Хінолінові та хіназолінові сполуки, фармацевтична композиція на їх основі, спосіб їх одержання (варіанти), проміжні сполуки (варіанти), спосіб лікування доброякісної гіперплазії простати








Формула / Реферат
1. Хінолінова або хіназолінова похідна формули I
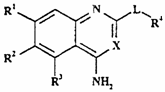 , (І)
, (І)
де
R1 являє собою C1-4-алкокси, необов'язково заміщену одним чи декількома атомами фтору;
R2 являє собою Н чи C1-6-алкокси, необов'язково заміщену одним чи декількома атомами фтору;
R3 являє собою 5- чи 6-членне гетероциклічне кільце, що містить, принаймні, один гетероатом, вибраний з N, О і S, причому цей цикл необов'язково заміщений одною чи декількома групами, вибраними з галогену, C1-4-алкокси, C1-4-алкілу і CF3;
R4 являє собою 4-, 5-, 6- чи 7-членне гетероциклічне кільце, що містить, принаймні, один гетероатом, вибраний з N, О і S, причому цей цикл необов'язково конденсований з бензольним кільцем, або з 5- чи 6-членним гетероциклічним кільцем, що містить, принаймні, один гетероатом, вибраний з N, О і S, ця циклічна система в цілому необов'язково заміщена одною чи декількома групами, незалежно вибраними з ОН, C1-4-алкілу, C1-4-алкокси, галогену, CONR8R9, SО2NR8R9, (CH2)bNR8R9 і NHSО2(C1-4-алкілу), і якщо S є членом циклічної системи, він може бути заміщений одним чи двома атомами кисню;
R8 і R9 незалежно являють собою Н або C1-4-алкіл, або разом з атомом N, до якого вони приєднані, можуть являти собою 5- чи 6-членне гетероциклічне кільце, що містить, принаймні, один гетероатом, вибраний з N, О і S;
b дорівнює 0, 1, 2 чи 3;
X являє собою СН або N; і
L відсутнє чи являє собою циклічну групу формули Iа
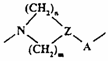 , (Iа)
, (Iа)
у якій N приєднаний до другої позиції хінолінового чи хіназолінового циклу;
А відсутнє чи являє собою СО або SО2;
Z являє собою СН або N;
m дорівнює 1 чи 2, і, крім того, якщо Z являє собою СН, воно може бути рівним 0; і
n дорівнює 1, 2 чи 3, за умови, що сума m і n дорівнює 2, 3, 4 чи 5;
або являє собою ланцюг формули Ib
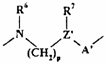 , (Ib)
, (Ib)
в якій N приєднаний до 2 позиції хінолінового чи хіназолінового циклу;
А’ і Z’ мають, відповідно, такі ж значення, як А і Z, описані вище;
R6 і R7 незалежно являють собою Н або C1-4-алкіл;
і
р дорівнює 1, 2 чи 3, і, крім того, якщо Z являє собою СН, воно може дорівнювати 0;
або її фармацевтично придатні солі.
2. Сполука за п. 1, де кожний з R1 і R2 являє собою метоксильну групу.
3. Сполука за п. 1 або п.2, де R3 являє собою 2-піридиніл або 2-піримідиніл.
4. Сполука за будь-яким з попередніх пунктів, де X являє собою N.
5. Сполука за будь-яким з попередніх пунктів, де L відсутнє.
6. Сполука за будь-яким з попередніх пунктів, де R4 включає в себе насичений 6-членний N-вмісний цикл, що конденсований з бензольним чи піридиновим кільцем.
7. Сполука за п. 6, де бензольне кільце заміщене NHSО2(C1-4-алкілом).
8. Сполука за п. 1 або її фармацевтично прийнятна сіль для використання як лікарського засобу.
9. Сполука за п. 1 або її фармацевтично прийнятна сіль як активний інгредієнт лікарського засобу для лікування доброякісної гіперплазії простати.
10. Фармацевтична композиція, що включає в себе сполуку формули I, як визначено в п. 1, або її фармацевтично прийнятну сіль, в суміші з фармацевтично прийнятним ад’ювантом, розріджувачем або носієм.
11. Спосіб лікування доброякісної гіперплазії простати, при якому вводять сполуку формули I, як визначено в п. 1, або її фармацевтично прийнятну сіль, пацієнту, який потребує такого лікування.
12. Спосіб одержання сполуки формули I, як визначено в п. 1, або її фармацевтично прийнятної солі, при якому у випадку, якщо Х являє собою СН, проводять циклізацію сполуки формули X
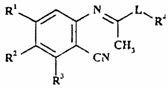 , (Х)
, (Х)
у якій R1-R4 і L - такі, як визначені в п. 1;
і, якщо бажано або необхідно, перетворення одержаної сполуки формули I на фармацевтично прийнятну сіль або навпаки.
13. Спосіб одержання сполуки формули I, як визначено в п. 1, або її фармацевтично прийнятної солі, при якому у випадку, якщо присутні А чи А’, а Z чи Z’ являють собою N, проводять взаємодію сполуки формули ХIIIа чи XIIIb, відповідно,
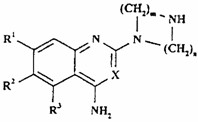 ,
,
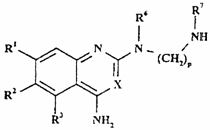 ,
,
(ХІІІа)
(XIIIb)
де R1-R3, R6, R7, X, m, n і р такі, як визначено у п. 1, зі сполукою формули XIV
![]() , (ХIV)
, (ХIV)
в якій R4 - такий, як визначено в п. 1, А’’ являє собою СО чи SO2 і Lg являє собою групу, що відходить,
і, якщо бажано або необхідно, перетворення одержаної сполуки формули I на фармацевтично прийнятну сіль або навпаки.
14. Спосіб одержання сполуки формули I, як визначено в п. 1, або її фармацевтично прийнятної солі, при якому проводять взаємодію сполуки формули XVIII
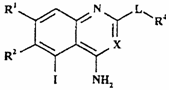 , (XVIII)
, (XVIII)
де R1, R2, R4, X і L - такі, як визначено в п. 1, зі сполукою формули XIX
R3-M, (XIX),
в якій R3 - такий, як визначено в п. 1, а М являє собою заміщений бор, цинк або олово, у присутності паладієвого каталізатора;
і, якщо бажано або необхідно, перетворення одержаної сполуки формули I на фармацевтично прийнятну сіль або навпаки.
15. Спосіб одержання сполуки формули I, як визначено в п. 1, або її фармацевтично прийнятної солі, при якому у випадку, якщо Х являє собою N, проводять взаємодію сполуки формули XХII
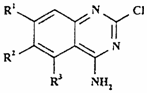 , (ХХІІ)
, (ХХІІ)
де R1-R3 - такі, як визначено в п. 1, зі сполукою формули ХХIIIа чи XXIIIb, відповідно,
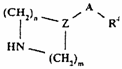 ,
,
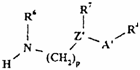 ,
,
(ХХІІІа)
(XXIIIb)
де R4, R6, R7, A, A', Z, Z', m, n і р - такі, як визначено у п. 1;
і, якщо бажано або необхідно, перетворення одержаної сполуки формули I на фармацевтично прийнятну сіль або навпаки.
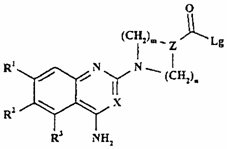
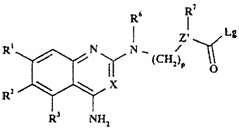
16. Спосіб одержання сполуки формули I, як визначено в п. 1, або її фармацевтично прийнятної солі, при якому у випадку, якщо А або А' являє собою СО, а R4 включає нуклеофільний атом азоту в гетероциклічному кільці, приєднаному до L, проводять взаємодію сполуки формули XXVIIIa або XXVIIIb, відповідно,
 ,
,
 ,
,
(XXVIIIa)
(XXVIIIb)
де R1-R3, R6, R7, X, Z, Z', m, n і р - такі, як визначено у п. 1, і Lg являє собою групу, що відходить, зі сполукою формули XXIX
HR4а, (XXIX)
де R4а являє собою групи, визначені у п. 1 за допомогою R4, які містять нуклеофільний атом азоту в циклі, причому цей атом азоту з'єднаний з Н;
і, якщо бажано або необхідно, перетворення одержаної сполуки формули I на фармацевтично прийнятну сіль або навпаки.
17. Спосіб одержання сполуки формули I, як визначено в п. 1, або її фармацевтично прийнятної солі, при якому проводять перетворення сполуки формули I, де L являє собою циклічну групу формули Іа, на відповідну сполуку формули I, де L являє собою ланцюг формули Ib, в якій кожен з R6 і R7 являють собою Н, шляхом впливу сильної основи;
і, якщо бажано або необхідно, перетворення одержаної сполуки формули I на фармацевтично прийнятну сіль або навпаки.
18. Спосіб одержання сполуки формули I, як визначено в п. 1, або її фармацевтично прийнятної солі, при якому у випадку, якщо А або А' відсутні, a Z або Z' являють собою N, проводять взаємодію сполуки формули XIIIa або XIIIb, як визначено вище, зі сполукою формули XXX
R4-Hal, (XXX)
де R4 - такий, як визначено в п. 1, і Hal являє собою атом галогену, приєднаний до циклу;
і, якщо бажано або необхідно, перетворення одержаної сполуки формули I на фармацевтично прийнятну сіль або навпаки.
19. Спосіб одержання сполуки формули I, як визначено в п. 1, або її фармацевтично прийнятної солі, при якому у випадку, якщо Х являє собою N, L відсутній, а R4 містить нуклеофільний атом азоту в гетероциклічному кільці, приєднаному до хінолінового або хіназолінового циклу, проводять взаємодію сполуки формули XXII, як визначено вище, зі сполукою формули XXIX, як визначено вище;
і, якщо бажано або необхідно, перетворення одержаної сполуки формули I на фармацевтично прийнятну сіль або навпаки.
20. Ортоімінобензонітрильна похідна формули X
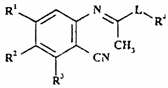 , (Х)
, (Х)
де R1-R4 і L - такі, як визначено в п. 1, яка є проміжною сполукою для одержання сполук формули І за пунктом 1.
21. Хінолінова або хіназолінова похідна формули ХIIIа
, (ХІІІа)
де R1-R3, R6, R7, X, m, n i p - такі, як визначено в п. 1, яка є проміжною сполукою для одержання сполук формули І за пунктом 1.
22. Хінолінова або хіназолінова похідна формули XIIIb
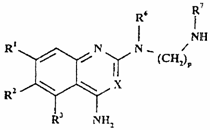 , (XIIIb)
, (XIIIb)
де R1-R3, R6, R7, X, m, n і р - такі, як визначено в п. 1, яка є проміжною сполукою для одержання сполук формули І за пунктом 1.
23. Хіназолінова похідна формули XXII
, (ХХІІ)
де R1-R3 - такі, як визначено в п. 1, яка є проміжною сполукою для одержання сполук формули І за пунктом 1.
24. Хінолінова або хіназолінова похідна формули XXVIIIa
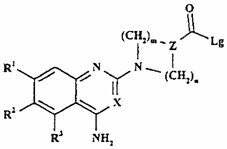 , (XXVIIIa)
, (XXVIIIa)
де R1-R3, R6, R7, X, Z, Z’, m, n і p - такі, як визначено в п. 1, яка є проміжною сполукою для одержання сполук формули І за пунктом 1.
25. Хінолінова або хіназолінова похідна формули XXVIIIb
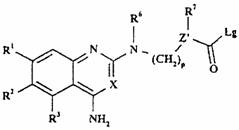 , (XXVIIIb)
, (XXVIIIb)
де R1-R3, R6, R7, X, m, n і p - такі, як визначено в п. 1, яка є проміжною сполукою для одержання сполук формули І за пунктом 1.
Текст
Даний винахід стосується нових сполук, які використовуються в терапії, особливо при лікуванні доброякісної гіперплазії простати. В міжнародній патентній заявці WO 89/05297 виявлено ряд заміщених хіназолінових сполук, які подані як інгібітори секреції шлункової кислоти. В міжнародній патентній заявці WO 97/23462(опублікованій після дати пріоритету даної заявки) виявлені хінолінові та хіназолінові сполуки, що мають 5-фенільний замісник. Ці сполуки показані при лікуванні доброякісної гіперплазії простати. Згідно з цим винаходом надається сполука формули І, де R1 являє собою C1-4 алкокси, необов’язково заміщену одним чи кількома атомами фтору; R2 являє собою H або C1-6 алкокси, необов’язково заміщену одним чи кількома атомами фтору; R3 являє собою 5- або 6-членне гетероциклічне кільце, що містить, принаймні, один гетероатом, вибраний з N, О і S, причому цей цикл необов'язково заміщений однією чи кількома групами, вибраними з галогену, СI-4 алкокси, СI-4 алкілу та CF3 ; R4 являє собою 4-, 5-, 6- або 7-членне гетероциклічне кільце, яке містить, принаймні один гетероатом, вибраний з N, О і S, причому цей цикл необов'язково конденсований з бензольним кільцем або з 5- чи 6членним гетероциклічним кільцем, що містить, принаймні, один гетероатом, вибраний з N, О і S, ця циклічна система в цілому необов'язково заміщена однією чи кількома групами, вибраними з ОН, СI-4 алкілу, СI-4 алкокси, галогену, CONR8R9 , SO 2NR8R9,(CH2)bNR8R9 і NHSО2(C1-4 алкілу), і якщо S є членом циклічної системи, він може бути заміщений одним або двома атомами кисню; R8 і R9 незалежно являють собою Н чи СI-4 алкіл, або разом з атомом N, до якого вони приєднані, можуть являти собою 5- або 6-членне гетероциклічне кільце, що містить принаймні один гетероатом, вибраний з N, О і S; b дорівнює 0, 1, 2 або 3; X являє собою СН або N; і L відсутній або являє собою циклічну гр упу формули Іа , в якій N приєднаний до другої позиції хінолінового або хіназолінового циклу; А відсутній або являє собою CO або SO2; Z являє собою СН або N; m дорівнює 1 або 2, і, крім того, якщо Z являє собою СН, воно може дорівнювати 0; і n дорівнює 1, 2 або 3, за умови, що сума m та n дорівнює 2, 3, 4 або 5; або являє собою ланцюг формули 1b, в якій N приєднаний до другої позиції хінолінового або хіназолінового циклу; А' і Z’ мають, відповідно, такі ж 3Начення, як А і Z, описані вище; R6 і R7 незалежно являють собою Н або СI-4 алкіл; і р дорівнює 1, 2 або 3, та, крім того, якщо Z являє собою СН, воно може дорівнювати 0; або їх фармацевтично прийнятні солі(у цьому документі назвиаються разом як "сполуки даного винаходу"). Фармацевтично прийнятні солі включають в себе адитивні кислотні солі, такі як гідрохлориди, гідроброміди та фосфати. Алкільні або алкоксильні групи, які R1-4 можуть представляти або включати, можуть бути з прямим ланцюгом, з розгалуженим ланцюгом, циклічними або їх комбінаціями. Більш прийнятно, R3 являє собою ароматичний цикл, наприклад, піридиніл, піримідиніл, тієніл, фураніл або оксазоліл. Гетероциклічні групи, які включають R4, можуть бути насиченими або ненасиченими. Однак більш прийнятно, щоб цикл, приєднаний до L, або, якщо L відсутній, до хінолінового або хіназолінового циклу, був насиченим. Сполуки даного винаходу можуть бути оптично активними. Зокрема, вони можуть виявляти атропоізомеризм навколо зв'язку, що з'єднує R з рештою молекули, якщо замісник R3 3Находиться в ортоположенні циклу. Даний винахід включає в себе оптичні ізомери сполук формули І і всі їх діастереоізомери. Більш прийнятні групи сполук, що можуть за3Начатись, включають ті, де: (a) R1 являє собою метоксильну груп у; (b) R2 являє собою метоксильну груп у; (c) R3 являє собою 2-піридинільну або 2-піримідинільну гр упу; (d) R4 включає насичений 6-членний N-вмісний цикл, що конденсований з бензольним або піридиновим циклом; наприклад, R4 може являти собою насичений 6-членний N-вмісний цикл, що конденсований з бензольним циклом, заміщеним NHSO2(C1-4 алкіл); (e) X являє собою N; (f) L відсутній. Згідно з даним винаходом також надається спосіб одержання сполуки даного винаходу, який включає: (а) якщо X являє собою СН, циклізацію сполуки формули X, у якій R1-4 та L такі, як ви3Начено вище; (b) якщо наявні А або A’, a Z чи Z' являють собою N, взаємодію сполуки формули ХІIIа або ХІIIb, відповідно, в якій R1-3, R6r R7, X, m, n і p такі, як ви3Начено вище, з сполукою формули XIV, в якій R4 такий, як ви3Начено вище, А’’ являє собою CO або SO2 і Lg являє собою гр упу, що відходить; (c) взаємодію сполуки формули XVIII, в якій R1, R2, R4, X і L такі, як ви3Начено вище, з сполукою формули XIX, R3-M (XIX) в якій R3 такий, як ви3Начено вище, а N являє собою заміщений бор, цинк чи олово, в присутності паладієвого каталізатора; (d) якщо X являє собою N, взаємодію сполуки формули XXII, в якій R1-3 такі, як ви3Начено вище, з сполукою формули ХХIIIа або ХХІІІb, відповідно, в якій R4, R6 R7 A, A', Z, Z' m, n та p такі, як ви3Начено вище; (e) якщо А або А1 являє собою CO, a R4 включає нуклеофільний атом азоту в гетероциклічному кільці, приєднаному до L взаємодію сполуки формули XXVIIIa або XXVIIIb, відповідно, в якій R1-3, R6, R7, X, Z, Z', m, n та р такі, як ви3Начено вище, і Lg являє собою групу, що відходить, з сполукою формули XXIX, HR4a (XXIX) в якій R4a являє собою групи, ви3Начені вище за допомогою R4, які містять нуклеофільний атом азоту в циклі, причому цей атом азоту з'єднаний з Н; (f) перетворення сполуки формули І, де L являє собою циклічну груп у формули Іа, на відповідну сполуку формули І, де L являє собою ланцюгфіг.формули Іb, в якій кожен з R6 і R7 являють собою Н, шляхом дії сильної основи; (g) якщо А або А' відсутній, а Z або Z’ являє собою N, взаємодію сполукифіг.формули ХIIIа або ХIIIb, як ви3Начено вище, з сполукою формули XXX, R4-Hal (XXX) в якій R4 такий, як ви3Начено вище, і Hal являє собою атом галогену, приєднаний до циклу; або (h) якщо X являє собою N, L відсутній, a R4 включає в себе нуклеофільний атом азоту в гетероциклічному кільці, приєднаному до хінолінового або хіназолінового циклу, взаємодію сполуки формули XXII, як ви3Начено вище, з сполукою формули XXIX, як ви3Начено вище; і, якщо бажано або необхідно, перетворення одержаної сполуки формули І на фармацевтично прийнятну сіль, або навпаки. У способі (а) циклізація може проводитись в присутності сильної основи(наприклад, діізопропіламіду літію) в розчиннику, що не справляє несприятливої дії на реакцію(наприклад, у тетрагідрофурані), при температурі, близької до кімнатної, і гаситись водою. Як варіант, циклізація може проводитись з використанням гідроксиду калію в такому розчиннику як DMSO, за підвищеної температури. Альтернативно, вона може проводитись з використанням хлориду цинку в розчиннику, що не справляє несприятливої дії на реакцію(наприклад, в тетрагідрофурані), при температурі кипіння розчинника із зворотним холодильником. У способі (b) підхожими групами, що відходять, є ОН і СІ. Якщо сполука формули XIV є карбоновою кислотою, реакція може проводитись в присутності підхожих конденсуючих агентів [наприклад, моногідрату 1-гідроксибензотриазолу, гідрохлориду 1-(3-диметиламінопропіл)-3-етилкарбодііміду та 4-метилморфоліну] в розчиннику, що не справляє несприятливої дії на реакцію(наприклад, в СН2СІ2), при температурі, близької до кімнатної. Якщо групою, що відходить, є СІ, реакція може проводитись в розчиннику, який не справляє несприятливої дії на реакцію(наприклад, в СН2СІ2), прибли3Но при 0°С. У способі (с) паладієвим каталізатором може бути тетракіс(трифенілфосфін)паладій. М може являти собою В(ОН)2, В(СН2СН2)2, Sn(CH 2CH2CH2CH3)3 або ZnCl. Реакція може проводитись в розчиннику, що не справляє несприятливої дії на реакцію(наприклад, якщо М являє собою В(ОН)2, в суміші толуолу, етанолу і 1М водного карбонату натрію) за підвищеної температури(наприклад, при температурі кипіння розчинника із зворотним холодильником). Якщо М являє собою ZnCl або заміщений Sn, йодид міді (І) може необов'язково використовуватись як співкаталізатор. У способі (d) реакція може проводитись в розчиннику, що не справляє несприятливої дії на реакцію(наприклад, в н-бутанолі) в присутності основи(наприклад, триетиламіну) за підвищеної температури(наприклад, при 100°С). У способі (e) підхожа група, що відходить, включає в себе СІ. Реакція може проводитись в розчиннику, що не справляє несприятливої дії на реакцію(наприклад, у ТГФ) в присутності основи(наприклад, триетиламіну) при кімнатній температурі. Реакція може також проводитись без виділення сполуки формули XXVIIIa або XXVIIIb шля хом взаємодії сполуки формули ХІІІа або ХІІІb з трифосгеном і сполукою формули XXIX. В цьому випадку групою, що відходить, є -СІ. Реакція може проводитись в розчиннику, що не справляє несприятливої дії на реакцію(наприклад, у СН 2Сl2), в присутності основи(наприклад, триетиламіну), при температурі, близької до кімнатної. У способі (f) підхожі сильні основи включають в себе діізопропіламід літію. Реакція може проводитись в розчиннику, що не справляє несприятливої дії на реакцію(наприклад, у ТГФ). У способі (g) реакція може проводитись в розчиннику, що не справляє несприятливої дії на реакцію(наприклад, в суміші н-BuOH і диметилацетаміду), в присутності основи(наприклад, триетиламіну), за підвищеної температури(наприклад, при 80°С). У способі (h) реакція може проводитись в розчиннику, що не справляє несприятливої дії на реакцію(наприклад, в суміші н-бутанолу і диметилацетаміду), в присутності основи(наприклад, триетиламіну), за підвищеної температури(наприклад, при 100°С). Сполуки формули X [див. спосіб(a)] можуть бути одержані шляхом взаємодії сполуки формули XI, в якій R1-3 такі, як ви3Начено вище, з комбінацією сполуки формули XII, в якій R4 і L такі, як ви3Начено вище, і оксихлориду фосфору в ди хлорметані при температурі кипіння розчинника із зворотним холодильником. Сполуки формул ХIIIа або ХIIIb [див. спосіб(b)], в яких X являє собою СН, можуть бути одержані з сполук формул XVa або XVb, відповідно, в яких R1-3, R6 R7 m, n та p такі, як ви3Начено вище, шляхом барботування газоподібного НСl через розчин сполуки в дихлорметані. Сполуки формул XVa або XVb можуть бути одержані з сполук формул XVIa або XVIbr відповідно, в яких R1-3, R6, R7, m, n та р такі, як ви3Начено вище, шляхом циклізації з використанням гідроксиду калію, за підвищеної температури(такої як 90°С), в ДМСО, або діізопропіламіду літію в розчиннику, що не справляє несприятливої дії на реакцію(наприклад, в тетрагідрофурані), при температурі, близької до кімнатної, і з гасінням водою. Сполуки формул XVIa або XVIb можуть бути одержані шляхом взаємодії сполуки формули XI, як ви3Начено вище, з сполукою формул XVIIa або XVIIb, відповідно, в яких R6, R7, m, n і p такі, як ви3Начено вище, за допомогою способу, описаного вище для одержання сполук формули X. Сполуки формул ХIIIа або ХІІІb, в яких X являє собою N, можуть бути одержані шляхом взаємодії сполуки формули XXII, в яких R1-3 такі, як ви3Начено вище, з сполукою формул ХХllа або ХХІІb, відповідно, в яких R6, R7, m, n та р такі, як ви3Начено вище, з використанням умов, за3Начених вище для способу (d), що його наведено вище. Сполуки формули XVIII [див. Спосіб (c)], де X являє собою СН, можуть бути одержані шляхом циклізації сполуки формули XX, в якій R1, R2, R4 , і L такі, як ви3Начено вище, з використанням умов реакції, за3Начених для способу (а), що його наведено вище. Сполуки формули XX можна одержати шляхом взаємодії сполуки формули XXI, в якій R1 і R2 такі, як ви3Начено вище, з сполукою формули XII, як ви3Начено вище, з використанням способу, описаного вище, для одержання сполук формули X. Сполуки формули XVIII, де X являє собою N, можуть одержуватись шляхом взаємодії сполуки формули XXVII, в якій R1 і R2 такі, як ви3Начено вище, з сполукою формул ХХlllа або ХХlllb, відповідно, як ви3Начено вище, з використанням умов реакції, за3Начених вище для способу (d). Сполуки формули XXII [див. Способи (d) і(h)] можуть одержуватись з сполуки формули XXl V, в якій R1-3 такі, як ви3Начено вище, шля хом взаємодії з РОСl3 і N,N-диметиланіліном, з подальшою обробкою аміаком. Сполуки формули XXIV можна одержати з сполуки формули XXV, в якій R1 і R2 такі, як ви3Начено вище, шляхом взаємодії з сполукою формули ХІХ як ви3Начено вище, з використанням умов реакції, описаних вище для способу (c). Сполуки формули XXV можуть бути одержані з сполук формули XXVI, де R1 і R2 такі, як ви3Начено вище, з використанням підхожих методів. Сполуки формули XXII можуть бути також одержані згідно зі схемою 1: Сполуки формули XXVIIIa або XXVIIIb [див. Спосіб (e)], в яких Lg являє собою СІ, можуть бути одержані з сполук формули ХІllа або Хlllb, відповідно, шляхом взаємодії з трифосгеном. Ця реакція може проводитись в розчиннику, що не справляє несприятливої дії на реакцію(наприклад, в СН2СІ2), в присутності основи(наприклад, триетиламіну), прибли3Но при -10°С. Сполуки формули X можуть бути також одержані шляхом взаємодії сполуки формули XX з сполукою формули XIX, з використанням умов, описаних для способу (c). Сполуки формул XI, XII, XIV, XVIIa, XVIIb, XIX, XXI, ХХІІа, XXIIb, ХХІllа, XXIIIb, XXVI, XXIX і XXX є або відомими, або доступними за допомогою відомих методів, як ілюстровано за допомогою прикладів. Проміжні сполуки формул X, Хlllа, ХІІІb, XXII, XXVIIIa і XXVIIIb складають наступний аспект даного винаходу. Для фахівців у цій галузі очевидно, що чутливі функціональні групи можуть бути захищені та захисні групи усунені в процесі синтезу сполуки даного винаходу. Це може бути здійснено за допомогою підхожих методів, наприклад, як описано в 'Protective Groups in Organic Synthesis' by T. W. Greene and P. G. M. Wuts, John Wiley and Sons Inc, 1991. Сполуки даного винаходу є корисними, тому що вони мають фармакологічну активність у тварин. Особливо ці сполуки є корисними при лікуванні ряду захворювань, включаючи гіпертонію, інфаркт міокарда, порушення ерекції у самців, гіперліпідемію, серцеву аритмію і доброякісну гіперплазію простати. Останнє захворювання представляє найбільший інтерес. Таким чином, згідно з іншим аспектом даного винаходу, надається спосіб лікування доброякісної гіперплазії простати, який включає в себе введення пацієнту, який страждає на таке захворювання, терапевтично ефективної кількості сполуки цього винаходу. Також пропонується використання сполук даного винаходу як лікарських препаратів, і використання сполук даного винаходу у виробництві ліків для лікування доброякісної гіперплазії простати. Сполуки даного винаходу можуть вводитись за допомогою будь-якого підхожого способу, наприклад, перорально, парентерально(наприклад, внутрішньовенно, трансдермально) або ректально. Необхідна щоденна доза як правило варіює залежно від конкретної сполуки, що використовується, конкретного стану при лікуванні та тяжкості цього стану. Однак, в основному загальна добова доза становить прибли3Но від 0,01 до 10мг/кг ваги тіла, і більш прийнятно прибли3Но від 0,05 до 1мг/кг, її вводять від 1 до 4 разів на день. Пероральне введення представляє особливий інтерес. Сполуки даного винаходу в основному вводять у вигляді підхожої фармацевтичної композиції. Таким чином, згідно з іншим аспектом даного винаходу, пропонується фармацевтична композиція, що включає, більш прийнятно, менше 50% за вагою сполуки даного винаходу в суміші з фармацевтично прийнятним ад'ювантом, розріджувачем або носієм. Фармацевтична композиція, більш прийнятно, перебуває у вигляді одиничної дози. Такі дози включають в себе тверді дозовані форми, наприклад, таблетки, пілюлі, капсули, порошки, гранули і супозиторії для перорального, парентерального або ректального введення; а також рідкі дозовані форми, наприклад, стерильні розчини або суспензії для парентерального введення, підхожим чином ароматизовані сиропи, ароматизовані емульсії з харчовими маслами, такими як бавовняна олія, кунжутна олія, кокосова олія та арахісова олія, а також еліксири і подібні фармацевтичні носії. Тверді композиції можуть бути одержані шляхом змішування активного інгредієнта з фармацевтичними носіями, наприклад, традиційними інгредієнтами для таблетування, такими як кукурудзяний крохмаль, лактоза, сахароза, сорбіт, тальк, стеаринова кислота, стеарат магнію, дикальцій фосфат, камеді та інші розріджувачі, наприклад, вода, для утворення гомогенної попередньо одержаної композиції, в якій активний інгредієнт однорідно диспергований таким чином, щоб його можна було легко поділити на рівно3Начно ефективні одиничні дозовані форми, що містять як правило прибли3Но від 0,1 до 500мг активного інгредієнта. Тверді дозовані форми можуть бути покриті оболонкою, або у інший спосіб приготовані для пролонгування дії композиції. Композиції винаходу можуть також містити сполуку з інгібіторною активністю щодо 5-α-редуктази людини [див. міжнародну патентну заявку WO 95/28397], або сполука цього винаходу може бути представлена у вигляді фармацевтичної упаковки, що також містить сполуку з інгібіторною активністю щодо 5-α-редуктази людини в якості комбінованого препарату для одночасного, роздільного або послідовного застосування. Сполуки даного винаходу можуть тестуватись у наведених нижче експериментах. Скорочувальні відповіді простати людини Тканину простати розрізають на поздовжні смуги(прибли3Но 3 x 2 x 10мм) і суспендують у бані для органів при залишковій напрузі в 1г в бікарбонатному розчині Кребса-Рингера такого складу(мМ): NaCl(119), КСl (4,7), CaCl2(2,5), КН2РО4(1,2), MgSO4(1,2), NaHCO3 (25), глюкоза (11), та газифікують сумішшю 95% О2 /5% СО2. Цей розчин також містить 10мм кокаїн і 10мм кортикостерон. Тканини піддають впливу збуджуючої дози (-)-норадреналіну(100мМ) та промивають протягом 45 хвилин. Ізометричні скорочення одержують у відповідь на кумулятивні додавання (-)-норадреналіну для одержання контрольних кривих в усі х тканинах. Наступну криву потім одержують в присутності або за відсутності антагоніста(інкубують протягом 2годин). 3Начення спорідненості антагоніста(рА2) ви3Начають, використовуючи єдин у концентрацію конкуруючого антагоніста, р А2 = -log[A]/(DR-l), де дозове співвідношення(DR), відносно до відповідного контролю, одержують за допомогою єдиної концентрації антагоніста [А], припускаючи, що конкурентний антагонізм і регресія за Шильдом близькі до єдності. Модель простатичного тиску і кров'яного тиску на анестезованому собаці Статевозрілих самців коротконогих гончих(з вагою тіла 12 - 15кг) анестезують пентобарбітоном натрію(30 - 50мг/кг в/в) і вставляють тра хейну канюлю. Подальшу анастезію підтримують за допомогою інфузії пентобарбітоном. Тварин респірують повітрям за допомогою респіратора Bird Mk8(Bird Corp., Palm Springs, CA, USA) для підтримання рівня газів у крові в інтервалі рО2 90 - 110мм Hg, pCO2 35 - 45мм Hg, pH 7,35 - 7,45. Температуру тіла підтримують при 36 - 37,5°С, використовуючи нагрітий операційний стіл. Катетери приміщують в ліву стегенну артерію для реєстрації кров'яного тиску і в ліву стегенну вен у для введення сполуки. Серцевий пульс реєструють через вивід II E.C.G. Лапаротомію проводять для введення канюль в обидві уретри для запобігання зміні об'єму рідини в сечовому міхурі. Серцевий катетер типу 7F(з шаровою насадкою об'ємом 1,5мл) вводять в сечовий міхур через уретру. Шарову насадку заповнюють повітрям і катетер виводять доки шарик не стає зануреним в простату, що підтверджується цифровим датчиком тиску. Тиск в шарику реєстр ують за допомогою перетворювача Друка. Тиск у простаті та гемодинамічні параметри реєструють за допомогою приладу Grass Polygraph(Grass Instruments, Quincy, Mass, U.S.A.) і дані реєструють в безперервному режимі з використанням мікрокомп'ютерної системи на основі Motorola 68000(Motorola Inc., Temple, AZ, U.S.A.). Сполуки змішують з PEG 300 і вводять внутрішньовенно через катетер в стегенну вену. Для одержання кривої залежності контрольна дозавідповідь реєструють відповіді на введення фенілефрину(в діапазоні 1-16мкг/кг внутрішньовенно в фіз. розчині)(дві контрольні криві для кожного експерименту). Сполуки вводять(з урахуванням сполуки основи) в діапазоні 10 - 300мкг/кг внутрішньовенно за 5 хвилин до одержання кривої від фенілефрину(одержують до максимальної дози, що дорівнює 128мкг/кг, в присутності тестової сполуки). Внаслідок α1-подібних дизритмічних властивостей фенілефрину абсолютні максимальні відповідні сигнали не були одержані, а взяті на рівні, що на 10% перевищує контрольний сигнал, одержаний від 16мкг/кг фенілефрину. Концентрації лікарських препаратів розраховують на основі співвідношення молярна вага сполуки/кг ваги тіла, таким чином дозволяючи проводити розрахунки по схемі "псевдо рА2" з використанням аналізу за Шильдом із застосуванням дозових співвідношень, одержаних по зсувам на кривих доза фенілефрину-відповідь. Перевага сполук даного винаходу полягає в тому, що вони є більш ефективними, мають більшу тривалість дії, більш широкий діапазон активності, є більш стабільними, мають менше побічних ефектів або є більш селективними(особливо, вони можуть справляти сприятливий ефект при лікуванні доброякісної гіперплазії простати, не викликаючи небажаних серцево-судинних ефектів, наприклад, тому що вони здатні бути селективними антагоністами підтипів cti-адренорецепторів рецепторів простати) або мають інші більш корисні властивості, ніж сполуки попереднього рівня техніки. Винахід ілюструється наступними прикладами, в яких можуть використовуватись такі скорочення: ВиОН = бутанол DMA = диметилацетамід DMF = диметилформамід DMPU = 1,3-диметил-3,4,5,6-тетрагідро-2(1H)-піримідон DMSO = диметилсульфоксид EDTA = етилендіамінтетраоцтова кислота EtOAc = е тилацетат EtOH = етанол год =година МеОН = метанол хвил = хвилина n-BuOH = н-бутанол p.s.i. = фунтів на кв. дюйм THF = тетрагідрофуран ТШХ = тонкошарова хроматографія Проміжна сполука 1 1-(т-бутилоксикарбоніл)-1,4-діазепан До розчину гомопіперазину(100г, 1,0моль) і триетиламіну(210мл, 152г, 1,5моль) в СН2СІ2(500мл) при 0°С додають розчин ди-(т-бутил)дикарбонату(195г, 0,89моль) в СН2СІ2(300мл). Суміш залишають нагріватися до кімнатної температури і перемішують протягом 18год, після чого СН2СІ2 упарюють за пониженого тиску. Одержаний залишок поділяють між ефіром і 2N лимонною кислотою та водний шар екстрагують ефіром(4x200мл). Водний шар підлуговують 2N водним NaOH і потім екстрагують СН2СІ2(4x400мл). Об'єднані СН2СІ2 екстракти промивають Н 2О(2х), насиченим сольовим розчином(1х) і висушують над MgSO4 . Упарювання за пониженого тиску супроводжується азеотропною перегонкою з СН2СІ2(4х), з одержанням названої у заголовку сполуки у вигляді жовтої воскоподібної твердої речовини(94,3г, 53%). Rf 0,25(СН2Сl2/МеОН/0,88 NH 3 90/10/1, об/об). MC m/z 201(МН+). Виявлено: С 58,86; Н, 10,03; N, 13,58; C 10H20N2 O2 0,05СН2Сl2 відповідає С, 59,02; Н, 9,91; N, 13,70%. Проміжна сполука 2 1-(т-бутилоксикарбоніл)-4-(4-морфолінкарбоніл)-1,4-діазепан Розчин проміжної сполуки 1(92,0г, 0,46моль) і триетиламіну(96,0мл, 69,7г, 0,69моль) в СН2СІ2(500мл) при 0°С обробляють краплями розчином 4-морфолінкарбоніл хлориду(64,0мл, 82,0г, 0,55моль) в СН2СІ2(100мл) і реакцію перемішують при кімнатній температурі в атмосфері N 2 протягом 18год. Реакційну суміш потім розбавляють СН2СІ2(400мл) і промивають 2N лимонною кислотою(3x400мл), насиченим сольовим розчином(1x500мл), висушують над MgSO4 і упарюють, одержуючи названу в заголовку сполуку у ви гляді не зовсім білої твердої речовини(141,7г, 98%). Rf 0,80(CH2Cl2/MeOH/0,88 NH3 90/10/1, об/об). MC m/z 314(МН+). Виявлено: С, 57,50; Н, 8,69; N, 13,41; C 15H27N3O 4 відповідає С, 57,50; Н, 8,69; N, 13,41%. Проміжна сполука 3 Гідрохлорид 1-(4-морфолінкарбоніл)-1,4-діазепану Розчин проміжної сполуки 2(140,0г, 0,44моль) в СН2Сl2/МеОН(1/1, об/об,600мл) при 0°С насичують газоподібним НСl і реакційну суміш перемішують при кімнатній температурі в атмосфері N2 протягом 18год, після чого реакційну суміш упарюють за пониженого тиску і суспендують в EtOAc, одержуючи після фільтрування білу гідроскопічну тверду речовину. Потім її очищають шляхом суспендування в ацетоні, фільтрування, промивання ефіром і висушування у вакуумі при 60°С, одержуючи названу в заголовку сполуку у вигляді безбарвної твердої речовини(99,0г, 90%). Rf 0,41(СН2Сl2/МеОН/0,88 NH3 84/14/2, об/об). MC m/z 214(МН+) Виявлено: С, 47,50; Н, 8,10; N, 16,55; C 10H19N3O2 HCl 0,2 Н2О відповідає С, 47,41; Н, 8,12; N, 16,59%. Проміжна сполука 4 1-Ацетил-4-(4-морфолінкарбоніл)-1,4-діазепан До розчину проміжної сполуки 3(50 г, 0,2моль) і триетиламіну(42мл, 30,5 г, 0,3моль) в СН2СI2(400мл) при 5°С додають оцтовий ангідрид(23мл, 24,9г, 0,24моль) краплями протягом 15 хвил, і реакцію потім перемішують протягом ще 2год при кімнатній температурі в атмосфері N2. Розведення за допомогою СН2СІ2(600мл) супроводжується промиванням насиченим водним розчином бікарбонату натрію(2x200мл)г і об'єднані водні шари екстрагують СН 2Сl2(1x100мл). СН2Сl2 шари об'єднують і промивають насиченим сольовим розчином, висушують над MgSO4 та упарюють, одержуючи світло-коричневе масло. Його розчиняють в СН 2СІ2(300мл) і обробляють триетиламіном(8мл, 5,8г, 0,06моль) і EtOH(5мл), перемішують протягом 1год при кімнатній температурі, потім промивають насиченим водним бікарбонатом натрію, і водний шар екстрагують СН 2СІ2(5 х). Об'єднані СН 2СІ2 шари висушують над MgSO4 і упарюють за пониженого тиску, одержуючи жовте масло, яке потім піддають азеотропній перегонці з СН2СІ2(4х), одержуючи названу в заголовку сполуку у вигляді жовтого масла(47,1г, 92%). Rf 0,45(СН2Сl2/МеОН/0,88 NH3 90/10/1, об/об). MC m/z 256(МН+). Виявлено: С, 52,62; Н, 8,18; N, 15,02; C 12H21N3O 3 0,ЗСН2Сl2 відповідає С, 52,61; Н, 7,75; N, 14,96%. Приклад 1 4-Аміно-6,7-диметокси-2-[4-(4-морфолінкарбоніл)-1,4-діазепан-1-іл]-5-(тіофен-3-іл)хінолін (а) 2-(3,4-диметоксифеніл)-4,4-диметил-Δ2 оксaзолін Названу у підзаголовку сполуку одержують з 3,4-диметоксибензойної кислоти згідно з способом Meyers et al, J. Org. Chem., 39,2787(1974). (b) 2-(3,4-диметокси-2-йодфеніл)-4,4-диметил-Δ2 -оксазолін н-Бутиллітій(2,5M в гексані, 8,9мл, 22,3ммоль) додають краплями до розчину продукту стадії(а)(4,2г, 17,8ммоль) в сухому е фірі(200мл) при 0°С і реакцію перемішують в атмосфері N 2 протягом 2год. Потім краплями додають розчин йоду(5,46г, 21,5ммоль) в ефірі(100мл) і реакцію залишають нагріватися до кімнатної температури протягом 1год. Реакційну суміш виливають в Н 2О, ефірний шар відділяють, промивають насиченим водним розчином тіосульфату натрію(їх), потім насиченим сольовим розчином(їх), потім висушують над MgSO4 та упарюють за пониженого тиску, одержуючи названу в підзаголовку сполуку у вигляді жовтого масла(5,2г, 80%). Rf 0,60(СН2Сl2/МеОН/0,88 NH 3 90/10/1, об/об). MC m/z 362 (c) 3,4-Диметокси-2-йодбензонітрил До розчину продукту стадії(b)(5,2г, 14,4ммоль) у піридині(30мл) додають оксихлорид фосфору(2,7мл, 4,4г, 28,8ммоль) і реакцію нагрівають до 85°С протягом 18год. Реакційну суміш о холоджують, поділяють між насиченим водним розчином карбонату натрію(300мл) і потім екстрагують ефіром(2x100мл). Ефірний шар промивають 2N НСl(2x75мл), потім Н2О(1х), висушують над MgSO4 та упарюють за пониженого тиску, одержуючи жовте масло. Його очищають шляхом суспендування в гексані і фільтрування, одержуючи названу в підзаголовку сполуку у вигляді не зовсім білої твердої речовини(2,82г, 68%). Rf 0,80(СН2Сl2/МеОН 95/5, об/об). MC m/z 307(МН+). Виявлено: С, 38,03; Н, 2,88; N, 4,64; C 9H8NO2 0,05 гексан відповідає С, 38,05; Н, 2,97; N, 4,77%. (d) 3,4-диметокси-2-йод-6-нітробензонітрил Нітронію тетрафторборат(1,73г, 13,0ммоль) додають порціями до розчину продукту стадії(c)(2,67г, 9,2ммоль) в ацетонітрилі(40мл) при 0°С. Реакцію перемішують протягом 30 хвил. в атмосфері N2 і потім виливають в насичений водний розчин бікарбонату натрію і екстрагують EtOAc(їх). Органічний шар промивають насиченим сольовим розчином(їх), висушують над MgSO4 та упарюють за пониженого тиску, одержуючи залишок, який суспендують в гексані і фільтрують, одержуючи названу в підзаголовку сполуку у вигляді не зовсім білої твердої речовини(2,51г, 82%). Rf 0,46(ЕtOАс/гексан 1/1, об/об), MC m/z 352(MNH 4). (e) 6-Аміно-3,4-димето кси-2-йо дбензонітрил До розчину продукту стадії(d)(3,50г, 0,01моль) в СН2Сl2(90мл) додають розчин дитіоніту натрію(20,11, 0,11моль) в Н2О(60мл). До одержаної суміші додають хлорид те тра-н-бутиламонію(1,45г, 5,24ммоль) і реакцію інтенсивно перемішують протягом 1,5год. Суміш поділяють між СН 2Сl2 і Н2О, органічний шар відділяють, висушують над MgSO4 і упарюють за пониженого тиску. Залишок поділяють між EtOAc та 2N НСl, потім водний шар підлуговують 2N водним NaOH і екстрагують EtOAc(3 х). Об'єднані органічні шари висушують над MgSO4 і упарюють за пониженого тиску, одержуючи залишок, який очищають на силікагелі, елююючи СН 2Сl2, одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(1,69г, 53%), Rf 0,55(ЕtOАс/гексан 1/1, об/об), MC m/z 322(MNH 4). (f) 3,4-диметокси-2-йод-6-{-[4-(4-мйрфолінкарбоніл)-1,4-діазепан-1-іл]етиліденаміно}бензонітрил Оксихлорид фосфору(0,6мл, 6,08ммоль) додають до розчину проміжної сполуки 4(2,82г, 11,0ммоль) в СН2Сl2(20мл) і реакцію перемішують протягом 20 хвил при кімнатній температурі. Потім додають продукт стадії(e)(1,68г, 5,52ммоль) і реакцію нагрівають із зворотним холодильником протягом 18год, після чого її охолоджують, виливають в лід, суміш підлуговують водним бікарбонатом натрію та продукт екстрагують EtOAc(3х). Об'єднані органічні екстракти висушують над MgSO4 і упарюють за пониженого тиску. Неочищений продукт очищають на силікагелі, елююючи СН2Сl2/МеОН(97/3, об/об), одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(2,60г, 87%). Rf 0,15(СН2Сl2). MC m/z 542(МН+). Виявлено: С, 46,00; Н, 5,17; N, 12,44; C 21H28N5O 4I 0,1×СН2Сl2 відповідає С, 46,08; Н, 5,17; N, 12,74%. (g) 4-Аміно-6,7-димето кси-5-йо д-2-[4-(4-морфолінкарбоніл)-1,4-діазепан-1-іл]хінолін Розчин продукту стадії(f)(2,0г, 3,7ммоль) в суміші ТГФ(50мл) і DMPU(10мл) охолоджують до -78°С і обробляють розчином діізопропіламіду літію в циклогексані(1,5М, 2,7мл) в атмосфері N2. Реакцію нагрівають до 0°С і перемішують впродовж 30хвил, після чого реакцію 3Нову охолоджують до -78°С і додають ще порцію діізопропіламіду літію в ТГФ(1,5М, 2,7мл). Реакційну суміш нагрівають до кімнатної температури і перемішують протягом 30 хвил, після чого її 3Нову охолоджують до -78°С і обробляють третьою порцією діізопропіламіду літію в ТГФ(1,5 М, 2,0мл). Реакцію 3Нову нагрівають до кімнатної температури та перемішують протягом 20хвил, після чого її гасять Н2О і екстрагують EtOAc(3х). Органічний шар промивають послідовно Н 2О і насиченим сольовим розчином, висушують над MgSO4 та упарюють за пониженого тиску. Неочищений продукт очищають на силікагелі, елююючи СН 2СІ2/МeОН(98/2, об/об). Названу в підзаголовку сполуку(1,30г, 65%) одержують у вигляді світло-коричневої твердої речовини. Rf -0,50(СН2СІ2/МеОН/0,88 NH3 90/10/1, об/об). MC m/z 542(MH+). Виявлено: С, 45,71; Н, 5,26; N, 12,44; C21H28N5 O4 I 0,25×СН2СІ2 відповідає С, 45,37; Н, 5,07; N, 12,46%. (b) 4-Аміно-6,7-диметокси-2-[4-(4-морфолінкарбоніл)-1,4-діазепан-1-іл]-5-(тіофен-3-іл)хінолін До розчину продукту стадії(g)(500мг, 0,92ммоль) в суміші толуолу(6мл) і EtOH(3мл) додають тіофен-3боронову кислоту(236мг, 1,85моль), тетракіс(трифенілфосфін)паладій(32мг, 0,03ммоль) і 1М водний розчин карбонату натрію(1мл) та реакційну суміш нагрівають із зворотним холодильником в атмосфері N2 протягом 18год. При охолоджуванні реакційну суміш розбавляють Н 2O екстрагують EtOAc(3х). Об'єднані органічні шари висушують над MgSO4 і упарюють за пониженого тиску. Продукт очищають на силікагелі, елююючи СН 2Сl2/МеОН/0,88 NН3(90/10/1, об/об), одержуючи названу в заголовку сполуку у вигляді безбарвної піни(230мг, 47%). Rf 0,50(СН2Сl2/МеОН/0,88 NH3 90/10/1, об/об). MC m/z 498(МН+). 1Н ЯМР(CDCl) δ 2,05(2Н, м), 3,13(4Н, м), 3,35(2Н, м), 3,50(3Н, с), 3,63(6Н, м), 3,71(2Н, м), 3,97(5Н, м), 4,30(2Н, шир. с), 5,76(1H, с), 7,10(2Н, м), 7,45(2Н, м). Виявлено: С, 57,90; Н, 6,19; N, 13,04; C25H31N5O4S 0,3СН2Сl2 відповідає С, 57,85; Н, 6,07; N, 13,32%. Приклад 2 4-Аміно-6,7-диметокси-2-[4-(4-морфолінекарбоніл)-1,4-діазепан-1-iл]-5-(тіофен-2-іл)хінолін Названу в заголовку сполуку одержують у спосіб прикладу 1(h) з сполуки прикладу 1(g) і тіофен-2 боронової кислоти. Неочищений продукт очищають на силікагелі, елююючи СН2Сl2/МеОН/0,88 NH3(90/10/1, об/об), одержуючи названу в заголовку сполук у(26%) у вигляді безбарвної піни. MC m/z 498(МН+). 1Н ЯМР(CDCl3) δ 2,05(2Н, м), 3,13(4Н, м), 3,32(2Н, м), 3,61(9Н, м), 3,74(2Н, м), 3,97(2Н, м), 4,00(3Н, с), 4,60(2Н, шир. с), 5,77(1H, с), 7,0-7,3(1H, шир. с), 7,06(1H, д),7,15(1H, дв. д),7,52(1H, д). Виявлено: С, 55,25; Н, 5,92; N, 12,63; C 25H31N5 O4S 0,7×СН2Сl2 відповідає С, 55,40; Н,5,86; N, 12,57%. Приклад 3 4-Аміно-6,7-диметокси-5-(2-фурил)-2-[4-(4-морфолінкарбоніл)-1,4-діазепан-1-іл]хінолін Названу в заголовку сполуку одержують у спосіб прикладу 1(h) з сполуки прикладу 1(g) і фуран-2боронової кислоти [Florentin et al., J. Heterocyclic Chem., 13, 1265(1976)]. Неочищений продукт очищають на силікагелі, елююючи СН 2Сl2/МеОН/0,88 NH3(90/10/1, об/об), з одержанням названої у заголовку сполуки(62%) у вигляді безбарвної піни. Rf 0,52(СН2Сl2/МеОН/0,88 NHs 90/10/1, об/об). MC m/z 482(МН+). 1 Н ЯМР(CDCl3) δ 2,06(2Н, м), 3,16(4Н, м), 3,37(2Н, м), 3,50(2Н, м), 3,60(7Н, м), 3,71(2Н, м), 3,97(2Н, м), 4,00(5Н, м), 5,80(1H, с), 6,50(1H, шир. с), 6,60(1Н, шир. с), 7,0-7,3(1H, шир. с), 7,62(1Н, шир. с). Виявлено: С, 60,36; Н,6,52; N, 13,46. C 25H31N5O 5 0,25×СН2Сl2 відповідає С, 60,29; Н,6,31; N, 13,92%. Приклад 4 4-Аміно-6,7-диметокси-5-(3-фурил)-2-[4-(4-морфолінкарбоніл)-1,4-діазепан-1-іл]хінолін Названу в заголовку сполуку одержують у спосіб прикладу 1(h) з сполуки прикладу 1(g) і фуран-3боронової кислоти [Florentin et al., J, Heterocyclic Chem., 13, 1265(1976)]. Неочищений продукт очищають на силікагелі, елююючи СН 2Сl2/МеОН/0,88 NH3(90/10/1, об/об), з одержанням названої у заголовку сполуки(60%) у ви гляді безбарвної піни. MC m/z 482(МН+). 1Н Я МР(CDCl3) δ 2,05(2Н, м), 3,13(4Н, м), 3,32(2Н, м), 3,55(3Н, с), 3,65(6Н, м), 3,74(2Н, м), 3,99(5Н, м), 4,55(2Н, шир. с), 5,77(1H, С), 6,50(1Н, с), 7,17,4(1Н, шир. с), 7,50(1Н, с), 7,60(1Н, с). Виявлено: С, 60,22; Н, 6,38; N, 13,76; C25H31N5O 5 0,25×СН2Сl2 відповідає С, 60,29; Н 6,31; N, 13,92%. Приклад 5 4-Аміно-6,7-диметокси-2-[4-(4-морфолінкарбоніл)-1,4-діазепан-1-іл]-5-(2-піридил)хінолін До розчину сполуки прикладу 1(g)(700мг, 1,29ммоль) в діоксані(15мл) додають 2-(три-нбутилстаніл)піридин(1,42г, 3,88ммоль), тетракіс(трифенілфосфін)-паладій(150мг, 0,13ммоль), йодид міді(І)(37мг, 0,19ммоль) і хлорид літію(271мг, 6,5ммоль), суміш нагрівають із зворотним холодильником в атмосфері N2 протягом 18год. При охолодженні реакційну суміш концентрують за пониженого тиску і залишок поділяють між 2N НСl і EtOAc. Водний шар промивають трьома додатковими порціями EtOAc і потім підлуговують 2N водним NaOH. Продукт потім екстрагують EtOAc(3x) висушують над MgSO4 і упарюють за пониженого тиску. Неочищений продукт очищають на силікагелі, елююючи СН2СІ2/МеОН/0,88 NH3(90/10/1, об/об), з одержанням названої у заголовку сполуки у вигляді безбарвної твердої речовини(210мг, 33%). Rf 0,23(СН2Сl2/МеОН/0,88 NH3 90/10/1, об/об). MC m/z 493(МН+). 1Н ЯМР(CDCl3) δ 2,05(2Н, м), 3,15(4Н, м), 3,32(2Н, м), 3,50(2Н, м), 3,55(3Н, с), 3,60(2Н, м), 3,68(4Н, м), 3,72(2Н, м), 3,94(2Н, м)г 4,00(3Н, с), 5,80(1H, с), 7,16(1Н, шир. с), 7,38(1H, м), 7,48(1Н, м), 7,60(1H, с), 8,74(1Н, шир. с). Виявлено: С, 60,89; Н, 6,41; N, 16,03; C 26H32N6O 4 0,3×СН2Сl2 відповідає С, 60,71; Н, 6,32; N, 16,14%. Приклад 6 4-Аміно-6,7-диметокси-2-[4-(4-морфолінкарбоніл)-1,4-діазепан-1-іл]-5-(тіофен-3-іл)хіназолін (а) 3,4-Диметокси-2-йодбензойна кислота Розчин сполуки прикладу 1(b)(115г, 0,32моль) в суміші 3N НСl(530мл) і EtOH(200мл) нагрівають із зворотним холодильником протягом 36год. При охолоджуванні продукт фільтрують, висушують на повітрі і потім промивають гексаном. Тверду речовину потім розчиняють в СН 2СІ2, висушують над МgSO4 та упарюють за пониженого тиску, одержуючи названу в підзаголовку сполуку у вигляді білої твердої речовини. Rf 0,38(EtOAc). MC m/z 309(МН+). (b) Eтиловий ефір 3,4-диметокси-2-йодбензойної кислоти До суспензії продукту стадії(а)(69,3г, 0,23моль) в СН2СІ2 при 0°С додають оксаліл хлорид(25мл, 0,27моль) і DMF(0,9мл, 11,3ммоль) і реакцію перемішують протягом 18год при кімнатній температурі. Потім в реакцію додають EtOH(20мл, 0,34моль), перемішують протягом ще 30 хвил, після чого її обробляють триетиламіном(78мл, 0,56моль). Реакційну суміш поділяють між СН 2СІ2 і Н2О. Органічний шар відділяють, промивають послідовно 2N НСl(3х) і насиченим сольовим розчином, висушують над MgSO4 та упарюють за пониженого тиску, одержуючи коричневе масло. Продукт очищають на силікагелі, елююючи СН2СІ2, з одержанням названої в підзаголовку сполуки у вигляді коричневого масла(30г, 39%). Rf 0,73(EtOAc). MC m/z 337(MH+). (c) Етиловий ефір 3,4-Диметокси-2-йод-6-нітробензойної кислоти Нітроній тетрафторборат(11г, 84ммоль) додають до розчину продукту стадії(b)(30г, 64ммоль) в ацетонітрилі(300мл) при 0°С і реакцію перемішують протягом 1,5год в атмосфері N2. Реакційну суміш потім розбавляють ефіром, підлуговують 2N водним NaOH і водний шар екстрагують ефіром(3х). Об'єднані органічні шари промивають насиченим сольовим розчином, висушують над MgSO4 та упарюють за пониженого тиску. Продукт очищають на силікагелі, елююючи гексан/EtOAc(85/15, об/об), з одержанням названої в підзаголовку сполуки у вигляді жовтої твердої речовини(21,3г, 87%), Rf 0,77(EtOAc). MC m/z 382(MH+). (d) E тиловий ефір 6-aміно-3,4-диметокси-2-йодбензойної кислоти Названу в підзаголовку сполуку одержують у спосіб прикладу 1(e) з продукту стадії(c). Зазначену в підзаголовку сполуку(84%) одержують у вигляді безбарвної твердої речовини. Rf 0,67(EtOAc). MC m/z 352(MH+). (e) 2,4-дигідрокси-6,7-диметокси-5-йодхіназолін Ціанат натрію(9г, 0,14моль) і трифтороцтову кислоту(11мл, 0,14моль) додають при перемішуванні до розчину продукту стадії (d) (12г, 34,2ммоль) в СН2СІ2 і перемішування продовжують протягом 18год. Реакційну суміш потім упарюють за пониженого тиску, додають Н2О і одержану тверду речовину фільтрують, промивають водою. Суспензію твердої речовини в Н 2О(50мл) обробляють гранульованим NaOH(10г) і суміш нагрівають до 60°С протягом 30хвил, після чого реакцію охолоджують, нейтралізують концентрованою НСl і одержану тверду речовину виділяють шляхом фільтрування, промивання Н2О та ефіром. Зазначену в підзаголовку сполуку одержують у вигляді безбарвної твердої речовини(8,4г, 71%). Rf 0,30(EtOAc). 1Н Я МР(D6-D MSO) δ 3,70(3Н, с), 3,94(3Н, с), 9,13(2Н, шир. с), 12,10(1Н, шир. с). (f) 2,4-Дигідрокси-6,7-диметокси-5-(тіофен-3-іл)хіназолін Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1 (h) з продукту стадії (e). Зазначену в підзаголовку сполуку(84%) одержують у вигляді блідо-жовтої твердої речовини. Rf 0,28(EtOAc). MC m/z 305(MH+). (g) 4-Аміно-2-хлор-6,7-диметокси-5-(тіофен-3-іл)хінaзолін Продукт стадії (f) додають до суміші оксихлориду фосфору(9млг 96ммоль) і N,N-диметаланіліну(1мл, 8ммоль) та реакційну суміш нагрівають до 110°С протягом 5год. При охолоджуванні реакційну суміш виливають в лід та поділяють між 2N НСl і ефіром. Органічний шар відділяють, промивають насиченим сольовим розчином і упарюють, одержуючи коричневе масло. Його приміщують в суміш СН 2СІ2(100мл) і МеОН(100мл), охолоджують до 0°С і насичують NH 3. Реакцію перемішують протягом 20год, насичують ще раз NH3 і перемішують протягом ще 5год. Реакційну суміш упарюють досуха за пониженого тиску і залишок розподіляють між EtOAc і 2N НСl. Органічний шар промивають насиченим сольовим розчином, висушують над MgSO4 та упарюють за пониженого тиску. Перетирають з метанолом і фільтрують, одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(255мг, 25%). Rf 0,78(EtOAc). MC m/z 322(MH+). (b) 4-Аміно-6,7-диметокси-2-[4-(4-морфолінкарбоніл)-1,4-діазепaн-1-іл]-5-(тіофен-3-іл)хінaзолін Суміш продукту стадії (g) (220мг, 0,68ммоль), триетиламіну(0,24мл, 1,7ммоль) і проміжної сполуки 3(250мг, 1,0ммоль) в н-бутанолі(50мл) нагрівають до 100°С в атмосфері N 2 протягом 5 днів. Після охолоджування реакційну суміш розподіляють між 2N водним NaOH і EtOAc. Органічний шар відділяють і промивають додатковими порціями 2N водного NaOH(2x)r потім насиченим сольовим розчином(2х). Після висушування над MgSO4 та упарювання за пониженого тиску, продукт перетирають з EtOAc, фільтр ують і перекристалізовують з толуолу, одержуючи названу в заголовку сполук у у вигляді безбарвної твердої речовини(33мг, 10%). Rf 0,08(EtOAc). MC m/z 499(MH+). 1H Я МР(CDCl3) δ 2,03(2Н, м), 3,18(4Н м), 3,35(2Н, м), 3,50(3Н, с)г 3,55(2Н, м), 3,65(4Н, м), 3,84(2Н, м), 3,99(5Н, м), 4,71(2Н, шир. с), 6,90(1Н, с), 7,12(1H, д), 7,30(1Н, д), 7,50(1Н, дв. д). Виявлено: С, 57,86; Н,6,03; N,16,45; C24H30N6O4S відповідає С, 57,82; Н, 6,07; N, 16,85%. Приклад 7 4-Аміно-6,7-диметокси-2-[4-(4-морфолінкарбоніл) -1,4-діазепан-1-іл]-5-(3-піридил)хіназолін (а) На трієва сіль 4-аміно-6,7-диметокси-2-гідрокси-3-йодхіназоліну Суспензію сполуки прикладу 1(e)(9,16г, 30ммоль) в СН2СІ2(200мл) обробляють ціанатом натрію(7,9г, 120ммоль) і трифтороцтовою кислотою(8,4мл, 105ммоль) краплями при кімнатній температурі в атмосфері N2 та реакцію перемішують протягом 60год. Суміш потім упарюють за пониженого тиску і одержану тверду речовину суспендують в суміші водного NaOH(20г в 150мл) і МеОН(200мл) і реакцію перемішують при кімнатній температурі протягом 1год. Одержаний оранжевий розчин потім упарюють за пониженого тиску для вилучення МеОН та одержану водну суспензію обробляють EtOAc, фільтрують і тверду речовину промивають послідовно Н2О(3х), ацетоном(3х) і ефіром, одержуючи названу в підзаголовку сполуку у вигляді блідо-жовтої твердої речовини(7,75г, 69%). Rf 0,53(CH2Cl2/MeOH/0,88 NH3 84/14/2, об/об). MC m/z 322(МН+). (b) 4-Аміно-2-хлор-6,7-диметокси-5-йодхіназолін DMF(1,8мл, 23,0ммоль) додають краплями до оксихлориду фосфору(5,4мл, 57,6ммоль) і потім додають продукт стадії (а)(4,0г, 11,5ммоль). Одержану суміш нагрівають до 90°С протягом 30хвил, після чого додають додаткову кількість(5мл) оксихлориду фосфор у та нагрівання продовжують протягом 18год. Реакційну суміш охолоджують і обережно виливають в суміш EtOAc(400мл) і Н2О(200мл), суміш нейтралізують водним бікарбонатом натрію і водний шар екстрагують ЕtOАс(2х). Об'єднані органічні шари об'єднують, промивають насиченим сольовим розчином, висушують над MgSO4 та упарюють за пониженого тиску, одержуючи коричневу тверду речовину. її суспендують в 2N водному NaOH(300мл), додають діоксан(100мл) і суміш нагрівають до 90°С при інтенсивному перемішування! протягом 2 хвил. При охолоджуванні тверду речовину відділяють і збирають шляхом фільтрування, промивають послідовно Н2О і ацетоном, та висушують у вакуумі при 60°С, одержуючи названу в підзаголовку сполуку у вигляді не зовсім білої твердої речовини(2,79г, 66%). Rf 0,76(EtOAc). MC m/z 366,368(MH+). (c) 4-Аміно-6,7-диметокси-5-йод-2-[4-(4-морфолінкарбоніл)-1,4-діазепан-1-іл]хіназолін Зазначену в підзаголовку сполуку одержують у спосіб прикладу 6(h) з продукту стадії(b). Зазначену в підзаголовку сполуку одержують з кількісним виходом у вигляді світло-коричневої піни. Rf 0,41(EtOAc). MC m/z 543(MH+). (d) 4-Аміно-6,7-диметокси-2-[4-(4-морфолінкарбоніл)-1,4-дія зепан-1 -іл]-5-(3 -піридил)хіназолін До розчину продукту стадії(c)(300мг, 0,55ммоль) в суміші THF(25мл) і Н2О(5мл) додають 3піридилдіетил боран(485мг, 3,3ммоль), тетракіс(трифенілфосфін)паладій(64мг, 0,055ммоль) і гідроксид калію(600мг, 10,7ммоль) та суміш нагрівають із зворотним холодильником протягом 18год в атмосфері N2. Після охолоджування реакційну суміш розподіляють між EtOAc і 2N водним NaOH, водний шар відділяють і екстрагують двома додатковими порціями EtOAc. Об'єднані органічні шари промивають насиченим сольовим розчином, висушують над MgSO 4 та упарюють за пониженого тиску, одержуючи піну. Продукт очищають на силікагелі, елююючи СН 2Сl2/МеОН(95/5, об/об), з одержанням названої у заголовку сполуки у вигляді безбарвної піни(42мг, 15%). Rf 0,10(СН2Сl2/МеОН 95/5, об/об). MC m/z 494(МН+). 1Н Я МР(CDCl3) δ 2,00(2Н, м), 3,18(4Н, м), 3,35(2Н, м), 3,50(3Н, с), 3,55(2Н, м), 3,67(4Н, м), 3,84(2Н, м), 3,97(2Н, м), 4,00(3Н, с), 4,48(2Н, шир. с), 6,97(1Н, с), 7,45(1H, м), 7,74(1H, м), 8,68(1H, м), 8,74(1Н, м). Виявлено: С, 59,85; Н, 6,42; N, 18,54; C 24H31N7 O4 0 ,2×EtOAc 0,5×Н2О відповідає С, 59,57; Н, 6,51; N, 18,85%. Приклад 8 4-Аміно-6,7-диметокси-2-[4-(4-морфолінкарбоніл)-1г4-діазепан-1-іл]-5-(2-піридил) -хіназолін Названу в заголовку сполуку одержують у спосіб прикладу 5 з сполуки прикладу 7(c). Продукт очищають на силікагелі, елююючи СН 2Сl2/МеОН(95/5, об/об), потім перетирають з гексан/ЕtOАс і перекристалізовують з толуолу, одержуючи названу в заголовку сполуку(19%) у ви гляді безбарвної твердої речовини. Rf 0,25(СН2СІ2/МeОН 95/5, об/об). MC m/z 494(МН+). 1Н ЯМР(CDCl3) 5 2,00(2Н, р), 3,15(4Н, т), 3,30(2Н, дв. д), 3,45-3,58(2Н, м), 3,50(3Н, с), 3,65(4Н, т), 3,82(2Н, т), 3,94(2Н, т), 3,97(3Н, с), 4,55(2Н, с), 6,94(1Н, с), 7,39(1H, м), 7,42(1Н, д), 7,82(1H, т), 8,77(1Н, д). Виявлено: С, 59,91; Н, 6,27, N, 19,23; C25H31N7 O4 0,5×Н2 О відповідає С, 59,75; Н, 6,42, N, 19,50%. Приклад 9 4-Аміно-6,7-диметокси-5-(2-піридил)-2-(5,6,7,8-тетрагідро-1,6-нафтирид-6-ил)хіназолін (а) 4-аміно-6,7-диметокси-5-йод-2-(5,6,7,8-тeтрагідро-1,6-нaфтирид-6-ил)хіназолін Зазначену в підзаголовку сполуку одержують у спосіб прикладу 6(h) з сполуки прикладу 7(b) та 5,6,7,8тетрагідро-1,6-нафтиридину [Shiozawa et al., Chem. Pharm. Bull, 32, 2522(1984)]. Зазначену в підзаголовку сполуку одержують з кількісним виходом у вигляді коричневої піни. Rf 0,35(СН2СI2/МеОН/0,88 NH3 90/10/1, об/об). MC m/z 464(МН+). (b) 4-аміно-6,7-диметокси-5-(2-піридил)-2-(5,6,7,8-тeтрагідро-1,6-нафтирид-6-ил)хіназолін Названу в заголовку сполуку одержують у спосіб прикладу 5 з продукту стадії(а). Продукт очищають на силікагелі, елююючи СН 2СI2/МеОН(98/2, об/об), одержуючи названу в заголовку сполуку(30%) у вигляді блідо-жовтої твердої речовини. Rf 0,13(СН2СІ2/МeОН 95/5, об/об). MC m/z 415(МН+). 1Н Я МР(CDCl3) δ 3,08(2Н, т), 3,52(3Н, с), 3,97(3Н, с), 4,20(2Н, т), 4,67(2Н, шир. с), 5,00(2Н, с), 7,03(1Н, с), 7,12(1Н, м), 7,40(1Н, м), 7,48(2Н, м), 7,84(1Н, подв. т), 8,40(1H, д), 8,78(1H, д). Виявлено: С, 65,17; Н, 5,27, N,19,64; C23H22N6 O2 0,5Н2 О відповідає С, 65,24; Н, 5,48, N, 19,84%. Приклад 10 4-Аміно-6,7-диметокси-5-(2-піримідил)-2-(5,6,7,8-тетрагідро-1,6-нафтирид-6-ил)хіназолін Названу в заголовку сполуку одержують у спосіб прикладу 5 з сполуки прикладу 9(а) і 2-(три-нбутилстаніл)піримідину [Sandosham et al., Tetrahedron, 50, 275(1994)]. Продукт очищають на силікагелі, елююючи СН 2СI2/МеОН(95/5, об/об), потім розподіляють між 2N НСI і EtOAc, промивають водний шар EtOAc(3х), підлуговують 2N водним NaOH і екстрагують EtOAc(3 х). Об'єднані органічні шари висушують над MgSO4 та упарюють за пониженого тиску, одержуючи названу в заголовку сполуку(21%) у вигляді блідо-жовтої твердої речовини. Rf 0,39(СН2СI2/МеОН 9/1, об/об). MC m/z 416(МН+). 1Н ЯМР(CDCI3) δ 3,06(2Н, т), 3,68(3Н, с), 3,98(3Н, с), 4,20(2Н, т), 4,61(2Н, шир. с), 5,00(2Н, с), 7,06(1Н, с), 7,13(1Н, м), 7,38(1Н, м), 7,50(1Н, д), 8,43(1Н, д), 8,92(2Н, д). Виявлено: С, 61,99; Н, 5,08, N 22,11; C22H28N7O2 0,15×CH2Cl2 0,1×EtOAc відповідає С, 61,98; H, 5,10, N, 22,44%. Приклад 11 4-Аміно-6,7-диметокси-5-(2-піримідил)-2-(5,6,7,8-тетрагідро-1,3,6-триазанафт-6-ил)хіназолін (a) 1-(т-бутилоксикaрбоніл)-3-N,N-диметилaмінометиліден)-4-піперидон DMF диметилацеталь(5,82мл, 0,044моль) додають до перемішуваного розчину 1l-boc-4-піперидону [Ashwood et al, J. Chem. Soc, Perkin 1, 641(1995)](8,73г, 0,044моль) в DMF(80мл) і реакційну суміш нагрівають до 80°С в атмосфері N2 протягом 18год. Після охолоджування DMF вилучають за пониженого тиску і залишок розподіляють між EtOAc і Н2О. Органічний шар промивають Н 2О і насиченим сольовим розчином, потім висушують над MgSO4 та упарюють, одержуючи названу в підзаголовку сполуку у вигляді твердої речовини(8,44г, 76%). Rf 0,33(СН2СI2/МеОН/0,88 NH 3 90/10/1, об/об). MC m/z 255(МН+). (b) 6-(1-Бутилоксикарбоніл)-(5,6,7,8- тетрaгідро-1,3,6-триазaнaфтaлін) Натрій(762мг, 0,033моль) додають до EtOH(150мл), потім формамідин ацетат(3,45г, 0,033моль) і реакцію перемішують при кімнатній температурі в атмосфері N2 протягом 30хвил. Потім додають розчин продукту стадії(а)(8,43г, 0,033моль) в EtOH(50мл) і реакцію нагрівають із зворотним холодильником протягом 18год, після чого суміш охолоджують та концентрують за пониженого тиску. Залишок розподіляють між EtOAc і Н2О. Органічний шар промивають насиченим сольовим розчином і висушують над MgSO4. Очи щають на силікагелі, елююючи СН 2Сl2/МеОН(96/4, об/об), з одержанням названої в підзаголовку сполуки у вигляді масла(5,09г 65%). Rf 0,57(СН2СI2/МеОН/0,88 NH 3 90/10/1, об/об). MC m/z 236 (c) 5,6,7,8-Тетрагідро-1,3,6-триазанафталін гідрохлорид НСI барботують через розчин продукту стадії(b)(4,80г, 0,020моль) в суміші МеОН і е фіру(50мл, 1/1, об/об) при 0°С до насичення. Потім суміш залишають нагріватися до кімнатної температури протягом 2год, після чого утворюється осад. Його відділяють шляхом декантації супернатанту розчину, промивання ефіром(2х) і висушування у вакуумі, одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(2,85г 81%). Rf 0,13(СН2СI2/МеОН/0,88 NH 3 90/10/1, об/об). MC m/z 136(МН+). (d) 4-Аміно-6,7-диметокси-5-йод-2-(5,6,7,8-тетрагідро-1,3,6-триазанафт-6-ил)хіназолін Зазначену в підзаголовку сполуку одержують у спосіб прикладу 6(h) з продукту стадії(b) і сполуки прикладу 7(b). Зазначену в підзаголовку сполуку(65%) одержують у вигляді безбарвної твердої речовини. Rf 0,52(СН2СI2/МеОН/0,88 NH 3 90/10/1, об/об). MC m/z 465(МН+). (e) 4-Аміно-6,7-диметокси-5-(2-піримідил)-2-(5,6,7,8-тетрагідро-1,3,6-триазaнафт-6-ил)хіназолін Названу в заголовку сполуку одержують у спосіб прикладу 5 з продукту стадії(d) і 2-(три-нбутилстаніл)піримідину. Продукт очищають на силікагелі, елююючи СН 2Cl2/МеОН(95/5, об/об), з одержанням названої у заголовку сполуки(15%) у вигляді безбарвної піни. Rf 0,30(CH2Cl2/MeOH 9/1, об/об). MC m/z 417(МН+). 1Н ЯМР(CDCI3) δ 3,03(2Н, т), 3,68(3Н, с), 4,00(2Н, м), 4,22(2Н, т), 4,47(2Н, шир. с), 5,00(2Н, с), 6,94(1Н, с), 7,07(1Н, с), 7,38(1Н, т), 8,55(1Н, с), 8,95(2Н, д), 9,00(1Н, с). Виявлено: С, 56,61; Н, 4,73, N, 24,84; C 21H20N8 O2 0 ,5×СН2СI2 відповідає С, 56,26; Н, 4,61, N, 24,42%. Приклад 12 4-Аміно-2-(7-аміносульфоніл-1,2,3г4-тетрагідроізохінол-2-іл)-6,7-диметокси-5-(2-піридил)хіназолін (a) 4-аміно-2-хлор-6,7-диметокси-5-(2-піридил)хіназолін До розчину сполуки прикладу 7(b)(1,0г, 2,7ммоль) в діоксані(20мл) додають 2-(три-нбутилстаніл)піридин(1,1г, 3,0ммоль), хлорид літію(1,5г, 35ммоль), тетракіс(трифенілфосфін)паладію(320мг, 0,27ммоль) та йодид міді(І)(78мг, 0,41ммоль) і реакцію нагрівають до 100°С протягом 2год. Після охолоджування реакційну суміш розподіляють між 2N НСI і EtOAc, водний шар додатково екстрагують EtOAc(3х), потім підлуговують 2N водним NaOH і екстрагують EtOAc(3 х). Об'єднані органічні шари висушують над MgSO4 та упарюють за пониженого тиску, одержуючи блідо-жовту тверду речовину. її суспендують в ефірі і фільтрують, одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини.(660мг, 76%). Rf 0,53(EtOAc). MC m/z 317,319(MH+). (b) 4-Аміно-2-(7-аміносульфотл-1,2,3,4-тeтрагідроізохінол-2-іл)-6,7-диметокси-5-(2-піридил)хіназолін До розчину продукту стадії(а)(250мг, 0,8ммоль) в н-бутанол/DMA(3:1, об/об, 8мл) додають гідрохлорид 1,2,3,4-тетрагідроізохінолін-7-сульфонаміду(300мг, 1,2ммоль) і триетиламін(0,33мл, 2,4ммоль), реакційну суміш нагрівають до 100°С в атмосфері N2 протягом 18год. Реакцію потім охолоджують, розподіляють між EtOAc і 2N водним розчином гідроксиду натрію, водний шар відділяють і екстрагують EtOAc, об'єднаний органічний шар промивають Н 2О, висушують над MgSO 4 та упарюють за пониженого тиску. Очищають на силікагелі, елююючи CH2Clz/MeOH/0,88 NH3(95/5/0,5, об/об), з одержанням масла, яке розчиняють в СН2Сl2/МеОН, і продукт висаджують гексаном, одержуючи, після фільтрування і висушування у вакуумі, названу в заголовку сполука у вигляді безбарвної твердої речовини(198мг, 50%). Rf 0,50(СН2СI2/МеОН/0,88 NH3 84/14/2, об/об). MC m/z 493(МН+). 1Н ЯМР(D6-DMSO) δ 0,90(2Н, м), 3,42(3Н, с), 3,94(3Н, с), 4,00(2Н, м), 4,94(2Н, с), 5,50(2Н, шир. с), 6,94(1Н, с), 7,26(2Н, с), 7,32(1H, д), 7,40-7,55(2Н, м), 7,55-7,68(2Н, м), 7,94(1Н, т), 8,71(1Н, д). Виявлено: С, 58,57; Н, 5,35, N, 15,75; C24H24N6O4S 0,4гексан 0,9×Н2О відповідає С, 58,97; Н, 5,73, N, 15,64%. Приклад 13 4-Аміно-6,7-диметокси-2-(2-ізоіндолініл) -5-(2-піридил)хіназолін Названу в заголовку сполуку одержують у спосіб прикладу 12(b) з сполуки прикладу 12(а) і гідрохлориду ізоіндоліну. Продукт очи щають на силікагелі, елююючи ЕtOАс, потім перетирають з СН 2СІ2 і ефіром, одержуючи названу в заголовку сполуку(51%) у вигляді безбарвної піни. Rf 0,42(EtOAc). MC m/z 400(MH+). 1H ЯМР(CDCI3) δ: 3,55(3Н, с), 4,03(3Н, с), 4,70(2Н, с), 4,97(4Н, с), 7,08(1Н, с), 7,21-7,50(6Н, м), 7,84(1Н, т), 8,88(1H, д). Виявлено: С, 65,15; Н, 5,21, N, 15,54; C23H21N5O2 0,4×СН2СI2 0,25×ефір відповідає С, 64,83; Н, 5,42, N, 15,50%, Приклад 14 4-Аміно-6,7-диметокси-5-(2-піридил)-2-(5,6,7,8-тетрагідро-1.3,6-триазанафт-6-ил)хіназолін Названу в заголовку сполуку одержують у спосіб прикладу 12(b) з сполуки прикладу 12(а) і сполуки прикладу 11(c). Продукт очищають на силікагелі, елююючи EtOAc/MeOH(95/5, об/об), потім перетирають з ефіром, одержуючи названу в заголовку сполуку(35%) у вигляді безбарвної твердої речовини. Rf 0,18(СН2СI2/МеОН/0,88 NH3 90/10/1, об/об). MC m/z 416(МН+). 1Н Я МР(CDCl3) δ: 3,03(2Н, т), 3,52(3Н, с), 4,00(3Н, с), 4,20(2Н, т), 4,74(2Н, с), 5,00(2Н, с), 7,03(1H, с), 7,40(1Н, м), 7,45(1Н, д), 7,84(1Н, т), 8,55(1Н, с), 8,78(1Н, д), 9,00(1Н, с). Виявлено: С, 60,94; Н, 5,13; N, 21,93; C22H21N7O2 0,3×СН2СI2 відповідає С, 60,74; Н, 4,94; N, 22,24%. Приклад 15 4-Аміно-6,7-диметокси-2-(7-метансульфонамідо-2,3,4,5-тетрагідро-1Н,3-бензазепін-3-іл)-5-(2піридил)хіназолін (а) 3-т-Бутилоксикарбоніл-7-нітро-2,3,4,5-тетрагідро-1Н,3-бензазепін До розчину 7-нітро-2-3,4,5-тетрагідро-1Н,3-бензазепіну [Pecherer et al.r J. Heterocyclic Chem. 8, 779(1971)](1,92г, 0,01моль) в СН2СI2(40мл) при 0°С додають краплями розчин ди-(т-бутил)дикарбонату в СН2СІ2 і реакцію залишають перемішуватись при кімнатній температурі протягом 18год. Реакційну суміш упарюють за пониженого тиску, одержуючи масло, яке приміщують в СН 2СІ2, промивають водним розчином карбонату натрію(3х), 1N HCl(3х) і насиченим сольовим розчином(2х). Органічний шар відділяють, висушують над MgSO4 та упарюють, одержуючи масло. Перетирають з гексаном, о держуючи названу в підзаго ловку сполуку у вигляді безбарвної твердо ї речовини(2,33г, 80%). Rf 0,8(СН2 СI2 /МеОН 95/ 5, об/об). Т. пл. 106-108°С. (b) 7-Аміно-3-т-бутилоксикарбоніл-2,3,4,5-тє трагідро-1H,3-бензазепін Розчин продукту стадії(а)(2,1г, 7,2ммоль) в суміші EtOAc(20мл) і МеОН(20мл) гідрогенують над паладієм на вугіллі(5% ваг/ваг, 100мг) при 345кПа(50p.s.i.) і кімнатній температурі протягом 3год. Після фільтрування фільтрат упарюють за пониженого тиску, одержуючи названу в підзаголовку сполуку у вигляді масла(2,0г, кількісно). Rf 0,7(СН2СI2/МеОН 9/1, об/об). Виявлено: С, 68,96; Н, 8,63; N, 10,33; C15H22N2 O2 відповідає С, 68,67; Н, 8,45; N, 10,68%. (с) 3-т-Бутилоксикарбоніл-7-метансульфонамідо-2,3,4,5-тетрагідро-1Н, 3-бензазепін Метансуль фонілхлорид(0,56мл, 7,3ммоль) додають краплями до розчину продукту стадії(b)(1,9г, 7,2ммоль) у піридині(40мл) при 0°С і одержаний оранжевий розчин залишають перемішуватись протягом 18год. Реакцію упарюють за пониженого тиску, одержуючи масло, яке розчиняють в СН 2СІ2 і екстрагують послідовно водним розчином бікарбонату натрію(3х) і насиченим сольовим розчином(3х). Органічний шар відділяють, висушують над MgSO4 та упарюють за пониженого тиску, одержуючи масло. Продукт очищають на силікагелі, елююючи СН 2Сl2/МеОН(95/5, об/об), потім перетирають з ефіром, одержуючи названу в підзаголовку сполуку у вигляді білої твердої речовини(1,2г, 49%). Rf 0,5(СН2Сl2/МеОН 95/5, об/об). Т. пл. 153 - 154°С. (d) Гідрохлорид 7-метансульфонамідо -2,3,4,5-тетрагідро-1Н,3-бензазепіну Зазначену в підзаголовку сполуку одержують з продукту стадії(d) відповідно до способу прикладу 11(c). Зазначену в підзаголовку сполуку(71%) одержують у вигляді безбарвної твердої речовини. Rf 0,25(СН2СI2/МеОН/0,88 NH 3 84/14/2, об/об). (e) 4-Аміно-6,7-димето кси-2-(7-метaнсульфонамідо-2,3,4,5-тетрагідро-1Н,3-бензазепін-3-іл)-5-(2піридил)хіназолін Названу в заголовку сполуку одержують у спосіб прикладу 12(b) з продукту стадії(d) і сполуки прикладу 12(а). Продукт очищають на силікагелі, елююючи CH2Cl2/MeOH/0,88 NH3(97/3/0,5, об/об) одержуючи названу в заголовку сполуку(40%) у вигляді безбарвної твердої речовини. Rf 0,31(EtOAc/MeOH 95/5, об/об). MC m/z 521(МН+). 1H Я МР(CDCI3) δ: 2,90(4Н, шир. м), 3,00(3Н, с), 3,53(3Н, с), 4,00(7Н, шир. м), 4,65(2Н, шир. с), 6,68(1H, шир. с), 6,96(2H, с), 7,03(1Н, с), 7,10(1Н, м), 7,42(1Н, м), 7,48(1Н, м), 7,95(1Н, т), 8,80(1H, д). Виявлено: С, 56,65; Н, 5,26; N, 14,66; C 26H28N6O4S 0,5×СН2СI2 відповідає С, 56,53; Н, 5,19; N, 14,93%. Приклад 16 4-Аміно-6,7-диметокси-2-[7-(4-морфолінсульфонамідо)-1,2,3,4-тетрагідроізохінолін-2-іл]-5-(2піридил)хіназолін (а) 7-(4-морфолінсульфонамідо)-1,2,3,4-тeтрагідроізохінолін До розчину 2-трифторацетил-1,2,3,4-тетрагідроізохінолін-7-сульфонілхлориду [Blank et al, J. Med. Chem., 23, 837(1980)] (1,0r, 3,3ммоль) в THF(40мл) додають морфолін(0,74мл, 8,5ммоль) і відразу одержують тонкий білий осад. Через 5хвил додають розчин карбонату натрію(1,7г, 16,5ммоль) в Н2О(20мл), потім суміш МеОН і Н2О(20мл, 1/1, об/об). Одержаний прозорий розчин перемішують протягом 18год при кімнатній температурі, після чого реакцію розподіляють між EtOAc і 2N водним NaOH, водний шар відділяють і екстрагують EtOAc(8 x). Об'єднані органічні шари промивають насиченим сольовим розчином, висушують над MgSO4 та упарюють, одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(650мг, 70%). Rf 0,50(СН2СI2/МеОН/0,88 NH 3 90/10/1, об/об). MC m/z 283(МН+). (b) 4-Аміно-6,7-диметокси-2-[7-(4-морфолінсульфонамідо)-1,2,3,4-тетрагідроізохінолін-2-іл]-5-(2піридил)хіназолін Названу в заголовку сполуку(40%) одержують у спосіб прикладу 12(b) з продукту стадії(а) і сполуки прикладу 12(a). MC m/z 563(МН+). 1Н ЯМР(CDCl3) δ: 3,00(6Н, м), 3,53(3Н, с), 3,73(4Н, м), 3,98(3Н, с), 4,12(2Н, т), 4,70(2Н, шир. с), 5,07(2Н, с), 7,05(1H, с), 7,32(1Н, д), 7,40(1Н, м), 7,48(1Н, д), 7,53(1Н, д), 7,60(1H, с), 7,84(1Н, т), 8,78(1H, d). Приклад 17 4-Аміно-6,7-диметокси-2-(2-метил-5,6,7г8-теграгідро-1,Зг6-триазанафт-6-ил)-5-(2-піридил)хіназолін (a) 6-(т-бутилоксикарбоніл)-2-метил-(5,6,7,8-тетрагідро-1,3,6- триазанафталін Розчин етоксиду натрію в EtОН одержують шляхом додання натрію(690мг, 30,0ммоль) до EtOH(75мл) і обробляють сполукою прикладу 11(а)(7,62г, 30,0ммоль) та гідрохлоридом ацетаміду(3,12г, 33,0ммоль). Реакцію нагрівають із зворотним холодильником протягом 18год, після чого її розподіляють між EtOAc і водним бікарбонатом натрію. Органічний шар відділяють, висушують над MgSO4 та упарюють за пониженого тиску, одержуючи масло. Продукт очищають на силікагелі, елююючи ЕtOАс/МеОН (98/2, об/об), з одержанням названої в підзаголовку сполуки у вигляді кристалічної твердої речовини(6,47г, 87%). Rf 0,31(EtOAc/MeOH 95/5, об/об). 1Н ЯМР(СОСІ3) δ: 1,48(9Н, с), 2,68(3Н, с), 2,92(2Н, м), 3,73(2Н, м), 4,57(2Н, с), 8,38(1Н, с). (b) Гідрохлорид 2-метил-5,6,7,8-тетрагідро-1,3,6-триазанафталіну Названу в підзаголовку сполуку одержують у спосіб прикладу 11(c) з продукту стадії(а). Продукт розподіляють між EtOAc і 2N водним NaОН та водний шар екстрагують кілька разів EtOAc. Об'єднані органічні шари висушують над MgSO4 та упарюють за пониженого тиску, одержуючи названу в підзаголовку сполуку(7%) у вигляді жовтого масла, яке кристалізується при стоянні. 1Н ЯМР(CDCI3) δ: 2,70(3Н, с), 2,90(2Н, т), 3,25(2Н, т), 4,00(2Н, с), 8,38(1Н, с). (c) 4-Аміно-6,7-диметокси-2-(2-метил-5,6,7,8-тетрагідро-1,3,6-триазанафт-6-ил)-5-(2-піридил)хіназолін Названу в заголовку сполуку одержують у спосіб прикладу 12(b) з продукту стадії(b) і сполуки прикладу 12(а). Продукт очищають на силікагелі, елююючи СН2СI2/МеОН(94/6, об/об), з одержанням названої у заголовку сполуки(32%) у вигляді безбарвної піни. Rf 0,33(СН2СI2/МеОН/0,88 NH3 90/10/1, об/об). MC m/z 430(МН+). 1H ЯМР(CDCl3) δ: 2,65(3Н, с), 2,97(2Н, м), 3,50(3Н, с), 3,98(3Н, с), 4,15(2Н, т), 4,73(2Н, шир. с), 4,93(2Н, шир. с), 7,00(1Н, с), 7,40(2H м), 7,82(1Н, т), 8,42(1Н, с), 8,75(1H, а). Виявлено: С, 56,65; H, 5,26: N,14,66; C 23H23N7O 2 0,3×СН2СІ2 відповідає С, 56,53; Н,5,19; N, 14,93%. Приклад 18 4-Аміно-6,7-диметокси-5-(2-піридил)-2-(5,6,7,8-тетрагідро-1,3,7-триазанафт-7-ил)-хіназолін (а) 1-Тритил-3-піперидон Тритилхлорид(13,1г, 47,10ммоль) додають до перемішуваної суспензії 3-піперидон гідрохлориду(5,79г, 42,7ммоль) і триетиламіну(14,9мл, 107ммоль) в СН2СІ2(100мл) та реакцію перемішують протягом 16год в атмосфері N2 при кімнатній температурі. Одержану суміш фільтрують і фільтрат промивають послідовно Н2О і 5% водною лимонною кислотою, висушують над MgSО4 та упарюють за пониженого тиску. Перетирають з пентаном, одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(4,8г, 33%). Rf 0,23(СН2СI2/пентан 2/3, об/об). 1Н ЯМР(CDCI3) δ: 2,05(2Н, м), 2,35(2Н, м), 2,45(2Н, м), 2,85(2Н, с), 7,06-7,55(15Н, м). (b) 4-(N,N-диметиламінометиліден)-1-тритил-3-піперидон Зазначену в підзаголовку сполуку одержують у спосіб прикладу 11(а) з продукту стадії(а). Кристалізують з ефіру, одержуючи названу в підзаголовку сполуку(52%) у вигляді безбарвної твердої речовини. Rf 0,23(СН2СІ2/пентан 2/3, об/об). 1Н ЯМР(CDCI3) δ: 2,35(2Н, т), 2,87(2Н, т), 2,97(2Н, с), 3,13(6Н, с), 7,13(3Н, м), 7,24(7Н, м), 7,50(6Н, м). (с) 7- тритил-5,6,7,8-тетрагідро-1,3,7-триазанафталін Зазначену в підзаголовку сполуку одержують у спосіб прикладу 11(b) з продукту стадії(b). Продукт очищають за допомогою хроматографії на силікагелі, елююючи СН2СI2/ефір(9/1, об/об), з одержанням зазначеної в підзаголовку сполуки(51%). Rf 0,33(СН2СI2/ефір 85/15, об/об). 1Н ЯМР(CDCІ3) δ: 2,60(2Н, т), 2,97(2Н, т), 3,58(2Н, с), 7,06-7,37(8Н, м), 7,52(7Н, м), 8,45(1H, с), 8,90(1Н, с). (d) Гідрохлорид 5,6-7-8-тетрагідро-1,3,7-триазанафталіну Зазначену в підзаголовку сполуку одержують у спосіб прикладу 11(c) з продукту стадії(c). Продукт кристалізують з суміші МеОН/ефір, одержуючи названу в підзаголовку сполуку(65%) у вигляді оранжевої гігроскопічної твердої речовини. 1Н ЯМР(D6-DMSO) δ: 3,06(2Н, м), 3,40(2Н, м), 4,26(2Н, с), 8,68(1Н, с), 9,00(1Н, с), 9,96(2Н, шир. с). (e) 4-Аміно-6,7-диметокси-5-(2-піридил)-2-(5,6,7,8-тетрагідро-1,3,7-триазанафт-7-ил)-хіназолін Названу в заголовку сполуку одержують у спосіб прикладу 12(b) з продукту стадії(d) і сполуки прикладу 12(а). Продукт очищають на силікагелі, елююючи СН2СI2/МеОН(95/5, об/об), з одержанням названої у заголовку сполуки(66%) у вигляді світло-коричневої твердої речовини. Rf 0,20(СН2СІ2/МеОН 95/5, об/об). MC m/z 416(МН+). 1Н ЯМР(CDCI3) δ: 2,92(2Н, т), 3 ,52(3Н, с), 4,00(3Н, с), 4,18(2Н, т), 4,70(2Н, шир. с), 5,18(2Н, шир. с), 7,05(1Н, с), 7,41(1Н, м), 7,43(1H, м), 7,83(1Н, т), 8,50(1H, с), 8,79(1H, д), 9,02(1H, с). Виявлено: С, 61,24; Н, 4,91; N, 22,35; C 22H21N7O 2 0,25×CH2Cl2 відповідає С, 61,20; Н, 4,96; N, 22,46%. Приклад 19 4-Аміно-6,7-диметокси-2-(5-метансульфонамідо-1,2,3,4-тетрагідроізохінол-2-іл)-5-(2-піридил)хіназолін (а) 5-Meтансульфонамідоізохінолін Метансуль фоніл хлорид(3,2мл, 42ммоль) додають до розчину 5-аміноізохіноліну(5,0г, 35ммоль) у піридині(40мл) і суміш залишають стояти протягом 72год. Реакційну суміш потім виливають у водну лимонну кислоту(10%,400мл) і екстрагують EtOAc(2 x 230мл). Органічний шар упарюють, одержуючи залишок, який очищають на силікагелі, елююючи СН 2СІ2/Ме ОН, з одержанням зазначеної в підзаголовку сполуки у вигляді твердої речовини(3,55г, 46%). Rf 0,03(СН2СI2/ефір 4/1, об/об). 1Н ЯМР(D6-DMSO) δ: 3,07(3Н, с), 7,68(1Н, т), 7,75(1H, д), 8,03(1H, д), 8,10(1Н, д), 8,54(1Н, д), 9,32(1Н, с), 9,79(1Н, шир. с). (b) Гідрохлорид 5-метансульфонамідо-1,2,3,4-тетрагідроізохіноліну Розчин продукту стадії(а)(3,50г, 15,7ммоль) в EtOH(205мл) обробляють діоксидом платини(1,5д) і 1N HCІ(15,7мл). Суміш гідрогенують при 414кПа(60p.s.і.) протягом 16год, після чого реакцію фільтрують. Фільтрат упарюють за пониженого тиску і перетирають з СН 2СІ2, одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини. Цей твердий залишок після фільтрування приміщують в МеОН/Н 2О(1/2 , об/об), суспензію фільтрують, промивають СН 2СІ2(3х) і фільтрат упарюють, одержуючи другий збір названої в підзаголовку сполуки(загальний вихід 3,45г, 84%). Rf 0,21(СН2СI2/МеОН/0,88 NH3 90/10/1, об/об). 1Н ЯМР(D6-D MSO) δ: 2,96-3,10(2Н, м), 3,31(3Н, м), 4,21(2Н, с), 7,12(1 Н, м), 7,26(2Н, м), 9,24(1H, с), 9,61(2Н, шир. с). (c) 4-Аміно-6,7-диметокси-2-(5-метансульфонамідо-1,2,3,4-тетрагідроізохінол-2-іл)-5-(2піридил)хінaзолін Названу в заголовку сполуку одержують у спосіб прикладу 12(b) з продукту стадії(b) і сполуки прикладу 12(а). Продукт очищають на силікагелі, елююючи СН2СI2/МеОН(95/5, об/об), з одержанням названої у заголовку сполуки(80%) у вигляді світло-коричневої твердої речовини. Rf 0,21(СН2СI2/МеОН 95/5, об/об). MC m/z 507(МН+). 1Н ЯМР(CDCI3) δ: 2,80(2Н, т), 3,02(3Н, с), 3,53(3Н, с), 4,00(3Н, с), 4,12(2Н, т), 4,67(2Н, шир. с), 4,97(2Н, с), 6,15(1Н, с), 7,03(1Н, с), 7,10(1Н, д), 7,21(1H, д), 7,32(1H, д), 7,42(1Н, м), 7,46(1Н, д), 7,84(1Н, т), 8,79(1Н, д). Виявлено: С, 55,09; Н, 4,90; N, 14,94 C25H26N6O4S 0,56×СН2СI2 відповідає С, 55,38; Н, 4,93; N, 15,16%. Приклад 20 4-Аміно-6,7-диметокси-2-[7-(1-піперазинсульфоніл)-1,2,3,4-тетрагідроізохінолін-2-іл]-5-(2піридил)хіназолін (а) 7-(4-т-Бутилоксикарбоніл-1 -піперазинсульфоніл)-1,2,3,4-тетрагідроізохінолін Зазначену в підзаголовку сполуку одержують у спосіб прикладу 16(а) з 1-тбутилоксикарбонілпіперазину і 2-трифтороацетил-1,2,3,4-тетрагідроізохінолін-7-сульфонілхлориду. Продукт очищають на силікагелі, елююючи СН 2СІ2/МеОН(93/7, об/об), з одержанням названої в підзаголовку сполуки(35%) у ви гляді безбарвної твердої речовини. Rf 0,56(СН2СІ2/МеОН 9/1, об/об). MC m/z 382(МН1). (b) 4-Аміно-6,7-диметокси-2-[7-(4-т-бу тило ксикарбоніл-1-піперазинсу льфоніл)-1,2,3,4 тетрагідроізо хінолін-2-іл]-5-(2-піридил)хіназолін Зазначену в підзаголовку сполуку одержують у спосіб прикладу 12(b) з продукту стадії(а) і сполуки прикладу 12(а). Продукт очищають на силікагелі, елююючи СН 2СІ2/МеОН(96/4, об/об), з одержанням названої в підзаголовку сполуки(69%) у вигляді безбарвної твердої речовини. Rf 0,25(СН2СІ2/МеОН 96/4, об/об). Тригідрохлорид 4-аміно-6,7-диметокси-2[7-(1-піперазинсульфоніл)-1,2,3,4-тетрагідроізохінолін-2-іл]-5(2-піридил)хіназоліну Названу в заголовку сполуку одержують у спосіб прикладу 11(c). Продукт перетирають з СН 2СІ2, одержуючи названу в заголовку сполуку(58%) у вигляді безбарвної твердої речовини. MC m/z 562(МН+). 1Н ЯМР(D6-DMSO) δ 3,03-3,25(11Н, m), 3,50(3Н, с), 4,00(3Н, с), 4,12(2Н, т), 5,17(2Н, шир. с), 5,58(1H, шир. с), 7,52-7,68(5Н, м), 7,94(1Н, с), 8,07(1Н, т), 8,62(1Н, шир. с), 8,80(1Н, д) 9,20(2Н, шир. с), 12,72(1Н, шир. с). Виявлено: С, 46,88; Н, 5,61; N, 13,68; C 28H31N7O 4S 3×НСI 2,5×Н2О відповідає С, 46,96; Н, 5,48; N, 13,69%. Приклад 21 4-Аміно-2-[5-(N,N-діетиламінометил)-1,2,3,4-тетрагідроізохінолін-2-іл]-6,7-диметокси-5-(2піридил)хіназолін (a) 5-(Трифторметансульфонато)ізохінолін Піридин(8,35мл, 0,10моль) додають до розчину 5-гідроксіізохіноліну(5,0г, 0,034моль) в СН2СІ2, цей розчин охолоджують до -40°C і додають краплями трифторметансульфоновий ангідрид(8,47мл, 0,052моль). Реакцію залишають нагріватися до кімнатної температури і перемішують протягом 18год, після чого додають Н 2О. Органічний шар відділяють, промивають насиченим сольовим розчином, висушують над MgSО4 та упарюють за пониженого тиску. Продукт очищають на силікагелі, елююючи СН2СІ2/МеОН(98/2, об/об), одержуючи названу в підзаголовку сполуку у вигляді твердої речовини(6,93г, 73%). Rf 0,70(СН2СI2/МеОН 9/1, об/об). (b) 5-(N,N-діетилкарбоксамідо)ізохінолін До розчину продукту стадії(а)(500мг, 1,8ммоль) в DMF(4мл) додають ацетат паладію(12мг, 0,054ммоль), трифенілфосфін(28мг, 0,11ммоль) і діетиламін(3,7мл, 36ммоль), та реакцію нагрівають при 60°С в атмосфері CO протягом 20год. Після охолоджування реакційну суміш розбавляють насиченим сольовим розчином і екстрагують EtOAc(3 х). Об'єднані органічні екстракти висушують над MgSO4 та упарюють за пониженого тиску. Продукт очищають на силікагелі, елююючи СН2СІ2/МеОН(97/3, об/об), одержуючи названу в підзаголовку сполуку у вигляді твердої речовини(220мг, 53%). Rf 0,45(СН2СI2/МеОН 9/1, об/об). MC m/z 229(МН+). (c) 5-(N,N-Діетилкарбоксамідо)-1,2,3,4-тeтрагідроізохінолін Зазначену в підзаголовку сполуку одержують у спосіб прикладу 19(b) з продукту стадії(b). Неочищений продукт розподіляють між СН 2СІ2 і водним розчином бікарбонату натрію та водний шар екстрагують додатковими порціями СН 2СІ2. Об'єднані органічні екстракти висушують над MgSO 4 і упарюють за пониженого тиску. Продукт очищають на силікагелі, елююючи СН2СI2/МеОН/0,88 NH3(90/10/1, об/об), одержуючи названу в підзаголовку сполуку(66%) у вигляді масла. Rf 0,09 NH3 90/10/1, об/об). MC m/z 233(МН+). (d) 5-(N,N-дieтиламінометил)-1,2,3,4-тетрагідроізохінолін Розчин борану в THF(1М, 18мл, 18,0ммоль) додають краплями до розчину продукту стадії(c)(1,39г, 6,0ммоль) в THF(20мл). Реакційну суміш нагрівають із зворотним холодильником протягом 18год в атмосфері N2, після чого реакційну суміш о холоджують, додають до суміші 2N НСІ/МеОН(1/1, об/об, 100мл) і перемішують протягом 2год. МеОН вилучають за пониженого тиску, реакційну суміш підлуговують до pH 10 і екстрагують СН 2СІ2(3х). Об'єднані органічні шари висушують над MgSO4 та упарюють за пониженого тиску, одержуючи названу в підзаголовку сполуку у вигляді масла(840мг, 64%). MC m/z 219(МН+). 1Н ЯМР(CDCI3) δ: 1,05(6Н, т), 2,40(1Н, шир. с), 2 ,52(4Н, q), 2,89(2Н, т), 3,20(2Н, т), 3,47(2Н, с), 4,06(2Н, с), 6,90(1Н, д), 7,09(1Н, т), 7,20(1H, d). (e) 4-аміно-2-[5-(N,N-діетиламінометил)-1,2,3,4-тетрагідроізохінолін-2-іл]-6,7-диметокси-5-(2піридил)хіназолін Названу в заголовку сполуку одержують у спосіб прикладу 12(b) з продукту стадії(d) і сполуки прикладу 12(а). Продукт очищають на силікагелі, елююючи СН 2СІ2/МеОН/0,88 NH3(90/10/1, об/об), одержуючи названу в заголовку сполук у(30%) у вигляді безбарвної піни. MC m/z 499(МН+). 1Н ЯМР(CDCl3) δ: 1,05(6Н, т), 2,53(4Н, q), 3,00(2Н, т), 3,50(3Н, с), 3,53(2Н, c), 4,00(3Н, с), 4,08(2Н, т), 4,73(2Н, шир. с), 4,98(2Н, с), 7,07,3(4Н, м), 7,40(2Н, м), 7,82(1Н, т), 8,77(1H, д). Виявлено: С, 68,36; Н, 6,71; N, 15,96; C 29H34N6O2 0,2×СН2СI2 відповідає С, 68,01; Н, 6,72; N, 16,30%. Приклад 22 4-Аміно-6,7-диметокси-2-(5-метансульфонамідо-1,2,3,4-тетрагідроізохінол-2-іл] -5-(2-піримідил)хінолін (а) 2-ацетил-(5-метансульфонамідо)-1,2,3,4-тетрагідроізохінолін До розчину сполуки прикладу 19(b)(2,87г, 10,9ммоль) в СН2СІ2 при 0°С додають оцтовий ангідрид(1,2мл, 13,1ммоль) і триетиламін(3,4мл, 24,0ммоль), реакцію перемішують при кімнатній температурі протягом 16год, після чого реакційну суміш розподіляють між EtOAc і водним розчином бікарбонату натрію, водну фазу екстрагують додатковими порціями EtOAc. Об'єднані органічні екстракти висушують над MgSO4 та упарюють, одержуючи масло. Його розчиняють в МеОН(15мл) і обробляють водним розчином карбонату натрію(7%, ваг/ваг, 15мл), суміш перемішують протягом 16год при кімнатній температурі, після чого МеОН вилучають за пониженого тиску, pH доводять до pH 8 2N водним розчином НСI і продукт екстрагують EtOAc(2 x). Об'єднані органічні екстракти висушують над MgSO 4 та упарюють, одержуючи масло, яке очищають на силікагелі, елююючи СН 2СІ2/МеОН(95/5, об/об), одержуючи продукт у вигляді масла(2,0г, 68%). Rf 0,20(СН2СI2/МеОН 9/5, об/об). MC m/z 269(МН+). (b) 3,4-Диметокси-2-йод-6-[1-(5-метансульфонамідо-1,2,3,4-тетрагідроізо хінол-2іл)етиліденаміно]бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(f) з продукту стадії(а) і сполуки прикладу 1(e). Продукт очищають на силікагелі, елююючи СН 2СI2/МеОН(98/2, об/об), одержуючи названу в підзаголовку сполуку(93%) у вигляді безбарвної піни. Rf 0,30(СН2СI2/МеОН 95/5, об/об). MC m/z 555(МН+). (c) 3,4-Диметокси-6-[1-(5-метансульфонамідо -1,2,3,4-тетрагідроізо хіно л-2-іл)етиліденаміно]-2-(2 піримідил)бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 5 з продукту стадії(b) і 2-(три-нбутилстаніл)піримідину. Продукт очи щають на силікагелі, елююючи СН 2СI2/МеОН(96/4, об/об), одержуючи названу в підзаголовку сполуку(32%) у вигляді піни. Rf 0,11(СН2СI2/МеОН 95/5, об/об). MC m/z 507(МН+). (d) 4-Аміно-6,7-диметокси-2-(5-метансульфонамідо-1,2,3,4-тетрагідроізо хінол-2-іл)-5-(2 піримідил)хіно лін Розчин хлориду цинку в THF(0,5М, 10,6мл, 5,3ммоль) додають до розчину продукту стадії(c)(180мг, 0,36ммоль) в THF(5мл) і реакційну суміш нагрівають із зворотним холодильником протягом 70год, після чого додають додаткову порцію хлориду цинку в THF(0,5 М, 3,5мл) і нагрівання продовжують із зворотним холодильником протягом додаткових 7год. Після охолоджування реакційну суміш розподіляють між СН 2СI2 і розчином EDTA в 2N водному NaOH. Органічний шар промивають насиченим сольовим розчином і висушують над MgSO4. Продукт очищають на силікагелі, елююючи СН 2СI2/МеОН/0,88 NH3(90/10/1, об/об), одержуючи названу в заголовку сполуку у вигляді безбарвної твердої речовини(38мг, 21%). Rf 0,28(СН2СI2/МеОН/0,88 NH3 90/10/1, об/об), MC m/z 507(МН+). 1Н ЯМР(СОСІ 3) δ: 2,83(2Н, м), 3,00(3Н, с), 3,64(3Н, с), 3,96(5Н, м), 4,46(2Н, шир. с), 4,77(2H, с), 6,07(1H, с), 7,0-7,2(1H, шир. с), 7,06(1H, с), 7,15(1Н, т), 7,22(1Н, д), 7,43(1Н, т), 7,50(1Н, шир. с)г 8,96(2Н, d). Приклад 23 4-Аміно-6-етокси-2-(5-метансульфонамідо-1,2,3,4-тетрагідроізохінол-2-іл)-7-метокси-5-(2піридил)хінолін (а) 3-гідрокси-2-йод-4-метокси-6-ні тробензонітрил До розчину сполуки прикладу 1(d)(10,0г, 30ммоль) в колідині(100мл) додають йодид літію(4,0г, 30ммоль) і реакцію перемішують при кімнатній температурі протягом 18год, що супроводжується нагріванням при 100°С протягом 1,5год. Після охолоджування реакційну суміш розподіляють між 2N водним NaOH і EtOAc, шари поділяють і продукт екстрагують 3 додатковими промиваннями EtOAc. Об'єднані органічні шари промивають 2N НСI(2х), висушують над MgSO 4 та упарюють за пониженого тиску. Неочищений продукт очищають на силікагелі, елююючи СН2СI2/МеОН(97/3, об/об), одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(6,96г, 73%). Rf 0,16(95/5, об/об). MC m/z 338(MNH4+). (b) 3-Етокси-2-йод-4-метокси-6-нітробензонітрил До розчину продукту стадії(а)(6,95г, 21,7ммоль) і бромоетану(1,78мл, 23,8ммоль) в DMF(70мл) додають карбонат калію(4,49г, 32,5ммоль) і реакцію нагрівають до 60°С протягом 18год. Після охолоджування реакційну суміш розподіляють між 2N водною НСI і EtOAc. Органічний шар відділяють, промивають Н2О, висушують над MgSO4 та упарюють. Неочищений продукт очищають на силікагелі, елююючи СН 2СI2/МеОН(97/3, об/об), одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(2,94г, 39%). Rf 0,68(СН2СI2/МеОН 95/5, об/об). MC m/z 366(MNH 4+). (c) 6-Аміно-3-етокси-2-йо д-4-мето ксибензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(e) з продукту стадії(b). Продукт очищають на силікагелі, елююючи СН 2СI2/МеОН(98/2, об/об), одержуючи названу в підзаголовку сполуку(72%) у вигляді безбарвної твердої речовини. Rf 0,11(СН2СI2/МеОН 95/5, об/об). MC m/z 336(MNH4+). (d) 3-Етокси-2-йод-6-[1-(5-метансу льфонамідо-1,2,3,4-тетрагідроізо хінол-2-іл)етиліденаміно]-4 метоксибензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(f) з продукту стадії(c) і сполуки прикладу 22(а). Продукт очищають на силікагелі, елююючи СН2СI2/МеОН(98/2, об/об), одержуючи названу в підзаголовку сполуку(53%) у вигляді безбарвної піни. Rf 0,14(СН2СI2/МеОН 95/5, об/об). MC m/z 569(МН+). (e) 3-Етокси-6-[1-(5-метансу льфонамідо-1,2,3,4-тетрагідроізо хінол-2-іл)етиліденаміно]-4-метокси-2 (2-піридил)бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 5 з продукту стадії(d) і 2-(три-нбутилстаніл)піридину. Продукт очищають на силікагелі, елююючи СН 2СI2/МеОН(94/6, об/об), одержуючи названу в підзаголовку сполуку(54%) у вигляді піни. Rf 0,14(СН2СI2/МеОН 95/5, об/об). MC m/z 520(MH +). (f) 4-Аміно-6-етокси-2-(5-метансульфонамідо-1,2,3,4-тетрагідроізохінол-2-іл)-7-метокси-5-(2тридил)хтолін Розчин продукту стадії(e)(1,14г, 2,19ммоль) в THF(10мл) охолоджують до -78°С і обробляють розчином діізопропіламіду літію в циклогексані(1,5М, 4,4мл, 6,6ммоль). Реакцію потім залишають нагріватися до кімнатної температури протягом 1год, розподіляють між EtOAc і Н2 О, та водний шар екстрагують 3 додатковими порціями EtOAc. Об'єднані органічні шари промивають насиченим сольовим розчином, висушують над MgSО4 та упарюють за пониженого тиску. Очищають на силікагелі, елююючи СН2СI2/МеОН/0,88 NH 3(93/7/1, об/об), потім перетирають з ефіром, одержуючи названу в заголовку сполуку у вигляді безбарвної піни(510мг, 45%). Rf 0,23(СН2СI2/МеОН/0,88 NH3 90/10/1, об/об). MC m/z 520(МН+). 1Н ЯМР(CDCІ3) δ: 0,92(3Н, т), 2,84(2Н, м), 3,00(3Н, с), 3,82(4Н, м), 3,98(3Н, с), 4,22(2Н, q), 4,77(2Н, с), 5,93(1Н, с), 7,07(1Н, м), 7,17(1Н, т), 7,20-7,35(2Н, м), 7,40(1H, т), 7,50(1Н, д), 7,81(1Н, т), 7,46(1H, д), 8,77(1H, д). Виявлено: С, 60,32; H 5,66; N, 12,60 C 27H29N5O4S O,25×CH2CІ2 відповідає С, 60,51; Н, 5,50; N, 12,95%. Приклад 24 4-Аміно-6,7-диметокси-5-(2-піридил)-2-(5,6,7,8-тетрагідро-1,6-нафтирид-6-ил)хінолін (а) 6-Ацетил-5,6,7,8- тетрагідро-1,6-нафтиридин До розчину 5,6,7,8-тетрагідро-1,6-нафтиридин(4,9г, 36,5ммоль) в СН2СІ2 при 0°С додають краплями триетиламін(6,1мл, 43,8ммоль) і ацетилхлорид(3,11мл, 43,8ммоль), і реакцію залишають нагріватися до кімнатної температури та перемішують протягом ще 18год. Реакційну суміш розподіляють між Н 2О і СН 2СI2, шари відділяють і водний шар екстрагують ще двічі СН 2СI 2. Об'єднані органічні шари висушують над MgSO4 та упарюють за пониженого тиску, одержуючи залишок, який очищають на силікагелі, елююючи EtOAc/MeOH/0,88 NH3(95/5/1, об/об). Одержують означену в підзаголовку сполуку(58%) у вигляді масла. Rf 0,60(CH2Cl2/MeOH/0,88 NH3 84/14/2, об/об). 1НЯМР(CDCI3) δ: 2,15(3Н, с), 3,04(2Н, м), 3,75 і 3,90(2Н,2хм), 4,60 і 4,70(2Н,2хс), 7,10(1Н, м), 7,42(1Н, м), 8,42(1H, м). (b) 3,4-Диметокси-2-йод-6-[1-(5,6,7,8-тетрагідро-1,6-нафтирид-6-ил)етиліденаміно]бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(f) з продукту стадії(а) і сполуки прикладу 1(e). Продукт очищають на силікагелі, елююючи EtOAc/MeOH(95/5, об/об), одержуючи названу в підзаголовку сполуку(80%) у вигляді блідо-жовтої твердої речовини. Rf 0,58(СН2СI2/МеОН/0,88 NH3 92/7/1, об/об). MC m/z 463(МН+). (c) 3,4-Диметокси-2-(2-піридил)-6-[1-(5,6,7,8-тетрагідро-1,6-нафтирид-6 ил)етиліденаміно]бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 5 з продукту стадії(b) і 2-(три-нбутилстаніл)піридину. Продукт очищають на силікагелі, елююючи EtOAc/MeOH/0,88 NH3(95/5/1, об/об)г одержуючи названу в підзаголовку сполуку(51%) у вигляді піни. Rf 0,26(EtOAc/MeOH/0,88 NH3 90/10/1, об/об). MC m/z 414(MH +). (d) 4-Аміно-6,7-диметокси-5-(2-піридил)-2-(5,6,7,8-тетрагідро-1,6-нафтирид-6-ил)хінолін Названу в заголовку сполуку одержують у спосіб прикладу 23(f) з продукту стадії(c). Продукт очищають на силікагелі, елююючи CH2Cl2/MeOH/0r88 NH3(96/3,5/0,5, об/об), потім перетирають з ефіром, одержуючи названу в заголовку сполуку(22%) у вигляді світло-коричневої твердої речовини. Rf 0,31(СН2СI2/МеОН/0,88 NH3 92/7/1, об/об). MC m/z 414(МН+). 1Н ЯМР(CDCI3) δ: 3,13(2Н, м), 3,52(3Н, с), 3,82(2Н, шир. с), 3,98(5Н, м), 4,83(2Н, с), 5,98(1H, с), 7,13(1Н, м), 7,22(1Н, шир. с), 7,38(1Н, м), 7,48(1Н, д), 7,53(1Н, м), 7,80(1Н, м), 8,43(1Н, д), 8,76(1Н, д). Виявлено: С, 67,74; Н, 6,26; N, 15,43 C24H23N5O2S 0,4×ефір 0,6×Н2О відповідає С, 67,86; Н, 6,07; N, 15,33%. Приклад 25 4-Аміно-6,7-диметокси-5-(2-піримідил)-2-(5,6,7,8-тетрагідро-1,6-нафтирид-6-ил)хінолін (а) 3-4-Диметокси-2-піримідил-6-[1-(5,6,7,8-тетрагідро-1,6-нафтирид-6-ил)етиліденаміно]бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 5 з продукту прикладу 24(b) і 2-(три-нбутилстаніл)піримідину. Продукт очи щають на силікагелі, елююючи СН 2СI2/МеОН(98/2, об/об), одержуючи названу в підзаголовку сполуку у вигляді піни(75%). R f 0,21(ефір). MC m/z 415(МН+). (b) 4-Аміно-6,7-димето кси-5-(2-піримідил)-2-(5,6,7,8-тетрагідро-1,6-нафтирид-6-ил)хіно лін Порошок гідроксиду калію(72мг, 1,29ммоль) додають до розчину продукту стадії(а)(530мг, 1,28ммоль) в DMSO(5мл). Реакційну суміш нагрівають до 95°С протягом 45хвил. Після охолоджування реакційну суміш виливають в лимонну кислоту і підлуговують 2N водним NaOH. Продукт потім екстрагують EtOAc(x4). Об'єднані органічні шари промивають Н 2О, насиченим сольовим розчином і висушують над MgSO4. Продукт очищають на силікагелі, елююючи СН 2СI2/МеОН/0,88 NH3(96/3,5/0,5, об/об). Продукт перетирають з ефіром, одержуючи названу в заголовку сполуку у вигляді оранжевої твердої речовини(91мг, 17%). Rf 0,11 СН2СI2/МеОН(95/5 об/об). MC m/z 415(МН+). 1Н ЯМР(CDCI3) δ: 3,10(2Н, т), Згб9(3Н, с), 3,79(2Н, с), 4,00(2Н, м), 4,05(3Н, с), 4,82(2Н, с), 6,01(1Н, с), 7,05(1Н, м), 7,40(1Н, т), 7,50(1Н, м), 8,40(1Н, м), 8,90(2Н, м). Приклад 26 4-Аміно-7-метокси-2-[4-(4-морфолінкарбоніл)-1,4-діазепан-1-іл]-5-(2-піридил)-6-(2,2,2трифторетокси)хінолін (a) Meтиловий ефір 3-гідрокси-4-метоксибензоиної кислоти До суспензії 3-гідрокси-метоксибензойної кислоти(33,63г, 0,2моль) в МеОН(500мл) додають концентровану сірчану кислоту(25мл) і реакційну суміш нагрівають із зворотним холодильником протягом 2год. Після охолоджування реакційну суміш концентрують за пониженого тиску до 100мл і залишок екстрагують EtOAc. Органічний шар промивають послідовно Н 2О(х2), насиченим водним бікарбонатом натрію і насиченим сольовим розчином, висушують над MgSO4 і упарюють за пониженого тиску, одержуючи названу в підзаголовку сполуку у вигляді безбарвних кристалів(33,0г, 91%). Rf 0,59(EtOAc). MC m/z 183(MH+). (b) 2,2-2-Трифторетилтрифторметансульфонат До розчину трифторметансульфонового ангідриду(80,4г, 0,3моль) в СН2СI2(50мл) додають краплями суміш 2,2,2-трифторетанолу(28,0г, 0,28моль) і триетиламіну(29,3г, 0,29моль) при -40°С протягом 45 хвил. Після додання реакційну суміш нагрівають до кімнатної температури, промивають послідовно Н 2О і насиченим водним бікарбонатом натрію і потім висушують над MgSO 4. Одержують названу в підзаголовку сполуку у ви гляді розчину в СН 2СI2, який негайно використовують на наступній стадії. Метиловий ефір 4-метокси-3-(2,2,2-трифторетокси)бензойної кислоти До розчину продукту стадії (а) (33,0г, 0,181моль) в суміші карбонату калію(41,4г, 0,3моль) і DMF(100мл) додають розчин продукту, одержаного на стадії (b) (65,0г, 0,28моль). Реакційну суміш перемішують при кімнатній температурі протягом 18год, після чого реакційну суміш упарюють за пониженого тиску. Залишок розподіляють між ефіром і Н 2О. Органічний шар промивають насиченим сольовим розчином, висушують над MgSO4 і упарюють за пониженого тиску. Неочищений продукт перетирають з гексаном, одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(42,55г, 93% з 2 стадій). Rf 0,47(СН2СI2). MC m/z 265(МН+). (d) 4-Метокси-3-(2,2,2-трифторетокси)бензойна кисло та До розчину продукту стадії(c)(42,25г, 0,16моль) в МеОН додають 2N водний NaOH(160мл, 0,32моль). Реакційну суміш перемішують при кімнатній температурі протягом 3год і потім при 50°С протягом 1год. Після охолоджування реакційну суміш упарюють за пониженого тиску. Залишок розподіляють між EtOAc і 2N НСI. Органічний шар висушують над MgSO4 до упарюють за пониженого тиску, одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(40,4г, 100%). Rf 0,13(гексан, ЕtOАс 1/1, об/об). MC m/z 251(МН+). (e) 4,4-Диметил-2-[4-метокси-3-(2,2,2-трифторетокси)феніл]-Δ2оксазолін До суспензії продукту стадії(d) (40,0г, 0,16моль) в СН2СІ2(200мл) і DMF(0,5мл) додають оксалілхлорид(40,6г, 0,32моль) при 0°С протягом 15хвил. Реакційну суміш перемішують при 0°С протягом 30хвил, нагрівають до кімнатної температурі протягом 1,5год і упарюють за пониженого тиску. Залишок потім знову розчиняють в СН 2СІ2(300мл) і додають протягом 15хвил. до розчину 2-аміно-2метилпропанолу(17,8г, 0,2моль) і триетиламіну(20,2г, 0,2моль) в СН2СI2(100мл) при 0°С. Реакційну суміш потім перемішують при кімнатній температурі протягом 30хвил, промивають послідовно 5% лимонною кислотою і розбавляють водним бікарбонатом натрію, висушують над MgSО 4 та упарюють за пониженого тиску до об'єму 200мл. Тіонілхлорид(21,4г, 0,18моль) додають краплями до розчину і одержану суміш перемішують протягом 1год. Продукт потім екстрагують Н 2О, потім 0,5N водним NaOH. Об'єднані водні фази підлуговують 2N водним NaOH і продукт екстрагують СН2СІ2(х2). Об'єднані органічні фази висушують над MgSO4 та упарюють за пониженого тиску одержуючи порцію неочищеного продукту. Первинний органічний екстракт потім струшують з 2N водним NaOH і продукт екстрагують СН 2СI2(х3). Об'єднані органічні екстракти висушують над MgSO4 і упарюють за пониженого тиску. Залишок перерозчиняють в ефірному розчині НСI(150мл), одержану білу тверду речовину фільтрують, підлуговують ще раз 2N водним NaOH і екстрагують СН 2СІ2(х3). Об'єднані органічні екстракти висушують над MgSO4 і упарюють за пониженого тиску, одержуючи другу порцію неочищеного продукту. Об'єднаний неочищений продукт очищають на силікагелі, елююючи СН 2СІ2/МеОН (95/5, об/об), одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(38,8г, 80%). Rf 0,54(EtOAc). MC m/z 304(MH +). (f) 4,4-Диметил-2-[2-йо д-4-мето кси-3-(2,2,2-трифторетокси)феніл]-Δ2 оксазолін Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(b) з продукту стадії(e). Неочищений продукт очищають на силікагелі, елююючи EtOAc/гексан(60/40, об/об). Продукт потім перетирають з ефіром, одержуючи названу в підзаголовку сполуку у вигляді оранжевої твердої речовини(53%). Rf 0,27(ефір/гексан 1/3, об/об). MC m/z 430(МН+). (g) 2-Йод-4-метокси-3-(2,2,2-трифторетокси)бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(c) з продукту стадії(f). Неочищений продукт перетирають зі сумішшю гексан/ефір(60/40, об/об), одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(96%). Rf 0,5(EtOAc, гексан 1/1, об/об). MC m/z 358(МН+). (b) 2-Йод-4-метокси-6-нітро-3-(2,2,2-трифторетокси)бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(d) з продукту стадії(g). Неочищену коричневу тверду речовину перетирають з ефіром, одержуючи названу в підзаголовку сполуку(72%). Rf 0,25(гексан/EtOAc 2/1, об/об). MC m/z 403(МН+). (і) 6-Аміно-2-йод-4-метокси-3-(2,2,2-трифторетокси)бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(e) з продукту стадії(h). Неочищений продукт промивають через основу з силікагелю, одержуючи названу в підзаголовку сполуку у вигляді оранжевої твердої речовини(70%). Rf 0,74(СН2СI2/МеОН/0,88 NH 3 93/7/1, об/об). MC m/z 373(МН+). (j) 2-Йод-4-метокси-6-{1-[4-(4-морфолінкарбоніл)-1,4-діазепан-1-іл]етиліденаміно}-3-(2,2,2трифторетокси)бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(f) з продукту стадії(і) та проміжної сполуки 4. Неочищений продукт очищають на силікагелі, елююючи СН2Сl2/МеОН(90,10, об/об), одержуючи названу в підзаголовку сполуку у вигляді оранжевого масла, яке перекристалізовують з EtOAc, з одержанням безбарвної твердої речовини(64%). Rf 0,12(EtOAc). MC m/z 610(MH +). (k) 4-метокси-6-{1-[4-(4-морфолінкарбоніл)-1,4-діазепан-1-іл]-етиліденаміно}-2-(2-піридил)-3-(2,2,2трифторетокси)бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 5 з продукту стадії(j) і 2-(три-нбутилстаніл)піридину. Неочищений продукт очищають на силікагелі, елююючи. СН2СІ2/МeОН(96/4, об/об), одержуючи названу в підзаголовку сполуку у вигляді жовтої піни(91%). Rf 0,43(СН2СI2/МеОН/0,88 NH3 93/7/1, об/об). MC m/z 561(МН+). (І) 4-Аміно-7-метокси-2-[4-(4-морфолінкарбоніл)-1,4-діaзепaн-1-iл]-5-(2-піридил)-6-(2,2,2трифторетокси)хінолін До розчину продукту стадії (к) (0,56г, 1ммоль) в THF(10мл) додають свіжоприготовлений діізопропіламід літію(4мл, 2ммоль) при -20°С. Реакційну суміш потім повільно нагрівають до кімнатної температури і перемішують протягом 20хвил, після чого її гасять Н2О і виливають в EtOAc. Органічний шар потім промивають 2N водним NaOH, потім насиченим сольовим розчином, висушують над MgSО4 та упарюють за пониженого тиску. Неочищений продукт очищають на силікагелі, елююючи СН2СI2/МеОН/0,88 NH3(90/10/1, об/об), одержуючи названу в заголовку сполуку у вигляді коричневої піни(0,39г, 70%). Rf 0,37(СН2СI2/МеОН/0,88 NH 3 93/7/1, об/об). MC m/z 561(МН+). 1 Н ЯМР(CDCІ3) δ: 2,00(2Н, м), 3,10(5Н, м), 3,30(2Н, м), 3,50-3,90(10Н, м), 3,95(3Н, с), 4,00(3Н, с), 5,80(1H, с), 7,10(1Н, шир. с), 7,39(1Н, м), 7,45(1Н, д), 7,80(1Н, м), 8,70(1H, д). Виявлено С, 51,49; Н, 5,72: N,14,35; C 27H31F 3N6О 4 0,33·EtOAc відповідає С, 51,69; Н, 5,71; N, 14,35%. Приклад 27 4-Аміно-7-метокси-5-(2-піримідиніл)-2-(5,6,7,8-тетрагідро-1,6-нафтирид-6-ил)-6-(2,2,2трифторетокси)хінолін (а) 2-йод-4-метокси-6-[1-(5,6,7,8-тетрагідро-1,6-нaфтирид-6-ил)етиліденaмін]-3-(2,2,2трифторетокси)бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(f) з продукту прикладу 26(і) і сполуки прикладу 24(а). Неочищений продукт очищають на силікагелі, елююючи EtOAc/MeOH(80/20, об/об), одержуючи названу в підзаголовку сполуку у вигляді безбарвної піни(70%). Rf 0,63(СН2СI2/МеОН/0,88 NH3 90/10/1, об/об). MC m/z 531(МН+). (b) 4-Метокси-2-(2-піримідил)-6-[1-(5,6,7,8-тетрагідро-1,6-нафтирид-6-ил)етиліденаміно]-3-(2,2,2трифторетокси)бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 5 з продукту стадії(а) і 2-(три-нбутилстаніл)піримідину. Неочищений продукт очищають на силікагелі, елююючи СН 2СІ2/МеОН/0,88 NН3(94/6/1, об/об), одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(49%). Rf 0,29(СН2СI2/МеОН/0,88 NH 3 93/7/1, об/об). MC m/z 483(МН+). (c) 4- Aміно-7-метокси-5-(2-піримідиніл)-2-(5,6,7,8-тeтрагідро-1,6-нaфтирид-6-ил)-6-(2,2,2трифторетокси)хінолін Названу в заголовку сполуку одержують у спосіб прикладу 25(b) з продукту стадії(b). Неочищений продукт очищають на силікагелі, елююючи СН 2СI2/МеОН(95/5, об/об), одержуючи названу в заголовку сполуку у ви гляді піни(8%). R f 0,07(СН2СI2/МеОН 95/5, об/об). MC m/z 483(МН+). 1 Н ЯМР(CDCl3) δ: 3,18(2Н, м), 3,80(2Н, шир. с), 4,00(5Н, м), 4,30(2Н, м), 4,90(2Н, с), 6,00(1Н, с), 7,10(1Н, м), 7,30(1Н, с), 7,40(1H, м), 7,50(1Н, д), 8,40(1Н, д), 8,90(2Н, д). Приклад 28 4-Аміно-6,7-диметокси-2-[4-(4-морфолінкарбоніл)-14-діазепан-1-іл]-5-(оксазол-2-іл)хінолін До розчину оксазолу(276мг, 4ммоль) в THF(15мл) додають краплями н-бутиллітій(1,6М в гексані, 2,75мл, 4,4ммоль) при -78°С в атмосфері азоту. Реакційну суміш перемішують при -78°С протягом 20хвил, потім до реакційної суміші додають розчин хлориду цинку(1,0М в ефірі, 12мл, 12ммоль) і одержаний розчин нагрівають до кімнатної температури. Додають сполуку прикладу 1(f) (1,05г, 1,94ммоль), потім тетракіс(трифенілфосфін)паладій (200мг). Реакційну суміш нагрівають із зворотним холодильником протягом 3год. Потім додають додаткову порцію оксазолцинкату, одержаного як описано вище за допомогою оксазолу(552мг, 8ммоль), н-бутиллітію(1,6М в гексані, 5,5мл, 8,8ммоль), розчин хлориду цинку(1,0М в ефір, 24мл, 24ммоль), потім тетракіс(трифенілфосфін)паладію (100мг). Реакційну суміш нагрівають із зворотним холодильником протягом 4год, після чого додають йодид міді (І) (100мг). Одержану суміш нагрівають із зворотним холодильником протягом 24год. Охолоджену реакційну суміш виливають в EtOAc і промивають водним розчином EDTA, підлуговують 2N водним NaOH і відділяють органічний шар, промивають насиченим сольовим розчином, висушують над MgSO 4 та упарюють за пониженого тиску. Неочищений продукт очищають на силікагелі, елююючи СН2СI2/МеОН/0,88 NH3(92/8/1, об/об), одержуючи названу в заголовку сполуку(87мг, 9%). Rf 0,46(СН2СI2/МеОН/0,88 NH3 93/7/1, об/об). MC m/z 483(МН+). 1 H ЯМР(CDCI3) δ: 2,10(2Н, м), 3,10(4Н, м), 3,35(2Н, м), 3,50-3,90(12 Н, кілька піків), 3,95(2Н, м), 4,00(3Н, с), 4,20(1Н, шир. с), 5,85(1Н, с), 7,35(1Н, с), 7,70(1H, м), 7,90(1H, с). Приклад 29 4-Аміно-6-7-диметокси-2-(2-метил-5,6,7,8-тетрагідро-1,6-нафтирид-6-ил)-5-(2-піридил)хінолін (а) 6-Ацетил-2-метил-5,6,7,8-тетрагідро-1,6-нафтиридин До розчину 2-метил-5,6,7,8-тетрагідро-1,6-нафтиридину [Shiozawa et al., Chem. Pharm. Bull., 32, 2522, (1984)] (2,73r, 0,0184моль) в СН2СI2(30мл) і триетиламіну(5,1мл, 0,0368моль) додають ацетилхлорид(1,57мл, 0,0221моль) при 0°С. Реакційну суміш перемішують протягом 24год при кімнатній температурі, після чого реакційну суміш промивають послідовно насиченим водним бікарбонатом натрію, Н2О, насиченим сольовим розчином, висушують над MgSO4 та упарюють за пониженого тиску, одержуючи названу в підзаголовку сполуку(3,27 г, 93%). Rf 0,5(СН2СI2/МеОН/0,88 NH 3 90/10/1, об/об). MC m/z 191(МН+). (b) 3,4-Диметокси-2-йод-6-[1-(2-метил-5,6,7,8-тeтрагідро-1,6-нафтирид-6-ил)етиліденаміно]бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(f) з продукту стадії (а) і сполуки прикладу 1(e). Неочищений продукт очищають на силікагелі, елююючи сумішшю EtOAc/гексан(96/4, об/об), одержуючи названу в підзаголовку сполуку у ви гляді піни(87%). Rf 0,42(EtOAc). MC m/z 477(MH +). (с) 3,4-Диметокси-6-[1-(2-метил-5,6,7,8-тетрагідро-1,6-нафтирид-6-ил)етиліденаміно]-2-(2пiридил)бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 5 з сполуки стадії (b) і 2-(три-нбутилстаніл)піридину. Неочищений продукт очищають на силікагелі, елююючи EtOAc/MeOH(97/3, об/об), одержуючи названу в підзаголовку сполуку у ви гляді піни(25%). Rf 0,29(EtOAc). MC m/z 428(MH +). (d) 4-Аміно-6,7-диметокси-2-(2-метил-5,6,7,8-тетрагідро-1,6-нафтирид-6-ил)-5-(2-піридил)хінолін Названу в заголовку сполуку одержують у спосіб прикладу 26(1) з продукту стадії (с). Неочищений продукт очищають на силікагелі, елююючи СН 2СІ2/МеОН/0,88 NH3(93/7/1, об/об), одержуючи названу в заголовку сполуку у вигляді піни(10%). R f 0,25(СН2СI2/МеОН/0,88 NH 3 90/10/1, об/об). MC m/z 233(МН+). 1 Н ЯМР(CDCI3) δ: 1,30(2Н, шир. с), 2,50(3Н, с), 3,10(2H, m) 3,59(3H, c), 3,85(2H, м), 3,95-4,00(6H, м), 4,80(2H, с), 6,00(1H, с), 7,00(1H, д), 7,20(1H, с), 7,40(1H, м), 7,45(1H, м), 7,80(1H, м), 8,75(1H, м). Приклад 30 4-Аміно-6,7-диметокси-2-(5-метокси-1,2,3,4-тетрагідроізохінол-2-іл)-5-(2-піридил)хінолін (а) 5-Метоксіізохінолін До розчину 5-гідроксиізохіноліну(10г, 69ммоль) в МеОН(100мл) додають розчин метоксиду натрію в метанолі(30% за вагою, 13,8мл, 72,4ммоль), потім фенілтриметиламонійхлорид(12,4г, 72,4ммоль). Реакційну суміш перемішують при кімнатній температурі протягом 2год, після чого її фільтрують і фільтрат упарюють за пониженого тиску, одержуючи масло, яке розчиняють в DMF(50мл). Реакційну суміш нагрівають із зворотним холодильником протягом 2год, після чого реакційну суміш упарюють за пониженого тиску. Одержані масло розподіляють між СН 2СІ2 і 1N водним NaOH. Органічний шар промивають двічі 1N водним NaOH, висушують над MgSO4 та упарюють за пониженого тиску. Неочищений продукт очищають на силікагелі, елююючи сумішшю EtOAc/гексан(1/1, об/об), одержуючи названу в підзаголовку сполуку у вигляді жовтого масла(6,11г, 56%). 1 Н ЯМР(CDCl3) δ: 4,05(3Н, с), 7,00(1H, д), 7,55(2Н, м), 8,02(1Н, д), 8,55(1Н, д), 9,22(1H, с). (b) 5-Meтокси-1,2,3,4-тетрагідроізохінолін До розчину продукту стадії (а) (6,11г, 384ммоль) в EtOH(200мл) додають оксид платини(0,611г), потім концентровану НСI(3,2мл, 38,4ммоль). Реакційну суміш гідрогенують при 345кПа(50p.s.і.) при кімнатній температурі протягом 4год, після чого каталізатор відфільтровують і промивають EtOH. Фільтрат упарюють за пониженого тиску, одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(7,27г, 95%). 1 Н ЯМР(D6-DMSO) δ: 2,80(2Н, м), 3,35(2Н, м), 3,80(3Н, с), 4,20(2Н, с), 6,80(1Н, д), 6,90(1Н, д), 7,20(1Н, т), 9,45(2Н, шир. с). (c) 2-Ацетил-5-метокси-1,2,З,4-тетрагідроізохінолін До розчину продукту стадії (b) (6,26г, 31,4ммоль) і триетиламіну(9,6мл, 69,0ммоль) в СН2СІ2(150мл) додають ацетилхлорид(2,7мл, 37,7ммоль) при 0°С протягом 15хвил. Реакційну суміш перемішують при кімнатній температурі протягом 18год, після чого розчин промивають послідовно і насиченим водним розчином бікарбонату натрію, висушують над MgSO 4 i упарюють за пониженого тиску. Неочищений продукт очищають на силікагелі, елююючи ЕЮАс, одержуючи названу в підзаголовку сполуку у вигляді оранжевого масла(6,07г, 94%). Rf 0,65(СН2СI2/МеОН/0,88 NH 3 90/10/1, об/об). MC m/z 206(МН+). (d) 3,4-Диметокси-2-йод-6-[1-(5-метокси-1,2,3,4-тетрагідроізохінол-2-іл)етиліденаміно]бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(f) з продукту стадії (c) і сполуки прикладу 1(e). Неочищений продукт очищають на силікагелі, елююючи СН2СІ2, одержуючи названу в підзаголовку сполуку у вигляді оранжевих кристалів(69%). Rf 0,77(СН2СI2/МеОН 95/5, об/об). MC m/z 492(MH+). (e) 3,4-Диметокси-6-[1-(5-метокси-1,2,3,4-тeтрагідроізохінол-2-іл)етиліденаміно]-2-(2піридил)бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 5 з продукту стадії (d) і 2-(три-нбутилстаніл)піридину. Неочищений продукт очищають на силікагелі, елююючи ефіром, одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(62%). Rf 0,73(СН2СІ2/МЕОН 90/10, об/об). MC m/z 443(МН+). (f) 4-Аміно-6,7-диметокси-2-(5-метокси-1,2,3,4-тетрагідро ізо хінол-2-іл)-5-(2-піридил)хінолін Названу в заголовку сполуку одержують у спосіб прикладу 25(b) з продукту стадії (e). Неочищений продукт очищають на силікагелі, елююючи СН 2С2/МеОН(95/5, об/об), одержуючи названу в заголовку сполуку у вигляді безбарвної твердої речовини(10%). Rf 0,5(СН2СI2/МеОН 90/10, об/об). MC m/z 443(МН+). 1 Н ЯМР(CDCI3) δ: 2,90(2Н, т), 3,55(3Н, с), 3,75-3,90(7Н, м), 4,00(3Н, с), 4,79(2Н, с), 5,95(1H, шир. с), 6,70(1H, д), 6,85(1Н, д), 7,19(1Н, т), 7,25(1Н, с), 7,39(1H, т), 7,45(1H, д), 7,80(1Н, т), 8,75(1Н, д). Виявлено С, 68,58; Н, 5,93; N, 12,66; C 26H26N4 O3 0 ,2ефір 0,6Н2О відповідає С, 68,76; Н, 6,29; N, 11,97%. Приклад 31 4-аміно-6,7-диметокси-2-(6,7-диметокси-1,2,3,4-тетрагідроізохінол-2-іл)-5-(2-піримідил)хінолін (а) 2-Ацетил-6-7-диметокси-1,2,3,4-тетрагідроізохінолін Зазначену в підзаголовку сполуку одержують у спосіб прикладу 30(c) з 6,7-диметокси-1,2,3,4тетрагідроізохінолін. Неочищений продукт очищають на силікагелі, елююючи ЕЮАс, одержуючи названу в підзаголовку сполуку у вигляді безбарвної твердої речовини(99%). Rf 0,15(EtOAc). 1Н ЯМР(d6-D MSO) δ: 2,05(3Н, с), 2,60-2,80(2Н, д), 3,55(2Н, м), 3,65(6Н, с), 4,25(2Н, д), 6,70(2Н, d). (b) 3,4-Диметокси-6-[1-(6,7-диметокси-1,2,3,4-тетрaгідроізохінол-2-іл)етиліденaміно]-2-йодбензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(f) з продукту стадії (а) і сполуки прикладу 1(e). Неочищений продукт очищають на силікагелі, елююючи СН2Сl2, одержуючи названу в підзаголовку сполуку(71%). Rf 0,74(EtOAc). MO m/z 522(MH +). (с) 3,4-Диметокси-6-[1-(6,7-диметокси-1,2,3,4-тетрагідроізохінол-2-іл)етиліденаміно]-2-(2піримідил)бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 5 з продукту стадії (b) і 2-(три-нбутилстаніл)піримідину. Очищають на силікагелі, одержуючи названу в підзаголовку сполуку(33%). R f 0,38(EtOAc). MC m/z 474(MH+). (d) 4-Аміно-6,7-диметокси-2-(6,7-диметокси-1,2,3,4-тетрагідроізохінол-2-іл)-5-(2-піримідил)хінолін Названу в заголовку сполуку одержують у спосіб прикладу 26(1) з продукту стадії (с). Неочищений продукт очищають на силікагелі, елююючи CH2Cl2/MeOH/0,88 NH3(95/5/0,5, об/об), одержуючи названу в заголовку сполуку у вигляді піни(29%). R f 0,16(СН2СI2/МеОН 95/5, об/об). MC m/z 474(МН+). 1 Н ЯМР(CDCl3) δ: 2,90(2Н, м), 3,70(5Н, с), 3,90(9Н, м), 4,00(3Н, с), 4,75(2Н, с), 6,65(1Н, с), 6,75(1H, с), 7,20(1H, с), 7,40(1H, т), 8,95(2Н, м). Приклад 32 4-Аміно-6,7-диметокси-2-(6,7-диметокси-1,2,3,4-тетрагідроізохінол-2-іл)-5-(2-піридил)хінолін (а) 4-Аміно-6,7-диметокси-2-(6,7-диметокси-1,2,3,4-тетрагідроізохінол-2-іл)-5-йодхінолін Зазначену в підзаголовку сполуку одержують у спосіб прикладу 1(g) з сполуки прикладу 31(b). Неочищений продукт очищають на силікагелі, елююючи сумішшю ЕЮАс/гексан(1/1, об/об), потім EtOAc, одержуючи названу в підзаголовку сполуку у вигляді не зовсім білої твердої речовини(67%). Rf 0,5(EtOAc). MC m/z 522(MH+). (b) 4-Аміно-6,7-диметокси-2-(6,7-диметокси-1,2,3,4-тетрагідроізохінол-2-іл)-5-(2-піридил)хінолін Названу в заголовку сполуку одержують у спосіб прикладу 5 з продукту стадії (а) і 2-(три-нбутилстаніл)піридину. Неочищений продукт очищають на силікагелі, елююючи CH2Cl2/MeOH/0,88 NH3(95/5/0,5, об/об), одержуючи названу в заголовку сполуку(20%). Rf 0,28(СН2СI2/МеОН 95/5, об/об). MC m/z 473(МН+). 1 Н ЯМР(CDCl3) δ: 2,85(2Н, т), 3,50(3Н, с), 3,70-3,90(10Н, м), 4,00(3Н, с), 4,70(2Н, с), 5,95(1H, с), 6,65(1Н, с), 6,70(1H, с), 7,20(1Н, с), 7,35(1H, т), 7,45(1Н, д), 7,80(1Н, д), 8,75(1Н, d). Приклад 33 4-Аміно-6,7-диметокси-2-[2-(4-морфолін)-5,6,7,8-тетрагідро-1,6-нафтирид-6-ил]-5-(2-піридил)хіназолін (а) 6-Бензил-3,4,5,6,7,8-гексагідро-1,6-нафтиридин-2он До розчину 1-бензил-4-піперидону(213г, 1,13моль) в толуолі(700мл) додають піролідин(190мл, 2,25моль), реакційну суміш закривають насадкою Дина-Старка і нагрівають до 150°С протягом 18год. Реакційну суміш о холоджують і упарюють за пониженого тиску, потім до залишку додають птолуолсульфонову кислоту(4,0г, 0,022моль), після чого акриламід(160г, 2,25моль). Реакційну суміш нагрівають при швидкому перемішуванні до 90°С протягом 1,5год і потім протягом ще 2год при 120°С. Охолоджену суміш потім фільтрують і одержану тверду речовину промивають ацетоном, потім ефіром. Маточні рідини об'єднують, упарюють за пониженого тиску і залишок розподіляють між EtOAc і Н2О. Органічний шар відділяють, висушують над MgSO4 та упарюють за пониженого тиску, одержуючи додаткову порцію твердої речовини. Тверду речовину об'єднують і нагрівають із зворотним холодильником з 4-толуолсуль фоновою кислотою(10г, 0,056моль) у діоксані(400мл) протягом 18год. При охолоджуванні одержують безбарвний кристалічний продукт, який фільтрують і промивають EtOAc, одержуючи названу в підзаголовку сполуку у вигляді безбарвних кристалів(176г, 65%). Rf 0,1(EtOAc). 1 Н ЯМР(CDCІ3) δ: 2,20(4Н, А), 2,50(2Н, т), 2,70(2Н, с), 3,00(2Н, с), 3,65(2Н, с), 7,20-7,45(5Н, м). (b) 6-Бензил-2-хлор-5,6,7,8-тетрагідро-1,6-нафтиридин До перемішуваної суспензії продукту стадії (а) (30г, 0 ,124моль) в толуолі(400мл) додають оксихлорид фосфор у(57,7мл, 0,619моль), потім тетрахлор-1,4-бензохінон(31,98г, 0,13моль). Реакційну суміш нагрівають із зворотним холодильником в атмосфері азоту протягом 18год, після чого толуол упарюють за пониженого тиску, залишок потім підлуговують 4N водним NaOH і продукт екстрагують ефіром(х3). Об'єднані органічні шари висушують над MgSО4 і упарюють за пониженого тиску. Неочищений продукт очищають на силікагелі, елююючи EtOAc, одержуючи названу в підзаголовку сполуку у вигляді твердої речовини(13,29г, 41%). Rf 0,8(EtOAc). MC m/z 259(MH +). (с) 2-Хлор-5,6,7,8-тетрагідро-1,6-нафтиридин До перемішуваного розчину продукту стадії (b) (13,28г, 0,0513моль) в толуолі(150мл) краплями додають 1-хлоретил хлорформат(5,54мл, 0,0513моль) при 0°С. Реакційну суміш нагрівають із зворотним холодильником протягом 2год. Після охолоджування толуол упарюють за пониженого тиску і залишок розподіляють між ЕtOАс/H 2О, органічний шар промивають послідовно 1N НСI і насиченим сольовим розчином, висушують над MgSO4 і упарюють за пониженого тиску. Одержаний залишок розчиняють в МеОН(150мл) і нагрівають із зворотним холодильником протягом 3год, після чого реакційну суміш упарюють за пониженого тиску і залишок розподіляють між СН 2СI2 і 2N водним NaOH, продукт екстрагують СН2СI2(х5). Об'єднані органічні шари висушують над MgSO4 та упарюють за пониженого тиску. Неочищений продукт очищають на силікагелі, елююючи СН2СI2/МеОН/0,88 NH3(90/10/1, об/об), одержуючи названу в підзаголовку сполуку(3,57г, 41%). Rf 0,25(СН2СI2/МеОН/0,88 NH3 90/10/1, об/об). MC m/z 169(МН+). (d) 2-Хлор-6-дифенілметил-5,6,7,8-тетрагідро-1,6-нафтиридин До розчину продукту стадії (c) (1,78г, 0,01моль) і триетиламіну(2,21мл, 0,016моль) в СН2СI2(20мл) додають дифенілхлорметан(2,13мл, 0,012моль). Реакційну суміш перемішують при кімнатній температурі протягом 20год і упарюють за пониженого тиску. Залишок розчиняють в DMA(20мл) і нагрівають до 100°С протягом 18год та охолоджують, розчин розбавляють СН 2СI2 і промивають послідовно насиченим водним розчином бікарбонату натрію, Н2О і насиченим сольовим розчином, потім висушують над MgSO4. Упарюють за пониженого тиску, одержуючи названу в підзаголовку сполуку у вигляді твердої речовини(1,01г, 30%). Rf 0,7(СН2СI2/МеОН/0,88 NH 3 90/10/1, об/об). MC m/z 335(МН+). (e) 6-Дифенілметил-2-(4-морфолін)-5,6,7,8-тетрагідро-1,6-нафтиридин До розчину морфоліну(0,62мл, 7,17ммоль) в THF(15мл) додають етилмагнійбромід(2,4мл, 7,17ммоль) при 0°С, реакційну суміш перемішують протягом 1год при кімнатній температурі, після чого додають розчин продукту стадії (d) (0,8г, 2,389ммоль) в THF(15мл), потім ацетилацетонат паладію (II) (0,073г, 0,239ммоль) і трифенілфосфін(0,125г, 0,478ммоль), та реакційну суміш нагрівають до 60°С протягом 18год. Після охолоджування розчин розподіляють між EtOAc і насиченим водним розчином хлориду амонію. Органічний шар відділяють, промивають послідовно Н2О, насиченим сольовим розчином, висушують над MgSO4 і упарюють за пониженого тиску. Неочищений продукт очищають на силікагелі, елююючи СН2СІ2/МеОН(97/3, об/об), одержуючи названу в підзаголовку сполуку(0,81г, 88%). Rf 0,63(СН2СI2/МеОН 95/5, об/об). MC m/z 386(МН+). (f) 2-(4-Морфолін)-5,6,7,8-тетрагідро-1,6-нафтиридин До розчину продукту стадії (e) (0,8г, 2,08ммоль) в МеОН, 1N HCІ(10/1, об/об, 33мл) додають 20% гідроксиду паладію на вугіллі(0,2г). Реакційну суміш гідрогенують при 345кПа(50p.s.і.) і 50°С протягом 56год, після чого каталізатор відфільтровують та промивають МеОН. Одержаний розчин упарюють за пониженого тиску і залишок розподіляють між СН 2СІ2 і насиченим водним розчином бікарбонату натрію. Продукт екстрагують СН 2СI2(х8), об'єднані органічні шари висушують над MgSO4 і упарюють за пониженого тиску. Неочищений продукт очищають на силікагелі, елююючи СН2СI2/МеОН/0,88 NH3(90/10/1, об/об), одержуючи названу в підзаголовку сполуку(0,13г, 28%). Rf 0,4(CH2Cl2/MeOH/0,88 NH390/10/1, об/об). MC m/z 220(МН+). (g) 4-Аміно-6,7-диметокси-2-[2-(4-морфолін)-5,6,7,8-тетрагідро-1,6-наф тирид-6-ил]-5-(2 піридил)хіназо лін Названу в заголовку сполуку одержують у спосіб прикладу 12(b) з сполуки прикладу 12(а) і продукту стадії (f). Неочищений продукт очищають на силікагелі, елююючи СН 2СI2/МеОН(95/5, об/об), одержуючи названу в заголовку сполуку у вигляді безбарвної піни(29%). Rf 0,37(СН2СІ2/Ме ОН/0,88 NH 3 90/10/1, об/об). MC m/z 450(MH+). 1 H ЯМР(CDCI3) δ: 1,30(2Н, с), 2,50(3Н, с), 3,10(2Н, м), 3,90-4,1(8Н, кілька піків), 4,80(2Н, с), 6,00(1Н, с), 7,00(1Н, д), 7,24(1Н, с), 7,40(2Н, м), 7,45(1Н, д), 7,80(1Н, т), 8,75(1Н, м). Приклад 34 4-Аміно-6,7-диметокси-2-(5-метансульфонамідо-1,2,3,4-тетрагідроізохінол-2-іл)-5-(2-піридил)хінолін (a) 3,4-диметокси-6-[1-(5-метансульфонамідо-1,2,З,4-тетрагідроізохінол-2-іл)етиліденаміно]-2-(2піридил)бензонітрил Зазначену в підзаголовку сполуку одержують у спосіб прикладу 5 з сполуки прикладу 22(b) і 2-(три-нбутилстаніл) піридину. Продукт очищають на силікагелі, елююючи СН 2СI2/МеОН(95/5, об/об), одержуючи названу в підзаголовку сполуку(45%) у вигляді піни. Rf 0,11(СН2Сl2/МеОН 95/5, об/об). (b) Гідро хлорид 4-аміно-6,7-диметокси-2-(5-метaнсульфонамідо-1,2,3,4-тетрaгідроізохінол-2-іл)-5-(2піридил)хінолiну Названу в заголовку сполуку одержують у спосіб прикладу 23(f) з продукту стадії (а). Продукт очищають на силікагелі, елююючи СН 2СI2/МеОН/0,88 NH3(90/10/1, об/об), потім обробляють надлишком ефірного розчину НСI, одержуючи названу в заголовку сполук у(10%) у вигляді безбарвної твердої речовини. Rf 0,21(СН2СІ2/МеОН/0,88 NH 3 93/7/1, об/об). MC m/z 506(МН+). 1 Н ЯМР(d6-DMSO) δ: 3,08(2Н, м), 3,48(3Н, с), 3,5-3,7(5Н, м), 3,80(2Н, м), 4,00(3Н, м), 4,78(2Н, с), 6,00(1H, шир. с), 6,19(1H, с), 7,20(1H, т), 7,28(2Н, м), 7,60(2Н, м), 7,90(1H, с), 8,01(1H, т), 8,77(1Н, д), 12,04(1Н, с). Виявлено С, 50,91; Н, 5,46; N, 10,89; C26H28C1N5O 4S 0,8·СН2СI2 Н2 О відповідає С, 51,26; Н, 5,07; N, 11,15%. Приклад 35 Сполуку прикладу 28 тестували в першому скринінгу, описаному вище("Скорочувальні відповіді простати людини") і виявили, що вона має значення рА2, що дорівнює 9,2.
ДивитисяДодаткова інформація
Назва патенту англійськоюQuinolin and quinazoline compounds, a pharmaceutical composition based thereon, a method for preparing thereof (alternatives), intermediate compounds (alternatives), a method for the treatment of benign prostatic hyperplasia.
Автори англійськоюFox, David, Nathan, Abraham
Назва патенту російськоюХинолиновые и хиназолиновы соединения, фармацевтическая композиция на их основе, способ их получения (варианты), промежуточные соединения (варианты), способ лечения доброкачественной гиперплазии простаты
Автори російськоюФокс Девид Натан Абрахам
МПК / Мітки
МПК: C07D 409/04, C07D 471/04, A61K 31/517, C07D 401/04, C07D 413/04, C07D 405/04, A61P 13/08, C07D 401/14, A61K 31/4725, A61K 31/519
Мітки: основі, гіперплазії, хіназолінові, варіанти, спосіб, проміжні, простати, композиція, одержання, фармацевтична, лікування, сполуки, хінолінові, доброякісної
Код посилання
<a href="https://ua.patents.su/23-62945-khinolinovi-ta-khinazolinovi-spoluki-farmacevtichna-kompoziciya-na-kh-osnovi-sposib-kh-oderzhannya-varianti-promizhni-spoluki-varianti-sposib-likuvannya-dobroyakisno-giperplazi-pro.html" target="_blank" rel="follow" title="База патентів України">Хінолінові та хіназолінові сполуки, фармацевтична композиція на їх основі, спосіб їх одержання (варіанти), проміжні сполуки (варіанти), спосіб лікування доброякісної гіперплазії простати</a>
Спіроазабіциклічні гетероциклічні сполуки, фармацевтична композиція на їх основі, спосіб лікування чи профілактики (варіанти), спосіб одержання сполуки (варіанти) та проміжні спіроазабіциклічні гетероциклічні с

Номер патенту: 61964
Опубліковано: 15.12.2003
Автори: Філліпс Ейфіон, Мек Роберт, Семас Саймон, Мейкор Джон
МПК: A61P 25/34, A61K 31/496, A61P 25/24, C07D 491/22, A61P 1/04, A61P 25/28, A61K 31/444, A61P 25/04, C07F 5/00, C07F 7/18, A61P 25/00, A61P 25/22, A61P 25/14, A61P 25/18, A61K 31/439, A61P 25/16, A61K 31/5377, A61K 31/695, A61P 43/00, A61K 31/4738
Мітки: фармацевтична, композиція, гетероциклічні, основі, профілактики, варіанти, лікування, спосіб, проміжні, спіроазабіциклічні, сполуки, одержання
Формула / Реферат:
1. Спіроазабіциклічні гетероциклічні сполуки формули І,де n дорівнює 0 чи 1,m дорівнює 0 чи 1,p дорівнює 0 чи 1,Х являє собою оксиген чи сульфур,Y являє собою CH, N чи NO,W являє собою оксиген, H2 чи F2,A являє собою N чи C(R2),G являє собою N чи C(R3),D являє собою N чи C(R4)за умови, що не більш ніж один з A, G та D являє собою нітроген, але принаймні один з Y, A,...
Спосіб одержання циталопраму, проміжні сполуки та фармацевтична композиція на його основі

Номер патенту: 62984
Опубліковано: 15.01.2004
Автори: Петерсен Ханс, Брегнедал Петер, Бьогесьо Клаус Петер
МПК: A61K 31/34, C07D 307/87
Мітки: сполуки, проміжні, спосіб, композиція, циталопраму, одержання, основі, фармацевтична
Формула / Реферат:
1. Спосіб одержання циталопраму, при якомуа) здійснюють реакцію сполуки формули IV, IVде R1 - це Н або С1-6-алкілкарбоніл, з реактивом Гриньяра 4-галогенфторофенілу,b) здійснюють реакцію отриманої сполуки формули V, Vде R1 має значення, як визначено вище, з...
Спосіб одержання циталопраму, проміжні сполуки та фармацевтична композиція на його основі

Номер патенту: 62985
Опубліковано: 15.01.2004
Автори: Петерсен Ханс, Бьогесьо Клаус Петер, Бек Соммер Мікаел
МПК: A61K 31/34, C07D 307/87, A61P 25/24
Мітки: одержання, сполуки, проміжні, основі, циталопраму, фармацевтична, спосіб, композиція
Формула / Реферат:
1. Спосіб одержання циталопраму, при якому а) відновлюють сполуку формули IV, IVде R1 - це CN, С1-6-алкілоксикарбоніл або С1-6-алкіламінокарбоніл, b) здійснюють замикання кільця в отриманій сполуці формули V, Vде R1 є таким, як визначено вище, з одержанням таким чином сполуки формули VI, VIде R1 є таким, як визначено вище;с) потім, якщо R1 є ціаногрупою, використовують сполуку...
Піридазинохіноліни, спосіб їх одержання (варіанти), проміжні сполуки, фармацевтична композиція (варіанти) для лікування або профілактики нейродегенеративних порушень та способи лікування

Номер патенту: 60291
Опубліковано: 15.10.2003
Автори: Девенпорт Тімоті Вейн, Бер Томас Майкл, МакКінні Джеффрі Алан, Спаркс Річард Брюс, Емпфілд Джеймс Рой
МПК: C07D 215/56, A61K 31/50, C07D 495/14, A61P 43/00, A61P 25/00, A61P 25/08, C07D 471/04, A61P 3/08, A61P 3/10, A61P 25/28, C07D 471/14
Мітки: порушень, варіанти, способи, лікування, профілактики, проміжні, одержання, композиція, нейродегенеративних, фармацевтична, спосіб, сполуки, піридазинохіноліни
Формула / Реферат:
1. Пиридазинохинолины формулы II или их фармацевтически приемлемые соли и таутомеры:,где:Z выбран из О, S или NH, или, когда кольцо В N-таутомеризовано, группа Z может быть выбрана из Н, ОН, SH или NH2,кольцо А выбрано из ортоконденсированного ароматического или гетероароматического пяти- или шестичленного кольца, выбранного из фенила,...
Хінолінові і хіноксалінові сполуки, фармацевтична композиція, спосіб інгібування (варіанти) та спосіб лікування (варіанти)

Номер патенту: 57790
Опубліковано: 15.07.2003
Автори: Персонс Пол Е., Майерс Майкл Р., Магвір Мартін П., Спада Альфред П.
МПК: A61K 31/498, C07D 453/00, C07D 403/04, A61P 17/06, A61P 43/00, C07D 215/38, A61P 19/08, A61P 9/10, C07D 215/20, A61P 9/00, A61K 31/5377, C07D 241/42, A61P 35/02, C07D 405/12, A61P 35/00, C07D 241/54, A61P 29/00, C07D 241/52, C07D 405/10, A61K 31/47, C07D 241/44
Мітки: лікування, хіноксалінові, варіанти, хінолінові, фармацевтична, спосіб, композиція, інгібування, сполуки
Формула / Реферат:
1. Хінолінові і хіноксалінові сполуки формули І:, (I)деХ означає L1 або L2Z2;L1 означає (CR3aR3b)rН або (CR3aR3b)m-Z3- (CR3’aR3’b)nН;L2 означає (CR3aR3b) p-Z4- (CR3’aR3’b)q або етеніл;Z1 означає СН або N;Z2 означає необов'язково заміщений циклоалкіл, необов'язково заміщений циклоалкеніл, необов'язково заміщений гетероцикліл або необов'язково заміщений гетероцикленіл;Z3 означає О, NR4, S,...
Попередній патент: Інерційний супермаховичний відцентровий сепаратор для розділення полідисперсних рідких систем
Наступний патент: Спосіб попереднього підігріву дуттєвого повітря
Випадковий патент: Спосіб приготування полегшеного тампонажного розчину