терапевтичний агент використовується в кількості, що робить комбінацію ефективною для інгібування протеази ВГС NS3 або для лікування інфекції ВГС і/або зменшення імовірності виникнення або тяжкості інфекції ВГС. (е) Комбінацію (d), у якій анти-ВГС агент являє собою антивірусний агент, вибраний із групи, яка складається з інгібіторів протеази ВГС і інгібіторів полімерази ВГС NS5B. (f) Спосіб інгібування протеази ВГС NS3 у суб’єкта, який цього потребує, що включає введення суб'єкту ефективної кількості сполуки формули (I). (g) Спосіб лікування інфекції ВГС і/або зменшення імовірності виникнення або тяжкості інфекції ВГС у суб’єкта, який цього потребує, що включає введення суб'єкту ефективної кількості сполуки формули (I). (h) Спосіб (g), у якому сполуку формули (I) вводять у комбінації з ефективною кількістю щонайменше одного другого терапевтичного агента, вибраного з групи, яка складається з антиВГС агентів, імуномодуляторів і протиінфекційних агентів. (і) Спосіб (h), у якому анти-ВГС агент являє собою антивірусний агент, вибраний із групи, яка складається з інгібіторів протеази ВГС і інгібіторів полімерази ВГС NS5B. (j) Спосіб інгібування протеази ВГС NS3 у суб’єкта, який цього потребує, що включає введення суб'єкту фармацевтичної композиції (a), (b) або (c) або комбінації (d) або (e). (k) Спосіб лікування інфекції ВГС і/або зменшення імовірності виникнення або тяжкості інфекції ВГС у суб’єкта, який цього потребує, що включає введення суб'єкту фармацевтичної композиції (a) (b), або (c) або комбінації (d) або (e). (l) сполуку формули (I) для застосування в медицині, для профілактики або лікування інфекції ВГС або для застосування (і), (ii) як лікарського засібу, або (iii) для готування лікарського засобу для: (a) інгібування протеази ВГС NS3 або (b) лікування інфекції ВГС і/або зменшення імовірності виникнення або тяжкості інфекції ВГС. У таких застосуваннях сполуки за даним винаходом необов'язково використовуються в комбінації з одним або декількома другими терапевтичними агентами, вибраними з анти-ВГС агентів, протиінфекційних агентів і імуномодуляторів. В усіх цих варіантах здійснення сполука необов'язкова використовується у вигляді фармацевтично прийнятної солі. Термін "або", як використовується тут, позначає альтернативні варіанти, що, при необхідності, можуть бути об'єднані. Таким чином, термін "або" включає кожну перераховану альтернативу окремо, а також їхню комбінацію, якщо така комбінація не є взаємовиключною. Посилання на сполуку також включає стабільні комплекси цієї сполуки, такі як стабільний гідрат. "Стабільна" сполука являє собою сполуку, що може бути отримана і виділена, і структура, і властивості якої залишаються або можуть залишатися без істотних змін протягом періоду часу, достатнього для можливості використовувати цю сполуку з описаною в даній заявці метою (наприклад, терапевтичне або профілактичне застосування суб'єктом). II. Застосування і композиції Термін "застосування" і його варіанти (наприклад, "уведення" сполуки) означає надання сполуки або проліків даної сполуки індивідууму, що потребує лікування. Якщо сполука за винаходом або її проліки надаються в комбінації з одним або декількома іншими активними агентами (наприклад, антивірусними агентами, придатними для лікування інфекції ВГС), і "застосування", і його варіанти варто інтерпретувати як такі, що включають паралельне і послідовне надання цієї сполуки або її солі й інших агентів. Сполуки за даним винаходом можна вводити у вигляді фармацевтично прийнятних солей. Термін "фармацевтично прийнятна сіль" належить до солі батьківської сполуки, що має 2 UA 100436 C2 5 10 15 20 25 30 35 40 45 50 55 60 активність і не є небажаною, ні біологічно, ні будь-яким іншим чином (наприклад, не є ні токсичною, ні будь-яким іншим чином шкідливою для того, хто її приймає). Прийнятні солі включають кислотно-адитивні солі, що, наприклад, можуть бути утворені шляхом змішування розчину сполуки з розчином фармацевтично прийнятної кислоти, такої як соляна кислота, сірчана кислота, оцтова кислота, трифтороцтова кислота або бензойна кислота. Сполуки, що несуть кислотний фрагмент, можуть бути змішані з придатними фармацевтично прийнятними солями для одержання, наприклад, солей лужного металу (наприклад, солей натрію або калію), солей лужноземельного металу (наприклад, солей кальцію або магнію), і солей, утворених прийнятними органічними лігандами, таких як солі четвертинного амонію. Крім того, при наявності кислотної (-COOH) групи або групи спирту, для зміни розчинності або характеристик гідролізу даної сполуки можна використовувати фармацевтично прийнятні ефіри. Термін "проліки", як використовується тут, охоплює неактивну форму лікарської речовини або сполуки, що перетворюється в активну форму лікарської речовини або сполуки під впливом ферментів, хімікатів або метаболічних процесів у тілі людини, якій її вводять. Термін "композиція", як використовується тут, охоплює продукт, що містить вказані компоненти, а також будь-який продукт, що отримується, безпосередньо або опосередковано, з об'єднання вказаних компонентів. "Фармацевтично прийнятний" означає, що компоненти фармацевтичної композиції повинні бути сумісними один з одним і нешкідливими для реципієнта. Термін "суб'єкт" (що альтернативно згадується тут як "пацієнт"), використовуваний у даній заявці, стосується тварини, переважно, ссавця, найбільш переважно, людини, що була об'єктом лікування, спостереження або експерименту. Термін "ефективна кількість" указує на кількість, достатню для прояву терапевтичного або профілактичного ефекту. Для пацієнта, інфікованого ВГС, ефективною кількістю є кількість, достатня для досягнення одного або декількох з наступних ефектів: зменшення здатності ВГС до реплікації, зменшення навантаження ВГС і посилення елімінації вірусу. Для пацієнта, не інфікованого ВГС, ефективною кількістю є кількість, достатня для досягнення одного або декількох з наступних ефектів: зниженої сприйнятливості до інфекції ВГС і ослабленої здатності інфікуючого вірусу викликати персистентну інфекцію у випадку хронічного захворювання. З метою інгібування протеази ВГС NS3 і лікування інфекції ВГС і/або зменшення імовірності виникнення або тяжкості симптомів інфекції ВГС сполуки за даним винаходом, необов'язково у вигляді солі, можуть уводитися таким чином, щоб виникав контакт активного агента з ділянкою його впливу. Сполуки можуть уводитися за допомогою звичайних засобів, доступних для застосування разом з фармацевтичними препаратами, або у вигляді окремих терапевтичних агентів або у комбінації з терапевтичними агентами. Вони можуть вводитися окремо, але звичайно вводяться з фармацевтичним носієм, вибраним у залежності від вибраного режиму введення і стандартної фармацевтичної практики. Сполуки можуть уводитися, наприклад, одним або декількома з наступних способів: перорально, парентерально (включаючи підшкірні ін'єкції, внутрішньовенні, внутрішньом'язові, інтрастернальні ін'єкції або техніку інфузії), за допомогою інгаляції (такої як у формі спрею) або ректально, у вигляді одиничних доз фармацевтичної композиції, що містить ефективну кількість сполуки і звичайні нетоксичні фармацевтично прийнятні носії, ад’юванти і розріджувачі. Рідкі препарати, прийнятні для перорального введення (наприклад, суспензії, сиропи, еліксири і т. п.), можуть бути приготовлені відповідно до методів, відомих у даній галузі, при цьому можна використовувати кожне зі звичайних середовищ, таких як вода, гліколі, олії, спирти і т. п. Тверді препарати, прийнятні для перорального введення (наприклад, порошки, пігулки, капсули і таблетки), можуть бути приготовлені відповідно до методів, відомих у даній галузі, при цьому можна використовувати такі тверді наповнювачі, як крохмалі, цукри, каолін, лубриканти, зв’язувальні агенти, дезінтегруючі агенти і т. п. Парентеральні композиції можна готувати відповідно до методів, відомих у даній галузі, при цьому як носій звичайно використовується стерильна вода і необов'язково інші компоненти, речовини, що сприяють розчинності. Ін’єктовані розчини можуть бути приготовлені відповідно до способів, відомих у даній галузі, де носій містить фізіологічний розчин, розчин або глюкози розчин, що містить суміш фізіологічного розчину і глюкози. Додаткові інструкції щодо способів, прийнятних для використання при готуванні фармацевтичних композицій за даним винаходом, і компонентів, прийнятних для використання в зазначених композиціях, можна знайти в Rеміngtоn's Pharmaceutical Sciences, 20th edition (ed.. A.R. Gennaro, Mack Publishing Co., 2000). Сполуки за даним винаходом можна вводити перорально дозою від 0,001 до 1000 мг/кг маси тіла ссавця (наприклад, людини) у добу у вигляді однієї дози або у вигляді невеликих доз. Один з діапазонів доз складає 0,01-500 мг/кг маси тіла на добу перорально у вигляді однієї дози або у 3 UA 100436 C2 5 10 15 20 25 30 35 40 45 50 55 60 вигляді невеликих доз. Інший діапазон доз складає 0,1-100 мг/кг маси тіла на добу перорально у вигляді однієї або дози у вигляді невеликих доз. Для перорального введення композиції можуть знаходитися у вигляді таблеток або капсул, що містять 1,0-500 мг активного компонента, зокрема, 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500 і 750 мг активного компонента для підбору дозування в залежності від симптомів для пацієнта, що знаходиться на лікуванні. Визначений рівень дози і частота прийому доз для будь-якого конкретного пацієнта можуть бути різними і будуть залежати від множини факторів, включаючи активність конкретної використовуваної сполуки, метаболічну стабільність і тривалість впливу цієї сполуки, вік, масу тіла, загальний стан здоров'я, стать, дієту, спосіб і час уведення, швидкість виведення, комбінацію лікарських речовин, тяжкість конкретного стану й основну терапію, що проводиться. III. Комбінована терапія Макроциклічні хіноксалінові сполуки, описані тут, можуть використовуватися в комбінованій терапії, що включає один або декілька додаткових терапевтичних агентів. Додаткові терапевтичні агенти також включають агенти, спрямовані на ВГС, спрямовані на агенти, що викликають інші захворювання, або включають агенти, що підсилюють імунну систему. Агенти, що підсилюють імунну систему, включають агенти, в основному посилюючі функцію імунної системи, і агенти, що викликають певну імунну відповідь проти ВГС. Додаткові терапевтичні агенти, спрямовані на ВГС, включають агенти, спрямовані на NS3, і агенти, спрямовані на інші активності ВГС, такі як NS5A і NS5B, і агенти, спрямовані на ті види активності клітини-хазяїна, що беруть участь у реплікації ВГС. Різні інгібітори ВГС описані в різних публікаціях. Макроциклічні сполуки, використовувані як інгібітори протеази ВГС, описані в WO 06/119061, WO 7/015785, WO 7/016441, WO 07/148135, WO 08/051475, WO 08/051477, WO 08/051514, WO 08/057209. Додаткові інгібітори протеази ВГС NS3 розкриті в публікаціях міжнародних заявок на патент WO 98/22496, WO 98/46630, WO 99/07733, WO 99/07734, WO 99/38888, WO 99/50230, WO 99/64442, WO 00/09543, WO 00/59929, WO 02/48116, WO 02/48172, британський патент GB 2337262 і патент США 6323180. Додаткові приклади терапевтичних агентів, що можуть бути присутніми у комбінації, включають рибавірин, левовірин, вірамідин, тимозин альфа-1, інтерферон-β, інтерферон-α, пегилований інтерферон-α (пегінтерферон-α), комбінацію інтерферону-α і рибавірину, комбінацію пегінтерферону-α і рибавірину, комбінацію інтерферону-α і левовірину і комбінацію пегінтерферону-α і левовірину. Інтерферон-α включає рекомбінантний інтерферон-α2a (такий як інтерферон РОФЕРОН, що випускається компанією Hoffmann-LaRoche, Nutley, Нью-Джерсі), пегілований інтерферон-α2a (PEGASYS), інтерферон-α2b (такий як інтерферон ІНТРОН-A, що випускається компанією Schering, Kenilworth, Нью-Джерсі), пегілований інтерферон-α2b (ПЕГІНТРОН), рекомбінантний консенсусний інтерферон (такий як інтерферон альфакон-1) і очищений продукт інтерферону-α. Рекомбінантний консенсусний інтерферон компанії Amgen має торгову назву ІНФЕРГЕН. Левовірин являє собою L-енантіомер рибавірину, що виявляє імуномодуляторну активність, аналогічну рибавірину. Вірамідин являє собою аналог рибавірину, розкритий у WO 01/60379. Окремі компоненти комбінації можна вводити окремо в різний час у ході лікування або одночасно у вигляді окремих форм або у вигляді однієї комбінації. Рибавірин, левовірин і вірамідин можуть виявляти анти-ВГС ефекти шляхом модуляції внутрішньоклітинних пулів гуанінових нуклеотидів через інгібування внутрішньоклітинного ферменту інозин-монофосфат-дегідрогенази (IMPDH). IMPDH являє собою фермент, що обмежує швидкість біосинтезу de novo гуанінових нуклеотидів у біосинтетичному шляху. Рибавірин здійснює швидке внутрішньоклітинне фосфорилування, а похідна монофосфату є інгібітором IMPDH. Таким чином, інгібування IMPDH є іншою корисною мішенню для виявлення інгібіторів реплікації ВГС. Таким чином, сполуки за даним винаходом також можна вводити в комбінації з інгібітором IMPDH, таким як VX-497, що розкритий у публікаціях міжнародних заявок на патент WO 97/41211 і WO 01/00622; іншим інгібітором IMPDH, як описано в WO 00/25780; або мікофенолат мофетилом. Див. A.C. Allison і E.M. Eugui, 44 (Suppl). Agents Action 165(1993). Для лікування інфекції ВГС сполуки за даним винаходом також можна вводити в комбінації з противірусним засобом амантадином (1-аміноадамантан). Для одержання всебічного опису цього агента див. J. Kirschbaum, 12 Anal. Profiles Drug Subs. 1-36(1983). Для лікування інфекції ВГС сполуки за даним винаходом також можна вводити в комбінації з противірусним засобом інгібітором полімерази R7128 (Roche). Для лікування інфекції ВГС сполуки за даним винаходом також можна комбінувати з противірусними 2'-C-розгалуженими рибонуклеозидами, розкритими в R.E. Harry-O'Kuru et al., 62 J. Org. Chem. 1754-59(1997); М.S. Wolfe et al., 36 Tet.Lett. 7611-14 (1995); патенті США 3480613; і публікаціях міжнародних заявок на патент WO 01/90121, WO 01/92282, WO 02/32920, WO 04/002999, WO 04/003000 і WO 04/002422; зміст, кожного з яких включений в даний опис у 4 UA 100436 C2 5 10 15 20 25 30 35 40 45 50 55 60 всій повноті як посилання. Такі 2'-C-розгалужені рибонуклеозиди включають, без обмежень, 2'C-метил-цитидин, 2'-C-метил-уридин, 2'-C-метил-аденозин, 2'-C-метил-гуанозин і 9-(2-C-метилβ-D-рибофуранозил)-2,6-диамінопурин і відповідний складний ефір амінокислоти і C-2'-, C-3'- і C-5'-гідроксилів рибози і відповідні складні ефіри необов'язково заміщеного циклічного 1,3пропандіолу з похідною 5'-фосфату. Для лікування інфекції ВГС сполуки за даним винаходом також можна комбінувати з іншими нуклеозидами, що мають анти-ВГС властивості, такі як розкрито в публікаціях міжнародних заявок на патент WO 02/51425, WO 01/79246, WO 02/32920, WO 02/48165 і WO 2005/003147 (включаючи R1656, (2'R)-2'-дезокси-2'-фтор-2'-C-метилцитидин, показаний у вигляді сполук 3-6 на стор.77); WO 01/68663; WO 99/43691; WO 02/18404 і WO2006/021341, і заявці на патент US 2005/0038240, включаючи 4'-азидо нуклеозиди, такі як R1626, 4'-азидоцитидин; публікаціях заявок на патент US 2002/0019363, US 2003/0236216, US 2004/0006007 і US 2004/0063658; і публікаціях міжнародних заявок на патент WO 02/100415, WO 03/026589, WO 03/026675, WO 03/093290, WO 04/011478, WO 04/013300 і WO 04/028481; зміст кожної з який включено в даний опис у всій повноті як посилання. Для лікування інфекції ВГС сполуки за даним винаходом також можна поєднувати з агентом, що є інгібітором полімерази ВГС NS5B. Такі інгібітори полімерази ВГС NS5B, що можуть використовуватися у вигляді комбінованої терапії, включають, без обмеження, інгібітори, описані в публікаціях міжнародних заявок на патент WO 02/057287, WO 02/057425, WO 03/068244, WO 2004/000858, WO 04/003138 і WO 2004/007512; патенті США 6777392 і публікації заявки на патент US 2004/0067901; зміст кожного з який включено в даний опис у всій повноті як посилання. Інші інгібітори полімерази ВГС включають, без обмеження, валопіцитабін (НМ 283; Idenix), і 2'-F-2'-бета-метилцитидин (див. також WO 2005/003147). В одному з варіантів здійснення нуклеозидні інгібітори полімерази ВГС NS5B, що використовуються в комбінації з інгібіторами протеази ВГС NS3 за даним винаходом, вибирають з наступних сполук: 4-аміно-7-(2-C-метил-β-D-арабінофуранозил)-7Н-піроло[2,3d]піримідин; 4-аміно-7-(2-C-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин; 4-метиламіно7-(2-C-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин; 4-диметиламіно-7-(2-C-метил-β-Dрибофуранозил)-7Н-піроло[2,3-d]піримідин; 4-циклопропіламіно-7-(2-C-метил-β-Dрибофуранозил)-7Н-піроло[2,3-d]піримідин; 4-аміно-7-(2-C-вініл-β-D-рибофуранозил)-7Нпіроло[2,3-d]піримідин; 4-аміно-7-(2-с-гідроксиметил-β-D-рибофуранозил)-7Н-піроло[2,3d]піримідин; 4-аміно-7-(2-с-фторметил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин; 4-аміно5-метил-7-(2-C-метил-(3-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин; 4-аміно-7-(2-C-метил-βD-рибофуранозил)-7Н-піроло[2,3-d]піримідин-5-карбоксильна кислота; 4-аміно-5-бром-7-(2-Cметил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин; 4-аміно-5-хлор-7-(2-C-метил-β-Dрибофуранозил)-7Н-піроло[2,3-d]піримідин; 4-аміно-5-фтор-7-(2-C-метил-β-D-рибофуранозил)7Н-піроло[2,3-d]піримідин; 2,4-діаміно-7-(2-C-метил-β-D-рибофуранозил)-7Н-піроло[2,3d]піримідин; 2-аміно-7-(2-з-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин; 2-аміно-4циклопропіламіно-7-(2-з-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин; 2-аміно-7-(2-Cметил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3Н)-он; 4-аміно-7-(2-C-етил-β-Dрибофуранозил)-7Н-піроло[2,3-d]піримідин; 4-аміно-7-(2-C,2-о-диметил-β-D-рибофуранозил)-7Нпіроло[2,3-d]піримідин; 7-(2-C-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3Н)-он; 2аміно-5-метил-7-(2-C,2-о-диметил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3Н)-он; 4аміно-7-(3-дезокси-2-C-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин; 4-аміно-7-(3дезокси-2-с-метил-β-D-арабінофуранозил)-7Н-піроло[2,3-d]піримідин; 4-аміно-2-фтор-7-(2-Cметил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин; 4-аміно-7-(3-C-метил-β-Dрибофуранозил)-7Н-піроло[2,3-d]піримідин; 4-аміно-7-(3-C-метил-β-D-ксилофуранозил)-7Нпіроло[2,3-d]піримідин; 4-аміно-7-(2,4-ди-C-метил-β-D-рибофуранозил)-7Н-піроло[2,3d]піримідин; 4-аміно-7-(3-дезокси-3-фтор-2-C-метил-β-D-ксилофуранозил)-7Н-піроло[2,3d]піримідин; і відповідні 5'-трифосфати; або їх фармацевтично прийнятні солі. Для лікування інфекції ВГС сполуки за даним винаходом також можна комбінувати з ненуклеозидними інгібіторами полімерази ВГС такими, як розкрито в публікаціях міжнародних заявок на патент WO 01/77091; WO 01/47883; WO 02/04425; WO 02/06246; WO 02/20497; WO 2005/016927 (зокрема, JTK003); зміст кожної з яких включений в даний опис у всій повноті як посилання; і HCV-796 (Viropharma Inc). В одному з варіантів здійснення ненуклеозидні інгібітори полімерази ВГС NS5B, що використовуються в комбінації з інгібіторами протеази ВГС NS3, вибирають з наступних сполук: 14-циклогексил-6-[2-(диметиламіно)етил]-7-оксо-5,6,7,8-тетрагідроіндоло[2,1a][2,5]бензодіазоцин-11-карбоксильна кислота; 14-циклогексил-6-(2-морфолін-4-ілетил)-5,6,7,8тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-карбоксильна кислота; 14-циклогексил-6-[2 5 UA 100436 C2 5 10 15 20 25 30 35 40 45 50 55 (диметиламіно)етил]-3-метокси-5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11карбоксильна кислота; 14-циклогексил-3-метокси-6-метил-5,6,7,8-тетрагідроіндоло[2,1a][2,5]бензодіазоцин-11-карбоксильна кислота; метил({[(14-циклогексил-3-метокси-6-метил5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-іл)карбоніл]аміно}сульфоніл)ацетат; ({[(14циклогексил-3-метокси-6-метил-5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-іл)карбоніл]аміно}сульфоніл)оцтова кислота; 14-циклогексил-N-[(диметиламіно)сульфоніл]-3метокси-6-метил-5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-карбоксамід; 3-хлор-14циклогексил-6-[2-(диметиламіно)етил]-7-оксо-5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин11-карбоксильна кислота; N'-(11-карбокси-14-циклогексил-7,8-дигідро-6Н-індоло[1,2e][1,5]бензоксазоцин-7-іл)-N,N-диметилетан-1,2-діаміній біс(трифторацетат); 14-циклогексил7,8-дигідро-6Н-індоло[1,2-e][1,5]бензоксазоцин-11-карбоксильна кислота; 14-циклогексил-6метил-7-оксо-5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-карбоксильна кислота; 14циклогексил-3-метокси-6-метил-7-оксо-5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11карбоксильна кислота; 14-циклогексил-6-[2-(диметиламіно)етил]-3-метокси-7-оксо-5,6,7,8тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-карбоксильна кислота; 14-циклогексил-6-[3(диметиламіно)пропіл]-7-оксо-5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-карбоксильна кислота; 14-циклогексил-7-оксо-6-(2-піперидин-1-ілетил)-5,6,7,8-тетрагідроіндоло[2,1a][2,5]бензодіазоцин-11-карбоксильна кислота; 14-циклогексил-6-(2-морфолін-4-ілетил)-7-оксо5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-карбоксильна кислота; 14-циклогексил-6-[2(диетиламіно)етил]-7-оксо-5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-карбоксильна кислота; 14-циклогексил-6-(1-метилпіперидин-4-іл)-7-оксо-5,6,7,8-тетрагідроіндоло[2,1a][2,5]бензодіазоцин-11-карбоксильна кислота; 14-циклогексил-N-[(диметиламіно)сульфоніл]-7оксо-6-(2-піперидин-1-ілетил)-5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-карбоксамід; 14-циклогексил-6-[2-(диметиламіно)етил]-N-[(диметиламіно)сульфоніл]-7-оксо-5,6,7,8тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-карбоксамід; 14-циклопентил-6-[2(диметиламіно)етил]-7-оксо-5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-карбоксильна кислота; 14-циклогексил-5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-карбоксильна кислота; 6-аліл-14-циклогексил-3-метокси-5,6,7,8-тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11карбоксильна кислота; 14-циклопентил-6-[2-(диметиламіно)етил]-5,6,7,8-тетрагідроіндоло[2,1a][2,5]бензодіазоцин-11-карбоксильна кислота; 14-циклогексил-6-[2-(диметиламіно)етил]-5,6,7,8тетрагідроіндоло[2,1-a][2,5]бензодіазоцин-11-карбоксильна кислота; 13-циклогексил-5-метил4,5,6,7-тетрагідроіндоло[3',2’:6,7][1,4]діазоцино[1,8-a]індол-10-карбоксильна кислота; 15циклогексил-6-[2-(диметиламіно)етил]-7-оксо-6,7,8,9-тетрагідро-5Н-індоло[2,1a][2,6]бензодіазонін-12-карбоксильна кислота; 15-циклогексил-8-оксо-6,7,8,9-тетрагідро-5Ніндоло[2,1-a][2,5]бензодіазонін-12-карбоксильна кислота; 13-циклогексил-6-оксо-6,7-дигідро-5Ніндоло[2,1-d][1,4]бензодіазепін-10-карбоксильна кислота; і їх фармацевтично прийнятні солі. IV. Оцінка сполук Описані тут сполуки можуть бути оцінені відносно різних видів активності, таких як здатність інгібувати активність ВГС NS3, активність реплікону ВГС і активність реплікації ВГС, використовуючи методи, відомі в даній галузі. (Див., наприклад, Carroll et al., J. Biol Chem. 278:11979-11984, 2003). Одним з таких видів аналізу є флуоресцентний аналіз протеази ВГС NS3 з часовим розрізненням (TRF), як описано нижче, а також у Mao et al., Anal. Biochem. 373:1-8, 2008 і в публікації міжнародної заявки на патент WO 2006/102087. Аналіз протеази NS3 може бути виконаний, наприклад, у буфері для аналізу з кінцевим об’ємом 100 мкл, що містить 50 мм HEPES (pН 7,5), 150 мм NaCl, 15% гліцерину, 0,15% тритону X-100, 10 мм DTT і 0,1% ПЕГ 8000. Протеазу NS3 і NS4A попередньо інкубують з різними концентраціями інгібіторів у ДМСО протягом 30 хвилин. Реакцію ініціюють додаванням TRF пептидного субстрату (кінцева концентрація 100 нм). NS3-опосередкований гідроліз субстрату завершують через 1 годину при кімнатній температурі, використовуючи 100 мкл 500 мМ MES (pН 5,5). Флуоресценцію продукту детектують за допомогою флуорофотометра або VICTOR V2, або FUSION (Perkin Elmer Life і Analytical Sciences) зі збудженням на 340 нм і випромінюванням на 615 нм із 400 мкс затримкою. Тестовані концентрації різних форм ферменту вибирають таким чином, щоб у результаті вийшло відношення сигналу до фону (S/B), що знаходиться в межах 10-30. Значення IC50 одержують, використовуючи стандартне чотирипараметричне припасування даних. Значення Ki виводять зі значень IC50 за допомогою наступної формули: IC50=Ki(1+[S]/КМ), Рівняння (1), де [S] - концентрація пептидного субстрату в реакції, а КМ - константа Міхаеліса. Див. P. Gallinari et al., 38 BIOCHEM. 5620-32(1999); P. Gallinari et al., 72 J. VIROL. 6758-69 (1998); М. Taliani et al., 240 ANAL. BIOCHEM. 60-67 (1996); Mao et al., Analytical Biochemistry 373: 1-8, 2008. 6 UA 100436 C2 5 10 15 20 V. Загальна схема одержання сполук Даний винахід також включає способи одержання сполук формули (I). Сполуки за даним винаходом можна легко одержати відповідно до наступних схем реакцій і прикладів, або шляхом їхньої модифікації, використовуючи легко доступні вихідні речовини, реактиви і звичайні способи синтезу. У цих реакціях також можна використовувати варіанти, що добре відомі фахівцям у даній галузі. Інші способи одержання сполук за винаходом є очевидними для фахівця з нижчеподаних схем реакції і прикладів. Якщо не зазначено інше, усі перемінні варто розуміти відповідно до приведеного вище визначення. Наступні схеми реакції і приклади служать тільки як ілюстрація винаходу і його практичного застосування. Каталізатори метатезису олефінів включають наступні різновиди на основі рутенію: F. Miller et al., 118 J. Am. Chem. Soc. 9606 (1996); G. Kingsbury et al., 121 J. Am. Chem.Soc. 791 (1999); H. Scholl et al., 1 Org. Lett. 953 (1999); публікація заявки на патент США US2002/0107138; K. Furstner et al., 64 J.Org.Chem. 8275 (1999). Користь цих каталізаторів у замкнутому циклі метатезису відома з літератури (наприклад, Trnka and Grubbs, 34 Acc. Chem. Res. 18 (2001). Наступні приклади служать тільки як ілюстрація винаходу і його практичного застосування. Приклади не слід розглядати як обмежуючі обсяг або суть винаходу. Список скорочень DCM/CH2Cl2 дихлорметан DCE 1,2 дихлоретан DIEA діізопропілетиламін DMF диметилформамід DMSO диметилсульфоксид Dppf дифенілфосфінофероцен Et2O діетилефір EtOAc етилацетат HATU О-(7-азабензотриазол-1-іл)-N,N,N',N'-тетраметилуроній гексафторфосфат HCl хлороводнева кислота TMSCl хлортриметилсилан TBAF тетра-бутиламоній фторид DMAP диметиламінопіридин MeCN ацетонітрил MeOH метанол Pd/C паладій на вугіллі TBTU O-бензотриазол-1-іл-N,N,N',N'-тетраметилуроній гексафторфосфат TFA трифтороцтова кислота THF тетрагідрофуран Флеш-хроматографія очищення за допомогою системи Biotage Horizon c використанням картриджа на основі силікагелю і визначеного градієнта рухомої фази ВЕРХ автоматизована масабо УФ-ініційована високоефективна рідинна хроматографія з використанням як рухомої фази градієнтів підкисленого MeCN і Н2О МГц Мега Герц Синтез проміжних сполук Проміжні сполуки A Проміжна Структура Назва Посил сполука # ання А1 (1R,2S)-1-аміно-NWang (циклопропілсульфоніл)-2et al., US вінілциклопропанкарбоксаміду 6,995,174 гідрохлорид Проміжна сполука В1: 3-метил-N-({[(1R,2R)-2-пент-4-ен-1-ілциклопропіл]окси}карбоніл)-Lвалін 7 UA 100436 C2 Стадія 1: [(1E)-гепта-1,6-дієн-1-ілокси](триметил)силан 5 10 15 20 25 30 35 40 45 Розчин (0,5 M) бутенілмагній броміду в THF (1,4 екв.) обробляли при -78°C, використовуючи Cu(I)Br.SMe2 (0,05 екв.) і HMPA (2,4 екв.). Суміш перемішували протягом 10 хвилин, потім розчин (1 M) акролеїну (1 екв.) і TMSCl (2 екв.) у THF додавали протягом більше 1 год.. таким чином, щоб внутрішня температура залишалася нижчою -68°C. Отриману суміш перемішували при -78°C протягом 2 год.., потім обробляли надлишком Et 3N і розбавляли гексаном. Після того як температура суміші досягла кімнатної, суміш обробляли невеликою кількістю H2O і фільтрували через целіт. Фільтрат промивали 10 разів, використовуючи H 2O, а потім розсіл. Органічний шар сушили, і леткі компоненти видаляли, а отриманий залишок дистилювали при зниженому тиску (20 мбар). При 80-86°C збирали фракцію, до складу якої входила названа 1 сполука (58%) у вигляді безбарвної рідини. H ЯМР (400 МГц, CDCl3) δ 6,19 (д, J=11,6 Гц, 1H), 5,85-5,75 (м, 1H), 5,02-4,92 (м, 3H), 2,08-2,02 (м, 2H), 1,94-1,88 (м, 2H), 1,46-1,38 (м, 2H), 0,18 (с, 9H). Стадія 2: транс-2-пент-4-ен-1-ілциклопропанол Розчин (0,45 M) попередньої сполуки в гексані обробляли розчином (15%) Et 2Zn (1,2 екв.) у толуолі, і отриманий розчин охолоджували в крижаній бані. Дийодметан (1,2 екв.) додавали по краплях, потім, перед тим як нагріти до 20°C, розчин перемішували протягом 1 год.. Додавали піридин (6 екв.), і кашку перемішували протягом 15 хв, потім виливали в петролейний ефір. Суміш фільтрували через целіт декілька разів доти, доки не одержали прозорий розчин. Цю суміш концентрували при 100 мбар, і розчин, що залишився, (що містить триметил{[(транс-2пент-4-ен-1-ілциклопропіл]окси}силан, толуол і піридин) додатково розбавляли THF. Суміш охолоджували до 0°C, потім по краплях додавали розчин (1M) TBAF (1,2 екв.) у THF. Через 10 хвилин залишали суміш для того, щоб вона нагрілася до 20°C, а потім ще через 1 год.. виливали в H2O. Водну фазу екстрагували за допомогою EtOAc, і об'єднані органічні екстракти промивали розсолом, а потім сушили. Після видалення летких компонентів одержували залишок, що очищали за допомогою флеш-хроматографії (елюент 0-66% Et2О/петролейний 1 ефір), одержуючи названу сполуку (71%) у вигляді безбарвної рідини. Н ЯМР (400 МГц, CDCl3) δ 5,85-5,75 (м, 1H), 5,00 (дд, J=17,1, 1,6 Гц, 1H), 4,94 (уш.д, J=10,4 Гц, 1H), 3,20 (очевидний дт, J=6,4, 2,5 Гц, 1H), 2,10-2,04 (м, 2H), 1,52-1,44 (м, 2H), 1,29-1,19 (м, 1H), 1,15-1,07 (м, 1H), 0,950,87 (м, 1H), 0,71-0,66 (м, 1H), 0,31 (очевидний кв., J=6,0 Гц, 1H). Стадія 3: метил 3-метил-N-(оксометилен)-L-валінат Розчин (0,39 M) метил 3-метил-L-валінату в суміші насиченого водного розчину NаНСО 3 і CH2Cl2 (2:1) охолоджували в крижаній бані і швидко перемішували. Суміш обробляли трифосгеном (0,45 екв.) у вигляді однієї порції, і отриману суміш перемішували протягом 0,5 год.. Реакційну суміш розбавляли CH2Cl2, і шари відділяли. Водну фазу екстрагували, використовуючи CH2Cl2, потім об'єднані органічні шари промивали розсолом і сушили. Після видалення розчинника одержали названу сполуку у вигляді прозорої масла, яке витримували 1 протягом 12 год. під вакуумом (0,1 мбар), потім відразу використовували на наступній стадії. H ЯМР (400 МГц, CDCl3) 3,79 (с, 3H), 3,75 (с, 1Н), 1,00 (с, 9H). Стадія 4: метил 3-метил-N-({[(1R,2R)-2-пент-4-ен-1-ілциклопропіл]окси}карбоніл)-L-валінат і метил 3-метил-N-({[(1S,2S)-2-пент-4-ен-1-ілциклопропіл]окси}карбоніл)-L-валінат 8 UA 100436 C2 5 10 15 20 Розчин (0,45 M) транс-2-пент-4-ен-1-ілциклопропанолу в толуолі обробляли метил 3-метилN-(оксометилен)-L-валінатом (1,1 екв.), а потім DMAP (1 екв.). Отриману суміш нагрівали зі зворотним холодильником протягом 12 год., потім охолоджували до 20°C. Додавали H 2O і EtOAc, і органічний шар відділяли і промивали 1N HCl, розсолом і сушили. Після видалення летких компонентів одержували залишок, що двічі очищали за допомогою флеш-хроматографії (елюент 0-30% Et2О/петролейний ефір). Перші фракції містили метил 3-метил-N-({[(1R,2R)-2+ пент-4-ен-1-ілциклопропіл]окси}карбоніл)-L-валінат (38%) у вигляді масла. MS(ES ) m/z 298 + (M+H) Більш пізні фракції містили метил 3-метил-N-({[(1S,2S)-2-пент-4-ен-1+ + ілциклопропіл]окси}карбоніл)-L-валінат (28%) у вигляді масла. MS(ES ) m/z 298 (M+H) Стадія 5: 3-метил-N-({[(1R,2R)-2-пент-4-ен-1-ілциклопропіл]окси}карбоніл)-L-валін Розчин (0,1 M) метил 3-метил-N-({[(1R,2R)-2-пент-4-ен-1-ілциклопропіл]окси}карбоніл)-Lвалінату в суміші МеОН/Н2О (2:1) обробляли LiOH.H2O (4 екв.) і потім нагрівали до 60°C протягом 4 год.. Суміш охолоджували і концентрували до половини об’єму, потім розбавляли, використовуючи EtOAc, і підкисляли водним розчином HCl (1N). Органічний шар відділяли і промивали розсолом, потім сушили. Після видалення летких компонентів одержали названу + + сполуку (98%) у вигляді масла. MS(ES ) m/z 284 (M+H) Проміжні сполуки С Проміжна сполука С1: метил (4R)-4[(3-хлор-7-метоксихіноксалін-2-іл)окси]-L-пролінату гідрохлорид Стадія 1: 6-метоксихіноксалін-2,3-діол 25 30 Суспензію 4-метоксибензен-1,2-діамін дигідрохлориду в діетилоксалаті (8 екв.) обробляли Et3N (2 екв.) і потім нагрівали до 150°C протягом 2 год.. Суміш охолоджували і фільтрували, потім зібрану тверду речовину промивали, використовуючи H2O і EtOH. Отриманий залишок + + сушили й одержували названу сполуку (69%). MS(ES ) m/z 193 (M+H) Стадія 2: 3-хлор-6-метоксихіноксалін-2-ол 35 Розчин (1,53 M) 6-метоксихіноксалін-2,3-діолу в DMF обробляли SOCl2 (1 екв.) і нагрівали до 110°C. Після 1,5 год. реакційну суміш охолоджували і наливали у водний розчин HCl (1 N). Отриманий осад фільтрували і промивали, використовуючи H 2O і Et2О. Висушена тверда речовина складалася в основному з названої сполуки у вигляді суміші з 6-мітоксихіноксалін-2,3 9 UA 100436 C2 діолом і 2,3-дихлор-6-метоксихіноксаліном. Цю речовину відразу використовували на наступній + + стадії. MS(ES ) m/z 211 (M+H) Стадія 3: 1-трет-бутил 2-метил(2S,4R)-4[(3-хлор-7-метоксихіноксалін-2-іл)окси]піролідин-1,2дикарбоксилат 5 10 15 20 25 30 Розчин (0,35 M) 3-хлор-6-метоксихіноксалін-2-олу в NMP обробляли Cs2CО3 (1,5 екв.) і 1трет-бутил 2-метил(2S,4S)-4-{[(4-бромфеніл)сульфоніл]окси}піролідин-1,2-дикарбоксилатом (1,1 екв.). Отриману суміш перемішували при 50°C протягом 18 год., потім додавали ще одну частину (0,1 екв.) 1-трет-бутил 2-метил(2S,4S)-4-{[(4-бромфеніл)сульфоніл]окси}піролідин-1,2дикарбоксилату. Після перемішування протягом 2 год. суміш охолоджували і розбавляли, використовуючи H2O і EtOAc. Органічні фази промивали водним розчином HCl (1 N), насиченим водним розчином NaHCО3 і розсолом. Висушену органічну фазу концентрували до одержання залишку, який очищали за допомогою флеш-хроматографії (0-60% EtOAc/петролейний ефір) і одержували названу сполуку (35% за два проходи) у вигляді твердої речовини. MS(ES+) m/z 438 (M+H)+ Стадія 4: метил(4R)-4-[(3-хлор-7-метоксихіноксалін-2-іл)окси]-L-пролінату гідрохлорид Розчин (0,62 M) 1-трет-бутилметил(2S,4R)-4-[(3-хлор-7-метоксихіноксалін-2іл)окси]піролідин-1,2-дикарбоксилату в CH2Cl2 обробляли розчином (4 M) HCl у діоксані (5 екв.). Суміш перемішували при 20°C протягом 2 год., потім обробляли розчином (4 M) HCl у діоксані (2 екв.). Через 5 год. після початку реакції було вирішено, що реакція завершилася, після чого суміш концентрували при зниженому тиску. Отриманий залишок розтирали в порошок з Et2О + + для одержання названої сполуки (95%) у вигляді твердої речовини. MS(ES ) m/z 338 (M+H) Приклад 1: Калій {[(1R,2S)-1-({[(1a,5S,8S,10R,22a)-5-трет-бутил-14-метокси-3,6-діоксо1,1,a,3,4,5,6,9,10,18,19,20,21,22,22a-тетрадекагідро-8H-7,10метанциклопропа[8,19][1,10,3,6]діоксадіазациклононадецино[11,12-b]хіноксалін-8іл]карбоніл}аміно)-2-вінілциклопропіл]карбоніл}(циклопропілсульфоніл)азанід Стадія 1: метил 3-метил-N-({[(1R,2R)-2-пент-4-ен-1-ілциклопропіл]окси}карбоніл)-L-валіл(4R)-4-[(3-хлор-7-метоксихіноксалін-2-іл)окси]-L-пролінат 10 UA 100436 C2 5 10 15 20 25 Розчин (0,2 M) метил (4R)-4-[(3-хлор-7-метоксихіноксалін-2-іл)окси]-L-пролінату гідрохлорид у DMF обробляли 3-метил-N-({[(1R,2R)-2-пент-4-ен-1-ілциклопропіл]окси}карбоніл)-L-валіном (1,1 екв.), DIEA (5 екв.) і HATU (1,2 екв.). Отриману суміш перемішували при 20°C протягом 5 год., потім розбавляли EtOAc. Органічний шар відділяли і промивали водним розчином HCl (1N), насиченим водним розчином NаНСО3 і розсолом. Висушену органічну фазу концентрували при зниженому тиску для одержання залишку, що очищали флеш-хроматографією (елюент 10+ 30% EtOAc/петролейний ефір), одержуючи названу сполуку (96%) у вигляді олії. MS(ES ) m/z + 604 (M+H) Стадія 2: метил 3-метил-N-({[(1R,2R)-2-пент-4-ен-1-ілциклопропіл]окси}карбоніл)-L-валіл(4R)-4-[(7-метокси-3-вінілхіноксалін-2-іл)окси]-L-пролінат Розчин (0,1 M) метил 3-метил-N-({[(1R,2R)-2-пент-4-ен-1-ілциклопропіл]окси}карбоніл)-Lваліл-(4R)-4-[(3-хлор-7-метоксихіноксалін-2-іл)окси]-L-пролінат у EtOH обробляли калій трифтор(вініл)боратом (1,5 екв.) і триетиламіном (1,5 екв.). Отриману суміш дегазували, потім додавали аддукт PdCl2(dppf)-CH2Cl2 (0,1 екв.). Суміш нагрівали зі зворотним холодильником протягом 1 год., потім охолоджували до кімнатної температури і розбавляли, використовуючи H2O і EtOAc. Органічну фазу відділяли, промивали H2O і розсолом, потім сушили. Після видалення летких компонентів одержували залишок, що очищали флеш-хроматографією (2030% EtOAc/петролейний ефір), одержуючи названу сполуку у вигляді жовтої піни, що відразу + + використовували на наступній стадії. MS(ES ) m/z 595 (M+H) Стадія 3: метил (1a,5S,8S,10R,18E,22a)-5-трет-бутил-14-метокси-3,6-діоксо1,1a,3,4,5,6,9,10,20,21,22,22a-додекагідро-8H-7,10метанциклопропа[18,19][1,10,3,6]діоксадіазациклононадецино[11,12-b]хіноксалін-8-карбоксилат Розчин (0,02 M) метил 3-метил-N-({[(1R,2R)-2-пент-4-ен-1-ілциклопропіл]окси}карбоніл)-Lваліл-(4R)-4-[(7-метокси-3-вінілхіноксалін-2-іл)окси]-L-пролінату в DCE нагрівали до 80°C, потім обробляли каталізатором Zhan 1 (0,15 екв.). Отриману суміш перемішували при 80°C протягом 1 год., потім охолоджували до кімнатної температури і концентрували при зниженому тиску. 11 UA 100436 C2 5 10 15 20 25 30 Отриманий залишок очищали флеш-хроматографією (20-50% EtOAc/петролейний ефір) і + одержували названу сполуку (25% за 2 проходи) у вигляді піни. MS(ES+) m/z 567 (M+H) Стадія 4: метил (1a,5S,8S,10R,22a)-5-трет-бутил-14-метокси-3,6-діоксо1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-тетрадекагідро-8H-7,10метанциклопропа[18,19][1,10,3,6]діоксадіазациклононадецино[11,12-b]хіноксалін-8-карбоксилат Розчин (0,05 M) метил(1a,5S,8S,10R,18E,22a)-5-трет-бутил-14-метокси-3,6-діоксо1,1a,3,4,5,6,9,10,20,21,22,22a-додекагідро-8H-7,10метанциклопропа[18,19][1,10,3,6]діоксадіазациклононадецино[11,12-b]хіноксалін-8-карбоксилату в суміші MeOH/діоксан (1:1) обробляли Pd/C (8 вага. %). Отриману суміш перемішували в атмосфері водню протягом 4 год.. Каталізатор відфільтровували, і фільтрат концентрували при + зниженому тиску, одержуючи названу сполуку (98%) у вигляді твердої речовини. MS(ES ) m/z + 569 (M+H) Стадія 5: (1a,5S,8S,10R,22a)-5-трет-бутил-14-метокси-3,6-діоксо1,1а,3,4,5,6,9,10,18,19,20,21,22,22a-тетрадекагідро-8H-7,10метанциклопропа[18,19][1,10,3,6]діоксадіазациклононадецино[11,12-b]хіноксалін-8карбоксильна кислота Розчин (0,1 M) метил(1a,5S,8S,10R,22a)-5-трет-бутил-14-метилокси-3,6-діоксо1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-тетрадекогідро-8H-7,10метанциклопропа[18,19][1,10,3,6]діоксадіазациклононадецино[11,12-b] хіноксалін-8. карбоксилату у суміші 1:1 H2O/THF обробляли LiOH H2O (3 екв.). Отриману суміш перемішували при 20°C протягом 18 год., підкисляли водним розчином HCl (0,2 M) і розбавляли EtOAc. Органічну фазу відділяли, промивали водним розчином HCl (0,2 M) і розсолом, потім сушили. Після видалення летких компонентів одержали названу сполуку (98%) у вигляді твердої + + речовини. MS(ES ) m/z 555 (M+H) Стадія 6: (1a,5S,8S,10R,22a)-5-трет-бутил-N-((1R,2S)-1{[(циклопропілсульфоніл)аміно]карбоніл}-2-вінілциклопропіл)-14-метокси-3,6-діоксо1,1а,3,4,5,6,9,10,18,19,20,21,22,22a-тетрадекагідро-8H-7,10метанциклопропа[18,19][1,10,3,6]діоксадіазациклононадецино[11,12-b]хіноксалін-8-карбоксамід 12 UA 100436 C2 5 10 15 20 25 30 Розчин (0,1 M) (1a,5S,8S,10R,22a)-5-трет-бутил-14-метокси-3,6-діоксо1,1а,3,4,5,6,9,10,18,19,20,21,22,22a-тетрадекагідро-8H-7,10метанциклопропа[18,19][1,10,3,6]діоксадіазациклононадецино[11,12-b]хіноксалін-8карбоксильної кислоти у CH2Cl2 обробляли (1R,2S)-1-{[(циклопропілсульфоніл)аміно]карбоніл}2-вінілциклопропанаміній хлоридом (1,3 екв.), DIEA (3 екв.), DMAP (1,5 екв.) і TBTU (1,45 екв.). Отриману суміш перемішували при 20°C протягом 18 год., а потім розбавляли EtOAc. Розчин промивали водним розчином HCl (0,2 M), насиченим водним розчином NaHCО 3 і розсолом. Органічні фази сушили і концентрували до одержання залишку, що очищали флешхроматографією (елюент 2,5% МеОН/СН2Сl2), одержуючи названу сполуку (89%) у вигляді 13 твердої речовини. C ЯМР (100 МГц, DMSO-d6) δ 172,32 170,63 169,04 159,86, 156,95 154,74 148,10 140,41, 133,55 (2 сигнали), 128,94 118,21 117,58, 105,89, 74,88, 59,75, 58,71, 55,68, 54,13, 54,01, 40,13, 34,49, 34,04, 33,76, 32,68, 30,71, 30,43, 28,55, 27,69, 27,28, 26,38, 21,98, 18,49, + + 10,67, 5,69, 5,46; MS(ES ) m/z 767 (M+H) Стадія 7: калій {[(1R,2S)-1-({[(1a,5S,8S,10R,22a)-5-трет-бутил-14-метокси-3,6-діоксо1,1а,3,4,5,6,9,10,18,19,20,21,22,22a-тетрадекагідро-8H-7,10метанциклопропа[18,19][1,10,3,6]діоксадіазациклононадецино[11,12-b]хіноксалін-8-іл]карбоніл}аміно)-2-вінілциклопропіл]карбоніл}(циклопропілсульфоніл)азанід Попередню речовину використовували в EtOH, і отриманий розчин (0,025 M) охолоджували до 0°C. Додавали розчин (0,02 M) трет-BuOK (1,5 екв.) у EtOH, що приводило до утворення осаду. Суміш перемішували при 20°C протягом 18 год., потім фільтруванням збирали тверду речовину. Цю речовину промивали EtOH і сушили, одержуючи названу сполуку (93%) у вигляді білої кристалічної твердої речовини. MS(ES+) m/z 767 (M+H)+ Приклад 2: Порівняння різних сполук Сполуку, описану в прикладі 1, порівнювали зі сполукою, розкритою у прикладах 110 і 118 WO 2008/057209. Результати показані нижче в таблицях 1 і 2. Як можна бачити з таблиць і обговорення результатів, виявилося, що сполука формули (I) має декілька корисних властивостей в порівнянні як зі сполукою прикладу 118 документа WO 2008/057209, так і сполукою прикладу 110 документа WO 2008/057209. 13 UA 100436 C2 Таблиця 1 Приклад 1 WO 2008/057209, приклад 118 WO 2008/057209, приклад 110 Структура NS 3/4A інгібіторна 1 активність (Ki) 1b Реплікативна 2 активність ЕС50 gt1b Плазмова AUC у щурів @ 25 mpk per 3 os Концентрація в печінці щура @ 24 год. (25 mpk 3 per os) Плазмова AUC у собак @ 25 mpk per 3 os Концентрація в печінці собаки @ 24 год. (25 mpk 3 per os) Ковалентне зв'язування 4 білка in vivo Фізичні 5 властивості
ДивитисяДодаткова інформація
Назва патенту англійськоюMacrocyclic quinoxaline compounds as hcv ns3 protease inhibitors
Автори англійськоюHarper, Steven, Summa, Vincenzo, Liverton, Nigel, J., Mccauley, John, A.
Назва патенту російськоюМакроциклические хиноксалиновые соединения как ингибиторы протеазы вгс ns3
Автори російськоюХарпер Стивен, Сумма Винченцо, Ливертон Найджел Дж., Макколи Джон А.
МПК / Мітки
МПК: A61K 38/06, A61P 31/14, C07K 5/08, C07K 5/10, C07K 5/12, A61K 38/08, A61K 38/07
Мітки: макроциклічні, хіноксалінові, сполуки, вгс, інгібітори, протеази
Код посилання
<a href="https://ua.patents.su/21-100436-makrociklichni-khinoksalinovi-spoluki-yak-ingibitori-proteazi-vgs-ns3.html" target="_blank" rel="follow" title="База патентів України">Макроциклічні хіноксалінові сполуки як інгібітори протеази вгс ns3</a>
Хінолінові і хіноксалінові сполуки як інгібітори тфр-р і/або lck тирозинкінази

Номер патенту: 59480
Опубліковано: 15.09.2003
Автори: Хе Вей, Спада Альфред П., Майерс Майкл Р.
МПК: A61P 19/00, C07D 405/12, A61P 11/00, A61M 29/00, A61P 17/06, A61P 35/00, C07D 215/20, C07D 241/44, C07D 215/38, A61P 9/10, A61P 9/00, A61P 43/00, A61K 31/50, A61K 31/498, A61K 31/47, A61F 2/06, C07D 215/22, A61P 29/00, A61P 1/18, A61P 35/02, C07D 241/42, A61P 13/12
Мітки: хіноксалінові, інгібітори, сполуки, тирозинкінази, тфр-р, хінолінові
Формула / Реферат:
1. Сполука формули І, (I)де Х представляє L1OH або L2Z2;L1 представляє (СR3aR3b)r або (СR3aR3b)m-Z3- (СR3’aR3’b)n;L2 представляє (СR3aR3b)р-Z4- (СR3’aR3’b)q або етеніл;Z1 представляє СН або N;Z2 представляє необов'язково заміщений гідроксициклоалкіл, необов'язково заміщений гідроксициклоалкеніл, необов'язково заміщений...
Макроциклічні сполуки як інгібітори вірусної реплікації

Номер патенту: 91677
Опубліковано: 25.08.2010
Автори: Венгловскі Стівен М., Блетт Лоренс М., Стенджел Пітер Д., Маддуру Мачендер Р., Джіанг Ютонг, Вуддард Бенджамін Т., (покійний), Ендрюс Стівен В., Кондроски Кевін Р., Джосі Джон Е., Догерти Джордж Е., Кеннеді Ейпріл Л.
МПК: C07D 487/00
Мітки: вірусної, інгібітори, сполуки, макроциклічні, реплікації
Формула / Реферат:
1. Сполука, що має формулу І, (І)де:Q являє собою центральне кільце, вибране з:де центральне кільце може бути незаміщеним або заміщеним Н, галогеном, ціано, нітро, гідрокси, С1-6алкілом, С3-7циклоалкілом, С4-10алкілциклоалкілом, С2-6алкенілом, С1-6алкокси, гідроксі-С1-6алкілом, С1-6алкілом, заміщеним С1-6алкілом, С1-6алкокси, заміщеним С1-6алкокси, С6 або С10арилом, піридилом, піримідилом, тієнілом,...
Сполуки та композиція як інгібітори активуючої канали протеази

Номер патенту: 95502
Опубліковано: 10.08.2011
Автори: Таллі Девід К., Спрейггон Глен, Чаттерджі Арнаб К., Бурсулая Бадрі, Відал Агнес
МПК: A61K 31/425, A61K 31/401, A61K 31/444, A61P 11/08, C07K 5/06, A61P 11/06, A61P 11/00, A61P 29/00
Мітки: композиція, сполуки, активуючої, інгібітори, протеази, каналі
Формула / Реферат:
1. Сполука Формули (1): (1)або її фармацевтично прийнятні солі, у якій O-(CR2)p-R2 є замісником у будь-якому положенні на кільці А; J є 5-12-членним моноциклічним або конденсованим карбоциклічним кільцем, гетероциклічним кільцем, яке включає атоми N, О та/або S; арильним або гетероарильним кільцем, за умови, що...
Сполуки 1-(d-циклопропілгліциніл)-4-(піперидин-4-іл)піперазину як інгібітори фактора xa серинової протеази

Номер патенту: 76849
Опубліковано: 15.09.2006
Автори: Інджел Гарі Лоуелл, Уайлі Майкл Роберт
МПК: C07D 211/58, A61K 31/496, A61P 7/02, C07C 235/52, C07C 57/00
Мітки: фактора, сполуки, протеази, інгібітори, серинової, 1-(d-циклопропілгліциніл)-4-(піперидин-4-іл)піперазину
Формула / Реферат:
1. Сполука формули (І), (I)де R - атом водню або атом фтору, чи її фармацевтично прийнятна сіль.2. Сполука за п. 1, де R - атом водню.3. Сполука за п. 2, вибрана з 1-(4-метоксибензоїл-D-циклопропілгліциніл)-4-(1-метилпіперидин-4-іл)піперазину і його солей з хлористоводневою, фумаровою та малеїновою кислотами.4. Сполука за п. 3, вибрана з...
Інгібітори протеази віл, фармацевтична композиція на їх основі та спосіб інгібування протеази реплікації віл

Номер патенту: 57772
Опубліковано: 15.07.2003
Автори: Кобаясі Такуо, Варні Майкл Д., Альбідзаті Кім Ф., Райх Зігфрід, Джанг Канін Е.
МПК: A61K 31/472, A61P 31/12, C07D 401/12, C07D 405/12, A61P 31/18, C07D 409/12, C07D 217/26
Мітки: протеази, інгібітори, спосіб, фармацевтична, віл, реплікації, композиція, основі, інгібування
Формула / Реферат:
1. Сполука формули (9):, 9де R та R' незалежно обирають з групи, яку утворюють Н, заміщена або незаміщена група алкіл-OR1, циклоалкільна група, заміщена С1-С6алкільною групою або групою С1-С6алкіл-ОН, гетероциклічна група, заміщена С1-С6алкільною групою або групою С1-С6алкіл-ОН, група алкіл-NR2R3 і група алкіл-S(Х)(Y)R4,у якихR1 - Н, заміщений або незаміщений алкіл або ацильна група,R2, R3 незалежно обрані...
Попередній патент: Фільтр, курильний виріб, що його містить, та спосіб забезпечення підвищеного рівня фільтрації диму зі збільшенням тяги
Наступний патент: Блок низької, середньої або високої напруги
Випадковий патент: Ангоб