Фармацевтичні композиції, що містять нікотинову кислоту та анатгоніст рецептора dp
Номер патенту: 89615
Опубліковано: 25.02.2010
Автори: Чен Кан, Меттерс Кетлін М., О'Нейл Гері, Уотерс Джерард







Формула / Реферат
![]()
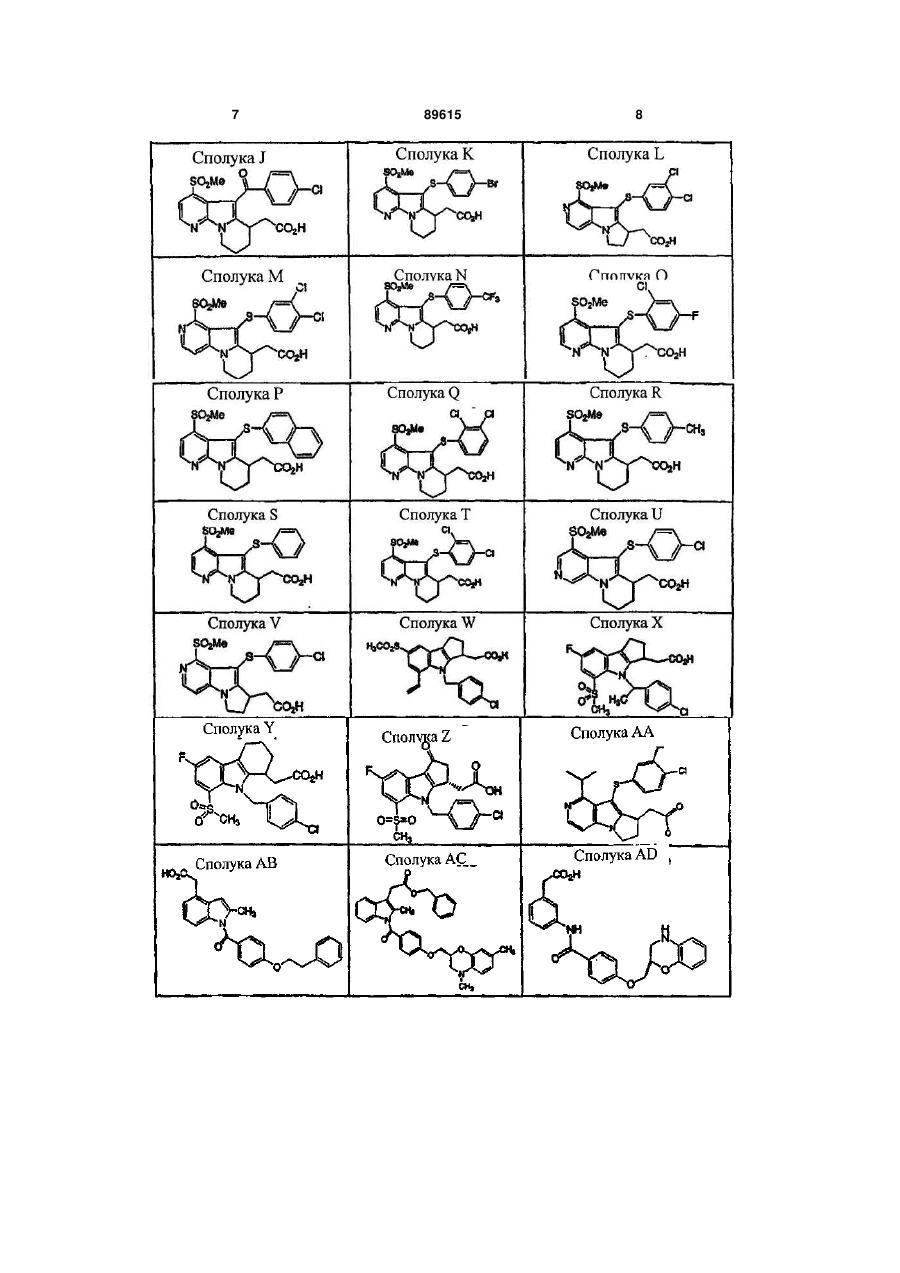
![]() 1. Фармацевтична композиція, яка містить нікотинову кислоту і сполуку формули Е:
1. Фармацевтична композиція, яка містить нікотинову кислоту і сполуку формули Е:
або їх фармацевтично прийнятну сіль або сольват, у поєднанні з фармацевтично прийнятним носієм.
2. Фармацевтична композиція, яка містить приблизно 1000 мг нікотинової кислоти і антагоніст DP формули Е або їх сіль або сольват, у поєднанні з фармацевтично прийнятним носієм.
3. Фармацевтична композиція за п. 2, в якій антагоніст DP є сполукою формули Е або його сіллю або сольватом і представлений у кількості, яка змінюється від приблизно 1 мг до приблизно 500 мг.
4. Фармацевтична композиція за будь-яким з пунктів 1-3 у формі таблетки або капсули.
5. Фармацевтична композиція за будь-яким з пунктів 1-4 у формі таблетки.
6. Фармацевтична композиція за п.5 у вигляді таблетки з уповільненим вивільненням.
7. Фармацевтична композиція, яка містить нікотинову кислоту і сполуку формули Е:
або її фармацевтично прийнятну сіль або сольват, і інгібітор редуктази HMG Co-A, вибраний з ловастатину, симвастатину, симвастатину з вільною дигідроксильованою кислотною групою, правастатину, флювастатину, аторвастатину, пітавастатину і розувастатину, в комбінації з фармацевтично прийнятним носієм.
8. Фармацевтична композиція за п.7, у якій інгібітор редуктази HMG Co-A являє собою аторвастатин.
9. Фармацевтична композиція за п.7, яка містить нікотинову кислоту і сполуку формули Е:
або її фармацевтично прийнятну сіль або сольват і симвастатин, у комбінації з фармацевтично прийнятним носієм.
Текст
1. Фармацевтична композиція, яка містить нікотинову кислоту і сполуку формули Е: 2 Cl або її фармацевтично прийнятну сіль або сольват і симвастатин, у комбінації з фармацевтично прийнятним носієм. 3 Ніацин, або нікотинова кислота (піридин-3карбонова кислота) являє собою лікарський засіб, широко відомий своїм ефектом підвищення рівня ліпопротеїнів високої щільності (HDL) у сироватці. Однак застосування нікотинової кислоти часто пов'язане з розширенням кровоносних посудин шкіри, що іноді називають гіперемією. Цей побічний ефект обумовлений залежним від нікотинової кислоти вивільненням у шкірі простагландину D2 і є настільки важким, що багато пацієнтів переривають лікування нікотиновою кислотою. Даний винахід належить до лікування атеросклерозу, дисліпідемій, діабету й пов'язаних з ними станів за допомогою введення нікотинової кислоти або іншого агоніста рецептора нікотинової кислоти в поєднанні із сполукою, що так знижує або усуває розширення кровоносних судин шкіри, яке відбувається в протилежному випадку, що лікування можна продовжувати без істотної гіперемії. У людини цього досягають за допомогою введення нікотинової кислоти або агоніста рецептора нікотинової кислоти і сполуки, що є антагоністом рецептора DP. Із простагландином D2 взаємодіють рецептори різних підтипів. Один рецептор простагландину D2 позначають як "DP", а інший рецептор простагландину D2 відомий як "CRTH2". Для запобігання, мінімізування або зниження гіперемії, що може відбуватися в протилежному випадку, у даному винаході використовують антагонізм до рецептора DP. Таким чином, однією із цілей даного винаходу є усунення або зниження істотної гіперемії (частоти й/або тяжкості) як побічного ефекту у процесі лікування в людини атеросклерозу, дисліпідемій, діабету й пов'язаних з ними станів з використанням нікотинової кислоти або іншого агоніста рецептора нікотинової кислоти. Іншою метою даного винаходу є забезпечення комбінованого лікування атеросклерозу, що знижує побічні ефекти в цілому. Ще однією метою є забезпечення фіксованої комбінації фармацевтичної композиції для перорального застосування. Ці та інші цілі очевидні з наданого тут опису винаходу. Представлено спосіб лікування атеросклерозу в пацієнта-людини, при необхідності такого лікуванні, що включає в себе одержання пацієнтом нікотинової кислоти або її солі або сольвату, або іншого агоніста рецептора нікотинової кислоти й антагоніста рецептора DP у кількостях, ефективних для лікування атеросклерозу при відсутності істотної гіперемії. Винахід ілюструють за допомогою фігур, що додаються тут, у яких: На Фіг.1 представлений графік, на якому показане інгібування сполукою D викликаного простагландином D2 розширення кровоносних судин у миші. На Фіг.2 представлений графік, на якому показане інгібування сполукою D викликаного нікотиновою кислотою розширення кровоносних судин у миші. 89615 4 На Фіг.3 представлений графік, на якому показане інгібування іншими вибраними сполуками викликаного нікотиновою кислотою розширення кровоносних судин у миші. Ніацин, або нікотинова кислота (піридин-3карбонова кислота) являє собою лікарський засіб, широко відомий своїм ефектом підвищення рівня ліпопротеїнів високої щільності (HDL) у сироватці, а також іншими сприятливими змінами ліпідного профілю (зниженням ліпопротеїнів дуже низької щільності (VLDL) і низької щільності (LDL), тригліцеридів, вільних жирних кислот (FFA) і ліпопротеїну(а) [Lp(a)]. При одержанні людиною в терапевтично ефективних дозах, таких як приблизно від 50мг до таких високих як приблизно 8 грамів на добу, нікотинова кислота підвищує рівень HDL. Однак нікотинова кислота часто викликає розширення кровоносних судин шкіри, що іноді називають гіперемією. Гіперемія звичайно викликає почервоніння шкіри, що супроводжується відчуттям тепла, сверблячкою або подразненням. Вона може бути винятково неприємна й настільки важка, що багато пацієнтів переривають лікування нікотиновою кислотою. Даний винахід належить до лікування, профілактики або зворотного розвитку атеросклерозу та інших захворювань і станів, тут описаних, нікотиновою кислотою або її сіллю або сольватом, або іншим агоністом рецептора нікотинової кислоти без істотної гіперемії. У людини цього досягають за допомогою введення нікотинової кислоти або її солі або сольвату, або іншого агоніста рецептора нікотинової кислоти й сполуки, що являє собою антагоніст рецептора DP, що запобігає, знижує або мінімізує частоту й/або тяжкість ефекту гіперемії. Існують щонайменше два рецептори, що взаємодіють із простагландином D2 і що позначаються як "DP" і "CRTH2". Даний винахід переважно належить до нікотинової кислоти або агоністів рецептора нікотинової кислоти, застосовуваним у сполученні з антагоністами рецептора DP. В одному з аспектів, що представляє інтерес, винахід належить до способу лікування атеросклерозу в пацієнта-людини, при необхідності такого лікування, що включає в себе одержання пацієнтом нікотинової кислоти або її солі або сольвату, або іншого агоніста рецептора нікотинової кислоти й антагоніста рецептора DP у кількостях, ефективних для лікування атеросклерозу при відсутності істотної гіперемії. В іншому аспекті, що представляє інтерес, винахід належить до способу підвищення рівня HDL у сироватці пацієнта-людини при необхідності такого лікування, що включає в себе одержання пацієнтом нікотинової кислоти або її солі або сольвату, або іншого агоніста рецептора нікотинової кислоти й антагоніста рецептора DP, де зазначене поєднання ефективне для підвищення рівня HDL у сироватці пацієнта при відсутності істотної гіперемії. В іншому аспекті, що представляє інтерес, винахід належить до способу лікування дисліпідемії у пацієнта-людини, при необхідності такого лікування, що включає в себе одержання пацієнтом ніко 5 тинової кислоти або її солі або сольвату, або іншого агоніста рецептора нікотинової кислоти й антагоніста рецептора DP у кількостях, ефективних для лікування дисліпідемії при відсутності істотної гіперемії. В іншому аспекті, що представляє інтерес, винахід належить до способу зниження рівнів VLDL або LDL у сироватці пацієнта-людини, при необхідності такого лікування, що включає в себе одержання пацієнтом нікотинової кислоти або її солі або сольвату, або іншого агоніста рецептора нікотинової кислоти й антагоніста рецептора DP у кількостях, ефективних для зниження рівнів VLDL або LDL у сироватці пацієнта при відсутності істотної гіперемії. В іншому аспекті, що представляє інтерес, винахід належить до способу зниження рівня тригліцеридів у сироватці пацієнта-людини, при необхідності такого лікування, що включає в себе одержання пацієнтом нікотинової кислоти або її солі або сольвату, або іншого агоніста рецептора нікотинової кислоти й антагоніста рецептора DP в кількостях, ефективних для зниження рівня тригліцеридів у сироватці пацієнта при відсутності істотної гіперемії. В іншому аспекті, що представляє інтерес, винахід належить до способу зниження рівня Lp(a) в сироватці пацієнта-людини, при необхідності такого лікування, що включає в себе одержання пацієнтом нікотинової кислоти або її солі або сольвату, або іншого агоніста рецептора нікотинової кислоти й антагоніста рецептора DP в кількостях, ефективних для зниження рівня Lp(a) у сироватці пацієнта при відсутності істотної гіперемії. Тут Lp(a) належить до ліпопротеїну(а). В аспекті, що представляє особливий інтерес, винахід належить до кожного з вищеописаних способів, де використовують нікотинову кислоту або її сіль або сольват. Більш конкретний інтерес являє собою застосування нікотинової кислоти. 89615 6 Крім того, у ще одному аспекті, що представляє інтерес, винахід належить до вибіркової модуляції рецептора DP антагоністом рецептора DP у кількостях, ефективних для зниження або запобігання ефекту гіперемії в пацієнта. В іншому аспекті, що представляє особливий інтерес, винахід належить до кожного з вищеописаних способів, де використовують нікотинову кислоту, і антагоніст рецептора DP викликає вибіркову модуляцію рецептора DP і не викликає істотної модуляції рецептора CRTH2. В іншому аспекті, що представляє особливий інтерес, винахід належить до способу лікування атеросклерозу, дисліпідемій, діабету або пов'язаних з ними станів у пацієнта-людини, при необхідності такого лікування, що включає в себе одержання пацієнтом нікотинової кислоти або її солі або сольвату, або іншого агоніста рецептора нікотинової кислоти й антагоніста рецептора DP, де зазначену комбінацію вводять в кількості, ефективній для лікування атеросклерозу, дисліпідемій, діабету або пов'язаного з ними стану при відсутності істотної гіперемії. У ще одному аспекті винахід належить до застосування сполуки антагоніста рецептора DP у поєднанні з нікотиновою кислотою або її сіллю або сольватом, або іншим агоністом рецептора нікотинової кислоти для лікування атеросклерозу в людини при відсутності істотної гіперемії. В іншому аспекті, що представляє особливий інтерес, винахід належить до вищеописаних способів, де антагоніст рецептора DP вибраний із групи, що включає в себе сполуки від А до AJ і їх фармацевтично прийнятні солі й сольвати. Приклади сполук, що особливо придатні для застосування як селективні антагоністи рецепторів DP і таких, що пригнічують ефект гіперемії, включають в себе наступні: 7 89615 8 9 а також їх фармацевтично прийнятні солі й сольвати. Тут атеросклероз належить до форми судинного захворювання, що характеризується накопиченням атеросклеротичних бляшок, що містять холестерин і ліпіди у внутрішній оболонці стінок великих і середніх артерій. Атеросклероз включає в себе судинні захворювання й стани, які розпізнаються лікарями, що практикують у відповідних галузях медицини, і зрозумілі їм. Терміни "атеросклероз" і "атеросклеротична хвороба" описують атеросклеротичне серцево-судинне захворювання, що включає в себе рестеноз внаслідок процедур реваскуляризації, коронарну хворобу серця (також відому як хвороба коронарних артерій або ішемічна хвороба серця), цереброваскулярну хворобу, включаючи мультиінфарктну деменцію, і хворобу периферичних судин, включаючи еректильну дисфункцію, оскільки всі вони являють собою клінічні прояви атеросклерозу. Термін "дисліпідемія", застосовуваний у загальноприйнятому розумінні, належить до рівнів, що відхиляються від норми, таких ліпідів плазми як HDL (низький), LDL (високий), VLDL (високий), тригліцериди (високий), ліпопротеїн(а) (високий), FFA (високий) та інших ліпідів сироватки або їх поєднань. Це може бути неускладнений стан або ланка конкретного відповідного захворювання або стану, такого як діабет (діабетична дисліпідемія), метаболічний синдром і т.п. Таким чином, у даний винахід включені неускладнені дисліпідемії, а також ті, що пов'язані з станами, що лежать в їх основі. Термін "пацієнт" включає в себе ссавців, особливо людину, які застосовують дані активні речовини для профілактики або лікування медичного стану. Введення пацієнту лікарських засобів включає в себе як введення самим пацієнтом, так і введення пацієнтові іншим індивідуумом. У пацієнта може бути необхідність у лікуванні вже наявного захворювання або медичного стану або бажання профілактичного лікування для запобігання або зниження ризику розвитку атеросклерозу. Під терміном "терапевтично ефективна кількість" розуміють кількість лікарського засобу, що 89615 10 викликає необхідну біологічну або медичну відповідь. Наприклад, нікотинову кислоту часто вводять у дозах приблизно від 50мг до приблизно 8 грамів щодоби. Терміни "профілактично ефективна кількість" і "кількість, ефективна для запобігання", відносяться до кількості лікарського засобу, що запобігає або знижує ризик розвитку біологічної або медичної події, якій потрібно запобігти. У багатьох випадках профілактично ефективна кількість - те саме, що й терапевтично ефективна кількість. Описаний тут винахід включає в себе введення сполук і композицій, описаних тут, для запобігання або зниження ризику розвитку або повторного розвитку, якщо існує така ймовірність, події ішемічної хвороби серця, цереброваскулярної події й/або переміжної кульгавості. Під явищами коронарної хвороби серця (CHD), мають на увазі, зокрема, смерть від CHD, інфаркт міокарда (тобто серцевий напад) і процедури коронарної реваскуляризації. Під цереброваскулярними явищами мають на увазі, зокрема, ішемічний або геморагічний інсульти (відомі також як гострі порушення мозкового кровообігу) і транзиторні ішемічні атаки. Переміжна кульгавість являє собою клінічний прояв хвороби периферичних посудин. Тут під терміном "подія атеросклеротичної хвороби" мають на увазі, зокрема, явища коронарної хвороби серця, цереброваскулярні явища й переміжну кульгавість. Передбачається, що індивідууми, які перенесли одну або більше несмертельних подій атеросклеротичної хвороби, являють собою індивідуумів, для яких існує ймовірність повторного розвитку таких подій. Відповідно, даний винахід також представляє спосіб запобігання або зниження ризику первинного або наступного розвитку явищ атеросклеротичної хвороби, що включає в себе введення пацієнтові, у випадку наявності ризику такої події, профілактично ефективної кількості сполук, описаних тут, із запобіганням або мінімізацією істотної гіперемії. Під час введення пацієнт може вже мати атеросклеротичну хворобу або ризик її розвитку. 11 Крім того, спосіб належить до запобігання або уповільнення формування нового осередка ураження або атеросклеротичної бляшки й запобіганню або уповільненню розвитку наявних осередків ураження або бляшок, а також зворотного розвитку наявних осередків ураження або бляшок із запобіганням або мінімізацією істотної гіперемії. Відповідно, в одному з аспектів даний винахід належить до способу зупинення або уповільнення розвитку атеросклерозу, а також зупинення або уповільнення розвитку атеросклеротичних бляшок, що включає в себе одержання пацієнтом, при необхідності такого лікування, терапевтично ефективної кількості кожного з описаних тут антагоністів рецептора DP у сполученні з нікотиновою кислотою або іншим агоністом рецептора нікотинової кислоти. Цей спосіб також належить до зупинення або уповільнення розвитку атеросклеротичних бляшок, що існують на момент початку даного лікування (тобто "наявних атеросклеротичних бляшок"), а також до зупинення або уповільнення утворення нових атеросклеротичних бляшок у пацієнтів з атеросклерозом. В іншому аспекті даний винахід належить до способу запобігання або зниження ризику розриву атеросклеротичної бляшки, що включає в себе введення пацієнтові, при необхідності такого лікування, профілактично ефективної кількості кожної з описаних тут сполук разом з нікотиновою кислотою або іншим агоністом рецептора нікотинової кислоти. Як застосовують тут, розрив належить до відриву бляшки, що може потрапити в кровоносні судини. У додатковому аспекті даний винахід належить до способу запобігання або зниження ризику розвитку атеросклерозу, що включає в себе одержання пацієнтом, при необхідності такого лікування, профілактично ефективної кількості описаних тут сполук. В іншому аспекті винахід належить до способу лікування або запобігання атеросклерозу, дисліпідеміям або пов'язаному з ними стану, що включає в себе попереднє лікування пацієнта-людини, при необхідності такого лікування, ефективною для інгібування або зниження гіперемії кількістю антагоніста рецептора DP, а потім лікування зазначеного пацієнта нікотиновою кислотою, її сіллю або сольватом, або іншим агоністом рецептора нікотинової кислоти в кількостях, ефективних для лікування або запобігання зазначеним атеросклерозу, дисліпідемії або пов'язаному з ними стану при відсутності істотної гіперемії. Крім того, в іншому аспекті винахід належить до вищеописаного способу, що додатково включає в себе попереднє лікування або лікування пацієнта інгібітором HMG Со-А-редуктази. В іншому аспекті винахід належить до способу лікування або запобігання вищевказаним станам, де інгібітор HMG Со-А-редуктази являє собою симвастатин. В одному з аспектів описані тут способи відносяться до застосування нікотинової кислоти або сполуки іншого агоніста рецептора нікотинової кислоти в кількості, ефективній для досягнення описаних тут результатів, і антагоніста рецептора DP, що вибірково регулює рецептор DP без істот 89615 12 ної регуляції рецептора CRTH2. Так, антагоніст рецептора DP має спорідненість до рецептора DP (тобто Kі), щонайменше приблизно в 10 разів більш високоу (чисельно меншою величиною Kі), ніж спорідненість до рецептора CRTH2. Будь-яка сполука, що вибірково взаємодіє з DP згідно із цими правилами, вважається "DP-селективною". Фраза "при відсутності істотної гіперемії" належить до побічного ефекту, що часто спостерігають при введенні нікотинової кислоти в терапевтичних кількостях. Гіперемічний ефект нікотинової кислоти звичайно стає менш частим і менш важким у міру розвитку в пацієнта толерантності до лікарського засобу в терапевтичних дозах, однак до деякої міри ефект гіперемії однаково є присутнім. Таким чином, "при відсутності істотної гіперемії" належить до зниження тяжкості гіперемії, коли вона виникає, або меншим, ніж відбувається в іншому випадку, подіям гіперемії. Переважно частоту виникнення гіперемії знижують щонайменше приблизно на третину, більш переважно, якщо частоту виникнення знижують наполовину, і найбільше переважно, якщо частоту виникнення гіперемії знижують приблизно на дві третини й більше. Подібним чином, тяжкість переважно знижують щонайменше приблизно на третину, більш переважно щонайменше наполовину, і найбільше переважно щонайменше приблизно на дві третини. Очевидно, що стовідсоткове зниження частоти виникнення й тяжкості гіперемії являє собою найбільш переважне, але не вимагається. Специфічні для будь-якого конкретного пацієнта рівні дози й режим введення дози залежать від різних факторів, включаючи вік пацієнта, масу його тіла, загальний стан здоров'я, стать, дієту, час введення, спосіб введення, швидкість виведення, комбінацію лікарських засобів і тяжкість стану пацієнта. Розгляд цих факторів входить у сферу дій звичайного клінічного лікаря-фахівця з метою визначення терапевтично ефективної або профілактично ефективної кількості дози, необхідної для запобігання, зворотного розвитку або зупинення розвитку стану. Передбачається, що описані тут сполуки будуть вводитися на щоденній основі протягом періоду, призначеного для лікування або запобігання відповідному медичному стану пацієнта, включаючи курс лікування, що триває місяці, роки або все життя пацієнта. З описаними тут сполуками можна вводити одну або більше додаткових активних речовин. Додаткові активна речовина або речовини можуть являти собою сполуки, що модифікують ліпіди або речовини, що мають інші фармацевтичні активності, або речовини, що мають як ліпід-модифікуючі ефекти, так і інші фармацевтичні активності. Приклади додаткових активних речовин, які можна застосовувати, містять, не обмежуючись ними, інгібітори HMG-CoA-редуктази, які, у свою чергу, містять статини у формі лактону або вільної дигідроксильованої кислоти і їх фармацевтично прийнятні солі й складні ефіри, включаючи, але не обмежуючись ними, ловастатин (див. патент США №4342767), симвастатин (див. патент СІЛА №4444784), симвастатин у формі вільної дигідроксильованої кислоти, особливо її амонієвої або ка 13 льцієвої солей, правастатин, особливо його натрієву сіль (див. патент США №4346227), флувастатин, особливо його натрієву сіль (див. патент США №5354772), аторвастатин, особливо його кальцієву сіль (див. патент США №5273995), пітавастатин, також відомий як NK-104 (див. номер міжнародної публікації РСТ WO 97/23200) і розувастатин, також відомий як ZD-4522, ® (CRESTOR ; див. патент США №5260440); інгібітори HMG-Co-A-синтази; інгібітори скваленепоксидази; інгібітори скваленсинтетази (також відомі як інгібітори скваленсинтази); інгібітори ацил-СоАхолестеринацилтрансферази (АСАТ), включаючи селективні інгібітори АСАТ-1 або АСАТ-2, а також інгібітори подвійної дії, АСАТ-1 і -2; інгібітори мікросомального білка-переносника тригліцеридів (МТР); інгібітори ендотеліальної ліпази; засоби, що зв'язують жовчні кислоти; засоби, що індукують рецептор LPL; інгібітори агрегації тромбоцитів, наприклад, антагоністи глікопротеїнового ІІb/IIIа рецептора фібриногену й аспірин; агоністи рецептора гамма-типу людини, що активуються пероксисомним проліфератором (PPAR ), включаючи сполуки, звичайно відомі як глітазони, наприклад, піоглітазон і росиглітазон, і включаючи сполуки, що входять у структурний клас, відомий як тіазолідиндіони, а також агоністи PPAR , що не входять у структурний клас тіазолідиндіонів; агоністи PPARα, такі як клофібрат, фенофібрат, включаючи мікронізований фенофібрат, і гемфіброзил; агоністи PPAR подвійної дії, α/ ; вітамін В6 (також відомий як піридоксин) і його фармацевтично прийнятні солі, такі як сіль НСІ; вітамін В12 (також відомий як ціанкобаламін); фолієва кислота або її фармацевтично прийнятні сіль або складний ефір, такі як натрієва сіль і метилглукамінова сіль; вітаміниантиоксиданти, такі як вітаміни С і Ε й бетакаротин; бета-блокатори; антагоністи ангіотензину І, такі як лозартан; інгібітори ангіотензинперетворюючого ферменту, такі як еналаприл і каптоприл; інгібітори реніну; блокатори кальцієвих каналів, такі як ніфедипін і дилтіазем; антагоністи ендотеліну; речовини, що підвищують експресію гена АВСА1; сполуки, які інгібують білок-переносник ефірів холестерину (СЕТР); сполуки, які інгібують білок-активатор 5-ліпоксигенази (FLAP); сполуки, які інгібують 5-ліпоксигеназу (5-LO); ліганди Хрецептора фарнезоїду (FXR), що включають в себе як антагоністи, так і агоністи; ліганди Хрецептора печінки типу альфа (LXR); ліганди LXRбета; сполуки дифосфонатів, такі як алендронат натрію; інгібітори циклооксигенази-2, такі як рофекоксиб і целекоксиб; і сполуки, що знижують судинне запалення. У даному винаході можна також застосовувати інгібітори всмоктування холестерину. Такі сполуки перешкоджають переміщенню холестерину із просвіту кишечнику в ентероцити стінки тонкого кишечнику, знижуючи в такий спосіб рівень холестерину в сироватці. Приклади інгібіторів всмоктування холестерину описані в патентах США №№5846966, 5631365, 5767115, 6133001, 5886171, 5856473, 5756470, 5739321, 5919672 і заявках РСТ №№WO 00/63703, WO 00/60107, WO 00/38725, WO 00/34240, WO 00/20623, WO 89615 14 97/45406, WO 97/16424, WO 97/16455, і WO 95/08532. Найбільш значимий інгібітор всмоктування холестерину являє собою езетиміб, також відомий як 1-(4-фторфеніл)-3(R)-[3(S)-(4фторфеніл)-3-гідроксипропіл)]-4(S)-(4гідроксифеніл)-2-азетидінон, описаний у патентах США №№5767115 і 5846966. Терапевтично ефективні кількості інгібіторів всмоктування холестерину містять дози приблизно від 0,01мг на кг маси тіла приблизно до 30мг на кг маси тіла на добу, переважно приблизно від 0,1мг/кг приблизно до 15мг/кг. Пацієнти, хворі на діабет, можуть одержувати сполуки, що використовуються в даному винаході, із традиційними діабетичними лікарськими засобами. Наприклад, пацієнт, хворий на діабет, якому проводять описане тут лікування, може також приймати інсулін або антидіабетичний лікарський засіб перорального застосування. Метформін являє собою один із прикладів такого анти діабетичного лікарського засобу перорального застосування. Інформація про дози Як застосовують тут, нікотинова кислота належить до піридин-3-карбонової кислоти. Однак у даному винаході застосовують також солі й сольвати нікотинової кислоти, і в даному винаході застосовні численні фармацевтично прийнятні солі й сольвати нікотинової кислоти. Застосовні, як описано тут, солі являють собою солі лужних металів, зокрема натрієві й калієві. Подібним чином, лужноземельні метали, зокрема кальцій і магній, утворюють застосовні тут солі. Також застосовні тут солі являють собою різні солі амінів, таких як амоній і сполуки заміщеного амонію. Подібним чином, в об'ємі даного винаходу застосовні сольватовані форми нікотинової кислоти. Приклади включають в себе напівгідрат, моно-, ди-, три- і півторагідрати. Особливий інтерес у даному винаході представляє застосування вільної кислоти, піридин-3карбонової кислоти. Як описано тут, для зниження або запобігання ефекту гіперемії в пацієнтів ссавців, особливо людини, застосовні антагоністи DP, у дозах в діапазоні від таких низьких, як приблизно 0,01мг/кг/добу, до таких високих, як приблизно 100мг/кг/добу, що вводять як разову або дробову добову дозу. Переважні дози, разові або дробові добові дози, приблизно від 0,1мг/добу до таких високих, як приблизно 1,0г/добу. Як описано тут, прийнятна разова або дробова добова доза нікотинової кислоти входить в діапазон від такої низької, як приблизно 50мг/добу, до такої високої, як приблизно 8г/добу. Спочатку можна застосовувати низькі дози, а для подальшого зниження ефекту гіперемії дози можна підвищувати. Дози агоністів рецептора нікотинової кислоти, відмінних від нікотинової кислоти, варіюють у широкому діапазоні. В основному застосовні для лікування атеросклерозу агоністи рецептора нікотинової кислоти одержують у кількостях у діапазоні від таких низьких, як приблизно 0,01мг/кг/добу, до таких високих, як приблизно 100мг/кг/добу, у разо 15 вій або дробовій дозах. Звичайна доза становить приблизно від 0,1мг/добу до приблизно 2г/добу. Сполуки, застосовувані в даному винаході, можна вводити за допомогою будь-якого загальноприйнятого способу введення. Переважний пероральний спосіб введення. Нікотинову кислоту, її сіль або сольват, або інший агоніст рецептора нікотинової кислоти й антагоніст DP можна вводити разом або послідовно в добових дозах, однократно або багаторазово, наприклад, два, три або чотири рази на добу, залишаючись в об'ємі винаходу. Якщо бажано особливо тривале вивільнення, таке, коли тривале вивільнення продукту характеризують профілем вивільнення, що виходить за межі 24 годин, дози можна вводити кожну наступну добу. Однак переважні однократні добові дози. Подібним чином, можна застосовувати введення доз ранком або ввечері. Фармацевтичні композиції Описані тут фармацевтичні композиції в основному включають в себе нікотинову кислоту або інший агоніст рецептора нікотинової кислоти, антагоніст рецептора DP і фармацевтично прийнятний носій. Приклади прийнятних композицій для перорального застосування включають в себе таблетки, капсули, пастилки, лікарські льодяники, суспензії, дисперсні порошки або гранули, емульсії, сиропи й еліксири. Приклади інгредієнтів носія включають в себе розріджувачі, зв'язувальні речовини, дезінтегруючі речовини, змащувальні речовини, підсолоджувачі, ароматизатори, барвники, консерванти й т.п. Приклади розріджувачів включають в себе, наприклад, карбонат кальцію, карбонат натрію, лактозу, фосфат кальцію й фосфат натрію. Приклади гранулярних речовин і дезінтегруючих речовин включають в себе кукурудзяний крохмаль і альгінову кислоту. Приклади зв'язувальних речовин включають в себе крохмаль, желатин і гуміарабік. Приклади змащувальних речовин включають в себе стеарат магнію, стеарат кальцію, стеаринову кислоту й тальк. Таблетки можуть бути не покритими або покритими оболонкою за допомогою відомих способів. Такі оболонки можуть затримувати розпад і, таким чином, всмоктування в шлунково-кишковому тракті, іза допомогою цього забезпечувати тривалу дію протягом більш тривалого періоду. В одному зі втілень винаходу нікотинову кислоту, її сіль або сольват, або інший агоніст рецептора нікотинової кислоти сполучають із антагоністом рецептора DP і носієм, утворюючи фіксовану комбінацію продукту. Ця фіксована комбінація продукту може являти собою таблетку або капсулу для перорального застосування. Більш конкретно, в іншому втіленні винаходу таблетку або капсулу для перорального застосування одержують, сполучаючи нікотинову кислоту або її сіль або сольват, або інший агоніст рецептора нікотинової кислоти (приблизно від 1 приблизно до 1000мг) і антагоніст рецептора DP (приблизно від 1 приблизно до 500мг) з фармацевтично прийнятним носієм. 89615 16 Тривале вивільнення протягом більш тривалого періоду може бути особливо важливе в підборі складу фармацевтичних композицій нікотинової кислоти. Особливо переважні таблетки тривалого вивільнення. Наприклад, можна застосовувати такі речовини, що створюють тимчасову затримку, як гліцерилмоностеарат або гліцерилдистеарат. Лікарські форми можуть бути також покриті оболонкою за допомогою способів, описаних у патентах США №№4256108; 4166452 і 4265874, утворюючи таблетки терапевтичної осмотичної дії з контрольованим вивільненням. Також доступні й входять сюди інші способи контрольованого вивільнення. Звичайні інгредієнти, застосовні для уповільнення вивільнення нікотинової кислоти з таблеток із тривалим вивільненням, включають в себе різні сполуки целюлози, такі як метилцелюлоза, етилцелюлоза, пропілцелюлоза, гідроксипропілцелюлоза, гідроксіетилцелюлоза, гідроксипропілметилцелюлоза, мікрокристалічна целюлоза, крохмаль і т.п. У складах лікарських засобів тривалого вивільнення також застосовні різні природні й синтетичні речовини. Приклади включають в себе альгінову кислоту й різні альгінати, полівінілпіролідон, трагакантову камедь, смолу плодоворіжкового дерева, гуарову камедь, желатин, різні довголанцюгові спирти, такі як цетиловий спирт і бджолиний віск. Особливий інтерес являють таблетки тривалого вивільнення, у яких застосовують нікотинову кислоту в поєднанні з одним або більше зазначеними вище сполуками целюлози, пресовані для утворення полімерної основи в таблетки тривалого вивільнення. Сполуку антагоніста DP можна включати в суміш перед пресуванням або покривати нею зовнішню поверхню основи. У втіленні, що представляє більший інтерес, поєднують нікотинову кислоту й основоутворювальну речовину й пресують, утворюючи ядро тривалого вивільнення, а сполуку антагоніста DP змішують із одною або більше покривних речовин і покривають зовнішню поверхню ядра. Ще більший інтерес являє собою вищеописана таблетка, яку додатково, але не обов'язково, покривають інгібітором HMG Со-А-редуктази, наприклад, симвастатином. Це конкретне здійснення містить у собі, таким чином, три активних речовини, інгібітор HMG Со-А-редуктази й антагоніст DP, вивільнення яких може в основному відбуватися при проковтуванні, і нікотинову кислоту, вивільнення якої може відбуватися протягом більш тривалого періоду, як описано вище. Звичайні тимчасові межі вивільнення для таблеток тривалого вивільнення згідно з даним винаходом входять у діапазон приблизно від 1 до настільки тривалих як приблизно 48 годин, переважно приблизно від 4 приблизно до 24 годин і більш переважно приблизно від 8 приблизно до 16 годин. Іншу тверду лікарську форму для перорального застосування складають тверді желатинові капсули. Такі капсули подібним чином, як описано вище, включають в себе активні речовини в суміші з речовинами-носіями. М'які желатинові капсули включають в себе активні речовини в суміші з во 17 дорозчинними розчинниками, такими як пропіленгліколь, поліетиленгліколь і етанол, або з оліями, такими як арахісова олія, вазелінова олія або маслинова олія. Також розглядають водні суспензії як такі, що містять активну речовину в суміші з ексципієнтами, прийнятними для одержання водних суспензій. Такі ексципієнти включають в себе суспендуючі речовини, наприклад, карбоксиметилцелюлозу натрію, метилцелюлозу, гідроксипропілметилцелюлозу, альгінат натрію, полівінілпіролідон, трагакантову камедь і гуміарабік; диспергуючі речовини або зволожувачі, наприклад, лецитин; консерванти, наприклад, етилабо н-пропіл-парагідроксибензоат; барвники; ароматизатори; підсолоджувачі й т.п. Суміш активних речовин з диспергуючою речовиною або зволожувачем, суспендуючою речовиною й одним або більше консервантами одержують за допомогою додавання води до дисперсних порошків і гранул, прийнятних для отримання водної суспензії. Приклади прийнятних диспергуючих речовин або зволожувачів і суспендуючих речовин уже наведені вище. Також у рецептуру можуть входити сиропи й еліксири. Фармацевтична композиція, що представляє особливий інтерес, являє собою таблетку тривалого вивільнення, що включає в себе нікотинову кислоту або її сіль або сольват і антагоніст рецептора DP у сполученні з фармацевтично прийнятним носієм. Інша фармацевтична композиція, що представляє особливий інтерес, являє собою таблетку тривалого вивільнення, що включає в себе нікотинову кислоту або її сіль або сольват, антагоніст рецептора DP і інгібітор HMG Со-А-редуктази в сполученні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє ще більший інтерес, являє собою таблетку тривалого вивільнення, що включає в себе нікотинову кислоту, антагоніст рецептора DP і симвастатин у поєднанні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє особливий інтерес, являє собою таблетку тривалого вивільнення, що включає в себе нікотинову кислоту й антагоніст рецептора DP, вибраний із групи, що включає в себе сполуки від А до AJ, у поєднанні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє більший інтерес, включає в себе нікотинову кислоту й сполуку як антагоніст рецептора DP, вибрану із групи, що включає в себе сполуки А, В, D, E, Χ, АА, AF, AG, АН, ΑΙ і AJ, у сполученні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє особливий інтерес, включає в себе нікотинову кислоту й сполуку А як антагоніст рецептора DP у сполученні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє більший інтерес, включає в себе нікотинову кислоту й сполуку В як антагоніст рецепто 89615 18 ра DP у сполученні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє більший інтерес, включає в себе нікотинову кислоту й сполуку D як антагоніст рецептора DP у сполученні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє більший інтерес, включає в себе нікотинову кислоту й сполуку Е як антагоніст рецептора DP у поєднанні із фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє більший інтерес, включає в себе нікотинову кислоту й сполуку X як антагоніст рецептора DP у поєднанні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє більший інтерес, включає в себе нікотинову кислоту й сполуку АА як антагоніст рецептора DP у поєднанні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє більший інтерес, включає в себе нікотинову кислоту й сполуку AF як антагоніст рецептора DP у поєднанні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє більший інтерес, включає в себе нікотинову кислоту й сполуку AG як антагоніст рецептора DP у поєднанні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє більший інтерес, включає в себе нікотинову кислоту й сполуку АН як антагоніст рецептора DP у поєднанні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє більший інтерес, включає в себе нікотинову кислоту й сполуку АІ як антагоніст рецептора DP у поєднанні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє більший інтерес, включає в себе нікотинову кислоту й сполуку AJ як антагоніст рецептора DP у поєднанні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що представляє ще більший інтерес, включає в себе нікотинову кислоту, одну із зазначених вище сполук антагоністів рецептора DP і симвастатин у поєднанні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що має більший інтерес, являє собою таблетку тривалого вивільнення, що включає в себе нікотинову кислоту, антагоніст рецептора DP, вибраний із групи, що включає в себе сполуки від А до AJ, і симвастатин у поєднанні з фармацевтично прийнятним носієм. Ще одна фармацевтична композиція, що має ще більший інтерес, являє собою таблетку тривалого вивільнення, що включає в себе нікотинову кислоту, антагоніст рецептора DP, вибраний із групи, що включає в себе сполуки А, В, D, Е, X, АА, AF, AG, АН, ΑΙ і AJ, і симвастатин у поєднанні з фармацевтично прийнятним носієм. 19 Термін "композиція", на додаток до опису вищевикладених фармацевтичних композицій, також описує будь-який продукт, що отримують, прямо або побічно, в результаті поєднання, комплексування або агрегації будь-яких двох і більше інгредієнтів, активного або ексципієнта, або в результаті дисоціації одного або більше інгредієнтів, або в результаті інших типів реакцій і взаємодій одного або більше інгредієнтів. Відповідно, фармацевтична композиція згідно з винаходом включає в себе будь-яку композицію, отриману за допомогою змішування або іншого об'єднання сполук, будь-якого додаткового активного інгредієнта (інгредієнтів) і фармацевтично прийнятних ексципієнтів. В іншому аспекті винахід належить до застосування нікотинової кислоти або її солі або сольвату, або іншого агоніста рецептора нікотинової кислоти й антагоніста DP для одержання лікарського засобу. Цей лікарський засіб застосовують, як описано тут. Більш конкретно, в іншому аспекті винахід належить до застосування нікотинової кислоти або її солі або сольвату, або іншого агоніста рецептора нікотинової кислоти, антагоніста DP та інгібітору HMG Со-А-редуктази, такого як симвастатин, для одержання лікарського засобу. Цей лікарський засіб застосовують, як описано тут. На додаток до нікотинової кислоти, що являє собою еталон агоніста рецептора нікотинової кислоти, описані численні агоністи рецептора нікотинової кислоти. Сполуки, що являють собою агоністи рецептора нікотинової кислоти, описані в наступних публікаціях: Lorenzen, A. et al. Molecular Pharmacology 59: 349-357 (2001), Lorenzen, A. et al. Biochemical Pharmacology 64: 645-648 (2002), Soga, T. et al. Biochemical and Biophysical Research Comm. 303: 364-369 (2003), Tunaru, S. et al. Nature Medicine 9: 352-355 (2003), Wise, A. et al. Journal of Biological Chemistry 278: 9869-9874 (2003), і Van Herk, T. et al Journal of Medicinal Chemistry 46: 3945-3951 (2003). Зазначено, що дані композиції й способи лікування включають в себе часткові агоністи рецептора нікотинової кислоти, такі як описані van Herk, et al. Крім того, рецептор нікотинової кислоти був встановлений і охарактеризований в WO02/084298A2, опублікованій 24 жовтня, 2002, і в Soga, Т. et al., Tunaru, S. et al. and Wise, A. et al. (посилання вище). Способи згідно з винаходом включають в себе численні опубліковані й застосовні сполукиантагоністи рецептора DP. Наприклад, антагоністи рецептора DP можна одержувати згідно з WO01/79169, опублікованою 25 жовтня, 2001, ЕР 1305286, опублікованою 2 травня, 2003, WO 02/094830, опублікованою 28 листопада, 2002, і WO 03/062200, опублікованою 31 липня, 2003. Сполуки АВ можна одержувати згідно з описом, зазначеним в WO 01/66520A1, опублікованій 13 вересня, 2001; сполуки АС можна одержувати згідно з описом, зазначеним в WO 03/022814А1, опублікованій 20 березня, 2003, і сполуки AD і АЕ можна одержувати згідно з описом, зазначеним в WO 03/078409, опублікованій 25 вересня, 2003. Інші 89615 20 представлені сполуки-антагоністи DP, застосовувані в даному винаході, можна одержувати згідно з прикладами, наведеними нижче. Приклад 1 [5-[(4-хлорфеніл)тіо]-4-(метилсульфоніл)6,7,8,9-тетрагідропіридо[3,2-b]індолізин-6іл]оцтова кислота (сполука G) Стадія 1 4-хлорнікотинальдегід Зазначену в заголовку сполуку одержували, як описано в F. Marsais et al., J. Heterocyclic Chem., 25, 81 (1988). Стадія 2 4-(метилтіо)нікотинальдегід До розчину NaSMe (9,5г, 135ммоль) у МеОН (250мл) додавали 4-хлорнікотинальдегід (13,5г, 94,4ммоль) зі стадії 1 у МеОН (250мл). Реакційну суміш витримували при 60°С 15 хвилин. Реакційну суміш виливали на NH4CI і ЕtOАс. Органічну фазу відокремлювали, промивали Н2О і сушили над Na2SO4. Потім, для одержання зазначеної в заголовку сполуки, сполуку очищали на силікагелі з 50% EtOAc у гексані. Стадія 3 метил(2Z)-2-азидо-3-[4(метилтіо)піридин-3-іл]проп-2-еноат До розчину 25% NaOMe у МеОН (16,9мл, 78ммоль) при -12°С додавали розчин 4(метилтіо)нікотинальдегіду (4,8г, 31ммоль) і метилазидоацетату (9,0г, 78ммоль) у МеОН (50мл). Проводили спостереження за внутрішньою температурою й підтримували її від -10°С до -12°С протягом додаткових 30 хвилин. Отриману суміш потім декілька годин перемішували на льодяній бані з наступним витримуванням на льодяній бані протягом ночі в холодній кімнаті. Потім суспензію виливали в суміш льоду й NH4Cl і після 10 хвилин перемішування завись фільтрували. Для отримання зазначеної в заголовку сполуки як твердої речовини бежевого кольору (7,4г), що включає в себе декілька солей, продукт промивали холодною Н2О і потім сушили у вакуумі. Потім сполуку очищають на силікагелі з EtOAc. Стадія 4 метил-4-(метилтіо)-1Н-піроло[2,3b]піридин-2-карбоксилат Суспензію сполуки зі стадії 3 (0,40г, 1,6ммоль) у ксилолі (16мл) повільно нагрівали до 140°С. Після періоду тривалістю 15 хвилин при 140°С жовтий розчин охолоджували при кімнатній температурі. Внаслідок можливості екзотермічного ефекту, викликаного утворенням азоту, повинні бути вжиті заходи обережності. Потім, для отримання зазначеної в заголовку сполуки, суспензію охолоджували до 0°С, фільтрували й промивали ксилолом. Стадія 5 етил-4-(метилтіо)-6-оксо-6,7,8,9тетрагідропіридо[3,2-b]індолізин-7-карбоксилат До розчину сполуки зі стадії 4 (0,35г, 1,6ммоль) в DMF (20мл) при 0°С додавали NaH (1,2екв.). Після періоду тривалістю 5 хвилин додавали nBu4NI (0,10г) і етил-4-бромбутират (0,40мл). Після періоду тривалістю 1 год. при кімнатній температурі реакційну суміш виливали на насичені 21 NH4CI і EtOAc. Органічну фазу відокремлювали, промивали Н2О і сушили над Na2SO4. Після випарювання неочищений продукт очищали флешхромотографією. Потім біс-складний ефір розчиняли в THF (7,0мл) і додавали при 0°С 1,06Μ розчин трет-бутилату калію (2,2мл) в THF. Після періоду тривалістю 1 год. при кімнатній температурі реакційну суміш виливали на насичені NH4Cl і EtOAc. Для одержання зазначеної в заголовку сполуки як суміші складних етилового й метилового ефірів органічну фазу відокремлювали, сушили над Na2SO4 і випарювали при зниженому тиску. Стадія 6 4-(метилтіо)-8,9-дигідропіридо[3,2b]індолізин-6(7Н)-он До сполуки зі стадії 5 (0,32г) додавали EtOH (8,0мл) і концентровану НСІ (2,0мл). Отриману суспензію кип'ятили 5 годин зі зворотним холодильником. Реакційну суміш розподіляли між EtOAc і Na2CO3. Для отримання зазначеної в заголовку сполуки відокремлювали й випарювали органічну фазу. Стадія 7 етил(2Е,2Z)-[4-(метилтіо)-8,9дигідропіридо[3,2-b]індолізин-6(7Н)-іліден]етаноат До розчину триетилфосфоноацетату (0,45г, 2,17ммоль) в DMF (12мл) додавали 80% NaH (0,06г, 2,00ммоль) і сполуку зі стадії 6 (0,22г, 1,00ммоль). Після періоду тривалістю 4 години при 55°С реакційну суміш виливали на насичений NH4CI і ЕtOАс. Органічну фазу відокремлювали й випарювали при зниженому тиску. Для отримання зазначеної в заголовку сполуки неочищений продукт очищали флеш-хроматографією. Стадія 8 етил[4-(метилтіо)-6,7,8,9тетрагідропіридо[3,2-b]індолізин-6-іл]ацетат Сполуку зі стадії 7 розчиняли в MeOH-THF, застосовуючи для розчинення нагрівання. До попереднього охолодженого розчину додавали при кімнатній температурі РtO2 і отриману суміш витримували 18 годин при атмосферному тиску водню. Реакційну суміш обережно фільтрували через целіт, застосовуючи СН2Сl2. Для одержання зазначеної в заголовку сполуки фільтрат випарювали при зниженому тиску. Альтернативно, сполуку зі стадії 7 можна гідрогенізувати за допомогою Pd(OH)2 в ЕtOАс при 40фунт/кв. дюйм Н2 протягом 18 годин. Стадія 9 етил[4-(метилсульфоніл)-6,7,8,9тетрагідропіридо[3,2-b]індолізин-6-іл]ацетат До сполуки зі стадії 8 (0,08г, 0,27ммоль) у МеОН (3,0мл) додавали Na2WO4 (0,10г) і 30% Н2О2 (600мкл). Після періоду тривалістю 1 година реакційну суміш розподіляли між Н2О й ЕtOАс. Органічну фазу промивали Н2О, відокремлювали й випарювали. Зазначену в заголовку сполуку очищали флеш-хромотографією. Стадія 10 етил[5-[(4-хлорфеніл)тіо]-4(метилсульфоніл)-6,7,8,9-тетрагідропіридо[3,2b]індолізин-6-іл]ацетат До розчину 4,4'-дихлордифенілдисульфіду (0,24г) в 1,2-дихлоретані (2,0мл) додавали SO2Cl2 (50мкл). До сполуки зі стадії 9 (0,05г) в DMF (2,0мл) додавали попередню суміш ( 180мкл). 1 Реакційну суміш піддавали аналізу H-ЯМР і витримували при кімнатній температурі до зникнення вихідної речовини. Реакційну суміш виливали на 89615 22 насичені NaHCO3 і ЕtOАс. Органічну фазу відокремлювали, випарювали, і зазначену в заголовку сполуку очищали флеш-хроматографією. Стадія 11 [5-[(4-хлорфеніл)тіо]-4(метилсульфоніл)-6,7,8,9-тетрагідропіридо[3,2b]індолізин-6-іл]оцтова кислота До сполуки зі стадії 10, розчиненій в 1/1 суміші THF-MeOH, додавали 1н NaOH. Після періоду тривалістю 18 годин при кімнатній температурі реакційну суміш розподіляли між насиченими NH4Cl і ЕtOАс. Для одержання зазначеної в заголовку сполуки органічну фазу відокремлювали, сушили над Na2SO4 і випарювали. 1 H-ЯМР (500МГц, ацетон-d6) δ 11,00 (уш. с, 1Н), 8,60 (д, 1Н), 7,80 (д, 1H), 7,20 (д, 2Н), 7,00 (д, 2Н), 4,65 (м, 1Н), 4,20 (м, 1Н), 3,75 (м, 1Н), 3,35 (с, 3Н), 2,80-2,10 (м, 6Н). Приклад 2 [5-[(4-хлорфеніл)тіо]-4-(метилтіо)-6,7,8,9тетрагідропіридо[3,2-b]індолізин-6-іл]оцтова кислота (сполука H) Зазначену в заголовку сполуку можна отримувати із сполуки зі стадії 8 прикладу 1, способом, подібним до описаного для стадій 10 і 11 прикладу 1. m/z418. Приклад 3 [5-[(3,4-дихлорфеніл)тіо]-4-(метилсульфоніл)6,7,8,9-тетрагідропіридо[3,2-b]індолізин-6іл]оцтова кислота (сполука І) Зазначену в заголовку сполуку одержували, як описано в прикладі 1, застосовуючи на стадії 10 біс-(3,4-дихлорфеніл)дисульфід. 1 H-ЯМР (500МГц, ацетон-d6) δ 8,55 (д, 1H), 7,85 (д, 1Н), 7,35 (д, 1Н), 7,15 (с, 1Н), 6,95 (д, 1Н), 4,60 (м, 1Н), 4,15 (м, 1H), 3,80 (м, 1Н), 3,40 (с, 3Н), 2,80-2,10 (м, 6Н). m/z 484. Енантіомери розділяли на колонці Chiralecel OD 25см 20мм, застосовуючи 30% ізопропанол, 17% етанол, 0,2% оцтову кислоту в гексані, з об'ємною швидкістю потоку 8мл/хв. Ступінь чистоти енантіомерів перевіряли на колонці Chiralecel OD 25см 4,6мм, застосовуючи 35% ізопропанол, 0,2% оцтову кислоту в гексані, з об'ємною швидкістю потоку 1,0мл/хв. Для більш рухливого енантіомеру Тr=9,7 хвилин, для менш рухливого енантіомеру Тr=11,1 хвилини. Приклад 4 23 89615 24 [5-(4-хлорбензоїл)-4-(метилсульфоніл)-6,7,8,9тетрагідропіридо[3,2-b]індолізин-6-іл]оцтова кислота (сполука J) Приклад 6 Спосіб 1 [9-[(3,4-дихлорфеніл)тіо]-1-(метилсульфоніл)7,8-дигідро-6Н-піридо[3,4-b]піролізин-8-іл]оцтова кислота (сполука L) Стадія 1 етил[5-(4-хлорбензоїл)-4-(метилтіо)6,7,8,9-тетрагідропіридо[3,2-b]індолізин-6-іл]ацетат До розчину 4-хлорбензоїлхлориду (0,30г, 1,7ммоль) в 1,2-дихлоретані (6,0мл) додавали АlСl3 (0,24г, 1,8ммоль). Після періоду тривалістю 5 хвилин до попередньої суміші додавали розчин етил[4-(метилтіо)-6,7,8,9-тетрагідропіридо[3,2b]індолізин-6-іл]ацетату зі стадії 8 прикладу 1, (0,15г, 0,47ммоль) в 1,2-дихлоретані (6,0мл). Після періоду тривалістю 4 години при 80°С реакційну суміш розподіляли між ЕtOАс і NaHCO3. Органічну фазу відокремлювали, сушили над Na2SO4 і випарювали. Зазначену в заголовку сполуку очищали флеш-хроматографією. Стадія 2 етил[5-(4-хлорбензоїл)-4(метилсульфоніл)-6,7,8,9-тетрагідропіридо[3,2b]індолізин-6-іл]ацетат До розчину етил[5-(4-хлорбензоїл)-4(метилтіо)-6,7,8,9-тетрагідропіридо[3,2-b]індолізин6-іл]ацетату (0,12г, 0,27ммоль) у МеОН (5,0мл) додавали Na2WO4 (0,1г) і 30% Н2О2 (300мкл). Реакційну суміш перемішували 1 годину при 55°С. Потім реакційну суміш розподіляли між Н2О і ЕtOАс. Органічну фазу промивали Н2О, сушили над Na2SO4 і випарювали. Зазначену в заголовку сполуку очищали флеш-хроматографією. Стадія 3 [5-(4-хлорбензоїл)-4(метилсульфоніл)-6,7,8,9-тетрагідропіридо[3,2b]індолізин-6-іл]оцтова кислота Для одержання зазначеної в заголовку сполуки етил[5-(4-хлорбензоїл)-4-(метилсульфоніл)6,7,8,9-тетрагідропіридо[3,2-b]індолізин-6-іл]ацетат обробляли, як описано для стадії 11 прикладу 1. 1 H-ЯМР (500МГц, ацетон-d6) δ 8,55 (д, 1Н), 7,90 (д, 2Н), 7,65 (д, 1H), 7,45 (д, 2Н), 4,55 (м, 1Н), 4,25 (м, 1Н), 3,45 (м, 1H), 3,20 (с, 3Н), 2,05-3,00 (м, 6Н). m/z 446. Приклад 5 [5-(4-бромфеніл)тіо]1-4-(метилсульфоніл)6,7,8,9-тетрагідропіридо[3,2-b]індолізин-6іл]оцтова кислота (сполука K) Зазначену в заголовку сполуку одержували, як описано в прикладі 1, застосовуючи 4,4'дибромдифенілдисульфід. 1 H-ЯМР (500МГц, ацетон-d6) δ 8,60 (д, 1Н), 7,80 (д, 1H), 7,35 (д, 2Н), 7,00 (д, 2Н), 4,65 (м, 1Н), 4,20 (м, 1H), 3,80 (м, 1Н), 3,35 (с, 3Н), 2,80-2,10 (м, 6Н). Стадія 1 2-(метилтіо)нікотинальдегід Зазначену в заголовку сполуку одержували з 2-бромнікотинальдегіду (А. Numata Synthesis 1999p. 306), як описано для стадії 2 прикладу 1, за винятком того, що розчин нагрівали 2 години при 55°С. Стадія 2 метил(2Z)-2-азидо-3-[2(метилтіо)піридин-3-іл]проп-2-eноат Зазначену в заголовку сполуку одержували, як описано для стадії 3 прикладу 1. Стадія 3 метил-4-(метилтіо)-1Н-піроло[3,2с]піридин-2-карбоксилат Розчин метил(2Z)-2-азидо-3-[2(метилтіо)піридин-3-іл]проп-2-еноату (1,00г, 4,00ммоль) у мезитилені (50мл) нагрівали при 160°С протягом періоду тривалістю 1 година. Для одержання зазначеної в заголовку сполуки реакційну суміш охолоджували до кімнатної температури, а потім до 0°С, осад фільтрували й промивали холодним мезитиленом. Стадія 4 метил-1-(метилтіо)-8-оксо-7,8дигідро-6Н-піридо[3,4-b]піролізин-7-карбоксилат До суспензії метил-4-(метилтіо)-1Н-піроло[3,2с]піридин-2-карбоксилату (0,30г, 1,35ммоль) в THF (3мл)-толуолі (12,0мл) додавали 1,06Μ розчин трет-бутилату калію (1,42мл/1,41ммоль) в THF і метилакрилат (300мкл). Отриману суміш нагрівали 18 годин при 80°С. Суміш розподіляли між ЕtOАс і NH4Cl і фільтрували через целіт. Для отримання зазначеної в заголовку сполуки органічну фазу відокремлювали, сушили над Na2SO4 і фільтрували. Стадія 5 1-(метилтіо)-6,7-дигідро-8Нпіридо[3,4-b]піролізин-8-он Метил-1-(метилтіо)-8-оксо-7,8-дигідро-6Нпіридо[3,4-b]піролізин-7-карбоксилат перетворювали в зазначену в заголовку сполуку, як описано для стадії 6 прикладу 1. Стадія 6 метил[8-гідрокси-1-(метилтіо)-7,8дигідро-6Н-піридо[3,4-b]піролізин-8-іл]ацетат Суміш 1-(метилтіо)-6,7-дигідро-8Н-піридо[3,4b]піролізин-8-ону (0,15г, 0,68ммоль), метилбромацетату (0,34мл), Zn-Cu (0,226г) в THF (3,0мл) піддавали протягом 2 годин впливу ультразвуком. Потім суміш нагрівали 5 хвилин при 60°С до завершення реакції. Реакційну суміш розподіляли між EtOAc і NH4Cl. Для отримання зазначеної в заголовку сполуки органічну фазу відокремлювали, сушили над Na2SO4, фільтрували й випарювали при зниженому тиску. Сполуку очищали флешхроматографією. 25 Стадія 7 метил[1-(метилтіо)-7,8-дигідро-6Нпіридо[3,4-b]піролізин-8-іл]ацетат До Nal (0,300г) в CH3CN (3,2мл) додавали TMSCl (0,266мл). Цю суміш додавали до суспензії метил[8-гідрокси-1-(метилтіо)-7,8-дигідро-6Нпіридо[3,4-b]піролізин-8-іл]ацетату (0,15г, 0,515ммоль) в CH3CN (1,5мл), на водяній бані. Після періоду тривалістю 0,5 години реакційну суміш розподіляли між EtOAc і NaHCO3. Органічну фазу відокремлювали, промивали тіосульфатом натрію, сушили над MgO4 і випарювали. Зазначену в заголовку сполуку очищали флешхроматографією. Стадія 8 метил[1-(метилсульфоніл)-7,8дигідро-6Н-піридо[3,4-b]піролізин-8-іл]ацетат Метил [1-(метилтіо)-7,8-дигідро-6Н-піридо[3,4b]піролізин-8-іл]ацетат перетворювали в зазначену в заголовку сполуку, як описано для стадії 9 прикладу 1. Стадія 9 [9-[(3,4-дихлорфеніл)тіо]-1(метилсульфоніл)-7,8-дигідро-6Н-піридо[3,4b]піролізин-8-іл]оцтова кислота Метил[1-(метилсульфоніл)-7,8-дигідро-6Нпіридо[3,4-b]піролізин-8-іл]ацетат перетворювали в зазначену в заголовку сполуку, як описано для стадій 10 і 11 прикладу 1, застосовуючи на стадії 10 біс-(3,4-дихлорфеніл)дисульфід. 1 H-ΗΜΡ (500МГц, ацетон-d6) δ 8,35 (д, 1Н), 7,80 (д, 1H), 7,35 (д, 1Н), 7,15 (с, 1H), 6,95 (д, 1H), 4,55 (м, 1H), 4,35 (м, 1Н), 3,90 (м, 1Н), 3,30 (с, 3Н), 3,15 (м, 1Н), 3,05 (м, 1Н), 2,80 (м, 1H), 2,50 (м, 1H). Приклад 6 Спосіб 2 [9-[(3,4-дихлорфеніл)тіо]-1-(метилсульфоніл)7,8-дигідро-6Н-піридо[3,4-b]піролізин-8-іл]оцтова кислота Стадія 1 1-(метилтіо)-7,8-дигідро-6Нпіридо[3,4-b]піролізин-8-ол До суспензії 1-(метилтіо)-6,7-дигідро-8Нпіридо[3,4-b]піролізин-8-ону зі стадії 5 способу 1 прикладу 6 (0,55г, 2,2ммоль) в ЕtOН (10мл)-ТНР (1мл) додавали NaBH4 (0,10г, 2,6ммоль) при 0°С. Після періоду тривалістю 30 хвилин при кімнатній температурі реакцію гасили додаванням ацетону. Розчинники випарювали при зниженому тиску й до залишку додавали EtOAC і Н2О. Органічну фазу відокремлювали, сушили над MgO4 і випарювали. Зазначену в заголовку сполуку промивали сумішшю ЕtOАс/гексан і фільтрували. Стадія 2 диметил-2-[1-(метилтіо)-7,8-дигідро6Н-піридо[3,4-b]піролізин-8-іл]малонат До суспензії 1-(метилтіо)-7,8-дигідро-6Нпіридо[3,4-b]піролізин-8-олу (0,54г, 2,1ммоль) в THF (10мл) при -78°С додавали 1Μ NaHMDS в THF (2,35мл, 2,4ммоль ) і дифенілхлорфосфат (0,53мл, 2,6ммоль). Після періоду тривалістю 30 хвилин додавали диметилмалонат (0,73мл, 6,4ммоль) і 1Μ NaHMDS в THF (6,8мл, 6,8ммоль). Реакційну суміш доводили до 0°С, а потім до кімнатної температури. Потім суміш розподіляли між ЕtOАс і NH4CI. Органічну фазу сушили над MgO4, фільтрували й випарювали. Зазначену в заголовку сполуку очищали флеш-хроматографією. Стадія 3 метил[1-(метилтіо)-7,8-дигідро-6Нпіридо[3,4-b]піролізин-8-іл]ацетат 89615 26 До суміші диметил-2-[1-(метилтіо)-7,8-дигідро6Н-піридо[3,4-b]піролізин-8-іл]малонату (0,59г, 2,17ммоль) і DMSO (4мл) додавали NaCl (0,45г) у Н2О (0,45мл). Після періоду тривалістю 18 годин при 150°С реакційну суміш розподіляли між ЕtOАс і Н2О. Органічну фазу відокремлювали, сушили над Na2SCO4 і випарювали. Зазначену в заголовку сполуку очищали флеш-хроматографією. Стадія 4 [9-[(3,4-дихлорфеніл)тіо]-1(метилсульфоніл)-7,8-дигідро-6Н-піридо[3,4b]піролізин-8-іл]оцтова кислота Зазначену в заголовку сполуку одержували з метил[1-(метилтіо)-7,8-дигідро-6Н-піридо[3,4b]піролізин-8-іл]ацетату, як описано для стадій з 8 по 9 способу 1 прикладу 6. Приклад 7 [10-[(3,4-дихлорфеніл)сульфоніл]-1(метилсульфоніл)-6,7,8,9-тетрагідропіридо[3,4b]індолізин-9-іл]оцтова кислота (сполука М) Стадія 1 етил[1-(метилсульфоніл)-6,7,8,9тетрагідропіридо[3,4-b]індолізин-9-іл]ацетат Зазначену в заголовку сполуку одержували із продукту стадії 3 прикладу 6, способом, подібним, як описано для стадій з 5 по 9 прикладу 1. Стадія 2 [10-[(3,4-дихлорфеніл)сульфаніл]-1(метилсульфоніл)-6,1,8,9-тетрагідропіридо[3,4b]індолізин-9-іл]оцтова кислота Продукт зі стадії 1 перетворювали в зазначену в заголовку сполуку способом, подібним до того, як описано для стадій 10-11 прикладу 1, застосовуючи на стадії 10 біс-(3,4дихлорфеніл)дисульфід. MS M+1=485. Приклад 8 (4-(метилсульфоніл)-5-{[4(трифторметил)феніл]тіо}-6,7,8,9тетрагідропіридо[3,2-b]індолізин-6-іл]оцтова кислота (сполука N) Зазначену в заголовку сполуку одержували, як описано в прикладі 1, застосовуючи біс-[4трифторметил)феніл]дисульфід. 1 H-ЯМР (500МГц, ацетон-d6) δ 8,55 (д, 1Н), 7,75 (д, 1H), 7,45 (д, 2Н), 7,15 (д, 2Н), 4,55 (м, 1H), 4,15 (м, 1Н), 3,80 (м, 1Н), 3,30 (с, 3Н), 2,80-2,10 (м, 6Н). m/z 513 (M+1). Приклад 9 [5-[(2-хлор-4-фторфеніл)тіо]-4(метилсульфоніл)-6,7,8,9-тетрагідропіридо[3,2b]індолізин-6-іл]оцтова кислота (сполука О) 27 Зазначену в заголовку сполуку одержували, як описано в прикладі 1, застосовуючи біс-[2-хлор-4фторфеніл]дисульфід. m/z 469 (M+1). Приклад 10 [4-(метилсульфоніл)-5-(2-нафтилтіо)-6,7,8,9тетрагідропіридо[3,2-b]індолізин-6-іл]оцтова кислота (сполука Р) Зазначену в заголовку сполуку одержували, як описано в прикладі 1, застосовуючи ди-[2нафтил]дисульфід. m/z 467 (M+1). Приклад 11 [5-[(2,3-дихлорфеніл)тіо]-4-(метилсульфоніл)6,7,8,9-тетрагідропіридо[3,2-b]індолізин-6іл]оцтова кислота (сполука Q) Зазначену в заголовку сполуку одержували, як описано в прикладі 1, застосовуючи біс-[2,3дихлорфеніл]дисульфід. 1 H-ЯМР (500МГц, ацетон-d6) δ 8,85 (д, 1Н), 7,80 (д, 1Н), 7,30 (д, 1Н), 7,00 (т, 1Н), 6,60 (д, 1H), 4,60 (м, 1H), 4,20 (м, 1Н), 3,80 (м, 1H), 3,40 (с, 3Н), 2,80-2,10 (м, 6Н). Приклад 12 [5-[(4-метилфеніл)тіо]-4-(метилсульфоніл)6,7,8,9-тетрагідропіридо[3,2-b]індолізин-6іл]оцтова кислота (сполука R) Зазначену в заголовку сполуку одержували, як описано в прикладі 1, застосовуючи птолілдисульфід. 1 H-ЯМР (500МГц, ацетон-d) δ 8,55 (д, 1H), 7,80 (д, 1H), 6,95 (м, 4Н), 4,60 (м, 1Н), 4,15 (м, 1Н), 3,80 (м, 1Н), 3,35 (с, 3Н), 2,80-2,10 (м, 6Н). Приклад 13 [4-(метилсульфоніл)-5-(фенілтіо)-6,7,8,9тетрагідропіридо[3,2-b]індолізин-6-іл]оцтова кислота (сполука S) 89615 28 Зазначену в заголовку сполуку одержували, як описано в прикладі 1, застосовуючи дифенілдисульфід. 1 H-ЯМР (500МГц, ацетон-d6) δ 8,55 (д, 1H), 7,80 (д, 1Н), 7,15-6,90 (м, 5Н), 4,60 (м, 1Н), 4,15 (м, 1Н), 3,75 (м, 1Н), 3,30 (с, 3Н), 2,80-2,10 (м, 6Н). Приклад 14 [5-[(2,4-дихлорфеніл)тіо]-4-(метилсульфоніл)6,7,8,9-тетрагідропіридо[3,2-b]індолізин-6іл]оцтова кислота (сполука Т) Зазначену в заголовку сполуку одержували, як описано в прикладі 1, застосовуючи біс-(2,4дихлорфеніл)дисульфід. Дисульфід одержували з 2,4-дихлортіофенілу, застосовуючи Вr2 в ефірі. 1 H-ЯМР (500МГц, ацетон-d6) δ 8,55 (д, 1Н), 7,85 (д, 1Н), 7,35 (с, 1Н), 7,00 (д, 1Н), 6,65 (д, 1Н), 4,55 (м, 1Н), 4,15 (м, 1Н), 3,80 (м, 1Н), 3,35 (с, 3Н), 2,80-2,10 (м, 6Н). Приклад 15 [5-[(4-хлорфеніл)тіо]-4-(метилсульфоніл)6,7,8,9-тетрагідропіридо[4,3-b]індолізин-6іл]оцтова кислота (сполука U) Зазначену в заголовку сполуку одержували, як описано в прикладі 1, з 3-хлорнікотинальдегіду (Heterocycles p.151, 1993), за винятком того, що кінцеве утворення циклу проводили додаванням азиду до декаліну при кип'ятінні зі зворотним холодильником. 1 H-ЯМР (500МГц, ацетон-d6) δ 9,20 (с, 1Н), 8,85 (с, 1H), 7,20 (д, 2Н), 7,00 (д, 2Н), 4,70 (м, 1Н), 4,30 (м, 1Н), 3,75 (м, 1Н), 3,35 (с, 3Н), 2,80-2,10 (м, 6Н). Приклад 16 [9-[(4-хлорфеніл)тіо]-1-(метилсульфоніл)-7,8дигідро-6Н-піридо[3,4-b]піролізин-8-іл]оцтова кислота (сполука V) Зазначену в заголовку сполуку одержували із продукту зі стадії 8 способу 1 прикладу 6, як описано в процедурах, зазначених для стадій 10 і 11 29 прикладу 1, застосовуючи на стадії 10 біс-(4хлорфеніл)дисульфід. 1 H-ЯМР (500МГц, ацетон-d6) δ 8,25-8,3 (м, 1H), 7,71-7,75 (м, 1Н), 7,12-7,17 (м, 2Н), 6,97-7,04 (м, 2Н), 4,45-4,51 (м, 1Н), 4,32-4,39 (м, 1Н), 3,73-3,80 (м, 1H), 3,29 (с, 3Н), 3,15-3,21 (м, 1Н), 2,99-3,08 (м, 1Н), 2,66-2,73 (м, 1H), 2,46-2,54 (м, 1H). Приклад 17 (-)-[(4-хлорбензил)-7-фтор-5-метансульфоніл)1,2,3,4-тетрагідроциклопента[b]індол-3-іл]оцтова кислота (сполука Е) Стадія 1 етиловий ефір (+/-)-(7-фтор-1,2,3,4тетрагідроциклопента[b]індол-3-іл)оцтової кислоти Розчин 10,00г 4-фтор-2-йоданіліну, 6,57г етил2-(2-оксоциклопентил)ацетату й 121мг птолуолсульфонової кислоти в 100мл бензолу кип'ятили зі зворотним холодильником з пасткою Діна-Старка в атмосфері N2 24 години. Після цього періоду часу бензол видаляли дистиляцією. Потім додавали 60мл DMF і дегазували розчин перед послідовним додаванням 19мл основи Hunig'a і 405мг Pd(OAc)2. Розчин нагрівали до 115°С 3 години, потім охолоджували до кімнатної температури. Для гасіння реакції додавали 300мл 1н НСІ і 200мл етилацетату й фільтрували суміш через целіт. Фази розділяли й кислотну фазу двічі екстрагували 200мл етилацетату. Органічні шари поєднували, промивали насиченим сольовим розчином, сушили над безводним Na2SO4, фільтрували через целіт і концентрували. Для одержання зазначеної в заголовку сполуки неочищену речовину додатково очищали флеш-хроматографією, проводячи елюювання 100% толуолом. 1 H-ЯМР (ацетон-d6) δ 9,76 (уш. с, 1Н), 7,34 (дд, 1H), 7,03 (д, 1Н), 6,78 (тд, 1Н), 4,14 (кв, 2Н), 3,57 (м, 1Н), 2,85-2,55 (м, 5Н), 2,15 (м, 1H), 1,22 (т, 3Н). Стадія 2 (+/-)-(7-фтор-1,2,3,4тетрагідроциклопента[b]індол-3-іл)оцтова кислота До розчину 1,24г складного ефіру зі стадії 1 в 14мл тетрагідрофурану (THF) при кімнатній температурі додавали послідовно 7мл МеОН і 7мл 2н NaOH. Після 2,5 годин реакційну суміш виливали в ділильну лійку, що містить етилацетат (EtOAc)/1н НСІ. Фази розділяли й кислотну фазу двічі екстрагували ЕtOАс. Для одержання неочищеного масла, що застосовували як таке на наступній стадії (ступінь чистоти >90%), органічні шари об'єднували, промивали насиченим сольовим розчином, суши 89615 30 ли над безводним Na2SCO4 і випарювали до сухого стану. 1 H-ЯМР (ацетон-d6) δ 10,90 (уш. с, 1H), 9,77 (уш с, 1Н), 7,34 (дд, 1Н), 7,04 (дд, 1Н), 6,79 (тд, 1H), 3,56 (м, 1Н), 2,90-2,50 (м, 5Н), 2,16 (м, 1H). MS (-APCI) m/z 232,2 (М-Н)-. Стадія 3 (+/-)-(5-бром-7-фтор-1,2,3,4тетрагідроциклопента[b]індол-3-іл)оцтова кислота До розчину 2,20г кислоти зі стадії 2 (ступінь чистоти >90%) в 30мл піридину додавали 6,85мл триброміду піридинію (ступінь чистоти 90%) при 40°С. Суспензію перемішували 10 хвилин при 0°С і нагрівали 30 хвилин до кімнатної температури. Потім розчинник видаляли в глибокому вакуумі без нагрівання. Неочищену речовину розчиняли в 40мл АсОН і до холодного розчину при 0°С додавали порційно 2,88г порошку Zn. Суспензію перемішували 15 хвилин при 15°С і нагрівали додатково 15 хвилин до кімнатної температури. У цей час реакційну суміш гасили додаванням 1 н НСІ і цю суміш виливали в ділильну лійку, що містить насичений сольовий розчин/ЕtOАс. Шари розділяли й органічний шар промивали водою, насиченим сольовим розчином, сушили над безводним Na2SO4 і концентрували. Цю речовину застосовували без додаткового очищення на наступній стадії. 1 H-ЯМР (ацетон-d6) δ 10,77 (уш. с, 1H), 9,84 (уш. с, 1Н), 7,09 (м, 2Н), 3,60 (м, 1Н), 2,95-2,65 (м, 4Н), 2,56 (дд, 1Н), 2,19 (м, 1H). Стадія 4 (+/-)-[5-бром-4-(4-хлорбензил)-7фтор-1,2,3,4-тетрагідроциклопента[b]індол-3іл]оцтова кислота До розчину 2,13г кислоти зі стадії 3 в 10мл THF додавали в надлишку розчин діазометану в ефірі до повного, як спостерігають на TLC, поглинання кислоти. Потім розчинники видаляли в глибокому вакуумі. До розчину отриманого в такий спосіб неочищеного складного метилового ефіру в 20мл DMF додавали 539мг суспензії NaH (60% у маслі) при -78°С. Суспензію перемішували 10 хвилин при 0°С, охолоджували знову до -78°С і обробляли 1,70г 4-хлорбензилброміду. Після 5 хвилин температуру піднімали до 0°С і суміш перемішували 20 хвилин. У цей час реакцію гасили додаванням 2мл АсОН і цю суміш виливали в ділильну лійку, що містить 1н НСІ/ЕtOАс. Шари розділяли й органічний шар промивали насиченим сольовим розчином, сушили над безводним Na2SCO4 і концентрували. Алкільовану речовину гідролізували, застосовуючи процедуру, описану для стадії 2. Для одержання зазначеної в заголовку сполуки неочищену речовину додатково очищали розтиранням у суміші EtOAc/гексан. 31 1 H-ЯМР (ацетон-d6) δ 10,70 (уш. с, 1Н), 7,31 (д, 2Н), 7,18 (д, 1H), 7,06 (д, 1H), 6,92 (д, 2Н), 5,90 (д, 1Н), 5,74 (д, 1Н), 3,61 (м, 1H), 3,00-2,70 (м, 3Н), 2,65 (дд, 1Н), 2,39 (дд, 1Н), 2,26 (м, 1H), MS (APCI) m/z 436,3, 434,5 (М-Н)-. Стадія 5 (+)-[5-бром-4-(4-хлорбензил)-7-фтор1,2,3,4-тетрагідроциклопента[b]індол-3-іл]оцтова кислота До розчину 2,35г кислоти зі стадії 4 в 130мл ЕtOН при 80°С додавали 780мкл (S)-(-)-1-(1нафтил)етиламіну. Розчин охолоджували до кімнатної температури й перемішували протягом ночі. Відновлену сіль (1,7г) знову перекристалізовували з 200мл ЕtOН. Після фільтрації отриману білу тверду сіль нейтралізували за допомогою 1н НСІ і продукт екстрагували EtOAc. Органічний шар промивали насиченим сольовим розчином, сушили над безводним Na2SO4 і концентрували. Для одержання зазначеного в заголовку енантіомеру речовину фільтрували через подушку SiO2, проводячи елюювання ЕtOАс. Час утримання для двох енантіомерів склав, відповідно, 7,5 хвилин і 9,4 хвилини [колонка ChiralPak AD, гексан/2пропанол/оцтова кислота (95:5:0,1)]. Надлишок більш полярного енантіомеру склав 98%. Надлишок енантіомеру =98%; час утримання =9,4 хвилини [колонка ChiralPak AD: 250 4,6мм, гексан/2-пропанол/оцтова кислота (75:25:0,1) ]; 21 [α]D - +39,2°(c 1,0, MeOH). Стадія 6 (-)-[4-(4-хлорбензил)-7-фтор-5(метансульфоніл)-1,2,3,4тетрагідроциклопента[b]індол-3-іл]оцтова кислота й натрієва сіль Спочатку кислоту зі стадії 5 (15,4г) естерифікували діазометаном. Сульфонілювання виконували змішуванням отриманого в такий спосіб складного ефіру з 16,3г натрієвої солі метансульфінової кислоти й 30,2г CuI(I) в Nметилпіролідиноні. Суспензію дегазували в потоці N2, нагрівали до 150°С і перемішували 3 години, потім охолоджували до кімнатної температури. Для гасіння реакції додавали 500мл етилацетату й 500мл гексану, і фільтрували суміш через подушку SiO2, проводячи елюювання ЕtOАс. Органічні фази концентрували. Неочищене масло розчиняли в ЕtOАс, промивали три рази водою й один раз насиченим сольовим розчином, сушили над безводним Na2SO4, фільтрували й концентрували. Для одержання 14г сульфонованого складного ефіру, що гідролізували, застосовуючи процедуру, описану для стадії 2, неочищену речовину додатково очищали флеш-хроматографією, проводячи елюювання в градієнті від 100% толуолу до 50% толуолу в ЕtOАс. Зазначену в заголовку сполуку одержували після двох послідовних перекристалізацій: ізопропілацетат/гептан і наступної СН2Сl2/гексан. 1 H-ЯМР (500МГц, ацетон-d6) δ 10,73 (уш. с, 1Н), 7,57 (д, 2Н, J=8,8Гц), 7,31 (м, 1H), 7,29 (м, 1H), 6,84 (д, 2Н, J=8,8Гц), 6,29 (д, 1H, JAB=17,8Гц), 5,79 89615 32 (д, 1H, JAB=17,8Гц), 3,43 (м, 1H), 2,98 (с, 3Н), 2,94 (м, 1Н), 2,85-2,65 (м, 3Н), 2,42 (дд, 1H, J1=16,1Гц, J2=10,3Гц), 2,27 (м, 1Н). 13С-ЯМР (125МГц, ацетонd6) δ 173,0, 156,5 (д, JCF=237Гц), 153,9, 139,2, 133,7, 133,3, 130,0 (д, JCF=8,9Гц), 129,6, 128,2, 127,5 (д, JCF=7,6Гц), 122,2 (д, JCF=4,2Гц), 112,3 (д, JCF=29,4Гц), 111,0 (д, JCF=22,6Гц), 50,8, 44,7, 38,6, 36,6, 36,5, 23,3. MS (-APCI) m/z 436,1, 434,1 (М-Н) . Надлишок енантіомеру =97%; час утримання =15,3 хвилини [колонка ChiralCel OD: 250 4,6мм, гексан/2-пропанол/етанол/оцтова кислота 21 (90:5:5:0,2)]; [α]D = -29,3° (с 1,0, МеОН). Мр 175,0°С. Натрієву сіль одержували обробкою 6,45г (14,80ммоль) вищевказаної сполуки кислоти в ЕtOН (100мл) 14,80мл водного розчину 1н NaOH. Органічний розчинник видаляли у вакуумі й неочищену тверду речовину розчиняли при кип'ятінні зі зворотним холодильником в 1,2л ізопропілового спирту. Кінцевий об'єм знижували до 500мл дистиляцією розчинника. Натрієву сіль кристалізували охолодженням до кімнатної температури. Для одержання зазначеної в заголовку сполуки як натрієвої солі кристалічну натрієву сіль суспендували в Н2О, заморожували на сухій льодяній ванні й ліофілізували в глибокому вакуумі. 1 H-ЯМР (500МГц, ДМСО-d6) δ 7,63 (дд, 1Н, J1=8,5Гц, J2=2,6Гц), 7,47 (дд, 1H, J1=9,7Гц, J2=2,6Гц), 7,33 (д, 2Н, J=8,4Гц), 6,70 (д, 2Н, J=8,4Гц), 6,06 (д, 1Н, JAB=17,9Гц), 5,76 (д, 1H, JAB=17,9 Гц ), 3,29 (м, 1H), 3,08 (с, 3Н), 2,80 (м, 1Н), 2,69 (м, 1Н), 2,55 (м, 1H), 2,18 (м, 2Н), 1,93 (дд, 1H, J1=14,4Гц, J2=9,7Гц). Приклад 17А Альтернативна процедура для (+/-)-[5-бром-4(4-хлорбензил)-7-фтор-1,2,3,4тетрагідроциклопента[b]індол-3-іл]оцтової кислоти (стадія 4 прикладу 17) Стадія 1 дициклогексиламінова (DCHA) сіль (+/-)-(7-фтор-1,2,3,4-тетрагідроциклопента[b]індол3-іл)оцтової кислоти 0,526Μ розчин 2-бром-4-фтораніліну в ксилолі разом з етил(2-оксоциклопентил)ацетатом (1,5екв.) і сірчаною кислотою (0,02екв.) кип'ятили зі зворотним холодильником 20 годин. Воду азеотропно видаляли в пристрої Діна-Старка. Реакційну суміш піддавали ЯМР аналізу й через 20 годин, в основному, спостерігали 80-85% перетворення в необхідну проміжну сполуку іміну. Реакційну суміш промивали 1Μ гідрокарбонатом натрію (0,2 об'єми) 15 хвилин і випарювали органічну фракцію. Концентрований розчин, що залишився, дистилювали у вакуумі (0,5мм рт.ст.). Залишки ксилолу дистилювали при 30°С, потім у діапазоні 50-110°С виділяли надлишок кетону й аніліну, що не прореагував; імін одержували у фракції 110-180°С як яснокоричневу прозору рідину зі ступенем чистоти 83%. Потім проміжну сполуку іміну додавали до дегазованої суміші ацетату калію (3екв.), тетра-nбутиламонійхлорид моногідрату (1екв.), ацетату паладію (0,03екв.) і Ν,Ν-диметилацетаміду (кінцева концентрація іміну =0,365М). Реакційну суміш нагрівали до 115°С 5 годин і дозволяли остудитися до кімнатної температури. Потім додавали 3н КОН 33 (3екв.) і суміш перемішували при кімнатній температурі 1 годину. Реакційну суміш розбавляли водою (1,0 об'єм), промивали толуолом (3 0,75 об'єми). Водну фазу підкисляли до рН1 за допомогою 3н НСІ і екстрагували трет-бутилметиловим ефіром (2 0,75 об'єми). Об'єднані органічні фракції промивали водою (0,75 об'єму). До прозорого ясно-коричневого розчину додавали дициклогексиламін (1екв.) і розчин перемішували при кімнатній температурі 16 годин. Для одержання зазначеної в заголовку сполуки сіль фільтрували, промивали етилацетатом, трет-бутилметиловим ефіром і дозволяли сушитися. Аналіз: 94 А%. 1 H-ЯМР (500МГц, CDCI3) δ 9,24 (с, 1Н), 7,167,08 (м, 2Н), 6,82 (т, 1H), 6,2 (уш., 2Н), 3,6-3,5 (м, 1H), 3,04-2,97 (м, 2Н), 2,88-2,70 (м, 3Н), 2,66 (дд, 1H), 2,45-2,37 (м, 1H), 2,13-2,05 (м, 2,05), 1,83 (д, 4Н), 1,67 (д, 2Н), 1,55-1,43 (м, 4Н), 1,33-1,11 (м, 6Н). Стадія 2 (+/-)-(5-бром-7-фтор-1,2,3,4тетрагідроциклопента[b]індол-3-іл)оцтова кислота Завись солі DCHA з вищевказаної стадії 1 у дихлорметані (0,241Μ розчин) охолоджували до температури в діапазоні від -20 до -15°С. Додавали за один цикл піридин (2екв.) і до суспензії додавали по краплях бром (2,5екв.) протягом від 30 до 45 хвилин, підтримуючи температуру в діапазоні від -20 до -15°С. (При додаванні приблизно 1/3 брому реакційна суміш гусла й було потрібно ефективне перемішування. Згодом, при додаванні приблизно 1/2 брому, суміш знову ставала "рідкою"). Після завершення додавання реакційну суміш додатково витримували одну годину при -15°С. Потім додавали протягом 5 хвилин оцтову кислоту (3,04екв.) і порційно додавали порошок цинку (3,04екв.). (Порцію цинку додавали при -15°С і суміш витримували приблизно 5 хвилин для пересвідчення в тім, що відбувся екзотермічний ефект (з -15°С до 10°С)). Цю процедуру повторювали приблизно з 5 порціями цинку протягом приблизно 30 хвилин. Коли екзотермічного ефекту більше не спостерігали, цинк, що залишився, додавали швидше. Вся процедура займала приблизно від 30 до 45 хвилин. Після завершення додавання порцію речовини нагрівали до кімнатної температури, витримували 1 годину й концентрували. Реакційну суміш переносили в трет-бутилметиловий ефір (МТВЕ, 0,8 об'єми) і додавали 10% водний розчин оцтової кислоти (0,8 об'єми). Суміш (кристалізації солей, наприклад, феназопіридину) витримували 1 годину при кімнатній температурі й фільтрували через деревну целюлозу. Подушку деревної целюлози промивали МТВЕ (приблизно 0,2 об'єми) і переносили фільтрат (двофазний, МТВЕ/водна фаза) в екстрактор. Органічну фазу промивали водою (0,8 об'єми). Екстракт МТВЕ концентрували й для кристалізації сполуки переносили в ізопропіловий спирт (IPА, 0,25 об'єми). Додавали воду (0,25 об'єми) і порцію речовини витримували 1 годину. Протягом 1 години додавали додатково воду (0,33 об'єми). Після завершення додавання води порцію 89615 34 речовини витримували додатково одну годину, фільтрували й промивали сумішшю IPА/вода в співвідношенні 30/70 (0,15 об'єми). Кристалічну бромокислоту сушили в сушильній шафі при +45°С. Стадія 3 (+/-)-[5-бром-4-(4-хлорбензил)-7фтор-1,2,3,4-тетрагідроциклопента[b]індол-3іл]оцтова кислота Бромокислоту зі стадії 2 розчиняли в диметилацетаміді (0,416Μ розчин) і додавали однією порцією карбонат цезію (2,5екв.). До суспензії додавали однією порцією 4-хлорбензилхлорид (2,5екв.) і порцію речовини нагрівали до 50°С 20 годин. Охолоджували порцію речовини до кімнатної температури й додавали протягом 5 хвилин гідроксид натрію 5н (4,00екв.) (температура підвищувалася до +40°С). Реакційну суміш витримували при 50°С приблизно 3 години, охолоджували до кімнатної температури й переносили в L екстрактор. Розчин розбавляли ізопропілацетатом (IPAc, 2 об'єми) і охолоджували до +15°С. Розчин підкисляли за допомогою 5н НСІ до рН~2. Шари розділяли й органічний шар промивали водою (2 2 об'єми). Розчин ІРАс концентрували й для кристалізації продукту переносили в ІРА (0,8 об'єму). Для одержання зазначеної в заголовку сполуки додавали протягом 2 годин воду (8л) і фільтрували порцію речовини. Порцію речовини можна сушити в сушильній шафі при +40°С 24 години. Приклад 18 (+/-)-(4-[1-(4-хлорфеніл)етил]-7-фтор-5метансульфоніл-1,2,3,4тетрагідроциклопента[b]індол-3-іл)оцтова кислота (сполука X) Зазначену в заголовку сполуку одержували згідно з описом, представленим у РСТ WO 03/062200, опублікованим 30 липня, 2003. Приклад 19 (+/-)-[9-(4-хлорбензил)-6-фторметансульфоніл2,3,4,9-тетрагідро-1Н-карбазол-1-іліоцтова кислота (сполука Y) Зазначену в заголовку сполуку одержували у відповідності із описом, представленим у РСТ WO 03/062200, опублікованим 30 липня, 2003. Приклад 20 [4-(4-хлорбензил)-7-фтор-5-метансульфоніл-1оксо-1,2,3,4-тетрагідроциклопента[b]індол-3іл]оцтова кислота (сполука Z) 35 Зазначену в заголовку сполуку одержували згідно з описом, представленим у РСТ WO 03/062200, опублікованим 30 липня, 2003. Приклад 21 {9-[(3,4-дихлорфеніл)тіо]-1-ізопропіл-7,8дигідро-6Н-піридо[3,4-b]піролізин-8-іл}оцтова кислота (енантіомер А і енантіомер В) (сполука АА) Стадія 1 2-хлорнікотинальдегід До розчину діізопропіламіну (110мл, 780ммоль) в THF (500мл) додавали при -40°С 2,5Μ розчин n-BuLi (300мл, 750ммоль) у гексані. Після 5 хвилин реакційну суміш охолоджували до 95°С, а потім послідовно додавали DMPU (15мл) і 2-хлорпіридин (50мл, 532ммоль). Отриману суміш потім нагрівали й перемішували при -78°С 4 години. Після цього періоду часу, перед додаванням DMF (70мл), жовту суспензію знову охолоджували до -95°С. Кінцеву реакційну суміш нагрівали до 78°С і перемішували при цій температурі 1,5 години. Реакційну суміш виливали в холодну водну НСІ (3н, 800мл) і перемішували 5 хвилин. Для доведення рН до 7,5 додавали концентрований водний NH4OH. Водний шар тричі екстрагували за допомогою ЕtOАс. Об'єднаний органічний шар промивали водним NH4Cl і насиченим сольовим розчином, сушили над безводним Na2SO4, фільтрували й концентрували. Для одержання зазначеної в заголовку сполуки як блідо-жовтої твердої речовини неочищену речовину додатково очищали на подушці силікагелю, проводячи елюювання в градієнті від 100% гексану до 100% ЕtOАс, і продукт кристалізували в холодному гексані. Стадія 2 метил(2Z)-2-азидо-3-(2-хлорпіридин3-іл)проп-2-еноат До розчину 25% NaOMe у МеОН (80мл, 349ммоль) додавали при -20°С розчин 2хлорнікотинальдегіду (20,0г, 139,9ммоль) і метилазидоацетат (32,2мл, 349,7ммоль) у МеОН (168мл). Проводили спостереження за внутрішньою температурою й підтримували її на рівні 20°С протягом додаткових 30 хвилин. Отриману суміш потім декілька годин перемішували на льодяній бані з наступним витримуванням на льодяній бані протягом ночі в холодній кімнаті. Потім суспензію виливали в суміш льоду й NH4Cl і після 10 хвилин перемішування завись фільтрували. Продукт промивали холодною Н2О і потім сушили у вакуумі. Неочищену речовину розчиняли в СН2Сl2 і 89615 36 додавали MgO4. Суспензію фільтрували через подушку силікагелю, промивали за допомогою СН2Сl2. Для одержання осаду бежевого кольору (20г) зазначеної в заголовку сполуки фільтрат концентрували при зниженому тиску. Стадія 3 метил-4-хлор-1H-піроло[3,2с]піридин-2-карбоксилат Розчин метил(2Z)-2-азидо-3-(2-хлорпіридин-3іл)проп-2-еноату (21м, 88ммоль) у мезитилені (880мл) кип'ятили зі зворотним холодильником протягом періоду тривалістю 1 година. Реакційну суміш охолоджували до кімнатної температури, а потім до 0°С, і осад фільтрували й промивали холодним гексаном. Для одержання зазначеної в заголовку сполуки як блідо-жовтої твердої речовини (13,2г) речовину перемішували в суміші 1:20 ЕtOАс/гексан протягом ночі й фільтрували. Стадія 4 метил-1-хлор-8-оксо-7,8-дигідро-6Нпіридо[3,4-b]піролізин-7-карбоксилат До суспензії метил-4-хлор-1Н-піроло[3,2с]піридин-2-карбоксилату (12,5м, 59ммоль) в THF (116мл)-толуолі (460мл) додавали 1,0Μ розчин трет-бутилату калію (64мл, 64ммоль) в THF і метилакрилат (55мл, 611ммоль). Отриману суміш нагрівали при 100°С 18 годин. Після цього часу суспензію охолоджували до кімнатної температури й виливали її в суміш насиченого водного NH4CI (400мл) і гексану (400мл). Для одержання зазначеної в заголовку сполуки тверді речовини відстоювали, фільтрували й промивали Н2О і гексаном. Стадія 5 1-хлор-6,7-дигідро-8Н-піридо[3,4b]піролізин-8-он До сполуки з попередньої стадії додавали ізопропанол (8,0мл) і концентровану НСІ (2,0мл) при нагріванні протягом 1 години при 100°С. Реакційну суміш розподіляли між ЕtOАс і Na2CO3. Для одержання зазначеної в заголовку сполуки відокремлювали й випарювали органічну фазу. Стадія 6 1-ізопропеніл-6,7-дигідро-8Нпіридо[3,4-b]піролізин-8-он До суміші 1-хлор-6,7дигідро-8Н-піридо[3,4-b]піролізин-8-ону (5,0м, 24,3ммоль), трис-(дибензиліденацетон)дипаладію(0) (1,0г, 1,09ммоль) і трифеніларсину (2,70г, 8,82ммоль) в DMF (100мл) додавали трибутилізопропенілолово (9,60г, 29,00ммоль). Отриману суміш дегазували й нагрівали при 78°С протягом періоду тривалістю 18 годин. Розчинник випарювали при зниженому тиску. До отриманої суміші додавали СН2СІ2 і целіт і, потім, фільтрували через целіт. Зазначену в заголовку сполуку очищали флеш-хроматографією (від 50% до 100% ЕtOАс у гексані). Стадія 7 етил(2Е)-(1-ізопропеніл-6,7-дигідро8Н-піридо[3,4-b]піролізин-8-іліден)етаноат До розчину 1-ізопропеніл-6,7-дигідро-8Нпіридо[3,4-b]піролізин-8-ону (0,60г, 2,8ммоль) і триетилфосфоноацетату (1,00г, 4,46ммоль) в THF (24мл) додавали при -78°С 80% NaH (0,12г, 4,00ммоль), реакційній суміші дозволяли нагріватися до 0°С, а потім до кімнатної температури. Реакційну суміш виливали в насичені NH4Cl і ЕtOАс. Органічну фазу відокремлювали, сушили над Na2SO4 і випарювали. Зазначену в заголовку сполуку очищали флеш-хроматографією (40% ЕtOАс у гексані). 37 Стадія 8 етил(1-ізопропіл-7,8-дигідро-6Нпіридо[3,4-b]піролізин-8-іл)ацетат До розчину етил(2Е)-(1-ізопропеніл-6,7дигідро-8Н-піридо[3,4-b]піролізин-8іліден)етаноату (0,40г, 1,4ммоль) у МеОН (20мл) додавали Pd(OH)2 (0,20г). Суміш перемішували 3 години при тиску Н2 1атм. Для одержання зазначеної в заголовку сполуки суміш фільтрували через целіт і випарювали. Стадія 9 етил{9-[(3,4-дихлорфеніл)тіо]-1ізопропіл-7,8-дигідро-6Н-піридо[3,4-b]піролізин-8іл}ацетат До розчину біс-(3,4-дихлорфеніл)дисульфіду (0,24г, 0,67ммоль) у СН2Сl2 (5,6мл) додавали SO2Cl2 (0,036мл). Отриману жовту суміш перемішували при кімнатній температурі 1 годину. Цей розчин додавали при 0°С до розчину етил(1ізопропіл-7,8-дигідро-6Н-піридо[3,4-b]піролізин-8іл)ацетату (0,15г, 0,52ммоль) в DMF (5,6мл). Після 1,5 години при 0°С реакційну суміш виливали в насичені NaHCO3 і ЕtOАс. Органічну фазу відокремлювали, сушили над Na2SO4 і випарювали. Зазначену в заголовку сполуку очищали флешхроматографією (від 30% до 40% ЕtOАс у гексані). Стадія 10 {9-[(3,4-дихлорфеніл)тіо]-1-ізопропіл7,8-дигідро-6Н-піридо[3,4-b]піролізин-8-іл}оцтової кислоти До розчину етил{9-[(3,4-дихлорфеніл)тіо]-1ізопропіл-7,8-дигідро-6Н-піридо[3,4-b]піролізин-8іл}ацетату (0,23г, 0,50ммоль) в THF (5мл) і МеОН (2,5мл) додавали 1,0Μ NaOH (1,5мл, 1,5ммоль). Після перемішування протягом 18 годин при кімнатній температурі додавали НОАс (0,25мл) і випарювали розчинник. Залишок поміщали в ЕtOАс/Н2О і промивали органічний шар Н2О й насиченим сольовим розчином. Після висушування (Na2SO4) розчин фільтрували й випарювали. Для одержання зазначеної в заголовку сполуки як білої твердої речовини залишок перемішували із сумішшю 1:1 ЕtOАстексан і фільтрували. 1 H-ЯМР (MeOH-d4) δ 1,14-1,26 (м, 6Н), 2,472,56 (м, 1Н), 2,56-2,64 (м, 1H), 2,94-3,05 (м, 2Н), 3,81-3,89 (м, 1Н), 4,22-4,30 (м, 1Н), 4,33-4,44 (м, 2Н), 6,93-6,99 (м, 1Н), 7,14-7,19 (м, 1H), 7,33-7,39 (м, 1H), 7,54-7,59 (м, 1Н), 8,16-8,21 (м, 1H). Продукт зі стадії 10 перетворювали в його складний метиловий ефір, застосовуючи CH2N2, і складний ефір піддавали поділу за допомогою HPLC у хіральній нерухомій фазі (колонка chiralcel OD 2 25см), проводячи елюювання в 12% 2пропанолі в гексані при об'ємній швидкості потоку 6мл/хв. Час утримання енантіомеру А (менш полярного) склав 31,9 хвилин, а час утримання енантіомеру В (більш полярного) склав 35,5 хвилин. Для одержання енантіомерів А і В зазначеної в заголовку сполуки і А, і В гідролізували, як зазначено для стадії 10 прикладу 17. Приклад 22 ((1R)-6-фтор-8-(метилсульфоніл)-9-{(1S)-1-[4(трифторметил)феніл]етил}-2,3,4,9-тетрагідро-1Нкарбазол-1-іл)оцтова кислота (сполука AJ) 89615 38 Стадія 1 2-(2-бром-4фторфеніл)гідразинійхлорид До суспензії 2-бром-4-фтораніліну в концентрованій НСІ (1,5М) повільно додавали при -10°С 10,0Μ водний розчин NaNO2 (1,1екв.). Суміш перемішували при 0°С 2,5 години. Потім, підтримуючи внутрішню температуру нижче 10°С, повільно додавали холодний (-30°С) розчин SnCl2 (3,8Μ) у концентрованій НСІ. Отриману суміш механічно перемішували 20 хвилин при 10°С, а потім 1 годину при кімнатній температурі. Густу суспензію фільтрували й твердий осад сушили на повітрі протягом ночі. Твердий осад ресуспендували в холодній НСІ і знову фільтрували. Для одержання зазначеної в заголовку сполуки як бежевої твердої речовини висушену речовину ресуспендували в Et2O, перемішували 10 хвилин, фільтрували й сушили на повітрі протягом ночі. Стадія 2 (+/-)-етил(8-бром-6-фтор-2,3,4,9тетрагідро-1Н-карбазол-1-іл)ацетат До суспензії сполуки зі стадії 1 (1екв.) в АсОН (0,5М) додавали етил(2-оксоциклогексил)ацетат (1екв.). Суміш перемішували при кип'ятінні зі зворотним холодильником 16 годин, охолоджували й видаляли АсОН випарюванням при зниженому тиску. Залишок розбавляли ЕtOАс і промивали водою й насиченим водою NaHCO3. Органічний шар сушили над Na2SO4 і концентрували. Потім залишок очищали на подушці силікагелю, проводячи елюювання толуолом. Для одержання зазначеної в заголовку сполуки як білої твердої речовини фільтрат концентрували й перемішували в гексані, а потім фільтрували. MS (+APCI) m/2 354,2 (М+Н)+. Стадія 3 (+/-)-етил[6-фтор-8-(метилсульфоніл)2,3,4,9-тетрагідро-1H-карбазол-1-іл]ацетат До розчину сполуки зі стадії 2 (1екв.) у безводному DMSO (0,28М) додавали метансульфінат натрію (3екв.) і йодид міді (3екв.). Барботували N2 через суміш 5 хвилин і потім перемішували реакційну суміш при 100°С в атмосфері N2. Після 12 годин додавали ще метансульфінат натрію (2екв.) і йодид міді (2екв.). Суміш перемішували додатково 12 годин при 100°С, охолоджували, розбавляли за допомогою ЕtOАс і для підкислення суміші додавали 1 н НСІ. Суспензію переміщували 30 хвилин і фільтрували через целіт. Фільтрат промивали водою, сушили над Na2SO4 і концентрували. Залишок фільтрували через подушку силікагелю, спочатку, для видалення неполярних домішок, проводячи елюювання толуолом, а потім сумішшю 2:1 гексан/EtOAc для елюювання необхідного продукту. Для одержання зазначеної в заголовку сполуки як блідо-жовтої твердої речовини фільтрат концентрували після елюювання сумішшю гексан/EtOAc. MS (-APCI) m/z 352,1 (М-Н)-. 39 Стадія 4 етил[(1R)-6-фтор-8(метилсульфоніл)-2,3,4,9-тетрагідро-1Н-карбазол1-іл]ацетат Суміш рацематів зі стадії 3 розділяли за допомогою препаративної HPLC на препаративній колонці chiralpak AD, проводячи елюювання сумішшю 15% іРrOН в гексані. На підставі активності кінцевого продукту за вказану в заголовку сполуку приймали більш полярний енантіомер (з більш тривалим часом утримання). Стадія 5 етил[(1R)-9-[(1S)-1-(4хлорфеніл)етил]-6-фтор-8-(метилсульфоніл)2,3,4,9-тетрагідро-1Н-карбазол-1-іл]ацетат До розчину сполуки зі стадії 4 (1екв.), трифенілфосфіну (1,5екв.) і (1R)-1-(4-хлорфеніл)етанолу (1,5екв., отриманого у відповідності із загальною процедурою, описаною в довідковому прикладі 1) в THF (0,175М) додавали протягом 10 хвилин розчин ди-трет-бутилазодикарбоксилату (2,1Μ у THF, 1,5екв.). Суміш перемішували при кімнатній температурі 2 години й концентрували. Для одержання необхідного продукту (ступінь чистоти ~90%), що застосовували як такий в наступній реакції, залишок очищали флеш-хроматографією на силікагелі, проводячи елюювання 7% ЕtOАс у толуолі. Стадія 6 [(1R)-9-[(1S)-1-(4-хлорфеніл)етил]-6фтор-8-(метилсульфоніл)-2,3,4,9-тетрагідро-1Нкарбазол-1-іл]оцтова кислота і [(1S)-9-[(1S)-1-(4хлорфеніл)етил]-6-фтор-8-(метилсульфоніл)2,3,4,9-тетрагідро-1Н-карбазол-1-іл]оцтова кислота До розчину сполуки зі стадії 5 у суміші THF і метанолу (0,1М) 2:1 додавали 1н водний LiOH (3екв.). Суміш перемішували при кімнатній температурі 2 години, додавали АсОН і видаляли розчинник випарюванням. Залишок переносили в суміш ЕtOАс/Н2О і органічний шар промивали насиченим сольовим розчином, сушили над Na2SO4, фільтрували й концентрували. Для одержання зазначеної в заголовку сполуки як білої твердої речовини залишок перемішували з 30% ЕtOАс у гексані, ресуспендували продукт у діетиловому ефірі й піддавали протягом 45 хвилин впливу ультразвуком, фільтрували й сушили в глибокому вакуумі при 50°С 24 години. MS (-АРСІ) m/z 462,1 (М-Н). Альтернативно, для одержання суміші 2 діастереомерів: eтил[(1R)-9-[(1S)-1-(4хлорфеніл)етил]-6-фтор-8-(метилсульфоніл)2,3,4,9-тетрагідро-1Н-карбазол-1-іл]ацетату й етил [(1S)-9-[(1S)-1-(4-хлорфеніл)етил]-6-фтор-8(метилсульфоніл)-2,3,4,9-тетрагідро-1H-карбазол1-іл]ацетату, у реакції алкілування на стадії 5 застосовували (+/-)-етил[6-фтор-8-(метилсульфоніл)2,3,4,9-тетрагідро-1Н-карбазол-1-іл]ацетат. Суміш діастереомерів розділяли за допомогою вибіркового гідролізу, застосовуючи для одержання необхідної [(1R)-9-[(1S)-1-(4-хлорфеніл)етил]-6-фтор-8(метилсульфоніл)-2,3,4,9-тетрагідро-1Н-карбазол1-іл]оцтової кислоти наступну процедуру. Розділення: Суміш діастереомерів етил[(1R)-9-[(1S)-1-(4хлорфеніл)етил]-6-фтор-8-(метилсульфоніл)2,3,4,9-тетрагідро-1Н-карбазол-1-іл]ацетату й етил[(1S)-9-[(1S)-1-(4-хлорфеніл)етил]-6-фтор-8 89615 40 (метилсульфоніл)-2,3,4,9-тетрагідро-1Н-карбазол1-іл]ацетату (1екв.) розчиняли у суміші 3,5/1 THF/MeOH (0,25Μ) і охолоджували при 0°С. Повільно додавали водний 1н LiOH (1екв.) і перемішували суміш при 0°С 12 годин або до майже повного гідролізу етил[(1R)-9-[(1S)-1-(4-хлорфеніл)етил]6-фтор-8-(метилсульфоніл)-2,3,4,9-тетрагідро-1Нкарбазол-1-іл]ацетату, тоді як гідроліз іншого діастереомеру в цих умовах проходив у незначному ступені. Додавали АсОН і видаляли розчинник випарюванням. Залишок переносили в суміш ЕtOАс/Н2О і органічний шар промивали насиченим сольовим розчином, сушили над Na2SO4, фільтрували й концентрували. Етил[(1S)-9-[(1S)-1-(4хлорфеніл)етил]-6-фтор-8-(метилсульфоніл)2,3,4,9-тетрагідро-1Н-карбазол-1-іл]ацетат і [(1R)9-[(1S)-1-(4-хлорфеніл)етил]-6-фтор-8(метилсульфоніл)-2,3,4,9-тетрагідро-1Н-карбазол1-іл]оцтову кислоту розділяли флешхроматографією, проводячи елюювання 40% ЕtOАс у гексані, що містить 1% АсОН, для одержання необхідної [(1R)-9-[(1S)-1-(4хлорфеніл)етил]-6-фтор-8-(метилсульфоніл)2,3,4,9-тетрагідро-1Н-карбазол-1-іл]оцтової кислоти з надлишком діастереомеру >90%, яку потім, для одержання необхідної сполуки в як білої твердої речовини з надлишком діастереомеру >95%, перемішували з 30% ЕtOАс у гексані. Стадія 7 метил[(1R)-6-фтор-8(метилсульфоніл)-2,3,4,9-тетрагідро-1H-карбазол1-іл]ацетат До розчину [(1R)-9-[(1S)-1-(4-хлорфеніл)етил]6-фтор-8-(метилсульфоніл)-2,3,4,9-тетрагідро-1Нкарбазол-1-іл]оцтової кислоти ([α]D=-226° у МеОН) у МеОН (0,1М) додавали 10% паладію у вуглецю (10% мас/мас). Барботували потік N2 через суміш протягом 5 хвилин. Реакційну суміш перемішували при кімнатній температурі в атмосфері Н2 (балон) 24 години й фільтрували через подушку целіту, проводячи елюювання СН2Сl2. Для отримання сполуки метил[(1R)-6-фтор-8-(метилсульфоніл)2,3,4,9-тетрагідро-1Н-карбазол-1-іл]ацетату розчинники видаляли випарюванням при зниженому тиску й залишок переносили в МеОН. Стадія 8 ((1R)-6-фтор-8-(метилсульфоніл)-9{(1S)-1-[4-(трифторметил)феніл]етил}-2,3,4,9тетрагідро-1Н-карбазол-1-іл)оцтова кислота (сполука AJ) До розчину сполуки зі стадії 7 (1екв.), трифенілфосфіну (1,5екв.) і (1R)-1-[4(трифторметил)феніл]етанолу (1,5екв.) в THF (0,2М) додавали протягом періоду тривалістю 20 хвилин розчин ди-трет-бутилазодикарбоксилату (1Μ у THF, 1,5екв.). Суміш перемішували при кімнатній температурі 2 години й концентрували. Для одержання метил((1R)-6-фтор-8(метилсульфоніл)-9-{(1S)-1-[4(трифторметил)феніл]етил}-2,3,4,9-тетрагідро-1H 41 карбазол-1-іл)ацетату (ступінь чистоти ~90%), що застосовували як такий в наступній реакції, залишок очищали флеш-хроматографією на силікагелі, проводячи елюювання 10% ЕtOАс у толуолі. До розчину вищевказаного складного ефіру (1екв.) у суміші 3,5/1 THF/MeOH (0,25Μ) повільно додавали при 0°С водний 1н LiOH (1екв.) і суміш перемішували при 0°С 16 годин або до майже повного гідролізу складного ефіру; швидкість гідролізу в цих умовах іншого, побічного діастереомеру, набагато нижче. Додавали АсОН і видаляли у вакуумі розчинник. Залишок переносили в суміш EtOAc/H2O і органічний шар промивали насиченим сольовим розчином, сушили над Na2SO4, фільтрували й концентрували. Для видалення складного метилового ефіру, що не прореагував, залишок фільтрували через подушку силікагелю, проводячи спочатку елюювання в суміші 10% ЕtOАс/толуол, а потім у суміші 60% EtOАс/толуол, що містить 1% АсОН. Для одержання зазначеної в заголовку сполуки як білої твердої , речовини з надлишками діастереомеру й енантіомеру >95% (перевіряли за допомогою хіральної HPLC) залишок перемішували із сумішшю 30% ЕtOАс/гексан і сушили в глибокому вакуумі при 50°С 16 годин. MS(-APCI) m/z 496,0 (М-Н)-. [α]D=-181° y MeOH. Біологія Сполуки, застосовувані в даному винаході, що функціонують як селективні антагоністи DP, звичайно мають спорідненість (Ki) до DP щонайменше приблизно в 10 разів більш високу (чисельно меншу величину Ki), ніж спорідненість (Ki) до рецепторів CRTH2. Звичайні антагоністи DP, застосовувані в даному винаході, мають щонайменше приблизно в 10 разів більшу селективність стосовно рецептора DP, ніж до рецептора CRTH2. Більш конкретно, селективний антагоніст рецептора DP має щонайменше приблизно в 100 разів більшу селективність до рецептора DP у порівнянні з рецептором CRTH2. Ще більш конкретно, сполука селективного антагоніста DP має щонайменше приблизно в 800-1000 разів більшу селективність до рецептора DP у порівнянні з рецептором CRTH2, тобто спорідненість (Kі) до рецептора DP щонайменше приблизно в 800-1000 разів більш високу, ніж спорідненість (Kі) до рецептора CRTH2. Як застосовують тут, сполука "вибірково регулює рецептор DP" означає, що сполука зв'язується й впливає як антагоніст на рецептор DP у концентрації, досяжній при терапевтичних дозах, але істотно не регулює рецептор CRTH2 у таких терапевтично досяжних концентраціях. Як застосовують тут, в основному, антагоністи DP мають спорідненість (Kі) до рецептора CRTH2 приблизно 0,5мкмоль/л або вище. Для інгібування ефекту гіперемії, що спостерігається при введенні нікотинової кислоти без таких селективних антагоністів DP, застосовні сполуки, що мають афінність зв'язування з рецептором CRTH2 приблизно 0,5мкмоль/л або вище й селективність до рецептора DP щонайменше приблизно в 10 разів більшу, ніж до CRTH2. Визначення спорідненості й селективності сполук до рекомбінантних рецепторів DP і CRTH2 людини 89615 42 Спорідненість і селективність сполук до рецепторів DP і CRTH2 визначали, застосовуючи аналізи радіолігандного зв'язування, як описано в Abramovitz M, et al. Biochem. Biophys. Acta (2000) 1483: 285-293, and Sawyer N, et al. Br. J. Pharmacol. (2002); 137: 1163-1172. Коротко, стабільні лінії клітин, які окремо експресують рецептори DP і CRTH2 людини, установлювали, застосовуючи клітини (позначені НЕK293Е лінії клітин) нирки ембріона людини (НЕK) 293EBNA (ядерний антиген вірусу Епштейна-Барра). Для визначення спорідненості й селективності сполук до рецепторів DP і CRTH2 застосовували отримані із цих рекомбінантних ліній клітин фракції мембрани в аналізах радіолігандного зв'язування при рівноважному конкуруванні. кДНК DP і CRTH2, що відповідають повнорозмірним кодуючим послідовностям, переносили у відповідні ділянки експресуючого вектора ссавця рСЕР 4 (Invitrogen) і експресували в клітинах НЕK293Е. Мембрани одержували за допомогою диференціального центрифугування (1000 g протягом 10 хвилин, потім 160000 g протягом 30 хвилин, усе при 4°С) після лізису клітин за допомогою пороутворення азотом при 800фунт/кв. дюйм протягом 30 хвилин на льоду в присутність інгібіторів протеази (2мМ AEBSF, 10мкМ Е-64, 100мкМ лейпептину і 0,05мг/мл пепстатину). Осади 160000 g ресуспендували в 10мМ HEPES/KOH (рН7,4), що містить 1мМ EDTA при приблизно від 5 до 10мг/мл білка, за допомогою гомогенізації Dounce (Dounce A; 10 ударів), заморожували в рідкому азоті й зберігали при -80°С. Аналізи зв'язування рецептора проводили в кінцевому об'ємі для інкубації 0,2мл в 10мМ HEPES/KOH (рН7,4), що містить 1мМ EDTA, 10мМ МnСl2 і 0,7нм [3H]PGD2 (200 Кі/ммоль). Реакцію ініціювали за допомогою додавання білка мембрани (приблизно 30мкг для DP і 10мкг для CRTH2) із фракції 160000 g. Ліганди додавали в диметилсульфоксиді (DMSO), що зберігали сталим, 1% (об./об.), у всіх інкубаціях. Неспецифічне зв'язування визначали в присутності 10мкМ нерадіоактивного PGD2. Інкубації проводили на мініорбітальному шейкері при кімнатній температурі 60 хвилин. Аналіз зв'язування зупиняли за допомогою швидкої фільтрації через 96-ямковий Unifliter GF/C (Canberra Packard), повторно змочуючи в застосовуваному для аналізу буфері для інкубації, без EDTA, (при 4°С), застосовуючи 96ямковий напівавтоматичний збирач клітин Tomtec Mach III. Фільтри промивали від 3 до 4мл того ж буфера, сушили 90 хвилин при 55°С і залишкову радіоактивність, пов'язану з окремими фільтрами, визначали вимірюванням активності сцинтиляційним способом з додаванням 50мкл Ultima Gold F (Canberra Packard), застосовуючи лічильник 1450 MicroBeta (Wallac). Максимальне специфічне зв'язування визначали як різницю між загальним зв'язуванням і неспецифічним зв'язуванням за відсутності конкурента. Специфічне зв'язування визначали при кожній концентрації сполуки й виражали як відсотки від максимального специфічного зв'язування. Сигмоїдальні криві рівноважного конкурування будували, виражаючи відсоток максимального специфічного 43 зв'язування як функцію аналізованої концентрації сполуки й аналізували за допомогою сконструйованого за особливими умовами пакета програмного забезпечення, застосовуючи симплекс-алгоритм нелінійного підбору кривої методом найменших квадратів, що використовує для визначення точки перегину (InPt) рівняння із чотирма параметрами. Афінність зв'язування аналізованої сполуки визначали, обчислюючи константу рівноваги інгібування (Ki) з рівняння Ki=InPt/1+([радіоліганд]/Kd) де Kd являє собою константу рівноваги дисоціації для взаємодії радіоліганд-рецептор. У випадку неможливості визначення InPt використовували ІС50 (тобто концентрацію аналізованої сполуки, необхідну для інгібування 50% максимального специфічного зв'язування). В основному, сполуки, застосовувані в даному винаході, мають Ki для рецептора DP приблизно від такої низької як 0,4нм до такої високої як приблизно 16,3нм. Подібним чином, сполуки, застосовувані в даному винаході, в основному, мають Ki для рецептора CRTH2 приблизно від такої низької як 180нм до такої високої як приблизно 22000нм. Дія сполук на розширення кровоносних судин у миші, що викликається нікотиновою кислотою Ефективність селективних антагоністів DP, описана тут, може бути показана при вимірюванні ефекту інгібування гіперемії із застосуванням мишачої моделі гіперемії людини, що викликається нікотиновою кислотою. Кровотік у вусі миші (ступінь розширення кровоносних судин, помітна складова гіперемії людини) вимірювали після введення нікотинової кислоти миші, якій проводили попереднє лікування індиферентною речовиною (як контроль) або антагоністом DP. Конкретно, у дослідженні використовували самців миші C57BL/6 (~25г). В кожній групі, що аналізувалася, оцінювали п'ять мишей. Нембутал розбавляли водою до 89615 44 кінцевої концентрації 5мг/мл і вводили 0,3мл/мишу за допомогою внутрішньочеревинної ін'єкції. Антагоністи DP розчиняли в 5% гідроксипропіл-βциклодекстрині до кінцевої концентрації 5мг/мл і сполуки вводили внутрішньочеревинно в об'ємі 0,2мл/мишу (~40мг/кг). Нікотинову кислоту розчиняли в 5% гідроксипропіл-β-циклодекстрині до кінцевої концентрації 12,5мг/мл. Вихідний розчин нікотинової кислоти доводили до рН7,4 за допомогою 2н NaOH і вводили 0,2мл/мишу за допомогою підшкірної ін'єкції (~100мг/кг). Кровопостачання шкірного покриву вуха миші оцінювали, застосовуючи лазерну доплерівську систему зображення кровотоку (PeriScan РІМII, Perimed, Sweden), кожні 30 секунд протягом 15 хвилин, починаючи за 5 хвилин до введення нікотинової кислоти. Обчислювали процентну зміну середньої величини кровотоку за період часу тривалістю 10 хвилин після введення індиферентної речовини або нікотинової кислоти й будували для кожної тварини графік залежності процентної зміни середньої величини кровотоку від часу. Потім для кожного графіка обчислювали площу під кривою (AUC) середньої величини кровотоку (% Δ хв.) і результати виражали в середній величині AUC+SEM для кожної групи. Сполука D пригнічувала розширення кровоносних судин у миші, що викликалося PGD-2 (Фіг.1). Аналізовані антагоністи DP пригнічували розширення кровоносних судин, що викликалося нікотиновою кислотою, у миші, дані для вибраних сполук надані на Фіг.2 і 3. Всі патенти, патентні заявки й публікації, що цитуються тут, наведені тут повністю як посилання. Хоча тут детально описані конкретні кращі втілення, численні альтернативні втілення розглядають як такі, що входять в обсяг винаходу. 45 Комп’ютерна верстка Т. Чепелева 89615 Підписне 46 Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюPharmaceutical compositions comprising nicotinic acid and dp receptor antagonist
Автори англійськоюCheng Kang, Waters Gerard, Metters Kathleen M., O'neill Gary
Назва патенту російськоюФармацевтические композиции, которые содержат никотиновую кислоту и антагонист рецептора dp
Автори російськоюЧен Кан, Уотерс Джерард, Меттерс Кетлин М., ОНейл Гери
МПК / Мітки
МПК: A61K 31/351, A61K 31/405, A61P 3/06, A61K 31/455, A61P 9/10
Мітки: анатгоніст, рецептора, містять, фармацевтичні, нікотинову, кислоту, композиції
Код посилання
<a href="https://ua.patents.su/23-89615-farmacevtichni-kompozici-shho-mistyat-nikotinovu-kislotu-ta-anatgonist-receptora-dp.html" target="_blank" rel="follow" title="База патентів України">Фармацевтичні композиції, що містять нікотинову кислоту та анатгоніст рецептора dp</a>
Химерне моноклональне антитіло, що розпізнає гангліозиди, які включають n-гліколільовану сіалову кислоту, лінія клітин, яка його продукує, та фармацевтичні композиції, що його містять

Номер патенту: 75393
Опубліковано: 17.04.2006
Автори: Матео Де Акоста Дель Ріо Крістіна Марія, Лопес Рекена Алехандро, Роке Наварро Лурдес Татіана, Ломбардеро Валладарес Хосефа
МПК: A61K 39/395, C12N 5/10, A61P 43/00, A61P 35/04, A61K 39/00, A61K 31/7032, C12P 21/08, C12N 1/15, A61P 37/00, C07K 16/30, C07K 16/00, A61P 35/00, A61K 31/739, C12N 1/19, C12N 5/20, C07K 16/42, C07K 16/46, C07K 16/18, C12N 1/21, C12N 15/09, A61P 37/04
Мітки: розпізнає, гангліозиди, моноклональне, фармацевтичні, містять, яка, химерне, антитіло, клітин, продукує, сіалову, лінія, n-гліколільовану, кислоту, включають, композиції
Формула / Реферат:
1. Химерне моноклональне антитіло, одержане із мишачого моноклонального антитіла MAb P3, що розпізнає гангліозиди, які включають N-гліколільовану сіалову кислоту, яке продукується лінією клітин гібридоми, депонованою у ЕСАСС під № 94113026, причому гіперваріабельні домени важкого та легкого ланцюгів згаданого антитіла мають такі послідовності:важкий ланцюг:CDR1: RYSVHCDR2: MIWGGGSTDYNSALKSCDR3:...
Похідні пурину, спосіб їх одержання і фармацевтичні композиції, що містять їх

Номер патенту: 68441
Опубліковано: 16.08.2004
Автор: Асслен Жан-Люк
МПК: C07D 473/16, C07D 473/26, A61K 31/52, A61K 31/522
Мітки: одержання, фармацевтичні, композиції, містять, спосіб, пурину, похідні
Формула / Реферат:
1. Сполука формули І:,в якійΖ означає двовалентний радикал -СН2-, -SO2-, -CO-, -COO-, -CONH- або -(CH2)2-NR3-;n означає ціле число 0 або 1;R1 вибирають з атома водню, радикалів арил, -СН2-арил, -SО2-арил, -СО-арил, гетероциклічний радикал, -СН2-гетероциклічний радикал, алкіл і -SО2-алкіл;R2 означає можливо заміщений...
Дифенілалкілтетрагідропіридини, спосіб їх одержання і фармацевтичні композиції, що їх містять

Номер патенту: 52708
Опубліковано: 15.01.2003
Автори: ФУРНЬЄ Жаклін, КАРДАМОН Розанна, ГУЗЗІ Умберто, БАРОНІ Марко
МПК: A61P 25/28, A61K 31/4418, A61K 31/444, C07D 401/04, C07D 211/70, C07D 277/38
Мітки: фармацевтичні, одержання, композиції, дифенілалкілтетрагідропіридини, містять, спосіб
Формула / Реферат:
1. Сполука формули (І) (I),в якій Y означає -СН- або -N-,R1 означає галоген, групу СF3, (С1-С4)алкіл або (С1-С4)алкоксигрупу,кожний з радикалів R2 та R3 означає водень або (С1-С3)алкіл, n дорівнює 0 або 1,Ph1 і Ph2 означають, кожний незалежно від іншого, незаміщену, монозаміщену або полізаміщену фенільну групу,а також її солі і...
L-арабінодисахариди антрациклінів, способи їх одержання та фармацевтичні композиції, що містять ці речовини

Номер патенту: 69446
Опубліковано: 15.09.2004
Автори: Чіполлоне Амалія, Береттоні Марко, Біджоні Маріо, Маджі Карло Альберто, Аніматі Фабіо
МПК: A61K 31/704, C07H 15/252, A61P 35/00
Мітки: антрациклінів, містять, фармацевтичні, способи, композиції, l-арабінодисахариди, речовини, одержання
Формула / Реферат:
1. L-арабінодисахариди антрациклінів загальної формули (І) (І)та їх фармацевтично прийнятні солі,деR - ОН;R1 - Η або ОН;R2 - Η або ОН, або NH2.2. Сполуки формули (І) за п. 1, які входять у групу, яка складається з таких сполук:і) 7-O-[2,6-дидеокси-4-O-(2,3,6-тридеоксі-3-аміно--L-арабіногексопіранозил)--L-ліксогексопіранозил]-4-деметоксидоксорубіцинонгідрохлорид (R=R1=OH,...
Фармацевтичні композиції, що містять a hmg coa інгібітор редуктази

Номер патенту: 77156
Опубліковано: 15.11.2006
Автори: Віджинс Норман Алфред, Крікмор Джозеф Річард
МПК: A61K 31/505, A61K 47/02, A61P 3/06
Мітки: фармацевтичні, редуктази, інгібітор, містять, композиції
Формула / Реферат:
1. Фармацевтична композиція, що містить (Е)-7-[4-(4-фторфеніл)-6-ізопропіл-2-[метил(метилсульфоніл)аміно]піримідин-5-іл]-(3R,5S)-3,5-дигідроксигепт-6-енову кислоту або її фармацевтично прийнятну сіль як активний інгредієнт і неорганічну сіль, в якій катіон є полівалентним, за умови, що аніон неорганічної солі не є фосфатом.2. Фармацевтична композиція за пунктом 1, в якій катіон неорганічної солі вибирають з кальцію, магнію, цинку,...
Попередній патент: Спосіб одержання кальцієвої солі розовастатину
Наступний патент: Гідролізат казеїну, спосіб його одержання та застосування
Випадковий патент: Пристрій для виконання пазів у кутових профілях