Хіральні біфосфінові сполуки, комплекси на їх основі, 4,16-дибром[2.2]парациклофани як проміжні сполуки для їх одержання та спосіб одержання хіральних біфосфінових сполук

















Формула / Реферат
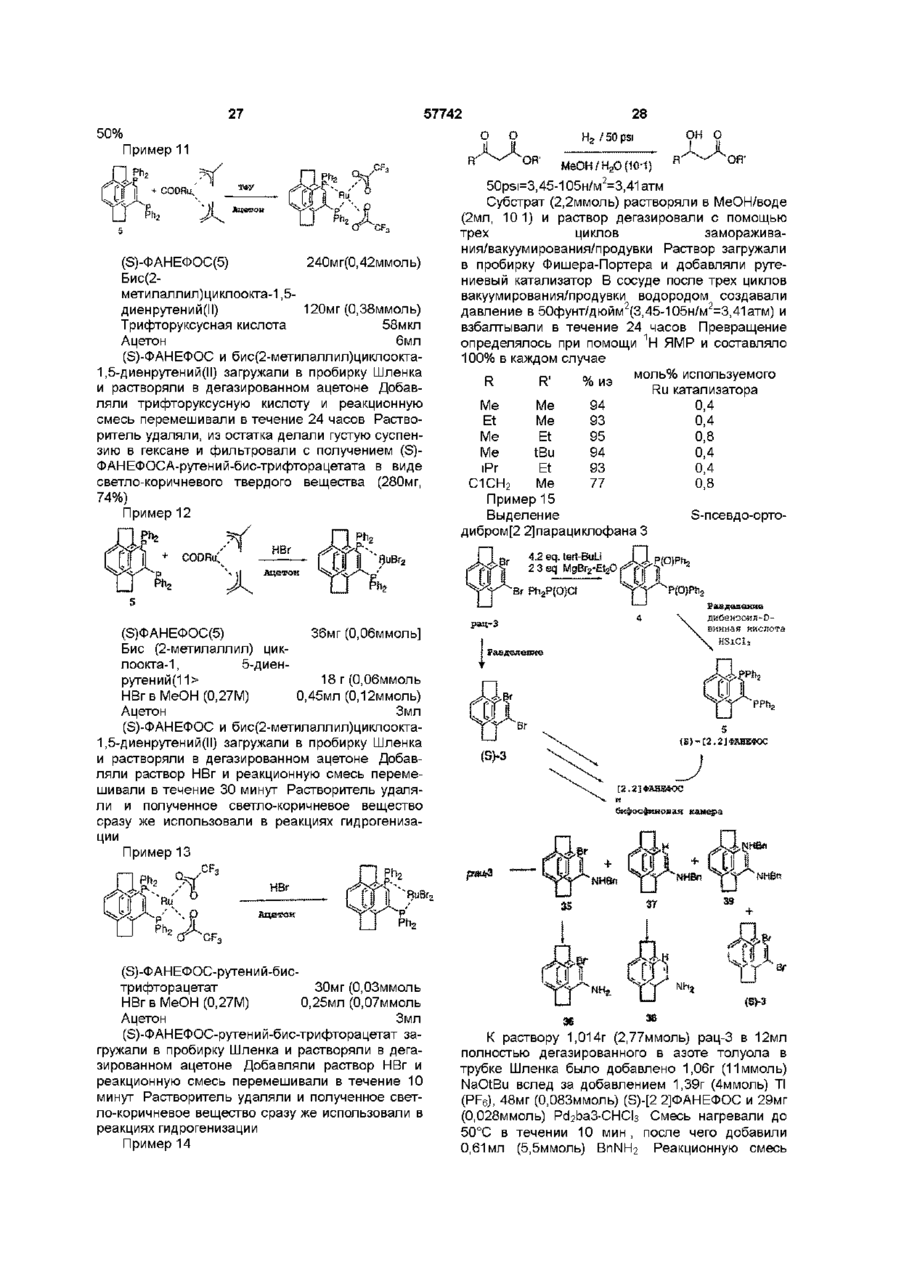
1. Соединение нижеприведенной формулы, представляющей собой хиральные бифосфины
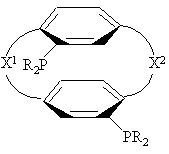 или
или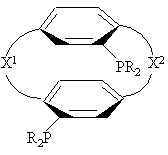 ,
,
где R представляет С1-4-алкил, С3-6-циклоалкил или арил, незамещенный или замещенный -F, -СН3, -СF3 или СН3О-, и
X1 и X2 связывают два R2P-замещенные бензола и независимо образуют 2-4-членную связывающую группу, состоящую из 2-4 атомов углерода и одного незамещенного или замещенного гетероатома, выбранных из О, S, SO, SO2 или 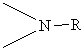 .
.
2. Соединение по пункту 1, в котором число атомов в связи X1 является таким же, как и число атомов в связи X2.
3. Соединение по пункту 2, формулы
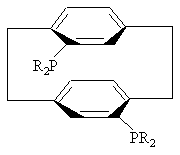 или
или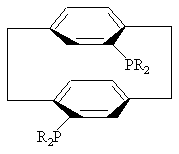 .
.
4. Соединение по пункту 3, формулы
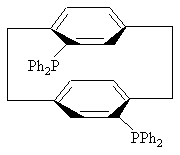 или
или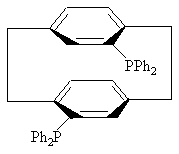 .
.
5. Комплекс (БИФОС)М®Ln,
где n является целым числом и равно 0, 1, 2, 3 или 4,
Μ представляет Rh, Ir, Ru или Pd,
L является лигандом, обратимо скоординированным для замены субстратом,
БИФОС является соединением формулы
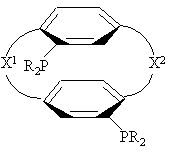 или,
или,
где R представляет С1-4алкил, С3-6циклоалкил или арил, незамещенный или замещенный -F, -СН3, -СF3 или СН3О-, и
X1 и X2 связывают два R2P-замещенных бензола и независимо образуют 2-4-членные связывающие группы, состоящие из 2-4 атомов углерода и до одного незамещенного или замещенного гетроатома, выбранного из О, S, SO, SO2 или .
6. Комплекс по пункту 5, в котором лиганд представляет собой
(а) норборнадиен,
(б) C1-4OH,
(в) NRlR2RЗ, а R1 и R2, и R3 независимо представляют Н, С1-4-алкил, С3-7-циклоалкил, арил, пиридил или тетрагидрофурил, или
(г) диен формулы
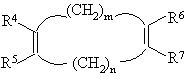 ,
,
где m представляет целое число и равно 1, 2, 3 или 4,
n представляет целое число и равно 0, 1, 2, 3 или 4, и R4, R5, R6 и R7 независимо представляют Η, C1-4-алкил,
(д) простой эфир формулы R1OR2 или циклический эфир формулы
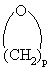 ,
,
где p является целым числом и равно 2, 3, 4 или 5,
(е) би-простой эфир формулы
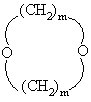 ,
,
где m определено выше,
(ж) биспирт формулы
HO-(CR8R9)p-OH,
где p как определено выше, а
R8 и R9 независимо представляют Н, C1-4-алкил или арил, или
(з) этилен.
7. Комплекс по пункту 5 или 6, где число атомов в связи X1 БИФОС является таким же, что и число атомов в связи X2 БИФОС.
8. Комплекс по пункту 7, в котором Μ представляет Rh или Ir, n равно 1, a L представляет циклооктадиен.
9. Комплекс по пункту 5 или 6 структуры
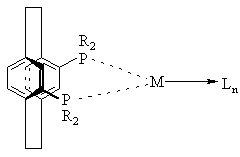 ,
,
где значения R, М и L определены выше.
10. Комплекс по пункту 5 или 6 структуры
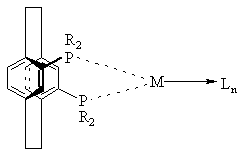 ,
,
где значения R, М и L определены выше.
11. Комплекс по пункту 10 структуры
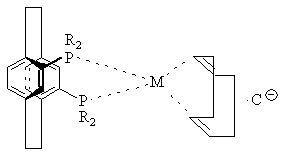 ,
,
где Μ представляет Rh или Ir,
С![]() представляет противоион,
представляет противоион,
а R определено выше.
12. Комплекс по пункту 9 структуры
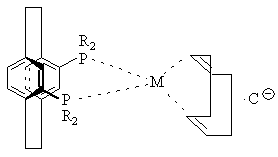 ,
,
где М представляет Rh или Ir,
С![]() представляет противоион,
представляет противоион,
а R определено выше.
13. Комплекс по пункту 5 или 6 структуры
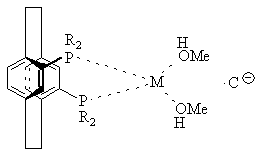 или
или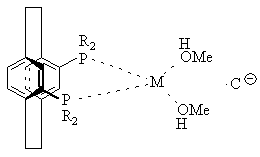 ,
,
где М представляет Rh или Ir,
С![]() представляет противоион,
представляет противоион,
а R определено выше.
14. Комплекс по пункту 5 структуры
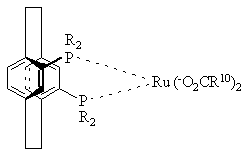 или
или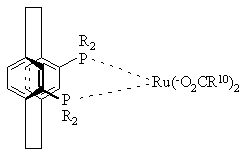 ,
,
где R10 представляет СF3 или СН3,
а R определено выше.
15. Комплекс по пункту 5 структуры
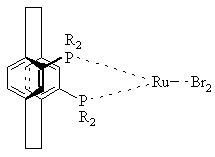 или
или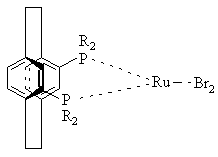 ,
,
где R определено выше.
16. Частично или полностью энантиометрно чистое соединение формулы
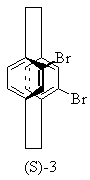 или
или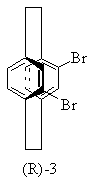 .
.
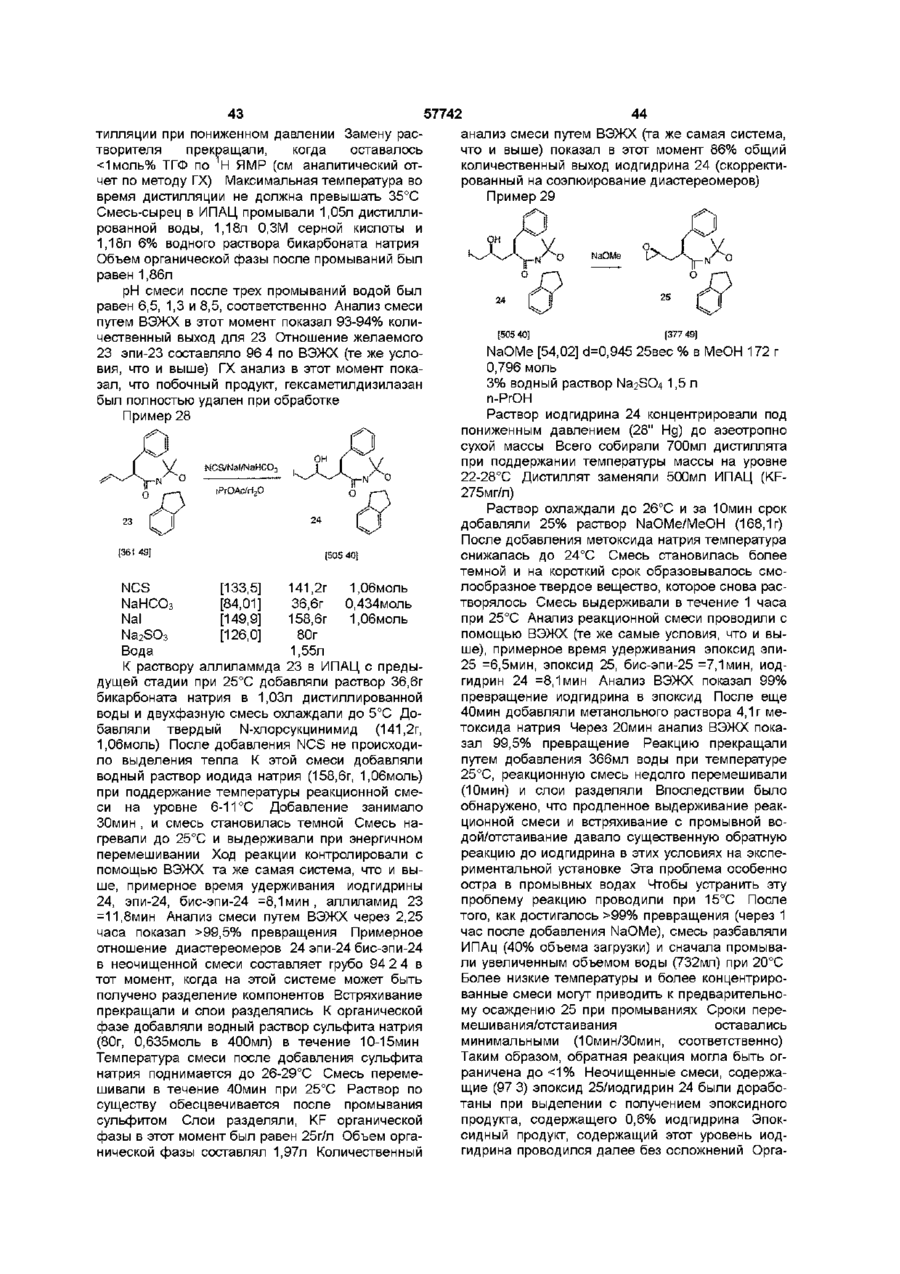
17.Способ получения хирального бифосфинового соединения (S)-40 или (R)-40
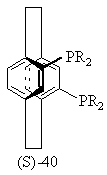 или
или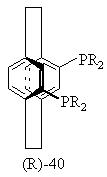 ,
,
где R представляет С1-4-алкил, С3-6-циклоалкил или арил, незамещенный или замещенный -F, -СН3 -СF3 или СН3О-, отличающийся тем, что включает следующие стадии:
(а) воздействуют на рацемическое фосфинильное соединение 41
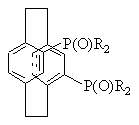
рац-41
с помощью разделяющего вещества для получения хирального соединения (S)-41 или (R)-41, и
(б) восстанавливают хиральное соединение (S)-41 или (R)-41 с получением хирального бифосфинового соединения (S)-40 или (R)-40
или.
18. Способ по пункту 17, отличающийся тем, что разделяющим веществом является дибензоил-L-винная кислота, а каждый R представляет собой фенил.
Текст
1 Соединение нижеприведенной формулы, представляющей собой хиральные бифосфины атомов в связи X 3 Соединение по пункту 2, формулы R.P или 4 Соединение по пункту 3, формулы РРЬ, Ph,p или 5 Комплекс (БИФОС)М-»1_п, где п является целым числом и равно 0, 1, 2, 3 или 4, М представляет Rh, Ir, Ru или Pd, L является лигандом, обратимо скоординированным для замены субстратом, БИФОС является соединением формулы О 1 ю 2 или где R представляет Сі 4-алкил, Сз б-циклоалкил или арил, незамещенный или замещенный -F, СНз, -CF3 или СНзО-, и X1 и X2 связывают два РгР-замещенные бензола и независимо образуют 2-4-членную связывающую группу, состоящую из 2-4 атомов углерода и одного незамещенного или замещенного гетероатома, выбранных из О, S, SO, SO2 или 2 Соединение по пункту 1, в котором число атомов в связи X1 является таким же, как и число или где R представляет Сі 4алкил, Сз бЦиклоалкил или арил, незамещенный или замещенный -F, -СНз, CF3 или СНзО-, и X1 и X2 связывают два R2P-3aMeL4eHHbix бензола и независимо образуют 2-4-членные связывающие группы, состоящие из 2-4 атомов углерода и до одного незамещенного или замещенного гетроатома, выбранного из О, S, SO, SO2 или 6 Комплекс по пункту 5, в котором лиганд представляет собой (а) норборнадиен, 57742 (б) Сі 4 0 Н , 2 3 1 2 3 (в) NR'R R , a R и R , и R независимо представляют Н, Сі 4-алкил, Сз 7-Циклоалкил, арил, пиридил или тетрагидрофурил, или (г) диен формулы -(СН2)п 4 R м с© представляет Rh или Ir, (СН 2 ) П где m представляет целое число и равно 1, 2, 3 или 4, п представляет целое число и равно 0, 1, 2, 3 или 4 5 6 7 4, и R , R , R и R независимо представляют Н, Сі 4-алкил, 1 2 (д) простой эфир формулы R OR или циклический эфир формулы представляет противоион, a R определено выше 12 Комплекс по пункту 9 структуры м где М представляет Rh или Ir, где р является целым числом и равно 2, 3, 4 или 5, (е) би-простой эфир формулы С ^ представляет противоион, a R определено выше 13 Комплекс по пункту 5 или 6 структуры н ОМе О О С© ОМе Н или где m определено выше, (ж) биспирт формулы HO-(CR8R9)P-OH, где р как определено выше, а R и R9 независимо представляют Н, Сі 4-алкил или арил, или (з) этилен 7 Комплекс по пункту 5 или 6, где число атомов в связи X1 БИФОС является таким же, что и число атомов в связи X2 БИФОС 8 Комплекс по пункту 7, в котором М представляет Rh или Ir, n равно 1, a L представляет циклооктадиен 9 Комплекс по пункту 5 или 6 структуры н ОМе ОМе н где М представляет Rh или Ir, С ^ представляет противоион, a R определено выше 14 Комплекс по пункту 5 структуры 10 Ru(O2CR )2 Ru(O2CR10)2 ИЛИ м где значения R, М и L определены выше 10 Комплекс по пункту 5 или 6 структуры м где значения R, М и L определены выше 11 Комплекс по пункту 10 структуры где R представляет CF3 или СНз, a R определено выше 15 Комплекс по пункту 5 структуры Ru—Вг, Ru — В г , ИЛИ где R определено выше 16 Частично или полностью энантиометрно чистое соединение формулы 57742 (R)-3 (S)-3 или 17 Способ получения хирального бифосфинового соединения (S)-40 или (R)-40 рац-41 с помощью разделяющего вещества для получения хирального соединения (S)-41 или (R)-41, и (б) восстанавливают хиральное соединение (S)-41 или (R)-41 с получением хирального бифосфинового соединения (S)-40 или (R)-40 PR, (SH0 (R)-40 и л и где R представляет Сі 4-алкил, Сз б-циклоалкил или арил, незамещенный или замещенный -F, СНз -CF3 или СНзО-, отличающийся тем, что включает следующие стадии (а) воздействуют на рацемическое фосфинильное соединение 41 Данная заявка родственна U S S N 60/019590, содержание которой включено здесь в виде ссылки Хиральные биосфины являются важным классом хиральных лиганд, которые используются для получения катализаторов для асимметричной гомогенной гидрогенизации, обменной гидрогенизации /гидросилирование, гидроборирование или гидроформилирование олефинов, кетонов, иминов, а также энантиоселективной изомеризации олефинов, соединения реагентов Гриньяна и органических галогенидов и арилирования олефинов, например, реакции Хека Хорошее краткое изложение этих реакций содержится в R Noyon, Asymmetric Catalysis in Organic Synthesis, John Wiley & Sons, 1994, и I Ojima, Catalytic Asymmetric Catalysis, VCH Publishers, 1994 Известные хиральные бифосфины представлены следующими классами бифосфины, полученные из природных продуктов, таких как ДНОП или НОРФОС, бифосфины с хиральным Р, такие как ДИПАМФ, замещенными 1, 2-би(фосфолано)бензольными лигандами (ДюФОС), аксиальные хиральные соединения соответственно замещенных бифенилов со стерическим торможением Наиболее известными и наиболее применимыми для синтеза представителями последнего класса, в частности, и известных хиральных бифосфинов в основном являются БИНАП (BINAP) и ТолБИНАП (TolBINAP) следующей структуры (S)-40 (RHO и л и 18 Способ по пункту 17, отличающийся тем, что разделяющим веществом является дибензоил-Lвинная кислота, а каждый R представляет собой фенил Afe Ph BINAP Af= pCH3-C6H4 TolBiNAP Асимметричные катализаторы на основе БИНАП часто дают большой избыток энантиомеров (ее) в ряде типов реакций К сожалению, ряд субстратов или реакций дают в результате нежелательно низкий избыток (ее), то есть должны применяться более громоздкие классические решающие проблему методы В данном изобретении описан новый набор бифосфиновых лиганд, которые являются соответственно замещенными циклофанами с плоскостной хиральностью Данное изобретение охватывает энантиомеры псевдо-ортозамещенные циклофаны В одном примере бифосфины данного изобретения образуют комплексы, используемые для синтеза хирального промежуточного соединения для лекарства для лечения СПИДКРИКСИВАНА® (торговое название Merck & Со , Inc) Хиральные бифосфины данного изобретения дают в качестве катализаторов желаемый высокий энантоимерный избыток в ряде асимметричных реакций 7 57742 Данное изобретение представляет новые хиральные бифосфины, соответственно используе 8 мые в качестве катализаторов асимметричных реакций ТАБЛИЦА АББРЕВИАТУР БИНАП Бок Бок-БФФМ 2,2'-бис (дифенилметилфосфино)-1,1'-бинафтил Трет-Бутилоксикарбонил (23,43)-трет-Бутил-4-(дифенилфосфино)-2-(дифенилфосфинометил)-2пирролидинкарбоксилат противоион, противоположно заряженный ион Бензилоксикарбонил 1,5-циклооктадиен (Р,Р)-2,3-О-изопропилиден-2,3-дигидрокси-1,4-бис-(дифенилфосфино)-бутан (Р,Р)-1,2-бис((о-метоксифенил)фенилфосфино)этан 1,2-Бис-((23,53)-2,5-диэтилфосфолано)бензол [(2Р,ЗР)-8,9,10-тринорборн-5-ен-2,3-диил]-бис(дифенилфосфин) 1,2-бис(дифенилфосфино) пропан 2,4-бис(дифенилфосфино) пентан Три флат С" Кбз Цод Диоп ДИФАМФ ДУФОС НОРФОС ПРОФОС СКЬЮФОС ТФО Псевдо-ортозамещенные хиральные бифосфины данного изобретения применимы для получения комплексов, которые действуют как катализаторы в ряде асимметричных реакций, включая асимметричную однородную гидрогенизацию, обменной гидрогенизации, гидросилилирование, гидроборирование или гидроформилирование олефинов, кетонов, иминов, а также энантиоселективной изомеризации олефинов, соединения реагентов Гриньяра и органических галогенидов и арилирования олефинов Описаны также способы получения и использования хиральных бифосфинов и их комплексов Данное изобретение охватывает соединения формул представляет Сі 4-алкил, где циклоалкил или арил, незамещенный или замещенный -F, -СНз, -CF3 или СНзО-, и Xі и X2 связывают дваРгР-замещенные бензола и независимо образуют 2-4-членную связывающую группу, состоящую из 2-4 атомов углерода и одного незамещенного или замещенного гетероатома, выбранного из О, S, SO, SO2 или >N-R Одно из воплощений данного изобретения ограничивается соединениями, в которых число атомов в связывающей группе X1 является таким же, как и число атомов в связывающей группе X2 Одним из предпочтительных воплощений является соединение 4,12-бис[дифенилфосфино]2,2-парациклофан, представленный здесь, как ФАНЕФОС РРЛ» Данное изобретение также включает комплекс для катализа ряда асимметричных реакций Комплекс данного изобретения представляет (BISPHOS)M-> Ln (БИФОС) где п является целым числом и равно 0, 1, 2, 3 или 4, М представляет Rh, Ir, Ru или Pd, L является лигандом, обратимо скоординированным для замены субстратом, БИФОС является соединением формул где R представляет Сі_4-алкил, Сз_єциклоалкил или арил, незамещенный или замещенный -F -СНз, -СРз или СНзО-, и X1 и X связывают два R2P-3aMeL4eHHbix бензола и независимо образуют 2-4-членные связывающие группы, состоящие из 2-4 атомов углерода и одного незамещенного или замещенного гетероатома, выбранного из О, S, SO, SO2 или Одно из воплощений данного изобретения ограничивается комплексом, в котором лиганд представляет собой (а) норборнадиен, (б) Сі 4ОН, (в) NR1R2R3, a R1 и R2 и R3 независимо представляют Н, Сі_4-алкил, Сз_7-циклоалкил, арил, пиридил или тетрагидрофурил, или (г) диен формулы где m представляет целое число и равно 1, 2, 3 или 4, п представляет целое число и равно 0, 1, 2, 3 или 4, и R4, R5, R6 и R7 независимо представляют Н, Сі_4-алкил, (д) простой эфир формулы R1OR2 или циклический эфир формулы -о где р является целым числом и равно 2, 3, 4 или 5, 57742 10 (е) биэфир формулы где m определено выше, (ж) биспирт формулы HO-(CR8R9)P-OH где р определено выше, а R и R независимо представляют Н, C-i_4алкил или арил, или (з) этилен Другое воплощение данного изобретения ограничивается комплексом, в котором число атомов в связывающей группе X1 БИСФОС является таким же, что и число атомов в связывающей группе X2 БИСФОС Другое воплощение данного изобретения ограничивается комплексом, в котором М представляет Rh или Ir, n равно 1, a L является циклооктадиеном Еще одно воплощение данного изобретения ограничивается комплексом следующего строения где R представляет CF3 или СНз Другое осуществление данного изобретения ограничивается комплексом "Ru—Вгг ,Ru--Br2 В еще одном аспекте данное изобретение частично или полностью представляет энантиометрически чистое соединение формулы Вг (SJ-3 (Щ-з предпочтительно, полностью энантиометрически чистое соединение формулы Еще одно осуществление данного изобретения ограничивается комплексом следующего строения Вг (SJ-3 (RJ-3 Еще одним воплощением данного изобретения является способ получения хирального бифосфинового соединения (S)-40 Другое воплощение данного изобретения ограничивается комплексом .0 где М представляет Rh или Іг В еще одном воплощении данного изобретения ограничивается комплексом £S)-40 где R представляет Сі_4-алкил, Сз_єциклоалкил или арил, незамещенный или замещенный -F, -СНз, -CF3 или СНзО-, включающий стадии (а) воздействия на рацемическое фосфинильное соединение 41 Р(О)Я2 где М представляет Rh или 1 г Другое воплощение данного изобретения ограничивается комплексом е н ОМе ,© где М представляет Rh или Іг Еще одно воплощение данного изобретения ограничено комплексом P(O)R2 с помощью разрешающего вещества для получения хирального (S)-41, и (б) восстановления хирального (S)-41 с получением хирального бифосфинового соединения (S)-40 57742 11 12 R ^ ^со2н Ri" "со 2 н 3 асимметричную гидрогенизацию при изохинолиновом синтезе, например, Кроме того, опытному специалисту ясно, что этот процесс может также использоваться для получения (Р)-хирального энантиомера ОМе ,ОМе ОМе PR, (RM0 Предпочтительно, разрешающим веществом, используемым в этом процессе, является дибензоил-І_-винная кислота, а каждый R является фенилом Для комплексов данного изобретения подходит широкий ряд лиганд Подходит любой лиганд, который обратимо координирует и может быть заменен субстратом, включая растворитель для реакционной смеси Соответствующие лиганды включают, но не ограничиваются ими, диены, такие как циклооктадиен или норборнадиен, низшие спирты, простые эфиры, циклические простые эфиры, биэфиры, биспирты и простые олефины, такие как этилен Один из предпочтительных лиганд является циклооктадиеном Металл М выбирается из Rh, Ir, Ru или Pd Комплексы, образованные по данному изобретению, включают хиральный бифосфин, металл (М) выбирается из Rh, Ir, Ru или Pd, один или более лиганд, и необязательно, противоион (Се) Соответствующие противоионы являются не нуклеофилами и включают, но не ограничиваются ими, OTfe, CIO 4 e , SbF 6 e или PF 6 e Понятно, что любая формула для комплекса в данном изобретении включает, если это нужно, противоион Чтобы проиллюстрировать использование хиральных бифосфинов и их комплексов, заявители представляют синтез хирального промежуточного соединения Бос .N. С1 N Cz b 4 асимметричную гидрогенизацию в ненасыщенных спиртах, например, 5 асимметричную гидрогенизацию при нехелатирующем замещении, например, о О 6 гидрогенизацию кетонов, например, о он NMe, ММе, 7 двойную гидрогенизацию 1,3- и 1,2-кетонов, например, 0 0 ОН ОН 8 энантиоселективную изомеризацию олефинов, например, Я' "*" NEt2 — •• " - г 9 асимметричную гидрогенизацию иминов, например, 10 асимметричное гидроборирование, например, он СОМНЕВи для синтеза известного эффективного ингибитора протеаз ВИЧ, КРИКСИВАНА®, соединение J в примерах ниже Псевдо-ортозамещенные бифосфины данного изобретения являются катализаторами в ряде известных реакций, включая, но не ограничиваясь ими, 1 хиральную гидрогенизацию енамидных структур, например, RjCON" И GO2H Н со г н 2 хиральную гидрогенизацию не енамидных структур, например, 11 асимметричную циклизацию олефиновых альдегидов, например, 12 арилирование олефинов, например, OTf О *О Q4 О 13 асимметричное алкилирование, например, 57742 13 СаНс СОгМе Na—j-NHAC CO2Me ОАс псездо-мета и пара МеОгС ^ Ac 14 аминирование арилгалогенидов (реакция Хартвига-Бюхвальда) R ' ~ NR.R, Pd катализатор X = Вг, I, CI, OTf Хотя бифосфин имеющий в основе [2 2] парациклофановую систему, дает превосходные асимметрические катализаторы, для некоторых субстратов требуются по-разному сконструированные комбинации металл/лиганд чтобы достичь высокого превращения при высоком ее(изб) Замечательным свойством [2,2]парациклофановой системы является ее высокая стойкость, усиленная тесной близостью 2 бензольных колец, и возможно ввести некоторую эластичность в эти системы путем увеличения размера кольца с [2 2] до систем [2 3],[3 3], [3 4], [4 4] и [2 4] Хотя увеличенный размер кольца в этих системах дает возможность некоторой конформационной эластичности, замещенные бензольные кольца все еще не способны вращаться, так что обсуждаемые материалы являются конфигурационно стабильными Кроме того, введение гетероатомов в мостик может привести к желаемым эффектам, таким как повышенная растворимость и различная полярность В нижеследующем описывается подход к синтезу этих циклофанов с увеличенным кольцом Бифосфины с плоскостной хиральностью В этом изобретении описан новый класс симметричных относительно С-2 бифосфиновых лиганд с общим строением I Это - парациклофан с углеродными мостиками и/или мостиками с гетероатомами Парациклофаны [2 2], [2 3], [3 3], [3 4], [2 4] и [4 4] - все конфигурационно стабильны і X Х = -(СН2)п-,-СНгОСМ2-, - С HgSOgC И2' Самый легкий подход к этим системам - через коммерчески доступный [2 2] парациклофан II, который бромируется с получением четырех изомерных дибромидов [см Reich and Cram, J Am Chem Soc 1969, 91, 3527](Схема І) Псевдопараизомер 4 кристаллизуется из гексанов, тогда как желаемый псевдо-ортоизомер IV получают с помощью хроматографии маточных жидкостей II псеадо-пара III поевдо-орто X V Включение дифенилфосфиногрупп достигается одним из трех путей на схеме 2 показана прямая замена Вг дифенилфосфиногруп-пой Хотя эти пути высокоэффективны, легкость окисления V в условиях реакции делает этот подход менее привлекательным СХЕМА 2 псееяо-opwo OR RizPLf NiClgdppe THF/ RT моля- и биоксидн РРП, Схеме 5 (ниже) представляет предпочтительный путь На схеме 5 после низкотемпературного литиирования с помощью BuLi, трансметаллирование с помощью МдВгг приводит к получению реактива Гриньяра Альтернативно реактив Гриньяра может быть получен непосредственно из Мд и чистого псевдоорто-дибромида IV Реактив Гриньяра затем приводят в реакцию с дифенилфосфорилхлоридом с получением бифосфиноксида IX Альтернативно, трансметаллирование до реактива Гриньяра может быть исключено путем непосредственного использования билитиевого соединения с добавлением дополнительных веществ в конце, например, тетраметилэтилендиамина На последней стадии фосфиноксид может быть восстановлен до фосфина при использовании стандартных условий, например, с помощью HS1CI3 или L1AIH4 Оптическое разделение может быть выполнено одним из трех путей при простейшем разделении хроматография псевдо-ортодибромида IV на хиральной стационарной фазе дает оптически чистый псевдо-орто-дибромид Например, энантиомеры IV легко разделяются на кристаллической триацетатцеллюлозе с использованием ЕЮН в качестве элюента Одинаково хорошее разделение получают с использованием коммерческих колонок Chiracell и Chiralpak Последующее прямое введение дифенилфосфиногруппы или двустадийное введение путем первоначального введения дифенилфосфиноксидной группы с последующим восстановлением, что таким образом дает оптически чистый бифосфиновый ліганд IV Альтернативно, получают рацемический бифосфиноксид IX Это вещество может быть разделено путем образования комплексов включения с хиральными веществами, например, бензоилвинной кислотой или N-бензилхинхонидиниевыми 57742 16 15 солями Разделяемый фосфиноксид отделяется лением субстрата делает возможным выполнение от разделяющего вещества и получается в оптигидрогенизации при пониженных температурах, чески чистом виде Восстановление фосфинокситаких низких, как примерно -45°С (схема 4а) да выполняется рядом путей, например, HS1CI3 с СХЕМА 4 а EtsH или, предпочтительно, одним HS1CI3 В третьих, оптически чистый псевдо-ортоH Via дибромид может быть получен из рацемического материала при использовании кинетического разMe МеОН AcHN деления Так, реакция рацемического материала с AcHN ~45°C первичным амином и натрий-трет-бутоксидом, ATM Нг катализируемая хиральным Pd-бифосфиновым 9 4 % и э {ее) комплексом приводит к преимущественной реакИз-за лабильности V была предпринята поции одного из энантиомеров, оставляя неиспольпытка разделения через более устойчивый бизованным оптически чистый IV Особенно привлефосфиноксид IX Воздействие на биреагент кательный Pd катализатор получается с Гриньяра РпгРОС1 давало IX (схема 5) хиральным лигандом V С этим лигандом один из Схема 5 представляет предпочтительный энантиомеров IV реагирует 3-5 раз также быстро, путь как и с другими, оставляя неиспользованный оптически чистый IV Добавление соли таллия поСХЕМА 5 вышает эту хиральную разрешающую способность до очень практичной 10-15-кратной степени Вг 1,4.2eq. fe/7-BuLt V(O)?\\ различия 2 Чтобы определить эффективность бифосфинового лиганда при катализируемой родием реакции гидрогенизации синтезировали комплекс VI Схема 3 Rh(COD)3-OT( •T Of PPh-, Когда VI добавляют к метанольному раствору а-ациламиноциннамовой кислоты VII и подают водород при ЮООфунт/дюйм (6,89-106н/м2, 682,18атм) получается производное фенилаланина VIII (схема 4) 2 4 2eq. МдВг г -ОЕі 2 P(O)Ph2 З Ph2POCi поевдо-орто рац-ХХ Смесь рац-ІХ разделяют, используя комплексы включения с хиральными веществами, например, бензоилвинную кислоту или солей Nбензилхинхонидиния Разделенный фосфиноксид выделяют из разделяющего вещества и получают оптически чистую форму Восстановление фосфиноксида выполняется рядом путей, например, HSiCIs с EtsN или, предпочтительно, одним HS1CI3 Бромирование мостика в радикальных условиях приводит к получению соединений XI и XII СХЕМА 6 СХЕМД й Ph 2 OP POPh, •T Of VI Ph. MeOH AcHN' "СОгН 1000 psi H2 CO2H VI! VIII Гидрогенизация предкатализатора VI в метаноле приводит к потере циклооктадиенового лиганда с получением Via Образование катализатора Via перед добав PhjOF Простое изменение стехиометрии при бромировании и с использованием 2 эквивалентов ВГ2 приводит к получению смеси би-бромированных продуктов XIII и XIV, которые снова трудно разделить (схема 7} 17 18 Первоначально спирт окисляют в соответствующий кетон с использованием условий Swern или ряда окислителей на основе металлов, таких как пиридиния дихромат или тетрапропиламмония перрутената Кетон затем подвергают расширению кольца с использованием одного из диазометана и триметилсилилдиазометана или условий Демьянова после первоначального добавления к кетону или нитрометана или цианида и его восстановления до аминометильной группы Получающийся в результате кетон с расширенным кольцом затем восстанавливается до соответствующей метиленовой группы Это лучше всепо осуществляется с использованием с использованием модификации Хуанг-Минлон классического восстановления Вольф-Кишнер (ДМСО, калийтрет-бутоксид, гидразин) или условий восстановления Клемменсона (Zn, разбавленная HCI) Получающийся в результате [2,3]парациклофан является хиральным, но не симметричным относительно С2 Восстановление двух кетонов, полученных из бибромидов со схемы 3, дает такую же С2 симметричную [3,3] систему для обоих исходных материалов, так что по причинам целесообразности смесь бибромидов может быть осуществлена через эту последовательность расширения кольца без разделения какой-либо из диастереометрических смесей, которые образуются во время последовательности реакций Как и перед этим фосфиноксиды прямо восстанавливаются до фосфинов с помощью SiHCb/EtsN и получающиеся в результате бифосфины перечислены в схеме 10 57742 С Е А 7 ХМ Вг 1 Ль.»™:™..^ I POPh, хш + xrv Простой гидролиз бромидов с помощью одного из NaOAc/HOAc с последующим воздействием основания или использованием солей серебра прямо приводит к получению соответствующих спиртов, которые могут быть легко разделены на силикагеле (схема 8) СХЕМА 8 С Е А 10 Х М Вг PfljOF xvn Ph,P Ph2OP xvm Последующие реакции выполняют с энантиомерно чистыми диастереомерами XV-XVIII, и они состоят из следующей последовательности классических реакций Они показаны в качестве примера только для соединения XIX на схеме 9, но эта последовательность реакций сохраняется и для всех подобных производных спиртов СХЕМА 9 РЬ,ОР' Ph,OP XIX POPh, Ph2GP POPh, PPh a Энантиомеры со схемы 5 затем используются для получения активных Rh, Ir, Ru или Pd катализаторов, которые описаны для [2,2]парациклофанбифосфина в примере 6, или использующихся процедур, подобных описанным в литературе для известных бифосфинов Подводя итог, последовательность классических реакций дает возможность, получения [2,3] и [3 3]парациклофанбифосфинов из легко доступных энантиометрически чистого [2 2]парациклофанби-фосфиноксида Опытному специалисту легко увидеть, что та же самая стратегия синтеза, состоящая из радикального бромирования мостика с последующим гидролизом бромида до спирта, окислением спирта до кетона, расширением кольца, и восстановление кетона до метиленовой группы дает [2,4]парациклофан, ко 19 20 57742 гда [2,3]парациклофан используется в качестве исходного материала, и [3,4]парациклофаны, когда исходным материалом для последовательности расширения кольца является [3,3]парациклофан Возможно дополнительное аналогичное расширение кольца для получения [4 4]системы, но дополнительное расширение кольца приводит к системам, которые конфигурационно стабильны только при низких температурах и поэтому их практическое использование ограничено (схема 11) СО2Ме POPh; Р1чг0Р' XX СХЕМА 11 СО 2 Ме XXI Ph,P Схема 12 легко завершается путем гидролиза сложного эфира до кислоты и кислоты затем разделяют классическим методом с помощью хирального амина Для этой цели подходят фенетиламин и бруцин Альтернативно, энантиомеры можно разделить на колонке для хиральной хроматографии После разделения различные химические превращения соседних карбоксиметоксигрупп приводят к получению хиральных но не С2 симметричных предшественников бифосфиновых катализаторов Например, приведение во взаимодействие кислоты с тетраацетатом свинца и хлоридом лития дает бихлорид XXII, а дегалогенирование затем приводит к получению незамещенного метиле-нового мостика См , например, J Am Chem Soc 89, 3078 (1967) и схему 13 СХЕМА 1 3 Ph,P Полезная исходная информация по простым незамещенным системам найдена, в J Am Chem Soc 88, 3513(1966) и J Am Chem Soc-*"88, 3667 (1966) Применение этих методов для получения разделенных энантиометрически чистых фосфиноксидов является новым и приводит к получению семейства хиральных бифосфинов, которые применимы для получения катализаторов для асимметрических каталитических реакций Несимметричные относительно С2 хиральные бифосфины альтернативный синтез Альтернативное, методически простое получение хирального, но не длиннее С2 симметричного [2 4] парациклофанбифосфина также начинается с [2 2]парациклофанбифосфиноксидаХХХ Расширение кольца незамещенного [2 2]парациклофана до [2 4]парациклофана описано в J Am Chem Soc 89, 3078 (1967) Так, нагревание X до примерно 200°С в диметилфумарате или диметилмалеате приводит к чистому образованию расширенной кольцевой системы в виде смеси XX и XXI Поскольку энантиометрически чистый X превращается в рацемат во время этого расширения кольца, при синтезе используется рацемический X, и необходимо разделение для XX или XXI (схема 12) Ph 3 OP Ph2OP XXI PhjOP ХХП Использование тетраацетата свинца в присутствии кислорода приводит к получению олефина XXIII См, например, Org React 1972, 19, 279 и схему 14 СХЕМА 14 PhjOP1 xxm 21 57742 Последующее восстановление фосфиноксидов до фосфина с помощью SiHCb/EtsN приводит к образованию хиральных фосфинов, которые используются для получения Rh, Ru, Ir и Pd катализаторов Хирапьные бифосфины с гетероатомными заместителями Включение гетероатомов в мостики в дополнение к чистым С мостикам в бифосфинпарацикo,s лофанах, описанных выше, приводит к улучшенной растворимости, полярности и незначительному изменению в предпочтительной конформации системы Так, С2 симметричный биокса[3,3]парациклофанбифосфин XXV получают путем бромирования известного незамещенного биокса [3 3]парациклофана с помощью катализатора, кислоты Льюиса (РеВгз) и Вгг, и полученную в результате смесь бромированных PhgOP соединений с кольцевым строением разделяют xxvn путем хроматографии на S1O2 и А12О3 Когда любая переменная (например, арил, X1, Хроматография дает чистую фракцию псевдоX2, R и т д ) встречается более чем один раз в ортодибромида XXIV, который трансформируется любой составляющей или в формуле I, ее опрев бифосфиноксид, как описано выше для деление является независимым от ее определе[2 2]парациклофан (t-Bul_i, MgBr2, Ph2POCI) Разния в каждом другом присутствии К тому же сочеделение полученного рацемата выполняется путания заместителей и/или переменных тем образования комплекса включения с дибензодопустимы, только если такие сочетания дают в илтартратом или путем хроматографии на результате стабильные соединения хиральной среде Затем разделенные фосфинокТак как он используется здесь, за исключенисиды восстанавливают, используя стандартные ем отмеченного особо, подразумевается, что терусловия (HS1CI3, EtsN) и полученные оптически мин "алкил" включает насыщенные алифатичечистые бифосфины XXV затем используются для ские углеводородные группы с прямой и получения асимметрических катализаторов с Rh, разветвленной цепью, имеющие определенное Ru, Ir и Pd (схема 15) число атомов углерода (Me представляет метил, Et представляет этил, Рг представляет пропил, Ви представляет бутил) Как он использован здесь, с СХЕМИ 15 исключениями, которые указаны, "арил" предназначен для обозначения фенила (Ph) или нафтила Соединения данного изобретения могут иметь асимметричные центры и встречаются в виде раxxrv цематов, рацемических смесей и в виде отдельных диастереомеров или энатиомеров, причем все изомерные формы включаются в данное изобретение Если конкретно не представлено иначе, данный энантиомер также обозначает энантиомер такого соединения или пары К тому же комбинации растворителей, заместителей и/или переменPh OP XXV ных допустимы только в том случае, если такие комбинации дают в результате стабильные соОкисление известного бисединения тиа[3 3]парациклофана XXVI с помощью Типичные экспериментальные методы с H2O2/Na2WO4 приводит к получению соответстиспользованием нового процесса представлены в вующего сульфона Последующее применение деталях ниже Эти методики являются только последовательности синтеза, описанной выше, примерами, но не ограничениями нового процесса включая катализируемое кислотой Льюиса бромиэтого изобретения рование кольца, образование билитиевого соединения и трансметаллирование до реактива ГриньПример 1 яра, реакцию с Ph^POCI, разделение на энантиомеры и восстановление фосфиноксидов псевдо-мета и до фосфинов дает в результате энантиометричепара ски чистое XVII См схему 16 Эти последние могут использоваться для получения катализаторов псевдо-пара псвядо-орто для асимметричной трансформации с Rh, Ru, Ir и 2 3 Pd 1 2 23 57742 ист Reich, H J and Cram, D J J Am Chem Soc 1969,91(13), 3527 [2 2]парациклофан 37,6г (0,180моль) Бром 58,3г (0,364моль) Метиленхлорид 110Омл Железо, порошок 0,60г Бром (15,5г) добавляли к перемешиваемой суспензии порошка железа в метиленхлориде (300мл) Через один час добавляли [2 2]парациклофан (37,6г) и метиленхлорид (800мл) и реакционную смесь нагревали с обратным холодильником По каплям в течение 3 часов добавляли остаток брома (42,8г) и нагревание продолжали в течение еще 4 часов Реакционную смесь промывали 10% водным раствором бисульфита (2x150мл), насыщенным раствором соли (1x150мл) и сушили (MgS04) Выпаривание растворителя давало не совсем белое твердое вещество (55,Ог, 83%, смесь четырех дибромидов) Твердое вещество растворяли в горячем хлороформе (500мл) и добавляли диэтиловый эфир (300мл) Твердые вещества отфильтровывали с получением псевдо-пара-дибромида 2 После охлаждения до 0°С получали второй выход псевдопара-дибромида 2 и объединяли с представленным выше (общий выход 15,2г, 23%) Маточные жидкости концентрировали, нагревали в гексанах и фильтровали Гексановые маточные жидкости концентрировали и подвергали хроматографии на силикагеле Псев-до-орто-дибромид 3 получали в виде белого твердого вещества (8,3г, 70% чистоты по ЖХ) Данные ЯМР для 2 и 3 находятся в согласии с литературными данными Пример 2 216Х Псевдо-пара -дибром[2 2]парациклофан 2 10,5г (0,029моль) Диметиловый эфир триэтиленгликоля 40мл Густую взвесь 2 в 40мл диметилового эфира триэтиленгликоля нагревали до 210°С в течение 18 часов После охлаждения твердые вещества отфильтровывали и снова проводили через вышеописанные условия После охлаждения твердые вещества отфильтровывали снова с получением псевдо-пара-дибромида 2 (900мг) Маточные жидкости от обеих реакций объединяли и растворитель удаляли путем дистилляции Фильтрование через слой силикагеля давало псевдо-ортодибромид 3 в виде белого твердого вещества (6,70г. 64%) Пример 3 24 Псевдо-орто-дибром[2 2]парациклофан 2 (-70% чистоты) 4,80г (9,2ммоль) трет-Бутиллитий (1,7М пентан) 32,4мл (55,1ммоль) Тетрагидрофуран (высушенный на ситах ЗА) 100мл Дифенилфосфиния хлорид 5,5мл (28,8 ммоль Магния бромиддиэтилэтерата 14,2г (55,0ммоль) Трет-Бутиллитий по каплям в течение одного часа добавляли к раствору 3 в тетрагидрофуране при -78°С Через 30 минут дополнительно добавляли магния бромид-диэтилэтерат и реакционной смеси позволяли дойти до комнатной температуры Добавляли дифенилфосфиния хлорид и через еще два часа реакционную смесь выливали в 2N НС1 (100 мл) Кислоту экстрагировали метиленхлоридом (3x100мл), органические слои объединяли, сушили (MgS04) и растворитель выпаривали Полученное твердое вещество нагревали в этилацетате/гексанах (2 3), охлаждали и фильтровали с получением 4 в виде белого твердого вещества (5,0г, 90%) Пример 4 СОгН l-ОВг | і N NaOH Псевдо-орто-бис (дифенилфосфинил)-[2 2Парациклофан 4 1,00r (1,64ммоль) Дибензоил-Ьвинной кислоты моногидрат 0,62г (1,6ммоль) Этилацетат 50мл Хлороформ 75мл Горячий раствор моногидрата дибензоил-Lвинной кислоты в этилацетате медленно добавляли к раствору 4 в хлороформе при 60°С Примерно одну треть растворителя удаляли и раствор охлаждали до комнатной температуры Через 18 часов твердое вещество отфильтровывали, растворяли в хлороформе (100мл) , промывали 1 N водным раствором гидроксида натрия (3x100мл), сушили (MgS04) и растворитель выпаривали с получением 4 в виде белого твердого вещества (380мг, 76%, 100% изб ,(ее) причем избыток энантиомера определяли с помощью суперкритической жидкостной хроматографии (СЖХ), используя хиральную OD-H колонку) Пример 5 PPh, 1.4.2eq.№f-BiiLf 2 4.2eq МдВг г О £ і г 3 Ph 2 POCi Псевдо-орто-бис (дифенилфосфинил)-[2 2]парациклофан 4 180мг (О.ЗОммоль) Трихлорсилан 1,0г (7,3ммоль) Ацетонитрил 10мл 25 57742 Трихлорсилан добавляли к густой суспензии 4 в ацетонитриле и реакционную смесь нагревали до 150°С в течение 5 часов Добавляли 20% водный раствор гидроксида натрия (100мл) и продукт экстрагировали в хлороформ (Зх50мл) После осушения (MgS04) растворитель выпаривали с получением псевдо-орто-бис(дифенил-фосфино)[2 2]парациклофана 5 в виде белого твердого вещества (140мг, 82%) Пример 6 Rh(COD)2-OT( PPh, •T Of DM C Псевдо-ортобис(дифенилфосфино)-[2 2] парациклофан 5 50мг (0,09ммоль Бис(1,5циклооктадиен)родий(І)42мг (0:,09ммоль трифторметансульфонат Дихлорметан 2мл Трет-Бутилметиловый эфир 2мл Дихлорметан добавляли к 5 и бис(1,5циклооктадиен)-родий(І)трифторметансульфонату и раствор перемешивали при комнатной температуре в течение 1 часа Дихлорметан удаляли и добавляли третбути л метиловый эфир Оранжевое твердое вещество 6 отфильтровывали и сушили в атмосфере азота (80мг, 87%) P h Пример 7 Л №.. ) AcHN'К •со 2 н 7 ш 2 Р-. **! ж'' S """ Ph'' AcHN МеОН СОЯН нг а-ацетамидоциннамовая кислота 1 10МГ (0,54ММОЛЬ) Родиевый кат 6 20мг (0,02ммоль) Метанол 2мл 6 добавляли к дегазированному раствору (аацетамидоциннамовой кислоты 7 в метаноле, и реакционную смесь гидрогенизировали в течение 18 часов при комнатной температуре Избыток энантиомера продукта определяли путем суперкритической жидкостной хроматографии с использованием хиральной ODH колонки 8 1Н ЯМР в согласии с литературными данными Давление Нг/фунтов/дюйм2 иэ (ее) 1000 52% 60 65% Пример 8 -OTf А AcHN СО,Н МеОН X AcHN . СО г Н 10 26 а-ацетамидоакриловая кислота 97мг (0,54ммоль Родиевый кат 6 18мг (0,02ммоль Метанол 20мл 6 добавляли к дегазированному раствору аацетамидакриловой кислоты 9 в метаноле и реакционную смесь дегидрогенизировали при 2 5 2 40фунт/дюйм (2,76-10 н/м , 2,73атм) в течение 18 часов при комнатной температуре Избыток энантиомера продукта определяли путем дериватизации соответствующего метилового сложного эфира 12 метанол удаляли под пониженным давлением и добавляли раствор диазометана (~0,6М в Et20) Через 15 минут диэтиловый эфир удаляли и избыток энантиомера определяли с помощью газовой хроматографии, используя колонку Chiracil-Val III (изотермальный 105°С, поток 10см/с, отношение разделения 140 1) (ее) иэ = 95%, 1Н ЯМР в согласии с литературными данными Пример 9 Р., •ОТ! Ар--'' II A H CO,Me cN и РИ г 6 AcHN МеОН 11 12 Метил-2ацетамидоакрилат 120мг (0,54ммоль) Родиевый кат 6 6мг (0,02ммоль) Метанол 20мл 6 добавляли к дегазированному раствору метил-2-ацетамидакрилата 11 в метаноле и реакционную смесь гидрогенизировали при атмосферном давлении в течение 2 часов при комнатной температуре Избыток энантиомера продукта определяли путем газовой хроматографии с использованием колонки Chiracil-Val III (изотермальный 105°С, поток Юсм/сек, отношение разделения 1 140 1) (ее) иэ = 99,8%, Н ЯМР в согласии с литературными данными Пример 10 'т: •T Of МеОН PH hN і4 4J 0 12А НА Пример 10 R R11 R12 иэ(%) 12А Конфиг б 1 Ph H A c 98а (83 ) R а 2 Me H A c 94 R 3 Ph H Bz 97а R б 4 H H C b z 91аб (78 ) R (а) Предкатализатор 6 восстанавливали при 23°C перед добавлением субстрата при -45°С (б) Предкатализатор 6 смешивали с субстратом перед добавлением Нг при 23°С (в) Превращение через 3 часа составляло 27 50% 57742 C .R Пример 11 ?•> н (3)-ФАНЕФОС(5) 240мг(0,42ммоль) Бис(2метилаллил)циклоокта-1,5диенрутений(И) 120мг (0,38ммоль) Три фтору ксусная кислота 58мкл Ацетон 6мл (SJ-ФАНЕФОС и бис(2-метилаллил)циклоокта1,5-диенрутений(11) загружали в пробирку Шленка и растворяли в дегазированном ацетоне Добавляли трифторуксусную кислоту и реакционную смесь перемешивали в течение 24 часов Растворитель удаляли, из остатка делали густую суспензию в гексане и фильтровали с получением (S)ФАНЕФОСА-рутений-бис-трифторацетата в виде светло-коричневого твердого вещества (280мг, 74%) Пример 12 CODRLL 28 О О OR' R OR1 Я MeOH/HaOfWi) 50psi=3,45-105н/м2=3,41 атм Субстрат (2,2ммоль) растворяли в МеОН/воде (2мл, 10 1) и раствор дегазировали с помощью трех циклов замораживания/вакуумирования/продувки Раствор загружали в пробирку Фишера-Портера и добавляли рутениевый катализатор В сосуде после трех циклов вакуумирования/продувки водородом создавали давление в 50фунт/дюйм2(3,45-105н/м2=3,41атм) и взбалтывали в течение 24 часов Превращение определялось при помощи 1Н ЯМР и составляло 100% в каждом случае моль% используемого R R1 % иэ Ru катализатора Me Me 94 0,4 Et Me 93 0,4 Me Et 95 0,8 Me tBu 94 0,4 iPr Et 93 0,4 C1CH2 Me 77 0,8 Пример 15 Выделение S-псевдо-ортодибром[2 2]парациклофана З ^ ^ ? с, 4.2 eq. tett-BuU 2 3 eq MgBra-BgO Br Ph2P(O)Ci (Б)ФАНЕФОС(5) 36мг (О.Обммоль] Бис (2-метилаллил) циклоокта-1, 5-диенрутений(11> 18 г (О.Обммоль НВг в МеОН (0.27М) 0,45мл (0,12ммоль) Ацетон Змл (SJ-ФАНЕФОС и бис(2-метилаллил)циклоокта1,5-диенрутений(11) загружали в пробирку Шленка и растворяли в дегазированном ацетоне Добавляли раствор НВг и реакционную смесь перемешивали в течение 30 минут Растворитель удаляли и полученное светло-коричневое вещество сразу же использовали в реакциях гидрогенизации Пример 13 ОН О Н г / 50 psi рац-3 Разделение 1 і РГШРЬ ^ ^ ^ P(O)Phg Разделение дкОензоил-Векниая кислота KSiClj (8)-[2.2]ФАНЕФОС (S}-3 [2.23ФАНЕФОС и бифосфиноаая камера НВг йцетон (З)-ФАНЕФОС-рутений-бистрифторацетат 30мг (О.ОЗммоль НВг в МеОН (0.27М) 0,25мл (0,07ммоль Ацетон Змл (З)-ФАНЕФОС-рутений-бис-трифторацетат загружали в пробирку Шленка и растворяли в дегазированном ацетоне Добавляли раствор НВг и реакционную смесь перемешивали в течение 10 минут Растворитель удаляли и полученное светло-коричневое вещество сразу же использовали в реакциях гидрогенизации Пример 14 К раствору 1,014г (2,77ммоль) рац-3 в 12мл полностью дегазированного в азоте толуола в трубке Шленка было добавлено 1,06г (11ммоль) NaOtBu вслед за добавлением 1,39г (4ммоль) ТІ (PF6), 48мг (0,083ммоль) (S)-[2 2]ФАНЕФОС и 29мг (0,028ммоль) РсігЬаЗ-СНСІз Смесь нагревали до 50°С в течении 10 мин , после чего добавили 0,61мл (5,5ммоль) BnNH2 Реакционную смесь 29 57742 перемешивали при 50°С в течение 10 часов и охлаждали путем добавления 5мл МеОН и 50мл ЕЮАС Сырая реакционная смесь фильтровалась через слой S1O2 для удаления солей ТІ (чрезвычайно ядовито) Фильтрат был обработан стандартным образом, а остаток 3 был отделен S1O2 хроматоргафией в виде белой пудры (0,214г, 42% выход) Определили, что (ее) составляет 93% (R)3 при этом использовали Hewlett Packard жидкосную хромотографию с колонной Chiralcel OD-H 30 Условия отделения ЗООбар ССЬ с модифицированным МеОН градиентом 4мин при 4%, затем выстаивание вплоть до 36% в течении 32мин, поток -1 мл/мин Время выдерживания (R)-3 22,6мин, (S)-3 25,5мин Известна абсолютная конфигурация (R)-3 и (S)-3 посредством корреляции с оксидом бисфосфина [2 2]ФАНЕФОС, в котором абсолютная конфигурация определена рентгеновской кристаллографией, как комплекс дибензоил-О-винной кислотой Таблица 1 Условия реакции a Pd БИНАП б Pd ФАНЕФОС в Pd ФАНЕФОС Юэкв BnNH2 г Pd ФАНЕФОС 2экв Вгд Pd ФАНЕФОС Юэкв NaOtBu е Pd ФАНЕФОС 2экв T1PF6 2ч 0,95 0 62 (34% иэ s=4) 4ч 0,91 0 39 (51% иэ s=3) 6ч 0,86 0 29 (70% иэ s=3) 8ч 0,82 (5%иэ(ее >s=2) 0 23 (74% иэ, s=3) 0 64 (36% иэ s=6) 0 56 (42% иэ s=5) 0 25 (45% иэ s=2) 0 17 (47% иэ, s=2) 0 58 (42% иэ s=6) 0 39 (65% иэ s=4) 0 27 (76% иэ s=3) 0 19 (84% иэ, s=3) 0 48 (41% иэ s=3) 0 32 (64% иэ s=3) 0 23 (74% иэ s=3) 0 12 (83% иэ, s=3) 0 84 (18% иэ, s=42 0, 70 (32% иэ, s=10) 0, 66 (41% иэ, s=13) Все реакции протекали в пробирке Шленка в атмосфере N2 с катализатором, полученным из моль% Pd2dba3-CHCl3 и Змоль% бифосфина, в тщательно дегазированном толуоле при 50°С при концентрации 0,2М Во всех реакциях использовались 2экв BnNH2 (за исключением в) и Зэкв NaOtBu (за исключением д) Дополнительно добавляли к г 2экв СНз(СН2)і7 г\ІтезВг, и 2экв TIPFe к е Реакции оценивали количественно, используя ВЭЖХ интеграцию реакционной смеси, содержащей 1-метилнафталин в качестве внутреннего стандарта Разделение псевдо-ортодибром[2 2]парациклофана (3) Рац-Псевдо-орто-дибром[2 2]парациклофан 3 (400мг) растворяли в этаноле и загружали на колонку (100мм диаметром х 300мм длиной) предварительно набитой триацетатцеллюлозой (поры 15-25мкм) в качестве адсорбента Колонку элюировали этанолом и собирали 150мл фракции Разделение контролировали с помощью суперкритической газовой хроматографии, используя колонку Chiracel OD-(H) Фракции выпаривали с получением энантиометрически чистого R и S псевдо-орто-дибром[2 2]парациклофана 3,180мг и 160мг соответственно Пример 16 PR, Было предпринято получение соединений, аналогичных [2 2]ФАНЕФОСУ таким же образом, что и получение [2 2]ФАНЕФОСА Синтез начинали с оптически чистого псевдо-орто-дибромида с последующим металлированием с помощью BuLi и необязательного металлирования до реактива Гриньяра с помощью МдВгг с последующим гашением с помощью R2POCI Последующее восста 0, 63 (45% из, s=12) новление приводило к получению бифосфинового лиганда Альтернативно, лиганды получали из рацемического псевдо-орто-дибромида, следуя тому же порядку синтеза и производя разделение на стадии фосфиноксида Были получены следующие лиганды R 4-метилфенил 4-метоксифенил 4-фторфенил 3,5-бис-(трифторметил)фенил 3,5-диметилфенил 3,5-диметил-4-метоксифенил циклогексил изопропил Соединения были получены с выходом 7586% Пример 17 Pd/C ВосгО 1 CONH tBu Вое 13 CONH tBu 14 Пиразин-2-третбутилкарбоксамид 13 15,00г(0,084моль Ди-трет-бутилдикарбонат 10% Pd/C 21,9г (0,1 моль) ЕЮН 150мл К раствору 13 в ЕЮН добавляли Pd/C Реакционную смесь гидрогенизировали в качалке Парра при 40°с и 35°С в течение 18 часов Катализатор отфильтровывали и осадок на фильтре промывали 100мл ЕЮН Растворитель меняли на ЕЮАс (~100мл) и при внесении затравки осаждался 14 в виде белых кристаллов (17,3г, 73% выход) 13С ЯМР (CDCI3) 165,1, 155,7, 130,0, 129,8, 81,3, 50,5, 41,5, 40,5, 29,2, 28,3 57742 31 Пример 18 Н BOG неї 8oc CONH tBu 14 15 Через густую суспензию 14 (18,58г, 0,066моль) в 200мл ЕЮАс пропускалиизбыток газообразного HCI при 10-15°С Полученную в результате густую суспензию выдерживали в течение ночи при 20°С и фильтровали Фильтрат промывали ЕЮАс и гексаном и сушили в потоке N2 с получением 1-2НСІ (16,42г. 98%) Пример 19 С..1 2 НСІ Cz-Ou b S ІІ N ' CONH tBu Cbz CONH tBu 15 16 Густую суспензию 15-2HCI(12,09г, 0,047моль) в 160мл ЕЮАс дегазировали в потоке N2 и охлаждали до 5°С Добавляли EtsN (16,5мл, 0,12моль) и М-(бензилоксикарбонилокси)-сукцинимид (12,35г, 0,05моль) и реакционную смесь перемешивали при 22°С в течение ночи Реакционную смесь промывали НгО, 5% лимонной кислотой, 5% МаНСОз и насыщенным раствором соли После высушивания (MgSO4) органическую фазу фильтровали через слой S1O2 и выпаривали Кристаллизация из ЕЮАс/циклогексана 10/90 давала 16(9,39г, 63% выход) Анал Вычислено для C17H23N3O3 С, 64,33, Н, 7,30, N, 13,24 Найдено С, 64,23, Н, 7,31, N, 13,17 Тпл 161-162°С Пример 20 Вое .N. Cbz CONH tSu 16 Сі CH B N O tu C N b z 1 7 К густой суспензии 16 (18,59г, 0,059моль) в 120мл изопропилацетата добавляли ВосгО (20мл, 0,12моль) и диизопропилэтиламин (1мл) При нагревании с обратным холодильником реакционная смесь превращалась в гомогенную и ее нагревали с обратным холодильником в течение 18 часов Реакционную смесь выпаривали и подвергали хроматографии (S1O2, ЕЮАс/гексан 50/50) с получением 17 в виде масла (24/5г, 100%) Кристаллизация из циклогексана/изопропилацетата 10/1 давала 17 в виде белого твердого вещества Анал Вычислено для C22H31N3O5 С, 63,29, Н, 7,48, N, 10,06 Найдено С, 63,30, Н, 7,40, N, 9,94 Тпл 99100°С Пример 21 •OTf 2 НСІ CONH tBu н 32 'N' C N t u B CbzO H MeOH a 17 C z C O HB b C NJu ' 18 Boc-Cbz Тетрагидропиразин-третбутилкарбоамид 17 433мг (1,04ммоль) Родиевый кат 6 20мг (0,02ммоль) Метанол 10мл Катализатор 6 добавляли к дегазированному раствору 17 в метаноле и реакционную смесь гид2 рогенизировали при давлении 40фунт/дюйм 5 2 (2,76-10 н/м , 2,73атм) в течение 18 часов при 40°С в аппарате для гидрогенизации Парра Избыток энантиомера продукта определяли путем суперкритической жидкостной хроматографии 65% иэ 1Н ЯМР 18 находилась в соответствии с литературными данными Пример 22 Вое Pd/C Вое N CONH tBu Cbz 18 сі N H 19 CONH (Ви (S) - СЬг-Вос-Пиперазин-2трет-бутилкарбоксамид 18 1,055г (2,52ммоль) Катализатор Реагітап 0,157г МеОН 16мл К раствору 18 в МеОН добавляли катализатор Реагітап Раствор гидрогенизировали при давлении 40фунт/дюйм2 (2,76-105н/м, 2,73атм) и 22°С ТСХ (ЕЮАс/гекс 50/50) показала завершение реакции Катализатор удаляли путем фильтрования и фильтрат выпаривали Добавляли циклогексан (5мл) и масло растворяли с помощью нагревания При охлаждении осаждался 19 и его отфильтровывали с получением после высушивания 0,7г (99%) 19 в виде белого порошка, [альфа]589-22°С (с=0,2, МеОН), т п л 107°С, 13С ЯМР (CDCI3) 170,1, 154,5, 79,8, 58,7, 50,6, 46,6, 43,6, 43,4, 28,6, 28,3 Пример 23 А Превращение инденоксида в цис-1-амино2-инданол Миллимоли Инденоксид 132 8,33 Ацетонит-рил 41 244 Вода 18 119,4 98 16,6 Конц H2S04 5N КОН 57 15 Dowex 50x4 смолы 1,9мэк/мл 15мл влажн 28,5мэк (Н+) Метанол 17 50мл 50 Материалы Мол вес Граммы или мл 1мл 10мл 2,15мл 0,92мл 3,0мл 33 57742 К одному мл инденоксида (8,33ммоль), растворенному в 10мл ацетонитрила добавляли 0,15мл воды (8,33ммоль) Смесь охлаждали до 05°С на ледяной бане По каплям добавляли концентрированную серную кислоту в то же время поддерживая температуру всей массы на уровне ниже 10°С Когда вся кислота была добавлена, температуре давали повыситься до 20-25°С Прозрачный раствор выдерживали в течение 30 минут К этой смеси добавляли 2мл воды и раствор нагревали в течение 30 минут Когда метилоксазолин полностью превращался в цисаминоинданол, реакционную смесь охлаждали до комнатной температуры Добавляли 5N раствор КОН (Змл, 15ммоль) Это составляет 90% теоретического количества по серной кислоте Раствор оставался кислым по лакмусу Если рН поднимается выше, происходит 2 реацилирование и выход аминоинданола снижается Белое твердое вещество (K2SO4) удаляли путем фильтрования При перемешивании добавляли смолу Dowex 15мл (увлажненную ацетонитрилом) Перемешанную смолу выдерживали в течение 15 минут и отбирали образец для ЖХ (разб х50) Когда пик на ЖХ для аминоинданола исчезал, смолу собирали путем фильтрования, промывали ацетонитрилом и затем метанолом Влажную смолу обрабатывали раствором 50мл IN NH3 в метаноле и густую суспензию перемешивали при комнатной температуре в течение 30 минут Смолу снова собирали фильтрованием и метанол/ЫНз сохраняли Добавляли другую загрузку IN ЫНз/МеОН (20мл) и смолу снова взбалтывали После удаления смолы растворы аминоинданола в метанол/ЫНз объединяли и концентрировали для удаления ІЧНз Анализ конечного раствора в МеОН выявлял 1,0г (81% выход) цис-1-амино-2-инданола, готового для разделяющего винную кислоту средства Б Получение рацемического инденоксида Инден (95%, 122мл) растворяли в метаноле (812мл)и ацетонитриле (348мл) и затем фильтровали Фильтрат разбавляли 0.05М двухосновным фосфатом натрия (116мл), затем доводили до рН 10,5 с помощью 1м водного раствора гидроксида натрия Водный раствор перекиси водорода (35%, 105мл) разбавляли водой (53мл) и добавляли в течение 3 часов, в то же время поддерживая температуру на уровне 25°С, а внутренний рН на 10,5 с помощью 1М водного раствора гидроксида натрия (всего 120мл) Через 6 часов добавляли 1М водный раствор метабисульфита натрия (26мл), в то же время, поддерживая рН выше 8,3 путем добавления 1М водного раствора NaOH (39мл) Добавляли воду (700мл) и смесь экстрагировали с помощью метиленхлорида (580мл и 300мл) Объединенные органические экстракты, содержащие инденоксид (117г) концентрировали до объема в 600мл В Получение (13,2Р)-инденоксида Субстрат, (13,2Р)-инденоксид получали по методу, описанному D J O'Donnell, et al , J Organic Chemistry, 43, 4540 (1978), включенного сюда для этих целей в виде ссылки 34 Г Получение цис-1-амино-2-инданола Инденоксид (117г), разбавленный до общего объема 600мл в метиленхлориде, разбавляли ацетонитрилом (600мл) и охлаждали до -20°С Затем добавляли метансульфоновую кислоту (114мл) Смесь нагревали до 25°С и выдерживали в течение 2 часов Добавляли воду (600мл) и смесь нагревали до 45°С в течение 5 часов Органическую фазу отделяли и водную фазу дополнительно нагревали с обратным холодильником в течение 4 часов с концентрированием до примерно 200г/л Раствор доводили до рН 12,5 с помощью 50% водного раствора гидроксида натрия, затем охлаждали до 5°С и фильтровали, сушили под пониженным давлением с получением цис-1амино-2-инданола Д Получение 13-амино-2Р-инданола (1S, 2Р)-инденоксид (85% иэ) (250г, 0,185моль) растворяли в хлорбензоле (300мл) и гептане (1200мл) и медленно добавляли к раствору метансульфоновой кислоты (250мл, 0,375моль) в ацетонитриле (1250мл) при температуре менее примерно -10°С Реакционную смесь нагревали до 22°С и выдерживали в течение 1,0 часа К смеси добавляли воду и концентрировали путем дистилляции до тех пор, пока внутренняя температура не достигнет 100°С Реакционную смесь нагревали до 100°С в течение 2-3 часов, затем охлаждали до комнатной температуры Добавляли хлорбензол (1000мл), смесь перемешивали, органическую фазу отделяли Оставшуюся водную фазу, содержащую 13-амино,2Р-инданол (85% иэ, 165г, 60%) доводили до рН 12 с помощью 50% водного раствора гидроксида натрия и продукт собирали фильтрованием и сушили под пониженным давлением при 40°С с получением IS-амино, 2Rинданола (85% иэ, 160г) Е Получение 1S-aMHHO-2R-HHflaHona (1S,2R)-HHfleHOKCHfl (85% иэ), (250г, 0,185моль) растворяли в хлорбензоле (300мл) и гептанах (1200мл) и медленно добавляли к раствору дымящей серной кислоты (21% SO3, 184мл) в ацетонитриле (1250мл) при температуре менее примерно -10°С Реакционную смесь нагревали до 22°С и выдерживали в течение 1,0 часа К смеси добавляли воду и концентрировали путем дистилляции до тех пор, пока внутренняя температура не достигала 100°С Реакционную смесь нагревали до 100°С в течение 2-3 часов, затем охлаждали до комнатной температуры Добавляли хлорбензол (1000мл), смесь перемешивали, органическую фазу отделяли Оставшуюся водную фазу, содержащую IS-амино,2R-HHflaHon (85% иэ, 205г, 74%), разбавляли равным объемом ацетонитрила Показатель рН доводили до 12,5 с помощью 50% водного раствора гидроксида натрия и органическую фазу отделяли Оставшуюся водную фазу экстрагировали дополнительным количеством ацетонитрила Объединенный ацетонитрильный экстракт концентрировали под пониженным давлением с получением 1S-aMHHO-2R-HHflaHona (85% иэ, 205г) Альтернативно, оставшуюся водную фазу, содержащую 1S-aMHHO-2R-HHflaHon (85% иэ, 205г, 74%) разбавляли равным объемом бутанола и рН доводили до 12,5 с помощью 50% водного раство 35 3 Отделение цис-1-амино-2-инданола Цис-1-амино-2-инданол (ЮОг) растворяли в метаноле (1500мл) и добавляли раствор L-винной кислоты (110г) в метаноле (1500мл) Смесь нагревали до 60°С и охлаждали до 20°С, фильтровали и сушили под пониженным давлением с получением соли 13-амино,2Р-инданола и L-винной кислоты в виде метанольного сольвата (88г) И Получение 13-Амино-2Р-инданола Метанольного сольвата соли 1S-aMHHO,2Rинданола и L-винной кислоты растворяли в воде (180мл) и нагревали до 55-60°С Раствор осветляли фильтрованием и рН доводили до 12,5 с помощью 50% водного раствора гидроксида натрия Смесь охлаждали до 0-5°С в течение 2 часов, затем выдерживали при этой температуре в течение 1 часа, фильтровали, промывали холодной водой и сушили под пониженным давлением при 40°С с получением 13-амино,2Р-инданола (100% иэ, 99% чистоты, 37г) К Превращение 1,2-инданола в цис-1-амино2-инданол Материалы Мол вес 36 нагревали до кипения в колбе с обратным холодильником на паровой бане длягидролиза оксазолина Когда Ic анализ показывал полный гидролиз, добавляли 1,6мл 5N КОН для нейтрализации серной кислоты Сульфат калия отфильтровывали из раствора Фильтрат исследовали на цис-аминоинданол и он содержал 196мг (66% от теоретического, что также на 75% корректируется по не прореагировавшему исходному материалу) Раствор пропускали через 10мл Dowex 50x4 (Н+) Выход с колонки проверяли на продукт Весь аминоинданол адсорбировался После промывания смолы метанолом продукт элюировали 1М раствором ІЧНз (сухой) Аммонизированный метанол концентрировали для удаления ІЧНз и исследовали конечный раствор аминоинданола, готового для разделения (175мг, или 59% от теоретического, без коррекции на не прореагировавший гликоль) 57742 pa гидроксида натрия и органическую фазу отделяли Органическую фазу промывали хлорбензолом Добавляли L-винную кислоту и воду удаляли путем дистилляции, чтобы выкристаллизовалась соль винной кислоты и аминоинданола Ж Использование бензонитрила Инденоксид (5г) растворяли в бензонитриле (50мл) при 25°С и добавляли серную кислоту (98%, 2,25мл) Смесь разбавляли 5М водным раствором гидроксида натрия (50мл) и экстрагировали метиленхлоридом Органические экстракты концентрировали под пониженным давлением с получением 5,03г оксазолина Граммы или мл 300мг 2,5мл 0,04мл 0,22мл 1,6мл 10мл Миллимоли 2 47,3 2 4 8,0 1, 2-индандиол 150 ацетонитрил 41 вода 18 серная кислота 98 5N КОН 57 Dowex 50х4(Н+) метанолам NH3) 30мл К 300мг индандиола, растворенного в Змл ацетонитрила, содержащего 0,04мл воды по каплям при 0-10°С добавляли концентрированную H2SO4 объемом 0,22мл После завершения добавления ледяную баню удаляли и загрузку нагревали до комнатной температуры После 30минутного выдерживания из прозрачного раствора отбирали образец для Ic исследования (разв х500) Когда весь гликоль израсходовался, раствор дополнительно обрабатывали водой и Л получение инданольных реагентов Соединения (±)-транс-2-бром-3-инданола получали методами S М Sutter et al , J Am Chem Soc, 62, 3473 (1940) и D R Dalton et al , J C S Chem Commun , 591 (1966) Соединения (+)транс-2-бром-1 -инданола и цис- и транс-1,2индандиолы получали методами М Imuta et al , J Org Chem , 43, 4540(1978) M Получение цис-1 -амино-2-инданола из транс-2-бром-1 -инданола Транс-2-бром-1-инданол (Юг, 46,9ммоль, разбавленный в 100мл ацетонитрила, содержащего 0,8мл воды) охлаждали до -5°С и добавляли концентрированную серную кислоту (5,2мл) Смесь выдерживали, в течение 1 часа, затем добавляли 5М водный раствор гидроксида калия, чтобы довести рН до 11 Реакционную смесь фильтровали, удаляя соли - сульфаты калия Водный ацетонитрильный фильтрат доводили до рН менее 2 с помощью серной кислоты и нагревали до 80-100°С, удаляя ацетонитрил путем дистилляции с получением водного раствора цис-аминоинданола Раствор концентрировали до объема 20мл, затем доводили до рН 12,5 с помощью гидроксида калия Выпадали кристаллы продукта, его отфильтровывали и сушили под пониженным давлением с получением цис-1-амино-2-инданола (4,25г) Н Получение цис-1 S-aMHHO-2R-HHflaHona из L4HC-(1S,2R)-HHflaHonn (1г) растворяли в ацетонитриле (10мл), охлаждали до 0°С и добавляли концентрированную серную кислоту (1,0мл) Смесь выдерживали в течение 40 минут с подогревом до 20°С Добавляли воду (0,8мл) и смесь нагревали до температуры кипения с обратным холодильником Добавляли водный 5М раствор гидроксида калия (1,6мл), чтобы довести рН до более 11 и полученное твердое вещество (сульфат калия) удаляли фильтрованием с получением водного раствора цис-13-амино-2К-инданола (0,79г, 66% выход) О Получение цис-1-амино-2-инданола из транс-1,2-индандиола Транс-1,2-индандиол (1,5г) растворяли в ацетонитриле (25мл) охлаждали до 0°С и добавляли концентрированную серную кислоту (1,1мл) 37 57742 Смесь постепенно нагревали до 20°С и выдерживали до 3 часов Добавляли воду (2мл) и смесь нагревали до кипения с обратным холодильником Добавляли водный раствор гидроксида натрия, чтобы довести рН до 12 Получающееся твердое вещество удаляли фильтрованием с получением водного ацетонитриль-ного раствора цис-1-амино2-инданола (1,02г, 63% выход) П Получение цис-1-амино-2-инданола из цис1,2-индандиола Цис-1,2-индандиол (1,0г) растворяли в ацетонитриле (20мл), охлаждали до -40°С и добавляли дымящую серную кислоту (21% SO3, 0,8мл) Смесь выдерживали в течение 1 часа с постепенным нагреванием до 0°С Добавляли воду и смесь нагревали до 80°С в течение 1 часа с получением водного раствора цис-1-амино-2-инданола Пример 24 Получение ацетонида 22 СОСІ NH ОН (-)-цис-1 -аминоиндан-2-ол (20)(99,7% вес 99,9% площади, >99,5% 900г 6,02моль карбоната натрия моногидрат 760г 6,13моль диэтоксиметан (ДЭМ) 56,3л 3-фенилпропионилхлорид (23) 1,05кг 6,23моль метансульфоновая кислота 18,6г 0,19моль 2-метоксипропен (95% по ГХ) 1,28л 13,3моль 5% вод н ы й раствор NaHCO3 10,8л вода 26,2л Густую суспензионную смесь состоящую из (-)цис-1-аминоиндан-2-ола (20,900г, 6,02моль) в 40л ДЭМ и водного раствора карбоната натрия (760г, 6,13моль, Na2CO3-H2O в 6,4л воды), в 100л реакторе с четырьмя входными отверстиями, оборудованном термопарным пробником, механической мешалкой, адаптером для ввода азота и барботером, нагревали до 46-47°С и выдерживали в течение 15 минут Реакционную смесь нагревали до 46-47°С и выдерживали в течение 15 минут, чтобы гарантировать растворение твердых веществ Водная фаза имела рН 11,5 Добавляли чистый 3фенилпропионилхлорид 23 (1/05кг, 6,23моль) в течение 2 часов при температуре от 47°С до 59°С Внутренняя температура повышалась с 47°С до 59°С во время добавления 23, в процессе добавления хлорангидрида кислоты из раствора выкристаллизовывался гидроксиамид 21 После завершения добавления реакционную смесь 38 выдерживали при 59°С в течение 0,5 часа и затем нагревали до 72°С, чтобы обеспечить растворение твердых веществ Температуру повышали до 72°С, чтобы растворить гидроксиамид, так чтобы можно было получить гомогенный образец для исследования путем ВЭЖХ и упростить разделение фракций фаз Развитие (ход) реакции контролировали путем анализа с помощью ВЭЖХ 60 40 ацетонитрил/5,0мМ каждого КН2РО4 и К2НРО4 Примерное время удерживания время удерживания(мин) идентичность 4,1 гидроксиамид 21 6,3 цис-аминоинданол 20 12,5 амид сложного эфира, побочный продукт После завершения добавления хлорангидрида кислоты и выдерживания в течение 0,5 часа при 72°С исследование реакционной смеси с помощью ВЭЖХ показало -0,6% площади 20, -0,2% площади побочного продукта, амида сложного эфира и 98,7% площади гидроксиамида Гидроксиамид 21 не был достаточно отделен при выделении ацетонида 22 Водную фазу отделяли и органическую фазу дважды промывали 4,5л воды Промытую органическую фазу концентрировали и сушили путем атмосферной азеотропной дистилляции Первоначальный объем -40 л концентрировали до 27л Все 16 литров свежего ДЭМ загружали в перегонный аппарат и загруженную партию концентрировали при 88°С-^89°С до 40л Высушенную густую ДЭМ суспензию гидроксиамида 21 обрабатывали 1,28л 2метоксипропена с последующим воздействием 18,6г МСК при 30°С Добавление МСК в отсутствие 2-метоксипропена приводило к образованию сложного эфира амина Эта примесь снова превращается в гидроксиамид 21 во время основной обработки в конце образования ацетонида рН 1,0мл образца, разбавленного 1,0мл воды, как установлено, был равен 2,8-3,0 Полученную смесь выдерживали при 39°С-40°С в течение 3 часов Образование ацетонида контролировали с помощью анализа путем ВЭЖХ, применяя те же самые условия, которые описаны выше в этом примере Примерное время удерживания время удерживания идентичность 4,1 6,9 9,0 12,5 гидроксиамид 21 метиленкеталь, примесь ацетонид 22 амид сложного эфира, побочный продукт Смесь выдерживали при 38-40°С до тех пор пока 21 не составит 99,5% пл по ВЭЖХ) Пример 26 Получение ацетонида 22 (растворитель изопропилацетат) (-)-цис-1-аминоиндан-2ол(20)(98,5вес %) 80г 535ммоль изопропилацетат (ИПАЦ) 1,2л вода 560мл 5N гидроксид натрия 116мл 580ммоль 3-фенилпропионилхлорид (23) 90,8г 539ммоль метансульфоновая кислота (ИСК) 1,1мл 17,0ммоль 2-метоксипропен (95% по 1,24моль ГХ) 119мл 5% вод н ы й раствор NaHCO3 950мл вода 400мл метил циклогексан 2,25л Смесь (-)-цис-1-аминоиндан-2-ола 20 (80г, 535ммоль) в 1,2л ИПАЦ и 560мл воды приводили во взаимодействие с 23 (90,8г, 539ммоль), в то же время рН поддерживали в интервале 8,0-10,5 при 42 41 57742 70-72°С с помощью 5N гидроксида натрия (116мл, 580 м моль) Ход реакции контролировали с помощью анализа ВЭЖХ 60 40 ацетонитрил/5,0мМ каждого из -15 to 20"C КН2РО4 и К2НРО4 Примерное время удерживания [120 96] время удерживания (мин) идентичность 4,1 гидроксиамид 21 6,3 цис-аминоинданол 20 12,5 амид сложного эфира, побочный продукт В конце реакции водную фазу отделяли, а органическую фазу промывали водой (400мл) при 72°С-73°С рН водной фазы и промывной воды составлял 8,1 и 7,9, соответственно Влажную фазу ИПАЦ сушили с помощью атмосферной дистилляции Загружали количество ИПАЦ, равное в Адетонид (22) общем 3,0л, чтобы снизить КФ в загрузке до [321,42](99,1 вес %) 200г 0,617моль 99% Анализ реакционной смеси проводили фильтрованием и промывали холодным метилс помощью ВЭЖХ Примерное время удерживациклогексаном (200мл) Промытый осадок сушили ния побочный продукт гидроксиацетонида под пониженным давлением (26" Нд) при 30°С с =5,3мин, этилбензол =5,6мин , ацетонид 22 получением 151 г ацетонида 22 (87,5%, >99,5% пл =6,6мин , аллилацетонид 23 =11,8мин , эпи-23 по ВЭЖХ) =13,3 мин Через 1 час в реакционной смеси достигалось >99,5% превращение Реакцию останавПример 27 ливали путем добавления раствора лимонной кислоты (35,7г, 0,186моль) в 186мл ТГФ Смесь выдерживали при 15°С в течение ЗОмин после добавления лимонной кислоты Смесь концентрировали при пониженном давлении (примерно 28" Нд) до примерно 30% первоначального объема при поддержании температуры в емкости на уровне 11-15°С и сборе 900мл дистиллята в охлаждаемом сухим льдом сепараторе Затем растворитель заменяли, используя в целом 2,7л изопропилацетата (ИПАЦ) при продолжении дис 43 тилляции при пониженном давлении Замену растворителя прекращали, когда оставалось 99,5% превращения Примерное отношение диастереомеров 24 эпи-24 бис-эпи-24 в неочищенной смеси составляет грубо 94 2 4 в тот момент, когда на этой системе может быть получено разделение компонентов Встряхивание прекращали и слои разделялись К органической фазе добавляли водный раствор сульфита натрия (80г, 0,635моль в 400мл) в течение 10-15мин Температура смеси после добавления сульфита натрия поднимается до 26-29°С Смесь перемешивали в течение 40мин при 25°С Раствор по существу обесцвечивается после промывания сульфитом Слои разделяли, KF органической фазы в этот момент был равен 25г/л Объем органической фазы составлял 1,97л Количественный NCS NaHCO3 Nal 2 3 [133,5] [84,01] [149,9] [126,0] О 24 NaOMs 25 £505 40] (377 49] NaOMe [54,02] d=0,945 25вес % в МеОН 172 г 0,796 моль 3% водный раствор Na2SO4 1,5 л п-РЮН Раствор иодгидрина 24 концентрировали под пониженным давлением (28" Нд) до азеотропно сухой массы Всего собирали 700мл дистиллята при поддержании температуры массы на уровне 22-28°С Дистиллят заменяли 500мл ИПАЦ (KF275 м г/л) Раствор охлаждали до 26°С и за Юмин срок добавляли 25% раствор NaOMe/MeOH (168,1г) После добавления метоксида натрия температура снижалась до 24°С Смесь становилась более темной и на короткий срок образовывалось смолообразное твердое вещество, которое снова растворялось Смесь выдерживали в течение 1 часа при 25°С Анализ реакционной смеси проводили с помощью ВЭЖХ (те же самые условия, что и выше), примерное время удерживания эпоксид эпи25 =6,5мин, эпоксид 25, бис-эпи-25 =7,1 мин, иодгидрин 24 =8,1 мин Анализ ВЭЖХ показал 99% превращение иодгидрина в эпоксид После еще 40мин добавляли метанольного раствора 4,1 г метоксида натрия Через 20мин анализ ВЭЖХ показал 99,5% превращение Реакцию прекращали путем добавления 366мл воды при температуре 25°С, реакционную смесь недолго перемешивали (Юмин) и слои разделяли Впоследствии было обнаружено, что продленное выдерживание реакционной смеси и встряхивание с промывной водой/отстаивание давало существенную обратную реакцию до иодгидрина в этих условиях на экспериментальной установке Эта проблема особенно остра в промывных водах Чтобы устранить эту проблему реакцию проводили при 15°С После того, как достигалось >99% превращения (через 1 час после добавления NaOMe), смесь разбавляли ИПАц (40% объема загрузки) и сначала промывали увеличенным объемом воды (732мл) при 20°С Более низкие температуры и более концентрированные смеси могут приводить к предварительному осаждению 25 при промываниях Сроки перемешивания/отстаивания оставались минимальными (Юмин/ЗОмин, соответственно) Таким образом, обратная реакция могла быть ограничена до < 1 % Неочищенные смеси, содержащие (97 3) эпоксид 25/иодгидрин 24 были доработаны при выделении с получением эпоксидного продукта, содержащего 0,6% иодгидрина Эпоксидный продукт, содержащий этот уровень иодгидрина проводился далее без осложнений Орга 45 Пример 30 Получение предпоследнего соединения 27 BocN NH S-BuNH^l [285 4] 25 46 2(Б)-третбутилкарбоксамид-4-NБок-пиперазин 19 (98,9вес %, 99,6% иэ) 159г 557ммоль эпоксид 25 (97,6вес %, 1,0%эпи-25) 200г 530ммоль метанол 1,06л НСІ (г) 194г 5,32моль 23% NaOH 740мл изопропилацетат 4,0л вода 700мл * скорректировано на вес % чистоты Твердый 2(3)-трет-бутилкарбоксамид-4-третбутоксикарбонил-пиперазин 3 (159г, 557ммоль) и эпоксид 25 (200г, 530моль) добавляли в 2л трехгорлую колбу, оборудованную механической мешалкой, конденсатором для кипячения с обратным холодильником, обогревающим кожухом, покрытой тефлоном термопарой и вводом для азота Добавляли метанол (756мл) и полученную суспензию нагревали до температуры кипения Через 40мин получали гомогенный раствор Внутренняя температура во время кипячения с обратным холодильником была равна 64-65°С Ход реакции контролировали с помощью анализа ВЭЖХ 60 40 ацетонитрил/ЮмМ (КН2РО4/К2НРО4) Примерное время удерживания Время удерживания(мин) идентичность 57742 ническую фазу промывали 3% водным раствором сульфата натрия (2х750мл) Объем органической фазы после промываний составлял 1,98л рН трех промывных вод был равен 10,7, 9,4 и 8,6, соответственно Анализ ВЭЖХ показал 86% общий количественный выход эпоксида 25 в этот момент (скорректирован для 4% соэлюирования бис-эпи25) Раствор эпоксида 25 в ИПАц концентрировали при пониженном давлении (28" Нд) до объема примерно 600мл при поддержании массы загрузки при 15-22°С Растворитель заменяли на п-РЮН путем добавления 750мл п-РЮН при концентрировании под пониженным давлением до объема в сосуде, равного примерно 500мл, поддерживая температуру массы загрузки на уровне 35°С во время концентрирования/замены растворителя может давать нпропиловый простой эфир как побочный продукт разложения, получающийся из эпоксида 25 Анализ состава растворителя с помощью 1Н ЯМР показал
ДивитисяДодаткова інформація
Назва патенту англійськоюChiral biphosphine compounds, complexes based thereon, 4,16-dibrome [2.2]paracyclophanes as intermediary compounds for preparing thereof and a process for preparing chiral biphosphine compounds
Назва патенту російськоюХиральные бифосфиновые соединения, комплексы на их основе, 4,16-дибром[2.2]парациклофаны как промежуточные соединения для их получения и способ получения хиральных бифосфиновых соединений
МПК / Мітки
МПК: C07D 241/24, C07F 9/50, C07D 241/06, C07D 241/04, C07F 9/655, C07F 9/6553, C07C 67/31, C07D 401/06
Мітки: комплекси, одержання, 4,16-дибром[2.2]парациклофани, хіральні, спосіб, сполуки, біфосфінові, основі, хіральних, сполук, проміжні, біфосфінових
Код посилання
<a href="https://ua.patents.su/25-57742-khiralni-bifosfinovi-spoluki-kompleksi-na-kh-osnovi-416-dibrom22paraciklofani-yak-promizhni-spoluki-dlya-kh-oderzhannya-ta-sposib-oderzhannya-khiralnikh-bifosfinovikh-spoluk.html" target="_blank" rel="follow" title="База патентів України">Хіральні біфосфінові сполуки, комплекси на їх основі, 4,16-дибром[2.2]парациклофани як проміжні сполуки для їх одержання та спосіб одержання хіральних біфосфінових сполук</a>
Спосіб одержання бензопіранових сполук, проміжні сполуки, спосіб одержання проміжних сполук
Номер патенту: 41904
Опубліковано: 15.10.2001
Автори: Джонсон Грехем, Новак Ванс, Гін Річард Грехем, Манн Індерджіт Сінг, Сміт Ніл
МПК: C07D 257/00, C07D 405/04
Мітки: одержання, бензопіранових, сполуки, спосіб, проміжні, сполук, проміжних
Формула / Реферат:
1. Способ получения бензопирановых соединений, общей формулы (I)в которой R1 представляет собойА представляет простую связь, Х - кислород, R2 - водород и R3 - водород, отличающийся тем, что осуществляют циклизацию соединения структуры (II)или его соли, гидрата или сольвата, причем в формуле R1, R2, R3, А и Х имеют значения, определенные для структуры (I), и при необходимости получают после этого соль,...
Спосіб одержання гетероциклічних сполук та проміжні сполуки для їх одержання

Номер патенту: 26678
Опубліковано: 12.11.1999
Автори: Норе Пентті Тапіо, Пюстюнен Ярмо Йохан, Хонканен Ерккі Юхані, Луіро Анне Марія, ПІППУРІ Айно Кюллікі, Хайкала Хеймо Олаві, ЛЕННБЕРГ Карі Калєві
МПК: A61P 9/12, C07D 403/12, C07D 413/12, A61K 31/50, A61K 31/502, C07D 417/12, C07D 237/04, C07D 253/00, A61K 31/53, C07D 237/14, A61P 9/04, A61P 9/08, C07D 237/32, A61K 31/535, C07D 273/00, A61K 31/54, C07D 401/12, C07D 285/16
Мітки: спосіб, одержання, гетероциклічних, сполук, проміжні, сполуки
Формула / Реферат:
1. Способ получения гетероциклических соединений формулы (I)в которой Het представляет одну из следующих групп:где R11, R13 и R14 представляют независимо водород или низшую алкильную группу;Z представляет собой S, О или NH;A представляет валентную связь, -CH-CH- или CH2-CH2-группу;R1 и R2 независимо представляют нитро, циано, галогеновую, амино, карбоксамидо, фенильную, бензоильную,...
Похідні еритроміцину, спосіб їх одержання, фармацевтична композиція на їх основі та аміни як проміжні сполуки
Номер патенту: 51618
Опубліковано: 16.12.2002
Автори: Шанто Жан-Франсуа, Гуен Д'Амбрієр Солянж, Дені Алексіс, Агурідас Константін, Ле Мартре Оділь
МПК: A61K 31/7048, C07H 17/08, A61K 31/04
Мітки: основі, похідні, спосіб, композиція, фармацевтична, еритроміцину, одержання, сполуки, проміжні, аміни
Формула / Реферат:
1. Производные эритромицина формулы (I):, (І)в которой R представляет радикал , в котором n представляет число 3, 4 или 5 и Ar представляет гетероциклический радикал, несущий, в случае необходимости, один или несколько заместителей, представляющих собой алкил, алкокси, трифторметокси или галоген, и выбираемый в группе...
Металоорганічні комплекси, що включають гетероциклічні карбени, проміжні сполуки та спосіб їх одержання

Номер патенту: 57142
Опубліковано: 16.06.2003
Автори: Гюрре Олівер, Бюрон Крістоф, Бертранд Гі, Горніц Хейнс, Бураттін Паоло, Штельціг Лутц
МПК: C07C 47/20, C08F 4/44, C07F 3/00, C08F 4/80, C07B 61/00, C07F 15/00, C07F 1/00, C08F 4/04, C07F 7/14, C07C 45/50, C07F 19/00, B01J 31/00, C08F 20/00, B01J 31/16
Мітки: металоорганічні, включають, проміжні, одержання, гетероциклічні, карбени, спосіб, сполуки, комплекси
Формула / Реферат:
1. Металоорганічні комплекси, що включають гетероциклічні карбени, які відрізняються тим, що відповідають загальній формулі (І)[(Z+X-)mMLn]Y (I),у якій:Z+ означає іон 1,2,4-триазолій-5-ілідену, у якому принаймні частина атомів кільця є заміщеною радикалами, що включають вуглеводневі залишки,L означає ліганд, який може бути іонним чи нейтральним,М означає метал, обраний з перехідних елементів груп...
Аналоги 15-дезоксиспергуаліну, спосіб їх одержання, проміжні сполуки, фармацевтична композиція на основі аналогів 15-дезоксипергуаліну

Номер патенту: 42798
Опубліковано: 15.11.2001
Автори: РЕНО Патріс, ЛЕБРЕТОН Люк, ДЮМА Крістін
МПК: C07C 279/14, C07C 277/00, C07C 279/24, C07C 215/00, A61P 33/02, C07C 279/12, A61P 37/06, C07C 271/20, A61K 31/27
Мітки: аналоги, проміжні, композиція, фармацевтична, 15-дезоксипергуаліну, 15-дезоксиспергуаліну, аналогів, основі, одержання, спосіб, сполуки
Формула / Реферат:
1. Аналоги 15-дезоксиспергуалина общей формулы (I)в которой:А обозначает группу -CO-NH- или группу -NH-CO-;R обозначает атом водорода или группу СН3;*С обозначает, если R не является атомом водорода, асимметрический углерод конфигурации (R, S) или конфигурации (R); и их соли присоединения.2. Соединение по п. 1 формулы (I), в которой R обозначает метильную группу и асимметрический углерод *С имеет...
Попередній патент: Висадко-садильна машина
Наступний патент: Основа для трансдермальної терапевтичної системи та трансдермальна терапевтична система
Випадковий патент: Пристрій для прямої сівби трав та піддернинного внесення добрив на луках