Спосіб хроматографічної вірус-інактивації
Номер патенту: 109254
Опубліковано: 10.08.2015
Автори: Гаврилюк Олена Сергіївна, Жукова Анастасія Іванівна, Гаврилюк Сергій Петрович, Волков Георгій Леонідович, Краснобрижа Євгенія Миколаївна




















Формула / Реферат
1. Спосіб хроматографічної вірус-інактивації розчину цільового білка, комплексу білків або пептиду, у якому
піддають кондиціюванню розчин цільового білка, комплексу білків або пептиду;
здійснюють аплікацію кондиційованого розчину цільового білка, комплексу білків або пептиду на хроматографічну колонку з адсорбентом зі зв′язуванням білка, комплексу білків або пептиду;
промивають адсорбент буфером аплікації;
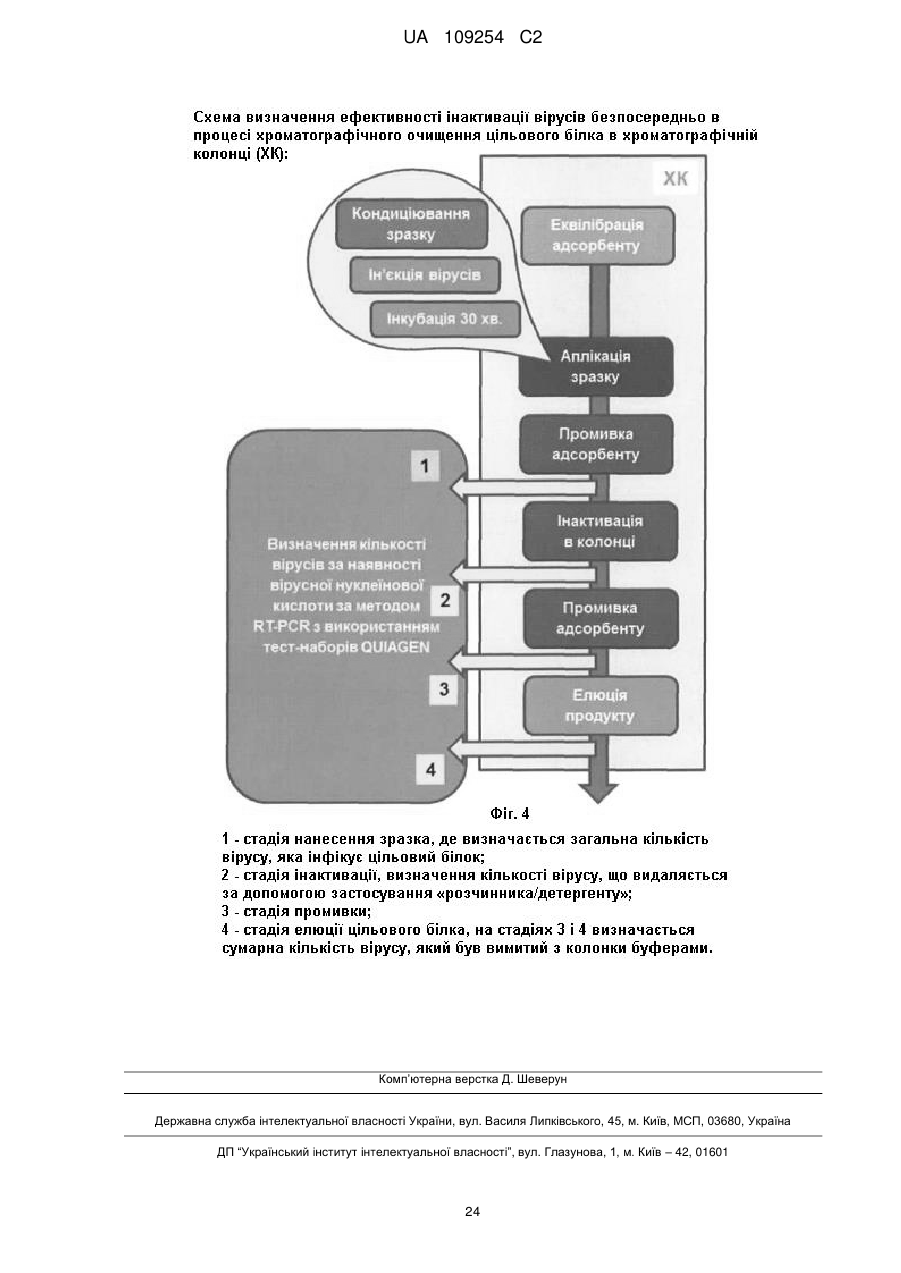
промивають адсорбент буфером інактивації вірусів, який містить розчинник і детергент у буфері аплікації;
відмивають адсорбент від розчинника/детергента та продуктів розпаду вірусів буфером аплікації; та
елююють розчин цільового білка, комплексу білків або пептиду з адсорбенту;
де зазначений адсорбент являє собою іонообмінний, афінний, обернено-фазовий, або адсорбент гідрофобної взаємодії.
2. Спосіб за п. 1, у якому адсорбент являє собою афінний адсорбент, що має динамічну ємність зв′язування не менше 15 мг білка×мл-1 адсорбенту, при швидкості протікання буфера від 10 до 50 мл×хв-1.
3. Спосіб за п. 1, у якому адсорбент являє собою іонообмінний сорбент, вибраний з аніоніту або катіоніту, з динамічною ємністю зв′язування білка від 50 мг×мл-1 адсорбенту, при швидкості протікання буфера від 50 до 500 cм×год.-1.
4. Спосіб за п. 1, у якому адсорбент являє собою обернено-фазовий адсорбент, якщо цільовий білок, комплекс білків або пептид є низькомолекулярним (МВ=1-30 кДа), та адсорбент являє собою іонообмінний, афінний або адсорбент гідрофобної взаємодії, якщо цільовий білок, комплекс білків або пептид має середню або високу молекулярну масу (МВ=20-≥4000 кДа).
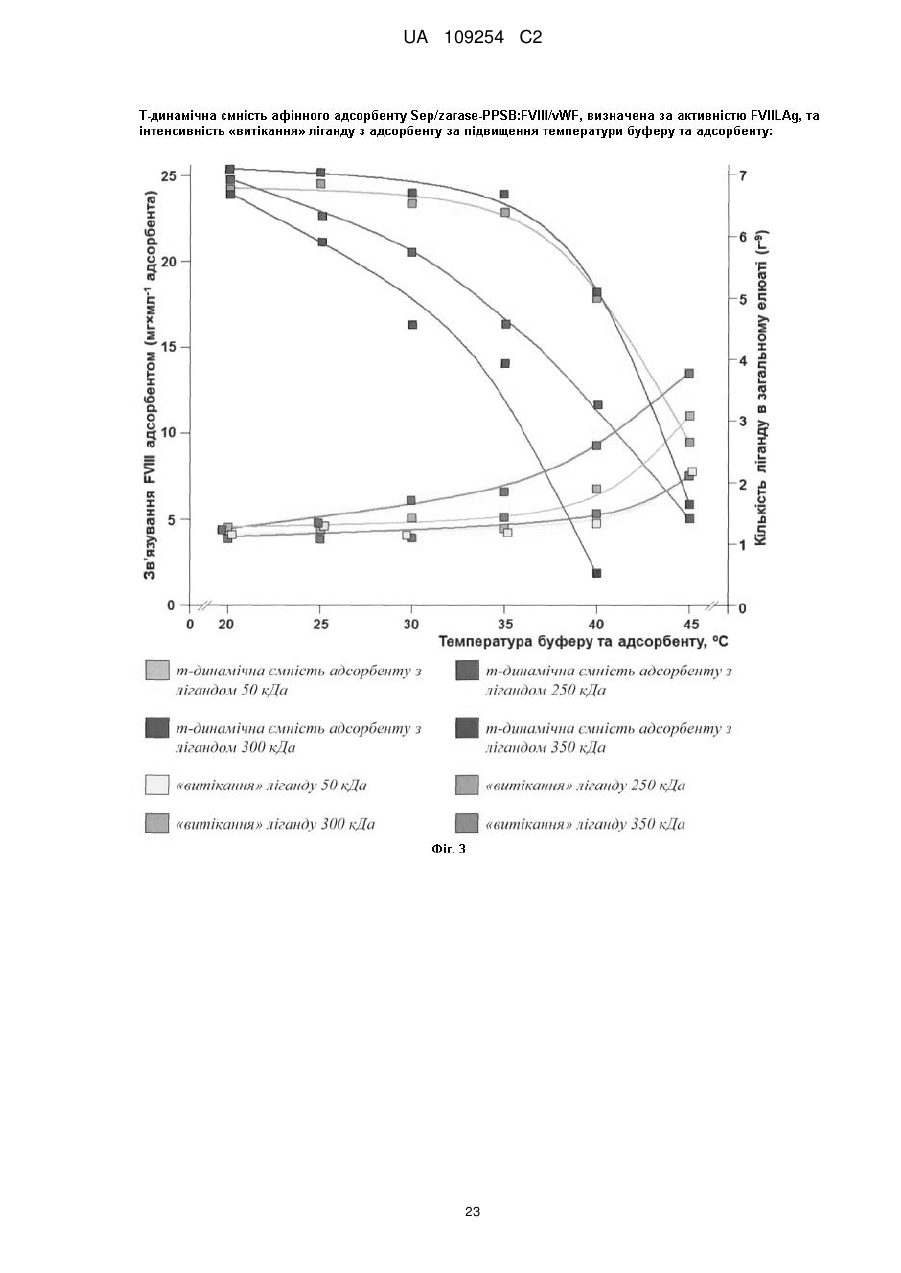
5. Спосіб за будь-яким з пп. 1, 2 або 4, у якому афінний адсорбент являє собою хімічно нейтральну, стійку до тиску в 5-10 бар полімерну матрицю з пришитим поліпептидним фрагментом або хімічною сполукою, яка специфічно взаємодіє з цільовим білком, комплексом білків або пептидом.
6. Спосіб за п. 5, у якому пришитий поліпептидний фрагмент є синтетичного, рекомбінантного або природного походження.
7. Спосіб за п. 5 або п. 6, у якому молекулярна маса пришитого поліпептидного фрагмента становить від 1 до 500 кДа.
8. Спосіб за будь-яким з пп. 1, 3 або 4, у якому адсорбент являє собою аніонообмінний адсорбент у випадку видалення денатурованих нуклеїнових кислот вірусів або їх фрагментів.
9. Спосіб за будь-яким з пп. 1 - 8, у якому інактивуючий буфер містить:
розчинник, вибраний з групи, що включає: ди- і триалкілфосфати, які містять алкільний ланцюг довжиною 1-10 вуглеводних атомів, такі як ди-n-пропілфосфат (DNPP) і три-n-бутилфосфат (TNBP); прості ефіри, такі як етиловий і пропіловий; складні ефіри, такі як амілацетат; алкільовані та гідроксильовані сполуки, такі як бутиловий ефір гідроксиланізолу (ВНА) і бутиловий ефір гідрокситолуолу (ВНТ); та їх суміші, необов′язково з додатковими органічними сполуками в концентрації 0,002-3,0 %;
детергент, вибраний з групи, що включає: оксіетильовані алкілфеноли, такі як тритони К-60, Х-45, Х-100, W-30; холат і деоксихолат натрію; сульфобетаїни, такі як додецилсульфонат натрію; додецилбензосульфонат натрію (Nacconol NR), N-додециламіноетансульфонова кислота, 2-сульфоетилолеат натрію (Igepon A); поліоксіетильовані похідні складних ефірів, такі як полісорбат 20 або твін 20, полісорбат 80 або твін 80; ефіри поліоксіетильованих жирних спиртів, такі як Brij 35; конденсат етиленоксиду і пропіленоксиду (Pluronic copolymers); оксіетильовані аміни, такі як Ethomeen; нонідет Р-40 (Nonidet P-40) і люброкс PХ (Lubrox PX); неіонні детергенти, такі як каприлат в концентрації 0,005-5,0 %.
10. Спосіб за будь-яким з пп. 1-9, у якому промивання адсорбенту буфером інактивації вірусів здійснюють в чотири стадії, де на першій стадії інактивації вірусів промивання здійснюють градієнтом буфера інактивації, концентрація якого у буфері аплікації зростає від 0 % до 100 %, об′ємом 5-10 Vc зі швидкістю 0,1-0,2 Vc×хв-1, на другій стадії - 10-20 Vc буфером інактивації зі швидкістю 0,1-0,2 Vc×хв-1, на третій стадії градієнтом буфера інактивації, концентрація якого у буфері аплікації знижується від 100 % до 0 %, об′ємом 5-10 Vc зі швидкістю 0,1-0,2 Vc×хв-1, на четвертій стадії - буфером аплікації об′ємом 10-20 Vc зі швидкістю 0,1-0,2 Vc×хв-1.
11. Спосіб за п. 10, у якому промивання адсорбенту на першій стадії інактивації вірусів здійснюють буфером інактивації з температурою, що зростає від 4-18 °С до 30-45 °С, зі швидкістю 0,25-0,50 °С×хв-1, на другій стадії - з температурою 30-45 °С, на третій стадії – з температурою, що знижується від 30-45 °С до 4-18 °С, зі швидкістю 0,25-0,50 °С×хв-1, на четвертій стадії - буфером аплікації з температурою 4-18 °С.
12. Спосіб за будь-яким з пп. 1-11, у якому хроматографічна колонка оснащена пристроєм для термостабілізації.
13. Спосіб за п. 12, у якому температуру хроматографічної колонки відтворюють відповідно до температури процесу інактивації вірусів і наступного відмивання адсорбенту.
14. Спосіб за п. 1, у якому адсорбент являє собою аніонообмінний адсорбент з максимальним зворотним тиском не менше 3 бар, динамічною ємністю зв′язування
БСА≥150 мг×мл-1 при швидкості потоку буфера через колонку
від 50 до 500 см×год.-1, типу WorkBeads 40 Q, температура інактивуючого буфера становить від 40 до 45 °С, при швидкості потоку через колонку 0,1-0,2 Vc×хв-1 та тривалості інактивації не менше 2,0 год.
Текст
Реферат: Винахід належить до способу хроматографічної вірус-інактивації розчину цільового білка, комплексу білків або пептиду, у якому піддають кондиціюванню розчин цільового білка, комплексу білків або пептиду, здійснюють аплікацію кондиційованого розчину цільового білка, UA 109254 C2 (12) UA 109254 C2 комплексу білків або пептиду на хроматографічну колонку з адсорбентом зі зв′язуванням білка, комплексу білків або пептиду, промивають адсорбент буфером аплікації, промивають адсорбент буфером інактивації вірусів, який містить розчинник і детергент у буфері аплікації, відмивають адсорбент від розчинника/детергента та продуктів розпаду вірусів буфером аплікації; та елююють розчин цільового білка, комплексу білків або пептиду з адсорбенту, де зазначений адсорбент являє собою іонообмінний, афінний, обернено-фазовий або адсорбент гідрофобної взаємодії. UA 109254 C2 5 10 15 20 25 30 35 40 45 50 55 Винахід належить до способу інактивації вірусів у білкових розчинах природного або штучного походження за допомогою застосування процесу обробки білків або пептидів, зв'язаних або іммобілізованих на хроматографічному адсорбенті безпосередньо у хроматографічній колонці розчином буфера, який містить розчинник і детергент. Особливістю цього процесу є використання фізичної властивості природних, синтетичних або рекомбінантних поліпептидів втрачати певну кількість ступенів свободи, тобто внутрішню рухомість фрагментів молекули при адсорбуванні на хроматографічному адсорбенті. Це, у свою чергу, істотно знижує здатність до необоротної денатурації поліпептидної молекули як наслідок присутності в буфері органічного розчинника і детергента в значимих для поліпептидів концентраціях, а також при підвищенні температури буфера. В результаті використання у процесі обробки органічного розчинника (переважно - ди- і триалкілфосфатів) і детергента (переважно -ефіри ангідридів сорбіту, що включають оксіетильовані алкілфеноли і тритони) поліпептиди значною мірою звільняються від вірусів, вкритих оболонкою (ВВО), наприклад, від вірусів гепатиту, вірусів імунодефіциту людини, аденовірусів, цитомегаловіруса та інших вірусних інфекцій, і, таким чином, вище перераховані білкові препарати стають повністю очищеними. Способи дезінфекції в біотехнології насамперед почали розробляти для видалення вірусної інфекції людини, яка найчастіше зосереджувалася у плазмі крові донорів. Дезінфектант був визначений як ″агент″, що звільняє від інфекції, яка виявляється, як правило, хімічною речовиною, що руйнує хвороботворні або інші згубні мікроорганізми або інактивує віруси» [1]. Існує багато різних методів дезінфекції, основною мішенню яких є специфічні віруси. Ці методи включають використання високого гідростатичного тиску, опромінення УФ-світлом, обробку перекисом водню, хлорним вапном й іншими компонентами, що містять хлор, глутаральдегідом, етанолом, іонами металів, сполуками, що містять четвертинний азот, їдким натром, карбонатом натрію і багатьма іншими речовинами [2]. Таку ж різноманітність методів розроблено і для проведення вірусної інактивації плазми донорів і її продуктів [3]. Показано, що обробка замороженої плазми флуоресцентним світлом протягом 1 години при освітленості 45000-60000 люкс у присутності 1 мкМ метиленового синього забезпечує високий рівень інактивації вірусів як для ВВО (для ВІЧ >6,7 log10), так і для вірусів без оболонки (ВБО), наприклад, аденовірусів (≥4,0 log10). Проте, такі ВБО, як віруси енцефаломіокардиту, гепатиту А, поліомієліту та свинячого парвовірусу залишалися неінактивованими [4-5]. Також у плазмі були активні проти ВІЧ-1 фоточутливі сполуки, що містили хлор [6-7], 3,3'-(1,4-нафтабоден)дипропіонат проти ВВО простого герпесу і герпесу свиней [8], в плазматичних фракціях і в концентраті фактора згортання VIII - псоралени (8-метоксипсорален і амінометил-триметилпсорален) проти ВВО гепатитів В і С, везикулярного стоматиту і лейкемії котів, ВБО синього гребня [9]. βПропіолактон у концентрації 0,1-0,25 % при низьких і середніх температурах (від 4 до 22 °С) і нейтрально-лужних значеннях рН (7,2-8,1) був ефективним у зниженні концентрації як ВВО, так і ВБО на 2,5-8,0 порядків у фракціях імуноглобулінів, альбуміну і фактора згортання IX [10-14]. Проте фактор згортання VIII виявився нестійким у присутності Р-пропіолактону [10]. Каприлова кислота в концентрації 12-40 мМ (25-40 °С, рН 5,1-5,4) протягом 60 хв. знижувала вміст ВВО (везикулярного стоматиту, діареї бика) у фракціях фактора згортання VIII, IgG і альбуміну не менше, чим на 4,4 log10 [15-16]. Опромінення потоком пульсуючого УФ-світла (254-320 нм) -2 потужністю 750-2000 Дж×см тонких плівок розчинів фібриногену [17], концентрату фактора VIII [18], альбуміну [19], імуноглобулінів [20] та інших білків [21] істотно знижувало кількість ВБО (на ≥4,6-6,5 log10) і ВВО (на ≥3,4-6,6 log10). Як було показано методом MALDI-TOF/TOF масспектрометрії фактором інактивації вірусів при опроміненні УФ-світлом є атомарний кисень, який окислює капсидні білки [21]. Пульсуюче світло широкого спектра[22-23] і гамма-промені [24] також були ефективні при вірус-інактивації. Деякі гідролітичні ферменти у поєднанні з високою (37 °С) або низькою (4 °С) температурою, зниженими значеннями рН (4-6), певним вмістом стабілізаторів (сахарози і NaCl) і заданим вмістом білка (6-16 %), істотно знижували концентрацію ВВО (на ≥2,4-6,0 log10) в розчинах очищених імуноглобулінів при тривалому процесі інактивації [25-27]. Крім того, низькі значення рН розчинів імуноглобулінів (4,0-4,5) протягом 7-21 діб при температурі 21 °С викликали інактивацію вірусів гепатиту С і діареї бика [28], а 0,7 М розчин аргініну в кислих значеннях рН був більше ефективним у процесі інактивації ВВО, ніж розчини 0,1-0,7 М цитрату або 0,1 М цитрату/0,6 М NaCl [29]. Процес обробки білкових розчинів «розчинником/детергентом» (РДП) почали використовувати більше 25 років назад, завдяки класичному патенту Neurath і Horowitz із Центру крові Нью-Йорка (США) [30] на спосіб неденатуруючої обробки біологічно активних білкових дериватів плазми крові донорів. Цей РДП продовжує до цього часу залишатися основним способом інактивації вірусів, з яким порівнюють нові методи. 1 UA 109254 C2 5 10 15 20 25 30 35 40 45 50 55 До 2000 р. було вже точно встановлено, що найбільше прийнятним співвідношенням РДП у процесі вірус-інактивації є 0,3-1 % три-n-бутил-фосфату (TNBP) і 1-2 % тритону Х-100 [31]. В найбільш важливих роботах того часу [32-36] було показано, що ВВО практично повністю інактивуються при використанні різних співвідношень розчинника і детергенту (TNBP/Na-холат, TNBP/тритону Х-100, TNBP/твін 80). Важливим залишається розумне поєднання концентрації агентів інактивації вірусів і максимальне збереження активності білків, що виробляються. В цьому випадку найбільш цікаву модель являє собою високомолекулярний комплекс коагуляційних факторів VIII і фон Віллебранда (FVIII/vWF). Ще в 2000 p. Kim I.S. і співавтори [34] показали, що суміш TNBP/тритону Х-100 в кінцевій концентрації 0,3 і 1,0 % (об'єм/об'єм), відповідно, при 22 °С і рН буфера 7,2 інактивувала ВІЧ-1 практично до значень нижньої межі методу виявлення протягом першої хвилини РДП, який йшов 1 годину. Вихід активності FVIII не обговорювався, але він, скоріше за все, був задовільним, так як зазначену розробку підприємство Greencross PD Corp. використовувало у виробництві антигемофілічного препарату «GreenMono». BurnoufT. і співавтори [37] виявили, що інкубація кріопреципітату в 2 % TNBP або 1 % TNBP/1 % тритоні Х-45 4 години при 37 °С не змінює активність FVIII/vWF. І при 90-93 %-ному виході активності FVIII/vWF і фібриногену вдається досягнути зниження концентрації вірусів >4,17, >4,73 і >4,72 для ВІЧ, вірусів діареї биків і псевдосказу, відповідно [18]. Staldrem M. і співробітники [38] для препарату «Wilate», що включає високоактивний -1 концентрат FVIII/vWF (122 МО×мг білка), встановили, що оптимізований РДП досить агресивний при інактивації ВВО і в той же час м'який відносно до збереження структури і функцій молекул факторів VIII і фон Віллебранда. Хоча було виявлено, що vWF після обробки розчинником-детергентом в основній масі складався всього лише з 10 мультимерів, це не позначилося на різкому зниженні виходу активності концентрату при наступному очищенні. Roberts P.L., вивчаючи вплив суміші 0,3 % TNBP/1 % тритон Х-100 на вірус Sinbisлихоманки, як моделі ВВО, показали, що помітну вірус-інактивацію (>4-6 log10) високо очищеного концентрату FVIII («Replenate») можна досягнути вже при температурі 4-5 °С і в 2 рази менших концентраціях розчинника та детергенту, але при тривалому терміні інкубації. Концентрація солей і рН буфера істотного впливу на ступінь інактивації не мала, вихід активності FVIII залишався задовільним для промислового виробництва [39]. Потім автору після заміни тритона Х-100 на полісорбат 20 вдалося збільшити до >5,0 log10 ефективність інактивації ВВО в тому ж концентраті при збереженні досить високої активності (препарат був зареєстрований під маркою «Optivate»). Проте, інактивації модельного вірусу коров'ячої віспи застосованим методом досягти не вдалося навіть при тривалому періоді інкубації [40]. Велика група авторів з різних фармацевтичних компаній під керівництвом Dichtelmuller H.О. у 2009 р. опублікувала підсумковий документ 20-ти річного вивчення застосування РДП на семи підприємствах, що входять в «Асоціацію терапії білками плазми» [41-42]. Більше 300 виробничих документів, що відображають умови і варіанти комбінацій розчинника (TNBP) і детергента (ТВІН 80, тритон Х-100, Na-холат, октоксінол-9) для різних продуктів, стали основою висновку, що ні клас продукту, ні концентрація білка, ні величина рН буфера не мають особливого значення для інактивації вірусів, критичною є тільки концентрація агентів інактивації - розчинника і детергента, а РДП - це промислово прийнятний метод інактивації широкого спектра ВВО, таких як ВІЧ-1,2, гепатитів В і С та ін. Менш схильний до денатурації концентрат фактора коагуляції IX також витримував обробку розчинником/детергентом при зниженні концентрації вірусів на 4,5, 6,1, 5,1 і 5,3 log10 для ВІЧ-1, вірусів везикулярного стоматиту, діареї бика та псевдосказу, відповідно. Але для ВБО (вірус гепатиту А) відмічали тільки незначне зниження концентрації (1,6 log10), і у цьому випадку була потрібна додаткова антивірусна обробка іншими методами [43]. Такі ж результати для комбінації TNBP/полісорбат 80 отримали й інші розробники [44-47]. Проте, спосіб давав можливість або отримати високий вихід фактора, або високу ефективність видалення вірусів. Вірус-інактивацію імуноглобулінів G успішно здійснювали вже традиційними сумішами TNBP/твін 80 [48-49], TNBP/тритон Х-100 [50] і каприлатом [51]. До того ж, автори розробок переконливо довели, що РДП знижував концентрацію ВВО (віруси бичачої діареї, запального ринотрахеїту, симплексного герпесу і везикулярного стоматиту), що виявляються як за кінцевим титром, так і шляхом вимірювання експресії вірусних білків [52] на 4-6 log10 [48-52], але не впливав на вміст та інфекційність ВБО [48, 52]. Метод також не змінював ні фізичних, ні біологічних властивостей імуноглобулінів [49, 53]. За останні два роки РДП був розроблений для вірус-інактивації факторів X і Ха [54], ХІа [55], інгібітора альфа-1-протеази [56]. При цьому продукти інактивації вірусів та інактивуючі агенти 2 UA 109254 C2 5 10 15 20 25 30 35 40 45 50 55 успішно видаляли з розчинів застосуванням одного хроматографічного етапу [44] або нанофільтрації [50]. Ще в 2000 р. було впевнено показано, що стандартний етап імуноафінної хроматографії знижує концентрацію вірусу поліомієліту (ВБО) на ~3 log10, a катіонообмінної - на 4-5 log10 [57]. Такі ж результати в 2005 були отримані і для хроматографії гідрофобної взаємодії [58]. Curtis S. [59] шляхом аніонообмінної хроматографії при рН 7,0-8,0, швидкості потоку буфера ≤500 -1 -1 см×год , динамічною ємністю 200 мг×мл і висоті шару адсорбенту 11 см, досягнув елімінації на >4,0 log10 як для вірусу лейкемії мишей (ВВО), так і для ВБО Simian 40 (мавпячий вірус 40). У більшості досліджень фактор зниження концентрації ВВО і ВБО шляхом хроматографії був ідентичний або перевищував такий при процесі пастеризації [60-61]. До того ж, видалення вірусів хроматографічним шляхом здійснювалося, скоріш за все, за рахунок простого механічного вимивання, так як помітної взаємодії вірусних частинок із зарядженим лігандом у присутності стандартних концентрацій солі не відмічалося [62]. Крім того, існує значна різниця в ефективності взаємодії протилежно заряджених цільового білка і ліганду або афінності білка і ліганду хроматографічної матриці, що спричиняє швидке вимивання вірусних частинок із хроматографічної колонки [63]. В доповіді Groner А. на конференції «PDA Virus & TSE Safety Forum» у червні 2011 p. підсумовано п'ятнадцятирічний період застосування різних методів рідинної хроматографії для зниження концентрації ВВО і ВБО для багатьох фармацевтичних білків, що виробляються. Наведений фактор зниження концентрації вірусів при застосуванні хроматографії, що розділяє за розміром, хроматографій гідрофобної взаємодії, афінної, іонообмінної складав ≥4,5-6,5, ≥2,7-4,2, ≤2,9-8,1 і ≥2,6-4,5, відповідно. При цьому цей фактор залежав від величини вірусної корпускули, температури буфера і швидкості його потоку, а також від висоти шару адсорбенту [63]. Із зазначеного вище видно, що ефективна інактивація ВВО і ВБО у білкових розчинах є значною проблемою при виробництві фармацевтичнопридатних білкових розчинів, і особливо при крупномасштабному виробництві, тому задачею винаходу було створення нового і ефективного способу вірус-інактивації, який забезпечував би високий вихід продукту без втрати його активності, та значний ступінь інактивації вірусів. Винахідники зробили припущення, що ефективна інактивація ВВО і ВБО у білкових розчинах може бути досягнута при: а) високій концентрації агентів інактивації - розчинника і детергенту, у випадку використання найбільш відомих TNBP/тритон Х-100 >0,3 % і >1,0 %, відповідно, б) високій температурі процесу інактивації, від 35 до 45 °С, в) достатній тривалості процесу інактивації, ≥2-3 год, г) використанні декількох кроків хроматографічного очищення білка з одночасним видаленням вірусів, інактивованих вірусів або їх фрагментів і агентів інактивації. Етапи (а), (б) і (в) при всій обережності їх застосування значною мірою є етапами, які викликають денатурацію білків, що особливо відображається на виході активності високомолекулярних білків, наприклад, таких як комплекс факторів коагуляції VIII і фон Віллебранда. Крім того, РДП практично не впливає на ВБО у білкових розчинах. Враховуючи, що схильність білка до незворотної денатурації багато в чому визначається доменною організацією його структури [64-66], фактично денатурація є інтегральною функцією кількості ступенів свободи білка [67-68]. Як відомо, у розчинах з низькою концентрацією сильних іонів кількість ступенів свободи специфічного білка досягає максимуму [69]. Після збільшення концентрації сильних аніонів (особливо полівалентних) їх взаємодія одночасно з різними сайтами білкової молекули обмежує ступінь її свободи [69]. Більше того, взаємодія білка з органічним або неорганічним матриксом меншої або більшої по величині молекули спричиняє наступну втрату ступенів свободи [70-71]. Наприклад, шаперони, «розпинаючи» білок всередині свого величезного матриксу, зводять кількість ступенів свободи білка практично до нуля, захищаючи цим його від можливої денатурації термальним шоком [72]. Відповідно, зазначена вище ціль винаходу була досягнута способом хроматографічної вірусінактивації цільового білка, комплексу білків або пептиду, у якому піддають кондиціюванню розчин цільового білка, комплексу білків або пептиду; потім здійснюють аплікацію кондиційованого розчину цільового білка, комплексу білків або пептиду на хроматографічну колонку з адсорбентом, зі зв'язуванням білка, комплексу білків або пептиду; після чого промивають адсорбент буфером аплікації; та промивають адсорбент буфером інактивації вірусів, який містить розчинник і детергент у буфері аплікації; згодом відмивають адсорбент від розчинника/детергенту та продуктів розпаду вірусів буфером аплікації; та елююють розчин цільового білка, комплексу білків або пептиду з адсорбенту. У додатковому варіанті реалізації винаходу адсорбент являє собою іонообмінний, афінний, обернено-фазовий або адсорбент гідрофобної взаємодії. 3 UA 109254 C2 5 10 15 20 25 30 35 40 45 50 55 60 У ще одному варіанті реалізації винаходу адсорбент являє собою обернено-фазовий адсорбент, якщо цільовий білок, комплекс білків або пептид є низькомолекулярним (МВ=1-30 кДа), та адсорбент являє собою іонообмінний, афінний або адсорбент гідрофобної взаємодії, якщо цільовий білок, комплекс білків або пептид має середню або високу молекулярну масу (МВ=20-≥4000 кДа). У додатковому варіанті реалізації винаходу афінний адсорбент являє собою хімічно нейтральну, стійку до тиску в 5-10 бар полімерну матрицю з пришитим поліпептидним фрагментом або хімічною сполукою, яка специфічно взаємодіє з цільовим білком, комплексом білків або пептидом. При цьому пришитий поліпептидний фрагмент може бути синтетичного, рекомбінантного або природного походження, та молекулярна маса пришитого поліпептидного фрагмента бажано становить приблизно від 1 до 500 кДа. Також бажано, щоб афінний -1 адсорбент мав динамічну ємність зв'язування не менше приблизно 15 мг білка×мл адсорбенту, -1 при швидкості протікання буфера приблизно від 10 до 50 мл×хв . У ще одному варіанті реалізації винаходу іонообмінний сорбент являє собою аніоніт або -1 катіоніт з динамічною ємністю зв'язування білка приблизно від 50 до 150 мг×мл адсорбенту, -1 при швидкості протікання буфера приблизно від 50 до 500 см×год. . При цьому бажано адсорбент являє собою аніонообмінний адсорбент у випадку видалення денатурованих нуклеїнових кислот вірусів або їх фрагментів. У кращому варіанті реалізації винаходу інактивуючий буфер містить: розчинник, вибраний з групи, що включає: ди- і триалкілфосфати, які містять алкільний ланцюг довжиною 1-10 вуглеводних атомів, такі як ди-n-пропілфосфат (DNPP) і три-n-бутилфосфат (TNBP); прості ефіри, такі як етиловий і пропіловий; складні ефіри, такі як амілацетат; алкільовані та гідроксильовані сполуки, такі як бутиловий ефір гідроксиланізолу (ВНА) і бутиловий ефір гідрокситолуолу (ВНТ); та їх суміші, необов'язково з додатковими органічними сполуками в концентрації 0,002-3,0 %; та детергент, вибраний з групи, що включає: оксіетильовані алкілфеноли, такі як тритони К-60, Х-45, Х-100, W-30; холат і деоксихолат натрію; сульфобетаїни, такі як додецилсульфонат натрію; додецилбензосульфонат натрію (Nacconol NR), N-додециламіноетансульфонова кислота, 2-сульфоетилолеат натрію (Igepon А); поліоксіетильовані похідні складних ефірів, такі як полісорбат 20 або твін 20, полісорбат 80 або твін 80; ефіри поліоксіетильованих жирних спиртів, такі як Brij 35; конденсат етиленоксиду і пропіленоксиду (Pluronic copolymers); оксіетильовані аміни, такі як Ethomeen; нонідет Р-40 (Nonidet Р-40) і люброкс РХ (Lubrox РХ); каприлат та інші неіонні детергенти в концентрації 0,005-5,0 %. У ще кращому варіанті реалізації винаходу промивку адсорбенту буфером інактивації вірусів здійснюють в чотири стадії, де на першій стадії інактивації вірусів промивання здійснюють градієнтом буфера інактивації, концентрація якого у буфері аплікації зростає від 0 % до 100 %, -1 об'ємом 5-10 Vc зі швидкістю 0,1-0,2 Vc×хв , на другій стадії - 10-20 Vc буфером інактивації зі -1 швидкістю 0,1-0,2 Vc×хв , на третій стадії градієнтом буфера інактивації, концентрація якого у -1 буфері аплікації знижується від 100 % до 0 %, об'ємом 5-10 Vc зі швидкістю 0,1-0,2 Vc×хв , на -1 четвертій стадії - буфером аплікації об'ємом 10-20 Vc зі швидкістю 0,1-0,2 Vc×хв . Додаткових переваг отримують, коли промивання адсорбенту на першій стадії інактивації вірусів здійснюють буфером інактивації з температурою, що зростає від 4-18 °С до 30-45 °С, зі -1 швидкістю 0,25-0,50 °С×хв , на другій стадії - з температурою 30-45 °С, на третій стадії - з -1 температурою, що знижується від 30-45 °С до 4-18 °С, зі швидкістю 0,25-0,50 ºС×хв , на четвертій стадії - буфером аплікації з температурою 4-18 °С. У ще одному варіанті реалізації винаходу хроматографічна колонка оснащена пристроєм для термостабілізації, наприклад, водяною «сорочкою». При цьому бажано температуру хроматографічної колонки відтворювати відповідно до температури процесу інактивації вірусів і наступної відмивки адсорбенту. У найкращому варіанті реалізації винаходу адсорбент являє собою аніонообмінний адсорбент з максимальним зворотним тиском не менше 3 бар, динамічною ємністю зв'язування -1 БСА ≥≥150 мг×мл при швидкості потоку буфера через колонку приблизно від 50 до 500 -1 см×год. , наприклад, типу WorkBeads 40 Q, температура інактивуючого буфера становить -1 приблизно від 40 до 45 °С, при швидкості потоку через колонку 0,1-0,2 Vc×хв та тривалості інактивації не менше 2,0 год. При розробці даного винаходу, винахідниками було встановлено, що для істотного зниження денатурації цільового білка у процесі вірус-інактивації необхідно обмежити кількість його ступенів свободи, що найбільш перспективно здійснити шляхом зв'язування молекули на хроматографічному адсорбенті, який має або активний іонний заряд, або ділянки гідрофобної, або афінної взаємодії. При цьому, чим більше на молекулі білка буде сайтів взаємодії з 4 UA 109254 C2 5 10 15 20 25 30 35 40 45 50 55 60 хроматографічним лігандом, тим меншу кількість ступенів свободи буде мати білок, тим нижчим буде рівень його денатурації при будь-яких умовах хроматографування. З метою організації відповідних умов необхідно так кондиціювати зразок цільового білка, який наноситься на адсорбент, і/або таким чином кондиціювати і/або модифікувати адсорбент, щоб взаємодія цільового білка з лігандом адсорбенту забезпечувала оборотне зв'язування по максимальній кількості білкових сайтів з лігандом. Модифікація адсорбенту здійснюється у випадку проведення РПД на афінному сорбенті і означає «пришивку» специфічного пептидного ліганду, який було б легко ідентифікувати і гарантовано відділити від цільового білка при його «стіканні» з колонки в процесі хроматографії. Для ефективного утримання цільового білка на іонообмінному ліганді при зростаючій температурі та гідрофобності буфера через ріст концентрацій розчинника і детергента необхідно вибирати іонообмінний адсорбент з -1 динамічною ємністю ≥150 мг білка×мл . У випадку використання для РПД обернено-фазової або хроматографії гідрофобної взаємодії підвищення температури буде компенсовано зростанням гідрофобності буфера, завдяки збільшенню концентрації розчинника. Таким чином, для утримання цільового білка на гідрофобному або обернено-фазовому ліганді адсорбенту -1 -1 вистачить величини динамічної ємності ~20-25 мг IgG×Mn або ~35-50 мг інсуліну×мл , відповідно. За таких умов адсорбент із зв'язаним на ньому цільовим білком можна буде промивати буфером, що містить розчинник і детергент у необхідних концентраціях, і має температуру 35-45 °С, достатньо тривалий час, що спричинить елімінацію ВВО. Тривале промивання буфером з підвищеною температурою і елюція цільового білкав РДП, наступна іонообмінна (іншого типу) і/або гідрофобної взаємодії, а також «полірувальна» хроматографія, що виключає за розміром, будуть сприяти ефективному видаленню ВБО. Це, у свою чергу, дасть можливість скоротити час пастеризації/теплової обробки цільового білка, що призведе до істотного підвищення його виходу. До того ж, хроматографічні методи будуть мати перевагу перед методами нанофільтрації [73], так як при хроматографії турбулентність буфера і, відповідно, денатурація білка, що виникає через це, буде значно нижчою [74]. Відповідно до даного винаходу білкові розчини природного або штучного походження, зокрема розчини цільового білка, комплексу білків або пептиду, можуть бути такими як: а) білкові препарати крові, що вміщують будь-яку фракцію, концентрат або дериват плазми крові, продукти із плазми чи фракцій плазми, які містять будь-які, лабільні і стабільні, білки та інші поліпептиди, наприклад імуноглобуліни, фактори згортання, інгібітори або активатори гемостазу; б) білкові розчини/препарати з інших рідин організму тварин або рослин, що вміщують вищі та нижчі, які містять будь-які, лабільні та стабільні, білки й інші поліпептиди; в) приготовані або отримані білкові розчини або препарати з інших тканин, органів або клітин організму тварин або рослин, що вміщують вищі та нижчі, які містять будь-які, лабільні та стабільні, білки та інші поліпептиди; г) білкові розчини або препарати, отримані з клітин, продуктів клітин, наприклад тілець включення і т.д., середовищ культивування, гідропонних середовищ і природних рідких середовищ існування біологічних організмів, що вміщують генетично модифіковані; д) білкові розчини або препарати, отримані шляхом хімічного синтезу поліпептидів, які, можливо, зазнали дії хімікатів, розчинів або дихання та іншого впливу оператора, що їх інфікувало, У цілому, спосіб, відповідно до винаходу, полягає в тому, що розчин цільового білка кондиціюють шляхом доведення значення концентрації загального білка, рН, іонної сили або кондуктивності розчину до необхідних для вибраного методу хроматографії так, щоб цільовий білок після нанесення розчину на адсорбент зв'язувався адсорбентом. Адсорбент, упакований в хроматографічну колонку з «термосорочкою», урівноважують відповідним буфером для налаштування властивостей адсорбенту «захоплювати» цільовий білок. Зразок наносять на адсорбент і промивають 5-10 Vc (об'ємами колонки) буфера, що урівноважує. Потім колонку починають промивати буфером інактивації (БІ) -буфером, що урівноважує, в якому заданий градієнт концентрації розчинника і детергента, наприклад TNBP і тритону Х-100. Градієнт задають звичайним чином: шляхом поступового рівномірного збільшення концентрації «БІ-Б» з кінцевою максимальною концентрацією агентів інактивації в «БІ-А» з нульовою концентрацією агентів інактивації. Кінцева концентрація розчинника і детергента в «БІ-Б» у процентному співвідношенні для афінної, катіонообмінної, аніонообмінної, зворотно-фазової та гідрофобної взаємодії може досягати співвідношення 0,5:1,5, 0,7:2, 1:3, 2:3 і 3:4, відповідно. При цьому, в буфері, що урівноважує, концентрація сильних одновалентних катіонів, виражена кондуктивністю розчину, не повинна перевищувати у зазначених випадках 5, 15, 15, 25 і 150 -1 -1 mS×хв . Швидкість градієнта звичайно задають у межах 5-10 Vс×год , тобто за 1 годину через 5 UA 109254 C2 5 10 15 20 25 30 35 40 45 50 55 60 колонку має пройти 5-10 об'ємів буфера, і за даний об'єм і час градієнт повинен вийти на максимум концентрації. Одночасно з цим задають температурний градієнт теплоносія, що зростає, в «сорочці» колонки і камері підігріву буферів інактивації «БІ-А» і «БІ-Б» шляхом їх переміщення в термостат, який забезпечує циркуляцію теплоносія в «сорочці» колонки. Градієнт температури задають у межах від 18 °С (робоча температура приміщення) до 30-45 °С -1 зі швидкістю, яка відповідає швидкості градієнта концентрації, тобто 12-27 °С×год . Таким чином, максимальний рівень концентрації розчинника/детергента в колонці співпадає з досягненням заданого максимуму температури адсорбенту в колонці і буферів інактивації. Якщо умови хроматографічного очищення потребують нижчої температури, наприклад, 4-8 °С, то для проведення вірус-інактивації по цьому способу необхідний лише термостат, здатний охолоджувати як колонку, так і буфери. При досягненні заданих максимальних рівнів концентрації агентів інактивації і температури, адсорбент в колонці продовжують промивати протягом 2-3 годин «БІ-Б» при температурі 30-45 °С, а потім задають негативні градієнти концентрації-температури і протягом 1 години повертають адсорбент в колонці до початкової температури і нульової концентрації агентів інактивації. Колонку додатково промивають 5-10 Vc -1 буфера, що урівноважує, зі швидкістю 0,2-0,5 Vс×год. при необхідній температурі до повного видалення слідів розчинника і детергента. Елюцію цільового білка здійснюють розробленим способом. Зазначений винахід додатково проілюстровано за допомогою фігур, які описують посилання і процес інактивації вірусів безпосередньо в хроматографічній колонці. Фіг. 1 зображує графік зміни кількісного зв'язування бичачого сироваткового альбуміну (БСА) аніонообмінними адсорбентами в залежності від швидкості потоку буфера через -1 хроматографічну колонку. При низькій швидкості потоку буфера (100-200 см×год. ) адсорбенти Q Sepharose High Performance (HP) i WorkBeads 40Q мають практично однакову динамічну -1 ємність ~100 мг×мл адсорбенту. Проте, при збільшенні швидкості потоку буфера ліганд Q Sepharose HP не встигає «захоплювати» альбумін, що протікає, з попередньою інтенсивністю, в -1 силу чого динамічна ємність його падає. При швидкості буфера 500 см×год. ємність цього -1 адсорбенту знижується майже в 2 рази до ~50 мг×мл адсорбенту. В той же час динамічна ємність WorkBeads 40Q фактично не змінюється у всьому виміряному діапазоні швидкостей буфера. В експерименті була використана колонка ХК 26/20 (GE Healthcare AB, Швеція, Уппсала), упакована 80 мл Q Sepharose HP (GE Healthcare AB, Швеція, Уппсала) або WorkBeads 40Q (BioWorks Sweden AB, Швеція, Бромма) на висоту шару 15 см. Колонку урівноважували 800 мл 0,02 М Na-ацетатного буфера, рН 6,5. Зразок альбуміну з -1 -1 концентрацією 25 мг×мл наносили в 40 мл буфера, що урівноважує, зі швидкістю 20 мл×хв , -1 адсорбент промивали 400 мл того ж буфера зі швидкістю 80 мл×хв . Елюцію альбуміну здійснювали 200 мл 0,50 М Na-ацетатного буфера, рН 9,0, за різних швидкостей потоку буфера -1 -1 (1,7-8,5 см×хв або 8,8-44 мл×хв ). Фіг. 2 зображує графік зміни кількісного зв'язування бичачого сироваткового альбуміну (БСА), лізоциму й інсуліну адсорбентами різного типу зв'язування у залежності від температурних умов процесу при постійній швидкості потоку буфера через хроматографічну колонку. При підвищенні температури процесу Т-динамічна (температурозалежна) ємність іонообмінних адсорбентів, як катіонообмінника SP Sepharose HP, так і аніонообменників Q Sepharose HP і WorkBeads 40Q, знижується. При цьому адсорбент WorkBeads 40Q показує помітне зниження Т-динамічної ємності (на ~10-12 %) тільки при температурах вище 45 °С, у той час як при таких температурах ємність Q Sepharose HP падає на ~80 %, a SP Sepharose HP взагалі припиняє зв'язувати білок. Зв'язування білка по принципах гідрофобної (Phenyl Sepharose HP) або зворотно-фазової (SOURCE 15RPC) взаємодій, обумовлено експонуванням на поверхні білка гідрофобних ділянок взаємодії з лігандом сорбенту за допомогою високих концентрацій солей або розчинника. Підвищення температури буфера спричиняє деяке підвищення експонування гідрофобних ділянок на поверхні білка, тому незначне збільшення Тдинамічної ємності обох адсорбентів у всьому діапазоні вимірювання температур знаходить своє пояснення. В експерименті була використана колонка ХК 26/20 (GE Healthcare AB, Швеція, Уппсала), упакована 80 мл Q Sepharose HP, SP Sepharose HP, Phenyl Sepharose HP (GE Healthcare AB, Швеція, Уппсала) або WorkBeads 40Q (Bio-Works Sweden AB, Швеція, Бромма) на висоту шару 15 см. Для експериментів з SOURCE 15RPC використовували готову набивну колонку RESOUCE 15RPC об'ємом 3 мл і висотою шару 10 см (GE Healthcare AB, Швеція, Уппсала). Для колонок, наповнених Q Sepharose HP і WorkBeads 40Q, умови хроматографування -1 наведені в описі ФІГ. 1. Швидкість потоку буфера при елюції складала 8 мл×хв . 6 UA 109254 C2 5 10 15 20 25 30 35 40 Колонку, наповнену SP Sepharose HP, урівноважували 800 мл 0,025 М гліцинового буфера, -1 рН 3,0, який містив 3 мМ ЕДТА і 0,1 % твін 80, зразок лізоциму з концентрацією 25 мг×мл -1 наносили в 40 мл буфера, що урівноважує, зі швидкістю 20 мл×хв , адсорбент промивали 400 -1 мл того ж буфера зі швидкістю 80 мл×хв , елюцію лізоциму здійснювали 200 мл 0,025 М Na-1 фосфатного буфера, рН 7,5, який містить 0,5 М NaCl, зі швидкістю потоку буфера 8 мл×хв . Колонку, наповнену Phenyl Sepharose HP, урівноважували 800 мл 0,050 М Nа2НРО4-буфера, -1 рН 7,0, що містить 2 М (NH4)2SO4, зразок лізоциму з концентрацією 25 мг×мл наносили в 40 мл -1 буфера, що урівноважує, зі швидкістю 20 мл×хв , адсорбент промивали 400 мл того ж буфера -1 зі швидкістю 80 мл×хв , елюцію лізоциму здійснювали 200 мл 0,050 М Na2HPO4 буфера, рН 7,0, -1 зі швидкістю потоку буфера 8 мл×хв . Колонку RESOURCE 15RPC урівноважували 30 мл 0,1 % розчином трифтороцтової кислоти -1 у воді, рН 2,0, зразок інсуліну з концентрацією 25 мг×мл наносили в 4-5 мл буфера, що -1 урівноважує, зі швидкістю 1 мл×хв , адсорбент промивали 15 мл того ж буфера зі швидкістю 1,5 -1 мл×хв , елюцію лізоциму здійснювали 6 мл 60 % ацетонітрилу в 0,1 % розчині трифтороцтової -1 кислоти у воді, рН 2,0, зі швидкістю потоку буфера 1 мл×хв . Фіг. 3 зображує собою графік зміни кількісного зв'язування фактора коагуляції VIII у комплексі з фактором фон Віллебранда (FVIII/vWF) синтезованим афінним адсорбентом з різною довжиною (MW 50, 250, 300 і 350 кДа) ліганду, а також інтенсивність «стікання» ліганду з адсорбенту в залежності від температурних умов процесу при постійній швидкості потоку буфера через хроматографічну колонку. Т-динамічна ємність синтезованих афінних адсорбентів при зміні температури мало залежить від довжини ліганду або від його молекулярної маси до 250 кДа, але швидко знижується для лігандів з масою 300 і 350 кДа. Зворотна залежність від величини ліганду спостерігається і для процесу його «стікання» з матриці адсорбенту при зростанні температури. Враховуючи практично однакові рівні «стікання» лігандів з молекулярною масою 50-250 кДа, для наступної роботи був обраний адсорбент з лігандом 250 кДа, у якого т-динамічна ємність -1 була на 10 % вище і складала для комплексу FVIII/vWF 27 мг×мл адсорбенту. Афінний адсорбент Sepharose-PPSB:FVIII/vWF (PPSB [75-76] - поліпептид, який специфічно зв'язує комплекс FVIII/vWF), синтезували на основі матриці Sepharose CL 4B (GE Healthcare AB, Швеція, Уппсала) і поліпептидів PPSB з різною молекулярною масою від 50 до 350 кДА згідно з рекомендаціями виробника. Колонки ХК 16/20 (GE Healthcare AB, Швеція, Уппсала) набивали 20 мл афінного адсорбенту на висоту шару 10 см, урівноважували 0,025 мМ трис-НСl буфером, рН 7,4, що містив 50 мМ -1 NaCl, зі швидкістю 2 мл×хв . На колонку наносили 20 мл зразка концентрату комплексу -1 факторів FVIII/vWF (25 мг×мл ). Для елюції використовували буфер, що урівноважує з рН 4,4, який містив 250 мМ NaCl. В елюйованих зразках вимірювали вміст FVIII:Ag (набір ELISA ASSERACHROM VIII:Ag, DiagnosticaStago S.A., Франція) і PPSB (IFA за допомогою антитіл проти поліпептиду). На Фіг. 4 представлена схема проведення інактивації ВВО і ВБО безпосередньо в процесі одного із етапів хроматографічного очищення цільового білка. Еквілібрацію адсорбенту, упакованого в хроматографічну колонку (ХК), здійснювали 5-10 Vc відповідного буфера. У зразок цільового білка, розчиненого в буфері, що урівноважує, ін'єктували модельні віруси відповідно до даних таблиці 1 і інкубували протягом 10-15 хв. при обережному перемішуванні. Табл. 1. Моделі вірусів, які були застосовані для визначення ефективності інактивації вірусів безпосередньо в хроматографічному процесі очищення і виділення цільового білка Розмір (нм) Ін'єкція в цільового білка 9 3,2×10 1,1×10* 8 8,4×10 8 l,8×10 Модель Вірус Оболонка/ геном Парвовірус собаки (CPV) Ентеровірус бика (BEV) Вірус гепатиту В качки (DHBV) Вірус вірусної діареї бика (BVDV) Вірус лейкемії миші типу С (MuLV) Вірус псевдосказу (PRV) В19 гепатит А гепатит В гепатит С Ні / ssДHК Ні / ssPHК Так / dsДНК Так / ssPHК 18-26 22-30 30-40 40-50 ВІЛ, ВЛТЛ Так / ssPHК 80-110 4,7×10 герпес Так / dsДНК 120-200 2,1×10 ВІЛ - вірус імунодефіциту людини, ВЛТЛ - Т-лімфотропний вірус людини, ssРНК/ssДНК - односпіральна форма рибо-/дезоксирибонуклеїнової кислоти, dsPHК/dsДHК - двоспіральна форма рибо-/дезоксирибонуклеїнової кислоти. 7 8 9 зразок UA 109254 C2 5 10 15 20 25 30 35 40 45 50 55 60 Потім зразок, інфікований одним з вірусів, наносили на афінний адсорбент, упакований в хроматографічну колонку (на схемі «ХК»), промивали 5-10 Vc буфера, що урівноважує, і в зібраному загальному об'ємі визначали вміст вірусів. Різницю в кількості вірусів, знайдених у зразку перед нанесенням на колонку, і в буфері, вимитому із колонки, приймали за вихідну величину кількості вірусів у зараженому білковому матеріалу QVвих. Потім цільовий білок, адсорбований в колонці, піддавали процесу інактивації вірусів (умови описані в Прикладах 1-6), промивці і елюції цільового білка з обов'язковим визначенням кількості вірусного матеріалу на кожному етапі - QVінк, QVвідм і QVбіл. Фактор зниження концентрації вірусів (ФЗК), тобто ступінь інактивації вираховували як логарифм відношення кількості вірусного матеріалу, виявленого на одній із стадій інактивації (QVінк, QVвідм і QVбіл), до кількості вихідного вірусного матеріалу (QVвих). Приклади реалізації винаходу В експериментальних роботах використовували набивні лабораторні колонки ХК 50/30, FineLINE Pilot 35 і хроматограф АКТAbasic 100 з програмним забезпеченням UNICORN 5,0 (GE Healthcare AB, Швеція, Уппсала). Колонки пакували, як вказано в інструкціях виробника, хроматографічними адсорбентами Q Sepharose High Performance, SP Sepharose High Performance, Phenyl Sepharose High Performance, SOURCE 15 RPC (GE Healthcare AB, Швеція, Уппсала), WorkBeads 40Q (Bio-Works Sweden AB, Бромма, Швеція) і афінним адсорбентом Sepharose-PPSB:FVIII/vWF на висоту шару адсорбенту 16 см. Цільові білки фактор коагуляції VIII - FVIII [77], IgG [78], церулоплазмін [79], лізоцим [80], 1-61 ФСК [81], фібрино(гено)літичний фермент [82] або їх концентрати з плазми крові донорів, коров'ячого молока, гідролізату стрептокінази, отрути щитомордника Agkistrodon blomhoffi ussuriensis отримували раніше описаними способами, їх активність/вміст вимірювали загальноприйнятими методами за допомогою IFA-наборів або стандартних флюоро/хромогенних реакцій [77-82]. Зразки білкових концентратів і отриманих фракцій відбирали і визначали концентрацію загального білка методом Бредфорда [83-84]. Процес вірус-інактивації методом «Розчинник-детергент» (РДП) здійснювали по схемі, наведеній на рисунку 4. В якості модельних ВВО/ВБО використовували віруси, представлені в Табл. 1. Концентрацію BVDV визначали методом RT-PCR з наборами «cador BVDV RT-PCR kit», BEV - «artus Enterovirus LC RT-PCR kit RUO», решти -«QuantiFast Pathogen+IC kit» (QIAGEN China (Shanghai) Co., Ltd., Китай, Шанхай). Вміст у цільовому білку після РДП розчинника визначали методом ГРХ, а детергенту HPLC. Приклад 1. Процес хроматографічної вірус-інактивації концентрату лізоциму молока за методом «розчинник/детергент». Колонку ХК 50/30 наповнювали 300 мл адсорбенту SP Sepharose High Performance на висоту 16 см згідно з рекомендаціями виробника й урівноважували 3 л 0,025 М гліцинового буфера, рН 3,0, що містив 3 мМ ЕДТА і 0,1 % твін 80 (БІ-А). Всі буфери і колонку перед процесом інактивації вірусів термостабілізували за допомогою термоциркулятора при 18 °С. -1 Інфікований вірусом (Табл. 1) зразок лізоциму з концентрацією 25 мг×мл наносили в 600 мл 1 буфера, що урівноважує, зі швидкістю 60 мл×хв" , адсорбент промивали 3 л того ж буфера зі -1 швидкістю 150 мл×хв . При проведенні інактивації вірусів автоматично створювався градієнтний буфер інактивації шляхом запрограмованого лінійного збільшення концентрації буфера, що урівноважує, який містив 0,7 % розчинника TNBP і 2 % детергента твін 80 (БІ-Б), в БІ-А від 0 до 100 % при -1 швидкості створення та нанесення на колонку 3000 мл×хв . БІ-А і БІ-Б, а також колонку в момент початку нанесення градієнта підігрівали від 18 до 30 °С за 1 годину, тобто градієнт -1 температури зростав зі швидкістю 12 °С×год . Таким чином, адсорбент зі зв'язаним лізоцимом промивали 3000 мл буфера градієнтами розчинника/детергента (до 0,7 % TNBP і 2 % твіну 80) і -1 температури (до 30 °С), що зростали. Адсорбент зі швидкістю 30 мл×хв промивали 3000 мл БІБ при 30 °С. Далі задавали зворотний градієнт температури і концентрації БІ-Б в БІ-А зі -1 швидкістю 3000 мл×год . Після досягнення температури 18 °С адсорбент промивали 3000 мл -1 буфера БІ-А зі швидкістю 60 мл×хв . Елюцію концентрату лізоциму здійснювали 600 мл 0,025 М -1 Na-фосфатного буфера, рН 7,5, що містив 0,5 М NaCl, зі швидкістю потоку буфера 60 мл×хв . Результати проведення РДП інактивації вірусів у концентраті лізоциму безпосередньо в процесі хроматографії представлені в Табл. 2. Дані таблиці 2 однозначно свідчать, що зв'язаний на хроматографічному адсорбенті лізоцим молока (у складі концентрату) піддавався тривалій (більше 2 годин) температурній обробці, 8 UA 109254 C2 5 проте його вміст (вихід) і активність після елюції практично не відрізнялася від вихідних значень. Разом з тим, істотно (на 6,9-7,9 log10) була знижена концентрація ВВО як за рахунок інактивації розчинником/детергентом (на 4,8-5,6 log10), так і за рахунок їх видалення на стадіях відмивки й елюції (≥1,9 log10). Інактивація за той же період часу ВБО була в 2 рази нижча, але додаткове видалення вірусів за рахунок відмивки (≥2,0 log10) призвело до достатньо високого сумарного фактора зниження концентрації (ФСК) вірусів - 3,7-4,5 log10. В отриманому цільовому білку використаними методами ні розчинник, ні детергент виявлені не були. Табл. 2. Результати хроматографічної вірус-інактивації концентрату лізоциму молока за методом «розчинник (0,7 % TNBP)/детергент (2 % твін 80)» за 30 °С на колонці ХК 50/30, наповненій 300 мл адсорбенту SP Sepharose HP Швидкість буфера на Вихід/Активність* -1 стадії, (Vc×хв ) цільового білка, (%) Аплікація, 18 °С 0,2 Відмивання, 18 °С 0,5 Інактивація-(+)град. 0,17 Відмивання, 30 °С 0,1 Інактивація-(-)град. 0,17 Відмивання, 18 °С 0,2 Елюція білка 0,2 98,87±3,04 97,33±4,17* Фактор зниження концентрації вірусів (log10) на стадії інактивації DHBV BVDV MuLV PRV CPV BEV 5,6 5,2 4,8 5,0 2,5 1,7 Фактор зниження концентрації вірусів (log10) на стадії відмивання DHBV BVDV MuLV PRV CPV BEV 2,1 1,8 2,1 1,7 2,1 2,0 Фактор зниження концентрації вірусів (log10) на стадії елюції цільового білка DHBV BVDV MuLV PRV CPV BEV 0,2 0,2 0,1 0,2 0,0 0,0 Фактор зниження концентрації вірусів (log10) сумарний DHBV BVDV MuLV PRV CPV BEV 7,9 7,2 7,0 6,9 4,5 3,7 Інактивація-(+)град. - стадія інактивації, градієнт розчинника/детергенту і температури наростає. 1нактивація-(-)град. - стадія інактивації, градієнт розчинника/детергента і температури знижується. 10 15 20 25 30 Приклад 2. Процес хроматографічної вірус-інактивації концентрату фібрино(гено)літичного ферменту (FgLE) із отрути щитомордника Agkistrodon blomhoffi ussuriensis за методом «розчинник/детергент». Колонку ХК 50/30 наповнювали 300 мл адсорбенту Q Sepharose High Performance на висоту 16 см згідно з рекомендаціями виробника і урівноважували 3 л 0,020 М імідазол-НСІ буфера, рН + -1 6,2, провідність по Na - 1 ,5 mS×cм (БІ-А). Всі буфери і колонку перед процесом інактивації вірусів термостабілізували за допомогою термоциркулятора при 18 °С. -1 Інфікований вірусом (Табл. 1) зразок FgLE з концентрацією 25 мг×мл наносили в 600 мл -1 буфера, що урівноважує, зі швидкістю 60 мл×хв , адсорбент промивали 3 л того ж буфера зі -1 швидкістю 150 мл×хв . При проведенні інактивації вірусів автоматично створювався градієнтний буфер інактивації шляхом запрограмованого лінійного збільшення концентрації буфера, що урівноважує, який містив 0,8 % розчинника TNBP і 2,4 % детергента тритона Х-100 (БІ-Б), в БІ-А від 0 до 100 % при швидкості створення і нанесення на колонку 3000 мл×год. БІ-А і БІ-Б, а також колонку в момент початку нанесення градієнта підігрівали від 18 до 33 °С за 1 годину, тобто градієнт температури -1 зростав зі швидкістю 15 °С×год . Таким чином, адсорбент зі зв'язаним FgLE промивали 3 л буфера, що мав градієнти розчинника/детергента (до 0,8 % TNBP і 2,4 % тритона Х-100) і -1 температури (до 33 °С), що зростали. Потім адсорбент зі швидкістю 30 мл×хв промивали 3000 мл БІ-Б при 33 °С. Після чого, задавали зворотний градієнт температури і концентрації БІ-Б в БІ-1 А зі швидкістю 3000 мл×год. . За досягнення температури 18 °С адсорбент промивали 1500 мл + -1 БІ-А і 1500 мл трис-НСl буфера, рН 8,2, що містив Na до провідності 31 mS×cм , зі швидкістю 9 UA 109254 C2 -1 5 10 15 60 мл×хв . Елюцію концентрату FgLE здійснювали 600 мл трис-НСІ буфера, рН 8,2, що містив + -1 -1 Na до провідності 56 mS×cм , зі швидкістю потоку буфера 60 мл×хв . Результати проведення РДП інактивації вірусів у концентраті FgLE безпосередньо в процесі хроматографії представлені в Табл. 3. Застосування позитивно зарядженого аніонообмінного адсорбенту сприяє зниженню концентрації як ВВО (~3,0 log10), так і ВБО (2,7-3,4 logio) У порівнянні з катіонообмінним адсорбентом (Табл. 2). Разом з приростом інактивації вірусів (5,0-6,0 log10) за рахунок збільшення концентрації розчинника/детергента і температури інактивації до 33 °С досягається сумарний ФЗК ВВО - 8,2-8,8 log10. Умови експериментів даного Прикладу жодного впливу на зміну ФЗК ВБО не здійснювали (1,8-2,2 log10), але значно збільшувався ФЗК для ВБО за рахунок їх видалення на стадіях відмивання і елюції (2,8-3,4 log10). Сумарний ФЗК для ВБО при даних умовах становив 5,2-5,6 log10. Вміст (вихід) і активність фібрино(гено)літичного ферменту після елюції його концентрату практично не відрізнялися від вихідних значень. В отриманому цільовому білку використаними методами ні розчинник, ні детергент виявлені не були. Табл. 3. Результати хроматографічної вірус-інактивації концентрату фібрино(гено)літичного ферменту з отрути щитомордника Agkistrodon hlomhoffx ussuriensis за методом «розчинник (1 % TNBP)/детергент (3 % тритон Х-100)» за 33 °С на колонці ХК 50/30, наповненій 300 мл адсорбенту Q Sepharose HP Швидкість буфера на Вихід/Активність* -1 стадії, (VC×XB ) цільового білка, (%) Аплікація, 18 °С 0,2 Відмивання, 18 °С 0,5 Інактивація-(+)град. 0,17 Відмивання, 33 °С 0,1 Інактивація-(-)град. 0,17 Відмивання, 18 °С 0,2 Елюція білка 0,2 99,39±6,72 98,61±5,53* Фактор зниження концентрації вірусів (log10) на стадії інактивації DHBV BVDV MuLV PRV CPV BEV 6,0 5,3 5,0 5,1 2,2 1,8 Фактор зниження концентрації вірусів (log10) на стадії відмивки DHBV BVDV MuLV PRV CPV BEV 2,7 2,5 3,1 3,0 3,4 2,7 Фактор зниження концентрації вірусів (log]0) на стадії елюції цільового білка DHBV BVDV MuLV PRV CPV BEV 0,1 0,1 0,1 0,2 0,0 0,1 Фактор зниження концентрації вірусів (log10) сумарний DHBV BVDV MuLV PRV CPV BEV 8,8 8,2 8,2 8,3 5,6 5,2 Інактивація-(+)град. - стадія інактивації, градієнт розчинника/детергенту і температури наростає. Інактивація-(-)град. - стадія інактивації, градієнт розчинника/детергенту і температури знижується. 20 25 30 Приклад 3. Процес хроматографічної вірус-інактивації концентрату імуноглобуліну G (IgG) із плазми крові донорів методом «розчинник/детергент». Колонку ХК 50/30 наповнювали 300 мл адсорбенту WorkBeads 40Q на висоту 16 см згідно з рекомендаціями виробника і урівноважували 3 л 0,020 М Na-ацетатного буфера, рН 6,5, + -1 провідність по Na - 1,4 mS×cм (БІ-А). Всі буфери і колонку перед процесом інактивації термостабілізували за допомогою термоциркулятора при 18 °С. -1 Інфікований вірусом (Табл. 1) зразок IgG з концентрацією 25 мг×мл наносили в 600 мл -1 буфера, що урівноважує, зі швидкістю 90 мл×хв , адсорбент промивали 3 л того ж буфера зі -1 швидкістю 150 мл×хв . При проведенні інактивації вірусів автоматично створювався градієнтний буфер інактивації шляхом запрограмованого лінійного збільшення концентрації буфера, що урівноважує, який містив 1 % розчинника TNBP і 3 % детергента твін 80 (БІ-Б), в БІ-А від 0 до 100 % при швидкості 10 UA 109254 C2 -1 5 10 створення і нанесення на колонку 3000 мл×год. . БІ-А, БІ-Б і колонку в момент початку нанесення градієнта підігрівали від 18 до 45 °С за 1 годину, тобто градієнт температури зростав -1 зі швидкістю 27 °С×год. . Таким чином, адсорбент зі зв'язаним IgG промивали 3 л буфера, який мав градієнти розчинника/детергенту (до 1 % TNBP і 3 % твін 80) і температури (до 45 °С), що 1 зростали. Потім адсорбент зі швидкістю 30 мл×хв" промивали 3000 мл БІ-Б при 45 °С і -1 задавали зворотний градієнт температури і концентрації БІ-Б в БІ-А зі швидкістю 3000 мл×год . -1 За досягнення температури 18 °С адсорбент промивали 3000 мл БІ-А зі швидкістю 90 мл×хв . Елюцію концентрату IgG здійснювали 900 мл 0,020 М Na-ацетатного буфера, рН 9,0, зі -1 швидкістю потоку буфера 30 мл×хв . Результати проведення РДП інактивації вірусів у концентраті IgG безпосередньо в процесі хроматографії представлені в Табл. 4. Табл. 4. Результати хроматографічної вірус-інактивації концентрату імуноглобуліну G з плазми крові донорів за методом «розчинник (1 % ТNВР)/детергент (3 % твін 80)» за 45 °С на колонці ХК 50/30, наповненій 300 мл адсорбенту WorkBeads 40Q Швидкість буфера на Вихід/Активність* -1 стадії, (VC×XB ) цільового білка, (%) Аплікація, 18 °С 0,3 Відмивання, 18 °С 0,5 Інактивація-(+)град. 0,17 Відмивання, 45 °С 0,1 Інактивація-(-)град. 0,17 Відмивка, 18 °С 0,3 Елюція білку 0,1 101,08±5,39 99,26±7,07* Фактор зниження концентрації вірусів (log10) на стадії інактивації DHBV BVDV MuLV PRV CPV BEV 6,6 6,2 5,7 6,1 2,5 2,1 Фактор зниження концентрації вірусів (log10) на стадії відмивки DHBV BVDV MuLV PRV CPV BEV 2,6 2,7 3,0 3,1 4,7 4,1 Фактор зниження концентрації вірусів (log10) на стадії елюції цільового білка DHBV BVDV MuLV PRV CPV BEV 0,2 0,3 0,1 0,1 0,1 0,2 Фактор зниження концентрації вірусів (log10) сумарний DHBV BVDV MuLV PRV CPV BEV 9,4 9,1 8,8 9,3 7,3 6,4 Інактивація-(+)град. - стадія інактивації, градієнт розчинника/детергенту і температури наростає. Інактивація-(-)град. - стадія інактивації, градієнт розчинника/детергенту і температури знижується. 15 20 25 30 Підвищення температури буфера інактивації з 33 до 45 °С сприяло підвищенню ФЗК для ВВО на 10-15 % і практично не впливало на ВБО. Рівень видалення ВБО за рахунок їх вимивання із позитивно зарядженого аніонообмінного адсорбенту буфером з високою температурою (45 °С) збільшувався на ≥50 %. Цей же процес для ВВО при відмиванні не залежав від температури буфера у межах 33-45 °С. Сумарний ФЗК при використанні буфера інактивації з температурою 45 °С зростав для ВВО на 0,6-1,0 log10, для ВБО на 1,2-1,7 log10 у порівнянні з РДП, в якому при інших рівних умовах температура інактивації була на 12 °С нижча (Табл. 4). В даних умовах сумарний ФЗК був у межах 8,8-9,4 і 6,4-7,3 log10 для ВВО і ВБО, відповідно. Такі показники є цілком достатніми для фактично повної інактивації як ВВО, так і ВБО, що інфікують білкові розчини природним шляхом. Вміст (вихід) і активність імуноглобулінів G після елюції їх концентрату практично не відрізнялися від вихідних значень. В отриманому цільовому білку використаними методами ні розчинник, ні детергент виявлені не були. Приклад 4. Процес хроматографічної вірус-інактивації концентрату церулоплазміну (СРт) із плазми крові донорів методом «розчинник/детергент». Колонку ХК 50/30 наповнювали 300 мл адсорбенту Phenyl Sepharose High Performance на висоту 16 см згідно з рекомендаціями виробника і урівноважували 3 л 0,020 М Na-фосфатного 11 UA 109254 C2 5 10 15 20 25 буфера, рН 7,2, що містив 1 М Na2SO4 (БІ-А). Всі буфери і колонку перед процесом інактивації вірусів термостабілізували за допомогою термоциркулятора при 18 °С. -1 Інфікований вірусом (Табл. 1) зразок СРm з концентрацією 25 мг×мл наносили в 100 мл -1 буфера, що урівноважує, зі швидкістю 90 мл×хв , адсорбент промивали 3 л того ж буфера зі -1 швидкістю 300 мл×хв . При проведенні інактивації вірусів автоматично створювався градієнтний буфер інактивації шляхом запрограмованого лінійного збільшення концентрації буфера, що урівноважує, який містив 3 % розчинника TNBP і 4 % детергенту тритон Х-100 (БІ-Б), в БІ-А від 0 до 100 % при -1 швидкості створення і нанесення на колонку 3000 мл×год . БІ-А, БІ-Б і колонку в момент початку нанесення градієнта підігрівали від 18 до 45 °С за 1 годину, тобто градієнт температури -1 зростав зі швидкістю 27 °С×год . Таким чином, адсорбент зі зв'язаним СРт промивали 3000 мл буфера, який мав градієнти розчинника/детергенту (до 3 % TNBP і 4 % тритона Х-100) і -1 температури (до 45 °С), що зростали. Адсорбент зі швидкістю 30 мл×хв промивали 3000 мл БІБ при 45 °С і задавали зворотний градієнт температури і концентрації БІ-Б в БІ-А зі швидкістю -1 3000 мл×год. . За досягнення температури 18 °С адсорбент промивали 3000 мл БІ-А зі -1 швидкістю 60 мл×хв . Елюцію концентрату СРт здійснювали 600 мл 0,020 М Na-фосфатного -1 буфера, рН 7,2, що містив 0,15 М Na2SO4 зі швидкістю буфера 60 мл×хв . Результати проведення РДП інактивації вірусів у концентраті СРm безпосередньо в процесі хроматографії представлені в Табл. 5. За додержання умов вірус-інактивації даного Прикладу ФЗК для ВВО і ВБО відповідали ФЗК Прикладу 3 і дорівнювали 8,5-9,2 та 6,0-7,2 log10, відповідно. Але приблизно 5 % цільового білка було втрачено за рахунок його дуже міцного зв'язування на адсорбенті, що підтвердила санітаризаційна відмивка Phenyl Sepharose High Performance за допомогою 0,5 М NaOH. В отриманому цільовому білку використаними методами ні розчинник, ні детергент визначені не були. Табл. 5. Результати хроматографічної вірус-інактивації концентрату церулоплазміну з плазми крові донорів за методом «розчинник (3 % TNBP)/детергент (4 % твін 80)» за 45 °С на колонці ХК 50/30, наповненій 300 мл адсорбенту Phenyl Sepharose HP Швидкість буфера на -1 стадії, (Vc×хв ) Аплікація, 18 °С 0,3 Відмивання, 18 °С 1,0 Інактивація-(+)град. 0,17 Відмивання, 45 °С 0,1 Інактивація-(-)град. 0,17 Відмивання, 18 °С 0,2 Елюція білка 0,2 Вихід/Активність* цільового білка, (%) 91,51±6,03 92,77±5,48* Фактор зниження концентрації вірусів (log10) на стадії інактивації DHBV BVDV MuLV PRV CPV BEV 6,4 6,4 5,7 6,0 2,2 1,9 Фактор зниження концентрації вірусів (log10) на стадії відмивання DHBV BVDV MuLV PRV CPV BEV 2,5 2,9 2,7 2,7 4,9 3,9 Фактор зниження концентрації вірусів (log10) на стадії елюції цільового білка DHBV BVDV MuLV PRV CPV BEV 0,2 0,1 0,1 0,2 0,1 0,2 Фактор зниження концентрації вірусів (log10) Сумарний DHBV BVDV MuLV PRV CPV BEV 9,1 9,2 8,5 8,9 7,2 6,0 Інактивація-(+) град. - стадія інактивації, градієнт розчинника/детергенту і температури наростає. Інактивація-(-)град. - стадія інактивації, градієнт розчинника/детергенту і температури знижується. 30 Приклад 5. Процес хроматографічної вірус-інактивації концентрату фрагмента стрептокінази 1-61 ФСК методом «розчинник/детергент». Колонку FineLINE Pilot 35 наповнювали 155 мл адсорбенту SOURCE 15RPC на висоту 16 см згідно з рекомендаціями виробника і урівноважували 1550 мл 0,1 % розчину трифтороцтової 12 UA 109254 C2 5 10 15 20 кислоти у воді, рН 2,0 (БІ-А). Всі буфери і колонку перед процесом інактивації термостабілізували за допомогою термоциркулятора при 18 °С. 1-61 -1 Інфікований вірусом (Табл. 1) зразок ФСК з концентрацією 25 мг×мл наносили в 200 мл -1 буфера, що урівноважує, зі швидкістю 50 мл×хв , адсорбент промивали 750 мл того ж буфера -1 зі швидкістю 75 мл×хв . При проведенні інактивації вірусів автоматично створювався градієнтний буфер інактивації шляхом запрограмованого лінійного збільшення концентрації буфера, що урівноважує, який містив 2 % розчинника TNBP і 3 % детергента тритон Х-100 (БІ-Б), в БІ-А від 0 до 100 % при -1 швидкості створення і нанесення на колонку 1500 мл×год . БІ-А, БІ-Б і колонку в момент початку нанесення градієнта підігрівали від 18 до 45 °С за 1 годину, тобто градієнт температури -1 1-61 зростав зі швидкістю 27 °С×год . Таким чином, адсорбент зі зв'язаним ФСК промивали 1500 мл буфера, який мав градієнти розчинника/детергента (до 2 % TNBP і 3 % тритона Х-100) і -1 температури (до 45 °С), що зростали. Адсорбент зі швидкістю 15 мл×хв промивали 1500 мл БІБ при 45 °С і задавали зворотний градієнт температури і концентрації БІ-Б в БІ-А зі швидкістю -1 1500 мл×год. За досягнення температури 18 °С адсорбент промивали 1500 мл БІ-А зі -1 1-61 швидкістю 75 мл×хв . Елюцію ФСК здійснювали 300 мл 60 % ацетонітрилу в 0,1 % розчині -1 трифтороцтової кислоти у воді, рН 2,0, зі швидкістю буфера 50 мл×хв . 1-61 Результати проведення РДП інактивації вірусів в концентраті ФСК безпосередньо в процесі хроматографії представлені в Табл. 6. Результати по вірус-інактивації, одержані в даному Прикладі 5 практично не відрізнялися від результатів Прикладу 4 як по сумарному ФЗК (8,9-9,2 та 6,1-6,9 log10 для ВВО і ВБО, відповідно), так і по виходу цільового білка (втрати приблизно 5 %). В отриманому цільовому білку використаними методами ні розчинник, ні детергент визначені не були. 25 Табл. 6. 1-61 Результати хроматографічної вірус-інактивації концентрату фрагмента стрептокінази ФСК за методом «розчинник (2 % TNBP)/детергент (3 % тритон Х-100)» за 45 °С на колонці FineLINE Pilot 35, наповненій 155 мл адсорбенту SOURCE 15RPC Швидкість буфера на Вихід/Активність* -1 стадії, (Vc×хв ) цільового білка, (%) Аплікація, 18 °С 0,3 Відмивання, 18 °С 0,5 Інактивація-(+)град. 0,17 Відмивання, 45 °С 0,1 Інактивація-(-)град. 0,17 Відмивання, 18 °С 0,5 Елюція білка 0,3 89,80±9,47 94,38±11,05* Фактор зниження концентрації вірусів (log10) на стадії інактивації DHBV BVDV MuLV PRV CPV BEV 6,2 6,1 6,1 6,0 2,3 1,9 Фактор зниження концентрації вірусів (log10) на стадії відмивання DHBV BVDV MuLV PRV CPV BEV 3,0 2,9 3,1 2,8 4,6 4,2 Фактор зниження концентрації вірусів (log10) на стадії елюції цільового білка DHBV BVDV MuLV PRV CPV BEV 0,0 0,0 0,0 0,1 0,0 0,0 Фактор зниження концентрації вірусів (log10) сумарний DHBV BVDV MuLV PRV CPV BEV 9,2 9,0 9,2 8,9 6,9 6,1 Інактивація-(+)град. - стадія інактивації, градієнт розчинника/детергенту і температури наростає. Інактивація-(-)град. — стадія інактивації, градієнт розчинника/детергента і температури знижується. 30 Приклад 6. Процес хроматографічної вірус-інактивації концентрату факторів коагуляції VIII і фон Віллебранда (FVIII/vWF) із плазми крові донорів за методом «розчинник/детергент». Колонку ХК 50/30 наповнювали 300 мл афінного адсорбенту Sepharose-PPSB:FVIII/vWF на висоту 16 см і урівноважували 3000 мл 0,025 мМ трис-НСl буфера, рН 7,4, що містив 50 мМ -1 NaCl (БІ-А), зі швидкістю 30 мл×хв . Всі буфери і колонку перед процесом інактивації вірусів термостабілізували за допомогою термоциркулятора при 8 °С. 13 UA 109254 C2 5 10 15 20 25 Інфікований вірусом (Табл. 1) зразок концентрату комплексу факторів FVIII/vWF з -1 1 концентрацією 25 мг×мл наносили в 100 мл буфера, що урівноважує, зі швидкістю 30 мл×хв - , -1 адсорбент промивали 3000 мл того ж буфера зі швидкістю 60 мл×хв . При проведенні інактивації вірусів автоматично створювався градієнтний буфер інактивації шляхом запрограмованого лінійного збільшення концентрації буфера, що урівноважує, який містив 0,5 % розчинника TNBP і 1,5 % детергента твін 80 (БІ-Б), в БІ-А від 0 до 100 % при -1 швидкості створення і нанесення на колонку 3000 мл×год. БІ-А, БІ-Б і колонку в момент початку нанесення градієнту підігрівали від 8 до 35 °С за 1 годину, тобто градієнт температури -1 зростав зі швидкістю 27 °С×год. Таким чином, адсорбент зі зв'язаним комплексом факторів FVIII/vWF промивали 3000 мл буфера, який мав градієнти розчинника/детергента (до 0,5 % TNBP і 1,5 % твін 80) і температури (до 35 °С), що зростали. Адсорбент промивали 6000 мл БІ-Б -1 при 35 °С зі швидкістю 30 мл×хв і задавали зворотні градієнти температури і концентрації БІ-Б -1 в БІ-А зі швидкістю 30 мл×год. За досягнення температури 8 °С адсорбент промивали 3000 мл -1 БІ-А зі швидкістю 30 мл×хв . Елюцію комплексу факторів FVIII/vWF здійснювали 600 мл 0,025 -1 мМ трис-НСl буфера, рН 4,4, що містив 250 мМ NaCl, зі швидкістю потоку буфера 30 мл×хв . Результати проведення РДП інактивації вірусів в концентраті комплексу факторів FVIII/vWF безпосередньо в процесі хроматографії представлені в Табл. 7. За рахунок зниження температури на 10 °С у порівнянні з попереднім прикладом (Табл. 6) ФЗК при інактивації зменшився на 0,8-1,2 log10. З іншого боку, за рахунок подовження часу РДП та відповідно об'єму буферів інактивації майже в 2 рази (4 год та 12 л проти 2,5 год. та 6 л у Прикладі 5) ФЗК відмивки для ВВО і ВБО зріс на 0,5-1,1 та 0,3-0,5 log10, відповідно. Тобто, сумарний рівень ФЗК майже не змінився у порівнянні з умовами Прикладів 3-5 (Табл. 4-6) при практично 100 % виході активності фактора VIII. В отриманому цільовому білку використаними методами ні розчинник, ні детергент визначені не були. Табл. 7. Результати хроматографічної вірус-інактивації концентрату факторів коагуляції VIII і фон Віллебранда (FVIII/vWF) з плазми крові донорів за методом «розчинник (0,5 % ТNВР)/детергент (1,5 % твій 80)» за 35 °С на колонці ХК 50/30, наповненій 300 мл афінного адсорбенту Sepharose-PPSB:FVIII/vWF Швидкість буфера на Вихід/Активність* -1 стадії, (Vc×хв ) цільового білка, (%) Аплікація, 18 °С 0,1 Відмивання, 18 °С 0,2 Інактивація-(+)град. 0,17 Відмивання, 45 °С 0,1 Інактивація-(-)град. 0,17 Відмивання, 18 °С 0,1 Елюція білка 0,1 101,51±4,97 99,69±7,31* Фактор зниження концентрації вірусів (logio) на стадії інактивації DHBV BVDV MuLV PRV CPV BEV 5,3 5,0 5,0 4,8 1,5 0,7 Фактор зниження концентрації вірусів (log10) на стадії відмивання DHBV BVDV MuLV PRV CPV BEV 3,7 3,8 3,6 3,9 4,9 4,7 Фактор зниження концентрації вірусів (log10) на стадії елюції цільового білка DHBV BVDV MuLV PRV CPV BEV 0,1 0,2 0,1 , 0,2 0,1 0,1 Фактор зниження концентрації вірусів (log10) сумарний DHBV BVDV MuLV PRV CPV BEV 9,1 9,0 8,7 8,9 6,5 5,5 Інактивація-(+)град. - стадія інактивації, градієнт розчинника/детергенту і температури наростає. Інактивація-(-)град. - стадія інактивації, градієнт розчинника/детергента і температури знижується. 30 Таким чином, Прикладами 1-6 показано, що інактивація вірусів методом «розчинник/детергент» в білкових концентратах безпосередньо в процесі хроматографічного отримання цільового білка, тобто, білка, який є зв'язаним на хроматографічному адсорбенті, складається з двох різних фаз, а саме 1) руйнування вірусної корпускули за допомогою 14 UA 109254 C2 5 10 15 20 25 30 35 40 45 50 55 розчинників і/або детергентів та 2) вимивання вірусного матеріалу з білкового концентрату разом з супутніми білками, які були зв'язані на адсорбенті менш міцно, ніж цільовий білок. Як відомо, руйнування розчинником/детергентом вірусів, покритих оболонкою, відбувається більш ефективно (на 3-4 log10), ніж вірусів без оболонки, що підтверджено даними Таблиць 2-7. ФЗК ВВО залежить від кількості і співвідношення розчинника і детергента в буфері інактивації, а також суттєво і пропорційно залежить від температури буфера інактивації та його об'єму, що був пропущений через колонку. Що стосується руйнування ВБО, то воно є досить низьким і практично не є залежним від концентрації розчинника і детергента, але має частково збільшення при підвищенні температури буфера інактивації (Табл. 2-7). Вимивання вірусів з цільового білка, який зв'язаний на адсорбенті, відбувається досить ефективно як для вірусів, вкритих оболонкою, так і для вірусів без оболонки. Таке вимивання залежить як від природи адсорбенту, від'ємно чи позитивно він заряджений, та пропорційно від температури та об'єму буфера, яким відмивають. Показано, що позитивно заряджені сильні аніонообмінні адсорбенти типу WorkBeads 40Q є найбільш ефективними при інактивації вірусів методом «розчинник/детергент» в білкових концентратах безпосередньо в процесі хроматографічного отримання цільового білка при температурі буфера інактивації 45 °С та часу інактивації не менш, ніж 2 години. Сумарно згадані етапи інактивації вірусів методом «розчинник/детергент» в білкових концентратах безпосередньо в процесі хроматографічного отримання цільового білка дають показники ФЗК вірусів, які є достатніми для фактично повної інактивації як ВВО, так і ВБО, що інфікують білкові розчини природним шляхом. Літературні джерела: st 1. Sofer G. Virus inactivation in the 1990s - and into the 21 Century. Part 2: Red blood cells and platelets // BioPharm. - 2002. - Vol. 15, No. 8. - P. 42-49. st 2. Sofer G. Virus inactivation in the 1990s - and into the 21 Century. Part 5: Desinfection // BioPharm International. - 2003. - Vol. 15, No. 8. - P. 42-49. st 3. Sofer G. Virus inactivation in the 1990s - and into the 21 Century. Part 3b: Plasma and plasma products (treatments other than heat or solvent/detergent) // BioPharm International. - 2002. - Vol. 15, No. 10. - P. 42-51. 4. Mohr H. Inactivation of viruses in human plasma methods // Enzymol. - 2000. - Vol. 319. - P. 207-216. 5. Mohr H., Lambrecht В., and Selz A. Photodynamic virus inactivation of blood components immunological investigations // Immunol. Invest. - 1995. - Vol. 24, No. 1, 2. - P. 73-85. 6. US Patent No. 6251644. Method for inactivating non-enveloped viral contaminants with a photosensitizer by increasing viral permeability to the photosensitizer. Int. C1. C12N 13/00, US C1. 435/173,3. Inv. - Sowemimo-Coker; Samuel O. Ass. - Baxter International, Inc. Round Lake, IL, USA. 26,06,2001. 7. Grandadam M., Ingrandb D., Hurauxa J.-M., Avelinec В., Delgadoc O., Vever-Bizetc C, Brault D. Photodynamic inactivation of cell-free HIV strains by a red-absorbing chlorin-type photosensitizer // J. Photochem. Photobiol. B. - 1995. - Vol. 1, No. 3. - P. 171-177. 8. Muller-Breitkreutz K., Mohra H., Brivibab K., Sies H. Inactivation of viruses by chemically and photochemically generated singlet molecular oxygen // J. Photochem. Photobiol. B. - 1995. - Vol. 30, No. 1. - P. 63-70. 9. Morel P., Lin L., Wiesehahn G., Corash L. Photochemical inactivation of viruses and bacteriophage in plasma and plasma fractions // Blood Cells. - 1992. - Vol. 18, No. 1. - P. 27-41, 4142. 10. Dichtelmuller H., Rudnick D., Breuer В., Ganshirt KH. Validation of virus inactivation and removal for the manufacturing procedure of two immunoglobulins and a 5 % serum protein solution treated with beta-propiolactone // Biologicals. - 1993. - Vol. 21, No. 3. - P. 259-268. 11. Lawrence S.A. Beta-Propiolactone: viral inactivation in vaccines and plasma products // PDA J. Pharm. Sci. Technol. - 2000. - Vol. 54, No. 3. - P. 209-217. 12. Scheidler A. Inactivation of viruses by beta-propiolactone in human cryo-poor plasma and IgG concentrates // Biologicals. - 1998. - Vol. 26, No. 2. - P. 135-144. 13. Norley S.G., Lower J., Kurth R. Insufficient inactivation of HIV-1 in human cryo poor plasma by beta-propiolactone: results from a highly accurate virus detection method // Biologicals. - 1993. - Vol. 21, No. 3. - P. 251-258. 14. Epstein J.S., Fricke W.A. Current safety of clotting factor concentrates // Arch. Pathol. Lab. Med. - 1990. - Vol. 114, No. 3. - P. 335-340. 15 UA 109254 C2 5 10 15 20 25 30 35 40 45 50 55 60 15. Korneyeva M., Hottaa J., Lebingb W., Rosenthala R.S., Franksc L., Petteway S.R., Jr. Enveloped virus inactivation by caprylate: a robust alternative to solvent/detergent treatment // Biologicals. - 2002. - Vol. 30, No. 2. - P. 153-162. 16. US Patent No. 5886154. Chromatographic method for high yield purification and viral inactivation of antibodies. Int. С1. С07К 16/6, US C1. 530/390,1. Inv. - Lebing W.R.; Aired P.; Lee D.C.; Paul H.-I. Ass. - same. 23,03,1999. 17. Marx G., Моu X., Freed R., Ben-Hur E., Yang C, Horowitz B. Protecting fibrinogen with rutin during UVC irradiation for viral inactivation // Photochem. Photobiol. - 1996. - Vol. 63, No. 4. - P. 541546. 18. Burnouf Т., Goubran H. A., Radosevich M., El-Ekiaby M. Preparation and viral inactivation of cryoprecipitate in blood banks in resource-limited countries // ISBT Science Series. - 2007. - Vol. 2, No. 2. - P. 121-128. 19. Hosseini M., Sawyer M., Schore C, Vu J., Trzepla-Nabagie K., Pina C, Smith W., LagunasSolar M., Osburn B.I. Inactivating adventitious viruses while preserving biological activity: treating fetal bovine serum with pulsed ultraviolet light // BioPharm. - 2002. - Vol. 15, No 12. - P. 35-40. 20. Schmidt S., Kauling J. Process and laboratory scale UV inactivation of viruses and bacteria using an innovative coiled tube reactor // Chem. Engineer. Technol. - 2007. - Vol. 30, No. 7. - P. 945950. 21. Wiggnton K.R., Menin L., Montoya J.P., Kohn T. Oxidation of virus protein during UV254 and singlet oxygen mediated inactivation // Environ. Sci. Technol. - 2010. - Vol. 44, No. 14. -P. 5437-5443. 22. Roberts P., Hope A. Virus inactivation by high intensity broad spectrum puls light // J. Virat Methods. - 2003. - Vol. 110, No. 1. - P. 61-65. 23. Cover W.H. Validation of broad spectrum pulsed light as a virus inactivation step. PDA/FDA Viral Clearance Forum, October 2001, Bethesda, MD, USA. P. 43-48. 24. Hersberger M., Nusbaumer N., Scholer A., Knopfli V., von Eckardstein A. Influence of practicable virus inactivation procedures on tests for frequently measured analytes in plasma // Clin. Chem. - 2004. - Vol. 50. - P. 944-946. 25. Omar A., Kempf C, Immelmann A., Rentsch M., Morgenthaler J.-J. Virus inactivation by pepsin treatment at pH 4 of IgG solutions: factors affecting the rate of virus inactivation // Transfusion. - 1996. - Vol. 36, No. 10. - P. 866-872. 26. Tanaka К., Sawatani Т., Dias G.A., Shigueoka E.M., Campos T.C.X.B., Nakao H.C., Arashiro F. High quality human immunoglobulin G purified from Cohn fractions by liquid chromatography // Braz. J. Med. Biol. Res. - 2000. - Vol. 33, No. 1. - P. 27-30. 27. Bumouf Т., Griffiths E., Padillac A., Seddikd S., Stephanoe M.A., Gutierrrez J.-M. Assessment of the viral safety of antivenoms fractionated from equine plasma // Biologicals. - 2004. - Vol. 32, No. 3. - P. 115-128. 28. Louie R.E. Galloway C.J., Dumas M.L., Wong M.F., Mitra G. Inactivation of hepatitis С virus in low pH intravenous immunoglobulin // Biologicals. - 1994. - Vol. 22, No. 1. P. 13-19. 29. Tsujimoto K., Uozaki M., Ikeda K., Yamazaki H., Utsunomiya H., Ichinose M, Koyama A.H., Arakawa T. Solvent-induced virus inactivation by acidic arginine solution // Int. J. Моl. Med. - 2010. Vol. 25, No. 3. - P. 433-437. 30. US Patent No. 4540573. Undenatured virus-free biologically active protein derivatives. Int. С1. А61К 39/00, US C1. 424/85. Inv. - Neurath A.R., Horowitz B. Ass. - New York Blood Center, Inc., New York, N.Y., USA. 09,10,1985. 31. Horowitz В., Prince A.M., Horowitz M.S., Watklevicz С Viral safety of solvent-detergent treated blood products // Dev. Biol. Stand. - 1993. Vol. 81. - P. 147-161. 32. US Patent No. 547954. Ultrapurification process for Factor VIII. Int. С1. А61К 35/16, US C1. 530/383. Inv. - Neslund G.G., Liu S.-L., Griffith M.J. Ass. - Baxter International Inc., Deerfield, Ill., USA. 11,28,1995. 33. Raut S., Giambattista M., Bevan S.A., Hubbard A.R., Barrowcliffe T.W., Laub R. Modification of factor VIII in therapeutic concentrates after virus inactivation by solvent-detergent and pasteurization // Thromb. Haemost. - 1998. - Vol. 80, No. 4. - P. 624-631. 34. Kim I.S., Choi Y.W., Woo H.S., Chang C.E., Lee S. Solvent/detergent inactivation and chromatographic removal of human immunodeficiency virus during the manufacturing of a high purity antihemophilic factor VIII concentrate // J. Microbiol. - 2000. - Vol. 38, No. 3. - P. 187-191. 35. Pamphilon D. Viral inactivation of fresh frozen plasma // Br. J. Haematol. - 2000. - Vol. 109, No. 4. P. 680-693. 36. Roberts P.L., Hart H. Comparison of the inactivation of canine and bovine parvovirus by freeze-drying and dry-heat treatment in two high purity factor VIII concentrates // Biologicals. - 2000. Vol. 28, No. 3. - P. 185-188. 16 UA 109254 C2 5 10 15 20 25 30 35 40 45 50 55 37. Burnouf Т., Goubran H. A., Radosevich M., Sayed M.A., Gorgy G., El-Ekiaby M. A minipool process for solvent-detergent treatment of cryoprecipitate at blood centers using a disposable bag system // Vox Sanguinis. - 2006. - Vol. 91, No. 1. - P. 56-62. 38. Stadler M., Graber G., Kannicht C, Biesert L., Radomski K.U. et al. Characterisation of a novel high-purity, double virus inactivated von Willebrand factor and factor VIII concentrate (Wilate) // Biologicals. - 2006. - Vol. 34, No. 4. - P. 281-288. 39. Roberts P.L. Virus inactivation by solvent/detergent treatment using Triton X-100 in a high purity factor VIII // Biologicals. - 2008. - Vol. 36, No. 5. - P. 330-335. 40. Roberts P.L, Lloyd D., Marshall P.J. Virus inactivation in a factor VIII/vWF concentrate using a solvent/detergent procedure based on polysorbate 20 // Biologicals. - 2009. - Vol. 37, No. 1. - P. 2631. 41. Dichtelmuller H.O., Beisert L., Fabbrizzi F., Gajardo R., Groner A. et al. Robustness of solvent/detergent treatment of plasma derivatives: a data collection from Plasma Protein Therapeutics Association member companies // Transfusion. - 2009. - Vol. 49, No. 9. - P. 1931-1943. 42. Octapharma - Web resource for von Willebrand disease healthcare professionals and patients site. «Wilate is a double virus inactivated vWF/FVIII complex» in Double virus inactivation // http://www.wilateusa.com/index.php?option=com_content&view=article&id=19&Itemid=49. 2011. 4 P. 43. Johnston A., Macgregor A., Borovec S., Hattark M., Stuckly K. et al. Inactivation and clearance of viruses during the manufacture of high purity factor IX // Biologicals. - 2000. - Vol. 28, No. 3. - P. 129-136. 44. Roberts P.L., Dunkerley C. Effect of manufacturing process parameters on virus inactivation by solvent-detergent treatment in a high-purity factor IX concentrate // Vox Sanguinis. - 2003. - Vol. 84, No. 3. - P. 170-175. 45. Grifols Biologicals Inc. Factor IX complex ROFILNINE SD solvent detergent treated // Los Angeles, CA, USA. http://pathology.columbia.edu/education/residents/current/cp/TM/components/profilnine.pdf. 2004. 2 P. 46. Shin J.S., Choi Y.W., Sung H.M., Ryu Y.-W., Kim I.S. Enhanced virus safety of a solvent/detergent-treated antihemophilic factor IX concentrate by dry-heat treatment // Biotech. Bioproc. Engineer. - 2006. - Vol. 11. - P. 19-25. 47. Kim I .S., Bae J.E., Sung H.M., Kang Y., Choi Y.W. Removal and inactivation of viruses during the manufacture of a high-purity antihemophilic factor IX from human plasma // Biotech. Bioproc. Engineer. - 2009. - Vol. 14, No. 6. - P. 716-724. 48. Pourmokhtar M., Dinarvand R., Mousavi Hosseini K., Rezvan H., Jalili M. A. Solvent-detergent treatment of IgM-enriched immunoglobulin // Dam. - 2003. - Vol. 11, No. 2. - P. 47-51. 49. Aghaie A., Pourfatollah A.A., Bathaie S.Z., Moazzeni S.M., Khorsand Mohammad Pour H., Banazadeh S. Preparation, purification and virus inactivation of intravenous immunoglobulin from human plasma // Hum Antobodies. - 2010. - Vol. 19, No. 1. - P. 1-6. 50. US Patent No. 7655233 B2. Optimal placement of a robust solvent/detergent process post viral ultrafiltration of an immune gamma globulin. Int. С1. А61K 39/00, US C1. 424/176,1. Inv. - Van Holten R.W.., Autenrieth S.M. Ass. - Ortho-Clinical Diagnostics, Inc., Rochester, NY, USA. 02,02,2010. 51. Korneyeva M., Hottaa J., Lebing W., Rosenthal R.S., Franks L., Petteway S.R., Jr. Enveloped virus inactivation by caprylate: a robust alternative to solvent-detergent treatment in plasma derived intermediates // Biologicals. - 2002. - Vol. 30, No. 2. - P. 153-162. 52. Aghaie A., Pourfatollah A.A., Bathaie S.Z., Moazzeni S.M., Khorsand Mohammad Pour H., Sharifi Z. Inactivation of virus in intravenous immunoglobulin G using solvent/detergent treatment and pasteurization // Hum Antobodies. - 2008. - Vol. 17, No. 3-4. - P. 79-84. 53. Nesterova N.V., Kurkina O.V., Samoilenko V.A., Skrynnyk M.M. Physico-chemical and biological properties of solvent/detergent treated immunoglobulin G preparations // Мікробіол. журн. 2009. - Т. 71, № 6. - c. 35-42. 54. US Patent Application Publication. Pub. No.: US 2010/0233149 A1. Methods for preparing factor X, activated factor X, inactivated factor X and inactivated factor Xa, and pharmaceutical compositions comprising same. Int. C1. A61K 38/48 (C12N 9/48, A61P 7/02), US C1. 424/94,64. Inv. Lloyd J., Feldman P. Ass. - Lloyd J., Feldman P., Hertforshire, Great Britain. 09,16,2010. 55. WO Patent, Int. Pub. No. WO 2011/032195 A1. Medicinal products for the treatment of blood coagulation disorders containing thrombin-free factor Xia concentrate. Int. С1. А61K 38/36, A61P 7/04. Inv. - Eibl J. Ass. - Bioproducts & Bio-engineering AG, Wien, Austria. 03,24,2011. 17 UA 109254 C2 5 10 15 20 25 30 35 40 45 50 55 60 56. Chao S.-F., Gall M., Hotta J.-A., Lang J., Lee D.C., Roth N.J. Effective and robust enveloped virus inactivation by a non-traditional solvent/detergent treatment step // US Respiratory Disease. 2010. - Vol. 6. - P. 40-46. 57. Darling A.J. Validation of the purification process for viral clearance evaluation in: Biopharmaceutical process validation / ed.: G. Sofer, D.W. Zabriskie, Marcel Dekker Publ., NY, USA. 2000. 382 P. 58. Viral clearance by chromatography: predicting optimal conditions and media lifespan in: Downstream, No. 31, 2005. Ed. GE Healthcare AB, Sweden. P. 10-11. 59. Curtis S. Generic/matrix evaluation of M-MulV and SV40 clearance by anion exchange th chromatography in flow-through mode. Oral presentation at IBC's 8 International conference on process validation for biological. March 7-8, 2005. San Diego, CA, USA. 60. Choi Y.W., Kim I.S. Viral clearance during the manufacturing of urokinase from human urine // Biotechnol. Bioproc. Engin. - 2008. - Vol. 13, No. 1. P. 25-32. 61. Adcock W.L., MacGregor A., Davies J.R., Hattarki M., Anderson D.A., Goss N.H. Chromatographic removal and heat inactivation of hepatitis A during manufacture of human albumin // Biotechnol. Appl Biochem. - 1998. - Vol. 28, No. 1. - P. 85-94. 62. Strauss D.M., Lute S., Tebaykina Z., Frey D.D. et al. Understanding the mechanism of virus removal by Q sepharose fast flow chromatography during the purification of CHO-cell derived biotherapeutics // Biotechnol. Bioeng. - 2009. - Vol. 104, No. 2. - P. 371-380. 63. Groner A. Virus removal by chromatography used for production of plasma-derived products // PDA Virus & TSE Safety Forum. June 27-30, 2011, Barcelona, Spain. 64. Privalov P.L. Thermodynamic problems of protein structure // Annu. Rev. Biophys. Biophys. Chem. - 1989. - Vol. 18. - P. 47-69. 65. Gorinstein S., Caspi A., Rosen A., Goshev I. et al. Structure characterization of human serum proteins in solution and dry state // J. Peptide Res. - 2002. Vol. 59. - P. 71-78. 66. Kleppe R., Haavik J. Different stabilities and denaturation pathways for structurally related aromatic amino acid hydroxylases // FEBS Lett. - 2004. - Vol. 565, No. 1-3. - P. 155-159. 67. Caldarelli G., De Los Rios P. Cold and warm denaturation of proteins // J. Biol. Physics. 2001. - Vol. 27, No. 2-3. - P. 229-241. 68. Olivares-Quiroza L., Garcia-Colin L.S. Protein's native state stability in a chemically induced denaturation mechanism // J. Theor. Biol. - 2007. - Vol. 246, No. 2. - P. 214-224. 69. Chang B.H., Bae Y.C. Salting-out in the aqueous single-protein solution: the effect of shape factor // Biophys. Chem. - 2003. - Vol. 104, No. 2. - P. 523-533. 70. Pace C.N., Fu H., Fryar K.L., Landua J. et ak. Contribution of hydrophobic interactions to protein stability // J. Моl. Biol. - 2011. - Vol. 408, No. 3. - P. 514-528. 71. Mchaourab H.S., Godar J.A., Stewart P.L. Structure and mechanism of protein stability sensors: chaperone activity of small heat shock proteins // Biochemistry. - 2009. - Vol. 48, No. 18. - P. 3828-3837. 72. Quan S., Koldewey P., Tapley Т., Kirsch N. et al. Genetic selection designed to stabilize proteins uncovers a chaperone called Spy // Nat. Struct. Моl. Biol. - 2011. - Vol. 18, No. 3. - P. 262269. 73. Hongo-Hirasaki Т., Yamagushi К., Yanagida К., Hayashida H., Ide S. Effects of varying virusspiking conditions on a virus-removal filter Planova 20N in a virus validation staudi of antibody solutions // Biotechnol. Prog. - 2011. - Vol. 27, No. 1. - P. 162-169. 74. Francis P., Haynes C.A. Scale-up of controlled-shear affinity filtration using computational fluid dynamics // Biotechnol. J. - 2009. Vol. 4, No. 5. - P. 665-673. 75. Lutters B.C.H., Meijers J.C.M., Derksen R.H.W.M., Arnout J., De Groot P.G. Dimers of p 2glycoprotein I mimic the in vitro effects of P2-glycoprotein I-anti-pVglycoprotein I antibody complexes // J. Biol. Chem. - 2001. Vol. 276, No. 5. - P. 3060-3067. 76. Munoz C.C., Campbell W., Constantinescu I., Gyongyossy-Issa M.I. Rational design of antithrombotic peptides to target the von Willebrand factor (vWf)-GPIb integrin interaction // J. Mol. Model - 2008. - Vol. 14, No. 12. - P. 1191-202. 77. Chromatographic production of human plasma proteins factor VIII, albumin and IgG / General Electric Company - GE Healthcare Amersham Biosciences AB, Uppsala, Sweden. Ed. Elanders Tofters, Sweden, 2005. P. 1-26. 78. Волков Г.Л. Технологія одержання імуноглобулінів. І. Технологічні аспекти очищення // Укр. біохім. ж. - 2005. - Т. 78, № 3. - c. 88-98. 79. Волков Г.Л., Гаврилюк СП., Скалка В.В., Гаврилюк Е.С. Метод АРС в пилотном производстве церулоплазміну як Приклад преимуществ современной промышленной хроматографії // Биофармацевтический ж. - 2009. - Т. 1, № 4. - c. 35-41. 18 UA 109254 C2 5 10 15 20 80. Волков Г.Л., Краснобрижая Е. Н., Гаврилюк С.П., Гаврилюк Е.С. Промышленная хроматографія білків з близкими физико-химическими свойствами. // Биофармацевтический ж. 2009. - Т. 1, № 4. - c. 20-34. 81. Mongolia Patent No. 3168. Manufacturing of activating plasminogen hydrolysis streptokinase fragment with molecular weight of 7 kDa. Int. С1. А61K 31/711Н. Inv. - Volkov G.L., Gavrylyuk S.P., Savchuk O.M. Ass. - Wisdoms Assets Holdings LLC, Mongolia, Ulaanbaatar. 15,07,2008, Bul. № 23. 82. Скалка В.В., Волков Г.Л., Краснобрижая Е.Н., Гаврилюк СП., Жукова А.И., Нарантуяа Нандинцэцэг, Чимгээ Туваансурэн, Одончимэг Ганболд, Болор Буянба-драх, Сумия Бямбасурэн, Понсандулам Дашням, Дармостук М.С., Гаврилюк Е.С. Получение і характеристика фібрино(гено)литического фермента із яда щитомордника рода Agkistrodon blomhoffi // Биофармацевтический ж. - 2010. - Т. 2, № 1. - c. 32-39. 83. Bradford M.M. A rapid and sensitive for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding // Anal. Biochem. - 1976. - Vol. 72. - P. 248-254. 84. Stoscheck C.M. Quantitation of protein // Methods Enzymol. - 1990. - Vol. 182. - P. 50-69. He дивлячись на те, що даний винахід описаний із зазначенням найкращих варіантів реалізації винаходу, зрозуміло, що ними він не обмежується. Для фахівця в даній галузі техніки відомо, що різні модифікації можуть бути описані, тому обсяг прав за даним винаходом обмежується тільки прикладеною формулою винаходу. Всі публікації, представлені в описі винаходу у вигляді посилань, включені в даний опис у всій їх повноті. ФОРМУЛА ВИНАХОДУ 25 30 35 40 45 50 55 60 1. Спосіб хроматографічної вірус-інактивації розчину цільового білка, комплексу білків або пептиду, у якому піддають кондиціюванню розчин цільового білка, комплексу білків або пептиду; здійснюють аплікацію кондиційованого розчину цільового білка, комплексу білків або пептиду на хроматографічну колонку з адсорбентом зі зв′язуванням білка, комплексу білків або пептиду; промивають адсорбент буфером аплікації; промивають адсорбент буфером інактивації вірусів, який містить розчинник і детергент у буфері аплікації; відмивають адсорбент від розчинника/детергента та продуктів розпаду вірусів буфером аплікації; та елююють розчин цільового білка, комплексу білків або пептиду з адсорбенту; де зазначений адсорбент являє собою іонообмінний, афінний, обернено-фазовий, або адсорбент гідрофобної взаємодії. 2. Спосіб за п. 1, у якому адсорбент являє собою афінний адсорбент, що має динамічну ємність -1 зв′язування не менше 15 мг білка×мл адсорбенту, при швидкості протікання буфера від 10 до -1 50 мл×хв . 3. Спосіб за п. 1, у якому адсорбент являє собою іонообмінний сорбент, вибраний з аніоніту або -1 катіоніту, з динамічною ємністю зв′язування білка від 50 мг×мл адсорбенту, при швидкості -1 протікання буфера від 50 до 500 cм×год. . 4. Спосіб за п. 1, у якому адсорбент являє собою обернено-фазовий адсорбент, якщо цільовий білок, комплекс білків або пептид є низькомолекулярним (МВ=1-30 кДа), та адсорбент являє собою іонообмінний, афінний або адсорбент гідрофобної взаємодії, якщо цільовий білок, комплекс білків або пептид має середню або високу молекулярну масу (МВ=20-≥4000 кДа). 5. Спосіб за будь-яким з пп. 1, 2 або 4, у якому афінний адсорбент являє собою хімічно нейтральну, стійку до тиску в 5-10 бар полімерну матрицю з пришитим поліпептидним фрагментом або хімічною сполукою, яка специфічно взаємодіє з цільовим білком, комплексом білків або пептидом. 6. Спосіб за п. 5, у якому пришитий поліпептидний фрагмент є синтетичного, рекомбінантного або природного походження. 7. Спосіб за п. 5 або п. 6, у якому молекулярна маса пришитого поліпептидного фрагмента становить від 1 до 500 кДа. 8. Спосіб за будь-яким з пп. 1, 3 або 4, у якому адсорбент являє собою аніонообмінний адсорбент у випадку видалення денатурованих нуклеїнових кислот вірусів або їх фрагментів. 9. Спосіб за будь-яким з пп. 1 - 8, у якому інактивуючий буфер містить: розчинник, вибраний з групи, що включає: ди- і триалкілфосфати, які містять алкільний ланцюг довжиною 1-10 вуглеводних атомів, такі як ди-n-пропілфосфат (DNPP) і три-n-бутилфосфат (TNBP); прості ефіри, такі як етиловий і пропіловий; складні ефіри, такі як амілацетат; 19 UA 109254 C2 5 10 15 20 25 30 алкільовані та гідроксильовані сполуки, такі як бутиловий ефір гідроксиланізолу (ВНА) і бутиловий ефір гідрокситолуолу (ВНТ); та їх суміші, необов′язково з додатковими органічними сполуками в концентрації 0,002-3,0 %; детергент, вибраний з групи, що включає: оксіетильовані алкілфеноли, такі як тритони К-60, Х45, Х-100, W-30; холат і деоксихолат натрію; сульфобетаїни, такі як додецилсульфонат натрію; додецилбензосульфонат натрію (Nacconol NR), N-додециламіноетансульфонова кислота, 2сульфоетилолеат натрію (Igepon A); поліоксіетильовані похідні складних ефірів, такі як полісорбат 20 або твін 20, полісорбат 80 або твін 80; ефіри поліоксіетильованих жирних спиртів, такі як Brij 35; конденсат етиленоксиду і пропіленоксиду (Pluronic copolymers); оксіетильовані аміни, такі як Ethomeen; нонідет Р-40 (Nonidet P-40) і люброкс PХ (Lubrox PX); неіонні детергенти, такі як каприлат в концентрації 0,005-5,0 %. 10. Спосіб за будь-яким з пп. 1-9, у якому промивання адсорбенту буфером інактивації вірусів здійснюють в чотири стадії, де на першій стадії інактивації вірусів промивання здійснюють градієнтом буфера інактивації, концентрація якого у буфері аплікації зростає від 0 % до 100 %, -1 об′ємом 5-10 Vc зі швидкістю 0,1-0,2 Vc×хв , на другій стадії - 10-20 Vc буфером інактивації зі -1 швидкістю 0,1-0,2 Vc×хв , на третій стадії градієнтом буфера інактивації, концентрація якого у -1 буфері аплікації знижується від 100 % до 0 %, об′ємом 5-10 Vc зі швидкістю 0,1-0,2 Vc×хв , на -1 четвертій стадії - буфером аплікації об′ємом 10-20 Vc зі швидкістю 0,1-0,2 Vc×хв . 11. Спосіб за п. 10, у якому промивання адсорбенту на першій стадії інактивації вірусів здійснюють буфером інактивації з температурою, що зростає від 4-18 °С до 30-45 °С, зі -1 швидкістю 0,25-0,50 °С×хв , на другій стадії - з температурою 30-45 °С, на третій стадії - з -1 температурою, що знижується від 30-45 °С до 4-18 °С, зі швидкістю 0,25-0,50 °С×хв , на четвертій стадії - буфером аплікації з температурою 4-18 °С. 12. Спосіб за будь-яким з пп. 1-11, у якому хроматографічна колонка оснащена пристроєм для термостабілізації. 13. Спосіб за п. 12, у якому температуру хроматографічної колонки відтворюють відповідно до температури процесу інактивації вірусів і наступного відмивання адсорбенту. 14. Спосіб за п. 1, у якому адсорбент являє собою аніонообмінний адсорбент з максимальним зворотним тиском не менше 3 бар, динамічною ємністю зв′язування -1 БСА≥150 мг×мл при швидкості потоку буфера через колонку -1 від 50 до 500 см×год. , типу WorkBeads 40 Q, температура інактивуючого буфера становить від -1 40 до 45 °С, при швидкості потоку через колонку 0,1-0,2 Vc×хв та тривалості інактивації не менше 2,0 год. 20 UA 109254 C2 21 UA 109254 C2 22 UA 109254 C2 23 UA 109254 C2 Комп’ютерна верстка Д. Шеверун Державна служба інтелектуальної власності України, вул. Василя Липківського, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 24
ДивитисяДодаткова інформація
МПК / Мітки
МПК: A61K 38/22, G01N 30/02, C07K 14/755, A61K 35/76
Мітки: спосіб, вірус-інактивації, хроматографічної
Код посилання
<a href="https://ua.patents.su/26-109254-sposib-khromatografichno-virus-inaktivaci.html" target="_blank" rel="follow" title="База патентів України">Спосіб хроматографічної вірус-інактивації</a>
Спосіб інактивації інфекційних властивостей гемаглютинуючих вірусів

Номер патенту: 92404
Опубліковано: 11.08.2014
Автори: Музика Денис Васильович, Стегній Борис Тимофійович, Алтахер Харітх Абдулла
МПК: C12N 7/04
Мітки: спосіб, властивостей, інактивації, інфекційних, вірусів, гемаглютинуючих
Формула / Реферат:
Спосіб інактивації інфекційних властивостей гемаглютинуючих вірусів, що включає інактивацію вірусів, який відрізняється тим, що одержують вірусовміщуючу рідину, центрифугують, доводять рН вірусної рідини до 7,4 та використовують як інактиванта бінарний етиленімін в кінцевій концентрації 1-3 %.
Спосіб інактивації вірусів при одержанні церулоплазміну

Номер патенту: 22953
Опубліковано: 25.04.2007
Автори: Куркіна Оксана Вікторівна, Скринник Максим Михайлович, Самойленко Вадим Анатолійович, Діденко Наталія Юріївна, Курищук Костянтин Васильович
МПК: A61K 35/16
Мітки: спосіб, церулоплазміну, інактивації, одержанні, вірусів
Формула / Реферат:
1. Спосіб інактивації вірусів при одержанні церулоплазміну, що включає очистку хроматографією розчину напівфабрикату церулоплазміну шляхом промивання ацетатним буферним розчином з наступною елюцією, який відрізняється тим, що попередньо до розчину напівфабрикату церулоплазміну додають 0,1 М ацетатний буферний розчин при рН 5,8, що містить 1 % мас. три-н-бутилфосфату і 1 % мас. полісорбату 80, суміш перемішують...
Спосіб інактивації лептоспірозного антигену

Номер патенту: 52982
Опубліковано: 15.01.2003
Автори: Кучерявенко Олексій Олександрович, Еверт Віктор Вікторович
МПК: C12N 1/20, A61K 39/02, C12N 1/36
Мітки: лептоспірозного, інактивації, спосіб, антигену
Формула / Реферат:
Спосіб інактивації лептоспірозного антигену, який включає внесення інактиватора в лептоспіровмісну суспензію з наступною її витримкою, який відрізняється тим, що як інактиватор використовують кристалвіолет в концентрації 0,05 % від об'єму лептоспіровмісної суспензії, а витримку здійснюють протягом 24 годин при температурі та рН 7,0 - 7,4.
Спосіб інактивації вірусів при одержанні імуноглобуліну

Номер патенту: 85734
Опубліковано: 25.02.2009
Автори: Куркіна Оксана Вікторівна, Самойленко Вадим Анатолійович, Діденко Наталія Юріївна, Курищук Костянтин Васильович, Скринник Максим Михайлович
МПК: C07K 1/18, C07K 16/06, C07K 1/36
Мітки: інактивації, спосіб, імуноглобуліну, одержанні, вірусів
Формула / Реферат:
1. Спосіб інактивації вірусів при одержанні імуноглобуліну, що включає очистку розчину імуноглобуліну, виділеного спиртовим фракціонуванням, який відрізняється тим, що при очистці розчин імуноглобуліну попередньо обробляють сольвент-детергентною сумішшю 0,05 М ацетатного буферного розчину при pH 5,5, що містить 1 % мас. три-н-бутилфосфату і 1 % мас. полісорбату 80, і обробку ведуть шляхом перемішування суміші протягом 12...16 годин...
Спосіб інактивації групоспецифічного антигену хламідій

Номер патенту: 46249
Опубліковано: 10.12.2009
Автори: Мартинюк Олександр Григорович, Недосєков Віталій Володимирович
МПК: A61K 39/118
Мітки: спосіб, хламідій, антигену, групоспецифічного, інактивації
Формула / Реферат:
Спосіб інактивації групоспецифічного антигену хламідій, що включає очищення, концентрування суспензії хламідій з наступною інактивацією, який відрізняється тим, що інактивацію проводять β-пропіолактоном в мінімальній концентрації 0,02 %, при експозиції 12 годин при температурі 20 С, в результаті чого антиген зберігає високу активність та специфічність.
Попередній патент: Асептичний пакет з отвором
Наступний патент: Циклічні інгібітори 11бета-гідроксистероїддегідрогенази 1
Випадковий патент: Система для вимірювання швидкості звуку