Похідні тетрагідрохіноліну
Номер патенту: 81442
Опубліковано: 10.01.2008
Автори: Тіммерс Корнеліс Маріус, Карстенс Віллем Фредерік Йога



















Формула / Реферат
1. Похідна тетрагідрохіноліну формули І
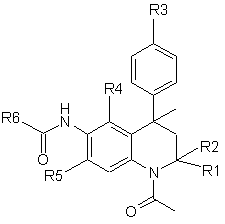 (I)
(I)
або її фармацевтичнo прийнятна сіль,
в якій
R1 і R2 - Н або Me;
R3 - Н, гідроксигрупа, (1-4С)алкокси, (ді)(1-4С)алкіламіно(2-4С)алкокси або (2-6)-гетероциклоалкіл(2-4С)алкокси;
R4 - Н, ОН, (1-4С)алкокси або R7 i R5 є Н, ОН, (1-4С)алкокси або R7;
за умови, що, якщо R4 - Н, то R5 не є Н, ОН або (1-4С)алкокси, і якщо R5 - Н, то R4 не є Н, ОН або (1-4С)алкокси;
R6 - (2-5С)гетероарил, (6С)арил, (3-8С)циклоалкіл, (2-6С)гетероциклоалкіл або (1-6С)-алкіл;
R7 - аміногрупа, (ді)(1-4С)алкіламіно, (6С)арилкарбоніламіно, (6С)арилкарбонілокси, (2-5С)гетероарилкарбоніламіно, (2-5С)гетероарилкарбонілокси, R8-(2-4C)aлкiлaмiнo, R8(2-4С)алкокси, R9-метиламіно або R9-метокси;
R8 - гідроксигрупа, аміногрупа, (1-4С)алкокси, (ді)(1-4С)алкіламіно, (2-6С)-гетероциклоалкіл, (2-6С)гетероциклоалкілкарбоніламіно, (ді)(1-4С)алкіламінокарбоніламіно, (1-4С)алкоксикарбоніламіно і
R9 - амінокарбоніл, (ді)(1-4С)алкіламінокарбоніл, (2-5С)гетероарил або (6С)арил.
2. Похідна тетрагідрохіноліну за п. 1, яка відрізняється тим, що в ній R6 - (2-5С)гетероарил, (6С)арил, (3-8С)циклоалкіл або (1-6С)алкіл.
3. Похідна тетрагідрохіноліну за п. 1 або п. 2, яка відрізняється тим, що в ній R7 -
(ді)(1-4С)алкіламіно, (2-5С)гетероарилкарбонілокси, R8-(2-4C)aлкoкcи, R9-метиламіно або R9-метокси.
4. Похідна тетрагідрохіноліну за будь-яким з пп. 1-3, яка відрізняється тим, що в ній R8 - аміногрупа, (ді)(1-4С)алкіламіно, (2-6С)гетероциклоалкіл або (2-6С)гетероциклоалкілкарбоніламіно.
5. Похідна тетрагідрохіноліну за будь-яким з пп. 1-4, яка відрізняється тим, що в ній R7 - (ді)(1-4С)алкіламіно, R8-(2-4C)алкокси, R9-метиламіно або R9-метокси.
6. Похідна тетрагідрохіноліну за будь-яким з пп. 1-5, яка відрізняється тим, що в ній R8 - аміногрупа, (ді)(1-4С)алкіламіно або (2-6С)гетероциклоалкіл.
7. Похідна тетрагідрохіноліну за будь-яким з пп. 1-6, яка відрізняється тим, що в ній R8 - (ді)(1-4С)алкіламіно або (2-6С)гетероциклоалкіл.
8. Похідна тетрагідрохіноліну за будь-яким з пп. 1-7, яка відрізняється тим, що в ній R7- (ді)(1-4С)алкіламіно, R8-(2-4C)алокси, R9-метиламіно або R9-метокси.
9. Похідна тетрагідрохіноліну за будь-яким з пп. 1-8, яка відрізняється тим, що в ній R6 - (2-5С)гетероарил або (6С)арил.
10. Похідна тетрагідрохіноліну за будь-яким з пп. 1-9, яка відрізняється тим, що в ній R6 - (4-5С)гетероарил або (6С)арил, а R9 - амінокарбоніл, (ді)(1-4С)алкіламінокарбоніл, (3-5С)гетероарил або (6С)арил.
11. Похідна тетрагідрохіноліну за будь-яким з пп. 1-10, яка відрізняється тим, що в ній R7 - (ді)(1-4С)алкіламіно, R8-етокси, R9-метиламіно або R9-метокси, а R9 - амінокарбоніл, (ді)(1-4С)алкіламінокарбоніл, (3-5С)гетероарил або (6С)арил.
12. Похідна тетрагідрохіноліну за будь-яким з пп. 1-11, яка відрізняється тим, що в ній R8 - (ді)(1-4С)алкіламіно, (4-5С)гетероциклоалкіл, а R9 - амінокарбоніл, (ді)(1-4С)алкіламінокарбоніл, (3-5С)гетероарил або (6С)арил.
13. Фармацевтична композиція, яка містить похідну тетрагідрохіноліну за будь-яким з пп. 1-12 і фармацевтичнo придатні допоміжні компоненти.
14. Похідна тетрагідрохіноліну за пп. 1-12, призначена для використання у терапії.
15. Використання похідної тетрагідрохіноліну за будь-яким з пп. 1-12 або її фармацевтично прийнятної солі або сольвату у виготовленні медикаменту, призначеного для регулювання плідності.
Текст
1. Похідна тетрагідрохіноліну формули І R3 C2 2 (19) 1 3 81442 4 9. Похідна тетрагідрохіноліну за будь-яким з пп. 18, яка відрізняється тим, що в ній R6 - (25С)гетероарил або (6С)арил. 10. Похідна тетрагідрохіноліну за будь-яким з пп. 1-9, яка відрізняється тим, що в ній R6 - (45С)гетероарил або (6С)арил, а R9 - амінокарбоніл, (ді)(1-4С)алкіламінокарбоніл, (3-5С)гетероарил або (6С)арил. 11. Похідна тетрагідрохіноліну за будь-яким з пп. 1-10, яка відрізняється тим, що в ній R7 - (ді)(14С)алкіламіно, R8-етокси, R9-метиламіно або R9метокси, а R9 амінокарбоніл, (ді)(14С)алкіламінокарбоніл, (3-5С)гетероарил або (6С)арил. 12. Похідна тетрагідрохіноліну за будь-яким з пп. 1-11, яка відрізняється тим, що в ній R8 - (ді)(14С)алкіламіно, (4-5С)гетероциклоалкіл, а R9 амінокарбоніл, (ді)(1-4С)алкіламінокарбоніл, (35С)гетероарил або (6С)арил. 13. Фармацевтична композиція, яка містить похідну тетрагідрохіноліну за будь-яким з пп. 1-12 і фармацевтичнo придатні допоміжні компоненти. 14. Похідна тетрагідрохіноліну за пп. 1-12, призначена для використання у терапії. 15. Використання похідної тетрагідрохіноліну за будь-яким з пп. 1-12 або її фармацевтично прийнятної солі або сольвату у виго товленні медикаменту, призначеного для регулювання плідності. Винахід стосується сполуки з модуляторною активністю до рецептора FSH, зокрема, похідної тетрагідрохіноліну, фармацевтичної композиції, що містить її, а також використання зазначеної сполуки у медичній терапії. Гонадотропіни грають важливу роль у різних тілесних функціях, включаючи метаболізм, регуляцію температури і процес репродукції. Гонадотропіни діють на специфічні гонадні типи клітин для ініціювання оваріальної і тестикулярної диференціації і стероїдо-генезу. Ппофізний гонадотропін FSH (гормон стимуляції фолікула), наприклад, грає центральну роль у стимуляції розвитку і дозрівання фолікула, a LH (гормон лютенеїзації) індукує овуляцію [Sharp, R.M. Clin Endocrinol. 33:787-807, 1990; Dorrington and Armstrong Recent Prog. Horm. Res. 35:301-342, 1979]. Сьогодні FSH застосовують клінічно, у комбінації з LH або hСГ, для оваріальної стимуляції, тобто гіперстимуляції для запліднення in vitro (TVF) і індукування овуляції у неплідних ановуляторних жінок [Insler, V., Int. J. Fertility 33:8597, 1988, Navot and Rosenwaks, J. Vitro Feit Embryo Transfer 5:3-13, 1988], а також у випадках недорозвитку статевої системи і неплідності у чоловіків. Гонадотропін FSH вивільняється з переднього гіпофізу під впливом гормону вивільнення гонадотропіну і естрогенів і з плаценти під час вагітності. У жінок FSH діє на яєчники, сприяючи розвитку фолікула, і є головним гормоном, що регулює секрецію естрогенів. У чоловіків FSH відповідає за цілісність сім'яносних канальців і діє на клітини Сертолі для підтримки гаметогенезу. Очищений FSH використовують клінічно для лікування неплідності жінок і деяких типів відсутності сперматогенезу у чоловіків. Гонадотропіни, призначені для терапії, можуть бути ізольовані з джерел людської сечі, але мають низьку чистоту [Morse et al, Amer. J. Reproduct. Immunol and Microbiology 17:143, 1988]. Їх також можна приготува ти як рекомбінантні гонадотропіни. Реко-мбінантний людський FSH є у продажу і його використовують для допомоги репродукції. [Olijveet al. Моl. Hum. Reprod. 2:371, 1996; Devroey et al. Lancet 339:1170, 1992]. Дії гормону FSH опосередковуються специфічним рецептором мембрани плазми, який є членом великої родини зв'язаних G-протеїном рецепторів. Ці рецептори складаються з єдиного поліпептиду з сімома трансмембранними доменами і можуть взаємодіяти з G-протеїном, викликаючи, наприклад, активування циклази аденілату. Рецептор FSH є високоспецифічним об'єктом у процесі росту оваріального фолікула і експресується виключно у яєчнику. Блокування цього рецептора або інгібування сигналізації, яка у нормальних умовах відбувається після опосередкованої активації рецептора FSH, порушує розвиток фолікула і, отже, овуляцію і плідність. Таким чином, низькомолекулярні антагоністи FSH можуть бути основою нових контрацептивів. Такі антагоністи FSH спричиняють розвиток зменшених фолікулів (без овуляції), але забезпечують достатню секрецію естрогену для запобігання шкідливим впливам, наприклад, на кісткову масу. З іншого боку, сполуки, що стимулюють активність рецептора FSH, можуть бути використані для імітації гонадотропічного ефекту природного ліганду. Винахід стосується приготування низькомолекулярних аналогів гормону, який селективно виявляє модуляторну активність до рецептора FSH. Сполуки винаходу можуть бути використані як (часткові) агоністи або (часткові) антагоністи рецептора FSH. Було виявлено, що описаний далі клас тетрагідрохінолінових сполук формули І або їх фармацевтично прийнятні солі мають FSHмодуляторну активність 5 81442 в якій R1, R2-H aбo Me; R3 - Η, гідроксигрупа, (1-4С)алкокси, (ди)(14С)алкіламіно(2-4С)алкокси або (2-6)гетероциклоалкіл(2-4С)алкокси; R4 - Н, ОН, (-МС)алкокси або R7; R5 - Н, ОН, (1-4С)алкокси або R7; за умови, що, якщо R4 - Н, R5 не є Н, ОН або (1-4С)алкокси і якщо R5 - Н, R4 нe є Н, ОН або (14С)алкокси; R6 (2-5С)гетероарил, (6С)арил, (38С)циклоалкіл, (2-6С)гетероциклоалкіл або (1-6С)алкіл; R7 аміногрупа, (ди)(1-4С)алкіламіно, (6С)арилкарбоніламіно, (6С)арилкарбонілокси, (25С)гетероарилкарбоніламіно, (25С)гетероарилкарбонілокси, Rs-(2-4С)алкіламіно, R8-(2-4С)алкокси, R9-метиламіно або R9-метокси; R8 - гідроксигрупа, аміногрупа, (1-4С)алкокси, (ди)(1-4С)алкіламіно, (2-6С)-гетероцикло-алкіл, (26С)гетероциклоалкілкарбоніламіно, (ди)(14С)алкіламінокарбоніламіно або (14С)алкоксикарбоніламіно і R9 амінокарбоніл, (ди)(14С)алкіламінокарбоніл, (2-5С)гетероарил або (6С)арил. R4 і R5 незалежно можуть бути вибрані з кожної з згаданих гр уп і можуть бути різними. Сполуки згідно з винаходом модулюють функцію рецептора FSH і можуть бути використані клінічно, як і природні FSH, якщо вони поводяться як агоністи, з тією перевагою, що вони мають змінені якості стабільності і можуть вводитись пацієнту в інший спосіб. Якщо вони блокують рецептор FSH, то можуть бути використані як контрацептивні агенти. Таким чином, модулятори рецептора FSH згідно з винаходом можуть бути використані для лікування неплідності, для контрацепції і для лікування таких гормоно-залежних розладів, як рак грудей, рак простати і ендометріоз. Наведені далі терміни мають визначені для них значення в описі і Формулі винаходу. Термін (1-4С)алкіл у цьому описі означає розгалужену або нерозгалужену алкільну гр упу, яка містить 1-4 атоми карбону, тобто метил, етил, пропіл, ізопропіл, бутил, втор-бутил і трет-бутил. Термін (1-6С)алкіл означає розгалужену або нерозгалужену алкільну гр упу, яка містить 1-6 атомів карбону, наприклад, метил, етил, пропіл, ізопропіл, бутил, втор-бутил, трет-бутил і гексил. Бажаними є (1-5С)алкільні групи, найбільш бажаними - (1-4С)алкільні. Термін (3-8С)циклоалкіл означає циклоалкільну груп у з 3-8 атомами карбону, а саме циклопропіл, циклобутил, циклопентил, циклогексил, циклогептил і циклооктил. Бажаними є (3-6С)циклоалкільні групи. Термін (2-6С)гетероциклоалкіл означає гетероциклоалкільну групу з 2-6, бажано, 3-5 атомами карбону і з щонайменше одним гетероатомом, вибраним з Ν, Ο і/або S, яка, якщо можливо, може бути приєднана через гетероатом або атом карбону. Бажаними гетероатомами є N або О. Бажаними гетероциклоалкільними групами 6 є піперидиніл, піперазиніл, морфолініл і піролідиніл. Термін (1-4С)алкокси означає алкоксигрупу з 1-4 атомами карбону, причому алкільний компонент є таким, що був визначений вище. Бажаними є (1-2С)алкоксигрупи. Термін (2-4С)алкокси означає алкоксигрупу з 2-4 атомами карбону, причому алкільний компонент є таким, що був визначений вище. Термін (ди)(1-4С)алкіламіно у цьому описі означає аміногрупу, моно- або дизаміщену алкільними групами, кожна з яких містить 1-4 атоми карбону і була визначена вище. Термін (6С)арил у цьому описі означає фенільну групу, яка, як варіант, може бути заміщеною одним або більше замісниками, вибраними з гідроксигрупи, аміногрупи, йоду, брому, хлор у, флуор у, нітрогрупи, дифлуорметилу, ціаногрупи, фенілу, (1-4С)алкілу, (1-4С)алкоксигрупи або (1-4С)(ди)алкіламіногрупи, алкілу, алкоксигрупи і діалкіламінових компонентів, таких, які були визначені вище, наприклад, фенілу, 3,5-дибромфенілу, 4-біфенілу, 3,5-ди хлорфенілу, 3-бром-6-метиламінофенілу, 3-хлор-2,6диметоксифенілу і 3,5-диметилфенілу. Термін (2-5С)гетероарил означає заміщену або незаміщену ароматичну груп у з 2-5 атомами карбону і з щонайменше одним гетероатомом, вибраним з Ν, Ο і/або S, наприклад, імідазоліл, піридил, піримідил, тієніл або фурил. Замісники на гетероарильній групі можуть бути вибрані з групи замісників, перелічених для (6С)арильної групи. Гетероари-льна група може бути приєднана через атом карбону або гетероатом, якщо це можливо. Бажаними гетероарильними групами тієніл, фурил і піридил. Термін ди(1-4С)алкіламіно(2-4С)алкокси у цьому описі означає (ди)алкіламіно групу, в якій алкільний компонент або алкільні компоненти містять кожний 1-4 атом карбону, приєднану через аміногрупу до алкільного компонента алкоксигрупи, і яка має 2-4 атоми карбону, в якій (ди)алкіламіногрупа і алкоксигрупа є такими, що були визначені вище. Термін (2-6С)гетероциклоалкіл(2-4С)алкокси у цьому описі означає гетероциклоалкільну групу з 2-6 атомами карбону, приєднану до алкільного компонента алкоксигрупи, яка має 2-4 атоми карбону, причому алкокси- і гетероциклоалкільна групи є такими, що були визначені вище. Термін (6C)арилкарбоніламіно у цьому описі означає фенільну груп у, як варіант, заміщену одним або більше замісниками, вибраними з групи замісників, перелічених для (6C)арильної групи, і приєднаними до карбонільного компонента карбоніламіногрупу, причому (6C)арильний компонент є таким, що був визначений вище. Термін (6C)арилкарбонілокси у цьому описі означає фенільну груп у, як варіант, заміщену одним або більше замісниками, вибраними з групи замісників перелічених для (6C)арильної групи, і приєднаними до карбонільного компонента карбоніламіногрупу, причому (6C)арильний компонент є таким, що був визначений вище. 7 81442 Термін (2-5С)гетероарилкарбоніламіно у цьому описі означає гетероарильну групу з 2-5 атомами карбону, як варіант, заміщену одним або більше замісниками, вибраними з групи замісників, перелічених для (6C)арильної групи, і приєднаними до карбонільного компонента карбоніламіногрупу, причому (6C)арильний компонент є таким, що був визначений вище. Термін (2-5С)гетероарилкарбонілокси у цьому описі означає гетероарильну групу з 2-5 атомами карбону, як варіант, заміщену одним або більше замісниками, вибраними з групи замісників, перелічених для (6C)арильної групи, і приєднаними до карбонільного компонента карбоніламіногрупу, причому (6C)арильний компонент є таким, що був визначений вище. Термін (2-6С)гетероциклоалкілкарбоніламіно у цьому описі означає гетероциклоалкільну групу з 2-6 атомами карбону, приєднану до карбонільного компонента карбоніламіногрупи; причому гетероциклоалкільна група є такою, що була визначена вище. Термін (ди)(1-4С)алкіламінокарбоніл у цьому описі означає (ди)алкіламіногрупу, в якій алкільні групи мають 1-4 атоми карбону, приєднані через аміногрупу до карбонільної групи, причому (ди)алкіламіногрупа є такою, що була визначена вище. Термін (ди)(1-4С)алкіламінокарбоніламіно у цьому описі означає (ди)алкіламіногрупу, в якій алкільна(і) група(и) мають 1-4 атоми карбону і приєднані через аміногрупу до карбонільного компонента карбоніламіногрупи, створюючи цим мочевин-ну функціональність, причому (ди)алкіл аміногрупа є такою, що була визначена вище. Термін (1-4С)алкоксикарбоніламіно у цьому описі означає алкоксигрупу з 1-4 атомами карбону, приєднану карбоїльного компонента карбоніламіногрупу, яка цим утворює карбаматну функціональність, причому алкоксигрупа є такою, що була визначена вище. Термін R8-(2-4С)алкіламіно у цьому описі означає групу R8 , приєднану до алкільного компонента (2-4С)алкіламіногрупи, яка є такою, що була визначена вище. Термін R8-(2-4С)алкокси у цьому описі означає груп у R8, приєднану до алкільного компонента (24С)алкоксигрупи, яка є такою, що була визначена вище. Термін R9-метиламіно у цьому описі означає груп у R9, приєднану до метильного компонента метиламіногрупи. Термін R9-метокси у цьому описі означає групу 9 R , приєднану до метильного компонента метоксигрупи. Термін "фармацевтично прийнятна сіль" стосується солей, які згідно з медичними міркуваннями, є придатними для використання у контакті з тканинами людини і нижчих тварин без токсичної дії, без створення подразнення, алергічних реакцій тощо і характеризуються розумним відношенням корисність/ризик. Фармацевтично прийнятні солі є добре відомими. Їх можна отримати під час кінцевої ізоляції і очищення сполук винаходу, або реакцією функції 8 вільної основи, якщо вона є, з придатною неорганічною кислотою, наприклад, гідрохлорною, фосфорною або сульфуровою, або з органічною кислотою, наприклад, аскорбіновою, лимонною, винною, молочною, малеїновою, малоновою, фумаровою, гліколевою, бурштиновою, пропіоновою, оцтовою, метансульфоновою, тощо. Кислотну функцію можна ввести в реакцію з органічною або неорганічною основою, наприклад, гідроксидом натрію, калію або літію. Винахід, таким чином, стосується сполук формули І, визначених вище Інше втілення винаходу стосується сполук формули І, де R3 - Н, гідроксигрупа або (14С)алкокси. Винахід також стосується сполук формули І, в якій R4 - Н, ОН або (1-4С)алкокси. Іншим втіленням винаходу є сполуки формули І, в якій R5 - ОН, (1-4С)алкокси або R7. Іншим втіленням винаходу є сполуки формули І, в якій R - (2-5С)гетероарил, (6С)арил, (38С)циклоалкіл або (1-6С)алкіл. В іншому аспекті винахід стосується сполук формули І, в якій R6 - (2-5С)гетероарил або (6С)арил. Згідно з ще одним аспектом, гетероарильна група у R6 має 4 або 5 атомів С. Винахід також стосується сполук формули І, в якій R7 (ди)(1-4С)алкіламіно, (2-5С)гетероарилкарбоніламіно, (25С)гетероарилкарбонілокси, R8-(2-4С)алкокси, R9метиламіно або R9-метокси. Іншим аспектом винаходу є сполуки формули І, в якій R7 -(ди)(1-4С)алкіламіно, (2-5С)гетероарилкарбонілокси, R8-(2-4C)алкокси, R9мeтилaмiнo або R9-метокси. Ще один аспект винаходу стосується сполук формули І, в якій R7 - (ди)(1-4С)-алкіламіно, R8-(24С)алкокси, R9-мeтилaмiнo або R9-метокси. В іншому аспекті винахід стосується сполук формули І, в якій R8-(2-4C)алкокси у R7 є R8етокси. У ще одному аспекті винахід стосується сполук формули І, в якій R8-(2-4С)алкіламіно у R7 є R5-етиламіно. Іншим втіленням винаходу є сполуки формули І, в якій R8 - аміногрупа, (ди)(1-4С)-алкіламіно, (26С)гетероциклоалкіл, (26С)гетероциклоалкілкарбоніламіно або (1-4С)алкокси-\карбоніламіно. Іншим втіленням винаходу є сполуки формули І, в якій R8 - аміногрупа, (ди)(1-4С)-алкіламіно, (26С)гетероциклоалкіл або (14С)алкоксикарбоніламіно. Ще одним втіленням винаходу є сполуки формули І, в якій R8 - аміно, (ди)(1-4С)-алкіламіно, (2-6С)гетероциклоалкіл або (26С)гетероциклоалкілкарбоніламіно. Винахід також стосується сполук формули І, в якій R8 - аміногрупа, (ди)(1-4С)-алкіламіно або (26С)гетероциклоалкіл. У ще одному аспекті винаходу гр упа R8 сполуки формули І є (ди)(1-4С)алкіламіно або (26С)гетероциклоалкілом. 9 81442 В іншому аспекті винахід стосується сполук формули І, в якій гетероциклоалкільна група у R8 має 4 або 5 атомів С. Згідно з іншим втіленням винаходу, R9 формули І є амінокарбонілом, (2-5С)гетероарилом або (6C)арилом. Згідно з ще одним втіленням винаходу, гетероарильна група R9 формули І має 3, 4 або 5 атомів С. Інший аспект винаходу стосується сполук, в яких всі конкретні визначення груп R1-R9, наведені вище, комбінуються у сполуці формули І. Далі описано способи приготування сполук винаходу. Сполуки винаходу, в яких R4 і R5 є (14С)алкокси, R1 і R2 є метилом, a R6 є такою, що була визначена вище, можна приготувати починаючи з належним чином заміщених анілінів загальної формули II, застосовуючи добре відому реакцію Скраупа, яка дає похідні 2,2,4-триметил1,2-дигідрохіноліну формули ІІІ-а. Опис циклоконденсації Скраупа можна знайти в літературі: [A. Knoevenagel, Chem. Ber. 54:1726, 1921; RX, Atkins and D.E. Bliss, J. Org. Chem. 43:1975, 1978; J.V. Johnson, B.S. Rauckman, DP. Baccanari and B. Roth., J, Med. Chem. 32:1942, 1989; W.C. Lin, S.-T. Huang and S.-T. Lin, J. Chin. Chem. Soc. 43:497, 1996; JJP. Edwards, S.J. West, K.B. Marschke, D.E. Mais, M. Gottardis and Т.K. Jones, J. Med, Chem. 41:303, 1998]. Цю реакцію звичайно проводять при підвищеній температурі в ацетоні або мезитилоксиді у присутності йоду або протоновмісної кислоти, наприклад, гідрохлорної або р-толуолсульфонової кислоти або водного гідрогенйодиду В іншому варіанті 1,2-дигідро-2,2,4триметилхіноліни формули ІІІ-а можна приготувати реакцією відповідного аніліну формули II з ацетоном у присутності MgSJ4, 4-третбутилкатехолу і йоду [L.G. Наmann, R.I. Higucbi, L Zhi, J.P. Edwards and X.-N. Wanr, J. Med. Chem, 41:623, 1998]. Згідно з іншою процедурою, реакцію можна проводити в ацетоні, використовуючи трифлати лантаніду (наприклад, трифлат скандію) 10 як каталізатори. У цьому випадку, реакцію проводять при кімнатній температурі або при підвищеній температурі, застосовуючи звичайне підігрівання або мікрохвильове опромінювання [М. Е. Theoclitou and L. A. Robinson, Tetrahedron Lett. 43:3907, 2002]. Вихідні матеріали можна придбати з комерційних джерел або приготувати. Сполуки формули ІІІ-b можна приготувати з NBoc-1,4-фенілендіаміну II реакцією з метилвінілкетоном. Належні циклізації описано у [патенті США 2686182 (Badische Anilin- & SodaFabrik Aktiengesellschaft)]. Подальше 1-Ν-ацетилування сполук формули Ill-a-b, в якій R1, R2 є такими, що були визначені вище, може бути здійснене в стандартних умовах. У типовому випадку, сполуки формули ІІІ-а-b гріють під зворотним холодильником в оцтовому ангідриді і вводять у реакцію у розчиннику, наприклад, ДХМ, ТГФ, толуолі або піридині, ацетилхлориді, у присутності основи, наприклад, Ν,Ν-діізопропілетиламіну, триетиламіну або гідриду натрію, і одержують похідні 1-N-ацетил-4метил-1,2-дигідрохіноліну формули IV-a-b. Відповідне N-ацилування каркасу дигідрохіноліну можна знайти у літературі: [G. Reddetien і A. Thurm, Chem. Ber. 65:1511, 1932; Zh. V. Shmyreva, Kh. S. Shikhaliev and E.B. Shpanir, Izv. Vyssh Uchebn. Zaved., Khim. Khim. Tekhnol. 31:45, 1988; Zh. V. Shmyreva, Кгод. S. Shikhaliev, L.P. Zalukaev, Y.A. Ivanov, Y.S. R yabokobylco and LE. Pokrovskaya, Zh. Obshch. Khim. 59:1391, 1989]. Введення необхідної заміщеної фенільної групи у позиції 4 каркасу гідрохіноліну можна здійснити алкілуванням Фриделя-Крафта анізолу сполуки загальної структури Vl-a-b для одержання сполуки загальної формули Vll-a-b. Цю реакцію звичайно проводять при підвищеній температурі в анізолі або у придатному інертному розчиннику, наприклад, гептані або гексані, з анізолом як реагентом і з каталізом кислотою Льюїса (наприклад, АІСІ3, АІВr3, FeCI3 або SnCI4). Опис алкілування Фриделя-Крафта 2,2,4-триметил-1,2дигідрохіноліном можна знайти у літературі: [В.А. Lugovik, L.G. Yudin andA.N. Kost, Dokl. Akad. Nauk SSSR, 170:340, 1966; B.A. Lugovik, L.G. Yudin, S.M. Vinogradova and A.N. Kost, Khim. Geterosikl. Soedin, 7:795, 1971]. В іншому варіанті аніліни загальної структури II можна ввести в реакцію з належним чином заміщеною похідною 1-метилстиролу і формальдегідом в ацетонітрилі при зовнішній або підвищеній температурі з отриманням сполуки загальної структури V-b. Відповідні циклізації описані в літературі: [LM. Mellor and G.D. Merriman, Tetrahedron, 51:6115, 1995]. Сполуки загальної структури V-a-b можна потім нітрувати регіоселективно у позиції 6 каркасу тетрагідрохіноліну і отримати сполуки загальної структури Vl-a-b. Цю реакцію звичайно проводять при температурах від -10°С кімнатної у ДХМ, використовуючи суміші азотної кислоти кислоти і оцтового ангідриду як нітрувального реагенту. В іншому варіанті, азотну кислоту можна додати до розчину сполуки загальної структури V-a-b у 11 81442 льодяній оцтовій кислоті або у суміші оцтової кислоти і ДХМ. Відповідні регіоселективні нітрування тетрагідрохінолінів описані у літературі: [В. Golankiewicz, Pol. J. Chem., 54:355, 1980; Zh. V. Shmyreva, Kh. S. SHkhaliev, L.P. Zalukaev, Y.A. Ivanov, Y.S. R yabokobylko і I.E. Pokrovskaya, Zh. Obshch. Khim. 59:1391, 1989]. Відновлення нітрогрупи сполук загальної структури Vl-a-b можна здійснити багатьма відомими методами відновлення ароматичних нітросполук, наприклад, гідрогенуванням, каталізованим перехідним металом, обробкою сульфідами, обробкою залізом або іншими металами і (слабою) кислотою, обробкою дихлоридом олова в кислотному середовищі тощо. Зокрема, відновлення нітрогруп сполуки загальної формули Vl-a-b можна здійснити обробкою цинковим порошком і оцтовою кислотою у ТГФ або 1,4-діоксані при температурі 0-100°С. Подальше 6-N-ацилування сполук формули Vll-a-b можна провести в стандартних умовах, відомих фахівцям, для одержання сполук загальної структури Ι-a-b Наприклад, сполуки формули VII вводять у реакцію у розчиннику, наприклад, ДХМ, ТГФ, толуолі або піридині з ацилгалогенідом (R6-C(O)-CI) або кислотним ангідридом (R6-C(O)-O-C(O)-R6) у присутності основи, наприклад, Ν,Ν-діізопропілетиламіну, триетиламіну, піридину або гідриду натрію з отриманням 6-N-ацилованої похідної 1,2,3,4тетрагідрохіноліну формули Ι-a-b. В іншому варіанті ацилування сполук загальної формули Vlla-b для отримання сполук загальної формули Ι-a-b можна провести через реакцію з відповідною карбоновою кислотою (R6-CO2H) у присутності сполучаючого реагенту, наприклад, тетрафлуорборату О-{бензотриазол-1-iл)N,N,N',N'-тетраметилуронiю (ТФТМ), гексафлуорфосфату О-(7-азабензотриазол-1-іл)N,N,N',N'-тетраметилуронію (ГФТМ) або гексафлуорфосфату бром-трипіролідинфосфонію (ГФФБПФ) і третинної основи, наприклад, Ν,Νдіізопропілетиламіну, у розчиннику, наприклад, Ν,Ν-ДМФ або ДХМ при кімнатній або підвищеній температурі. Сполуки винаходу, в яких R3=Н , ОН або (14С)алкокси, R4=ОН , R5=ОН або (1-4С)алкокси і R1, R2 і R6 є такими, що були визначені вище, можна приготувати деметилуванням сполук загальної 12 формули l-c-d. Реакції деметилування ароматичних метилових етерів є добре відомими. У типових випадках деметилування проводять, вводячи в реакцію сполуки формули VH-a-b з ВВr3 в інертному розчиннику, наприклад, ДХМ, при температурах від низької до зовнішньої, і отримують деметиловані сполуки загальної формули VHI-a-b. В іншому варіанті, деметилування здійснюється реакцією сполук формули VH-a-b з комплексом BF3Me2S при зовнішній температурі. Рівень деметилування можна контролювати через температуру реакції і кількість деметилувапьного реагенту. Взагалі отримують суміші моно-, ди- і, якщо потрібно, тригідроксисполук загальної формули І-е-і, які можна розділити хроматографією. Взагалі реакція деметилування проходить з невисоким рівнем селективності, переважно на позиції 5 каркасу тетрагідрохіноліну. Швидкість реакції деметилування (деалкілування) сполук загальної формули l-c-d становить 5-ОМе > 4-(р-ОАІk-феніл) > 7-ОМе. Сполуки винаходу в яких R3 є Η або (функціоналізованою) алкоксигрупою, a R4 і/або R5 є (функціоналізованими) алкоксигрупами або ацилоксигрупами, можна приготувати реалкілуванням або ацилуванням гідроксигруп сполук загальної формул І-е-і (функціоналізованими) алкілгалогенідами (наприклад, хлоретилпіролідином) або ацилгалогенідами (наприклад, 2-фуроїлхлорид або метилхлорформатом), відповідно, в стандартних умовах. Сполуки винаходу в яких R4=Н, R5 приєднана до каркасу тетрагідрохіноліну через атом нітрогену, a R1, R2 і R6 є такими, що були визначені вище, можна приготува ти, починаючи з М-Вос-1,4фенілендіаміну (VIII). Послідовність реакцій: (а) реакція Скраупа, (b) ацетилування і (с) алкілування Фриделя-Крафта бензолу або заміщеного бензолу (див. вище) дає сполуки загальної формули Х-а. Слід відзначити, що захисна група Вос відщеплюється в реакційних умовах реакції Фриделя-Крафта. В іншому варіанті N-Вос-1,4-фенілендіамін можна обробити метилвінілкетоном з подальшим ацетилуванням і реакцією Фриделя-Крафта, як це було описано, з отриманням сполук загальної формули Х-b. 13 81442 14 таким же заміщенням можна знайти у літературі [див., наприклад, S. Н. Reich., Μ. Α. Μ. Fuhry, D. Nguyen, Μ. J. Pino et. al, J. Med. Chem. 35: 847, 1992; A. Ivobe, M. Uchida, K. Kamata, Y. Hotei, H. Kusama, H. Harada, Chem. Pharm. Bull. 49: 822, 2001]. Умови нітрування є подібними описаним вище. Згідно з іншою процедурою, сполуки загальної формули Х-b можуть бути отримані, починаючи з часткового відновлення 4-метилхіноліну (XI) комплексом ВН3-ТГФ і біс(2метоксиетокси)алюмодигідридом натрію з отриманням 4-метил-1,2-дигідрохіноліну і з подальшим ацетилуванням, описаним вище, для одержання сполуки XII. Відновлення відповідних хінолінів до 1,2-дигідрохінолініве описано у літературі [див, наприклад, D. Roberts and J. A. Joule, J. Org. Chem. 62:568, 1997; R. F. Heier, L. A. Dolak, J. N. Duncan, D. K. H yslop, M. F. Upton, I. J. Martin, M. A. Mauragis, M. F. Piercey, N. F. Nichols, P. J. K. D. Schreur, M. W. Smith and M. W. Moon, J, Med. Chem. 40: 639, 1997]. Реакція ФриделяКрафта між XII і бензолом або відповідним заміщеним бензолом дає сполуки загальної формули XIII, які можуть бути перетворені у сполуки загальної формули Х-b регіоселективним 6-нітруванням і відновленням до відповідної похідної 6-аміногрупи в описаних вище умовах. Реакції регіоселективного нітрування на таких каркасах були описані у літературі [див., наприклад,: Zh. V. Shmyreva et a!., J. Gen. Chem. USSR (Engl. Transl.) 59: 1234, 1989]. Сполуки загальної формули X-a-b можуть бути потім захищені відомою 9флуоренілметилоксикарбонільною групою (група Fmoc) [див., наприклад, Т. W. Greene and P. M. Wuts, Protective Groups in organic synthesis (3rd ed., John Wiley & Sons, Inc., 1999, p.506)]. Захист звичайно здійснюють, використовуючи FmocCI у ТГФ з піридиновою основою. Регіоселективне нітрування у позиції 7 тетрагідрохінолінового каркасу сполук загальної формули XIV-a-b з подальшим відновленням нітрогрупи (див. вище) дає похідні 7-аміно-1,2,3,4тетрагідрохіноліну загальної формули XV-a-b. Регіоселективні нітрування на належних позиціях з Відновлювальне алкілування аміногрупи у позиції 7 похідної тетрагідрохіноліну загальної формули XV-a-b з використанням належним чином заміщених альдегідів і придатного відновлювального агента (наприклад, ціаноборгідриду або триацетоксибор-гідриду натрію) у придатному розчиннику, наприклад, метанолі або Ν,Ν-ДМФ дає утворення сполук загальної формули XVIa-b. Взагалі, формальдегід дає переважне утворення 7-диметиламінової похідної тетрагідрохіноліну (D=Ε=Me), а інші альдегіди дають переважне утворення моноалкілованих сполук загальної формули XVIab (D=Η, Ε=(функціоналізований) алкіл). Відновлювальні алкілування ароматичних амінів є добре відомими. Стандартне відщеплення захисної групи Fmoc з використанням піперидину у ДХМ дає 6-амінову похідну тетрагідрохіноліну загальної формули XVII-a-b, яку можна ацилувати у позиції 6, як було описано вище, з отриманням сполуки винаходу загальної формули I-j-k. Згідно з іншою процедурою, аміногрупа у позиції 7 похідної тетрагідрохіноліну загальної формули XV-a-b може бути ацилована (гетеро)арилкарбоновими кислотами (G-COaH) або ацилхлоридами (G-C(O)-CI), як було описано вище. У подальших операціях така ж стратегія зняття захисту-ацилування (зняття захисту 6-NFmoc і ацилування з отриманням 6-NH2), описана вище, дає сполуки винаходу загальної формули l1-m. 15 81442 16 Сполуки винаходу, в яких R4 і R5 приєднані через атом нітрогену до тетрагідрохінолінового каркасу, можна приготувати з сполук загальної формули XXIV, де PG є нітрогенною захисною групою, наприклад, Вос, ацетилом, метилкарбаматом або Fmoc, описаною вище реакцією, наприклад, реакцією Скраупа або циклоконденсацією метилвінілкетоном, Ι-Νацетилуванням, відщепленням захисної групи, Nалкілуванням, реакцією Фриделя-Крафта, нітруванням, нітровідновленням і ацилуванням (див. ви ще). Сполуки винаходу, в яких R4=Н, R 5 приєднана до тетрагідрохінолінового каркасу через атом оксигену, a R1, R2 і R6 є такими, що були визначені вище, можна приготувати, починаючи з 2-метокси4-нітроаніліну (XVII). Послідовність реакцій: (а) захист Fmoc для отримання XIX, (b) відновлення нітрогрупи для отримання XX з подальшою (с) регіоселективною реакцією Скраупа, (d) ацетилування і (e) зняття захисту Fmoc, описане вище, дає сполуки загальної формули ХХІ-а. Сполуки загальної формули ΧΧΙ-b можна отримати обробкою сполуки XX метилвінілкетоном в описаних вище умовах для перетворення сполук формули II у ІІІ-b з подальшим Ι-Ν-ацетилуванням і зняттям захисту Fmoc, описаним вище. Подальше перетворення сполук загальної формули XXI у XXIII можна провести ацилуванням 6-аміногруп, використовуючи належний ацилувальний агент, наприклад, ацилхлорид R6C(O)-CI, з подальшою реакцію Фриделя-Крафта з бензолом або належною похідною бензолу в описаних вище умовах. В кислотних умовах Льюїса відбувається одночасне деметилування реакцією Фриделя-Крафта функції 7-ОМе у сполуках загальної формули XXII. Отримана у такий спосіб вільна група 7-ОН загальної формули XXIII може бути реалкілована або ацилована (функціоналізованими) алкілгалогенідами (наприклад, хлоретилпіролідином) або ацилгалогенідів (наприклад, 2-фуроїлхлориду або метилхлорформату), відповідно, у стандартних умовах для отримання сполук загальної формули І-n-о (Ефункціонапізований алкіл , ацил або = карбамат). Реакції Скраупа з ацетоном або мезитилоксидом на сполуках загальної формули XXV можуть дати два різні регіоізомерні продукти загальної формули XXVI-a і XXVIl-а, відповідно. Перетворення сполук загальної формули XXV з використанням метилвінілкетону в описаних вище умовах може дати регіоізомерні продукти загальної формули XXVI-b і XXVII-b, відповідно. Взагалі ці регіоізомерні дигідрохіноліни можуть бути розділені хроматографією (силікагель, РХВР) або кристалізацією і перетворені у сполуки винаходу, як це було описано вище. Сполуки винаходу, в яких R3 , можна Н = приготувати відновленням 7-деоксигенованої сполуки загальної формули XXVIII або XXXI (L і/або Μ є належним (заміщеним) алкілом, ацилалкілоксикарбонілом або алкіламінокарбонілом) шляхом селективного 7-Oтрифлатування і подальшого відновлення групи 7OTf (Tf=три флуорметилсульфоніл). Бажані сполуки загальної формули XXXI можна одержати з похідних загальної формули XXVII в описаних вище умовах. Реакцію (регіоселективного) трифлатування можна провести в контрольованих умовах, використовуючи Tf 2N-феніл і Ν,Νдіізопропілетиламін у ДМФ при кімнатній 17 81442 температурі. Взагалі відбувається переважне трифлатування групи 7-ОН. Подальшу відновлення можна провести, використовуючи суміші трифенілфосфіну, триетиламіну, мурашиної кислоти і ацетату паладію(ІІ) [див., наприклад, К.А. Parker, Q. Ding, Tetrahedron 56:10249, 2000]. Перетворення отриманих у такий спосіб сполук загальної формули XXX або XXXII в описаних вище умовах дає сполуки загальної формули l-p-q, де R5=Н. Деякі сполуки винаходу, які можуть мати форму вільної основи, можна ізолювати з реакційної суміші у формі фармацевтично прийнятної солі. Такі солі можна також отримати, обробляючи вільну основу формули І органічною або неорганічною кислотою, наприклад, гідрохлоридною, гідробромідною, гідройодидною, сульфуровою, фосфорною, оцтовою, пропіоновою, гліколевою, малеїновою, малоновою, метансульфоновою, фумаровою, бурштиновою, винною, лимонною, бензойною і аскорбіновою. Сполуки винаходу містять щонайменше один хіральний атом карбону і тому можуть бути отримані як чисті енантіомери або як суміш енантіомерів, або як суміш діастереомерів. Способи отримання чистих енантіомерів є добре відомими, наприклад, кристалізація солей, отриманих з оптично чистих кислот і рацемічної суміші або хроматографією з застосуванням хіральних колонок. Для діастереомерів можуть бути використання прямофазні або зворотнофазні колонки. Сполуки винаходу можуть утворювати гідрати або сольвати. Відомо, що заряджені сполуки утворюють гідрати, здатні до ліофілізації водою, або утворюють сольватовані форми при концентрації у розчині з належним органічним розчинником. Сполуки згідно з винаходом включають гідрати або сольвати перелічених сполук. Для селекції активних сполук випробування при 10'5М має показати активність, на 20% вищу за максимальну активність FSH як еталона. Іншим критерієм може бути значення ІК50, яке має бути менше 10-5М, бажано, менше 10-7М. Фахівцю зрозуміло, що бажані значення ІК50 залежать від сполуки, що проходить тестування. Наприклад, сполука з ІК50 менше 10-5М взагалі розглядається як кандидат для вибору як ліки. Бажано, щоб це значення було менше 10-7М. Однак, сполука з вищим ІК50, але селективна до конкретного рецептора, може бути кращим кандидатом. Методи визначення зв'язування рецептора, а також аналізів in vitro і in vivo для визначення біологічної активності гонадотропінів є добре відомими. Взагалі експресований рецептор контактує з сполукою, що випробується, і зв'язування або стимуляція або інгібування функціональної реакції виміряється. Для вимірювання функціональної реакції ізольовану ДНК, що кодує ген рецептора FSH, бажано, людського, експресують у придатній хазяйській клітині. Такою клітиною може бути клітина яєчника китайського хом'яка, але 18 придатними є і інші клітини. Бажано, щоб клітини були від ссавця [Jia et al, MoLEndocrin., 5:759-776, 1991]. Способи конструювання ліній клітин, що експресують рекомбінантний FSH є добре відомими [Sambrook et al., Molecular Cloning: Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, latest edition]. Експресії рецептора досягають експресуванням ДНК, що кодує бажаний протеїн. Техніка спрямованого на сайт мутагенезу, лігування додаткових послідовностей, PCR і конструювання придатної системи експресії є відомими. Частини або ціла ДНК, що кодують бажаний протеїн, можуть бути сконструйовані синтетично, з використанням стандартної твердофазної техніки, бажано, з включенням рестрикційних сайтів для полегшення лігування. До кодуючої ДНК можуть бути додані належні контрольні елементи для транскрипції і трансляції включеної кодової послідовності. Як відомо, існують системи експресії, сумісні з різноманітними хазяями, включаючи прокаріоти, наприклад, бактерії, і еукаріоти, наприклад, дріжджі, клітини рослин, комах, птахів тощо. Клітини, що експресують рецептор, вводять у контакт з сполукою, що випробується, для спостереження зв'язування або стимуляції, або інгібування функціональної реакції. В іншому варіанті для вимірювання зв'язування сполукою можуть бути використані ізольовані мембрани клітин, що містять експресований рецептор. Для вимірювання зв'язування можуть бути використані радіоактивно або флюоресцентно мічені сполуки Можна також провести конкурентний аналіз зв'язування. Інші аналізи включають скринінг для сполуки агоніста рецептора FSH через визначення стимуляції опосередкованої рецептором акумуляції сАМР. Такий метод включає експресування рецептора на поверхні хазяйської клітини і піддання клітини дії сполуки, що випробується. Після цього виміряють кількість сАМР. Рі вень сАМР може бути знижений або підвищений залежно від інгібуючої або стимулюючої дії сполуки на рецептор. Скринінг для антагоністів рецептора FSH включає інкубацію клітин, що експресують рецептор FSH, при належних межах концентрації сполуки, що випробується, і при фіксованій субмаксимально ефективній концентрації FSH (тобто такої, що викликає приблизно 80% від максимальної стимуляції накопичення сАМР за відсутності сполуки, що випробується). З кривих залежності ефективності від концентрації для кожної сполуки, що проходить випробування, можна визначити значення ІК50 і процент інгібування індукованого FSH накопичення сАМР. Як еталон можна використати рекомбінантний FSH людини. На додаток до прямого вимірювання, наприклад, рівнів сАМР у експонованих клітинах, можна використати лінії клітин, які на додаток до трансфекції ДНК, що кодує рецептор, також трансфековані другою ДНК, що кодує ген 19 81442 рецептора, експресія якого реагує на рівень сАМР. Такі гени можуть бути сАМР-індуцібельними або можуть бути сконструйовані таким чином, що вони стануть новими приєднаними чутливими до сАМР елементами. Взагалі, експресія генів-репортерів можна контролювати будь-яким реактивним елементом, здатним реагувати на зміну рівнів сАМР. Придатними генами-репортерами є, наприклад, гени, що кодують Р-галактозидазу, лужну фосфатазу, світлякову люциферазу і протехін зеленої флюоресценції. Принципи такого трансактиваційного аналізу є добре відомими і описані, наприклад, у [Stratowa, C.H., Himmler, А. і Czernilofsky, A.P., (1995) Curr. Opm-Biotechnol. 6:574]. Винахід стосується також фармацевтичної композиції, яка включає похідну тетрагідрохіноліну або її фармацевтично прийнятні солі загальної формули І у суміші з фармацевтично прийнятними допоміжними компонентами і, як варіант, з іншими терапевтичними агентами. Допоміжні компоненти мають бути "прийнятними", тобто сумісними з іншими інгредієнтами композиції, і нешкідливими для реципієнтів. Композиції включають такі, що є придатними для орального, під'язичного, підшкірного, внутрішньовенного, внутрішньом'язового, локального або ректального введення пацієнту тощо, всі в одиничних дозованих формах. Для орального введення активний інгредієнт може бути репрезентований дискретними одиницями, наприклад, таблетками, капсулами, порошками, гранулами, розчинами, суспензіями тощо. Для парентерального введення фармацевтична композиція згідно з винаходом може бути презентована в одиничних дозах або у багатодозових контейнерах, наприклад, рідини для ін'єкцій у заздалегідь визначеній кількості, наприклад, у герметичних флаконах і ампулах, і можуть також зберігатись висушеними замороженням (ліофілізованими), потребуючи лише додання стерильного рідкого носія, наприклад, води, перед використанням. У суміші з такими фармацевтично прийнятними допоміжними компонентами, наприклад, описаними у [стандартному посібнику Gennaro, A.R. et a!., Remington: The Science and Practice of Pharmacy (20th Edition., Lippincott Williams & Wilkins, 2000, Part 5: Pharmaceutical Manufacturing)], активний агент може бути спресований у тверді дозовані одиниці, наприклад, пігулки, таблетки, або введений в капсули або супозиторії. З фармацевтично прийнятними рідинами активний агент може бути застосований як рідка композиція, наприклад, як рецептура для ін'єкцій у формі розчин, суспензії, емульсії або уприскування, наприклад, носового. Для виготовлення твердих дозованих одиниць можуть бути використані звичайні добавки, наприклад, наповнювачі, забарвлювачі, полімерні зв'язуючі агенти тощо. Взагалі може бути використана будь-яка фармацевтично прийнятна добавка, яка не порушує функцій активної сполуки. Придатними носіями, з якими активний агент 20 згідно з винаходом може бути введений як тверда композиція лактоза, крохмаль, похідні целюлози тощо, або їх суміші у належних кількостях. Для парентерального введення можуть бути використані водні суспензії, ізотонічні сольові розчини і стерильні розчини для ін'єкцій, які містять фармацевтично прийнятні диспергуючі агенти і/або зволожуючі агенти, наприклад, пропіленгліколь або бутиленгліколь. Винахід також включає описану вище фармацевтичну композицію у комбінації з пакувальним засобом, придатним для цієї композиції, і з інструкцією з використання композиції. Похідні тетрагідрохіноліну згідно з винаходом можна вводити також у формі фармацевтичних засобів, призначених для імплантації, які складаються з серцевини з активних матеріалів у мембрані, здатній регулювати вивільнення. Такі імплантати застосовують підшкірно або локально, і вони вивільняють активний інгредієнт з приблизно постійною швидкістю протягом тривалого часу, наприклад, тижнями і навіть роками. Способи приготування фармацевтичних засобів, призначених для імплантації, є відомими [див., наприклад, Європейський патенті 0303306 (AKZO Nobel N.V.)]. Точна доза і режим введення активного інгредієнта або фармацевтичної композиції залежать від бажаної терапевтичної дії (лікування, неплідності, контрацепція) і можуть бути різними для різних сполук, способів введення, віку і стану пацієнта, якому вводять медикамент. Взагалі парентеральне введення потребує менших доз, ніж інші способи введення, більш залежні від абсорбції. Бажана доза для людини становить 0,0001-25мг на кг маси тіла. Бажана доза може бути одиничною або розділена на декілька субдоз, які вводять з належними інтервалами протягом дня, або для пацієнтів-жінок - з денними інтервалами протягом менструального циклу. Дозування і режим введення можуть бути різними для чоловіків і жінок. Отже, сполуки згідно з винаходом можуть бути використані у терапії. Подальшим аспектом винаходу є використання похідної тетрагідрохіноліну загальної формули І для виготовлення медикаменту, призначеного для використання у лікуванні розладів, залежних від шляхів опосередкування рецептором FSH. Пацієнту, що потребує лікування, може бути введена належна кількість сполуки згідно з винаходом. Іншим аспектом винаходу є використання похідної тетрагідрохіноліну загальної формули І у виготовленні медикаменту, призначеного для контролю плідності. Ще одним аспектом винаходу є використання похідної тетрагідрохіноліну загальної формули І у виготовленні медикаменту, призначеного для використання у лікуванні неплідності. Іншим аспектом винаходу є використання похідної тетрагідрохіноліну загальної формули І у виготовленні медикаменту, призначеного для запобігання плідності. 21 81442 Сполуки згідно з винаходом можуть бути також використані для лікування гормоно-залежних розладів, наприклад, раку грудей, раку простати і ендометріозу. Винахід ілюструється наведеними далі прикладами. Приклади Загальні зауваження У прикладах використані такі абревіатури: ДМА = Ν,Ν-диметиланілін, ДІПЕА = Ν,Νдіізопропілетиламін; ТФК = трифлуороцтова кислота, ДтБАК - ди-трет-бутил-азодикарбоксилат; ТФТМ = тетрафлуорборат О-бензотриазол-1-ілΝ,Ν,Ν',Ν'-тетраметилуронію; ГФТМ = гексафлуорфосфат О-(7-азабензотриазол-1-іл)КІЧМ,ІЧ'-тетраметилуронію; Fmoc = 9флуоренілоксикарбоніл; Fmoc-CI = 9флуоренілметоксикарбонілхлорид; ДМФ = Ν,Νдиметилформамід; ДМАП 4диметиламінопіридин; ТГФ = тетрагідрофуран. Найменування кінцевих продуктів у прикладах побудовані з використанням Beit-stein Autonom program (version: 2,02,119). Якщо не визначено інше, всі кінцеві продукти прикладів ліофілізовані з сумішей вода/1,4-діоксан або вода/ацетонітрил. Якщо сполука є сіллю НСІ або ТФК, відповідні кислоти були додані у належній кількості до розчинника перед ліофілізацією. Для визначення часу утримання використано такі аналітичні методи РХВР (рідинна хроматографія високого розрізнення): Метод 1: Колонка: 5мкм Luna C-18(2) 150x4,6мм; потік: 1мл/хвил.; виявлення: 210нм; темп, колонки 40°С: розчинник A: CH3CN/H2O=1/9 (об'єм/об'єм); розчинник В: CH3CN; розчинник С: 0,1Μ водний розчин трифлуороцтової кислот; градієнт: розчинник А/В/С=від 65/30/5 до 10/85/5 (об'єм/об'єм/об'єм) 30,00хвил., потім постійно протягом ще 10,00хвил. при А/В/С=10/85/5 (об'єм/об'єм/об'єм). Метод 2: ідентичний методу 1, за винятком градієнта. Градієнт: розчинник А/В/С=від 75/20/5 до 15/80/5 (об'єм/об'єм/об'єм) 30,00хвил., потім постійно протягом ще 10,00хвил. при А/В/С=15/80/5 (об'єм/об'єм/об'єм). Метод 3: Колонка: 3мкм Luna C-18(2) 100x2мм; потік: 0,25мл/хвил.; виявлення: 210нм; темп, колонка: 40°С; розчинник А: Н2О; розчинник В: CH3CN; розчинник С: 50мм фосфатний буфер, рН 2,1; градієнт: розчинник А/В/С=від 70/20/10 до 10/80/10 (об'єм/об'єм/об'єм) 30,00хвил., потім постійно протягом ще 10,00хвил. при А/В/С=10/80/10 (об'єм/об'єм/об'єм). Метод 4: Колонка: 3мкм Luna C-18(2) 100x2мм; потік: 0,25мл/хвил.; виявлення: 210нм; температура колонки: 40°С; розчинник А: Н2О; розчинник В: CH3CN; градієнт: розчинник А/В=від 75/25 до 0/100 (об'єм/об'єм) 20,00хвил., потім постійно протягом ще 10,00хвил. при А/В=0/100 (об'єм/об'єм). Метод 5: Колонка: 3мкм Luna C-18(2) 100x2мм; потік: 0,25мл/хвил.; вявлення: 210нм; температура колонки: 40°С; розчинник А: Н2О; розчинник В: CH3CN; розчинник С: 50мм фосфатний буфер, рН 22 2,1; градієнт: розчинник А/В/С=від 70/20/10 до 10/80/10 (об'єм/об'єм/об'єм) 20,00хвил., потім постійно протягом ще 10,00хвил. при А/В/С=10/80/10 (об'єм/об'єм/об'єм). Метод 6: ідентичний методу 1, за винятком градієнта; градієнт: розчинник А/В/С=від 35/60/5 до 10/85/5 (об'єм/об'єм/об'єм) 30,00хвил., потім постійно протягом ще 10,00хвил. при А/В/С=10/85/5 (об'єм/об'єм/об'єм). Для очищення застосовуються такі методи очищення препаративною РХВР: Метод А: Колонка = Luna C-18. Градієнт: 0,1%-на трифлуороцтова кислота у H2O/CH3CN (9/1, o6'eM/o6'eM)/CH3CN = від 100/0 до 0/100 (об'єм/об'єм) 30-45хвил., залежно від легкості сепарації. Виявлення: 210нм. Фракції збирають і концентрують (частково) in vacuo. Метод В: Колонка = Luna C-18. Градієнт: H2O/CH3CN (9/1, об'єм/об'єм)/СН3СN=від 80/20 до 0/100 (об'єм/об'єм) 30-45хвил., залежно від легкості сепарації. Виявлення: 210нм. Приклад 1 N-[1-ацетил-5,7-диметокси-4-(4метоксифеніл)2,2,4-триметил-1,2,3,4тетрагідрохінолін-6-іл]-3-хлор-2,6диметоксибензамід (а). 5,7-диметокси-2,2,4-триметил-1,2дигідрохінолін Розчин 3,5-диметоксианіліну (50г) в ацетоні (800мл) додають краплями до суміші MgSO4 (100г) і Sc(OTa)3 (8,0г) у 1л ацетону при кімнатній температурі. Через 5год. додають ще Sc(OTf)3 (3,2г) і реакційну суміш перемішують до зникнення вихідного матеріалу. Після фільтрування, ацетон частково випарюють in vacuo, викликаючи кристалізацію бажаної сполуки, яку збирають фільтруванням. Вихід - 22г після сушіння in vacuo. Залишок маточної рідини концентрують in vacuo і залишок очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм) і отримують ще 19,4г бажаної сполуки. Вихід: 42г. (b) 1-ацетил-5,7-диметокси-2,2,4-триметил-1,2дигідрохінолін Суміш сполуки Прикладу 1а (42г) і оцтового ангідриду (100мл) перемішують при 100°С 20год. Реакційну суміш вливають у 500мл льоду-води з перемішуванням. Твердий осад збирають фільтруванням і сушать in vacuo при 40°С 2 дні. Залишкову коричневу тверду речовину можна використовува ти сирою у подальшому синтезі. Вихід: 45г. (с) 1-ацетил-5.7-диметокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4етрагідрохінолін Суміш сполуки Прикладу 1b (30г) і АІСІ3 (44г) в анізолі (500мл) перемішують при 50°С 18 год. Реакційну суміш о холоджують (0°С), гасять водою і додають етилацетат. Суміш перемішують протягом ночі. Органічний шар відокремлюють, сушать над MgSO4, фільтр ують і концентрують in vacuo. Залишок хроматографують на силікагелі з елюентом гептан/етилацетат=8/2 (об'єм/об'єм). Вихід: 15г. 23 81442 (d) 1-ацетил-5,7-диметокси-4-(4метоксифеніл)-2,2,4-триметил-6-нітро-1,2,3,4тетрагідрохінолін Розчин оцтового ангідриду (450мкл) у паруючій азотній кислоті (22,5мл) додають краплями до розчину сполуки Прикладу 1с (15г) у СН2СІ2 (500мл) при 0°С. Після цього реакційну суміш перемішують при кімнатній температурі 3год. Додають воду і органічний шар відокремлюють, сушать над MgSO4, фільтрують і концентрують in vacuo. Залишок кристалізують з етанол і отримують бажану сполуку як кристалічну тверду речовину. Вихід: 10г. (e) 1 -ацетил-6-аміно-5,7-диметокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4тетрагідрохінолін Розчин сполуки Прикладу 1d (11,75г) і оцтової кислоти (15,5мл) у ТГФ (600мл) охолоджують до 0°С. Порціями додають цинковий порошок (36г) і льодяну ванну видаляють. Температуру швидко підвищують до 30°С, після чого реакційну суміш залишають охолонути до кімнатної температури. Надлишок цинку видаляють фільтруванням і до фільтрату додають СН 2СІ2 і насичений водний розчин Na2CO3. Органічний шар відокремлюють, сушать над MgSO4, фільтр ують і концентрують in vacuo. Продукт використовують сирим. Вихід: 10,9г. (f) N-[1-ацетил-5,7-диметокси-4-(4метоксифеніл)2.2,4-триметил-1,2,3.4тетрагідрохінолін-6-іл]-3-хлор-2,6диметоксибензамід Загальна процедура А: До розчину сполуки Прикладу 1е (100мг), 3-хлор-2,6диметоксибензойної кислоти (60мг) і ДІПЕА (132мкл) у СН 2СІ2 (2мл) додають ГФТМ (143мг) при кімнатній температурі. Якщо реакція не завершується через 18год., додають ще ГФТМ і ДІПЕА. Після завершення реакції додають насичений водний розчин NaHCO3, органічний шар відокремлюють, сушать (MgSO 4) і концентрують in vacuo. Бажану сполуку очищають препаративною РХВР (метод А). Вихід: 87мг. MS-ESI: [М+Н]+=597,4 РХВР: Rt=17,98хвил. (метод 1). Приклад 2 [1-ацетил-5,7-диметокси-4-(4-метоксифеніл)2,2,4-триметил-1,2,3,4-тетрагідрохінолін-6-іл]-амід 4,5-диметилфуран-2-карбонової кислоти Загальна процедура В: До розчину сполуки Прикладу 1е (800мг), 4,5-диметилфуран-2карбонової кислоти (308мг) і ДМА (768мкл) у ДМФ (10мл) додають ГФТМ (1,1г) при кімнатній температурі. Якщо реакція не завершується через 18год., реакційну суміш нагрівають до 50°С. Після завершення реакції додають воду і етилацетат, органічний шар відокремлюють, сушать (MgSO 4) і концентрують in vacuo. Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм). Вихід: 444мг. MS-ESI: [М+Н]+=521,4 РХВР: Rt=16,96хвил. (метод 1). Приклад 3 24 [1-ацетил-5,7-диметокси-4-(4-метоксифеніл)2,2,4-триметил-1,2,3,4-тетрагідрохінолін-6-іл]-амід 5-бромтіофен-2-карбонової кислоти Згідно з загальною процедурою В, сполуку Прикладу 1е (800мг), ацилують 5-бромтіофен-2карбоновою кислотою (456мг), ДМА (768мкл) і ГФТМ (1,1г) у СН2СІ2 (10мл). Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм). Вихід: 1,0г. MS-ESI: [М+Н]+=589,2; РХВР: Rt=18,90хвил. (метод 2). Приклад 4 [1-ацетил-5,7-диметокси-4-(4-метокси-феніл)2,2,4-триметил-1,2,3,4-тетрагідрохінолін-6-іл]-амід Загальна процедура С: До розчину сполуки Прикладу 1е (800мг) і 4-біфенілкарбонілхлориду (475мг) у СН2СІ2 (10мл) додають ДМА (768мкл) при кімнатній температурі. Реакційну суміш перемішують до зникнення вихідного матеріалу, після чого додають воду. Органічний шар відокремлюють, сушать (MgSO 4) і концентрують in vacuo. Бажану сполуку очи щають хроматографією на силікагелі з елюентом гептан/етилацетат=1/0=>0/1 (об'єм/об'єм). Вихід: 678мг. MS-ESI: [M+H]+=579,4; РХВР: Rt=26,19хвил. (метод 2). Приклад 5 [1-ацетил-5-гідрокси-7-метокси-4-(4-(4метоксифеніл)-2,2.4-триметил-1,2,3,4тетрагідрохінолін-6-іл]-амід фуран-2-карбонової кислоти (a) [1-ацетил-5,7-диметокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4тетрагідрохінолін-6-іл]-амід фуран-2-карбонової кислоти Згідно з загальною процедурою С, сполуку Прикладу 1е (800мг) ацилують 2-фуроїлхлоридом (217мкл) і ДМА (768мкл) у СН 2СІ2 (10мл). Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм). Вихід: 896мг. (b) [1-ацетил-5-гідрокси-7-метокси-4-(4метоксифеніл)-2.2,4-триметил-1,2,3,4тетрагідрохінолін-6-іл]-амід фуран-2-карбонової кислоти Загальна процедура D: розчин сполуки Прикладу 5а (50мг) у СН2СІ2 (4мл) охолоджують до -78°С в атмосфері Ν 2. Додають краплями бортрибромід (28мкл) і після цього реакційну суміш залишають повільно нагрітись до кімнатної температури. Реакцію гасять водою і додають СН2СІ2. Органічний шар відокремлюють, сушать (MgSO4) і концентрують in vacuo. Бажану сполуку очищають препаративною РХВР (метод А). У даному випадку взагалі утворюються суміші сполук з різним рівнем деметилування, які можна розділити препаративною РХВР. Вихід: 9,1мг; MS-ESI: [М+Н]+=479,4; РХВР: Rt=23,40хвил. (метод 2). Приклад 6 N-[1-ацетил-5-гідрокси-7-метокси-4-(4метоксифеніл)-2.2.4-триметил-1,2,3.4-тетрагідрохінолін-6-іл]-3,5-дихлорбензамід 25 81442 (a) N-[1-ацетил-5,7-диметокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4-тетрагідрохінолін-6-іл]-3,5-дихлорбензамід Згідно з загальною процедурою С, сполуку Прикладу 1е (800мг) ацилують 3,5дихлорбензоїлхлоридом (460мг) і ДМА (768мкл) у СН2СІ2 (10мл). Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм). Вихід: 1,03г. (b) Ν-[1-ацетил-5-гідрокси-7-метокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4тетрагідрохінолін-6-іл]-3,5-дихлорбензамід Згідно з загальною процедурою D, сполуку Прикладу 6а (50мг) обробляють бор-трибромідом (24мкл) у СН2СІ2 (4мл). Бажану сполуку очи щають препаративною РХВР. (метод А). Вихід: 9,6мг; MS-ESI: [М+Н]+=557,2; РХВР: Rt=23,40хвил. (метод 2). Приклад 7 [1-ацетил-5-гідрокси-7-метокси-4-(4-метоксифеніл)-2.2.4-триметил-1,2б3б4-тетрагідрохінолін6-іл]-амід 5-хлортіофен-2-карбонової кислоти (а) [1-ацетил-5,7-диметокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4тетрагідрохінолін-6-іл]-амід 5-хлортіоФен-2карбонової кислоти Згідно з загальною процедурою В, сполуку Прикладу 1е (800мг) ацилують 5-хлортіофен-2карбоновою кислотою (456мг), ДМА (768рі) і ГФТМ (1,1г) у СН2СІ2 (10мл). Бажану сполуку очи щають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм). Вихід: 1,0г. (b) [1-ацетил-5-гідрокси-7-метокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4тетрагідрохінолін-6-іл]-амід 5-хлортіофен-2карбонової кислоти Згідно з загальною процедурою D, сполуку Прикладу 7а (200мг) обробляють бортрибромідом (350мкл) у СН2СІ2 (25мл) при температурі не вище -30°С. Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етилацетат=1/0=>0/1 (об'єм/об'єм), і потім препаративною РХВР (метод А). Вихід: 35мг; MS-ESI: [М+Н]+=529,2; РХВР: Rt=28,24хвил. (метод 2). Приклад 8 [1-ацетил-5-гідрокси-7-метокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4тетрагідрохінолін-6-іл]-амід біфеніл-4-карбонової кислоти Згідно з загальною процедурою D, сполуку Прикладу 4 (50мг) обробляють бортрибромідом (100мкл) у СН2СІ2 (4мл) при температурі не вище 0°С. Реакційна суміш також містить продукт Прикладу 10. Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм). Вихід: 43мг; MS-ESI: [М+Н]+=565,4; РХВР: Rt=32,53хвил. (метод 2). Приклад 9 [1-ацетил-5,7-дигідрокси-4-(4-гідроксифеніл)2,2,4-триметил-1,2,3,4-тетрагідрохінолін-6-іл]-амід біфеніл-4-карбонової кислоти 26 Згідно з загальною процедурою D, сполуку Прикладу 4 (50мг) обробляють бортрибромідом (100мкл) у СН2СІ2 (4мл) при температурі не вище 15°С. Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм). Вихід: 33мг; MS-ESI: [M+H]+=537,4; РХВР: Rt=24,16хвил. (метод 2). Приклад 10 [1-ацетил-5-гідрокси-4-(4-гідроксифеніл)-7метокси-2,2,4-триметил-1,2,3,4-тетрагідрохінолін6-іл]-амід біфеніл-4-карбонової кислоти Згідно з загальною процедурою D, сполуку Прикладу 4 (400мг) обробляють бортрибромідом (800мкл) у СН2СІ2 (25мл) при температурі не вище 0°С. Бажану сполуку (побічний продукт Прикладу 8) очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм). + Вихід: 50мг; MS-ESI: [М+Н] =551,4; РХВР: Rt=27,58хвил. (метод 2). Приклад 11 [1-ацетил-5-гідрокси-7-метокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4тетрагідрохінолін-6-іл]-амід 4,5-диметилфуран.-2карбонової кислоти Згідно з загальною процедурою D, сполуку Прикладу 2 (200мг) обробляють бортрибромідом (336мкл) у СН2СІ2 (25мл) при температурі не вище -78°С. Бажану сполуку очищають препаративною РХВР (метод А). Вихід: 51мг; MS-ESI: [M+H]+=507,4; РХВР: Rt=24,32хвил. (метод 1). Приклад 12 N-[1-ацетил-5-гідрокси-4-(4-гідроксифеніл)-7метокси-2,3,4-триметил-1,2,3,4-тетрагідрохінолін6-іл]-3,5-дихлорбензамід Згідно з загальною процедурою D, сполуку Прикладу 6а (75мг) обробляють бортрибромідом (38мкл) у СН 2СІ2 (5мл). Бажану сполуку очи щають препаративною РХВР(методА). Вихід: 11мг; MS-ESI: [М+Н]+=543,4; РХВР: Rt=25,66хвил. (метод 2). Приклад 13 N-[1-ацетил-5-гідрокси-7-метокси~4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4-тетрагідрохінолін-6-іл]-3,5-диметилбензамід (а) Ν-[1-ацетил-5.7-диметокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4тетрагідрохінолін-6-іл]-3,5-диметилбензамід Згідно з загальною процедурою В, сполуку Прикладу 1е (800мг), ацилують 3,5диметилбензойною кислотою (330мг), ДМА (768мкл) і ГФТМ (1,1г) У СН2СІ2 (10мл). Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм). Вихід: 1,18г. (b) N-[1-ацетил-5-гідрокси-7-метокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4тетрагідрохінолін-6-іл]-3,5-диметилбензамід Згідно з загальною процедурою D, сполуку Прикладу 13а (300мг) обробляють бортрибромідом (513мкл) у СН2СІ2 (25мл) при 27 81442 температурі не вище -40°С. Бажану сполуку очищають препаративною РХВР (метод А). Вихід: 41мг; MS-ESI: [М+Н]+=517,4; РХВР: Rt=13,89хвил. (метод 3). Приклад 14 N-[1-ацетил-5-гідрокси-7-метокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4тетрагідрохінолін-6-іл]-3,5-дибромбензамід (а) N-[1-ацетил-5,7-диметокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4,тетрагідрохінолін-6-іл]-3,5-дибромбензамід Згідно з загальною процедурою В, сполуку Прикладу 1е (800мг), ацилують 3,5дибромобензойною кислотою (616мг), ДМА (768рі) і ГФТМ (1,1г) у ДМФ (10мл). Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм). Вихід: 900мг. (b) Ν-[1-ацетил-5-гідрокси-7-метокси-4-(4метоксифеніл)-2,2,4-триметил-1,2,3,4тетрагідрохінолін-6-іл]-3,5-дибромбензамід Згідно з загальною процедурою D, сполуку Прикладу 14а (300мг) обробляють бортрибромідом (639мкл) у СН2СІ2 (25мл), при температурі не вище -60°С. Бажану сполуку очищають препаративною РХВР (метод В). Вихід: 28мг; MS-ESI: [М+Н]+=647,2; РХВР: Rt=16,29хвил. (метод 3). Приклад 15 [1-ацетил-2,2,4-триметил-7-(2-морфолін-4ілетокси)-4-феніл-1,2,3,4-тетрагідрохінолін-6-іл]амід біфеніл-4-карбонової кислоти (а) 1-Fmос-2-метокси-4-нітроанілін Розчин 2-метокси-4-нітроаніліну (3,0г) і піридину (1,6мл) у ТГФ (30мл) охолоджують до 0°С. Додають порціями FmocCI (5,07г), після цього видаляють льодяну ванну і суміш перемішують 5год. ТГФ видаляють in vacuo і залишок розчиняють у СН2СІ2 (175мл). Додають метанол (прибл. 100мл) і частково видаляють СН 2СІ2 in vacuo до утворення осаду. Суміш відстоюють 1год., після чого кристали збирають фільтруванням і сушать in vacuo, отримуючи бажану сполуку. Вихід: 6,32г; MS-ESI: [М+Н]+=391,2 (b) 9-флуоренілметил-N-(2-метокси-4амінофеніл)карбамат Загальна процедура Е: суміш сполуки, описаної у Прикладі 15а (6,07г), оцтової кислоти (8,9мл) і ТГФ (150мл) охолоджують до 0°С. Порціями додають цинковий порошок (20,4г) і льодяну ванну видаляють. Після повільного досягнення температури 10°С її швидко підвищують до 45°С. Потім реакційну суміш залишають охолонути до кімнатної температури, надлишок цинку видаляють фільтруванням і додають велику кількість СН 2СІ2 (прибл. 500мл). Суміш промивають насиченим водним NaHCO3 (3x200мл) і розсолом (1x200мл). Органічний шар відокремлюють, суша ть (MgSO4), фільтрують і концентрують in vacua до утворення осаду. С уміш відстоюють протягом ночі при 0°С, після чого кристали збирають фільтруванням і сушать in vaciio, отримуючи бажану сполуку. 28 Вихід: 4,45г. (с) 9Н-флуорен-9-ілметиловий естер (7метокси-2,2,4-триметил-1,2-дигідрохінолін-6-іл)карбамової кислоти Суміш сполуки Прикладу 15b (4,45г), І2 (157мг), MgSO4 (7,4г), 4-трет-бутил-бутилкатехол (61мг) і ацетон (прибл. 350мл) гріють під зворотним холодильником протягом 5год. MgSO 4 видаляють фільтруванням і фільтрат концентрують in vacuo. Бажану сполуку отримують хроматографією на силікагелі з елюентом гептан/етил ацетат=9/1=>7/3 (об'єм/об'єм) Вихід: 4,24г. (d) 9Н-флуорен-9-ілметиловий естер (1ацетил-7-метокси-2,2,4-триметил-1,2дигідрохінолін-6-іл)-карбамової кислоти До розчину сполуки Прикладу 15с (4,24г) у піридині (25мл) і СН2СІ2 (25мл) додають ДМАР (прибл. 20мг). Повільно додають ацетилхлорид (2,0мл) у СН 2СІ2 (20мл). Після цього суміш розбавляють СН 2СІ2 (прибл. 100мл) і промивають водою (3x100мл), 0,1М водною НСІ (3x100мл), 0,5М водною НСІ (1 x100мл) і розсолом (1x100мл). Органічний шар сушать (MgSO 4) і концентрують in vacuo. Бажану сполуку отримують хроматографією на силікагелі з елюентом гептан/етил ацетат=9/1=>7/3 (об'єм/об'єм). Вихід: 3,91г. (e) 1-ацетил-6-аміно-7-метокси-2,2,4триметил-1,2-дигідрохінолін Піперидин (8,0мл) додають до розчину сполуки Прикладу 15d (3,91г) у СН 2СІ2 (80мл). Через 1,5год., реакційну суміш концентрують in vacuo I бажану сполуку отримують хроматографією на силікагелі з елюентом гептан/етил ацетат=9/1=>7/3 (об'єм/об'єм). Вихід: 2,2г. (f) (1-ацетил-7-метокси-2.2.4-триметил-1,2дигідрохінолін-6-іл)-амід біфеніл-4-карбонової кислоти Загальна процедура F: До суміші сполуки Прикладу 15е (2,2г), толуолу (45мл) і піридину (5мл) додають 4-біфенілкарбонілхлорид (2,21г). Якщо реакція не завершується через 3год. при кімнатній температурі, додають ще 4-біфеніл карбоніл хлориду (2,0г). Перемішують ще 30 хвил., після чого реакційну суміш концентрують in vacuo. Залишок вносять в етилацетат (прибл. 100мл) і промивають насиченим водним NaHCO3 (100мл), 1М водною НСІ (3x100мл) і розсолом (100мл). Органічний шар сушать (MgSO 4) і концентрують in vacuo. До залишку додають СН 2СІ2 (прибл. 50мл), тверду речовину видаляють фільтруванням і викидають. Фільтрат концентрують in vacuo і бажану сполуку отримують хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>1/1 (об'єм/об'єм). Вихід: 3,1г. (g) (1-ацетил-7-гідрокси-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-амід біфеніл4-карбонової кислоти Загальна процедура G: До розчину сполуки Прикладу 15f (3,1г) у бензолі (100мл) додають трихлорид алюмінію (5,6г) і реакційну суміш перемішують 20год. при кімнатній температурі. 29 81442 Реакцію гасять Н 2О (прибл. 100мл) і рН суміші коригують до рН 8 доданням 2М водного NaOH з енергійним перемішуванням. Додають етилацетат (прибл. 300мл) і органічний шар промивають Н 2О (2x150мл) і розсолом (1x150мл), сушать (MgSO4) і концентрують in vacuo, одержуючи продукт, який використовують без очищення. Вихід:3,5г. (h) [1-ацетил-2,2,4-триметил-7-(2-молфолін-4ілетокси)-4-феніл-1,2,3,4-тетрагідрохінолін-6-іл]амід біфеніл-4-карбонової кислоти Загальна процедура Н: суміш сполуки Прикладу 15г (70мг), N-(2-хлоретил)морфолінгідрохлорид (31мг), Cs 2CO3 і ДМФ (3мл) перемішують при 50°С, до зникнення вихідного матеріалу. Реакційну суміш розбавляють етилацетатом (15мл) і додають воду (прибл. 15мл). Органічний шар промивають водою (3x15мл), відокремлюють, сушать (MgSO4), фільтрують і концентрують in vacuo. Бажану сполуку отримують як відповідну сіль НСІ ліофілізацієюз суміші 1,4-діоксану і водовмісної НСІ. Вихід: 63мг (сіль НСІ); MS-ESI: [М+Н]+=618,6; РХВР: Rt=19,49хвил. (метод 4). Приклад 16 (1-ацетил-7-диметилкарбамоїлметокси-2,2,4триметил-4-феніл-1,2,3,4-тетрагідрохінолін-6-іл)амід біфеніл-4-карбонової кислоти Згідно з загальною процедурою Н, сполуку Прикладу 15g (79мг) алкілують 2-хлор-Ν,Νдиметилацетамідом (23мг) і Cs 2CO3 (255мг) у ДМФ (2мл). Бажану сполуку очищають кристалізацією з CH3CN. Вихід: 15мг; MS-ESI: [М+Н]+ -590,6; РХВР: Rt=23,58хвил. (метод 5). Приклад 17 [1-ацетил-2.2.4-триметил-4-феніл-7-(3піперидин-1-іл-пропокси)-1,2,3,4-тетрагідрохінолін6-іл]-амід біфеніл-4-карбонової кислоти Згідно з загальною процедурою Н, сполуку Прикладу 15g (79мг) алкілують N-(3хлорпропіл)піперидингідрохлоридом (37,4мг) і Cs 2CO3 (255мг) у ДМФ (2мл). Бажану сполуку очищають кристалізацією CH3CN. Вихід: 83мг (сіль НСІ); MS-ESI: [М+Н]+=630,8; HPLG: R t=15,49хвил. (метод 5). Приклад 18 [1-ацетил-2,2,4-триметил-4-феніл-7-(піридин2-ілметокси)-1,2,3,4-тетрагідрохінолін-6-іл]-амід біфеніл-4-карбонової кислоти Згідно з загальною процедурою Н, сполуку Прикладу 15g (79мг) алкілують 2піколілхлоридгідрохлоридом (31мг) і Cs 2CO3 (255мг) у ДМФ (2мл). Бажану сполуку очи щають кристалізацією з CH3CN. Вихід: 32мг (сіль НСІ); MS-ESI: [М+Н]+=596,6; РХВР: Rt=22,41хвил. (метод 6). Приклад 19 [1-ацетил-2,2,4-триметил-4-феніл-7-(піридин3-ілметокси)-1,2,3,4-тетрагідрохінолін-6-іл]-амід біфеніл-4-карбонової кислоти Згідно з загальною процедурою Н, сполуку Прикладу 15g (79мг) алкілують 3піколілхлоридгідрохлоридом (31мг) і Cs 2CO3 30 (255мг) у ДМФ (2мл). Бажану сполуку очи щають кристалізацією з CH3CN. Вихід: 36мг (сіль НСІ); MS-ESI: [М+Н]+=596,6; РХВР: Rt=19,70хвил. (метод 6). Приклад 20 [1-ацетил-2,2,4-триметил-4-феніл-7-(піридин4-ілметокси)-1,2,3,4-тетрагідрохінолін-6-іл]-амід біфеніл-4-карбонової кислоти Згідно з загальною процедурою Н, сполуку Прикладу 15g (79мг) алкілують 4піколілхлоридгідрохлоридом (31мг) і Cs 2CO3 (255мг) у ДМФ (2мл). Бажану сполуку очи щають кристалізацією з CH3CN. Вихід: 31мг (сіль НСІ); MS-ESI: [М+Н]+=596,4; РХВР: Rt=17,09хвил. (метод 6). Приклад 21 [1-ацетил-7-(2-димвтиламіноетокси)-2,2,4триметил-4-феніл-1,2,3,4-тетрагідрохінолін-6-іл]амід біфеніл-4-карбонової кислоти Згідно з загальною процедурою Н, сполуку Прикладу 15g (79мг) алкілують гідрохлоридом 2диметиламіноетилхлориду (27мг) і Cs 2CO3 (255мг) у ДМФ (2мл). Бажану сполуку очищають кристалізацією з CH3CN. Вихід: 55мг (сіль НСІ); MS-ESI: [М+Н]+=576,6; РХВР: Rt=14,94хвил. (метод 5). Приклад 22 (1-ацетил-7-карбамоїлметокси-2,2,4-триметил4-феніл-1,2,3,4-тетрагідрохінолін-6-іл)-амід біфеніл-4-карбонової кислоти Згідно з загальною процедурою Н, сполуку Прикладу 15g (79мг) алкілують 2-хлорацетамідом (18мг) і Cs 2CO3 (255мг) у ДМФ (2мл). Бажану сполуку очищають препаративною РХВР (метод А). Вихід: 60,2мг; MS-ESI: [М+Н]+=562,4; РХВР: Rt=20,47хвил. (метод 5). Приклад 23 (3-{1-ацетил-6-[(біфеніл-4-карбоніламіно]2,2,4-триметил-4-феніл-1,2,3,4-тетрагідрохінолін7-ілокси}-пропіл)-амід морфолін-4-карбонової кислоти Згідно з загальною процедурою Н, сполуку, описану у Прикладі 15g (79мг) алкілують (3хлорпропіл)амідом морфолін-4-карбонової кислоти (40мг) і Cs 2CO3 (255мг) у ДМФ (2мл). Бажану сполуку очи щають препаративною РХВР (метод А). Вихід: 52,4мг; MS-ESI: [M+B]+=675,6; РХВР: Rt=22,31хвил. (метод 5). Приклад 24 2-ацетил-6-(3,5-дибромбензоїламіно)-2,2,4триметил-4-феніл-1,2,3,4-тетрагідрохінолін-7іловий естер фуран-2-карбонової кислоти (а) N-(1-ацетил-7-метокси-2,2,4-триметил-1,2дигідрохінолін-6-iл)-3,5-дибромбензамід Згідно з загальною процедурою F, сполуку Прикладу 15е (1,0г) ацилують 3,5дибромобензоїлхлоридом (1,72г) у толуолі (9мл) і піридині (1мл). Бажану сполуку отримують хроматографією на силікагелі з елюентом гептан/етил ацетат=8/2 (об'єм/об'єм). Вихід: 1,3г. 31 81442 (b) N-(1-aцeтил-7-гiдpoкcи-2,2,4-тpимeтил-4фeнiл-1,2,3,4-тeтpaгiдpoxiнoлiн-6-iл)-3,5дибромбензамід Згідно з загальною процедурою G, сполуку Прикладу 24а (1,3г) перемішують з АІСІ3 (1,0г) у бензолі (50мл). Продукт використовують без очищення. Вихід: 1,39г. (с) 1-ацетил-6-(3,5-дибромбензоїламіно)-2,2,4триметил-4-феніл-1,2,3,4-тетрагідрохінолін-7-іл естер фуран-2-карбонової кислоти Суміш сполуки Прикладу 24b (100мг), фуроїлхлориду (16мкл), ДІПЕА (60мкл) і СН 2СІ2 (5мл) перемішують при кімнатній температурі до зникнення вихідного матеріалу. Додають воду і органічний шар відокремлюють, промивають розсолом, сушать (MgSO3) і концентрують in vacuo. Бажану сполуку очищають препаративною РХВР (метод А). Вихід: 47мг; MS-ESI: [М+Н]+=681,2; РХВР: Rt=31,6мм (метод 2). Приклад 25 N-[1-ацетил-7-(2-аміноетокси)-2,2,4-триметил4-феніл-1,2,3,4-тетрагідрохінолін-6-іл]-3,5дибромбензамід Загальна процедура І: суміш сполуки Прикладу 24b (100мг), трет-бутил-N-(2гідроксиетил)карбамату (29мг), ДтБАК (79мг), ДІПЕА (60мкл) і надлишок полімерно зв'язаного трифенілфосфіну у СН2СІ 2 (5мл) перемішують при кімнатній температурі до зникнення вихідного матеріалу. Реакційну суміш фільтрують і промивають водою і розсолом. Органічний шар відокремлюють, сушать (MgSO 4) і концентрують in vacuo. Сирий продукт вносять CH3CN (прибл. 1мл) і додають декілька крапель ТФК для сприяння відщепленню трет-бутилкарбамату. Бажану сполуку очищають препаративною РХВР (метод А). Вихід: 17мг (сіль ТФК); MS-ESI: [М+Н]+=630,2; РХВР: Rt=15,6хвил. (метод 2). Приклад 26 Трет-бутиловий естер {2-[1-ацетил-6-(3,5диметилбензоїламіно)-2,2,4-триметил-4-феніл1,2,3,4-тетрагідрохінолін-7-ілокси]-етил}карбамової кислоти (a) N-(1-ацетил-7-метокси-2,2,4-триметил-1,2дигідрохінолін-6-іл)-3,5-диметилбензамід Згідно з загальною процедурою F, сполуку Прикладу 15е (1,0г) ацилують 3,5диметилбензоїлхлоридом (0,97г) у толуолі (9мл) і піридині (1мл). Бажану сполуку отримують хроматографією на силікагелі з елюентом гептан/етил ацетат=8/2 об'єм/об'єм). Вихід: 1,1г. (b) N-(1-ацетил-7-гідрокси-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-3,5диметилбензамід Згідно з загальною процедурою G, сполуку Прикладу 26а (1,1г) перемішують з АІСІ3 (1,0г) у бензолі (50мл). Продукт використовують без очищення. Вихід: 1,3г. (c) Трет-бутиловий естер {2-[1-ацетил-6-(3.5диметилбензоїламіно)-2,2,4-триметил-4-феніл 32 1,2,3,4-тетрагідрохінолін-7-ілокси]-етил}карбамової кислоти Згідно з загальною процедурою І, сполук у Прикладу 26b (100мг) алкілують трет-бутил-N-(2гідроксиетил)карбаматом (37мг), ДтБАК (101мг), ДІПЕА (77мкл) і надлишком полімерно зв'язаного трифенілфосфіну у СН2СІ2 (5мл). У цьому випадку трет-бутилкарбамат не відщеплюють і одержують бажаний продукт після препаративної РХВР (метод А) і ліофілізації. Вихід: 38мг; MS-ESI: [М+Н]+=600,4; РХВР: Rt=33,1хвил. (метод 2). Приклад 27 N-[1-ацетил-7-(фуран-2-ілметокси)-2,2,4триметил-4-феніл-1,2,3,4-тетрагідрохінолін-6-іл]3,5-диметилбензамід Згідно з загальною процедурою І, сполук у Прикладу 26b (100мг) алкілують 2(гідроксиметил)фураном (21мкл), ДтБАК (101мг), ДІПЕА (77мкл) і надлишком полімерно зв'язаного трифенілфосфіну у СН2СІ 2 (5мл). Бажаний продукт очищають препаративною РХВР (метод А), з подальшою ліофілізацією. Вихід: 16мг; MS-ESI: [М+Н]+=537,4; РХВР: Rt=32,8хвил. (метод 2). Приклад 28 N-[1-ацетил-2,2,4-триметил-4-феніл-7(піридин-4-ілметокси)-1,2,3,4-тетрагідрохінолін-6іл)-3,5-дихлорбензамід (a) N-(1-ацетил-7-метокси-2,2.4-триметил-1,2дигідрохінолін-6-іл)-3,5-дихлорбензамід Згідно з загальною процедурою F, сполуку Прикладу 15е (1,0г) ацилують 3,5дихлорбензоїлхлоридом (1,2г) у толуолі (9мл) і піридині (1мл). Бажану сполуку отримують хроматографією на силікагелі з елюентом гептан/етил ацетат=8/2 (об'єм/об'єм). Вихід: 1,47г. (b) N-(1-ацетил-7-гідрокси-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-3,5дихлорбензамід Згідно з загальною процедурою G, сполуку Прикладу 28а (1,47г) перемішують з АІСІ3 (1,5г) у бензолі (75мл). Продукт використовують без очищення. Вихід: 1,51г. (c) Ν-[1-ацетил-2,2,4-триметил-4-феніл-7(піридин4-ілметокси)-1,2,3,4-тетрагідрохінолін-6іл]-3,5-дихлорбензамід Згідно з загальною процедурою Н, сполуку Прикладу 28b (100мг) алкілують 4піколілхлоридгідрохлоридом (36мг) і Cs 2CO3 (255мг) у суміші ДМФ (1мл) і СН2СІ2 (4мл). Бажану сполуку отримують як відповідну сіль ТФК після препаративної РХВР (метод А), з подальшою ліофілізацією. Вихід: 35мг (сіль ТФК); MS-ESI: [M+H]+=588,4; РХВР: Rt=18,0хвил. (метод 2). Приклад 29 Ν-[1-ацетил-2,2,4-триметил-4-феніл-7-(2піролідин-1-ілетокси)-1,2,3,4-тетрагідрохінолін-6іл]-3,5-диметилбензамід Загальна процедура J: суміш сполуки Прикладу 26b (100мг), 2хлоретилпіролідингідрохлориду (41мг) і ДІПЕА 33 81442 (77мкл) у СН2СІ2 (5мл) перемішують при кімнатній температурі до зникнення вихідного матеріалу. Додають воду і органічний шар відокремлюють, промивають розсолом, суша ть (MgSO 4) і концентрують in vacuo. Очищення препаративною РХВР з подальшою ліофілізацією дає бажану сполуку як відповідну сіль ТФК. Вихід: 104мг (сіль ТФК); MS-ESI: [М+Н]+=554,4; РХВР: Rt=15,2хвил. (метод 2). Приклад 30 N-[1-ацетил-2,2,4-триметил-7-(5метилізоксазол-3-ілметокси)-4-феніл-1,2,3,4тетрагідрохінолін-6-іл]-3,5-диметилбензамід Згідно з загальною процедурою J, сполуку Прикладу 26b (100мг) алкілують (хлорметил)-5метилізооксазолом (32мг) і ДІПЕА (77мкл) у СН3СІ2 (5мл). Очищення препаративною РХВР (метод А) з подальшою ліофілізацією дає бажану сполуку. Вихід: 41мг; MS-ESI: [М+Н]+=552,4; РХВР: Rt=31,3хвил. (метод 2). Приклад 31 N-[1-ацетил-7-(2-диметиламiноетокси)-2,2,4триметил-4-феніл-1,2,3,4-тетрагідрохінолін-6-іл]3,5-диметилбензамід: сполука з ТФК Згідно з загальною процедурою J, сполуку Прикладу 26b (100мг) алкілують гідрохлоридом Ν,Ν-діетиламіноетилхлориду (42мг) і ДІПЕА (77мкл) у СН2СІ2 (5мл). Очищення препаративною РХВР (метод А) з подальшою ліофілізацією дає бажану сполуку як відповідну сіль ТФК. Вихід: 43мг (сіль ТФК); MS-ESI: [M+H]+=556,4; РХВР: Rt=15,2хвил. (метод 2). Приклад 32 N-[1-ацетил-2,2,4-триметил-4-феніл-7(піридин-4-ілметокси)-1,2,3,4-тетрагідрохінолін-6іл]-5-бром-2-метиламінобензамід (а) N-(1-ацетил-7-метокси-2,2,4-триметил-4феніл-7-(піридин-ілметокси)-1,2,3,4тетрагідрохінолін-6-іл)-5-бром-2метиламінобензамід Згідно з загальною процедурою F, сполуку Прикладу 15е (1,0г) ацилують 5-бром-2метиламінобензоїлхлоридом (1,43г) у толуолі (9мл) і піридині (1мл). Бажану сполуку отримують хроматографією на силікагелі з елюентом гептан/етил ацетат=8/2 (об'єм/об'єм). Вихід: 595мг (b) N-(1-ацетил-7-гідрокси-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-5-бром-2метиламінобензамід Згідно з загальною процедурою G, сполуку Прикладу 32а (595мг) перемішують з АІСІ 3 (0,75г) у бензолі (50мл). Продукт використовують без очищення. Вихід: 437мг (с) N-[1-ацетил-2,2,4-триметил-4-феніл-7(піридин-4-ілметокси)-1,2,3,4-тетрагідрохінолін-6іл]-5-бром-2-метиламінобензамід Згідно з загальною процедурою Н, сполуку Прикладу 32b (44мг) алкілують 4піколілхлоридгідрохлоридом (15мг) і Cs 2CO3 (прибл. 100мг) у суміші ДМФ (1мл) і СН 2СІ2 (4мл). Бажану сполуку отримують як відповідну сіль ТФК після препаративної РХВР (метод В). 34 Вихід: 18мг (сіль ТФК): MS-ESI: [М+Н]+=629,4; РХВР: Rt=18,1хвил. (метод 2). Приклад 33 (1-ацетил-7-диметиламіно-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-амід фуран-2карбонової кислоти (а) трет-бутиловий естер (2,2,4-триметил-1,2дигідрохінолін-6-іл)-карбамової кислоти Суміш N-Вос-1,4-фенілендіаміну (75г), MgSO 4 (216г), 4-трет-бутилкатехолу (1,8г) і йоду (4,7г) у безводному ацетону (600мл) витримують під зворотним холодильником 20год. MgSO4 видаляють фільтруванням і фільтрат концентрують in vacuo. Залишок хроматографують на короткій пробці силікагелю з елюентом гептан/етил ацетат=8/2 (об'єм/об'єм) і отримують продукт у вигляді коричневого масла. Вихід: 41г. (b) Трет-бутиловий естер (1-ацетил-2,2,4триметил-1,2-дигідрохінолін-6-іл)-карбамової кислоти Розчин сполуки Прикладу 33а (41г) у піридині (200мл) і СН 22СІ2 (200мл) охолоджують до 0°С. Краплями додають ацетилхлорид (21мл) у СН 2СІ2 (50мл), після чого суміш перемішують 3 год. при кімнатній температурі. Додають етилацетат (2л) і Н2О (2л) і органічний шар відокремлюють, сушать (MgSO4) і концентрують in vacuo. Бажану сполуку отримують кристалізацією з етилацетату. Вихід: 23г. (c) 1-ацетил-6-аміно-2,2,4-триметил-4-феніл1,2,3,4-тетрагідрохінолін Згідно з загальною процедурою G, сполуку Прикладу 33b (33,3г) перемішують з АІСІ3 (40,4г) у бензолі (700мл). Продукт очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=8/2 (об'єм/об'єм). Вихід: 22,4г. (d) 9-Флуорєнілмєтиловий естер (1-ацетил2,2,4-триметил-4-феніл-1,2,3,4-тетрагідрохінолін6-іл)-карбамової кислоти Піридин (6,4мл) додають до розчину сполуки Прикладу 33с (22,4г) у ТГФ (300мл) і одержану суміш охолоджують до 0°С. Додають краплями розчин FmocCI (20,7г) у ТГФ (100мл) і після цього суміш перемішують 1год. при кімнатній температурі. Реакційну суміш концентрують in vacuo і додають етилацетат (800мл) і 0,3М водну НСІ (500мл). Органічний шар відокремлюють і промивають 0,3М НСІ (2x500мл), Н2О (500мл) і розсолом (500мл), з подальшим сушінням (MgSO4) і концентруванням in vacuo. Продукт використовують без очищення. Вихід: 43г. (e) 9-Флуоренілетиловий естер (1-ацетил2,2,4-триметил-7-нітро-4-феніл-1,2,3,4тетрагідрохінолін-6-іл)-карбамової кислоти Паруючу азотну кислоту (3,07мл) додають краплями протягом 10хвил. до суміші сполуки Прикладу 33d (43г) і оцтової кислоти (230мл) у СН2СІ2 (230мл). Реакційну суміш перемішують до повного перетворення вихідного матеріалу, після чого додають Н 2О (150мл). Водний шар відокремлюють і екстрагують СН 2СІ2 (150мл). Об'єднані органічні шари промивають насиченим 35 81442 водним NaHCO3 (3x200мл) і розсолом (200мл), сушать над MgSO4) і фільтрують. Додають метанол (прибл. 200мл) і обережно видаляють СН2СІ2 in vacuo, після чого суміш відстоюють при кімнатній температурі протягом ночі. Яскравожовті кристали збирають фільтруванням і сушать (MgSO 4) in vacuo. Вихід: 29,3г. (f) 9-Флуоренілетиловий естер (1-ацетил-7аміно-2,2,4-триметил-4-феніл-1,2,3,4тетрагідрохінолін-6-іл)-карбамової кислоти Згідно з загальною процедурою Е, сполуку Прикладу 33е (20г) відновлюють, використовуючи цинковий порошок (45г) і оцтову кислоту (20мл) у ТГФ (прибл. 600мл) і отримують продукт, який використовують без очищення. Вихід: 21г. (g) 9-флуоренілметиловий естер (1-ацетил-7диметиламіно-2,2,4-триметил-4-феніл-1,2,3,4тетрагідрохінолін-6-іл)-карбамової кислоти Водний розчин формальдегіду (37%, 3,8мл) додають до розчину сполуки Прикладу 33f (12г), оцтової кислоти (15,7мл) і ціаноборгідриду натрію (2,9г) у метанолі (200мл), що викликає екзотермічну реакцію і утворення білого осаду. Додають МеОН для підсилення перемішування. Після перемішування протягом 15хвил.,осад збирають фільтруванням і промивають МеОН/Н 2О=1 /1 (об'єм/об'єм). Фільтрат частково концентрують і одержують тверду речовину, яку також збирають. Об'єднані тверді речовини рекристалізують з СН 2СІ2/МеОН і отримують диметиловану сполуку. Вихід: 9,7г. (h) 1 -ацетил-6-аміно-7-диметиламіно-2,2,4триметил-4-феніл-1,2,3,4-тетрагідрохінолін Піперидин (7,7мл) додають до розчину сполуки Прикладу 33g (4,5г) у СН2СІ2 (70мл). Через 24год. реакційну суміш розбавляють СН 2СІ2 (100мл) і промивають 0,5 Μ водною НСІ (2x150мл), водою (100мл) і розсолом (1мл). Органічний шар сушать (MgSO 4) і розбавляють до об'єму 200мл. Цей розчин бажаної сполуки (прибл. 13,8мг/мл) використовують у подальших реакціях. (і) (1-ацетил-7-диметиламіно-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-амід фуран-2карбонової кислоти Триетиламін (38мкл) і 2-фуроїлхлорид (27мкл) додають до розчину сполуки Прикладу 33h (96,6мг) у СН2СІ2 (10мл) і отриману суміш перемішують до повного перетворення вихідного матеріалу. Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм) Вихід: 5,5мг. MS-ESI: [М+Н]+=446,2; РХВР: Rt=19,02хвил. (метод 2). Приклад 34 (1-ацетил-7-диметиламіно-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-амід 5метилтіофен-2-карбонової кислоти Згідно з загальною процедурою А, сполук у Прикладу 33h (96,6мг) ацилують 5-метилтіофен-2карбоновою кислотою (39,1мг), ГФТМ (157мг) і ДІПЕА (239мкл) у СН 2СІ2 (10мл). Бажану сполуку очищають препаративною РХВР (метод В). 36 Вихід: 35,5мг; MS-ESI: [М+Н]+=476,2; РХВР: Rt=21,26хвил. (метод 2). Приклад 35 (1-ацетил-7-диметиламіно-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-амід біфеніл4-карбонової кислоти Згідно з загальною процедурою А, сполук у Прикладу 33h (96,6мг) ацилують 4біфенілкарбоновою кислотою (54,4мг), ГФТМ (157мг) і ДІПЕА (239мкл) у СН 2СІ2 (10мл). Бажану сполуку очищають препаративною РХВР (метод В). Вихід: 31,5мг; MS-ESI: [M+H]+=532,4; РХВР: Rt=24,92хвил. (метод 2). Приклад 36 N-(1-ацетил-7-диметиламiно-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-3,5дибромбензамід Згідно з загальною процедурою А, сполук у Прикладу 33h (96,6мг) ацилують 3,5дибромобензойною кислотою (77мг), ГФТМ (157мг) і ДІПЕА (239мкл) у СН 2СІ2 (10мл). Бажану сполуку очищають кристалізацією з СН 2СІ2/CH3CN. Вихід: 24,3мг; MS-ESI: [M+H]+=614,2; РХВР: Rt=27,71хвил. (метод 2). Приклад 37 (1-ацетил-7-диметиламіно-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-амід циклопентанкарбонової кислоти Згідно з загальною процедурою А, сполук у Прикладу 33h (137мкл) ацилують циклопентанкарбоновою кислотою (128мкл), ГФТМ (224мг) і ДІПЕА (400мкл) у СН 2СІ2 (10мл). Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм). Вихід: 148мг; MS-ESI: [М+Н]+=448,4; РХВР: Rt=12,93хвил. (метод 1). Приклад 38 N-(1-ацетил-7-диметиламіно-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-зобутирамід Згідно з загальною процедурою А, сполук у Прикладу 33h (137мг) ацилують ізобутировою кислотою (110мкл), ГФТМ (224мг) і ДІПЕА (400мкл) СН2СІ2 (10мл). Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм). Вихід: 43мг; MS-ESI: [М+Н]+=422,4; РХВР: Rt=9,99хвил. (метод 1). Приклад 39 (1-ацетил-7-фуран-2-ілкарбоніламіно-2,2,4триметил-4-феніл-1,2,3,4-тетрагідрохінолін-6-іл)амід фуран-2-карбонової кислоти До розчину сполуки Прикладу 33f (150мг) і триетиламіну (43мкл) у СН2СІ2 (1мл) додають 2фуроїлхлорид (30мкл). Після повного поглинання вихідного матеріалу, додають 1М водну НСІ, органічний шар відокремлюють, додають піперидин (1мл) і суміш перемішують протягом ночі. Реакційну суміш промивають 1М водною НСІ, органічний шар відокремлюють, сушать (MgSO 4) і концентрують in vacuo. Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=1/0=>0/1 (об'єм/об'єм), і потім препаративною РХВР (метод А). 37 81442 Вихід: 18мг; MS-ESI: [М+Н]+=512,4; РХВР: Rt=19,92хвил. (метод 2). Приклад 40 (1-ацетил-2,2,4-триметил-4-феніл-7пропіламіно-1,2,3,4-тетрагідрохінолін-6-іл)-амід 5метилтіофен-2-карбонової кислоти (a) 1-ацетил-6-аміно-2,2,4-триметил-4-феніл-7пропіламіно-1,2,3,4-тетрагідрохінолін Загальна процедура K: До суміші сполуки Прикладу 33f (750мг), оцтової кислоти (953мкл), ціаноборгідриду натрію (135мг) і МеОН (10мл) додають пропіональдегід (94,2мкл). Суміш перемішують 18год., додають воду і осад збирають фільтруванням. Осад вносять у СН2СІ2 (10мл), додають піперидин (1мл) і суміш перемішують 18год., потім промивають 1М водною НСІ, органічний шар відокремлюють і розбавляють до об'єму 50мл. Цей розчин використовують у подальших реакціях. (b) (1-ацетил-2,2,4-триметил-4-феніл-7пропіламіно-1,2,3,4-тетрагідрохінолін-6-іл)-амід 5метил-тіофен-2-карбонової кислоти Згідно з загальною процедурою А, сполук у Прикладу 40а (100мг) ацилують 5-метилтіофен-2карбоновою кислотою (39,1мг), ГФТМ (157мг) і ДІПЕА (239мкл у СН2СІ2 (10мл). Бажану сполуку очищають препаративною РХВР і ліофілізують. Вихід: 18,3мг; MS-ESI: [М+Н]+=490,4; РХВР: Rt=23,96хвил. (метод 2). Приклад 41 (1-ацетил-7-етиламіно-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-амід біфеніл4-карбонової кислоти (a) 1-ацетил-6-аміно-7-етиламіно-2,2,4триметил-4-феніл-1,2,3,4-тетрагідрохінолін Згідно з загальною процедурою K, сполуку Прикладу 33f (750мг) алкілують ацетальдегідом (73,3мкл), видаляють захист піперидином (1мл) і після проходження реакції отримують розбавлений розчин бажаної сполуки у СН 2СІ2. (b) (1-ацетил-7-етиламіно-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-амід біфеніл4-карбонової кислоти Згідно з загальною процедурою А, сполук у Прикладу 41а (100мг) ацилують 4біфенілкарбоновою кислотою (54,4мг), ГФТМ (157мг) і ДІПЕА (239мкл) у СН 2СІ2 (10мл). Бажану сполуку очищають препаративною РХВР і ліофілізують. Вихід: 9,8мг; MS-ESI: [М+Н]+=532,4; РХВР: Rt=25,55хвил. (метод 2). Приклад 42 {1-ацетил-2,2,4-триметил-4-феніл-7-[(піридин4-ілметил)аміно]-1,2,3,4-тетрагідрохінолін-6-іл}амід 5-метил-тіоФен-2-карбонової кислоти (а) 1-ацетил-6-аміно-2.2.4-триметиилфеніл-7[(піридин-4-ілметил)-аміно]-1,2,3,4тетрагідрохінолін Згідно з загальною процедурою К, сполуку Прикладу 33f (750мг) алкілують 4піридинкарбоксальдегідом (125мкл) (b {1-ацетил-2,2,4-триметил-4-феніл-7[(піридин-4-ілметил)-аміно]-1,2,3,4тетрагідрохінолін-6-іл}-амід 5-метилтіоФен-2карбонової кислоти 38 Згідно з загальною процедурою А, сполук у Прикладу 42а (114мг) ацилують 5-метилтіофен-2карбоновою кислотою (39,1мг), ГФТМ (157мг) і ДІПЕА (239мкл) у СН2СІ2 (10мл). Бажану сполуку очищають препаративною РХВР і ліофілізують. Вихід: 51мг; MS-ESI: [М+Н]+=539,4; РХВР: Rt=13,19хвил. (метод 2). Приклад 43 (1-ацетил-2,2,4-триметил-4-феніл-7-[(піридин3-ілметил)-аміно]-1,2,3,4-тетрагідрохінолін-6-іл}амід 5-метилтіоФен-2-карбонової кислоти (а) 1-ацетил-6-аміно-2,2,4-триметил-4-феніл-7[(піридин-3-ілметил)-аміно]-1,2,3,4тетрагідрохінолін Згідно з загальною процедурою K, сполуку Прикладу 33f (750мг) алкілують 3піридинкарбоксальдегідом (125мкл), видаляють захист піперидином (1мл) і після проходження реакції отримують розбавлений розчин бажаної сполуки у СН 2СІ2. (b) {1-ацетил-2,2,4-триметил-4-феніл-7[(піридин-3-ілметил)-аміно]-1,2,3,4тетрагідрохінолін-6-іл}-амід 5-метилтіоФен-2карбонової кислоти Згідно з загальною процедурою А, сполук у Прикладу 43 (114мг) ацилують 5-метилтіофен-2карбоновою кислотою (39,1мг), ГФТМ (157мг) і ДІПЕА (239мкл) у СН2СІ2 (10мл). Бажану сполуку очищають препаративною РХВР і ліофілізують. Вихід: 44мг; MS-ESI: [М+Н]+=539,4; РХВР: Rt=13,45хвил. (метод 2). Приклад 44 N-(1-ацетил-7-ізобутиламіно-2,2,4триметилфеніл-1,2,3,4-тетрагідрохінолін-6-іл)-3,5дибромбензамід (a) 1-ацетил-6-аміно-7-ізобутиламіно-2,2,4триметил-4-феніл-1,2,3,4-тетрагідрохінолін Згідно з загальною процедурою K, сполуку Прикладу 33f (750мг) алкілують ізобутиральдегідом (119мкл), видаляють захист піперидином (1мл) і після проходження реакції отримують розбавлений розчин бажаної сполуки у СН 2СІ2. (b) N-(1-ацетил-7-ізобутиламіно-2,2,4триметил-4-феніл-1,2,3,4-тетрагідрохінолін-6-іл)3,5-дибромбензамід Згідно з загальною процедурою А, сполук у Прикладу 44а (114мг) ацилують 3,5дибромобензойною кислотою (77мг), ГФТМ (157мг) і ДІПЕА (239мкл) у СН 2СІ2 (10мл). Бажану сполуку очищають препаративною РХВР і ліофілізують. Вихід: 54мг; MS-ESI: [М+Н]+=642,4; РХВР: Rt=29,47хвил. (метод 2). Приклад 45 (1-ацетил-7-бензиламіно-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-амід біфеніл4-карбонової кислоти (a) 1-ацетил-6-аміно-7-бензиламіно-2,2,4триметил-4-феніл-1,2,3,4-тетрагідрохінолін Згідно з загальною процедурою K, сполуку Прикладу 33f (750мг) алкілують бензальдегідом (133мкл) і видаляють захист піперидином (1мл) і після проходження реакції отримують розбавлений розчин бажаної сполуки у СН 2СІ2. 39 81442 (b) (1-ацетил-7-бензиламіно-2,2,4-триметил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-амід біфеніл4-карбонової кислоти Згідно з загальною процедурою А, сполук у Прикладу 45а (113мг) ацилують 4біфенілкарбоновою кислотою (54,4мг), ГФТМ (157мг) і ДІПЕА (239мкл) СН2СІ2 (10мл). Бажану сполуку очищають препаративною РХВР і ліофілізують. Вихід: 114мг: MS-ESI: [M+H]+=594,4; РХВР: Rt=26,46хвил. (метод 2). Приклад 46 (1-ацетил-7-бензиламіно-2,2,4-триметил-4феніл-1,2,3,4-)-амід 5-метилтіофен-2-карбонової кислоти Згідно з загальною процедурою А, сполук у Прикладу 45а (113мг) ацилують 5-метилтіофен-2карбоновою кислотою (39,1мг), ГФТМ (157мг) і ДІПЕА (239мкл) у СН 2СІ2 (10мл). Бажану сполуку очищають препаративною РХВР і ліофілізують. Вихід: 107мг; MS-ESI: [М+Н]+=538,4; РХВР: Rt=18,59хвил. (метод 2). Приклад 47 N-{1-ацетил-2,2,4-триметил-4-феніл-7[(піридин-3-ілметил)-аміно]-1,2,3,4тетрагідрохінолін-6-іл}-3,5-дибромбензамід Згідно з загальною процедурою А, сполук у Прикладу 43а (114мг) ацилують 3,5дибромобензойною кислотою (77мг), ГФТМ (157мг) і ДІПЕА (239мкл) у СН 2СІ2 (10мл). Бажану сполуку очищають препаративною РХВР і ліофілізують. Вихід: 41мг; MS-ESI: [М+Н]+=677,2; РХВР: Rt=14,88хвил. (метод 2). Приклад 48 N-(1-ацетил-7-диметиламiно-4-метил-4-феніл1,2,3.4-тетрагідрохінолін-6-іл)-3,5дибромобензамід (а) 4-метил-1,2-дигідрохінолін Розчин лепідину (10,0г) ТГФ охолоджують до 78°С і додають розчин ВН 3-ТГФ у ТГФ (1 М, 70мл). Через 2год., додають розчин біс(2метоксиетокси)алюмодигідриду натрію у толуолі (3,5М, 40мл) і реакційну суміш перемішують ще 2год. Додають воду і суміш розбавляють етилацетатом. Органічний шар відокремлюють, сушать (MgSO4) і частково концентрують in vacuo для кристалізації бажаної сполуки. Кристали збирають фільтруванням і після сушіння in vacuo отримують 3,5г. Залишок маточної рідини концентрують in vacuo і залишок очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=0/1=>1/0 ((об'єм/об'єм), отримуючи ще 4,4г бажаної сполуки. Вихід: 7,9г. b) 1-ацетил-4-метил-1,2-дигідрохінолін Згідно з загальною процедурою С, сполуку Прикладу 48а (7,9г) ацилують ацетилхлоридом (11,8мл) і ДМА (34мл) у СН 2СІ2 (50мл) при 0°С. Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=6/4. Вихід: 8,8г. (с). 1-ацетил-4-метил-4-феніл-1,2,3,4тетрагідрохінолін 40 Згідно з загальною процедурою G, сполуку Прикладу 48b (8,8г) перемішують з АІСІ3 (18,8г) у бензолі (250мл). Продукт використовують без очищення. Вихід: 12,0г. (d) 1-ацетил-4-метил-6-нітро-4-феніл-1,2,3,4тетрагідрохінолін До розчину сполуки Прикладу 48с (5,0г) і оцтового ангідриду (189мкл) у СН 2СІ2 (50мл) додають краплями паруючу HNO3 (9,4мл). Після завершення реакції додають воду і органічний шар промивають розсолом, відділяють і концентрують in vacuo. Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=6/4 (об'єм/об'єм). Вихід: 3,86г. (e) 1 -ацетил-6-аміно-4-метил-4-феніл-1,2,3,4тетрагідрохінолін Згідно з загальною процедурою Е, сполуку Прикладу 48d (3,86г) відновлюють, використовуючи цинковий порошок (16г) і оцтову кислоту (7мл) у ТГФ (прибл. 250мл) і отримують продукт, який використовують сирим. Вихід: 2,2г. (f) 9-Флуоренілметиловий естер (1-ацетил-4метил-4-феніл-1,2,3,4-тетрагідрохінолін-6-іл)карбамової кислоти Піридин (314мкл) додають до розчину сполуки Прикладу 48е (1,0г) у ТГФ (10мл) і суміш охолоджують до 0°С. Додають FmocCI (1,01г) і суміш перемішують 18год. при кімнатній температурі, після чого реакційну суміш концентрують in vacuo. Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=6/4 (об'єм/об'єм). Вихід: 950мг. (q) 9-Флуоренілметиловий естер (1-ацетил-4метил-7-нітро-4-феніл-1,2,3,4-тетрагідрохінолін-6іл)-карбамової кислоти До розчину сполуки Прикладу 48f (850мг) і оцтового ангідриду (17мкл) у СН 2СІ2 (5мл) додають краплями паруючу HNO3 (842мкл). Після завершення реакції додають воду і органічний шар промивають розсолом, відокремлюють і концентрують in vacuo. Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=9/1=>0/1 (об'єм/об'єм). Вихід: 714мг. (h) 9-Флуоренілметиловий естер (1-ацетил-7аміно-4-метил-4-феніл-1,2,3,4-тетрагідрохінолін-6іл)-карбамової кислоти Згідно з загальною процедурою Е, сполуку Прикладу 48g (2,37г) відновлюють, використовуючи цинковий порошок (5,6г) і оцтову кислоту (2,4мл) у ТГФ (прибл. 50мл), і отримують продукт, який використовують сирим. Вихід: 2,66г. (і) 9-Флуорєнілмєтиловий естер (1-ацетил-7диметиламіно-4-метил-4-феніл-1,2,3,4тетрагідрохінолін-6-іл)-карбамової кислоти Водний розчин формальдегіду (37%, 650мкл) додають до розчину сполуки Прикладу 48h (2,66г), оцтової кислоти (3,1мл) і ціаноборгідриду натрію (232мг) у метанолі (50мл), і суміш перемішують 18год. Реакційну суміш концентрують in vacuo, 41 81442 залишок вносять в етилацетат і промивають водою і розсолом. Органічний шар відокремлюють, сушать (MgSO4) і концентрують in vacuo. Бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=9/1=>0/1 (об'єм/об'єм). Вихід: 1,0г. (і) 1-ацетил-6-аміно-7-диметиламіно-4-метил4-феніл-1,2,3,4-тетрагідрохінолін Піперидин (1,8мл) додають до розчину сполуки Прикладу 48і (1г) у СН 2СІ2 (20мл) і суміш перемішують до зникнення вихідного матеріалу. Реакційну суміш концентрують in vacuo, і бажану сполуку очищають хроматографією на силікагелі з елюентом гептан/етил ацетат=9/1=>0/1 (об'єм/об'єм). Вихід: 370мг. (k) N-(1-ацетил-7-диметиламіно-4-метил-4феніл-1,2,3,4-тетрагідрохінолін-6-іл)-3,5дибромбензамід Згідно з загальною процедурою А, сполук у Прикладу 48j (74мг) ацилують 3,5дибромобензойною кислотою (70мг), ГФТМ (131мг) і ДІПЕА (120мкл) у СН 2СІ2 (3мл). Бажану сполуку очищають препаративною РХВР і ліофілізують. Вихід: 82мг: MS-ESI: [М+Н]+=586,2; РХВР: Rt=22,40хвил. (метод 2). Приклад 49 (1-ацетил-7-диметиламіно-4-метил-4-феніл1,2,3,4-тетрагідрохінолін-6-іл)-амід 5-бромтіофен2-карбонової кислоти Згідно з загальною процедурою А, сполук у Прикладу 48j (74мг) ацилують 5-бромтіофен-2карбоновою кислотою (52мг), ГФТМ (131мг) і ДІПЕА (120мкл) у СН 2СІ2 (3мл). Бажану сполуку очищають препаративною РХВР і ліофілізують. Вихід: 69мг; MS-ESI: [М+Н]+=514,2; РХВР: Rt=17,01хвил. (метод 2). Приклад 50 (1-ацетил-7-диметиламіно-4-метил-4-феніл1,2,3,4-тетрагідрохінолін-6-іл)-амід 5-хлортіофен2-карбонової кислоти Згідно з загальною процедурою А, сполук у Прикладу 48j (74мг) ацилують 5-хлортіофен-2карбоновою кислотою (52мг), ГФТМ (131мг) і ДІПЕА (120мкл) у СН 2СІ2 (3мл). Бажану сполуку очищають препаративною РХВР і ліофілізують. Вихід: 81мг; MS-ESI: [M+H]+=468,2; РХВР: Rt=17,49хвил. (метод 2). Приклад 51 Біоактивність CHQ-FSH in vitro Активність сполук до FSH була випробувана у клітинах яєчника китайського хом'яка (СНО), стабільно трансфекованих рецептором FSH людини і співтрансфекованих чутливим до сАМР елементом (CRE)/пpoмотером, що спрямовує експресію гена-репортера світлякової люциферази. Зв'язування ліганда з з'єднаним Gs рецептором FSH підвищує сАМР, що у свою чергу індукує трансактивацію конструкції репортера люциферази. Для виявлення антагоністичних властивостей додавали (10mU/мл, rec-hFSH) рекомбінантний FSH у концентрації, що забезпечує приблизно 80% максимальної 42 стимуляції акумуляції сАМР за відсутності сполуки, яку випробували. Сигнал люциферази квантували за допомогою люмінесцентного лічильника. Були обчислені значення ІК50 для сполуки, яку випробували (концентрацію сполуки, яка викликала половину максимальної стимуляції або зниження). Для цього була використана комп'ютерна програма GraphPad PRISM, version 3,0 (GraphPad software Inc., San Diego). Сполуки всіх прикладі показали ІК50 менше 105 М як у агоністичному, так і антагоністичному аналізі, або в обох. Сполуки прикладів 5-8, 10-14, 16, 18-20, 33-35, 37, 38, 41 і 45-50 показали ІК50 менше 10-7М у щонайменше одному аналізі.
ДивитисяДодаткова інформація
Назва патенту англійськоюTetrahydroquinoline derivatives
Автори англійськоюTimmers Cornelis Marius
Назва патенту російськоюПроизводные тетрагидрохинолина
Автори російськоюТиммерс Корнелис Мариус
МПК / Мітки
МПК: C07D 401/12, C07D 413/12, C07D 215/38, C07D 405/12, A61P 5/08, C07D 409/12, C07D 409/14
Мітки: похідні, тетрагідрохіноліну
Код посилання
<a href="https://ua.patents.su/21-81442-pokhidni-tetragidrokhinolinu.html" target="_blank" rel="follow" title="База патентів України">Похідні тетрагідрохіноліну</a>
Похідні тетрагідрохіноліну, фармацевтична композиція на їх основі та їх використання

Номер патенту: 80303
Опубліковано: 10.09.2007
Автори: Карстенс Віллем Фредерік Йоган, Тіммерс Корнеліс Маріус
МПК: C07D 409/12, A61P 5/08, C07D 401/12, C07D 215/38
Мітки: похідні, фармацевтична, використання, основі, тетрагідрохіноліну, композиція
Формула / Реферат:
1. Похідна тетрагідрохіноліну формули І (I)або її фармацевтично прийнятна сіль, в якійR1, R2 є H, Me;R3 - (2-6С)гетероциклоалкіл(1-4С)алкіл, (2-5С)гетероарил(1-4С)алкіл, (6С)арил(1-4С)-алкіл, (1-4С)(ді)алкіламінокарбоніламіно(2-4С)алкіл, (2-6С)гетероциклоалкілкарбоніламіно(2-4С)алкіл, R5-(2-4C)алкіл або R5-кapбoнiл(1-4C)aлкiл;R4 -...
Похідні 3-деоксидесмікозину та спосіб їх одержання

Номер патенту: 66927
Опубліковано: 15.06.2004
Автори: Павловіч Дражен, Дєрек Марко, НАРАНДЬЯ Амалія, ЛОПОТАР Невенка
МПК: A61K 31/7048, C07H 17/08, A61P 31/00, A61P 31/04
Мітки: похідні, одержання, спосіб, 3-деоксидесмікозину
Формула / Реферат:
1. Похідні 3-деоксі-3-оксодесмікозину формули І, (I)деR являє собою СНО або СН(ОСН3)2, R1 і R2 означають Н або ацетил, R3 являє собою Н або ОН, R4 являє собою N(CH3)2 або N-O(СН3)2, лінія - - - являє собою одинарний або подвійний зв'язок, лінія ....... являє собою або подвійний чи одинарний зв'язок, і лініяявляє собою подвійний чи одинарний зв'язок, і похідні 3-деокси-2,3-дидегідродесмікозину формули II,...
Похідні морфоліну, спосіб їх одержання та фармацевтичні препарати, що містять вказані похідні

Номер патенту: 73931
Опубліковано: 17.10.2005
Автори: ЕМОН-АЛЬТ Ксав'є, Проєтто Вінченцо, Гейоль Патрік, Дюку Жан Філіп
МПК: A61P 25/10, A61P 17/00, A61P 1/08, C07D 413/06, A61P 37/00, A61P 13/00, A61P 25/00, A61P 11/06, A61P 25/22, A61P 25/24, A61P 1/04, A61P 13/02, C07D 265/30, A61P 1/00, A61P 25/06, A61P 11/00, A61K 31/5377, A61P 29/00, A61P 29/02, A61P 9/00
Мітки: спосіб, фармацевтичні, одержання, вказані, морфоліну, похідні, містять, препарати
Формула / Реферат:
1. Сполука формули (I): (I),в якій Аr являє собою феніл, монозаміщений або дизаміщений атомом галогену; (С1-С3)алкіл;Χ являє собою групу R2-N=; групу R2-CH=;R1 являє собою атом хлору, атом брому, (С1-С3)алкіл або трифлуорметил;R2 являє собою (С1-С6)алкіл; (С3-С6)циклоалкіл; групу –CR4R5CONR6R7;R3 являє собою групу...
Похідні 2,3-бензодіазепіну

Номер патенту: 72261
Опубліковано: 15.02.2005
Автори: Гіглер Габор, Блажко Габор, Мартонне Марко Бернадетт, Сабо Геза, Раткаі Зольтан, Едьєд Андраш, ШІМІГ Дьюла, Шімо Аннамарія, Тіханьі Карой, Леваі Дьєрдь, Грефф Зольтан, БАРКОЦІ Йожеф
МПК: A61P 25/22, A61P 1/08, A61P 25/00, A61P 25/30, A61P 25/18, A61P 25/16, A61P 25/08, C07D 491/04, A61P 25/04, A61P 25/06, A61P 25/28, C07D 491/056, A61K 31/551, A61P 21/00, A61P 13/00
Мітки: 2,3-бензодіазепіну, похідні
Формула / Реферат:
1. Сполуки загальної формули, ІдеR1 означає метил, форміл, карбокси, ціано, -CH=NOH, -CH=NNHCONH2 або -NR5R6, деR5 і R6 незалежно один від одного являють собою водень або нижчий алкіл, або утворюють разом з атомом азоту, до якого вони приєднані, 5- або 6-членне, насичене або ненасичене гетероциклічне кільце, котре необов'язково містить один або більше додаткових атомів азоту, сірки і/або кисню;R2 -нітро або...
Спосіб одержання 4-карбоксіаміно-2-заміщеного-1,2,3,4-тетрагідрохіноліну

Номер патенту: 65615
Опубліковано: 15.04.2004
Автори: Деймон Девід Бернс, Даггер Роберт Вейн
МПК: A61K 31/4706, C07B 61/00, C07D 215/42, A61P 3/06, A61P 9/10
Мітки: спосіб, одержання, 4-карбоксіаміно-2-заміщеного-1,2,3,4-тетрагідрохіноліну
Формула / Реферат:
1. 4-толуолсульфонат етилового естеру 4-(3,5-біс-трифторметилбензиламіно)-2-етил-6-трифторметил-3,4-дигідро-2Н-хінолін-1-карбонової кислоти.2. Стереоізомер етилового естеру (–)-(2R,4S)-4-(3,5-біс-трифторметилбензиламіно)-2-етил-6-трифторметил-3,4-дигідро-2Н-хінолін-1-карбонової кислоти або його солі.3. Стереоізомер за п. 2, який відрізняється тим, що сіль являє собою 4-толуолсульфонат.4. Сіль (–)-ди-п-толуїл-L-винної...
Попередній патент: Пристрій для відкривання та закривання кришок розвантажувальних люків бункерного вагона
Наступний патент: Система змащення підшипникової опори ковзання
Випадковий патент: Спосіб визначення кількості життєздатних яєць у яйцекладці