М’які капсули, що містять палоносетрону гідрохлорид, які мають поліпшену стабільність і біологічну доступність
Номер патенту: 97653
Опубліковано: 12.03.2012
Автори: БОНАДЕО Даніеле, Бралья Енріко, Кальдерарі Джорджо, Бралья Ріккардо







Формула / Реферат
1. М'яка желатинова капсула для перорального введення, що містить:
a) м'яку желатинову зовнішню оболонку, яка має проникність для кисню, меншу ніж приблизно 1,0х10-3 мл∙см/(см2∙24 год.∙атм); і
b) ліпофільну рідку композицію внутрішньої начинки, що містить:
і) більше ніж приблизно 50 % мас. одного або декількох ліпофільних компонентів;
(іі) від приблизно 1 до приблизно 20 % мас. води, перемішаної або гомогенізованої у вказаних одному або декількох ліпофільних компонентах;
(ііі) від приблизно 0,05 до приблизно 2,0 мг палоносетрону у вигляді палоносетрону гідрохлориду, солюбілізованого або диспергованого у вказаній воді; і
iv) поверхнево-активну речовину,
де вказана капсула при пероральному прийомі натще демонструє фармакокінетику, яка біологічно еквівалентна препарату, що має більш ніж 95 % абсолютну біологічну доступність, причому біологічна еквівалентність встановлюється за допомогою 90 % довірчого інтервалу для AUC, який знаходиться у межах між 80 і 125 %.
2. Капсула за п. 1, в якій вказана композиція внутрішньої начинки містить:
a) від 0,5 до 1,0 мг палоносетрону у вигляді палоносетрону гідрохлориду; і
b) солюбілізуючу ефективну кількість рідини, що містить ліпофільний наповнювач і воду.
3. М'яка желатинова капсула за п. 1, що містить гліцерин у вказаній зовнішній оболонці і вказану композицію внутрішньої начинки.
4. М'яка желатинова капсула за п. 1, в якій:
a) вказана композиція внутрішньої начинки додатково містить антиоксидант або відновлювальний агент;
b) вказаний палоносетрон містить менше ніж приблизно 1 % мас. (3S)-3-[(3aS)-l-оксо-2,3,3а,4,5,6-гексагідро-1Н-бензо[dе]ізохінолін-2-іл]-1-азоніабіцикло[2,2,2]октан-1-олату.
5. Капсула за п. 1, що демонструє фармакокінетику, при пероральному прийомі натще, яка є біологічно еквівалентною препарату, що має більш ніж 95 % абсолютну біологічну доступність, і Сmax від 800 до 820 нг/л, де біологічна еквівалентність встановлюється за допомогою:
a) 90 % довірчого інтервалу для AUC, який знаходиться у межах між 80 і 125 %, і
b) 90 % довірчого інтервалу для Сmax, який знаходиться у межах між 80 і 125 %.
6. Капсула за п. 1, в якій вказана композиція внутрішньої начинки містить кисень у кількості, яка опосередковує не більше ніж приблизно 3,0 % мас. окиснювальної деградації, коли вказана дозована форма зберігається протягом трьох місяців при 40 °С і 75 % RH.
7. Капсула за п. 1, в якій не менше ніж приблизно 75 % вказаного палоносетрону або його фармацевтично прийнятної солі розчиняється через 45 хвилин, коли досліджується у пристрої для розчинення типу II з лопатевою мішалкою відповідно до Фармакопеї США, при 75 об/хв і 37 °С, в 500 мл 0,01 н. НСl.
8. Капсула за п. 1, в якій не менше ніж приблизно 75 % вказаного палоносетрону або його фармацевтично прийнятної солі розчиняється через 30 хвилин, коли досліджується у пристрої для розчинення типу II з лопатевою мішалкою відповідно до Фармакопеї США, при 75 об/хв і 37 °С, в 500 мл 0,01 н. НСl.
9. Капсула за п. 1, в якій вказана оболонка має проникність для кисню, меншу ніж приблизно 1,0х10-4 мл∙см/(см2∙24 год.∙атм).
10. Капсула за п. 1, в якій вказана внутрішня начинка містить від 0,5 до 4 % мас. поверхнево-активної речовини.
11. Дозована форма у вигляді м'якої капсули, заповненої рідиною, для перорального введення, що містить:
a) зовнішню оболонку, що має проникність для кисню, меншу ніж приблизно 1,0х10-3 мл∙см/(см2∙24 год.∙атм); і
b) композицію внутрішньої начинки, що містить від приблизно 0,05 до приблизно 2,0 мг палоносетрону або його фармацевтично прийнятної солі, де вказаний палоносетрон або його фармацевтично прийнятна сіль містить (3S)-3-[(3aS)-l-оксо-2,3,3а,4,5,6-гексагідро-1Н-бензо[dе]ізохінолін-2-іл]-1-азоніабіцикло[2,2,2]октан-1-олат або його фармацевтично прийнятну сіль у кількості, меншій ніж 1,0 % мас. відносно маси вказаного палоносетрону,
де вказана композиція внутрішньої начинки містить кисень у кількості, яка опосередковує не більше ніж приблизно 3,0 % мас. окиснювальної деградації вказаного палоносетрону або його фармацевтично прийнятної солі, коли вказана дозована форма зберігається три місяці або більше при 40 °С і 75 % RH.
12. Капсула за п. 11, яка демонструє фармакокінетику при пероральному прийомі натще, яка є біологічно еквівалентною препарату, що має більш ніж 90 % абсолютну біологічну доступність, де біологічна еквівалентність встановлюється за допомогою 90 % довірчого інтервалу для AUC, який знаходиться у межах між 80 і 125 %.
13. Капсула за п. 11, в якій вказана композиція внутрішньої начинки містить від приблизно 1 до приблизно 20 % мас. води.
14. Капсула за п. 11, що демонструє фармакокінетику при пероральному прийомі натще, яка є біологічно еквівалентною препарату, що має більш ніж 95 % абсолютну біологічну доступність, і Сmax від 800 до 820 нг/л, де біологічна еквівалентність встановлюється за допомогою:
a) 90 % довірчого інтервалу для AUC, який знаходиться у межах між 80 і 125 %, і
b) 90 % довірчого інтервалу для Сmax, який знаходиться у межах між 80 і 125 %.
15. Капсула за п. 11, в якій не менше ніж приблизно 75 % вказаного палоносетрону або його фармацевтично прийнятної солі розчиняється через 45 хвилин, коли досліджується у пристрої для розчинення типу II з лопатевою мішалкою відповідно до Фармакопеї США, при 75 об/хв і 37 °С, у 500 мл 0,01 н. НСl.
16. Капсула за п. 11, в якій вказана оболонка має проникність для кисню, меншу ніж приблизно 1,0х10-4 мл∙см/(см2∙24 год.∙атм).
17. Капсула за п. 11, в якій:
a) вказана композиція внутрішньої начинки містить гліцерин; і
b) вказана оболонка містить гліцерин.
18. Капсула за п. 11, в якій вказана оболонка містить желатин, целюлозу, крохмаль або НРМС.
19. Спосіб оптимізації біологічної доступності і стабільності палоносетрону у желатиновій капсулі, що містить палоносетрон, що включає в себе:
a) створення м'якої желатинової зовнішньої оболонки, що має проникність для кисню, меншу ніж приблизно 1,0x10-3 мл∙см/(см2∙24 год.∙атм); і
b) одержання композиції начинки за допомогою стадій, що включають в себе:
і) одержання від приблизно 0,05 до приблизно 2,0 мг палоносетрону у вигляді палоносетрону гідрохлориду, де вказаний палоносетрон містить (3S)-3-[(3aS)-l-оксо-2,3,3а,4,5,6-гексагідро-1Н-бензо[dе]ізохінолін-2-іл]-1-азоніабіцикло[2,2,2]октан-1-олат у кількості, меншій ніж 3,0 % мас.;
іі) розчинення або диспергування вказаного палоносетрону у воді з утворенням водного преміксу;
ііі) змішування вказаного водного преміксу з одним або декількома ліпофільними наповнювачами, при масовому співвідношенні водного преміксу та ліпофільних наповнювачів, меншому ніж 30:70, з утворенням перемішаної або гомогенної композиції ліпофільної начинки;
iv) змішування поверхнево-активної речовини з вказаною водою, вказаним водним преміксом або вказаною композицією начинки; і
v) балансування кількостей поверхнево-активної речовини і води у вказаній композиції начинки для полегшення біологічної доступності палоносетрону з вказаної желатинової капсули при пероральному прийомі і для зведення до мінімуму ступеня деградації палоносетрону; і
c) заповнення вказаної зовнішньої оболонки вказаною композицією начинки.
20. Спосіб за п. 19, в якому вказана композиція начинки містить від приблизно 0,1 до приблизно 10,0 % мас. поверхнево-активної речовини і від приблизно 0,1 до приблизно 20 % мас. води.
21. Спосіб за п. 19, в якому вказана композиція начинки містить від приблизно 0,5 до приблизно 4 % мас. поверхнево-активної речовини і від приблизно 1 до приблизно 10 % мас. води.
22. Спосіб за п. 19, в якому вказана зовнішня оболонка додатково містить гліцерин, що додатково включає в себе змішування вказаного водного преміксу з гліцерином, до або після утворення вказаної композиції ліпофільної начинки.
23. Спосіб одержання желатинової капсули за будь-яким з пп. 1-18, яка містить палоносетрон, що має знижену кількість домішок і продуктів деградації, опосередкованих киснем, що включає в себе:
а) змішування палоносетрону гідрохлориду і одного або декількох фармацевтично прийнятних наповнювачів з утворенням суміші;
b) переробку вказаної суміші з одержанням желатинової капсули; і
c) дослідження однієї або декількох вказаних желатинових капсул на одну або декілька сполук, споріднених з палоносетроном, вибраних з (3S)-3-[(3aS)-l-оксо-2,3,3а,4,5,6-гексагідро-1Н-бензо[dе]ізохінолін-2-іл]-1-азоніабіцикло[2,2,2]октан-1-олату, 2-[(3S)-1-азабіцикло[2,2,2]окт-3-ил]-2,4,5,6-тетрагідро-1Н-бензо[dе]ізохінолін-1-ону гідрохлориду і (3aR)-2-[(S)-l-азабіцикло[2,2,2]окт-3-ил]-2,3,3a,4,5,6-гексагідро-1-оксо-1Н-бенз[dе]ізохіноліну гідрохлориду або їх гідрохлоридної солі.
24. Спосіб за п. 23, що включає в себе дослідження на (3S)-3-[(3aS)-l-оксо-2,3,3а,4,5,6-гексагідро-1Н-бензо[dе]ізохінолін-2-іл]-1-азоніабіцикло[2,2,2]октан-1-олат або його гідрохлоридну сіль.
25. Спосіб за п. 23, що включає в себе дослідження на 2-[(3S)-1-азабіцикло[2,2,2]окт-3-ил]-2,4,5,6-тетрагідро-1Н-бензо[dе]ізохінолін-1-ону гідрохлорид або його гідрохлоридну сіль.
26. Спосіб за п. 23, що включає в себе дослідження на (3aR)-2-[(S)-l-азабіцикло[2,2,2]окт-3-ил]-2,3,3a,4,5,6-гексагідро-1-оксо-1Н-бенз[dе]ізохіноліну гідрохлорид або його гідрохлоридну сіль.
27. Спосіб за п. 23, що додатково включає в себе дослідження вказаного палоносетрону гідрохлориду або вказаної кінцевої дозованої форми на одну або декілька сполук, вибраних з (3S)-3-(1-oкco-2,4,5,6-тетpaгiдpo-1Н-бeнзo[de]-iзoxiнoлiн-2-іл)-1-азоніабіцикло[2,2,2]октан-1-олату, (3аR)-2-[(R)-1-азабіцикло[2,2,2]окт-3-ил]-2,3,3a,4,5,6-гeкcaгiдpo-1-oкco-1H-бенз[de]iзoxiнoлiну гідрохлориду, (3aS)-2-[(R)-1-aзaбiциклo[2,2,2]oкт-3-ил]-2,3,3a,4,5,6-гекcaгiдpо-1-oкco-1H-бeнз[de]iзoxiнoлiну гідрохлориду або (3аS)-2-[(S)-1-азабіцикло[2,2,2]окт-3-ил]-2,3,3а,4,5,6-гексагідро-1-oкco-1H-бeнз[de]iзoxiнoлiну гідрохлориду або їх гідрохлоридної солі.
Текст
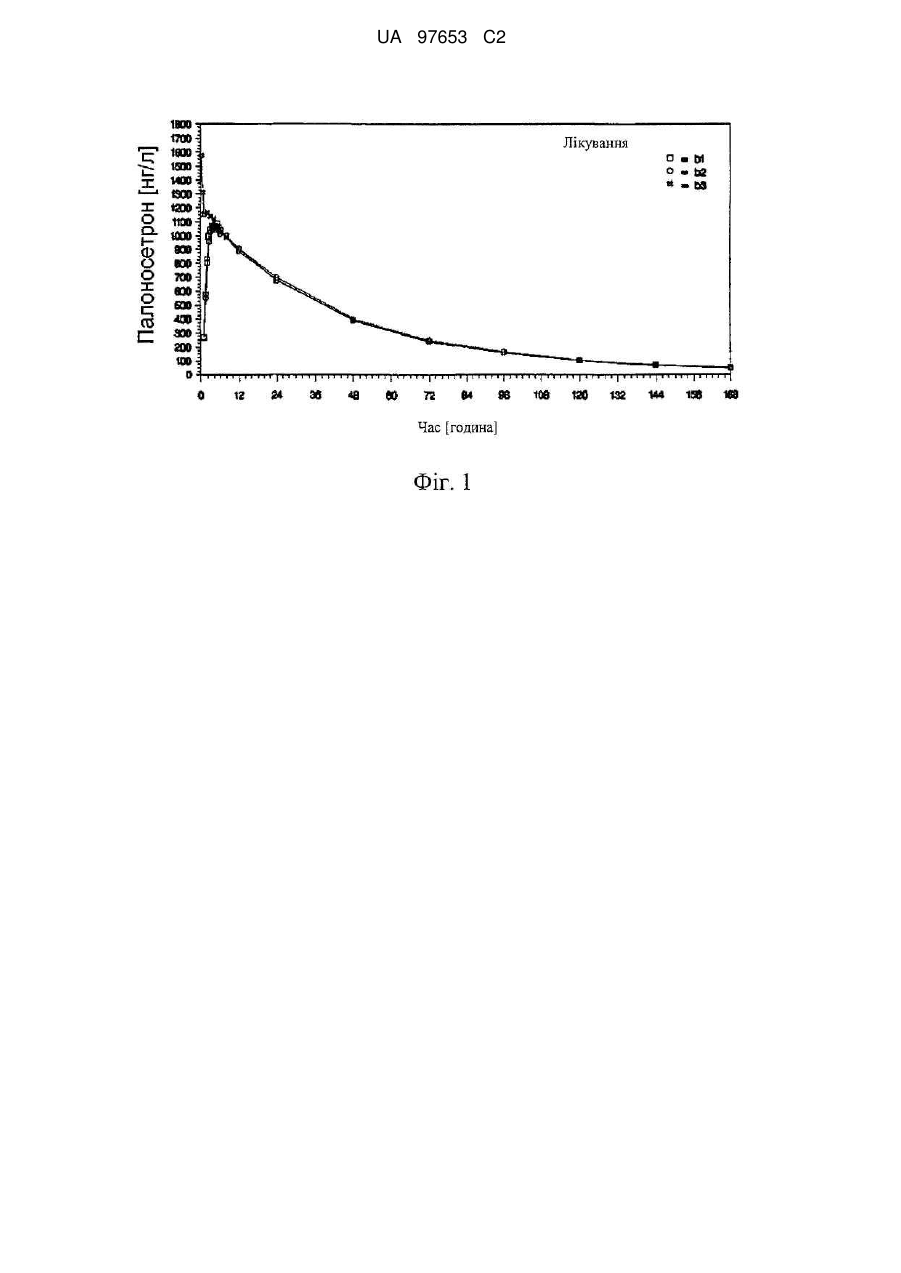
Реферат: Винахід належить до галузі фармацевтики і стосується м’яких желатинових капсул, дозованих форм для перорального введення та способу їх одержання, які містять палоносетрон гідрохлорид. Винахід також стосується способу оптимізації біологічної доступності та стабільності палоносетрону у желатиновій капсулі. UA 97653 C2 (12) UA 97653 C2 UA 97653 C2 5 10 15 20 25 30 35 40 45 50 Дана заявка заявляє про пріоритет попередньої заявки на патент Сполучених Штатів № 60/854342, зареєстрованої 24 жовтня 2006 року (термін дії закінчився). Даний винахід стосується палоносетрону і, зокрема, твердих пероральних дозованих форм палоносетрону гідрохлориду, які відповідають строгим вимогам стабільності при зберіганні в упакованому вигляді. Побічні впливи нудоти і блювання при протираковій хіміотерапії і радіаційній терапії являють собою широко поширену і довготривалу проблему. Можливо менш відомими, але не менш важливими, є післяопераційна нудота і блювання, які можуть мати фізіологічні механізми, споріднені з впливами, що спостерігаються при хіміотерапії. Палоносетрону гідрохлорид одержаний нещодавно як дуже ефективний засіб проти нудоти і проти блювання для використання разом з еметогенною протираковою хіміотерапією. (Macciocchi, А., et al., "А Phase II dose-ranging study to assesses single intravenous doses of palonosetron for the prevention of highly emetogenic chemotherapy-induced nausea and vomiting", Proc. Am. Soc. Clin. Oncol., 2002; Abstract 1480). Палоносетрон також запобігає післяопераційним нудоті і блюванню. (Chelly, J., et al., "Oral RS-25259 prevents postoperative nausea and vomiting following laparoscopic surgery", Anesthesiol., 85(Suppl. 21): abstract no.3 (1996)). Способи лікування нудоти і блювання, що викликаються хіміотерапією, (СINV) та нудоти і блювання, що викликаються радіацією, (RINV) за допомогою палоносетрону описуються у публікації PCT заявки на Міжнародний патент WO 2004/045615 від Helsinn Healthcare SA. Способи лікування післяопераційної нудоти і блювання (PONV) за допомогою палоносетрону описані у публікації PCT 2004/073714, також від Helsinn Healthcare SA. Палоносетрон є селективним, показує високу спорідненість як антагоніст з попередником рецептора 5-гідроксилтриптаміну 3 (рецептор 5-HT3) і показує низьку спорідненість з іншими рецепторами, такими як допамінові рецептори (Wong, E.H.F., et al., "The interaction of RS 25259197, а potent and selective antagonist, with 5-HT3 receptors, in vitro", Br. J. Pharmacol, 114:851-859 (1995); Eglen, R.M., et al, "Pharmacological characterization of RS 25259-197, а potent and selective antagonist, with 5-HT3 receptors, in vivo", Br. J. Pharmacol, 114:860-866 (1995)). Палоносетрон являє собою синтетичну сполуку, що існує як єдиний ізомер і вводиться як гідрохлоридна сіль, як представлено у наведеній нижче структурній формулі: Офіційне хімічне найменування для лікарського засобу являє собою (3aS)-2-[(S)-1азабіцикло[2,2,2]окт-3-ил]-2,3,3a, 4,5,6-гексагідро-1-оксо-1H-бенз[de]ізохіноліну гідрохлорид (CAS № 119904-90-4); його емпірична формула являє собою C19H24N2O·HCl, і його молекулярна маса дорівнює 332,87. Способи синтезу сполуки описані у патентах США №№ 5202333 і 5510486. Палоносетрону гідрохлорид продається у Сполучених Штатах як стерильна рідина для ін'єкцій, як ALOXI®, MGI Pharma і Helsinn Healthcare SA. Рідина для внутрішньовенних ін'єкцій є прозорою, безбарвною, непірогенною, знаходиться в ізотонічному, буферному розчині. Стабільний ізотонічний розчин палоносетрону для ін'єкцій описується у публікації PCT заявки на Міжнародний патент WO 2004/067005, Helsinn. Незважаючи на численні клінічні вигоди і переваги цього препарату для внутрішньовенних ін'єкцій, як правило, спостерігається, що системи доставки лікарських засобів за допомогою ін'єкцій доставляють конкретні проблеми стосовно часу життя при зберіганні і стабільності активного агента при зберіганні. Вони також є незручними, коли вводяться самостійно, і мають підвищений ризик забруднення і людської помилки. Таким чином, можливість пероральної доставки для палоносетрону, особливо у твердій формі, була б особливо привабливою. Способи поліпшення стабільності і часу зберігання в упаковці для препаратів палоносетрону також були б бажаними. Розробляються м'які гелеві капсули для палоносетрону, які демонструють чудову біологічну доступність при пероральному прийомі і стабільність, коли зберігаються протягом тривалих періодів часу. Зовнішня оболонка капсули базується на желатині і внутрішня начинка капсули являє собою суцільну ліпофільну внутрішню фазу, яка містить палоносетрон, розчинений у водному компоненті, що перемішується або гомогенізується у ліпофільній фазі за допомогою 1 UA 97653 C2 5 10 15 20 25 30 35 40 45 50 55 60 мінімальних кількостей поверхнево-активної речовини. Препарат представляє елегантне рішення для усунення несумісності, що спостерігається скрізь, між: - водною начинкою і стабільністю желатину; - деградацією поверхнево-активної речовини і палоносетрону; і - стабільністю палоносетрону і концентрацією палоносетрону. З цієї причини, у першому головному варіанті здійснення, даний винахід передбачає м'яку желатинову капсулу для перорального введення, що містить: (a) зовнішню оболонку з м'якого -3 2 желатину, яка має проникність для кисню, меншу, ніж приблизно 1,0×10 мл·см/(см ·24 год.·атм); і (b) композицію внутрішньої начинки з ліпофільної рідини, що містить: (i) більше, ніж приблизно 50 % мас. одного або декількох ліпофільних компонентів; (ii) від приблизно 1 до приблизно 20 % мас. води, перемішаної або гомогенізованої у вказаному одному або декількох ліпофільних компонентах; (iii) від приблизно 0,05 до приблизно 2,0 мг палоносетрону у вигляді палоносетрону гідрохлориду, солюбілізованого або диспергованого у вказаній воді; і (iv) від приблизно 0,5 до приблизно 5 % мас. поверхнево-активної речовини. Розробляються також препарати і способи одержання, які можуть визначатися за кількістю або за концентрацією палоносетрону у дозованій формі і за побічними продуктами деградації у дозованій формі. Один з таких побічних продуктів деградації являє собою продукт деградації, опосередковуваної киснем, і згадується тут як "Cpd1". Також розробляються дозовані форми палоносетрону, включаючи способи одержання, з підвищеною стабільністю, завдяки їх захисту від кисню і опосередковуваної киснем деградації. На основі цих відкриттів і розробок, розробляються дозовані форми, які можуть визначатися за однією або декількома з наведених нижче фізичних ознак: - оболонка або покриття, які по суті непроникні для кисню; - використання рідкої начинки всередині оболонки капсули, що переважно містить воду; - мінімальний вміст кисню у рідкій начинці; - хімічні засоби для запобігання окиснювальній деградації; - стійка до вологості упаковка, яка є стійкою до проникнення кисню; і/або - використання навколишнього середовища, збідненого киснем, при одержанні дозованих форм. Ці дозовані форми мають чудову стабільність протягом тривалих періодів часу, чудову стійкість до окиснювальної деградації і чудову біологічну доступність при пероральному прийомі. Ці дозовані форми можуть використовуватися при лікуванні будь-якого захворювання, для якого палоносетрон має клінічне застосування, але переважно вони використовуються для лікування блювання. У другому головному варіанті здійснення, з цієї причини, даний винахід передбачає дозовану форму капсули для перорального введення, що містить: (a) зовнішню оболонку, яка -3 2 має проникність для кисню, меншу, ніж приблизно 1,0×10 мл·см/(см ·24 год.·атм); і (b) композицію внутрішньої начинки, що містить: від приблизно 0,05 до приблизно 2,0 мг палоносетрону, у вигляді палоносетрону гідрохлориду, де вказаний палоносетрон містить Cpd1 у кількості, меншій, ніж 1,0 % мас.; при цьому не більше, ніж 5,0 % мас. вказаного палоносетрону гідрохлориду деградує, коли вказана дозована форма зберігається три місяці або більше при 40 °C і 75 % RH (відносній вологості). Зрозуміло, даний винахід міг би здійснюватися з використанням дозованих форм, інших, ніж капсули, і інший варіант здійснення даного винаходу передбачає тверду пероральну дозовану форму, що містить: (a) зовнішню оболонку або покриття, що має проникність для кисню, меншу, -3 2 ніж приблизно 1,0×10 мл·см/(см ·24 год.·атм); і (b) композицію внутрішньої начинки, що містить: від приблизно 0,05 до приблизно 2,0 мг палоносетрону, у вигляді палоносетрону гідрохлориду, де вказаний палоносетрон містить Cpd1 у кількості, меншій, ніж 1,0 % мас.; де не більше, ніж 5,0 % мас. вказаного палоносетрону гідрохлориду деградує, коли вказана дозована форма зберігається протягом трьох місяців або більше при 40 °C і 75 % RH. Також розробляються способи одержання дозованих форм палоносетрону, які мають знижену кількість домішок і продуктів деградації, опосередковуваної киснем, і дозованих форм палоносетрону, які одержують за допомогою цих способів. Таким чином, ще в одному варіанті здійснення даний винахід передбачає спосіб одержання набору дозованих форм палоносетрону, які мають знижену кількість домішок і продуктів деградації, опосередковуваної киснем, що включає в себе (a) змішування палоносетрону гідрохлориду і одного або декількох фармацевтично прийнятних наповнювачів з утворенням суміші; (b) переробку вказаної суміші у вигляді множини кінцевих дозованих форм і (с) дослідження однієї або декількох з вказаних кінцевих дозованих форм на Cpd1. Цей спосіб може здійснюватися за допомогою будь-якої дозованої форми, включаючи капсулу, гелькап або ампулу, заповнену рідиною. 2 UA 97653 C2 5 10 15 20 25 30 35 40 45 50 55 Додаткові переваги даного винаходу будуть частково представлені в описі, який йде далі, а частково стануть очевидні з опису, або вони можуть бути вивчені при здійсненні даного винаходу. Переваги даного винаходу будуть реалізовані і одержані за допомогою елементів і поєднань, конкретно описаних у доданій формулі винаходу. Необхідно зрозуміти, що як попередній загальний опис, так і наведений далі докладний опис, є тільки ілюстративними і пояснювальними, а не такими, що обмежують даний винахід, як описано у формулі винаходу. Фігура 1 являє собою графік фармакокінетики, що спостерігається у пацієнтів людей при дослідженнях біологічної еквівалентності, де b1 являє собою лікування за допомогою клінічного препарату А, b2 являє собою лікування за допомогою комерційного препарату В і b3 являє собою лікування за допомогою Aloxi®, внутрішньовенно. Фігура 2 являє собою графік фармакокінетики, що спостерігається у пацієнтів людей при дослідженнях біологічної еквівалентності, де b1 являє собою клінічний препарат А, а b2 являє собою комерційний препарат В. Обидві фігури показують середньоарифметичні концентрації палоносетрону у плазмі (нг/мл) в залежності від часу (год.) у лінійному масштабі (n=33). Докладний опис винаходу Даний винахід може бути легше зрозумілий за допомогою посилань на наведений далі докладний опис переважних варіантів здійснення даного винаходу і прикладів, що містяться тут. Визначення і використання термінів Як використовується у даному описі та у формулі винаходу, які йдуть далі, форми однини включають в себе посилання на множину, якщо тільки контекст не диктує чітко іншого. Так, наприклад, посилання на "інгредієнт" включає в себе суміші інгредієнтів, посилання на "активний фармацевтичний агент" включає в себе більше одного активного фармацевтичного агента, і тому подібне. "Лікування" або "вилікування" захворювання включає в себе (1) запобігання виникненню захворювання у тварини, яка може бути схильна до захворювання, але ще не відчуває або не проявляє симптомів захворювання, (2) уповільнення захворювання, тобто припинення його розвитку, або (3) ослаблення захворювання, тобто здійснення регресії захворювання. Як тут використовується, безпосереднє навколишнє середовище стосується навколишнього середовища, яке безпосередньо оточує елемент або спосіб, як правило, газового середовища, з яким елемент або спосіб знаходяться у контакті і сполученні. "Блювання" для цілей даної заявки буде мати значення, яке ширше, ніж звичайне словникове визначення, і включає в себе не тільки блювання, але також нудоту і позиви до блювання. "Помірно еметогенна хіміотерапія" стосується хіміотерапії, при якій еметогенний потенціал є порівнянним або еквівалентним з еметогенним потенціалом карбоплатину, цисплатину 250 мг/м , "У вищій мірі еметогенна хіміотерапія" стосується хіміотерапії, при якій еметогенний 2 потенціал є порівнянним або еквівалентним з еметогенним потенціалом цисплатину >60 мг/м , 2 циклофосфаміду >1500 мг/м або дакарбазину. "Фармацевтично прийнятний" означає, що він є придатним для використання при одержанні фармацевтичної композиції, яка є загалом безпечною, нетоксичною і не є небажаною, ні біологічно, ні в іншому значенні, і включає в себе те, що вона є прийнятною для ветеринарного застосування, а також для фармацевтичного застосування на людях. "Терапевтично ефективна кількість" означає ту кількість, яка, коли вводиться тварині для лікування захворювання, є достатньою для здійснення такого лікування захворювання. "de minimis" кількість кисню стосується такої кількості кисню, яка дає можливість для деградації не більше, ніж приблизно 0,5, 1,0, 1,5, 2,0, 2,5 або 3,0 % мас. вказаного палоносетрону (переважно визначають за деградацією до Cpd1), коли зберігають при кімнатній температурі за умов навколишнього середовища протягом шести, дванадцяти, вісімнадцяти, двадцяти чотирьох, тридцяти або тридцяти шести місяців. Стабільність при зберіганні у упаковці, для цілей даного винаходу, вимірюється за допомогою зберігання дозованої форми в її упаковці при 40 °C, при відносній вологості 75 % або за умов навколишнього середовища протягом трьох, шести, дванадцяти, вісімнадцяти, двадцяти чотирьох, тридцяти або тридцяти шести місяців. Стабільний препарат являє собою такий, в якому не більше, ніж приблизно 0,5, 1,0, 1,5, 2,0, 2,5, 3,0, або 5,0 % мас. палоносетрону у дозованій формі деградує (переважно визначають за деградацією до одного або декількох продуктів деградації, описаних тут). 3 UA 97653 C2 5 10 15 20 25 30 35 40 45 50 55 Коли діапазони наводяться за допомогою указання нижньої межі діапазону окремо від верхньої межі діапазону, необхідно зрозуміти, що діапазон може визначатися за допомогою селективного об'єднання будь-якої зі змінних нижньої межі з будь-якою зі змінних верхньої межі, які є математично можливими. Коли тут використовується термін "приблизно" або "ca.", він буде компенсувати розкид, допустимий для фармацевтичної промисловості і властивий фармацевтичним продуктам, такий як відмінності у міцності продукту через розкид при одержанні і обумовлену часом деградацію продукту. Термін робить можливим будь-який розкид, який у практиці фармацевтичних препаратів дав би можливість для оцінки продукту як такого, що вважається біологічно еквівалентним стосовно згадуваної сили впливу продукту, що заявляється. Термін "абсолютна біологічна доступність" стосується доступності активного лікарського засобу при системній циркуляції після введення, відмінного від внутрішньовенного (тобто, після перорального, ректального, трансдермального, підшкірного введення). Для визначення абсолютної біологічної доступності лікарського засобу повинні здійснюватися фармакокінетичні дослідження для одержання графіку залежності концентрації лікарського засобу у плазмі від часу для лікарського засобу, як після внутрішньовенного (IV), так і після введення, відмінного від внутрішньовенного. Абсолютна біологічна доступність являє собою скориговану на дозу площу під кривою (AUC) для введення, відмінного від внутрішньовенного, поділену на AUC для внутрішньовенного введення. Про препарат говориться, що він є біологічно еквівалентним у термінах абсолютної біологічної доступності еталонному препарату, коли є встановлений 90 % довірчий інтервал для AUC(0-∞), що знаходиться у межах між 80 % і 125 %, по відношенню до рівня біологічної доступності для еталонного препарату. Коли тут наводяться фармакокінетичні параметри (тобто T max, абсолютна біологічна доступність і тому подібне), буде зрозуміло, що вони можуть стосуватися середньої медіанної або індивідуальної фармакокінетики, що спостерігається, і що у формулі винаходу передбачається середня фармакокінетика, якщо не стверджується протилежне. Фармакокінетичний параметр також буде розумітися як такий, що спостерігається у голодному стані, якщо не стверджується протилежне. Обговорення Як розглянуто вище, даний винахід передбачає тверді пероральні дозовані форми, які мають поліпшену стабільність і стійкість до окиснювальної деградації, що базуються на декількох технологіях приготування, включаючи використання покриття або оболонки, яка є по суті непроникною для кисню, або використання ліпофільної рідкої начинки, що має воду, гомогенізовану або перемішану з нею. У першому головному варіанті здійснення даний винахід передбачає тверду пероральну дозовану форму, що містить: (a) зовнішню оболонку або -3 2 покриття, що має проникність для кисню, меншу, ніж приблизно 1,0×10 мл·см/(см ·24 год.·атм); і (b) композицію внутрішньої начинки, що містить: від приблизно 0,05 до приблизно 2,0 мг палоносетрону у вигляді палоносетрону гідрохлориду, де вказаний палоносетрон містить Cpd1 у кількості, меншій, ніж 1,0 % мас.; де вказана дозована форма демонструє стабільність при зберіганні в упаковці, що переважно визначається так, що не більше, ніж 5,0 % мас. вказаного палоносетрону гідрохлориду деградує, коли вказана дозована форма зберігається три місяці або більше при 40 °C і 75 % RH. Даний винахід додатково передбачає спосіб лікування блювання, що включає в себе пероральне введення пацієнту, який страждає від блювання або має ризик постраждати від блювання, дозованої форми за даним винаходом. Даний винахід може здійснюватися за допомогою будь-якого типу твердої пероральної дозованої форми, що визначається як будь-яка дозована форма, яка вводиться пероральним шляхом і заковтується, включаючи, наприклад, капсулу або гелькап (тобто капсулу, заповнену рідиною). У переважному варіанті здійснення дозована форма являє собою капсулу, а ще у більш переважному варіанті здійснення дозована форма являє собою гелькап, заповнений рідиною. Що не являла б собою дозована форма, вона переважно має зовнішню оболонку або покриття, яке має мінімальну проникність для кисню. У переважних варіантах здійснення даного винаходу, покриття або оболонка має проникність для кисню, яка менша, ніж приблизно 1,0×10 3 -4 -4 -5 -5 2 , 5,0×10 , 1,0×10 , 5,0×10 або навіть 2,0×10 мл∙см/(см ∙24 год.∙атм). Переважна дозована форма для даного винаходу являє собою капсулу, що має зовнішню оболонку, яка розчиняється у шлункових рідинах. Заповнена рідиною капсула, що переважно містить воду, є особливо переважною завдяки однорідності вмісту і дози при роботі з рідинами і можливості зведення до мінімуму експонування для кисню при одержанні дозованої форми і зберіганні дозованої форми протягом тривалих періодів часу. 4 UA 97653 C2 5 10 15 20 25 30 35 40 45 50 55 60 Серед доступних зовнішніх оболонок, м'яка зовнішня оболонка являє собою переважну структуру оболонки, завдяки її здатності утримувати рідини і протистояти проходженню кисню. Переважні матеріали для зовнішньої оболонки "гелькап" включають в себе, наприклад, желатин, целюлозу, крохмаль або HPMC (гідроксипропілметилцелюлозу). У переважному варіанті здійснення оболонка містить желатин і, необов'язково, один або декілька наповнювачів для оболонки, вибраних з гліцерину, сорбітолу і двоокису титану. Рідка композиція, що заповнює капсулу, переважно є (1) ліпофільною і (2) присутньою як безперервна рідка фаза (тобто така, в якій рідкі компоненти є або перемішаними, або повністю гомогенізованими/емульсифікованими). Суцільна фаза є переважною завдяки простоті обробки і однорідності композиції. Рідка начинка містить основу наповнювача і активний агент, рівномірно розподілений у рідкій начинці. Крім того, активний агент переважно є розчиненим або диспергованим як мікроемульсія в основі наповнювача. Загальна маса композиції начинки переважно є більшою, ніж приблизно 50, 75 або 100 мг, і переважно є меншою, ніж приблизно 500, 250, 200 або 150 мг, найбільш переважно знаходиться у межах від приблизно 100 до приблизно 150 мг. Рідка начинка переважно складається в основному з одного або декількох ліпофільних компонентів у кількості від приблизно 50 % мас. до приблизно 99 % мас., переважно від приблизно 75 % мас. до приблизно 98 % мас. Переважні ліпофільні компоненти включають в себе, наприклад, моно- і дигліцериди жирних кислот, зокрема, включаючи в себе моно- і дигліцериди каприлової/капринової кислоти. Рідка начинка може також містити гліцерин, переважно у кількості від приблизно 1 до приблизно 15 % мас., більш переважно від приблизно 2 до приблизно 10 % мас. В одному з переважних варіантів здійснення, композиція, як оболонки, так і внутрішньої начинки, містить гліцерин. В іншому переважному варіанті здійснення рідка начинка містить 0,25, 0,35 мг або більше палоносетрону у вигляді палоносетрону гідрохлориду (тобто 0,50 або 0,75 мг); солюбілізованого при солюбілізуючій ефективній кількості рідини, що містить ліпофільний наповнювач і воду. Композиція начинки може містити різні засоби для полегшення переходу палоносетрону з дозованої форми у шлунково-кишкові рідини шлунково-кишкового тракту, так щоб палоносетрон міг легше поглинатися у кровотоці. Наприклад, композиція рідкої начинки може містити поверхнево-активну речовину, оптимально, у кількості від приблизно 0,1 % мас. до приблизно 6 % мас., від приблизно 0,5 % мас. до приблизно 5 % мас. або від приблизно 1,0 % мас. до приблизно 3,0 % мас. Композиція рідкої начинки переважно містить більше, ніж 0,1, 0,5 або 1,0 % мас. поверхнево-активної речовини, і менше, ніж 10, 8, 5,4, або навіть 4 % мас. поверхнево-активної речовини. Особливо переважна поверхнево-активна речовина являє собою полігліцерилолеат. Альтернативно або на додаток до цього, засоби перенесення для заповненої рідиною капсули можуть включати в себе воду, яка утворює одну фазу або мікроемульсію з усіма іншими рідкими інгредієнтами в основі наповнювача. Композиція рідкої начинки переважно містить від приблизно 0,05 % мас. до приблизно 30 % мас. води, від приблизно 1 % мас. до приблизно 20 % мас. води, або від приблизно 2 % мас. до приблизно 10 % мас. води. Рідка начинка переважно містить більше, ніж 0,1, 0,5 або 1,0 % мас. води, і менше, ніж 20, 15, 10, 8 або 5 % мас. води. Крім того, основа наповнювача може містити один або декілька хімічних агентів для запобігання опосередковуваній киснем деградації палоносетрону у дозованій формі. Наприклад, основа наповнювача може містити хелатуючий агент, такий як етилендіамінтетраоцтова кислота (EDTA), антиоксидант, такий як бутилований гідроксіанізол (BHA), або відновлювальний агент, у кількості, що знаходиться у межах від приблизно 0,005 % мас. до приблизно 2,0 % мас., більш переважно від приблизно 0,01 % мас. до приблизно 1,0 % мас. або від приблизно 0,05 % мас. до приблизно 0,5 % мас. У переважному варіанті здійснення основа наповнювача містить антиоксидант. Активний агент, який переважно являє собою палоносетрону гідрохлорид, переважно присутній у композиції начинки у кількості, що знаходиться у межах від приблизно 0,01 % мас. до приблизно 10,0 % мас., від приблизно 0,05 % мас. до приблизно 5,0 % мас. або від приблизно 0,1 % мас. до приблизно 2,0 % мас. Альтернативно, виявлені особливо стабільні препарати, де концентрація палоносетрону перевищує 0,3 % мас., переважно, він знаходиться при концентрації не більше, ніж приблизно 1 % мас. Особливо важливою особливістю композиції внутрішньої начинки, яка є переважною у будьякому з варіантів здійснення даного винаходу, незалежно від дозованої форми або типу начинки, або способу одержання, є мінімальний вміст кисню. У переважному варіанті здійснення композиція внутрішньої начинки містить кисень у такій кількості, при якій деградує не більше, ніж 5 UA 97653 C2 5 10 15 20 25 30 35 40 45 50 приблизно 3,0 % мас., 2,5 % мас., 2,0 % мас., 1,5 % мас., 1,0 % мас. або 0,5 % мас. вказаного палоносетрону, коли дозована форма зберігається у режимах дослідження стабільності при зберіганні в упаковці, наприклад, протягом трьох місяців при 40 °C і 75 % RH. Цю кількість переважно вимірюють за допомогою кількості Cpd1 у композиціях. Інша важлива ознака препаратів за даним винаходом являє собою їх фармакокінетику. Визначено, що дозовані форми за даним винаходом мають абсолютну біологічну доступність приблизно 100 %, у межах біологічної еквівалентності. Таким чином, наприклад, у той час як 0,75 мг ін'єкція палоносетрону дає середню AUC(0-∞) ca. 58285 (нг∙год./л), 0,75 мг гелькап дає середню AUC(0-∞) ca. 57403 (нг·год./л). На противагу цьому, середня C max для 0,75 мг гелькапу становить приблизно 1224 нг/л, у той час як 0,75 мг ін'єкція дає середню C max приблизно 1665 нг/л. 0,50 мг гелькап, як показано, дає середню AUC(0-∞) ca. 38176 (нг·год./л), і середню Cmax приблизно 810 нг/л, тим самим демонструючи фармакокінетику, пропорційну дозі. У різних варіантах здійснення, отже, дозована форма за даним винаходом дає більшу, ніж 90, 95 або навіть 98 % абсолютну біологічну доступність, як середньоарифметичне значення, знову ж, у межах біологічної еквівалентності. Альтернативно або на додаток до цього, 50 мг гелькап дає середню Cmax від приблизно 700 до приблизно 950 нг/мл, або від приблизно 750 до приблизно 875 нг/мл. У найбільш переважному варіанті здійснення, 50 мг гелькап дає Cmax від 800 до 820 нг/л, переважно у межах біологічної еквівалентності. Оскільки дозовані форми за даним винаходом демонструють фармакокінетику, пропорційну дозі, буде зрозуміло, що ці значення Cmax можуть стандартизуватися на основі сили впливу дозованої форми, і що значення Cmax можуть приписуватися альтернативним значенням сили впливу на основі такої стандартизації. Ще одна важлива ознака дозованих форм за даним винаходом, яка також є переважною у будь-якому з варіантів здійснення даного винаходу, стосується розчинення дозованої форми, а у переважному варіанті здійснення не менше, ніж приблизно 75 % палоносетрону у дозованій формі розчиняється через 30 або 45 хвилин, коли досліджується у пристрої для розчинення типу II з лопатевою мішалкою відповідно до Фармакопеї США, при 75 об./хв. і 37 °C, у 500 мл, 0,01 н. HCl. Ще одна ознака дозованих форм за даним винаходом, яка також є переважною у будьякому з варіантів здійснення даного винаходу, незалежно від дозованої форми або типу начинки, або способу одержання, полягає у тому, що дозована форма піддається деградації палоносетрону не більше, ніж 5 % мас., 3 % мас. або 2 % мас., коли дозована форма в її стійкій до вологості упаковці експонується для навколишнього середовища 25 °C і 60 % RH або 40 °C і 75 % RH, протягом періодів, що дорівнюють або перевищують 3 місяці, 6 місяців, 9 місяців або навіть один рік. Палоносетрону гідрохлорид і споріднені сполуки Палоносетрон, що використовується у даному винаході, може являти собою палоносетрон як основу або фармацевтично прийнятну сіль, але переважно являє собою палоносетрону гідрохлорид. На додаток до цього, палоносетрон переважно присутній у кількості, що знаходиться у межах від приблизно 0,02 мг до приблизно 10 мг на дозовану форму, більш переважно від приблизно 0,05 мг або 0,15 мг до приблизно 2 мг на дозовану форму, і ще більш переважно від приблизно 0,2 мг до приблизно 1,0 мг на дозовану форму, по відношенню до маси основи, коли присутній як фармацевтично прийнятна сіль. Особливо переважні дози являють собою 0,25 мг, 0,50 мг і 0,75 мг палоносетрону або його солі по відношенню до маси основи. Особливо стабільні препарати виявлені за допомогою використання кількостей палоносетрону у рідких гелькапах, більших, ніж приблизно 0,25 мг, 0,35 мг або 0,45 мг, переважно менших, ніж приблизно 2,0 мг. Палоносетрону гідрохлорид, що використовується для одержання дозованої форми або міститься у кінцевій дозованій формі, може також відрізнятися присутністю різних споріднених з палоносетроном сполук, включаючи сполуки Cpd3, Cpd2 і/або Cpd1, як описується за допомогою наведених далі хімічних структур: 6 UA 97653 C2 5 10 15 20 25 Сполуки Cpd2 і Cpd3, як правило, присутні, індивідуально або у поєднанні по відношенню до палоносетрону гідрохлориду, у кількостях, менших, ніж 1,0 % мас., 0,75 % мас. або 0,5 % мас., і/або більших, ніж приблизно 0,05 % мас., 0,075 % мас. або 0,1 % мас. Cpd2 і Cpd3 можуть вимірюватися у дозованій формі або у сирому матеріалі палоносетрону, що використовується для одержання дозованої форми. Сполука Cpd1, як правило, присутня, індивідуально по відношенню до палоносетрону гідрохлориду, у кількості, більшій, ніж приблизно 0,05 % мас., 0,1 % мас. або 0,2 % мас., і/або меншій, ніж приблизно 3,0 % мас., 2,5 % мас., 2,0 % мас., 1,5 % мас., 1,0 % мас. або 0,5 % мас. Cpd1 переважно вимірюється у дозованій формі, оскільки це міра опосередковуваної киснем деградації. В одному з переважних варіантів здійснення дозовані форми визначаються за стабільністю, при якій не більше, ніж приблизно 5,0 % мас., 4,0 % мас., 3,0 % мас., 2,5 % мас., 2,0 % мас., 1,5 % мас., 1,0 % мас. або 0,5 % мас. сполуки Cpd1 утворюється, коли дозована форма в її упаковці, стійкій до вологості, експонується для навколишнього середовища при 25 °C і 60 % RH або при 40 °C і 75 % RH протягом періодів, що дорівнюють або перевищують 3 місяці, 6 місяців, 9 місяців або навіть один рік. З цієї причини, в іншому варіанті здійснення даний винахід передбачає тверду пероральну дозовану форму, що містить: (a) від приблизно 0,05 до приблизно 2,0 мг палоносетрону або його фармацевтично прийнятної солі; (b) один або декілька фармацевтично прийнятних наповнювачів; (с) Cpd1 у кількості, меншій, ніж 3,0 % мас. по відношенню до маси палоносетрону. В іншому варіанті здійснення даний винахід передбачає тверду пероральну дозовану форму, що містить: (a) від приблизно 0,05 до приблизно 2,0 мг палоносетрону або його фармацевтично прийнятної солі; (b) один або декілька фармацевтично прийнятних наповнювачів; (с) Cpd2 або Cpd3, у кількості, меншій, ніж 1,0 % мас., по відношенню до маси палоносетрону або його фармацевтично прийнятної солі. У будь-якому з цих варіантів здійснення дозована форма може необов'язково містити засоби для запобігання опосередковуваній киснем деградації вказаного палоносетрону. Інші споріднені з палоносетроном сполуки, які можуть бути присутніми у композиціях, включають в себе Cpd4, Cpd5, Cpd6 і Cpd7, як зображено нижче: 7 UA 97653 C2 5 10 15 20 25 30 35 40 Способи одержання Даний винахід також передбачає способи одержання дозованих форм палоносетрону. Таким чином, ще в одному варіанті здійснення даний винахід передбачає спосіб одержання набору дозованих форм палоносетрону, що мають знижену кількість домішок і продуктів деградації, опосередковуваної киснем, що включає в себе (a) змішування палоносетрону гідрохлориду і одного або декількох фармацевтично прийнятних наповнювачів з утворенням суміші; (b) переробку вказаної суміші у вигляді множини кінцевих дозованих форм і (с) дослідження однієї або декількох з вказаних кінцевих дозованих форм на одну або декілька сполук, споріднених з палоносетроном, вибраних з Cpd2, Cpd1 і Cpd3. "Переробка" стосується стадій, що використовуються для одержання фармацевтичного препарату і кінцевої дозованої форми з визначеного набору інгредієнтів, і виключає способи хімічного синтезу інгредієнтів, що використовуються у препараті. Цей варіант здійснення поширюється на всі дозовані форми палоносетрону, включаючи ампули зі стандартними одиничними дозами, заповненими палоносетроном, наприклад, разом зі стерильною рідиною для ін'єкцій. Таким чином, наприклад, даний винахід може поширюватися на способи заповнення ампул або контейнерів для стандартних одиничних доз стерильними розчинами палоносетрону для ін'єкцій, переважно у водних середовищах, і переважно приготованих, як описано у заявці на Міжнародний патент WO 2004/067005, Calderari et al. У цьому контексті "ампула" означає малий герметичний контейнер медичного препарату, який використовують тільки один раз, і який включає в себе скляні ампули, що розламуються і не розламуються, пластикові ампули, що розламуються, мініатюрні скляночки з кришкою на різі і будь-який інший тип контейнера з розміром, придатним для вміщення тільки однієї стандартної одиничної дози палоносетрону (як правило, приблизно 5 мл). Інший варіант здійснення охоплює баланс, що досягається за допомогою препаратів за даним винаходом, по відношенню до біологічної доступності і стабільності, і у цьому варіанті здійснення даний винахід передбачає спосіб оптимізації біологічної доступності і стабільності палоносетрону у желатиновій капсулі палоносетрону, що включає в себе: (a) створення м'якої желатинової зовнішньої оболонки, що має проникність для кисню, меншу, ніж приблизно 1,0×10 3 2 мл∙см/(см ∙24 год.∙атм); і (b) одержання композиції начинки за допомогою стадій, що включають в себе: (i) створення від приблизно 0,05 до приблизно 2,0 мг палоносетрону у вигляді палоносетрону гідрохлориду, де вказаний палоносетрон містить Cpd1 у кількості, меншій, ніж 1,0 % мас. по відношенню до маси вказаного палоносетрону; (ii) розчинення або диспергування вказаного палоносетрону у воді з утворенням водного преміксу; (iii) змішування вказаного водного преміксу з одним або декількома ліпофільними наповнювачами, при масовому співвідношенні водного преміксу до ліпофільних наповнювачів, меншому, ніж 50:50, 40:60, 30:70 або 20:80, з утворенням перемішаної або гомогенної ліпофільної композиції начинки; (iv) змішування поверхнево-активної речовини з вказаною водою, вказаним водним преміксом або вказаною композицією начинки, і (v) балансування кількостей поверхневоактивної речовини і води у вказаній композиції начинки для полегшення біологічної доступності палоносетрону з вказаної желатинової капсули при пероральному прийомі і для зведення до мінімуму ступеню деградації палоносетрону; і (с) заповнення вказаної зовнішньої оболонки вказаною композицією начинки. 8 UA 97653 C2 5 10 15 20 25 30 35 40 45 50 55 60 Ще один спосіб за даним винаходом включає в себе спосіб пакування дозованої форми палоносетрону, що включає в себе: (a) створення пустої оболонки; і (b) заповнення вказаного контейнера у формі оболонки композицією начинки у безпосередньому навколишньому середовищі, збідненому киснем, де вказана композиція начинки містить: (i) визначену кількість композиції активного інгредієнта, що містить палоносетрон або його фармацевтично прийнятну сіль; і (ii) фармацевтично прийнятний наповнювач. "Навколишнє середовище, збіднене киснем" переважно являє собою середовище, що визначається за допомогою вмісту кисню менше, ніж приблизно 10 % кисню, 5 % кисню або навіть 1 % або 0,1 % кисню (за масою або за об'ємом). У ще більш переважному варіанті здійснення способи одержання або пакування дозованих форм за даним винаходом здійснюють в атмосфері азоту або з продуванням азотом у збагаченому азотом навколишньому середовищі, що містить більше, ніж приблизно 90 %, 95 % або 98 % азоту (за масою або за об'ємом). В іншому конкретному варіанті здійснення спосіб визначається варіабельністю активного інгредієнта серед дозованих форм, при цьому передбачається спосіб одержання множини твердих пероральних дозованих форм, що включає в себе: (a) створення оболонки капсули; (b) заповнення вказаної оболонки композицією начинки, що містить: (i) визначену кількість палоносетрону або його фармацевтично прийнятної солі і (ii) фармацевтично прийнятний наповнювач; і (с) повторення стадій (a) і (b) додатково один або декілька разів, де вказана визначена кількість має розкид від однієї капсули до іншої, менший, ніж приблизно 3, 2, 1, 0,5 або 0,1 % мас. У будь-якому з вказаних вище варіантів здійснення спосіб одержання може також додатково включати в себе пакування вказаної дозованої форми або множини дозованих форм у стійкий до вологості герметичний контейнер. Матеріал, що використовується для одержання стійкого до вологості герметичного контейнера, переважно має проникність для кисню меншу, ніж -2 -3 -4 -5 2 приблизно 1,0×10 , 1,0×10 , 1,0×10 , або навіть 5,0×10 мл∙см/(см ∙24 год.∙атм). Альтернативно або на додаток до цього, упаковка може характеризуватися як "щільний контейнер" відповідно до стандартів, описаних у Фармакопеї США (тобто не більше одного з десяти досліджуваних контейнерів перевищують проникність для вологості 100 мг на день на л, і жоден не перевищує 200 мг на день на мл). Крім того, контейнер може визначатися за кількістю вологості, яку він дозволяє поглинути дозованим формам за даним винаходом під час зберігання. Наприклад, у різних переважних варіантах здійснення контейнер запобігає поглинанню вказаними дозами більш ніж 1,0, 0,1 або навіть 0,05 % мас. вологості через три місяці при зберіганні при 40 °C і 75 % відносної вологості. Блістерна упаковка є особливо переважним способом пакування. М'які желатинові капсули Фармацевтичні композиції з рідкою серцевиною за даним винаходом інкапсулюють у м'яку желатинову оболонку, описану нижче. Желатин являє собою переважний компонент м'яких желатинових оболонок за даним винаходом. Вихідний желатиновий матеріал може бути одержаний за допомогою часткового гідролізу колагенового матеріалу, такого як шкіра, білі сполучні тканини або кістки тварин. Желатиновий матеріал може класифікуватися як желатин типу А, який одержують за допомогою кислотної обробки свинячої шкіри і який демонструє ізоелектричну точку у межах між pH 7 і pH 9; і желатин типу В, який одержують за допомогою лужної обробки кісток і шкір тварин (корів) і який демонструє ізоелектричну точку у межах між pH 4,7 і pH 5,2. Суміші желатину типу А і типу В можуть використовуватися для одержання желатину з необхідними характеристиками в'язкості і міцності за Блумом для одержання капсули. Желатин, придатний для одержання капсули, є комерційно доступним від Sigma Chemical Company, St. Louis, Mo. Стосовно загального опису желатину і капсул на основі th желатину, див. Remington's Pharmaceutical Sciences, 16 ed., Mack Publishing Company, Easton, Pa (1980), page 1245 і pages 1576-1582; і патент США № 4935243, Borkan et at., виданий 19 червня 1990 року; ці два джерела включені сюди як посилання у всій їх повноті. М'які желатинові оболонки можуть містити від приблизно 20 % до приблизно 60 % желатину. Желатин може бути типу А або типу В, або являти собою їх суміш, з числами Блума, що знаходяться у межах від приблизно 60 до приблизно 300. М'які желатинові оболонки можуть також містити пластифікатор. Придатні для використання пластифікатори включають в себе гліцерин, сорбітан, сорбітол або подібні низькомолекулярні поліоли і їх суміші. Переважний пластифікатор, придатний для використання у даному винаході, являє собою гліцерин. М'які желатинові оболонки за даним винаходом можуть також містити воду. Не обмежуючись теорією, вода, як передбачається, допомагає у швидкому розчиненні або розриві м'якої желатинової оболонки при контакті з шлунково-кишковими рідинами, що зустрічаються в організмі. 9 UA 97653 C2 5 10 15 20 25 30 35 40 45 50 55 60 М'які желатинові капсули і способи інкапсулювання описані у P.K. Wilkinson et al., "Softgels: Manufacturing Considerations", Drugs and Pharmaceutical Sciences, 41 (Specialized Drug Delivery Systems), P. Tyle, Ed. (Marcel Dekker, Inc., New York, 1990) pp.409-449; F.S. Horn et at., "Capsules, Soft", Encyclopedia of Pharmaceutical Technology, vol. 2, J. Swarbrick and J.C. Boylan, eds. (Marcel Dekker, Inc., New York, 1990) pp. 269-284; M.S. Patel et at, "Advances in Softgel Formulation Technology", Manufacturing Chemist, vol. 60, no. 7, pp. 26-28 (July 1989); M.S. Patel et al., "Softgel Technology", Manufacturing Chemist, vol. 60, no. 8, pp. 47-49 (August 1989); R.F. Jimerson, "Softgel (Soft Gelatin Capsule) Update", Drug Development and Industrial Pharmacy (Interphex '86 Conference), vol. 12, no. 8 & 9, pp. 1133-1144 (1986); і W.R. Ebert, "Soft Elastic Gelatin Capsules: А Unique Dosage Form", Pharmaceutical Technology, vol. 1, no. 5, pp. 44-50 (1977); ці джерела включені сюди як посилання у всій їх повноті. Одержана м'яка желатинова капсула є розчинною у воді і у шлунково-кишковій рідині. При заковтуванні капсули желатинова оболонка швидко розчиняється або розривається у шлунково-кишковому тракті, тим самим вводячи активні фармацевтичні препарати з рідкої серцевини в організм. Способи лікування В інших варіантах здійснення даний винахід передбачає способи лікування блювання за допомогою введення однієї або декількох дозованих форм, описаних тут. Блювання може являти собою блювання у гострій фазі (тобто блювання, що відбувається у межах приблизно 24 годин після події, що викликає блювання) або уповільнене блювання (тобто блювання, що відбувається після гострої фази, але у межах семи, шести, п'яти або чотирьох днів після події, що викликає блювання). Блювання може складати нудоту і блювання, що викликається хіміотерапією ("CINV"), від помірно еметогенної або у вищій мірі еметогенної хіміотерапії, нудоту і блювання, що викликається радіаційною терапією ("RTNV"), або післяопераційну нудоту і блювання ("PONV"). Дослідження біологічної еквівалентності Коли про продукт кажуть, що він демонструє конкретний фармакокінетичний параметр "у межах біологічної еквівалентності" буде зрозуміло, що продукт є біологічно еквівалентним лікарському засобу, що досліджується при використанні дослідження біологічної еквівалентності, описаного тут. Дослідження біологічної еквівалентності, як правило, потребує дослідження in vivo на людях, у яких концентрація активного інгредієнта або активного залишку і, коли треба, його активного метаболіту (метаболітів) у цільній крові, плазмі, сироватці або іншій відповідній біологічній рідині вимірюється як функція часу. Біологічна еквівалентність ("BE"), що визначається як відносна біологічна доступність ("BA"), включає в себе порівняння між досліджуваним і еталонним продуктом лікарського засобу. Хоча BA і BE тісно пов'язані, порівняння BE звичайно базуються на (1) критерії, (2) довірчому інтервалі для критерію і (3) заданій межі BE. Стандарт при конструюванні досліджень BE in vivo базується на введенні або однієї, або множини доз досліджуваного і еталонного продуктів здоровим суб'єктам в окремих випадках, при випадковому призначенні двох можливих послідовностей введення лікарського продукту. Статистичний аналіз фармакокінетичних заходів, таких як площа під кривою (AUC) і пікова концентрація (Cmax), переважно базується на так званій "процедурі двосторонніх тестів", щоб визначити, чи є середні значення фармакокінетичних заходів, що визначаються після введення досліджуваного і еталонного продуктів, порівнянними. Цей підхід називають середньою біологічною еквівалентністю і він включає в себе обчислення 90 % довірчого інтервалу для співвідношення середніх значень (геометричних середніх по популяції) для заходів, для досліджуваного і еталонного продуктів. Для встановлення BE, обчислений довірчий інтервал повинен потрапити у межу BE, тобто 80-125 % для співвідношення середніх значень для продукту. Таким чином, наприклад, кажуть, що біологічна еквівалентність встановлена для даного набору обставин за допомогою 90 % довірчого інтервалу для AUC, який знаходиться у межах між 80 % і 125 %, і 90 % довірчого інтервалу для Cmax, який знаходиться у межах між 80 % і 125 %. Додаткові деталі стосовно процедур BE можна знайти в інструкції, що видається FDA (адміністрація США з лікарських препаратів і харчових продуктів), липень 1992 року, озаглавленій "Statistical Procedures for Bioequivalence Studies Using а Standard Two-Treatment Crossover Design", зміст якої включається сюди як посилання. Приклади Наведені далі приклади наводяться з тим, щоб забезпечити фахівців у даній галузі повним поясненням і описом того, як одержують і оцінюють сполуки, заявлені тут, і як передбачається, є чистими ілюстраціями даного винаходу і не призначаються для обмеження меж того, що автори розглядають як їх винахід. Здійснюються спроби забезпечити точність по відношенню до чисел 10 UA 97653 C2 5 (наприклад, кількостей, температури і тому подібного), але деякі помилки і відхилення повинні братися до уваги. Якщо не стверджується інше, частки являють собою масові частки, температура наводиться у°С або знаходиться при кімнатній температурі, і тиск є атмосферним або знаходиться у межах, близьких до нього. Приклад 1 - Репрезентативне приготування гелькапу Таблиця 1 описує репрезентативні препарати твердої пероральної дозованої форми гелькапу, що містить 0,25, 0,50 і 0,75 мг палоносетрону. Таблиця 1 Репрезентативне приготування гелькапу Найменування інгредієнтів 0,25 мг Активний лікарський засіб Палоносетрон HCl Наповнювачі Очищена вода Гліцерин, безводний Бутилований гідроксіанізол (BHA) Полігліцерилолеат (Plurol Oleique CC 497) Моно- і дигліцериди каприлової/капринової кислоти (Capmul MCM) Азот Теоретична маса начинки Оболонка желатинової капсули, #3, овальна (Cardinal Health) a відповідає 0,25 мг вільної основи b відповідає 0,50 мг вільної основи c відповідає 0,75 мг вільної основи * Препарат A (клінічне завантаження) ** Препарат B (комерційне завантаження) 10 15 20 25 Формула (мг на капсулу) 0,50 мг 0,75 мг a b c 0,28 0,56 0,84 5,57 6,40 0,13 6,65* (1,66)** 113,97* (118,96)** --133,00 мг 5,57 6,40 0,13 6,65* (1,66)** 113,69* (118,68)** --133,00 мг 5,57 6,40 0,13 6,65* (1,66)** 113,41* (118,40)** --133,00 мг 1 капсула 1 капсула 1 капсула Приклад 2 - Протокол одержання Спосіб компаундування включає в себе приготування двох окремих сумішей, побічної суміші, що містить активний інгредієнт, гліцерин і воду, і головної суміші, що містить інші наповнювачі. Спосіб починають з двох окремих сумішей, які потім об'єднують, щоб скласти кінцевий розчин начинки для інкапсулювання. Розчин начинки витримують в атмосфері азоту протягом фаз компаундування та інкапсулювання. Приклад 3 - Протокол репрезентативного дослідження розчинення Ілюстративний спосіб розчинення для пероральних капсул з палоносетроном, 0,25 мг, 0,50 мг і 0,75 мг використовує Пристрій 2 USP (лопатева мішалка) при 75 об./хв. в 500 мл 0,01 н. HCl при температурі розчинення 37,0±0,5 °C. Критерій прийнятності являє собою "не менше, ніж 75 % через 45 хвилин". Зважують індивідуально шість м'яких гелевих капсул. М'які гелеві капсули вміщують у кожну ємність і відбирають зразки через 15, 30, 45 і 60 хвилин. Відбір через 15, 30, 60 хвилин здійснюють тільки для інформації. Розчини зразка дістають і фільтрують через проточні фільтри у пробірки або флакони для ВЕРХ. Зразки аналізують з використанням системи ВЕРХ з УФдетектором. Таблиця 2 Умови розчинення Пристрій USP Середовище Температура Швидкість обертання Час відбору зразків 2 лопатевих мішалки 0,01 н. HCl, 500 мл 37±0,5 °C 75 об./хв. 45 хвилин 15, 30 і 60 хвилин (тільки для інформації) 11 UA 97653 C2 Продовження таблиці 2 3 мл (або 1-1,5 мл, коли збирають безпосередньо у флакони для ВЕРХ) 500 мл середовища для розчинення Об'єм зразків Об'єм 5 Приклад 4- Хімічна і фізична стабільність Таблиця 3 представляє результати дослідження хімічної і фізичної стабільності для 0,75 мг м'яких гелевих препаратів палоносетрону, про які повідомлялося у Прикладі 1, упакованих у блістерну упаковку 2 × 5 (формування: LM 15088, фольга: Reynolds 701). Таблиця 3 Хімічна і фізична стабільність Аналіз палоносетрону Дослідження % в заCpd3 Cpd2 Cpdl 90.0лежності 110.0% від t0 0.50 % 0.50 % 3.0 % Приблизний опис Вихідні значення 25 °C/60 % r.h. 3 місяці 6 місяців 9 місяців 12 місяців 40 °C/75 % r.h. 1 місяць 3 місяці 6 місяців 10 Речовини, споріднені з палоносетроном Дослідження розчинення (% розчиненого) 45 хв. 15 хв. 30 хв. 60 хв. тільки тільки тільки NLT для для для 75 % інформ. інформ. інформ. 98.5 70.4 97.7 98.9 97.7 100.0 0.28 0.28 0.20 97.6 96.5 96.0 93.7 99.9 98.8 98.3 95.9 0.23 0.26 0.23 0.22 0.26 0.27 0.27 0.26 0.23 0.46 0.6 0.6 99.2 97.6 99.0 96.8 97.6 87.4 80.5 81.9 99.1 97.2 98.9 96.5 98.8 97.7 99.1 96.7 97.2 97.5 96.2 99.5 99.8 98.5 0.36 0.24 0.26 0.25 0.26 0.27 0.56 0.65 0.68 99.9 97.3 96.9 92,3 79.9 52.5 100.5 97.0 96.7 100.1 97.5 97.0 Приклад 5 - Хімічна і фізична стабільність Таблиця 4 представляє результати дослідження хімічної і фізичної стабільності для 0,50 мг м'яких гелевих препаратів палоносетрону, про які повідомлялося у Прикладі 1, упакованих у блістерну упаковку 2×5 (формування: LM 15088, фольга: Reynolds 701). Таблиця 4 Хімічна і фізична стабільність Дослідження Приблизний опис вихідні значення 25 °C/60 % r.h. 3 місяці 6 місяців 9 місяців 12 місяців 40 °C/75 % r.h. Аналіз палоносетрону Речовини, споріднені з палоносетроном Дослідження розчинення (% розчиненого) 45 хв. 15 хв. 30 хв. 60 хв. % в заCpd3 Cpd2 Cpdl 90.0тільки тільки тільки лежності NLT 110.0% для для для від t0
ДивитисяДодаткова інформація
Назва патенту англійськоюSoft capsules comprising palonosetron hydrochloride having improved stability and bioavailability
Автори англійськоюBonadeo Daniele, Calderari Giorgio, Braglia Enrico, Braglia Riccardo
Назва патенту російськоюМягкие капсулы, содержащие палоносетрона гидрохлорид, которые имеют улучшенную стабильность и биологическую доступность
Автори російськоюБонадео Даниэле, Кальдерари Джорджо, Бралья Энрико, Бралья Риккардо
МПК / Мітки
МПК: A61K 9/66, A61K 31/473, A61K 9/48
Мітки: мають, м'які, поліпшену, стабільність, містять, гідрохлорид, капсули, біологічну, доступність, палоносетрону
Код посилання
<a href="https://ua.patents.su/21-97653-myaki-kapsuli-shho-mistyat-palonosetronu-gidrokhlorid-yaki-mayut-polipshenu-stabilnist-i-biologichnu-dostupnist.html" target="_blank" rel="follow" title="База патентів України">М’які капсули, що містять палоносетрону гідрохлорид, які мають поліпшену стабільність і біологічну доступність</a>
Пептиди, які мають високу біологічну активність, та фармацевтична композиція на їх основі

Номер патенту: 32523
Опубліковано: 15.02.2001
Автори: Сейверт Свен, Суханєк Ернест, Грабаревич Желько, Місе Степан, Петек Мар'ян, Мільднер Боріс, Дувняк Марко, Сікірич Предраг, Ротквич Іво, Туркович Бранко, Удовічич Іван
МПК: A61P 25/24, A61K 38/12, A61P 25/16, A61P 1/04, C07K 7/06, A61P 31/12, C07K 7/08, A61K 38/08, A61K 38/10
Мітки: мають, фармацевтична, активність, біологічну, композиція, високу, пептиди, основі
Текст:
...мужские особи альбино крыс /по 10 особей для каждого опыта, весом 200-250 г/. Крысы с кожными ранами индивидуально содержались в отдельных клетках. Каждому животному было сделано под легкой анестезией два пореза, каждый длиной 3 см, расположенных параллельно по обе стороны спины на расстоянии1,5 см от хребта. Одна ранка затем была защиплена двумя хирургическими скобами, а другая оставлена незалеченной. Пептид N 4, растворенный в...
Похідні дипіридодіазепіну та їх гідрати, що мають біологічну активність, зокрема гальмівну дію на зворотну транскриптазу вірусу hiv-1

Номер патенту: 34420
Опубліковано: 15.03.2001
Автори: Енгель Вольфхард, Еберлайн Вольфганг, Харгрейв Карл, Шмідт Гюнтер, Труммлітц Гюнтер
МПК: C07D 471/14
Мітки: активність, зокрема, гідрати, похідні, дипіридодіазепіну, біологічну, мають, транскриптазу, гальмівну, вірусу, дію, hiv-1, зворотну
Текст:
...раствора быстро добавляют 9,2 г 3-аминокарбонил-2-хлор-6-метил-4-(трифторметил)пиридина, причём температуру держат ниже 5°С. Полученную смесь размешивают до растворения 3(аминокарбонил)пиридина (менее 30 минут). Затем смесь нагревают при температуре 75°С в течение 30 минут. После охлаждения до комнатной температуры 3-аминопиридинсоединение экстрагируют этилацетатом, сушат над сульфатом магния, фильтруют и упаривают. Получают 4,9 г...
Гранули, що містять гідрохлорид венлафаксину, та спосіб їх виготовлення

Номер патенту: 82268
Опубліковано: 25.03.2008
Автори: Моріц Естер, Корбелі Тібор, Божо Агнеш, Фекете Паль
МПК: A61K 47/38, A61K 47/02, A61K 31/137, A61P 25/18, A61K 9/16
Мітки: гранули, містять, гідрохлорид, виготовлення, спосіб, венлафаксину
Формула / Реферат:
1. Гранули форми, близької до сферичної, що включають фармацевтично активний гідрохлорид венлафаксину, де зазначені гранули містять (щодо загальної маси) максимум 80 % по масі гідрохлориду венлафаксину, від 10 до 60 % по масі хлориду натрію і/або хлориду калію, від 10 до 60 % по масі мікрокристалічної целюлози і, необов’язково, інші фармацевтично прийнятні ексципієнти і/або добавки, що сприяють грануляції.2. Гранули за п. 1, що мають...
Похідні тетрагідрокарбазолу, що мають покращену біологічну дію та покращену розчинність, як ліганди g-білок сполучених рецепторів (gpcr)

Номер патенту: 89963
Опубліковано: 25.03.2010
Автори: Гюнтер Екхард, Кюне Рональд, Герлах Маттіас, Пауліні Клаус, Бааснер Сільке, Полімеропоулос Еммануель, Зодерхалл Арвід, Шмідт Петер
МПК: A61P 43/00, C07D 209/82, A61K 31/403, A61P 35/00, A61P 15/00
Мітки: сполучених, похідні, рецепторів, gpcr, мають, g-білок, розчинність, покращену, тетрагідрокарбазолу, дію, біологічну, ліганди
Формула / Реферат:
1. Сполука тетрагідрокарбазолу формули (І):, (I)у якій:X1 являє собою S, О або S+-O-,Х2 та Х3 незалежно один від іншого являють собою О або гемінально прикріплені Н2,R1 та R2 незалежно один від іншого вибирають з групи, що включає -Н, арильний, алкільний та арилалкільний радикали, які необов'язково заміщені у алкільній та/або арильній...
Рідкі фармацевтичні композиції палоносетрону

Номер патенту: 90449
Опубліковано: 11.05.2010
Автори: БОНАДЕО Даніеле, Кальдерарі Джорджо, Канелла Роберта, Бралья Енріко, Бралья Ріккардо
МПК: A61K 9/08, A61P 1/08, A61K 31/473
Мітки: композиції, палоносетрону, рідкі, фармацевтичні
Формула / Реферат:
1. Фармацевтично стабільний ізотонічний внутрішньовенний розчин палоносетрону гідрохлориду для профілактики або зменшення блювання, що включає:a) від 0,03 мг/мл до 0,2 мг/мл палоносетрону гідрохлориду в розрахунку на масу основи палоносетрону; іb) стерильний фармацевтично прийнятний водний носій, що містить маніт в кількості, ефективній для регуляції тонічності, при рН від 4,0 до 6,0, іс) EDTA в кількості від 0,005 мг/мл...
Попередній патент: Джбан для фільтрування рідин
Наступний патент: Дозувальна головка з поворотним ковпачком
Випадковий патент: Пристрій для волочіння дроту