Спосіб отримання похідних пірипіропену
Номер патенту: 108612
Опубліковано: 25.05.2015
Автори: Мітомі Масаакі, ФУКУДА Йосімаса, Куріхара Кеніті, Андо Такасі, Гото Кіміхіко, Мінова Нобуто, Наканісі Нозому, Ватанабе Такасі






Формула / Реферат
1. Спосіб отримання сполуки С, представленої формулою С:
Хімічна формула 1
 ,
,
де R представляє прямий ланцюг, розгалужений ланцюг або циклічний С2-6алкілкарбоніл, за умови, що, коли алкільний фрагмент в алкілкарбонільній групі являє собою розгалужений ланцюг або циклічну групу, R представляє С3-6алкілкарбоніл, що включає:
селективне ацилювання гідроксильних груп в 1-положенні і 11-положенні сполуки В1, представленої формулою В1:
Хімічна формула 2
 ,
,
ацилюючим агентом в кількості від 2,0 до 5,0 еквівалентів в розрахунку від сполуки В1 через одну-три стадії в присутності або за відсутності основи в апротонному полярному органічному розчиннику, що вибраний з диметилсульфоксиду, N,N-диметилацетаміду, ацетонітрилу, N-метил-2-піролідинону, N-метил-2-піперазинону, N,N-диметил-2-імідазолідинону.
2. Спосіб за п. 1, де сполуку С отримують за допомогою ацилювання гідроксильних груп в 1-положенні і 11-положенні сполуки В1 через одну стадію.
3. Спосіб за п. 1, який включає в себе отримання сполуки С за допомогою ацилювання через дві стадії, що складаються зі стадій:
ацилювання гідроксильної групи в 11-положенні сполуки В1 ацилюючим агентом з отриманням сполуки В2, представленої формулою В2:
Хімічна формула 3
 ,
,
де R такий, як визначено в формулі С в п. 1; і
додаткового ацилювання гідроксильної групи в 1-положенні сполуки В2.
4. Спосіб за п. 1, який включає в себе отримання сполуки С за допомогою ацилювання через три стадії, що складаються зі стадій:
ацилювання гідроксильної групи в 11-положенні сполуки В1 з отриманням сполуки В2, представленої формулою В2:
Хімічна формула 4
,
де R такий, як визначено в формулі С, в п. 1;
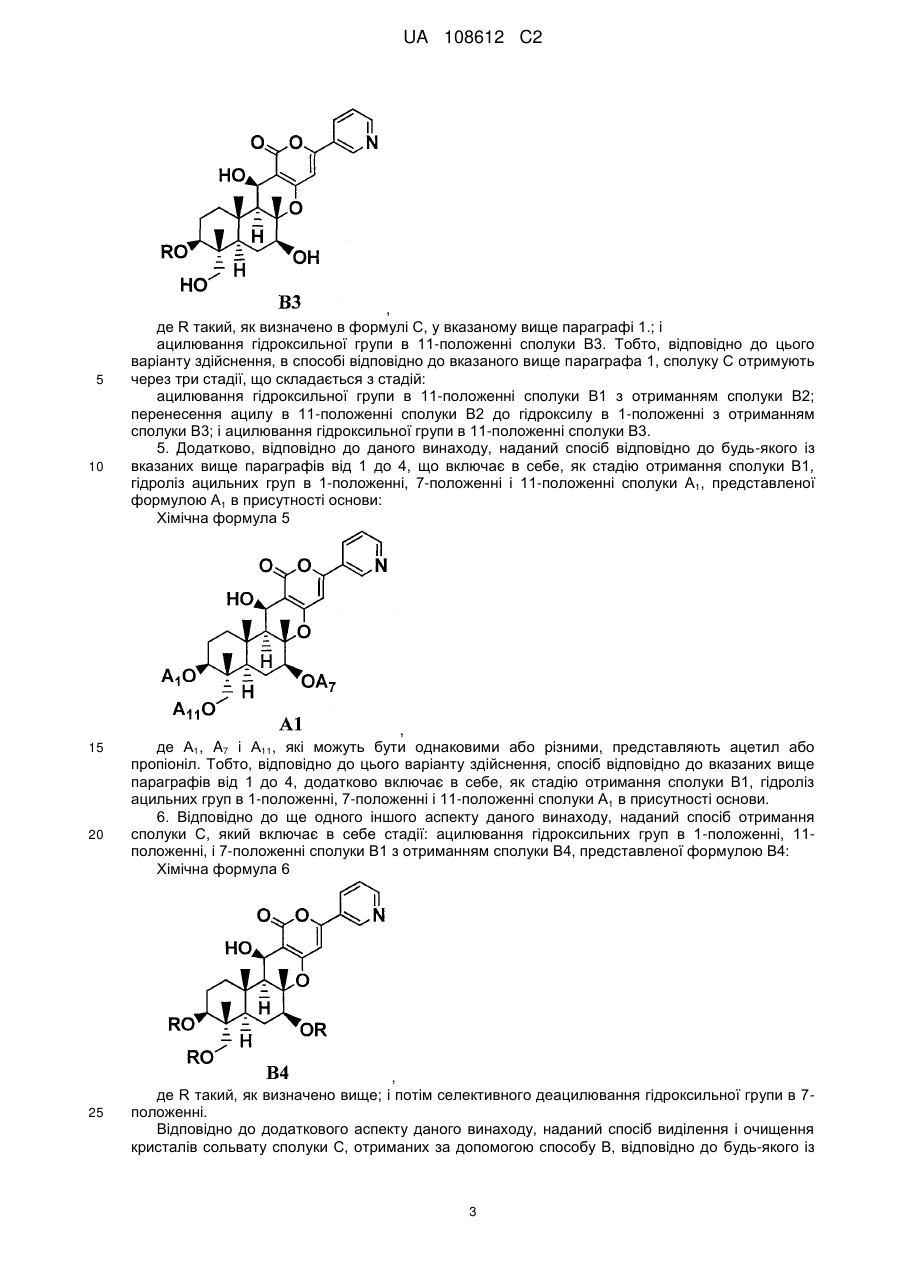
перенесення ацилу в 11-положенні сполуки В2 до гідроксилу в 1-положенні з отриманням сполуки В3, представленої формулою В3:
Хімічна формула 5
 ,
,
де R такий, як визначено в формулі С в п. 1; і
ацилювання гідроксильної групи в 11-положенні сполуки В3.
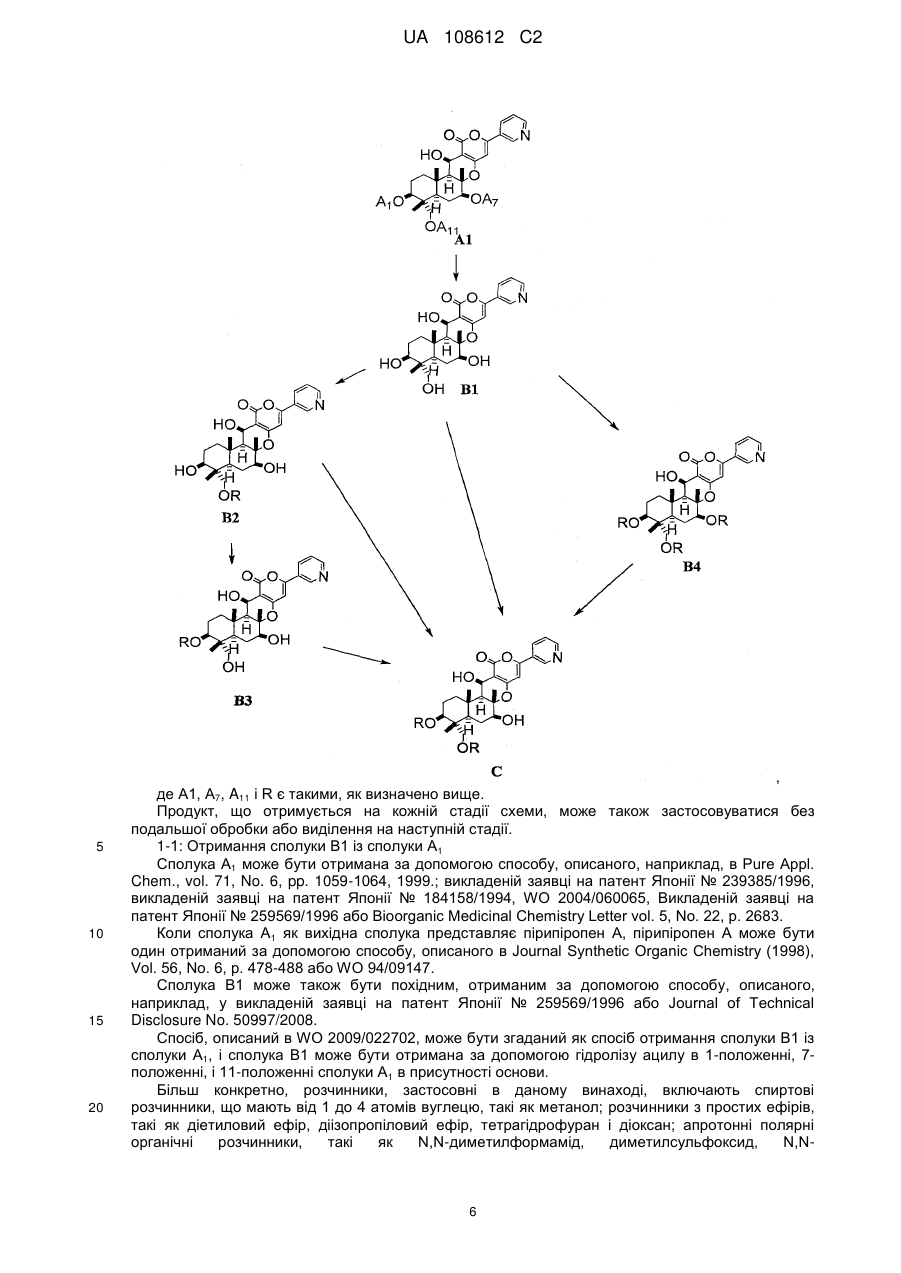
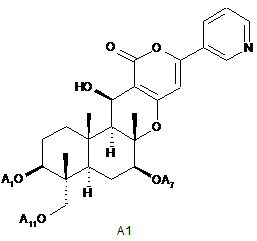
5. Спосіб за п. 1, який додатково включає в себе, як стадію отримання сполуки В1, гідроліз ацильних груп в 1-положенні, 7-положенні і 11-положенні сполуки А1, представленої формулою А1:
Хімічна формула 6
 ,
,
де А1, А7 і А11, які можуть бути однаковими або різними, представляють ацетил або пропіоніл, в присутності основи.
6. Спосіб за п. 1, де ацилювання здійснюють за відсутності основи.
7. Спосіб за п. 1, де основа, що використовується при ацилюванні гідроксилу в 1-положенні і/або 11-положенні сполуки В1, являє собою 2,4,6-колідин або 2,6-лутидин.
8. Спосіб за п. 3, де основу використовують на стадії отримання сполуки В2 і на стадії додаткового ацилювання гідроксилу в 1-положенні сполуки В2, кількість основи, що застосовують на стадії отримання сполуки В2, становить від 1,0 до 3,0 еквівалентів в розрахунку від сполуки В1, загальна кількість основи, що застосовують на стадії отримання сполуки В2, і основи, що застосовують на стадії додаткового ацилювання гідроксилу в 1-положенні сполуки В2, становить від 2,0 до 4,5 еквівалентів.
9. Спосіб за п. 3, де ацилюючий агент використовують на стадії отримання сполуки В2 і на стадії додаткового ацилювання гідроксилу в 1-положенні сполуки В2, кількість ацилюючого агента, що використовують на стадії отримання сполуки В2, становить від 1,0 до 3,5 еквівалентів в розрахунку від сполуки В1, загальна кількість ацилюючого агента, що застосовують на стадії отримання сполуки В2, і ацилюючого агента, що застосовують на стадії додаткового ацилювання гідроксилу в 1-положенні сполуки В2, становить від 2,0 до 4,5 еквівалентів.
10. Спосіб за будь-яким з пп. 1-9, де R представляє циклопропанкарбоніл.
11. Спосіб за п. 4, де стадію отримання сполуки В3 із сполуки В2 здійснюють в присутності основи.
12. Спосіб за будь-яким з пп. 1-10, який додатково включає в себе стадію виділення і очищення сполуки С з розчину реакції, що містить сполуку С, за допомогою кристалізації.
13. Спосіб за будь-яким з пп. 1-12,
що додатково включає стадію:
(a) екстракції реакційного розчину, що містить сполуку С, органічним розчинником, вибраним з групи, яка складається з метилацетату, етилацетату, бутилацетату, толуолу, хлорбензолу, хлороформу, дихлорметану, діетилового ефіру, діізопропілового ефіру, тетрагідрофурану і діоксану, і концентрування екстракту після або без осушування;
(b) упарювання реакційного розчину, що містить сполуку С, досуха з отриманням неочищеного продукту і потім розчинення неочищеного продукту в органічному розчиннику, вибраному з групи, яка складається з метилацетату, етилацетату, бутилацетату, толуолу, хлорбензолу, хлороформу, дихлорметану, діетилового ефіру, діізопропілового ефіру, тетрагідрофурану, діоксану, метанолу і етанолу, при кімнатній температурі або при нагріванні, або
(с) упарювання реакційного розчину, що містить сполуку С, досуха з отриманням неочищеного продукту, розчинення неочищеного продукту в органічному розчиннику, вибраному з групи, яка складається з метилацетату, етилацетату, бутилацетату, толуолу, хлорбензолу, хлороформу, дихлорметану, діетилового ефіру, діізопропілового ефіру, тетрагідрофурану, діоксану, метанолу і етанолу, при кімнатній температурі або принагріванні і додавання слабкого розчинника, вибраного з групи, яка складається з гептану, гексану і циклогексану, до розчину.
14. Застосування сполуки В2, представленої формулою В2:
Хімічна формула 9
,
де R такий, як визначено в формулі С, для отримання сполуки С,
представленої формулою С:
Хімічна формула 8

де R представляє прямий ланцюг, розгалужений ланцюг або циклічний С2-6алкілкарбоніл, за умови, що, коли алкільний фрагмент в алкілкарбонільній групі являє собою розгалужений ланцюг або циклічну групу, R представляє С3-6алкілкарбоніл.
15. Застосування сполуки В3, представленої формулою В3:
Хімічна формула 11
для отримання сполуки С, представленої формулою С:
Хімічна формула 10
де R представляє прямий ланцюг, розгалужений ланцюг або циклічний С2-6алкілкарбоніл, за умови, що, коли алкільний фрагмент в алкілкарбонільній групі являє собою розгалужений ланцюг або циклічну групу, R представляє С3-6алкілкарбоніл.
Текст
Реферат: Розкритий спосіб ефективного отримання похідних пірипіропену, що мають ацилокси в 1положенні і 11-положенні і гідроксил в 7-положенні. Спосіб включає в себе селективне ацилювання гідроксилу в 1-положенні і 11-положенні сполуки В1, представленої формулою В1, через одну-три стадії ацилюючим агентом в присутності або за відсутності основи. UA 108612 C2 (12) UA 108612 C2 UA 108612 C2 5 10 15 20 25 30 35 40 45 Перехресне посилання на споріднену заявку Дана заявка на патент є заявкою, по якій запитується пріоритет по попередніх патентних заявках Японії, заявці на патент Японії № 116305/2009 (дата подачі: 13 травня 2009 р.) і заявці на патент Японії № 44416/2010 (дата подачі: 1 березня 2010 р.). Повні розкриття попередніх заявок включені в даний документ за допомогою посилання. Передумови створення винаходу Галузь техніки, до якої належить винахід Даний винахід стосується способу отримання похідних пірипіропену, застосовних як засоби боротьби з сільськогосподарськими шкідниками, і, більш конкретно, отримання похідних пірипіропену, які мають ацилокси в його 1-положенні і 11-положенні і гідроксил в 7-положенні. Попередній рівень техніки Похідні пірипіропену, що мають ацилокси в його 1-положенні і 11-положенні і гідроксил в 7положенні, являють собою сполуки, які ефективні в боротьбі проти сільськогосподарських шкідників, як описано в WO 2006/129714. WO 2006/129714 і викладена заявка на патент Японії № 259569/1996 розкривають спосіб отримання похідних пірипіропену, що мають ацилокси в його 1-положенні і 11-положенні і гідроксил в 7-положенні. Відповідно до способу отримання, похідні пірипіропену очищають або виділяють з множини продуктів, отриманих за допомогою неселективного гідролізу ацилокси з використанням 1,7,11-триацилоксисполуки як вихідної сполуки. Додатково, викладена заявка на патент Японії № 259569/1996 описує застосування комбінації захисних груп для синтезу похідних пірипіропену. Journal of Antibiotics Vol. 49, No. 11, р. 1149 (1996), Bioorganic Medicinal Chemistry Letter Vol. 5, No. 22, р. 2683 (1995), і викладена заявка на патент Японії № 269065/1996 розкривають приклад синтезу, в якому вводять ацил в 7-положення за допомогою використання захисної групи. WO 2009/022702 розкриває спосіб отримання 1,11-діацил-7-деацетилпірипіропену з 1,7,11тридеацетилпірипіропену з використанням захисної групи. Похідні пірипіропену, що мають ацилокси в 1-положенні і 11-положенні і гідроксил в 7положенні до цього часу отримували через множину стадій з використанням неселективного гідролізу 1,7,11-триацилоксисполуки і з використанням захисної групи. Відповідно, при отриманні похідних пірипіропену в промисловому масштабі, бажаними є додаткове поліпшення ефективності при отриманні, наприклад, через зниження виробничих витрат, поліпшення, виходу, спрощення очищення і виділення або зниження числа стадій. Короткий виклад суті винаходу Автори даного винаходу досягли успіху при отриманні застосовної 1,11-діацилоксисполуки, що розглядається, через прискорений спосіб за допомогою селективного ацилювання, або безпосереднього або постадійного, гідроксилу в 1-положенні і 11-положенні тридеацилсполуки (викладена заявка на патент Японії № 259569/1996 і Journal of Technical Disclosure No. 500997/2008), що легко отримується з пірипіропену (природної речовини) і його аналога (Pure Appl. Chem., vol. 71, No. 6, pp.1059-1064, 1999; WO 94/09147; викладена заявка на патент Японії № 239385/1996, викладена заявка на патент Японії № 259569/1996, Bioorganic Medicinal Chemistry Letter Vol. 5, No. 22, р. 2683 (1995); і WO 2004/060065), що привело до завершення даного винаходу. 1. Відповідно до даного винаходу, наданий спосіб отримання сполуки С, представленої формулою С: Хімічна формула 1 , де R представляє прямий ланцюг, розгалужений ланцюг або циклічний C 2-6алкілкарбоніл, за умови, що, коли алкільний фрагмент в алкілкарбонільній групі являє собою розгалужений ланцюг або циклічну групу, R представляє С3-6алкілкарбоніл, що включає: 1 UA 108612 C2 селективне ацилювання, через одну-три стадії, гідроксильних груп в 1-положенні і 11положенні сполуки B1, представленої формулою B1: Хімічна формула 2 5 10 15 20 25 30 ацилюючим агентом в присутності або за відсутності основи. 2. Відповідно до даного винаходу, наданий спосіб, відповідно до вказаного вище параграфа 1, який відрізняється тим, що сполуку С ацилюють із сполуки B1 через єдину стадію. Тобто відповідно до цього варіанту здійснення, в способі, відповідно до вказаного вище параграфа 1, сполуку С отримують за допомогою ацилювання гідроксильних груп в 1-положенні і 11положенні сполуки B1 через єдину стадію. 3. Відповідно до даного винаходу, наданий спосіб відповідно до вказаного вище параграфа 1, який відрізняється тим, що ацилювання здійснюють двома стадіями, що складаються із стадій: ацилювання гідроксильної групи в 11-положенні сполуки B1 ацилюючим агентом з отриманням сполуки B2, представленої формулою B2: Хімічна формула 3 , де R такий, як визначено в формулі С у вказаному вище параграфі 1; і додаткового ацилювання гідроксильної групи в 1-положенні сполуки B2. Тобто, відповідно до цього варіанту здійснення, в способі відповідно до вказаного вище параграфа 1, сполуку С отримують за допомогою ацилювання через дві стадії, що складаються зі стадій: ацилювання гідроксильної групи в 11-положенні сполуки B1 ацилюючим агентом з отриманням сполуки B2; і додаткового ацилювання гідроксильної групи в 1-положенні сполуки B2. 4. Відповідно до ще одного аспекту даного винаходу, наданий спосіб відповідно до вказаного вище параграфа 1, який відрізняється тим, що ацилювання здійснюють в три стадії, що складаються зі стадій: ацилювання гідроксильної групи в 11-положенні сполуки B1 з отриманням сполуки B2; перенесення ацилу в 11-положенні сполуки B2 до гідроксилу в 1положенні з отриманням сполуки B3, представленої формулою B3: Хімічна формула 4 2 UA 108612 C2 5 10 15 20 25 , де R такий, як визначено в формулі С, у вказаному вище параграфі 1.; і ацилювання гідроксильної групи в 11-положенні сполуки B3. Тобто, відповідно до цього варіанту здійснення, в способі відповідно до вказаного вище параграфа 1, сполуку С отримують через три стадії, що складається з стадій: ацилювання гідроксильної групи в 11-положенні сполуки B1 з отриманням сполуки B2; перенесення ацилу в 11-положенні сполуки B2 до гідроксилу в 1-положенні з отриманням сполуки B3; і ацилювання гідроксильної групи в 11-положенні сполуки B3. 5. Додатково, відповідно до даного винаходу, наданий спосіб відповідно до будь-якого із вказаних вище параграфів від 1 до 4, що включає в себе, як стадію отримання сполуки B1, гідроліз ацильних груп в 1-положенні, 7-положенні і 11-положенні сполуки A1, представленої формулою A1 в присутності основи: Хімічна формула 5 , де A1, A7 і A11, які можуть бути однаковими або різними, представляють ацетил або пропіоніл. Тобто, відповідно до цього варіанту здійснення, спосіб відповідно до вказаних вище параграфів від 1 до 4, додатково включає в себе, як стадію отримання сполуки B1, гідроліз ацильних груп в 1-положенні, 7-положенні і 11-положенні сполуки A1 в присутності основи. 6. Відповідно до ще одного іншого аспекту даного винаходу, наданий спосіб отримання сполуки С, який включає в себе стадії: ацилювання гідроксильних груп в 1-положенні, 11положенні, і 7-положенні сполуки B1 з отриманням сполуки B4, представленої формулою B4: Хімічна формула 6 , де R такий, як визначено вище; і потім селективного деацилювання гідроксильної групи в 7положенні. Відповідно до додаткового аспекту даного винаходу, наданий спосіб виділення і очищення кристалів сольвату сполуки С, отриманих за допомогою способу В, відповідно до будь-якого із 3 UA 108612 C2 5 10 15 20 25 30 35 40 45 50 55 вказаних вище параграфів від 1 до 5, що включає в себе: додавання прийнятного розчинника до неочищеного продукту сполуки С, отриманого за допомогою концентрування реакційного розчину, що містить сполуку С, отриманого за допомогою будь-якого із вказаних вище параграфів від 1 до 5, при зниженому тиску; концентрування етилацетатного екстракту реакційного розчину, що містить сполуку С, отриману за допомогою способу відповідно до будьякого із вказаних вище параграфів від 1 до 5; або додаткове додавання вибраного розчинника до концентрату для осадження кристалів сольвату сполуки С. Відповідно до ще одного аспекту даного винаходу, наданий спосіб виділення і очищення кристалів сольвату сполуки С, що включає в себе стадії: (a) екстракції реакційного розчину, що містить сполуку С, органічним розчинником, вибраним з групи, яка складається з метилацетату, етилацетату, бутилацетату, толуолу, хлорбензолу, хлороформу, дихлорметану, діетилового ефіру, діізопропілового ефіру, тетрагідрофурану і діоксану, і концентрування екстракту після або без сушіння; (b) упарювання реакційного розчину, що містить сполуку С, досуха з отриманням неочищеного продукту і потім розчинення неочищеного продукту в органічному розчиннику, вибраному з групи, яка складається з метилацетату, етилацетату, бутилацетату, толуолу, хлорбензолу, хлороформу, дихлорметану, діетилового ефіру, діізопропілового ефіру, тетрагідрофурану, діоксану, метанолу і етанолу при кімнатній температурі або при нагріванні; або (с) упарювання реакційного розчину, що містить сполуку С, досуха з отриманням неочищеного продукту, розчинення неочищеного продукту в органічному розчиннику, вибраному з групи, яка складається з метилацетату, етилацетату, бутилацетату, толуолу, хлорбензолу, хлороформу, дихлорметану, діетилового ефіру, діізопропілового ефіру, тетрагідрофурану, діоксану, метанолу і етанолу при кімнатній температурі або при нагріванні, і додавання слабкого розчинника, вибраного з групи, яка складається з гептану, гексану і циклогексану, до розчину. У переважному варіанті здійснення даного винаходу, вказана стадія (a) повинна бути стадією (a') екстракції реакційного розчину, що містить сполуку С, етилацетатом, і концентрування екстракту після або без осушування. У ще одному переважному варіанті здійснення даного винаходу, вказана стадія (b) повинна бути стадією (b') упарювання реакційного розчину, що містить сполуку С, досуха з отриманням неочищеного продукту і потім розчинення неочищеного продукту в етилацетаті при кімнатній температурі або при нагріванні. У ще одному переважному варіанті здійснення даного винаходу, вказана стадія (с) повинна бути стадією (c') упарювання реакційного розчину, що містить сполуку С, досуха з отриманням неочищеного продукту, розчинення неочищеного продукту в етилацетаті при кімнатній температурі або при нагріванні, і додавання гексану до розчину. Відповідно до ще одного іншого аспекту даного винаходу, наданий спосіб отримання сполуки С із сполуки B1 відповідно до будь-якого із вказаних вище параграфів від 1 до 5, який включає в себе стадію виділення і очищення сполуки за допомогою кристалізації з реакційного розчину, що містить сполуку C. Тобто відповідно до цього варіанту здійснення, спосіб відповідно до будь-якого із вказаних вище параграфів від 1 до 5 додатково включає в себе стадію виділення і очищення сполуки С за допомогою кристалізації з реакційного розчину, що містить сполуку C. Відповідно до даного винаходу, похідні пірипіропену, які мають ацилокси в 1положенні і 11-положенні і гідроксил в 7-положенні, і є застосовними як засоби знищення сільськогосподарських комах-шкідників, можуть бути ефективно отримані через короткий спосіб. Короткий опис креслень Фіг. 1 являє собою порошкову рентнгенограму, виміряну для кристалів етилацетатного сольвату 1,11-ди-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А. Докладний опис винаходу Спосіб отримання Термін "алкіл", як використовується в цьому документі, як замісник або частина замісника означає алкіл, тобто з прямим ланцюгом, розгалуженим ланцюгом або циклічною групою або типу їх комбінації, якщо не указано інакше. Символ "Ca-b", приєднаний до замісника, як використовується в даному документі, означає, що число атомів вуглецю, які містяться в заміснику, як використовується в цьому документі, дорівнює від а до b. Додатково, "Ca-b" в "Ca-bалкілкарбоніл" означає, що число атомів вуглецю в алкільному фрагменті, виключаючи атом вуглецю в карбонільному фрагменті дорівнює від а до b. Конкретними прикладами прямоланцюгового, з розгалуженим ланцюгом або циклічним C 26алкілкарбонілу, представленим R, за умови, що, коли алкільний фрагмент в алкілкарбонільній групі являє собою розгалужений ланцюг або циклічну групу, R представляє C 3-6алкілкарбоніл, включають циклопропанкарбоніл і пропіоніл. 4 UA 108612 C2 5 10 15 20 25 30 35 40 45 Відповідно до ще одного переважного варіанту здійснення даного винаходу, в способі відповідно до будь-якого із вказаних вище параграфів від 1 до 5, ацилювання здійснюють за відсутності основи. Відповідно до переважного варіанту здійснення даного винаходу, в способі відповідно до будь-якого із вказаних вище параграфів від 1 до 5, основа, що використовується при ацилюванні гідроксилу в 1-положенні і 11-положенні сполуки B1, являє собою 2,4,6-колідин або 2,6-лутидин. Відповідно до ще одного переважного варіанту здійснення даного винаходу, в способі відповідно до вказаного вище параграфа 2, ацилюючий агент використовують в кількості від 2,0 до 5,0 еквівалентів в розрахунку від сполуки B1. Відповідно до додаткового переважного варіанту здійснення даного винаходу, спосіб відповідно до вказаного вище параграфа 3, відрізняється тим, що розчинник, який застосовується на стадії отримання сполуки B2, відрізняється від розчинника, що застосовується на стадії додаткового ацилювання гідроксилу в 1-положенні сполуки B2. Відповідно до ще одного переважного варіанту здійснення даного винаходу, спосіб, відповідно до вказаного вище параграфа 4, відрізняється тим, що стадію отримання сполуки B3 із сполуки B2 здійснюють в присутності основи. Відповідно до ще одного переважного варіанту здійснення даного винаходу, спосіб відповідно до вказаного вище параграфа 4, відрізняється тим, що стадію отримання сполуки B3 із сполуки B2 здійснюють в присутності 1,8-діазабіцикло[5.4.0]ундека-7-ену (DBU) як основи. Відповідно до додаткового переважного варіанту здійснення даного винаходу, C 26алкілкарбоніл, представлений R, є циклічним C3-6алкілкарбонілом, більш переважно, циклопропанкарбонілом. Відповідно до переважного варіанту здійснення даного винаходу, в способі відповідно до вказаного вище параграфа 3, основу застосовують на стадії отримання сполуки B2 і на стадії додаткового ацилювання гідроксилу в 1-положенні сполуки B2, кількість основи отримання сполуки, що застосовується на стадії B2, складає від 1,0 до 3,0 еквівалентів, в розрахунку від сполуки B1, загальна кількість основи, що застосовується на стадії отримання сполуки B2 і основи, що застосовується на стадії додаткового ацилювання гідроксилу в 1-положенні сполуки B2, складає від 2,0 до 4,5 еквівалентів, більш переважно від 2,0 до 3,0 еквівалентів. Відповідно до переважного варіанту здійснення даного винаходу, в способі, відповідно до будь-якого із вказаних вище параграфів від 1 до 4, ацилюючий агент використовують в кількості від 2,0 до 5,0 еквівалентів в розрахунку від сполуки B1. Відповідно до переважного варіанту здійснення даного винаходу, в способі, відповідно до вказаного вище параграфа 3, ацилюючий агент використовують на стадії отримання сполуки B2 і на стадії додаткового ацилювання гідроксилу в 1-положенні сполуки B2, кількість ацилюючого агента, що використовується на стадії отримання сполуки B2, складає від 1,0 до 3,5 еквівалентів в розрахунку від сполуки B1, загальна кількість ацилюючого агента, що застосовується на стадії отримання сполуки B2 і ацилюючого агента, що застосовується на стадії додаткового ацилювання гідроксилу в 1-положенні сполуки B2, складає від 2,0 до 4,5 еквівалентів. Відповідно до ще одного переважного варіанту здійснення даного винаходу, надане застосування сполуки B2 як проміжної сполуки при отриманні сполуки С із сполуки B1. Тобто у варіанті здійснення надане застосування сполуки B2 при отриманні сполуки С. Відповідно до ще одного переважного варіанту здійснення даного винаходу, надане застосування сполуки B2 і сполуки B3 як проміжної сполуки при отриманні сполуки С із сполуки B1. Тобто в цьому варіанті здійснення надане застосування сполуки B3 при отриманні сполуки С. Даний винахід буде описаний більш детально відповідно до наступної схеми. Хімічна формула 7 5 UA 108612 C2 5 10 15 20 , де А1, A7, A11 і R є такими, як визначено вище. Продукт, що отримується на кожній стадії схеми, може також застосовуватися без подальшої обробки або виділення на наступній стадії. 1-1: Отримання сполуки B1 із сполуки A1 Сполука A1 може бути отримана за допомогою способу, описаного, наприклад, в Pure Appl. Chem., vol. 71, No. 6, pp. 1059-1064, 1999.; викладеній заявці на патент Японії № 239385/1996, викладеній заявці на патент Японії № 184158/1994, WO 2004/060065, Викладеній заявці на патент Японії № 259569/1996 або Bioorganic Medicinal Chemistry Letter vol. 5, No. 22, р. 2683. Коли сполука A1 як вихідна сполука представляє пірипіропен А, пірипіропен А може бути один отриманий за допомогою способу, описаного в Journal Synthetic Organic Chemistry (1998), Vol. 56, No. 6, р. 478-488 або WO 94/09147. Сполука B1 може також бути похідним, отриманим за допомогою способу, описаного, наприклад, у викладеній заявці на патент Японії № 259569/1996 або Journal of Technical Disclosure No. 50997/2008. Спосіб, описаний в WO 2009/022702, може бути згаданий як спосіб отримання сполуки B1 із сполуки A1, і сполука B1 може бути отримана за допомогою гідролізу ацилу в 1-положенні, 7положенні, і 11-положенні сполуки A1 в присутності основи. Більш конкретно, розчинники, застосовні в даному винаході, включають спиртові розчинники, що мають від 1 до 4 атомів вуглецю, такі як метанол; розчинники з простих ефірів, такі як діетиловий ефір, діізопропіловий ефір, тетрагідрофуран і діоксан; апротонні полярні органічні розчинники, такі як N,N-диметилформамід, диметилсульфоксид, N,N 6 UA 108612 C2 5 10 15 20 25 30 35 40 45 50 55 60 диметилацетамід, і ацетонітрил; галогеновані розчинники, такі як дихлорметан і хлороформ; або вода; і змішані розчинники, складені з двох або більше з цих розчинників. Основи, застосовні в даному винаході, включають неорганічні основи, такі як карбонат натрію, карбонат калію, гідрокарбонат натрію, гідрокарбонат калію, гідроксид натрію, гідроксид калію, гідрид натрію, гідрид калію, ціанід натрію, ціанід калію, гідроксид магнію, гідроксид кальцію, гідроксид літію і гідроксид барію; алкоксиди лужних металів, такі як метоксид натрію, етоксид натрію і трет-бутоксид калію; алкоксиди лужноземельних металів; і органічні основи, такі як 1,8-діазабіцикло[5.4.0]ундека-7-ен, 1,5-діазабіцикло[4.3.0]нона-5-ен, триетиламін, діізопропілетиламін, піридин, гідразин і гуанідин. Переважними є 1,8-діазабіцикло[5.4.0]ундека7-ен, 1,5-діазабіцикло[4.3.0]нона-5-ен, карбонат натрію, карбонат калію, гідрокарбонат натрію, гідрокарбонат калію, гідроксид натрію і гідроксид калію. Кількість застосовуваної основи складає переважно від 0,01 до 4,5 еквівалентів в розрахунку від кількості сполуки A1. Температура реакції складає переважно від -20°С до 50°С. Час реакції складає переважно від 0,5 години до 72 годин. 2-1: Отримання сполуки С із сполуки B1 (1) Стадія отримання сполуки С безпосередньо із сполуки B1 Розчинники, застосовні в способі отримання сполуки С із сполуки B1 у вказаному вище параграфі 2, включають розчинники з простих ефірів, такі як діетиловий ефір, діізопропіловий ефір, тетрагідрофуран і діоксан; апротонні полярні органічні розчинники, такі як N,Nдиметилформамід, диметилсульфоксид, N,N-диметилацетамід, ацетонітрил, N-метил-2піролідинон, N-метил-2-піперазинон і N,N-диметил-2-імідазолідинон; галогеновані розчинники, такі як дихлорметан і хлороформ; або ароматичні вуглеводневі розчинники, такі як толуол; і змішані розчинники, складені з двох або більше з цих розчинників. Переважними є апротонні полярні органічні розчинники. Більш переважними є N-метил-2-піролідинон і N,N-диметил-2імідазолідинон. Особливо переважним є N-метил-2 піролідинон. Спосіб відповідно до вказаного вище параграфа 2., переважно здійснюють за відсутності основи. Однак, коли спосіб здійснюють в присутності основи, приклади застосовних основ включають неорганічні основи, такі як карбонат натрію, карбонат калію, гідрокарбонат натрію, гідрокарбонат калію, гідроксид натрію, гідроксид калію, гідрид натрію, гідрид калію, ціанід натрію, ціанід калію, гідроксид магнію, гідроксид кальцію, гідроксид літію, і гідроксид барію; і органічні основи, такі як 1,8-діазабіцикло[5.4.0]ундека-7-ен, 1,5-діазабіцикло[4.3.0]нона-5-ен, триетиламін, діізопропілетиламін, піридин, гуанідин, лутидин, колідин, 2,2’-біпіридил, трифеніламін, хінолін, N,N-диметиланілін, і N,N-діетиланілін. Переважними є піридин, 2,6лутидин, 2,4,6-колідин, 2,2'-біпіридил, трифеніламін, N,N-диметиланілін, N,N-діетиланілін і т. п. Більш переважними є 2,6-лутидин, 2,4,6-колідин, трифеніламін, N,N-диметиланілін, і N,Nдіетиланілін. Особливо переважними є 2,6-лутидин і 2,4,6-колідин. Коли застосовують основу, кількість основи складає переважно від 2,0 до 4,5 еквівалентів, більш переважно від 2,0 до 3,0 еквівалентів, в розрахунку від кількості сполуки B1. Група R може бути введена в 1-положення і 11-положення з використанням ROH, RCl, (R)2O або змішаного кислотного ангідриду, переважно, RCl або (R) 2О, як ацилюючого агент, що відповідає R, що розглядається. Реакція може здійснюватися в присутності або за відсутності основи або з використанням конденсуючого агента, такого як дициклогексилкарбодіімід, гідрохлорид l-етил-3-(3-диметиламінопропіл)карбодііміду, карбонілдіімідазол, дипіридилдисульфід, діімідазолілдисульфід, 1,3,5-трихлорбензоїлхлорид, 1,3,5-трихлорбензоїл ангідрид, PyBop або PyBrop. Переважно, реакцію здійснюють з використанням RCl або (R) 2О в присутності або за відсутності основи. Більш переважні ацилюючі агенти включають циклопропанкарбонілхлорид, бутирилхлорид, і ангідрид циклопропанкарбонової кислоти. Кількість застосовуваного ацилюючого агента складає переважно від 2,0 до 5,0 еквівалентів, більш переважно від 2,2 до 4,5 еквівалентів, в розрахунку від кількості сполуки B1. Цю кількість застосовують відразу або двома-п'ятьма розділеними частинами. Температура реакції складає переважно від -20°С до 50°С, більш переважно від -10°С до 50°С, ще більш переважно від -10°С до кімнатної температури. Час реакції складає переважно від 0,1 години до 7 днів, більш переважно від 3 годин до 4 днів. Відповідно до цього способу, сполука С може бути отримана із сполуки B1 через єдину стадію при виході не менше ніж 40%. (2) Стадія отримання сполуки B2 із сполуки B1 Розчинники, застосовні в способі отримання сполуки B2 із сполуки B1 у вказаному вище параграфі 3, або 4, включають розчинники з простих ефірів, такі як діетиловий ефір, діізопропіловий ефір, тетрагідрофуран, і діоксан; апротонні полярні органічні розчинники, такі як 7 UA 108612 C2 5 10 15 20 25 30 35 40 45 50 55 N,N-диметилформамід, диметилсульфоксид, N,N-диметилацетамід, ацетонітрил, N-метил-2піролідинон, N-метил-2-піперазинон і N,N-диметил-2-імідазолідинон; галогеновані розчинники, такі як дихлорметан і хлороформ; або ароматичні вуглеводневі розчинники, такі як толуол; і змішані розчинники, складені з двох або більше з цих розчинників. Переважними є апротонні полярні органічні розчинники. Особливо переважним є N-метил-2-піролідинон. Реакція може здійснюватися без застосування основи. Однак коли застосовують основу, приклади застосовних основ включають неорганічні основи, такі як карбонат натрію, карбонат калію, гідрокарбонат натрію, гідрокарбонат калію, гідроксид натрію, гідроксид калію, гідрид натрію, гідрид калію, ціанід натрію, ціанід калію, гідроксид магнію, гідроксид кальцію, гідроксид літію і гідроксид барію; і органічні основи, такі як 1,8-діазабіцикло[5.4.0]ундека-7-ен, 1,5діазабіцикло[4.3.0]нона-5-ен, триетиламін, діізопропілетиламін, піридин, гуанідин, лутидин, колідин, 2,2'-біпіридил, трифеніламін, хінолін, N,N-диметиланілін і N,N-діетиланілін. Переважними є піридин, 2,6-лутидин, 2,4,6-колідин, 2,2’-біпіридил, трифеніламін, N,Nдиметиланілін, N,N-діетиланілін і т. п. Більш переважними є триетиламін, 2,6-лутидин, 2,4,6колідин, трифеніламін, N,N-диметиланілін і N,N-діетиланілін. Особливо переважними є триетиламін і 2,6-лутидин. ROH, RCl, (R)2О або змішаний кислотний ангідрид, переважно, RCl або (R) 2O, застосовують як ацилюючий агент для введення як групи R. Реакція може здійснюватися в присутності або за відсутності основи або з використанням конденсуючого агента, такого як дициклогексилкарбодіімід, гідрохлорид l-етил-3-(3-диметиламінопропіл)карбодііміду, карбонілдіімідазол, дипіридилдисульфід, діімідазолілдисульфід, 1,3,5-трихлорбензоїлхлорид, 1,3,5-трихлорбензоїлангідрид, PyBop або PyBrop. Переважно, реакцію здійснюють з використанням RCl або (R)2O в присутності або за відсутності основи. Більш переважні ацилюючі агенти включають циклопропанкарбонілхлорид і ангідрид циклопропанкарбонової кислоти. Кількість застосовуваного ацилюючого агента складає переважно від 1,0 до 3,5 еквівалентів, більш переважно від 1,1 до 3,0 еквівалентів, в розрахунку від кількості сполуки B1. Коли застосовують основу, кількість основи складає переважно від 1,0 до 3,0 еквівалентів, більш переважно від 1,1 до 2,5 еквівалентів, в розрахунку від кількості сполуки B1. Температура реакції складає переважно від -20°С до 50°С, більш переважно від -10°С до 50°С. Час реакції складає переважно від 0,1 годину до 7 днів, більш переважно від 45 хв до 48 годин. (3) Стадія отримання сполуки С із сполуки B2 Розчинники, застосовні в способі отримання сполуки С із сполуки B2 у вказаному вище параграфі 3, включають розчинники з простих ефірів, такі як діетиловий ефір, діізопропіловий ефір, тетрагідрофуран і діоксан; апротонні полярні органічні розчинники, такі як N,Nдиметилформамід, диметилсульфоксид, N,N-диметилацетамід, ацетонітрил, N-метил-2піролідинон, N-метил-2-піперазинон і N,N-диметил-2-імідазолідинон; галогеновані розчинники, такі як дихлорметан і хлороформ; або ароматичні вуглеводневі розчинники, такі як толуол; і змішані розчинники, складені з двох або більше з цих розчинників. Переважними є апротонні полярні органічні розчинники. Особливо переважним є N-метил-2-піролідинон. Реакція може здійснюватися без застосування основи. Однак коли застосовують основу, приклади застосовних основ включають неорганічні основи, такі як карбонат натрію, карбонат калію, гідрокарбонат натрію, гідрокарбонат калію, гідроксид натрію, гідроксид калію, гідрид натрію, гідрид калію, ціанід натрію, ціанід калію, гідроксид магнію, гідроксид кальцію, гідроксид літію, і гідроксид барію; і органічні основи, такі як 1,8-діазабіцикло[5.4.0]ундека-7-ен, 1,5діазабіцикло[4.3.0]нона-5-ен, триетиламін, діізопропілетиламін, піридин, гуанідин, лутидин, колідин, 2,2’-біпіридил, трифеніламін, хінолін, N,N-диметиланілін, і N,N-діетиланілін. Переважними є піридин, 2,6-лутидин, 2,4,6-колідин, 2,2’-біпіридил, трифеніламін, N,Nдиметиланілін, N,N-діетиланілін і т. п. Більш переважними є триетиламін, 2,6-лутидин, 2,4,6колідин, трифеніламін, N,N-диметиланілін і N,N-діетиланілін. Особливо переважними є триетиламін і 2,6-лутидин. ROH, RCl, (R)2O або змішаний кислотний ангідрид, переважно RCl або (R) 2O, застосовують як ацилюючий агент для введення як групи R. Реакція може здійснюватися в присутності або за відсутності основи або з використанням конденсуючого агента, такого як дициклогексилкарбодіімід, гідрохлорид l-етил-3-(3-диметиламінопропіл)карбодііміду, карбонілдіімідазол, дипіридилдисульфід, діімідазолілдисульфід, 1,3,5-трихлорбензоїлхлорид, 1,3,5-трихлорбензоїл ангідрид, PyBop або PyBrop. Переважно, реакцію здійснюють з використанням RCl або (R)2O в присутності або за відсутності основи. 8 UA 108612 C2 5 10 15 20 25 30 35 40 45 50 55 60 Більш переважні ацилюючі агенти включають циклопропанкарбонілхлорид і ангідрид циклопропанкарбонової кислоти. Коли застосовують основу, кількість основи складає переважно від 0,1 до 5,0 еквівалентів, більш переважно від 0,1 до 3,0 еквівалентів в розрахунку від кількості сполуки B2. У більш переважному варіанті здійснення, загальна кількість основи, що використовується на цій стадії і на стадії, описаній у вказаному вище параграфі (2) складає від 2,0 до 4,5 еквівалентів, більш переважно від 2,0 до 3,0 еквівалентів. Кількість використовуваного ацилюючого агента складає переважно від 1,0 до 3,0 еквівалентів в розрахунку від кількості сполуки B1, більш переважно від 2,0 до 4,5 еквівалентів з урахуванням загальної кількості ацилюючого агента, що використовується на цій стадії і на стадії, описаній у вказаному вище параграфі (2). Температура реакції складає переважно від -20°С до 60°С. Час реакції складає переважно від 0,1 години до 7 днів. Ця стадія може також здійснюватися безперервно без відбору продукту, отриманого на стадії, описаній у вказаному вище параграфі (2). (4) Стадія отримання сполуки B3 із сполуки B2 Розчинники, застосовні в способі отримання сполуки B3 із сполуки B2 у вказаному вище параграфі 4, включають розчинники з простих ефірів, такі як діетиловий ефір, діізопропіловий ефір, тетрагідрофуран і діоксан; апротонні полярні органічні розчинники, такі як N,Nдиметилформамід, диметилсульфоксид, N,N-диметилацетамід, ацетонітрил, N-метил-2піролідинон, N-метил-2-піперазинон і N,N-диметил-2-імідазолідинон; галогеновані розчинники, такі, як дихлорметан і хлороформ; або ароматичні вуглеводневі розчинники, такі як толуол, хлорбензол і дихлорбензол; і змішані розчинники, складені з двох або більше з цих розчинників. Переважними є апротонні полярні органічні розчинники. Основи, застосовні в даному винаході, включають неорганічні основи, такі як карбонат натрію, карбонат калію, гідрокарбонат натрію, гідрокарбонат калію, карбонат цезію, гідроксид натрію, гідроксид калію, гідрид натрію, гідрид калію, ціанід натрію, ціанід калію, гідроксид магнію, гідроксид кальцію, гідроксид літію, гідроксид барію, і трет-бутоксид калію; і органічні основи, такі як 1,8-діазабіцикло[5.4.0]ундека-7-ен, 1,5-діазабіцикло[4.3.0]нона-5-ен, триетиламін, діізопропілетиламін, піридин, гуанідин, лутидин, колідин, хінолін, N,N-диметиланілін, N,Nдіетиланілін, фосфазен. Переважними є карбонат калію, карбонат цезію, трет-бутоксид калію, 1,8-діазабіцикло[5.4.0]ундека-7-ен і 1,5-діазабіцикло[4.3.0]нона-5-ен і т. п. Більш переважним є 1,8-діазабіцикло[5.4.0]ундека-7-ен і 1,5-діазабіцикло[4.3.0]нона-5-ен. Кількість застосовуваної основи складає переважно від 0,1 до 3,0 еквівалентів, більш переважно від 0,1 до 2,0 еквівалентів, в розрахунку від кількості сполуки B2. Температура реакції складає переважно від 0°С до 150°С. Час реакції складає переважно від 0,1 годину до 7 днів. (5) Стадія отримання сполуки С із сполуки B3 Розчинники, застосовні в способі отримання сполуки С із сполуки B3 у вказаному вище параграфі 4, включають розчинники з простих ефірів, такі як діетиловий ефір, діізопропіловий ефір, тетрагідрофуран і діоксан; апротонні полярні органічні розчинники, такі як N,Nдиметилформамід, диметилсульфоксид, N,N-диметилацетамід, ацетонітрил, N-метил-2піролідинон, N-метил-2-піперазинон і N,N-диметил-2-імідазолідинон; галогеновані розчинники, такі як дихлорметан і хлороформ; або ароматичні вуглеводневі розчинники, такі як толуол; і змішані розчинники, складені з двох або більше з цих розчинників. Переважними є апротонні полярні органічні розчинники. Особливо переважним є N-метил-2-піролідинон. Реакція може здійснюватися без застосування основи. Однак, коли застосовують основу, приклади застосовних основ включають неорганічні основи, такі як карбонат натрію, карбонат калію, гідрокарбонат натрію, гідрокарбонат калію, гідроксид натрію, гідроксид калію, гідрид натрію, гідрид калію, ціанід натрію, ціанід калію, гідроксид магнію, гідроксид кальцію, гідроксид літію, і гідроксид барію; і органічні основи, такі як 1,8-діазабіцикло[5.4.0]ундека-7-ен, 1,5діазабіцикло[4.3.0]нона-5-ен, триетиламін, діізопропілетиламін, піридин, гуанідин, лутидин, колідин, 2,2’-біпіридил, трифеніламін, хінолін, N,N-диметиланілін і N,N-діетиланілін. Переважними є піридин, 2,6-лутидин, 2,4,6-колідин, 2,2’-біпіридил, трифеніламін, N,Nдиметиланілін, N,N-діетиланілін і т. п. Більш переважними є 2,6-лутидин, 2,4,6-колідин, трифеніламін, N,N-диметиланілін, і N,N-діетиланілін. ROH, RCl, (R)2O або змішаний кислотний ангідрид, переважно, RCl або (R) 2O, застосовують як ацилюючий агент для введення R як групи. Реакція може здійснюватися в присутності або за відсутності основи або з використанням конденсуючого агента, такого як дициклогексилкарбодіімід, гідрохлорид l-етил-3-(3-диметиламінопропіл)карбодііміду, 9 UA 108612 C2 5 10 15 20 25 30 35 40 45 50 55 60 карбонілдіімідазол, дипіридилдисульфід, діімідазолілдисульфід, 1,3,5-трихлорбензоїлхлорид, 1,3,5-трихлорбензоїлангідрид, PyBop або PyBrop. Переважно, реакцію здійснюють з використанням RCl або (R)2O в присутності або за відсутності основи. Більш переважні ацилюючі агенти включають циклопропанкарбонілхлорид і ангідрид циклопропанкарбонової кислоти. Коли застосовують основу, кількість основи складає переважно від 1,0 до 3,0 еквівалентів в розрахунку від кількості сполуки B2. Кількість застосовуваного ацилюючого агента складає переважно від 1,0 до 2,5 еквівалентів в розрахунку від кількості сполуки B1. Температура реакції складає переважно від -20°С до 60°С. Час реакції складає переважно від 0,1 години до 7 днів. (6) Спосіб очищення і виділення сполуки С з неочищеного продукту Спосіб отримання сполуки С за допомогою кристалізації переважно згадають як спосіб очищення і виділення сполуки С з реакційного розчину або неочищеного продукту сполуки С, отриманого в способі, описаному у вказаному вище параграфі (1), (3) або (5). Кристали можуть бути отримані у вигляді кристалів сольвату, що містять розчинник, включений в кристалічні грати. Альтернативно, сполука С, що не містить якого-небудь розчинника або води, може бути отримана за допомогою сушіння кристалів сольвату або за допомогою отримання осадів, наприклад, за допомогою розчинення кристалів сольвату в метанолі і додавання води до розчину, збираючи осади фільтрацією, і сушіння зібраних осадів за допомогою нагрівання при зниженому тиску. Відповідно до переважного варіанту здійснення для отримання кристалів сполуки С, наданий спосіб, який включає в себе екстракцію реакційного розчину, що містить сполуку С, отриману за допомогою способу відповідно до будь-якого із вказаних вище параграфів від 1 до 5, органічним розчинником, вибраним з групи, яка складається з метилацетату, етилацетату, бутилацетату, толуолу, хлорбензолу, хлороформу, дихлорметану і простого ефіру, концентрування екстракту після або без сушіння і, в цьому стані, забезпечення протікання кристалізації, або спосіб, який включає в себе упарювання реакційного розчину, що містить сполуку С, досуха з отриманням неочищеного продукту, розчинення неочищеного продукту в органічному розчиннику, вибраному з групи, яка складається з метилацетату, етилацетату, бутилацетату, толуолу, хлорбензолу, хлороформу, дихлорметану, ефіру, метанолу, і етанолу, при кімнатній температурі або при нагріванні, і додавання слабкого розчинника, вибраного з групи, яка складається з гептану, гексану і циклогексану, до розчину, щоб викликати кристалізацію. Простий ефір, що застосовується в способі, переважно вибирають з діетилового ефіру, діізопропілового ефіру, тетрагідрофурану і діоксану. Більш конкретний приклад способу отримання кристалів сполуки С включає в себе: або стадію додавання розчинника до реакційного розчину, видалення розчинника за допомогою перегонки з отриманням неочищеного продукту, і додавання етилацетату до неочищеного продукту, або стадію концентрування етилацетатного екстракту реакційного розчину; і виділення етилацетатних сольватів кристалів після відстоювання при кімнатній температурі або необов'язково після нагрівання. Якщо необхідно, пентан, гексан або циклогексан, переважно, гексан, додають до етилацетатному екстракту або концентрату етилацетатного екстракту для отримання етилацетатних сольватів кристалів. Сполука С може бути отримана за допомогою розчинення етилацетатних сольватів кристалів в метанолі, додавання води до розчину, збирання отриманих в результаті осадів за допомогою фільтрації, і сушіння зібраних осадів за допомогою нагрівання при зниженому тиску. 2-2: Отримання сполуки С із сполуки B1 через сполуку B4 Стадія отримання сполуки B4 із сполуки B1 в способі, вказаному у вказаному вище параграфі 6, може також здійснюватися за відсутності розчинника. Однак, коли стадію здійснюють в присутності розчинника, приклади застосовних розчинників включають кетонові розчинники, такі як ацетон і діетилкетон; розчинники з простих ефірів, такі як діетиловий ефір, діізопропіловий ефір і тетрагідрофуран; складноефірні розчинники, такі як етилацетат і бутилацетат; апротонні полярні органічні розчинники, такі як N,N-диметилформамід, N,Nдиметилацетамід, диметилсульфоксид, ацетонітрил, N-метил-2-піролідинон і N-метил-2піперазинон; галогеновані вуглеводневі розчинники, такі як дихлорметан і хлороформ; або ароматичні вуглеводневі розчинники, такі як толуол; і змішані розчинники, складені з двох або більше таких розчинників. ROН, RCl, (R)2O, або змішану кислотну ангідрид можна згадати як ацилюючий агент, для введення групи R. Ацилуючий агент являє собою переважно RCl або (R) 2O. Реакція може здійснюватися в присутності або за відсутності основи або з використанням конденсуючого 10 UA 108612 C2 5 10 15 20 25 30 35 агента, такого як дициклогексилкарбодіімід, l-етил-3-(3-диметиламінопропіл)карбодіімід гідрохлорид, карбонілдіімідазол, дипіридилдисульфід, діімідазолілдисульфід, 1,3,5трихлорбензоїлхлорид, 1,3,5-трихлорбензоїл ангідрид, PyBop або PyBrop. Основи, застосовні в даному винаході, включають карбонат натрію, карбонат калію, гідрид натрію, трет-бутоксид калію, метоксид натрію, етоксид натрію, піридин, лутидин, 4диметиламінопіридин, імідазол, 1,8-діазабіцикло[5.4.0]ундека-7-ен, 1,5-діазабіцикло[4.3.0]нона5-ен, триетиламін і діізопропілетиламін. Температура реакції складає переважно від -20°С до 50°С. Час реакції складає переважно від 0,5 години до 48 годин. Розчинники, застосовні на стадії отримання сполуки С із сполуки B4, в способі, вказаному у вказаному вище параграфі 6., включають спиртові розчинники, що мають від 1 до 4 атомів вуглецю, такі як метанол; розчинники з простих ефірів, такі як діетиловий ефір, діізопропіловий ефір, тетрагідрофуран і діоксан; апротонні полярні органічні розчинники, такі як N,Nдиметилформамід, диметилсульфоксид, N,N-диметилацетамід, ацетонітрил, N-метил-2піролідинон і N-метил-2-піперазинон; галогеновані розчинники, такі як дихлорметан і хлороформ; або воду; і змішані розчинники, складені з двох або більше з цих розчинників. Основи, застосовні в даному винаході, включають неорганічні основи, такі як карбонат натрію, карбонат калію, гідрокарбонат натрію, гідрокарбонат калію, гідроксид натрію, гідроксид калію, гідрид натрію, гідрид калію, ціанід натрію, ціанід калію, гідроксид магнію, гідроксид кальцію, гідроксид літію і гідроксид барію; алкоксиди лужних металів, такі як метоксид натрію, етоксид натрію і трет-бутоксид калію; алкоксиди лужноземельних металів; і органічні основи, такі як 1,8-діазабіцикло[5.4.0]ундека-7-ен, 1,5-діазабіцикло[4.3.0]нона-5-ен, триетиламін, діізопропілетиламін, піридин, гідразин і гуанідин. Переважними є 1,8-діазабіцикло[5.4.0]ундека7-ен, 1,5-діазабіцикло[4.3.0]нона-5-ен, карбонат натрію, карбонат калію, гідрокарбонат натрію, гідрокарбонат калію, гідроксид натрію і гідроксид калію. Кількість використовуваної основи становить переважно від 0,01 до 24 еквівалентів в розрахунку від кількості сполуки B4. Температура реакції складає переважно від -20°С до 50°С. час реакції складає переважно від 0,5 години до 14 днів. Приклади Даний винахід додатково ілюструється за допомогою наступних прикладів, які не мають на увазі обмеження винаходу. Чистота, описана в Експериментальних прикладах, означає процентну частку площі речовини, що розглядається, виміряну при наступних умовах ВЕРХ, якщо не визначено інакше. Умови вимірювання для ВЕРХ Колонка: Inertsil ODS-2 або ODS-4 (5 мкм); 4,6 мм діаметр × 150 мм. (ODS-2 застосовували в прикладах 1-13, а ODS-4 застосовували в прикладах 14-20,) Температура колонки: 30°С Рухома фаза: Вода-ацетонітрил Умови для рухомої фази: Як показано в таблиці 1 нижче 40 Таблиця 1 Час (хв.) Вода (%) Ацетонітрил (%) 45 50 55 0 80 20 1 80 20 9 40 60 17 10 90 20 10 90 21,01 80 20 30 80 20 Об'ємна швидкість потоку: 1,0 мл/хв Довжина хвилі детектування: УФ 320 нм Приклад 1 Синтез_11-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (1,00 г), синтезований відповідно до способу, описаного в WO2006/129714, суспендували в 5 мл N-метил-2-піролідинону, до суспензії додавали 0,55 мл (2,2 еквівалента) 2,6-лутидину, і 0,44 мл (2,2 еквівалента) циклопропанкарбонілхлориду додавали по краплях до суспензії при кімнатній температурі. Після одного часу додавання по краплях, реакційний розчин додавали по краплях до 200 мл води. Суміш перемішували протягом 5 година, і отриманий в результаті осад потім збирали за допомогою фільтрації, промивали водою і сушили з отриманням 0,816 г порошку, що складається, в основному, з 11-Oциклопропанкарбоніл-1,7,11-тридеацетилпірипіропену A. Окремо, до фільтрату додавали 25 г хлориду натрію, і суміш екстрагували 20 мл етилацетату. Етилацетатний шар промивали водою, етилацетат видаляли за допомогою перегонки, і залишок сушили з отриманням 0,27 г 11 UA 108612 C2 5 10 15 20 25 30 35 40 45 50 55 пінистої речовини що складається, в основному, з 11-O-циклопропанкарбоніл-1,7,11тридеацетилпірипіропену А. Порошок і пінисту речовину об'єднували разом, з подальшою хроматографією на силікагелі (100 мл силікагелю С-60 виробництво Merck Ltd.; етилацетатметанол (50:1 (об/об); об'ємна швидкість потоку 10 мл/хв) з отриманням 532 мг 11-Oциклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (вихід: 46,3%) (чистота: 95,6%). 1 FAB-MС; m/z 526 (M+Н)+; H-ЯМР (CDCl3) 2,15 (1H, дт, J=3,4, 9,5 Гц), 2,42 (1H, ушир. с), 2,96 (1H, с), 3,41 (1H, дд, J=5,1, 11,0 Гц), 3,75 (1H, д, J=11,9 Гц), 3,83 (1H, дд, J=4,9, 11,9 Гц), 4,29 (1H, д, J=11,7 Гц), 5,00 (1H, д, J=3,2 Гц), 6,52 (1H, с), 7,42 (1H, дд, J=4,9, 8,1 Гц), 8,11 (1H, дт, J=2,0, 8,3 Гц), 8,69 (1H, дд, J=1,3, 4,8 Гц), 9,00 (1H, д, J=1,7 Гц). Приклад 2 Синтез 11-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (1,00 г) суспендували в 5 мл N-метил-2-піролідинону, 0,50 мл (2,0 еквівалента) 2,6-лутидину додавали до суспензії, і 0,33 мл (1,7 еквівалента) циклопропанкарбонілхлориду додавали по краплях до суспензії при кімнатній температурі. Після 45 хв покрапельного додавання, реакційний розчин додавали по краплях до 100 мл води. До них додавали хлорид натрію (5 г), і суміш перемішували протягом ночі. Отриманий в результаті осад потім збирали за допомогою фільтрації, промивали водою, і сушили з отриманням 1,053 г порошку що складається, в основному, з 11-O-циклопропанкарбоніл-1,7,11тридеацетилпірипіропену A. Порошок (526 мг; половинна кількість) очищали за допомогою хроматографії на силікагелі (100 мл силікагелю С-60N (40-50 мкм) виробництво KANTO CHEMICAL CO., INC.; етилацетат-метанол (50:1 (об/об); об'ємна швидкість потоку 5 мл/хв) з отриманням 366 мг 11-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (вихід: 63,7%) (чистота: 95,1%). Приклад 3 Синтез 1,11-ди-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (1,00 г) суспендували в 5 мл N-метил-2-піролідинону, 0,76 мл (2,6 еквівалента) 2,4,6-колідину додавали до суспензії, і суміш додавали по краплях до 0,50 мл (2,5 еквівалента) циклопропанкарбонілхлориду при кімнатній температурі. Забезпечували протікання реакції протягом 8,5 години. Реакційний розчин потім додавали по краплях до 200 мл води. Суміш перемішували протягом ночі, і отриманий в результаті осад потім збирали за допомогою фільтрації і сушили з отриманням 1,135 г порошку, що складається, в основному, з 1,11-ди-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену A. Окремо, 25 г хлориду натрію додавали до фільтрату, і суміш екстрагували 20 мл етилацетату. Етилацетатний шар промивали водою, етилацетат видаляли за допомогою перегонки, і залишок сушили з отриманням 0,12 г пінистої речовини, що складається, в основному, з 1,11-ди-Оциклопропанкарбоніл-1,7,11-тридеацетилпірипіропену A. Порошок і пінисту речовину об'єднували разом, з подальшою хроматографією на силікагелі (150 мл силікагелю С-60 виробництво Merck Ltd.; тільки етилацетат; об'ємна швидкість потоку 10 мл/хв) з отриманням 743 мг 1,11-ди-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (вихід: 57,2%) (чистота: 80,8%). Для сполука отриманої таким чином, вимірюють FAB-МС і 1Н-ЯМР, і, в результаті, було виявлено, що дані відповідали даним для сполуки 261, описаної в W02006/129714. + 1 FAB-MС; m/z 594 (M+Н) ; H-ЯМР (CDCl3) 3,75 (1H, д, J=12,0 Гц), 3,79 (1H, дд, J=4,6, 11,7 Гц), 3,87 (1H, д, J=11,7 Гц), 4,82 (1H, дд, J=4,9, 11,2 Гц), 4,99 (1H, с), 6,52 (1H, с), 7,42 (1H, дд, J=4,8, 7,9 Гц), 8,10 (1H, д, J=7,8 Гц), 8,69 (1H, д, J=3,9 Гц), 9,00 (1H, с). Приклад 4 Синтез 1,11-ди-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (1,00 г) суспендували в 4 мл N-метил-2-піролідинону, 0,75 мл (3,0 еквіваленти) 2,6-лутидину додавали до суспензії, і 0,54 мл (2,7 5 еквіваленти) циклопропанкарбонілхлориду додавали по краплях до суспензії при кімнатній температурі. Забезпечували протікання реакції протягом трьох годин. Реакційний розчин додавали по краплях до 100 мл води. Суміш перемішували протягом двох годин, і до неї потім додавали 10 г хлориду натрію. Суміш потім перемішували протягом ночі, і отриманий в результаті осад збирали за допомогою фільтрації, промивали водою, і сушили з отриманням 1,276 г порошку що складається, в основному, з 1,11-ди-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А. 1,11-ди-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропен А, отриманий таким чином, очищали за допомогою хроматографії на силікагелі (силікагель С-60 виробництво Merck Ltd.; 50 мл для першого разу, 150 мл в зібраних основних фракціях для другого разу, і тільки етилацетат; об'ємна швидкість потоку 5 мл/хв) з отриманням 576 мг 1,11-ди-O 12 UA 108612 C2 5 10 15 20 25 30 35 40 45 50 55 60 циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (вихід: 44,4%) (чистота: 88,6%) і 115 мг (вихід: 8,8%) (чистота: 74,9%). Приклад 5 Синтез 1,11-ди-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (500 мг) суспендували в 2,5 мл N-метил-2-піролідинону і 0,25 мл (2,5 еквіваленти) циклопропанкарбонілхлориду додавали по краплях до суспензії при кімнатній температурі. Забезпечували протікання реакції протягом 24 годин. Реакційний розчин додавали по краплях до 50 мл води. Суміш доводили до pH 7,5 за допомогою додавання 8% водного бікарбонату натрію. До неї потім додавали хлорид натрію (5 г), і суміш перемішували протягом ночі. Отриманий в результаті осад потім збирали за допомогою фільтрації і промивали водою з отриманням порошку. Порошок сушили з отриманням 604 мг порошку, що складається, в основному, з 1,11-ди-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А. 1,11-ди-Оциклопропанкарбоніл-1,7,11-тридеацетилпірипіропен А, отриманий таким чином, очищали за допомогою хроматографії на силікагелі (100 мл силікагелю С-60N виробництво KANTO CHEMICAL CO., INC.; тільки етилацетат; об'ємна швидкість потоку 5 мл/хв) з отриманням 338 мг 1,11-ди-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (вихід: 52,0%) (чистота: 93,2%). Приклад 6 Синтез 1,11-ди-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (500 мг) суспендували в 2,5 мл N-метил-2-піролідинону, суспензію охолоджували до 0°С, і 0,15 мл (1,5 еквівалента) циклопропанкарбонілхлориду додавали до неї по краплях. Суміш перемішували при 0°С протягом 20 годин, і 0,1 мл (1,0 еквівалент) циклопропанкарбонілхлориду потім додавали додатково. Суміш перемішували протягом 66 годин, і 0,1 мл (1,0 еквівалент) циклопропанкарбонілхлориду далі додавали додатково. Суміш перемішували протягом 95 годин і додавали по краплях до 50 мл води з льодом. Суміш доводили до pH 7,5 за допомогою додавання 8% водного бікарбонату натрію. Потім до неї додавали хлорид натрію (5 г), і суміш перемішували. Отриманий в результаті осад потім збирали за допомогою фільтрації і промивали водою. Фільтрат екстрагували етилацетатом, і етилацетатний шар потім промивали насиченим сольовим розчином і сушили над безводним сульфатом магнію. Розчинник потім видаляли за допомогою перегонки при зниженому тиску. Залишок і осад об'єднували разом, з подальшим очищенням за допомогою хроматографії на силікагелі (150 мл силікагелю С-60N виробництво KANTO CHEMICAL CO., INC.; тільки етилацетат; об'ємна швидкість потоку 5 мл/хв) з отриманням 396 мг 1,11-ди-Oциклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (вихід:60,9%) (чистота: 95,3%). Приклад 7 Синтез 1,11-ди-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 11-О-Циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (200 мг, чистота: 95,6%), отриманий в Прикладі 1, суспендували в 1,0 мл N-метил-2-піролідинону, і 0,06 мл (1,5 еквівалента) циклопропанкарбонілхлориду додавали по краплях до суспензії при кімнатній температурі. Забезпечували протікання реакції протягом 21,5 години, і 20 мл води додавали до реакційного розчину. Суміш доводили до pH 7,5 за допомогою додавання 8% водного бікарбонату натрію, і 10 мл етилацетату і до неї додавали 3 г хлориду натрію. Суміш екстрагували і потім промивали водою. Етилацетат (10 мл) далі додавали до водного шару, і суміш екстрагували. Екстракт потім промивали водою і об'єднували з етилацетатним шаром, отриманим вище. Етилацетат видаляли за допомогою перегонки при зниженому тиску з отриманням порошку (295 мг) що складається, в основному, з 1,11-ди-О-циклопропанкарбоніл1,7,11-тридеацетилпірипіропену A. Порошок очищали за допомогою хроматографії на силікагелі (100 мл силікагелю С-60N виробництво KANTO CHEMICAL CO., INC.; тільки етилацетат; об'ємна швидкість потоку 5 мл/хв) з отриманням 119 мг 1,11-ди-O-циклопропанкарбоніл-1,7,11тридеацетилпірипіропену А (вихід: 55,0%) (чистота: 96,5%). Приклад 8 Синтез 1,7,11-три-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (500 мг) суспендували в 2,5 мл N-метил-2-піролідинону, 0,44 мл (5 екв.) піридин додавали до суспензії, і 0,45 мл (4,5 екв.) циклопропанкарбонілхлориду додавали по краплях до суспензії при кімнатній температурі. Забезпечували протікання реакції протягом 1,5 години. Реакційний розчин додавали по краплях до 50 мл води. Суміш перемішували протягом трьох годин, і потім до неї додавали 5 г хлориду натрію. Потім, реакційний розчин перемішували протягом 1,5 години, і отриманий в результаті осад потім збирали за допомогою фільтрації і промивали водою. Порошок, отриманий таким чином, сушили з отриманням 721 мг 1,7,11-три-O-циклопропанкарбоніл-1,7,11 13 UA 108612 C2 5 10 15 20 25 30 35 40 45 50 55 60 тридеацетилпірипіропену А у вигляді порошку (вихід: 99,4%) (чистота: 89,6%). Для сполуки, отриманої таким чином, вимірювали FAB-МС і 1Н-ЯМР, і, в результаті, було виявлено, що дані відповідали сполуці 218, описаній в WO 2006/129714. 1 FAB-MС; m/z 662 (M+Н)+; H-ЯМР (CDC13) 2,89 (1H, с), 3,72 (1H, д, J=11,7 Гц), 3,82 (1H, д, J=11,7 Гц), 4,79 (1H, дд, J=4,9, 11,5 Гц), 5,01 (1H, ушир. с), 5,02 (1H, дд, J=4,9, 11,2 Гц), 6,46 (1H, с), 7,41 (1H, дд, J=4,8, 7,9 Гц), 8,10 (1H, дт, J=1,7, 6,4 Гц), 8,69 (1H, ушир. с), 9,02 (1H, с) Приклад 9 Синтез 1,11-О-дициклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-О-Трициклопропанкарбоніл-1,7,11-тридеацетилпірипіропен А (1,0 г), синтезований в прикладі 8, розчиняли в 95% розчині водного метанолу (30 мл), і до нього додавали третбутоксид калію (85 мг) додавали при кімнатній температурі. Суміш перемішували при цій температурі протягом 16 годин, і до неї потім додавали оцтову кислоту. Метанол видаляли за допомогою перегонки при зниженому тиску, і залишок екстрагували хлороформом. Хлороформний шар промивали насиченим сольовим розчином і сушили над безводним сульфатом магнію. Розчинник потім видаляли за допомогою перегонки при зниженому тиску з отриманням неочищеного продукту 1,11-O-дициклопропанкарбоніл-1,7,11тридеацетилпірипіропену А (724 мг, чистота: 50%). Неочищений продукт очищали за допомогою колонкової хроматографії на силікагелі (Merck силікагель 60F254 0,5 мм; гексан:ацетон = 10:5,5) з отриманням 1,11-О-дициклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (370 мг, вихід: 41%). Приклад 10 Синтез 1,11-O-дициклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (спосіб з використанням кристалізації) 1,7,11-О-трициклопропанкарбоніл-1,7,11-тридеацетилпірипіропен А (4 г), синтезований в прикладі 8, розчиняли за допомогою нагрівання в метанолі (100 мл), і до нього додавали карбонат калію (420 мг) при кімнатній температурі. Суміш перемішували при цій температурі протягом 6 годин, до неї додавали оцтову кислоту (370 мг) і воду (100 мл), і суміші давали відстоятися протягом 23 годин. Осаджену вихідну речовину видаляли за допомогою фільтрації, потім додавали воду (50 мл), і суміш давали відстоятися протягом 20 годин. Метанол видаляли за допомогою перегонки при зниженому тиску, і залишку давали відстоятися протягом 7 годин. У результаті, 1,11-O-дициклопропанкарбоніл-1,7,11-тридеацетилпірипіропен А був осаджений, і обложений 1,11-O-дициклопропанкарбоніл-1,7,11-тридеацетилпірипіропен А збирали за допомогою фільтрації (900 мг, вихід: 25,1%, чистота: 81%). Приклад 11 Синтез 11-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропен А 1,7,11-Тридеацетилпірипіропен А (4,53 г) суспендували в 22,5 г N-метил-2-піролідинону, і до суспензії додавали 1,51 г (1,51 еквіваленти) триетиламіну і 2,25 г ангідриду (1,47 еквіваленти) циклопропанкарбонової кислоти, і суміш нагрівали при перемішуванні при 60°С протягом 23 годин. Потім, нагріту суміш концентрували при зниженому тиску при температурі бані, що дорівнює 70°С. Воду (10 мл) додавали до масла, отриманого таким чином, для отвердження. Тверду речовину промивали тричі 10 мл води і збирали за допомогою фільтрації. Порошок, отриманий таким чином, промивали 5 мл води і сушили при зниженому тиску при 40°С протягом одного дня з отриманням 11-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (4,73 г, вихід: 91,4%, чистота: 76,2%). Приклад 12 Синтез 1-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 11-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропен А (199,7 мг, чистота: 95,6%), отриманий тим же чином, що в прикладі 1, суспендували в 2,0 мл хлорбензолу. DBU (0,02 мл, приблизно 0,4 еквіваленти) додавали до суспензії, і суміш нагрівали при перемішуванні при 80°С протягом 9 годин. Потім, реакційний розчин поступово охолоджували до кімнатної температури і перемішували при кімнатній температурі протягом двох днів. До нього додавали етилацетат (20 мл) і 5 мл води, і органічний шар відділяли і концентрували при зниженому тиску. Кристали осаджували в такому стані, що хлорбензол залишався в системі. Відповідно, кристали збирали за допомогою фільтрації і промивали толуолом. Кристали сушили при зниженому тиску при 60°С протягом ночі з отриманням 1-O-циклопропанкарбоніл-1,7,11тридеацетилпірипіропену А (153,4 мг, вихід: 76,8%, чистота: 94,5%). Приклад 13 Синтез 1,11-ди-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1-О-Циклопропанкарбоніл-1,7,11-тридеацетилпірипіропен А (500 мг) суспендували в 3,0 мл N-метил-2-піролідинону, і суспензію додавали по краплях до 0,10 мл (1,0 еквіваленти) 14 UA 108612 C2 5 10 15 20 25 30 35 40 45 50 55 60 циклопропанкарбонілхлориду при 0°С. Забезпечували протікання реакції протягом одного дня, і до неї додавали 0,025 мл (0,25 еквівалента) циклопропанкарбонілхлориду. Далі, через 41 годину від додавання циклопропанкарбонілхлориду, 1,0 мл N-метил-2-піролідинону і 0,025 мл (0,25 еквіваленти) циклопропанкарбонілхлориду додавали до реакційного розчину, і забезпечували протікання реакції протягом 65 годин. Реакційний розчин потім виливали в 30 мл води з льодом і 50 мл етилацетату. Суміш нейтралізували 8% водного бікарбонату натрію, до неї додавали 3 г хлориду натрію, і суміш перемішували, з подальшим розділенням. Органічний шар промивали двічі 10 мл води, і розчинник видаляли за допомогою перегонки при зниженому тиску. Порошок (678 мг), отриманий таким чином, піддавали хроматографії на силікагелі (силікагель С-60 (80 мл) виробництво Merck Ltd.; етилацетат-метанол (50:1 (об/об)) для витягання 1,11-ди-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (479 мг, вихід: 83,3%, чистота: 95,2%) і 51 мг (10,2%) 1-О-циклопропанкарбоніл-1,7,11тридеацетилпірипіропену А. Приклад 14 Синтез 1,11-ди-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (1,00 г) суспендували в 7,0 мл N-метил-2-піролідинону, суспензію охолоджували до 0°С, і 0,4 мл (2,0 еквівалента) циклопропанкарбонілхлориду додавали по краплях до суспензії. Потім, 0,1 мл (0,5 еквівалента) циклопропанкарбонілхлориду додатково додавали до неї по краплях при 0°С, після проходження кожного з періодів, що дорівнюють 7 годинам, 23 годинам і 26 годинам, від завершення покрапельного додавання. Через 4 дні від додавання по краплях, реакційний розчин виливали в 50 мл етилацетату і 50 мл води з льодом. Далі, суміш нейтралізували 0,7 г бікарбонату натрію і до неї додавали 8% водного бікарбонату натрію і 5,0 г хлориду натрію. Суміш перемішували і їй давали можливість відстоятися, з подальшим розділенням. Органічний шар промивали двічі 20 мл води і концентрували при зниженому тиску. Етилацетат (8,0 мл) додавали до пінистого порошку, отриманого таким чином суміш нагрівали до 60°С, і до неї додавали 8,0 мл н-гексану. Суміш охолоджували до 50°С, і додавали дуже малу кількість затравкових кристалів. Після осадження кристалів, додавали 2,0 мл н-гексану, і суміш перемішували протягом ночі. Кристали збирали за допомогою фільтрації, і зібрані кристали промивали 10 мл н-гексан-етилацетат (1:1 (об/об)). Кристали, отримані таким чином, сушили протягом ночі при 60°С з отриманням 1,11-ди-O-циклопропанкарбоніл-1,7,11тридеацетилпірипіропену А (787 мг, вихід: 60,5%, чистота: 87,5%). Приклад 15 Синтез 1,11-ди-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (10,0 г) суспендували в 40,0 мл N-метил-2-піролідинону, суспензію охолоджували до 0°С, і 3,0 мл (1,5 еквівалента) циклопропанкарбонілхлориду додавали по краплях до суспензії. Потім, до неї додатково додавали циклопропанкарбонілхлорид по краплях при 0°С після проходження кожного з періодів, що дорівнює 6 годинам (2,0 мл, 1,0 еквівалента), 24 годинам (1,0 мл, 0,5 еквівалента), 32 годинам (0,50 мл, 0,25 еквівалента), і 48 годинам (1,0 мл, 0,5 еквівалента) від завершення покрапельного додавання. Додатково, додавали N-метил-2піролідинон після проходження 6 годин (20,0 мл) і 48 годин (10,0 мл) від завершення покрапельного додавання. Через 96 годин від покрапельного додавання, реакційний розчин виливали в 100 мл етилацетату і 200 мл води з льодом і перемішували, з подальшим розділенням. Етилацетат (170 мл) додавали до водного шару, суміш далі нейтралізували 10,1 г бікарбонату натрію, і до неї додавали 20,0 г хлориду натрію. Суміш перемішували і їй давали можливість відстоятися з подальшим розділенням. Органічний шар промивали однократно 50 мл 5% сольовим розчином і двічі 30 мл води і концентрували при зниженому тиску. Етилацетат додавали до залишку до повного об'єму, що дорівнює 110 мл. Суміш далі нагрівали до 60°С і до неї додавали 100,0 мл н-гексану. Суміш охолоджували до 50°С, і додавали дуже малу кількість затравкових кристалів. Після тригодинного осадження кристалів, додавали 20 мл н-гексану, і суміш перемішували протягом двох днів. Кристали збирали за допомогою фільтрації, і зібрані кристали промивали 50 мл суміші н-гексан-етилацетат (1:1 (об/об)). Кристали, отримані таким чином, сушили при 60°С протягом одного дня з отриманням 1,11-ди-O-циклопропанкарбоніл1,7,11-тридеацетилпірипіропену А (8,83 г, масовий вихід: 67,9%, чистота: 86,4%). Потім, частина масою 8,70 г від 8,83 г 1,11-ди-O-циклопропанкарбоніл-1,7,11тридеацетилпірипіропену А розчиняли в 43,5 мл метанолу, і 29,0 мл води додавали до неї при кімнатній температурі. У результаті, розчин ставав молочним і, отже, його нагрівали до 30°С. До нього додавали метанол (1,0 мл) і додавали дуже малу кількість затравкових кристалів. Після 15 UA 108612 C2 5 10 15 20 25 30 35 40 45 50 55 60 осадження кристалів, змішаний розчин, що складається з 15,0 мл води і 3,0 мл метанолу, додавали двома розділеними порціями. Суміш перемішували при кімнатній температурі протягом ночі і відфільтровувати. Кристали промивали змішаним розчином, що складається з 16,0 мл води і 4,0 мл метанолу. Кристали сушили при 80°С при зниженому тиску з отриманням 1,11-ди-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (6,22 г, масовий вихід всього: 48,5%, чистота: 94,5%). Приклад 16 Синтез 1,11-ди-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (10,0 г) суспендували в 40,0 мл N-метил-2-піролідинону, суспензію охолоджували до 0°С, і до суспензії додавали по краплях 7,0 мл (3,5 еквівалента) циклопропанкарбонілхлориду. Потім, забезпечили протікання реакції при 0°С протягом 53 годин, і реакційний розчин виливали в 50 мл етилацетату і 80 мл води з льодом. Суміш перемішували при 7°С або нижче, з подальшим розділенням. Етилацетат (мл) додавали до водного шару, і суміш перемішували з подальшим розділенням. Етилацетат (100 мл) додавали до водного шару, отриманого таким чином, і суміш нейтралізували 72 мл 1н гідроксиду натрію, і невеликою кількістю 8% водного бікарбонату натрію. Хлорид натрію (15,0 г) додавали до суміші при від 10 до 15°С, і суміш перемішували, і їй давали можливість відстоятися з подальшим розділенням. Органічний шар промивали однократно 30 мл 5% сольовим розчином і двічі 30 мл води, і суміш концентрували при зниженому тиску. Етилацетат (20 мл) додавали до залишку, суміш нагрівали до 60°С, і до неї додавали 14 мл н-гексану. У результаті, розчин ставав молочним, і, тому, 4,0 мл етилацетату додавали для розчинення. Розчин потім охолоджували до 50°С, і додавали дуже малу кількість затравкових кристалів. Після проходження 1,5 години від осадження кристалів, додавали 10 мл н-гексану, і суміш перемішували протягом ночі. Кристали збирали за допомогою фільтрації і промивали 30 мл суміші н-гексан-етилацетат (1:1 (об/об)). Кристали сушили при 80°С протягом двох днів з отриманням 1,11-ди-O-циклопропанкарбоніл-1,7,11тридеацетилпірипіропену А (8,48 г, масовий вихід: 65,2%, чистота: 83,4%). Етилацетатний розчин, отриманий при подальшій обробці, нейтралізували, промивали насиченим сольовим розчином і водою і сушили при зниженому тиску. Окремо, фільтрат, отриманий при збиранні кристалів, і промивання концентрували і сушили. Ці дві речовини, отримані таким чином, об'єднували разом (5,71 г), і суміш розчиняли в метанолі (30,0 мл). Потім, 5,16 мл 5н розчину гідроксиду натрію додавали по краплях при кімнатній температурі. 5N розчин гідроксиду натрію (2,0 мл) далі додавали по краплях після проходження 1,5 години від додавання по краплях 5н розчину гідроксиду натрію. Суміш перемішували при кімнатній температурі протягом 18 годин, відфільтровувати, і промивали 22 мл суміші метанол-вода (1:1 (об/об)). Кристали, отримані таким чином, сушили при 80°С протягом одного дня з отриманням вихідної речовини, тобто, 1,7,11-тридеацетилпірипіропену А (1,96 г, витягання: 19,6%, чистота: 94,5%). Коли витягання було враховане, вихід 1,11-ди-О-циклопропанкарбоніл-1,7,11тридеацетилпірипіропену А становив 81,0%. Приклад 17 Синтез 1,11-ди-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (10,0 г) суспендували в 40,0 мл N-метил-2-піролідинону, суспензію охолоджували до 3°С, і 7,0 мл (3,5 еквівалента) циклопропанкарбонілхлориду додавали по краплях до суспензії. Потім забезпечували протікання реакції при 0°С протягом 48 годин, і реакційний розчин виливали в 50 мл етилацетату і 80 мл води з льодом. Суміш перемішували при 10°С або нижче, з подальшим розділенням. Етилацетат (100 мл) додавали до водного шару, суміш нейтралізували 25 5н гідроксиду натрію і невеликою кількістю 8% водного бікарбонату натрію, і до неї додавали 8 г хлориду натрію при від 10 до 15°С. Суміш перемішували для розчинення, і їй давали можливість відстоятися, з подальшим розділенням. Органічний шар промивали однократно 30 мл 5% сольового розчину і двічі 30 мл води, концентрували до 40 мл при зниженому тиску і перемішували при кімнатній температурі протягом 5 годин для осадження кристалів. Потім, додавали 20 мл н-гексану протягом періоду, що дорівнює двом годинам, і суміш перемішували протягом ночі. Кристали відфільтровувати і промивали 30 мл суміші н-гексан-етилацетат (1:1 (об/об)). Кристали сушили при кімнатній температурі при зниженому тиску протягом 30 хв з отриманням 7,31 г кристалів 1,11-ди-Oциклопропанкарбоніл-1,7,11-тридеацетилпірипіропену A. ЯМР-спектр (прилад: Lambda-400, розчинник: CDCl3, відношення між інтегральним значенням для двох протонів CH 3COOCH2CH3 при 4,12 і інтегральне значення для одного протона 1,11-ди-O-циклопропанкарбоніл-1,7,11 16 UA 108612 C2 5 10 15 20 25 30 35 40 45 50 55 60 тридеацетилпірипіропену А) кристалів, отриманих таким чином, показав, що вміст етилацетату становив 0,96 моль в розрахунку від 1,0 моля 1,11-ди-O-циклопропанкарбоніл-1,7,11тридеацетилпірипіропену А (масовий вихід (у вигляді етилацетатного сольвату): 49,1%, чистота: 88,9%). Дифракційна рентгенограма порошку кристалів мала наступні значення. Дифракційна рентгенограма порошку Прилад: RINT 2200 (виробництво Rigaku Denki Co., Ltd.) Умови вимірювання: рентгенівське джерело: CuK/40 кВ/20 мА, зазор для зразка: 0,020, швидкість сканування: 0,500/хв, зазор сканування: 2/, і діапазон сканування: від 3,0 до 40,0. Характеристичні піки виявлялися при наступних дифракційних кутах [2 ()]. Дифракційні кути (2 ): 7,4±0,1°, 12,0±0,1, 17,0±0,1, 18,3±0,1, і 19,1±0,1. Дифракційна рентгенограма порошку показана на фіг. 1. Приклад 18 Синтез 1,11-ди-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (10,0 г) суспендували в 40,0 мл N-метил-2-піролідинону, суспензію охолоджували до 0°С, і 3,0 мл (1,5 еквівалента) циклопропанкарбонілхлориду додавали по краплях до суспензії. Після проходження 4 годин від додавання по краплях, до неї по краплях додавали циклопропанкарбонілхлорид (2,0 мл (1,0 еквівалент)) при 0°С. Забезпечували протікання реакції при 0°С протягом 69 годин, і реакційний розчин потім виливали в 100 мл етилацетату і 120 мл води з льодом і перемішували, з подальшим розділенням. Етилацетат (100 мл) додавали до водного шару, суміш далі нейтралізували 9,5 г бікарбонату натрію, і до неї додавали 8,0 г хлориду натрію. Суміш перемішували і їй давали можливість відстоятися з подальшим розділенням. Органічний шар промивали однократно 30 мл 5% сольового розчину і двічі 30 мл води, і суміш концентрували при зниженому тиску. Етилацетат (35,0 мл) додавали до залишку, і суміш потім перемішували при кімнатній температурі протягом 1,5 години. Потім, до неї по краплях додавали 35,0 мл н-гексану протягом періоду, що дорівнює двом годинам. Суміш перемішували при кімнатній температурі протягом ночі. Осаджені кристали потім збирали за допомогою фільтрації, промивали 30 мл суміші нгексан-етилацетат (1:1 (об/об)), і сушили при зниженому тиску протягом чотирьох годин з отриманням 9,39 г кристалів, що містять 1,11-ди-O-циклопропанкарбоніл-1,7,11тридеацетилпірипіропен A. Кристали, отримані таким чином, аналізували за допомогою методу, описаного в прикладі 17, і було виявлено, що вони містять 1 моль етилацетату в розрахунку від 1,0 моля 1,11-ди-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (масовий вихід (у вигляді етилацетатного сольвату):63,0%) (чистота: 85,5%). Дифракційна рентгенограма порошку кристалів відповідала приведеній в прикладі 17. Етилацетатний сольват (порція в 8,00 г в 9,39 г) 1,11-ди-О-циклопропанкарбоніл-1,7,11тридеацетилпірипіропену А, отриманий таким чином розчиняли в 16,0 мл метанолу. Розчин нагрівали до 35°С, і додавали 10,0 мл води. У результаті, розчин ставав молочним, і, тому, до нього додавали 1,0 мл метанолу. Через одну годину від додавання метанолу, суміш охолоджували до 25°С, і змішаний розчин, який складається з 16,8 мл води і 7,2 мл метанолу додавали до неї по краплях при від 20 до 25°С протягом періоду, що дорівнює двом годинам. Суміш перемішували при кімнатній температурі протягом ночі. Отриманий в результаті осад збирали за допомогою фільтрації і промивали змішаним розчином, що складається з 7,0 мл води і 3,0 мл метанолу. Зразок (500 мг) екстрагували з твердої речовини, отриманої таким чином, і частину, що залишилася, сушили при 80°С при зниженому тиску з отриманням 5,68 г 1,11-ди-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (6,01 г, з урахуванням кількості екстрагованого зразка) (загальний масовий вихід від 1,7,11-тридеацетилпірипіропену А: 54,2%) (чистота: 92,3%). Приклад 19 Синтез 1,11-ди-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (20,0 г) суспендували в 80,0 мл N-метил-2-піролідинону, суспензію охолоджували до -10°С, і 12,0 мл (3,0 еквівалента) циклопропанкарбонілхлориду додавали по краплях до суспензії. Забезпечували протікання реакції при -10°С протягом 4 годин, і 4,0 мл (1,0 еквівалент) циклопропанкарбонілхлориду додавали до неї по краплях додатково. Потім забезпечували протікання реакції при -10°С протягом 72 годин, і реакційний розчин виливали в 200 мл етилацетату і 180 мл 8% водного бікарбонату натрію при 5°С або нижче. Суміш нейтралізували 20 мл 8% водного бікарбонату натрію, потім додавали 20 мл 15% насиченого сольового розчину, і суміш перемішували при 10°С з подальшим розділенням. Органічний шар промивали тричі 60 мл води і концентрували до 60 мл при зниженому тиску. Потім до неї додавали 100 мл етилацетату, і суміш концентрували до 80 мл при зниженому 17 UA 108612 C2 5 10 15 20 25 30 35 40 45 50 55 60 тиску. Суміш перемішували при кімнатній температурі протягом ночі, і осаджені кристали потім збирали за допомогою фільтрації і промивали змішаним розчином, що складається з 10 млнгексану і 20 мл етилацетату. Кристали, отримані таким чином, сушили при зниженому тиску при 80°С протягом ночі з отриманням 17,80 г 1,11-ди-O-циклопропанкарбоніл-1,7,11тридеацетилпірипіропену A. Кристали аналізували за допомогою методу, описаного в Прикладі 17, і було виявлено, що вони містять 0,75 моль етилацетату в розрахунку від 1,0 моля 1,11-диО-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (масовий вихід: 61,8% (у вигляді етилацетатного сольвату)) (чистота: 87,5%). Приклад 20 Синтез 1,11-ди-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (50,0 г) суспендували в 200 мл N-метил-2-піролідинону, суспензію охолоджували до -10°С, і 15,0 мл (1,5 еквівалента) циклопропанкарбонілхлориду додавали по краплях до суспензії. Потім, циклопропанкарбонілхлорид додавали по краплях до суміші, в кількості, що дорівнює 15,0 мл (1,5 еквівалента) 3 години після додавання по краплях і в кількості, що дорівнює 10,0 мл (1,0 еквівалент) 5 годин після додавання по краплях при -10°С. Забезпечували протікання реакції при -10°С протягом 72 годин. Реакційний розчин потім виливали в 500 мл етилацетату і 500 мл 8% водного бікарбонату натрію при 5°С або нижче. Суміш нейтралізували невеликою кількістю 8% водного бікарбонату натрію, і до неї додавали 300 мл 15% насиченого сольового розчину при 10°С або вище з подальшим розділенням. Органічний шар промивали тричі 100 мл води і концентрували до 150 мл при зниженому тиску. Потім до неї додавали 250 мл етилацетату, і суміш знову концентрували до 200 мл при зниженому тиску. До неї додавали етилацетат (50 мл), і суміш перемішували при кімнатній температурі протягом ночі. Осаджені кристали збирали за допомогою фільтрації і промивали 80 мл етилацетату. Кристали, отримані таким чином, сушили при зниженому тиску при 50°С протягом двох годин з отриманням 44,90 г кристалів, що містять 1,11-ди-О-циклопропанкарбоніл-1,7,11тридеацетилпірипіропен A. Кристали аналізували за допомогою методу, описаного в прикладі 17, і було виявлено, що вони містять 0,99 моля етилацетату в розрахунку від 1,0 моля 1,11-диO-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (масовий вихід: 60,2% (у вигляді етилацетатного сольвату)) (чистота: 87,5%). Приклад 21 Синтез 1,11-ди-О-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А 1,7,11-Тридеацетилпірипіропен А (50,0 г) суспендували в 200 мл N-метил-2-піролідинону, суспензію охолоджували до -10°С, і 15,0 мл (1,5 еквівалента) циклопропанкарбонілхлориду додавали по краплях до суспензії. Потім, циклопропанкарбонілхлорид додавали по краплях до суміші, в кількості, що дорівнює 15,0 мл (1,5 еквівалента) 3 години після додавання по краплях і в кількості, що дорівнює 10,0 мл (1,0 еквівалент) через 5 годин після додавання по краплях при 10°С. Забезпечували протікання реакції при -10°С протягом 75 годин. Реакційний розчин потім виливали в змішаний розчин, що складається з 500 мл етилацетату, 500 мл води з льодом, і 40,0 г бікарбонату натрію при 5°С або нижче. Суміш нейтралізували невеликою кількістю 8% водного бікарбонату натрію і 300 мл 15% сольового розчину додавали при 10ºС або вище, з подальшим розділенням. Органічний шар промивали тричі 150 мл води і концентрували до 100 мл при зниженому тиску. Потім до неї додавали 200 мл етилацетату, і суміш знову концентрували до 150 мл при зниженому тиску. Додатково, потім до неї додавали 50 мл етилацетату, і суміш перемішували при кімнатній температурі протягом ночі. Осадженні кристали збирали за допомогою фільтрації і промивали 60 мл етилацетату. Кристали, отримані таким чином, сушили при зниженому тиску при 40°С протягом однієї години і сушили при кімнатній температурі протягом двох годин з отриманням 49,10 г кристалів, що містять 1,11-диO-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А (масовий вихід: 65,8% (у вигляді етилацетатного сольвату)) (чистота: 84,7%). Кристали аналізували таким же чином, як в прикладі 17, і було виявлено, що вони містять 0,98 моль етилацетату в розрахунку від 1,0 моля 1,11-ди-О-циклопропанкарбоніл-1,7,11-10 тридеацетилпірипіропену А. Порцію масою 24,0 г кристалів, отриманих таким чином, суспендували в 48,0 мл етилацетату, і суспензію перемішували при 70°С протягом однієї години і перемішували при кімнатній температурі протягом ночі. Потім, реакційний розчин відфільтровувати, з подальшим промиванням 30 мл етилацетату. Промитий продукт сушили при кімнатній температурі протягом 5 годин з отриманням 20,54 г продукту, що розглядається (масовий вихід: 56,4% (у вигляді етилацетатного сольвату; загальний вихід від 1,7,11-тридеацетилпірипіропену А) (чистота: 93,2%). 18 UA 108612 C2 5 10 15 20 Кристали, отримані таким чином, містили 1,00 моль етилацетату в розрахунку від 1,0 моля 1,11-ди-O-циклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А. Приклад 22 1,11-О-дициклопропанкарбоніл-1,7,11-тридеацетил-пірипіропен А синтезували з 1,7,11-Отрициклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А, синтезованого в прикладі 8, при реагентах, розчинниках, часі і температурних умовах, описаних в таблиці 3 нижче. Після завершення реакції, реакційний розчин аналізували за допомогою високоефективної рідинної хроматографії при наступних аналітичних умовах для визначення кількості 1,11-Oдициклопропанкарбоніл-1,7,11-тридеацетилпірипіропену А, отриманого в реакційному розчині. Результати показані в таблиці 3. Аналітичні умови Детектор: детектор поглинання в ультрафіолетовому діапазоні або детектор з фотодіодною матрицею (довжина хвилі вимірювання: 254 нм) Колонка: CAPCELL PAK C18; 2,0 мм вн. діам. (150 мм внутрішній діаметр; 5 мкм Температура колонки: 40°С Рухома фаза А: Вода Рухома фаза В: Ацетонітрил для рідинної хроматографії Подача рухомої фази: Градієнт концентрації регулюється за допомогою зміни відношень між рухомою фазою А і рухомою фазою В таким чином. Об'ємна швидкість потоку: 0,2 мл/хв Умови для рухомої фази: Як показано в таблиці 2 нижче Таблиця 2 Рухома фаза (об. %) 70 70(0 0 Час після введення (хв) 0 до 1 хв 1 до 20 хв 20 до 24 хв Рухома фаза (об. %) 30 30(100 100 Таблиця 3 Реагент (число еквівалентів) DBU (1,1) DBN (1,1) Na2CO3 (1,1) K2CO3 (0,5) t-BuOK (0,5) KHCO3 (1=>24) NaHCO3 (1=>24) 0,05M NaOMe (1,0) 1M NaOH (1,0) 0,01M NaOMe (1,0) K2CO3 (2=>14) Cs2CO3 (2,0) 0,1M LiOH (1,0) 0,1M CsOH (1,0) Cs2CO3 (0,1) K2CO3 (0,2) K2CO3 (0,5) Cs2CO3 (0,5) Розчинник Час Темп. MeOH-H2O (4:1) MeOH-H2O (4:1) MeOH-H2O (9:1) MeOH-H2O (19:1) MeOH-H2O (19:1) MeOH-H2O (4:1) MeOH-H2O (4:1) MeOH MeOH MeOH MeOH MeOH MeOH-H2O (9:1) MeOH-H2O(9:1) MeOH-THF (3:2) MeOH-CHCl3 (3:2) MeOH MeOH 21 год 15 год 15 год 16 год 16 год 14 днів 14 днів 2 год 2 год 2 дні 6 год 24 год 19 год 19 год 15 год 45 год 13 год 13 год к.т. к.т. к.т. к.т. к.т. к.т. к.т. 50°С 50°С к.т. к.т. к.т. к.т. к.т. к.т. к.т. к.т. к.т. 25 ФОРМУЛА ВИНАХОДУ 1. Спосіб отримання сполуки С, представленої формулою С: Хімічна формула 1 19 Площа % 48% 50% 47% 45% 42% 46% 49% 50% 50% 33% 32% 34% 31% 44% 45% Вихід виділення 39% 45% 37% 38% 41% UA 108612 C2 O O N HO O H RO OH H RO 5 C , де R представляє прямий ланцюг, розгалужений ланцюг або циклічний С 2-6алкілкарбоніл, за умови, що, коли алкільний фрагмент в алкілкарбонільній групі являє собою розгалужений ланцюг або циклічну групу, R представляє С3-6алкілкарбоніл, що включає: селективне ацилювання гідроксильних груп в 1-положенні і 11-положенні сполуки В1, представленої формулою В1: Хімічна формула 2 O O N HO O H HO OH H HO 10 15 B1 , ацилюючим агентом в кількості від 2,0 до 5,0 еквівалентів в розрахунку від сполуки В1 через одну-три стадії в присутності або за відсутності основи в апротонному полярному органічному розчиннику, що вибраний з диметилсульфоксиду, N,N-диметилацетаміду, ацетонітрилу, Nметил-2-піролідинону, N-метил-2-піперазинону, N,N-диметил-2-імідазолідинону. 2. Спосіб за п. 1, де сполуку С отримують за допомогою ацилювання гідроксильних груп в 1положенні і 11-положенні сполуки В1 через одну стадію. 3. Спосіб за п. 1, який включає в себе отримання сполуки С за допомогою ацилювання через дві стадії, що складаються зі стадій: ацилювання гідроксильної групи в 11-положенні сполуки В1 ацилюючим агентом з отриманням сполуки В2, представленої формулою В2: Хімічна формула 3 20 UA 108612 C2 O O N HO O H HO OH H RO 5 B2 , де R такий, як визначено в формулі С в п. 1; і додаткового ацилювання гідроксильної групи в 1-положенні сполуки В2. 4. Спосіб за п. 1, який включає в себе отримання сполуки С за допомогою ацилювання через три стадії, що складаються зі стадій: ацилювання гідроксильної групи в 11-положенні сполуки В1 з отриманням сполуки В2, представленої формулою В2: Хімічна формула 4 O O N HO O H HO OH H RO 10 B2 , де R такий, як визначено в формулі С, в п. 1; перенесення ацилу в 11-положенні сполуки В2 до гідроксилу в 1-положенні з отриманням сполуки В3, представленої формулою В3: Хімічна формула 5 O O N HO O H RO OH H HO 15 B3 , де R такий, як визначено в формулі С в п. 1; і ацилювання гідроксильної групи в 11-положенні сполуки В3. 21 UA 108612 C2 5. Спосіб за п. 1, який додатково включає в себе, як стадію отримання сполуки В1, гідроліз ацильних груп в 1-положенні, 7-положенні і 11-положенні сполуки А1, представленої формулою А1: Хімічна формула 6 O O N HO O H A1O A11O 5 10 15 20 25 30 35 40 45 OA7 H A1 , де А1, А7 і А11, які можуть бути однаковими або різними, представляють ацетил або пропіоніл, в присутності основи. 6. Спосіб за п. 1, де ацилювання здійснюють за відсутності основи. 7. Спосіб за п. 1, де основа, що використовується при ацилюванні гідроксилу в 1-положенні і/або 11-положенні сполуки В1, являє собою 2,4,6-колідин або 2,6-лутидин. 8. Спосіб за п. 3, де основу використовують на стадії отримання сполуки В2 і на стадії додаткового ацилювання гідроксилу в 1-положенні сполуки В2, кількість основи, що застосовують на стадії отримання сполуки В2, становить від 1,0 до 3,0 еквівалентів в розрахунку від сполуки В1, загальна кількість основи, що застосовують на стадії отримання сполуки В2, і основи, що застосовують на стадії додаткового ацилювання гідроксилу в 1положенні сполуки В2, становить від 2,0 до 4,5 еквівалентів. 9. Спосіб за п. 3, де ацилюючий агент використовують на стадії отримання сполуки В2 і на стадії додаткового ацилювання гідроксилу в 1-положенні сполуки В2, кількість ацилюючого агента, що використовують на стадії отримання сполуки В2, становить від 1,0 до 3,5 еквівалентів в розрахунку від сполуки В1, загальна кількість ацилюючого агента, що застосовують на стадії отримання сполуки В2, і ацилюючого агента, що застосовують на стадії додаткового ацилювання гідроксилу в 1-положенні сполуки В2, становить від 2,0 до 4,5 еквівалентів. 10. Спосіб за будь-яким з пп. 1-9, де R представляє циклопропанкарбоніл. 11. Спосіб за п. 4, де стадію отримання сполуки В3 із сполуки В2 здійснюють в присутності основи. 12. Спосіб за будь-яким з пп. 1-10, який додатково включає в себе стадію виділення і очищення сполуки С з розчину реакції, що містить сполуку С, за допомогою кристалізації. 13. Спосіб за будь-яким з пп. 1-12, що додатково включає стадію: (a) екстракції реакційного розчину, що містить сполуку С, органічним розчинником, вибраним з групи, яка складається з метилацетату, етилацетату, бутилацетату, толуолу, хлорбензолу, хлороформу, дихлорметану, діетилового ефіру, діізопропілового ефіру, тетрагідрофурану і діоксану, і концентрування екстракту після або без осушування; (b) упарювання реакційного розчину, що містить сполуку С, досуха з отриманням неочищеного продукту і потім розчинення неочищеного продукту в органічному розчиннику, вибраному з групи, яка складається з метилацетату, етилацетату, бутилацетату, толуолу, хлорбензолу, хлороформу, дихлорметану, діетилового ефіру, діізопропілового ефіру, тетрагідрофурану, діоксану, метанолу і етанолу, при кімнатній температурі або при нагріванні, або (с) упарювання реакційного розчину, що містить сполуку С, досуха з отриманням неочищеного продукту, розчинення неочищеного продукту в органічному розчиннику, вибраному з групи, яка складається з метилацетату, етилацетату, бутилацетату, толуолу, хлорбензолу, хлороформу, дихлорметану, діетилового ефіру, діізопропілового ефіру, тетрагідрофурану, діоксану, метанолу і етанолу, при кімнатній температурі або при нагріванні і додавання слабкого розчинника, вибраного з групи, яка складається з гептану, гексану і циклогексану, до розчину. 14. Застосування сполуки В2, представленої формулою В2: Хімічна формула 9 22 UA 108612 C2 O O N HO O H HO OH H RO B2 , де R такий, як визначено в формулі С, для отримання сполуки С, представленої формулою С: Хімічна формула 8 O O N HO O H RO OH H RO 5 10 C де R представляє прямий ланцюг, розгалужений ланцюг або циклічний С 2-6алкілкарбоніл, за умови, що, коли алкільний фрагмент в алкілкарбонільній групі являє собою розгалужений ланцюг або циклічну групу, R представляє С3-6алкілкарбоніл. 15. Застосування сполуки В3, представленої формулою В3: Хімічна формула 11 O O N HO O H RO OH H HO B3 для отримання сполуки С, представленої формулою С: Хімічна формула 10 23 UA 108612 C2 O O N HO O H RO OH H RO C де R представляє прямий ланцюг, розгалужений ланцюг або циклічний С2-6алкілкарбоніл, за умови, що, коли алкільний фрагмент в алкілкарбонільній групі являє собою розгалужений ланцюг або циклічну групу, R представляє С3-6алкілкарбоніл. Комп’ютерна верстка А. Крулевський Державна служба інтелектуальної власності України, вул. Василя Липківського, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 24
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for producing pyripyropene derivative
Автори англійськоюFukuda, Yoshimasa, Ando, Takashi, Goto, Kimihiko, Nakanishi, Nozomu, Watanabe, Takashi, Kurihara, Kenichi, Minowa, Nobuto, Mitomi, Masaaki
Автори російськоюФукуда Йосимаса, Андо Такаси, Гото Кимихико, Наканиси Нозому, Ватанабе Такаси, Курихара Кенити, Минова Нобуто, Митоми Масааки
МПК / Мітки
МПК: C07B 61/00, C07D 405/04
Мітки: отримання, пірипіропену, похідних, спосіб
Код посилання
<a href="https://ua.patents.su/26-108612-sposib-otrimannya-pokhidnikh-piripiropenu.html" target="_blank" rel="follow" title="База патентів України">Спосіб отримання похідних пірипіропену</a>
Спосіб отримання (4-гідроксі-6-оксотетрагідропіран-2-іл)ацетонітрилу і його похідних

Номер патенту: 83662
Опубліковано: 11.08.2008
Автори: Волберг Міхель, Серейніг Наташа, Бустен Вільхельмус Хюбертус Йозеф, Мінк Даніел
МПК: A61P 3/00, A61K 31/40, C07D 309/00, A61P 3/06
Мітки: 4-гідроксі-6-оксотетрагідропіран-2-іл)ацетонітрилу, отримання, спосіб, похідних
Формула / Реферат:
1. Спосіб отримання сполуки формули 1, (1)в якому сполука формули 2, (2)де X означає відхідну групу, реагує з ціанід-іоном у воді і в якому рН далі знижують до значень 0-5.2. Спосіб за п. 1, в якому концентрація ціанід-іону складає принаймні 1 моль на літр.3....
Спосіб отримання [s-(r,s)]-b-[[[1-[1-оксо-3-(4-піперидиніл)пропіл]-3-піперидиніл]карбоніл]аміно]-3-піридинпропанової кислоти та її похідних

Номер патенту: 67832
Опубліковано: 15.07.2004
Автори: Юстус Міхаель, Меріанофф Цинтіа А., Росслер Армін, Віллані Франк Джон Джр, Кохен Юдит Х., Вен Хрістіан, Шродер Фрідтоф Хармен
МПК: A61K 31/444, A61P 9/10, C07B 61/00, C07D 401/14, C07D 213/55, C07D 211/60
Мітки: похідних, s-(r,s)]-b-[[[1-[1-оксо-3-(4-піперидиніл)пропіл]-3-піперидиніл]карбоніл]аміно]-3-піридинпропанової, кислоти, спосіб, отримання
Формула / Реферат:
1. Спосіб отримання сполуки формули (І):, (I)де R1 та R2 незалежно вибрані з групи, що містить атом водню, нижчий алкіл та атом галогену, який відрізняється тим, що проводять реакцію солі формули (II) (II)з сіллю формули (ІІІ):
Спосіб отримання похідних 3,6-дигідро-2н-тіопірану

Номер патенту: 63943
Опубліковано: 25.10.2011
Автори: Бучкевич Ірина Романівна, Фігурка Оксана Михайлівна, Мусянович Ростислав Ярославович, Стасевич Марина Володимирівна, Новіков Володимир Павлович, Платонов Микола Олександрович
МПК: C07C 61/00, C07C 321/00, C07C 323/00
Мітки: спосіб, отримання, похідних, 3,6-дигідро-2н-тіопірану
Формула / Реферат:
Спосіб отримання похідних 3,6-дигідро-2Н-тіопірану, що включає циклізацію тіокетону з диметилбутадієном в апротонному розчиннику, який відрізняється тим, що тіокетон попередньо одержують взаємодією триетиламіну з 2-заміщеним-3-сульфенілхлорид-1,4-нафтохіноном формули,а циклізацію здійснюють при температурі -5
Спосіб отримання похідних 1-(2-галогенбіфеніл-4-іл)-циклопропанкарбонової кислоти

Номер патенту: 106619
Опубліковано: 25.09.2014
Автори: Ре Марко, Піветті Фаусто, Форнаретто Марія Джіоя
МПК: C07C 253/14, C07C 51/09, C07C 253/30, C07C 255/46, C07C 255/35, C07C 61/00
Мітки: спосіб, 1-(2-галогенбіфеніл-4-іл)-циклопропанкарбонової, похідних, отримання, кислоти
Формула / Реферат:
1. Спосіб отримання сполуки формули (ІА) (ІА)або її фармацевтично прийнятної солі, де R - одна або більше груп, незалежно вибраних з атомів галогену; в якому здійснюють наступні стадії: (і) здійснюють взаємодію сполуки формули (IV)
Проміжні сполуки для отримання похідних феноксіоцтової кислоти і спосіб їх одержання

Номер патенту: 79498
Опубліковано: 25.06.2007
Автори: Кобаясі Дзуніті, Танака Нобуюкі, Мукаяма Харунобу, Тамаі Тецуро, Харада Хірому, Акахане Сатосі, Ісікава Такехіро
МПК: C07C 217/60, C07C 43/315, C07C 67/31, C07C 43/178, C07C 69/712, C07C 69/736
Мітки: отримання, проміжні, кислоти, сполуки, одержання, спосіб, феноксіоцтової, похідних
Формула / Реферат:
1. Сполука, представлена загальною формулою (І):, (I)де кожний з R1 і R2 незалежно являє собою нижчу алкільну групу.2. Сполука за п. 1, де R1 і R2 являють собою етильну групу.3. Спосіб отримання сполуки, представленої загальною формулою (І):, (I)де кожний з R1 і...
Попередній патент: Спосіб виготовлення металевої смуги з покриттям із покращеним зовнішнім виглядом
Наступний патент: Застосування інгібіторів сукцинатдегідрогенази для підвищення стійкості рослин або частин рослин проти абіотичного стресу
Випадковий патент: Морозиво "сонячне"