Конденсовані біциклічні або трициклічні амінокислоти
Номер патенту: 74879
Опубліковано: 15.02.2006
Автори: Уільямс Софі Керолайн, Браянс Джастін Стівен, Блейкмор Девід Клайв








Формула / Реферат
1. Сполука будь-якої формули І – XXV
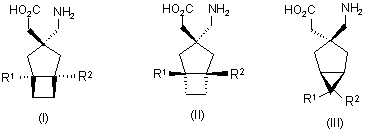

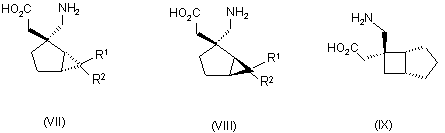
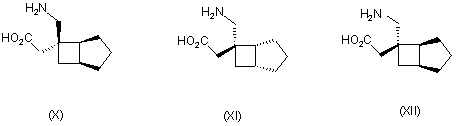
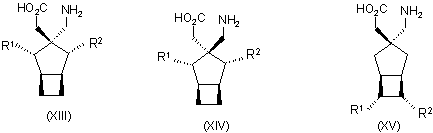
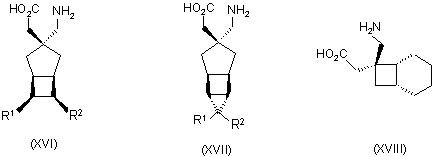
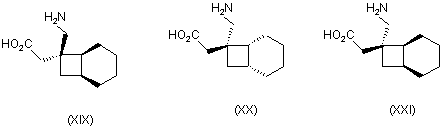
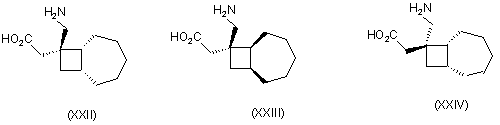
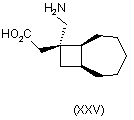 ,
,
де R1 та R2 кожний незалежно вибраний з водню, лінійного або розгалуженого алкілу з 1-6 атомами вуглецю, циклоалкілу з 3-6 атомами вуглецю, фенілу та бензилу, за умови, що, за виключенням випадку циклооктанової сполуки формули (XVII), R1 та R2 одночасно не означають водень, або її фармацевтично прийнятна сіль або сольват, або її пролікарська форма.
2. Сполука за п.1, в якій R1 та R2 обидва означають метил.
3. Сполука за п.1 або 2 або її сіль, сольват або її пролікарська форма, вибрана з групи, до складу якої входять:
((1R,5S)-3-амінометил-1,5-диметилбіцикло[3.2.0]гепт-3-ил)оцтова кислота;
((1S,5R)-3-амінометил-1,5-диметилбіцикло[3.2.0]гепт-3-ил)оцтова кислота;
((1R,5S)-3-амінометил-6,6-диметилбіцикло[3.1.0]гекс-3-ил)оцтова кислота;
((1S,5R)-3-амінометил-6,6-диметилбіцикло[3.1.0]гекс-3-ил)оцтова кислота;
((1S,2S,5R)-2-амінометил-6,6-диметилбіцикло[3.1.0]гекс-2-ил)оцтова кислота;
((1R,2S,5S)-2-амінометил-6,6-диметилбіцикло[3.1.0]гекс-2-ил)оцтова кислота;
((1S,2R,5R)-2-амінометил-6,6-диметилбіцикло[3.1.0]гекс-2-ил)оцтова кислота;
((1R,2R,5S)-2-амінометил-6,6-диметилбіцикло[3.1.0]гекс-2-ил)оцтова кислота;
((1R,5R,6S)-6-амінометилбіцикло[3.2.0]гепт-6-ил)оцтова кислота;
((1S,5S,6S)-6-амінометилбіцикло[3.2.0]гепт-6-ил)оцтова кислота;
((1R,5R,6R)-6-амінометилбіцикло[3.2.0]гепт-6-ил)оцтова кислота;
((1S,5S,6R)-6-амінометилбіцикло[3.2.0]гепт-6-ил)оцтова кислота;
цис-(1S,2R,4S,5R)-3-амінометил-2,4-диметилбіцикло[3.2.0]гепт-3-ил)оцтова кислота;
транс-((1S,2R,4S,5R)-3-амінометил-2,4-диметилбіцикло[3.2.0]гепт-3-ил)оцтова кислота;
((1S,5R,6S,7R)-3-амінометил-6,7-диметилбіцикло[3.2.0]гепт-3-ил)оцтова кислота;
((1S,5R,6R,7S)-3-амінометил-6,7-диметилбіцикло[3.2.0]гепт-3-ил)оцтова кислота;
((1R,2S,5S)-7-амінометил-3,3-диметилтрицикло[3.3.0.0]окт-7-ил)оцтова кислота;
((1R,6R,7S)-7-амінометилбіцикло[4.2.0]окт-7-ил)оцтова кислота;
((1S,6S,7S)-7-амінометилбіцикло[4.2.0]окт-7-ил)оцтова кислота;
((1R,6R,7R)-7-амінометилбіцикло[4.2.0]окт-7-ил)оцтова кислота;
((1S,6S,7R)-7-амінометилбіцикло[4.2.0]окт-7-ил)оцтова кислота;
((1R,7R,8S)-8-амінометилбіцикло[5.2.0]нон-8-іл)оцтова кислота;
((1S,7S,8S)-8-амінометилбіцикло[5.2.0]нон-8-іл)оцтова кислота;
((1R,7R,8R)-8-амiнoмeтилбiциклo[5.2.0]нoн-8-iл)oцтoвa кислота та
((1S,7S,8R)-8-амінометилбіцикло[5.2.0]нон-8-іл)оцтова кислота.
4. Сполука за будь-яким з пп. 1-3 або її сіль, сольват або її пролікарська форма, вибрана з групи, до складу якої входять:
[(1R,5R,6S)-6-(амінометил)біцикло[3.2.0]гепт-6-ил]оцтова кислота;
[(1S,5S,6R)-6-(амінометил)біцикло[3.2.0]гепт-6-ил]оцтова кислота;
[(1RS,5RS,6RS)-6-(амінометил)біцикло[3.2.0]гепт-6-ил]оцтова кислота;
[(1RS,6RS,7SR)-7-(амінoмeтил)бiциклo[4.2.0]oкт-7-ил]oцтoвa кислота; та
[(1RS,6RS,7RS)-7-(амінометил)біцикло[4.2.0]окт-7-ил]оцтова кислота.
5. Сполука за будь-яким з пп. 1-4 або її сіль, сольват або її пролікарська форма, яка є [(1R,5R,6S)-6-(амінометил)біцикло[3.2.0]гепт-6-ил]оцтовою кислотою.
6. Фармацевтична композиція, яка містить терапевтично ефективну кількість сполуки за будь-яким з пп. 1-5 та фармацевтично прийнятний носій.
7. Спосіб лікування захворювання, вибраного з епілепсії, нападів непритомності, гіпокінезії, краніальних розладів, нейродегенеративних розладів, депресії, неспокою, паніки, болю, синдрому подразнення кишечнику, розладів сну, остеоартриту, ревматоїдного артриту, нейропатологічних розладів, вісцерального болю, функціональних розладів кишечнику, запальних захворювань кишечнику, болю, пов'язаного з дисменореєю, пельвікального болю, циститу та панкреатиту, який полягає в уведенні ссавцю, який потребує згаданого лікування, терапевтично ефективної кількості сполуки за будь-яким з пп. 1-5.
8. Спосіб за п. 7, в якому захворюванням є нейропатичний біль.
9. Спосіб одержання сполуки за будь-яким з пп. 1-5, який складається з наступних стадій:
(і) кислотний гідроліз відповідної ізоціанатної похідної/похідної (С1-С6)алкілового естеру карбонової кислоти;
(іі) гідроліз відповідного циклічного лактаму;
(ііі) відновлення відповідної нітро/кислотної похідної, яка може бути необов'язково ненасиченою; або
(iv) відновлення відповідної нітропохідної/похідної бензил- або дифенілметилового естеру, які можуть бути необов'язково ненасиченими.
Текст
. му розчиннику, наприклад, тетрагідрофурані, 1,4Спосіб D: діоксані, н-гептані, толуолі, діетиловому етері або трет-бутилметиловому етері при температурі від 100°С до 110°С з одержанням ціаноестеру формули (52). (b) Ціаногрупу ціаноестеру (52) видаляють за допомогою основи, наприклад, гідроксиду калію, гідроксиду натрію, гідроксиду літію або гідроксиду цезію, в розчиннику, наприклад, етиленгліколі, 2метоксиетиловому етері, 1,4-діоксані або діетиленгліколі. Суміш перемішують при температурі від 25°С до 250°С з одержанням карбонової кислоти формули (53). (c) Карбоксильну групу кислоти (53) захищають перетворенням в її алкіловий естер з 1-6 ато(a) На першій стадії захищення карбоксильної мами вуглецю в алкільному залишку, наприклад, в групи кислоти (53), її перетворюють в хлорид (58) метиловий естер (54). Для цього кислоту (53) мошляхом взаємодії при температурі від -40°С до жуть додавати: 110°С, наприклад, з оксалілхлоридом або тіонілхдо суміші йодометану в розчиннику, вибранолоридом в апротонному органічному розчиннику, му з дихлорметану, хлороформу, тетрагідрофуранаприклад, дихлорметані, хлороформі, діетиловону, толуолу або 1,4-діоксану, до якої додають осму етері, толуолі або mpe/π-бутилметиловому нову, таку як, 1,8-діазабіцикло[5.4.0]ундец-7-ен етері, до якого додають 0.01мол.% до 10мол.% (DBU), триетиламін або 1,5-діазабіцикло[4.3.0]нонΝ,Ν-диметилформаміду (DMF). 5-ен (DBN), і перемішують при температурі від (b) Хлорангідрид (58) перетворюють в його 40°С до 110°С; або трет-бутиловий естер, наприклад, шляхом взаєдо суміші метанолу та концентрованої кисломодії з трет-бутиловим спиртом в апротонному ти, такої як, сірчана кислота або хлороводнева органічному розчиннику, наприклад, дихлорметані, кислота, при температурі в межах від 0°С до хлороформі, діетиловому етері, толуолі або трет100°С; або бутилметиловому етері, до якого додають Ν,Νдо триметилсилілдіазометану та метанолу в діізопропілетиламін (DІPEA) або триетиламін. Реабензолі або толуолі при температурі від -40°С до кційну суміш перемішують при температурі від 100°С; або 40°С до 110°С з одержанням естеру формули (59). до діазометану в розчиннику, такому як, бен(c) Фенільну групу естеру (59) окислюють до зол, толуол, дихлорметан, при температурі від карбоксильної групи взаємодією з перйодатом 29 74879 30 натрію та хлоридом рутенію (ίίί) в суміші тетрахломагнійхлоридом або -бромідом, або 2риду вуглецю або етилацетату та ацетонітрилу, до бутенілмагнійхлоридом та діалкілцинком, таким як, якої додають воду. Суміш перемішують при темдиметилцинк, або сіллю міді (l), такою як, йодид пературі від 40°С до 80°С з одержанням карбономіді (l) або ціанід міді (l), в сухому органічному розвої кислоти (60). чиннику, наприклад, тетрагідрофурані, 1,4(d) Карбоксильну групу кислоти (60) перетводіоксані, н-гептані, толуолі, діетиловому етері або рюють в естерну групу шляхом додавання: трет-бутилметиловому етері, при температурі від до суміші йодометану в розчиннику, вибрано100°С до 110°С з одержанням ненасиченого адиму з дихлорметану, хлороформу, тетрагідрофуративного продукту формули (65). ну, толуолу або 1,4-діоксану, до якої додають ос(b) Ціаногрупу адитивного продукту (65) виданову, таку як, 1,8-діазабіцикло[5.4.0]ундец-7-ен ляють шляхом взаємодії з основою, наприклад, (DBU), триетиламін або 1,5-діазабіцикло[4.3.0]нонгідроксидом калію, гідроксидом натрію, гідрокси5-ен (DBN), і перемішують при температурі від дом літію або гідроксидом цезію, в органічному 40°С до 110°С з одержанням естеру формули (61); розчиннику, вибраному з етиленгліколю, 2або метоксиетилового етеру, 1,4-діоксану або діетиледо суміші метанолу та концентрованої кислонгліколю. Реакційну суміш перемішують при темти, такої як, сірчана кислота або хлороводнева пературі від 25°С до 250°С, отримуючи карбонову кислота, при температурі в межах від 0°С до кислоту формули (66). 100°С; або (с) Карбоксильну групу кислоти (66) перетводо триметилсилілдіазометану та метанолу в рюють в естерну групу додаванням: бензолі або толуолі при температурі від -40°С до до суміші йодометану в розчиннику, вибрано100°С; або му з дихлорметану, хлороформу, тетрагідрофурадо діазометану в розчиннику, такому як, бенну, толуолу або 1,4-діоксану, до якої додають осзол, толуол, дихлорметан, при температурі від нову, таку як, 1,8-діазабіцикло[5.4.0]ундец-7-ен 40°С до 40°С. (DBU), триетиламін або 1,5-діазабіцикло[4.3.0]нон(е) трет-Бутоксигрупу видаляють з діестеру 5-ен (DBN), і перемішують при температурі від (61) взаємодією з трифтороцтовою кислотою в 40°С до 110°С з одержанням естеру формули (67); розчиннику, наприклад, дихлорметані, хлорофорабо мі, 1,4-діоксані, тетрагідрофурані, діетиловому до суміші метанолу та концентрованої кислоетері або трет-бутилметиловому етері. Реакційну ти, такої як, сірчана кислота або хлороводнева суміш перемішують при температурі від -40°С до кислота, при температурі в межах від 0°С до 110°С з одержанням карбонової кислоти формули 100°С; або (62). до триметилсилілдіазометану та метанолу в (f) Естерну групу кислоти (62) перетворюють в бензолі або толуолі при температурі від -40°С до ізоціанат (63) шляхом додавання: 100°С; або до суміші основи, вибраної з триетиламіну або до діазометану в розчиннику, такому як, бендіізопропілетиламіну, та розчинника, вибраного з зол, толуол, дихлорметан, при температурі від толуолу, бензолу, ксилолів, тетрагідрофурану, 40°С до 40°С. діетилового етеру або н-гептану, до яких додають (d) Ненасичену групу естеру (67) окислюють дифенілфосфорилазид (DPPA) і перемішують при перйодатом натрію та хлоридом рутенію (lll) в сутемпературі від 0°С до 150°С; або міші тетрахлориду вуглецю або етилацетату та до етилхлороформіату або ізобутилхлорофоацетонітрилу, до яких додають воду. Суміш перерміату та основи, такої як, триетиламін або діізопмішують при температурі від 40°С до 80°С з одерропілетиламін, в тетрагідрофурані або ацетоні, жанням карбонової кислоти формули (68). або діетиловому етері при температурі від -40°С (e) Карбонову кислоту (68) перетворюють в до 78°С з наступним додаванням азиду натрію у амінокислоту (69), як згадувалось в способі С воді та тетрагідрофурані або ацетоні, після чого Вищенаведені кетони можуть також бути педодають толуол або бензол і кип'ятять зі зворотретворені в амінокислоті з використанням одного ним холодильником. із загальних способів F-Г, як це показано нижче (g) Одночасним гідролізом ізоціанатної та шедля кетону групи (9). стерної груп сполуки (63), наприклад, за допомоСпосіб F гою водної хлороводневої кислоти з концентрацією від 0.01 Μ до 12Μ в присутності або у відсутності розчинника, такого як, 1,4-діоксан, оцтова кислота або вода, одержують амінокислоту (64). Спосіб Е: (a) Ціаноестер (47) піддають взаємодії з аліл (a) Кетон перетворюють в нітроестер (70) згідно з методиками, описаними вище. (b) Нітроестер (70) гідролізують придатною основою, такою як, водний гідроксид натрію з одержанням нітрокислоти (71), яку відновлюють шляхом гідрування, наприклад, Н2 з каталізатором паладій-на-вугіллі в придатному розчиннику, такому як, етанол, з одержанням амінокислоти (72). 31 Спосіб G (a) Ненасичений естер (73), в якому R означає бензил або дифенілметил, може бути отриманий з кетону відповідно до будь-якого із загальних способів, описаних вище. (b) Нітроестер (74) перетворюють в амінокислоту (75) відновленням шляхом каталітичного гідрування в придатному розчиннику. Сполуки винаходу альтернативно можуть бути одержані з відомих ненасичених модифікацій кетону типу (8), як це наведено далі в способах Η та l: Спосіб Η (а) Кетон (76) перетворюють в ненасичений нітроестер (78) згідно із загальними способами, описаними раніше. (b) Нітроестер (78) гідролізують придатною основою, такою як, водний гідроксид натрію з одержанням нітрокислоти (79), яку відновлюють шляхом гідрування, наприклад, Н2 з каталізатором паладій-на-вугіллі в придатному розчиннику, такому як, етанол, з одержанням амінокислоти (80). Спосіб l (a) Ненасичений нітроестер (82) може бути одержаний з кетону (76) у відповідності із способами, загалом описаними раніше. (b) Нітроестер (82) перетворюють в амінокислоту (83) відновленням шляхом каталітичного гідрування в придатному розчиннику. Фармацевтично прийнятна сіль сполуки винаходу може бути легко одержана змішуванням розчинів сполуки винаходу та необхідної кислоти або основи, в залежності від обставин. Сіль може бути осаджена з розчину та зібрана фільтрацією або може бути виділена випарюванням розчинника. Посилаючись на загальні методики, описані вище, фахівцю легко буде зрозуміти, що у випадку наявності захисних груп, вони загалом будуть взаємозамінними з іншими захисними групами подібної природи, наприклад, коли описується, що кислотна група захищається етильною групою, вона може бути легко замінена будь-якою придатною алкільною групою, наприклад, С1-6балкільною групою. Фахівцю буде зрозумілим, що конкретні стадії в загальних способах, представлених тут вище, можуть бути відповідним чином скомбіновані будьяким іншим способом, не показаним тут, з метою одержання сполуки відповідно до даного винаходу. 74879 32 Таким чином, в короткому викладі, винахід охоплює: (і) сполуку формули l-XXV або и фармацевтично прийнятну сіль, сольват, поліморфну або пролікарську форму; (іі) спосіб одержання сполуки формули l-XXV або її фармацевтично прийнятної солі, сольвату, поліморфної або пролікарської форми; (ііі) фармацевтичну композицію, яка включає сполуку формули l-XXV або її фармацевтично прийнятну сіль, сольват, поліморфну або пролікарську форму, разом із фармацевтично прийнятним екціпієнтом, розбавником або носієм; (iv) сполуку формули l-XXV або її фармацевтично прийнятну сіль, сольват, поліморфну форму, пролікарську форму або їх композицію для використання як медикаменту; (ν) застосування сполуки формули l-XXV або її фармацевтично прийнятної солі, сольвату, поліморфної форми, пролікарської форми або їх композиції для виробництва медикаменту для лікування будь-яких станів, згаданих вище; (vi) застосування сполуки формули І-XXV або її фармацевтично прийнятної солі, сольвату, поліморфної форми, пролікарської форми або їх композиції для виробництва медикаменту для лікування будь-яких станів, згаданих вище; (vіі) спосіб лікування у ссавця будь-якого із станів, згаданих вище, у тому числі лікування згаданого ссавця ефективною кількістю сполуки формули І-XXV або її фармацевтично прийнятної солі, сольвату, поліморфної форми, про лікарської форми або композиції; (viii) нові проміжні сполуки формул (Іа), (4а)(7а), (70), (71), (73), (74), (77)-(79), (81) або (82); (іх) спосіб лікування будь-якого з станів, згаданих вище, який полягає в уведенні пацієнту, який потребує такого лікування, одночасно, окремо або послідовно комбінації сполуки формули ί-XXV та додаткового агенту знеболювання; (х) застосування комбінації сполуки формули ІXXV та додаткового терапевтичного агенту для виробництва медикаменту для лікування будьякого стану, згаданого вище; і (хі) продукт, який містить сполуку формули ІXXV та додатковий терапевтичний агент, як комбінований препарат для одночасного, окремого або послідовного застосування в лікуванні будь-якого із станів, згаданих раніше. Даний винахід ілюструється наступними необмежувальними прикладами та проміжними сполуками. Приклад 1 [(1R,5R,6S)-6-(Амінометил)біцикло[3.2.0]гепт6-yl]оцтової кислоти гідрохлорид Iзоціанат з препаративного прикладу 9 (приблизно 9.33ммоль) та 6Ν соляна кислота (30мл) кип’ятили протягом 18 годин. Суміш залишали охолоджуватися, розбавляли водою (60мл) та екстрагували дихлорметаном (2 50мл). Водну фазу концентрували при зниженому тиску з одержанням 33 74879 34 твердої речовини жовтого кольору, яку промивали мивали lПС (5мл). Осад на фільтрі повторно суетилацетатом та ацетонітрилом з одержанням спендували в lПС (15мл), кип'ятили та охолоджу0.92г вказаної у заголовку сполуки у вигляді твервали до температури навколишнього середовища. дої речовини білого кольору. Суспензію фільтрували, залишок промивали ІПС 1 H-ЯМР (400МГц, d6-DMSO): δ=7.94 (3Н, шс), (5мл) і висушували у вакуумі при 40°С до постійної 3.15 (1Н, д), 3.07 (1Н, д), 2.72 (1Н, кв), 2.46 (1Н, м), ваги з одержанням вказаної у заголовку сполуки 2.42 (1Н, д), 2.33 (1Н, д), 1.98 (1Н, м), 1.80-1.64 вигляді твердої кристалічної речовини (1.4г). Тем(2Н, м), 1.59 (1Н, м), 1.48-1.28 (3Н, м), 1.23(1Н, дд). пература плавлення (Perkin Eimer DSC7): 208°С. НРМС (ХІАТ): m/z [(MH-HCl)+] 184. Приклад 2 РХМС (Prodigy ODS3 (3мк), колонка Гідрохлорид [(1S,5S,6R)-6(Амінометил)біцикло[3.2.0]гепт-6-ил]оцтової кис150мм 4.6мм (внутр.діам.), 20-100% ацетонітрил лоти +0.1% мурашина кислота) час утримування=4.34 хвилини, 100% чистота. [α]D(с=0.127 в метанолі)=-12.4° Мікроаналіз: Знайдено: С, 54.64; Η, 8.19; Ν, 6.42. C10H17NO2·HCl розраховано С, 54.67; Η, 8.26; Ν, 6.38%. Ізоціанат з препаративного прикладу 12 (приТемпература плавлення (Perkin Elmer DSC7): близно 11.0ммоль) та 6N соляну кислоту (30мл) 198°С. кип'ятили протягом 16 годин. Суміш залишали Альтернативно: охолоджуватися, розбавляли водою (100мл) і ексПриклад 1А трагували дихлорметаном (2 50мл). Водну фазу Гідрохлорид [(1R,5R,6S)-6концентрували при зниженому тиску з одержанням (амінометил)біцикло[3.2.0]гепт-6-ил]оцтової киствердої речовини жовтого кольору і промивали лоти етилацетатом та ацетонітрилом з одержанням Нітрокислоту препаративного прикладу 32 0.94г вказаної у заголовку сполуки у вигляді твер(2.0г; 9.4ммоль) в (або 1:1 IПС:Н2О або 1:1 дої речовини білого кольору. 1 MeCN:H2O (40мл; 20мл/г) гідрували, використовуН-ЯМР (400МГц, d6-DMSO): δ=7.94 (3Н, шс), ючи 10% Pd/C (0.2г; 0.1г/г) при 50°С та 60 psi про3.15 (1Н, д), 3.07 (1Н, д), 2.72 (1Н, кв), 2.46 (1Н, м), тягом 18 годин. Реакційну суміш фільтрували че2.42 (1Н, д), 2.33 (1Н, д), 1.98 (1Н, м), 1.80-1.64 рез Celite і осад на фільтрі промивали 1:1 lПС:Н2О (2Н, м), 1.59 (1Н, м), 1.48-1.28 (3Н, м), 1.23(1Н, дд). або 1:1 MeCN:H2O (20мл). Об'єднані фільтрати та НРМС (XІAT): m/z [(MH-HCt)+]184. промивні розчини концентрували у вакуумі та азеPXMC (Prodigy ODS3 (3мк), колонка отропували до сухого стану з додатковою кількістю 150мм 4.6мм (внутр. діам.), 20-100% ацетонітрил lПС або MeCN, отримуючи вказану у заголовку +0.1% мурашина кислота) час утримування сполуку у вигляді твердої кристалічної речовини =4.34хвил., 100% чистота. білого кольору (1.52г). [α]D(с=0.35 в метанолі) =+13.0° Приклад 1В Приклад 3 Гідрохлорид [(1R,5R,6S)-6Гідрохлорид [(1RS,5RS,6RS)-6(амінометил)біцикло[3.2.0]гепт-6-ил]оцтової кис(амінометил)біцикло[3.2.0]гепт-6-ил]оцтової кислоти лоти Лактам препаративного прикладу 33 (4.70г, 28.44ммоль) та соляну кислоту (57мл 6N розчину) кип'ятили разом протягом 6 годин. Суміш залишали охолоджуватися і потім розбавляли водою (60мл). Водний шар промивали дихлорметаномом (2 100мл), фільтрували і потім випарювали при Ізоціанат з препаративного прикладу 17 (призниженому тиску. Одержану тверду речовину жовблизно 2.79ммоль) та 6N соляну кислоту (15мл) туватого кольору розтирали з етилацетатом та кип'ятили протягом 18 годин. Суміш залишали перекристалізовували, використовуючи суміш ацеохолоджуватися, розбавляли водою (60мл) та екстонітрил:вода 1:1 з одержанням вказаної у заголотрагували дихлорметаном (3 50мл). Водну фазу вку сполуки (4.51г). концентрували при зниженому тиску з одержанням Приклад 1С твердої речовини жовтого кольору, яку промивали [(1R,5R,6S)-6-(Амінометил)біцикло[3.2.0]гептетилацетатом та ацетонітрилом з одержанням 6-ил)]оцтова кислота (Цвіттер-іон) 0.45г вказаної у заголовку сполуки у вигляді тверГідрохлорид амінокислоти Прикладу 1 (2.2г) дої речовини білого кольору. 1 розчиняли в 7.25мл Н2О (3.3мл/г). Доводили pH Н-ЯМР (400МГц, d6-DMSO): δ=7.84 (3Н, шс), розчину до 7.5, спочатку за допомогою 1.6мл вод2.92 (1Н, д), 2.85 (1Н, д), 2.75 (1Н, т), 2.69 (1Н, д), ного розчину NaOH, а в кінці декількома каплями 2.59 (1Н, д), 2.39 (1Н, т), 1.81-1.62 (4Н, м), 1.410.1N водного NaOH. Осаджений цвіттер-іон пере1.30 (4Н, м). мішували протягом 8 годин при температурі 8°С, НРМС (ХІАТ): m/z [(MH-HCІ)+]184 суспензію фільтрували і залишок промивали льоPXMC (Prodigy ODS3 (3мк), колонка дяною холодною водою (6мл). Змочуваний водою 150мм 4.6мм (внутр. діам.), 20-100% ацетонітрил осад, що утворився на фільтрі, суспендували в +0.1% мурашина кислота) час утримування lПС (15мл) та кип'ятили протягом 10 хвилин. Після =4.27хвил., 99.8% чистота. охолодження до температури навколишнього сеПриклад 4 редовища, суспензію фільтрували і залишок про 35 74879 36 Гідрохлорид [(1RS,6RS,7SR)-712.1г вказаної у заголовку сполуки у вигляді без(амінометил)біцикло[4.2.0]окт-7-ил]оцтової кислоти барвного масла. -1 mах(плівка)/см 1777. 1 Н-ЯМР (400МГц, CDCl3): δ=3.54 (1Н, м), 3.19 (1Н, ддд), 2.88 (1Н, м), 2.49 (1Н, ддд), 2.04 (1Н, м), 1.91-1.49 (5Н, м). Лактам з препаративного прикладу 22 (3.20г, Препаративний приклад 1А 17.9ммоль) нагрівали до кипіння в 1,4-діоксані (1R,5R)-Біцикло[3.2.0]гептан-6-он (15мл) та 6N НСI (50мл). Через 4 години суміш охолоджували до кімнатної температури та промивали дихлорметаном (2 30мл). Водну фазу збирали і розчинник видаляли у вакуумі. Залишок розтирали з етилацетатом і одержану тверду реРозчин (1R,5R)-біцикло[3.2.0]гепт-2-ен-6-ону човину збирали фільтрацією і висушували у ваку(50.0г; 462ммоль) в ЕtOАс (375мл) гідрували, виумі з одержанням 2.74г вказаної у заголовку спокористовуючи 5% Pd/C (5.0г) з 50% вологістю при луки у вигляді твердої речовини білого кольору. 60psi протягом 8 годин при температурі навколиш1 Н-ЯМР (400МГц, D2O): 3.24 (2Н, м), 2.58 (2Н, нього середовища. Реакційну суміш фільтрували с), 2.39 (1Н, м), 2.03 (1Н, м), 1.76 (2Н, м), 1.59-1.10 через Celite, і фільтрат концентрували у вакуумі з (7Н, м), 0.96 (1Н, м). одержанням 41.3г вказаної у заголовку сполуки у НРМС (ΦIΑΤ): m/z [(MH-HCI)+]198. вигляді безбарвного масла. 1 Приклад 5 Н-ЯМР (400МГц, CDCl3): δ=3.55 (1H, м), 3.20 Гідрохлорид [(1RS,6RS,7RS)-7(1Н, м), 2.90 (1Н, м), 2.50 (1Н, м), 2.0-1.5 (6Н, м). 1 (амінометил)біцикло[4.2.0]окт-7-ил]оцтової кислоти Посилання: ЕР0074856 Препаративний приклад 2 Етил (2Ε/Ζ)-(1RS,5RS)-біцикло[3.2.0]гепт-6иліден(ціано)етаноат Дифенілфосфорилазид (0.43мл, 1.98ммоль) додавали при перемішуванні до розчину триетиламіну (0.28мл, 2.03ммоль) та кислоти препаративного прикладу 29 (0.47г, 1.96ммоль приблизно) в толуолі (15мл) при кімнатній температурі у атмосфері азоту. Суміш перемішували протягом 16 годин і потім нагрівали до 35°С протягом 1 години. Суміш залишали охолоджуватися, розбавляли етилацетатом (60мл), промивали насиченим водним розчином гідрокарбонату натрію (2 100мл), насиченим розчином солі і висушували (МgSО4). Розчинник видаляли при зниженому тиску і одержане масло жовтого кольору нагрівали до кипіння в 6Ν НСl (20мл). Через 18 годин суміш охолоджували до кімнатної температури і промивали дихлорметаном (2 60мл) та діетиловим етером (60мл). Водну фазу збирали і розчинник видаляли у вакуумі. Залишок розтирали з етилацетатом і одержану тверду речовину збирали фільтрацією і висушували у вакуумі з одержанням 0.304г вказаної у заголовку сполуки у вигляді твердої речовини білого кольору. 1 H-ЯМР (400 МГц, d6-DMSO): 3.04 (1Н, д), 2.99 (1Н, д), 2.68 (1Н, д), 2.62 (1Н, д), 1.98 (1Н, м), 1.83 (1Н, т), 1.69-1.28 (9Н, м), 1.00 (1Н, м). НРМС (XlAT): m/z [(MH-HC)+]198. Препаративний приклад 1 (1RS,5RS)-Біцикло[3.2.0]гептан-6-он До розчину біцикло[3.2.0]гепт-2-ен-6-ону (12мл, 111.3ммоль) в етилацетаті (100мл) додавали паладій (1г, 10% ваг./ваг. на вугіллі) і суміш гідрували протягом 6 годин при 30°С і 483кПа (70p.s.i). Реакційну суміш фільтрували і розчинник видаляли при зниженому тиску з одержанням Кетон з препаративного прикладу 1 (22.4г, 204.1ммоль), етилціаноацетат (21.7мл, 204.1ммоль), ацетат амонію (15.7г, 204.1ммоль) та льодяну оцтову кислоту (11.7мл, 204.1ммоль) кип'ятили в толуолі (220мл): використовуючи насадку Dean-Stark. Через 8 годин суміш залишали охолоджуватися і розбавляли етилацетатом (300мл), промивали водою (3 150мл), насиченим розчином солі і висушували (MgSO4). Розчинник випарювали при зниженому тиску. Залишок хроматографували (SiO2, гептан/етилацетат, від 95:5 до 7:3) з одержанням 30г 6:4 суміші ізомерів вказаної у заголовку сполуки у вигляді твердої речовини жовтого кольору. -1 mах(плівка)/см 2225, 1725, 1640. 1 Н-ЯМР (400МГц, CDCl3): δ (основний і3омер) =4.26 (2Н, м), 3.64 (1Н, м), 3.36 (1Н, ддд), 2.96 (1Н, м), 2.70 (1Н, дт), 2.11 (1Н, м), (1.92-1.58, 5Н, м), 1.32 (3Н, м); δ (другорядний ізомер)=4.26 (2Н, м), 3.85 (1Н, м), 3.15 (1Н, ддд), 2.96 (1Н, м), 2.52 (1Н, дт, J20.0, 4.4), 2.02 (1Н, м), (1.92-1.58, 5Н, м), 1.32 (3Н, м). НРМС (XlAT): m/z [M-H] 204. Препаративний приклад 3 Етил [(1RS,5RS,6RS)-6бензилбіцикло[3.2.0]гепт-6-ил](ціано)ацетат Ціаноестер з препаративного прикладу 2 (10.0г, 48.7ммоль) в ТГФ (60мл) при перемішуванні додавали протягом 1 години до розчину бензилмагнійхлориду (78мл 1М розчину в етері, 78ммоль) в ТГФ (100мл) при -78°С в атмосфері аргону. Після 37 74879 38 перемішування протягом 2 годин при цій же тембільна фаза: 90% гексан, 10% ІПС з вмістом 0.5% пературі, реакцію зупиняли додаванням насиченоТФК)]: Час утримування=5.1хвил. (94% ее). го розчину хлориду амонію (40мл). Суміш залиша[α]D(с=1.13 в хлороформі)=-20.2° ли нагріватися до кімнатної температури і Другу частину солі переносили в дихлорметан, додавали розбавлену соляну кислоту (150мл). промивали розбавленою соляною кислотою, насиВодний шар екстрагували етилацетатом ченим розчином солі і висушували (MgSO4) з одержанням додаткових 5г кислоти 86% ее. (3 100мл). Об'єднані органічні шари промивали Аналогічним чином отримували: насиченим розчином солі, висушували (MgSО4) і Препаративний приклад 6 розчинник випарювали при зниженому тиску з [(1S,5S,6R)-6-бензилбіцикло[3.2.0]гепт-6одержанням вказаної у заголовку сполуки як суміил]оцтова кислота ші діастереоізомерів у вигляді масла жовтого кольору, яке використовували без очищення на наступній стадії. max(плівка)/см-1 2247,1741. НРМС (XlAT): m/z [M-H] 296. Препаративний приклад 4 [(1RS,5RS,6SR)-6-бензилбіцикло[3.2.0]гепт-6перекристалізацією солі, утвореної додаванил]оцтова кислота ням (5)-(-)-α-метилбензиламіну. HPLC [Chiralcel OD 250 4.6мм колонка (Мобільна фаза: 90% гексан, 10% lПС з вмістом 0.5% ТФК)]: Час утримування=4.2 хвилин (95% ее). [α]D(с=1.0 в хлороформі)=+17.3° Суміш діастереомерних ціаноестерів з препаПрепаративний приклад 7 ративного прикладу 3 (20.3г, 68.4ммоль) та гідрокМетил [(1R,5R,6S)-6-бензилбіцикло[3.2.0]гептсиду калію (23.0г, 410.4ммоль) нагрівали до 160°С 6-ил]ацетат в етиленгліколі (350мл) протягом 38 годин. Після цього суміш залишали охолоджуватися і обережно додавали соляну кислоту (300мл). Суміш екстрагували етилацетатом (3 200мл) і об'єднані органічні фракції промивали насиченим розчином солі, висушували (MgSО4) і розчинник видаляли при Триметилсилілдіазометан (17.7мл 2М розчину зниженому тиску. Залишок хроматографували в гексані, 35.4ммоль) при перемішуванні додавали (SіO2, гептан/етилацетат, 8:2) з одержанням 14.6г по краплям до розчину кислоти препаративного вказаної у заголовку сполуки як рацемічної суміші прикладу 5 (7.85г, 32.1ммоль) в суміші толуолу діастереомерів у вигляді твердої речовини білого (90мл) та метанолу (22.5мл) при 0°С у атмосфері кольору. аргону. Суміш залишали нагріватися до кімнатної -1 max(плівка)/см 3344, 1704. температури і перемішували протягом я 4 годин. 1 Н-ЯМР (400МГц, CDCІ3): δ=7.31-7.22 (5Η, м), Розчинник видаляли при зниженому тиску і зали3.02 (1Н, д), 2.97 (1Н, д), 2.64 (2Н, м), 2.34 (1Н, д), шок переносили в етилацетат (150мл), промивали 2.24 (1Н, д), 2.13 (1Н, м), 1.84-1.59 (3Н, м), 1.50насиченим розчином гідрокарбонату натрію 1.32 (4Н, м). (150мл), розбавленою соляною кислотою (100мл), НРМС (ΦІΑΤ): m/z [M-H] 243. насиченим розчином солі і висушували (МgSО4). Препаративний приклад 5 Розчинник видаляли при зниженому тиску Зали[(1R,5R,6S)-6-бензилбіцикло[3.2.0]гепт-6шок хроматографували (SiO2, гептан/етилацетат, ил]оцтова кислота 9:1) з одержанням 7.0г вказаної у заголовку сполуки у вигляді безбарвного масла. 1 max(плівка)/см 1736. 1 Н-ЯМР (400МГц, CDCl3): δ=7.28-7.21 (5Η, м), 3.67 (3Н, с), 2.97 (1Н, д), 2.92 (1Н, д), 2.65-2.60 (2Н, м), 2.26 (1Н, д), 2.18 (1Н, д), 2.08 (1Н, м), 1.82-1.52 (В)-(+)-а-Метилбензиламін (6.67г, 55ммоль) (3Н, м), 1.48-1.22 (4Н, м). додавали при перемішуванні до розчину рацемічНРМС (XlAT): m/z [MH+]259. ної суміші кислоти з препаративного прикладу 4 [α]D(с=0.11 в метанолі)=-24.1°. (24г, 98.2ммоль), розчиненої в етилацетаті. З розПрепаративний приклад 8 чину осаджувалася кисла сіль у вигляді твердої [(1R,5R,6S)-6-(2-Метокси-2речовини білого кольору. Цю сіль тричі перекрисоксоетил)біцикло[3.2.0]гепт-6-ил]оцтова кислота талізовували з етилацетату з одержанням 8.5г кислої солі. Подальшою перекристалізацією залишку отримували додаткову кількість 8.5г кислої солі. Першу частину отриманої солі переносили в дихлорметан, промивали розбавленою соляною Естер з препаративного прикладу 7 (7.0г, кислотою, насиченим розчином солі і висушували 27.1ммоль) та перйодат натрію (81.1г, (МgSО4). Розчинник випарювали при зниженому 379.3ммоль) перемішували разом в етилацетаті тиску з одержанням 5.0г вказаної у заголовку спо(100мл), ацетонітрилі (100мл) та воді (150мл) пролуки у вигляді твердої речовини білого кольору. тягом 5 хвилин. Суміш охолоджували до 0°С і до HPLC [Chiraiceд OD, колонка 250 4.6мм (Мореакційної суміші додавали гідрат хлориду рутенію 39 74879 40 (lll) (0.11г, 0.54ммоль). Реакційну суміш залишали (2Н, м), 2.26 (1Н, д), 2.18 (1Н, д), 2.08 (1Н, м), 1.82нагріватися до кімнатної температури і перемішу1.52 (3Н, м), 1.48-1.22 (4Н, м); вали протягом 24 годин. Додавали діетиловий [α]D (с=0.11 в метанолі)=+23.1° етер (150мл) і суміш перемішували протягом 40 Препаративний приклад 11 хвилин. Додавали розбавлену соляну кислоту [(1S,5S,6R)-6-(2-метокси-2(200мл) до суміші, яку потім екстрагували етилаоксоетил)біцикло[3.2.0]гепт-6-ил]оцтова кислота цетатом (3 100мл). Об'єднані органічні ; фракції промивали насиченим розчином тіосульфату натрію, насиченим розчином солі, висушували (MgSO4) і розчинник випарювали при зниженому тиску з одержанням вказаної у заголовку сполуки у вигляді масла жовтого кольору. Естер з препаративного прикладу 10 (7.0г, -1 max(плівка)/см 1733, 1715. 27.1ммоль) та перйодат натрію (81.1г, 1 Н-ЯМР (400МГц, CDCІ3): δ=3.65 (3Н, с), 2.82379.3ммоль) перемішували разом в суміш етила2.76 (3Н, м), 2.55-2.49 (3Н, м), 2.05 (1Н, м), 1.81 цетату (100мл), ацетонітрилу (100мл) та води (1Н, м), 1.73-1.69 (2Н, м), 1.49-1.28 (4Н, м). (150мл) протягом 5 хвилин. Суміш охолоджували HPMC (ΦІΑΤ): m/z [M-H] 225. до 0°С і до реакційної суміші додавали гідрат хлоПрепаративний приклад 9 риду рутенію (III) (0.11г, 0.54ммоль). Реакційну Метил [(1R,5R,6S)-6суміш залишали нагріватися до кімнатної темпе(Iзоціанатометил)біцикло[3.2.0]гепт-6-ил]ацетат ратури і перемішували протягом 24 годин. Додавали діетиловий етер (150мл) і суміш перемішували протягом 40 хвилин. Додавали до суміші розбавлену соляну кислоту (200мл) і потім екстрагували суміш етилацетатоом (3 100мл). Об'єднані органічні фракції промивали насиченим розчином Дифенілфосфорилазид (8.45мл, 39.2ммоль) тіосульфату натрію, насиченим розчином солі, додавали при перемішуванні до розчину триетивисушували (MgSO4) і розчинник випарювали при ламіну (5.6мл, 40.4ммоль) та кислоти з препаратизниженому тиску з одержанням вказаної у заголовного прикладу 8 (8.78г, 38.8ммоль) в толуолі вку сполуки у вигляді масла жовтого кольору. (80мл) при кімнатній температурі у атмосфері азо1 H-ЯМР (400МГц; CDCI3): δ=3.65 (3Н, с), 2.82ту. Суміш перемішували протягом 3 годин і потім 2.76 (3Н, м), 2.55-2.49 (3Н, м), 2.05 (1Н, м), 1.81 нагрівали до 35°С протягом 1.5 годин. Суміш за(1Н, м), 1.73-1.69 (2Н, м), 1.49-1.28 (4Н, м). лишали охолоджуватися, розбавляли етилацетаПрепаративний приклад 12 том (150мл), промивали насиченим водним розчиМетил [(1S,5S,6R)-6ном гідрокарбонату натрію (150мл), насиченим (iзоціанатометил)біцикло[3.2.0]гепт-6-ил]ацетат розчином солі і висушували (MgSО4). Розчинник видаляли при зниженому тиску з одержанням 8.7г вказаної у заголовку сполуки у вигляді масла жовтого кольору. -1 max(плівка)/см 2265, 2171, 1733. Препаративний приклад 10 Дифенілфосфорилазид (2.4мл, 11.1ммоль) Метил [(1S,5S,6R)-6-бензилбіцикло[3.2.0]гептдодавали при перемішуванні до розчину триети6-ил]ацетат ламіну (1.6мл, 11.4ммоль) та кислоти з препаративного прикладу 11 (11.0ммоль приблизно) в толуолі (30мл) при кімнатній температурі в атмосфері азоту. Суміш кип'ятили протягом 2 годин. Суміш залишали охолоджуватися, розбавляли етилацетатом (150мл), промивали насиченим водним розДо розчину кислоти з препаративного приклачином гідрокарбонату натрію (2 150мл), насичеду 6 (2.77г, 11.3ммоль) в суміші з толуолом (30мл) ним розчином солі і висушували (MgSO4). та метанолом (7.5мл) при 0°С в атмосфері аргону Розчинник видаляли при зниженому тиску з одердодавали при перемішуванні по краплинам тримежанням вказаної у заголовку сполуки у вигляді тилсилілдіазометан (5.7мл 2М розчин в гексані, масла жовтого кольору. 11.4ммоль). Суміш залишали нагріватися до кім-1 max(плівка)/см 2265, 2151, 1734. натної температури і перемішували протягом 4 Препаративний приклад 13 годин. Розчинник видаляли при зниженому тиску і трет-Бутил [(1RS,5RS,6SR)-6залишок переносили в етилацетат (100мл), пробензилбіцикло[3.2.0]гепт-6-ил]ацетат мивали насиченим розчином гідрокарбонату натрію (100мл), розбавленою соляною кислотою (100мл), насиченим розчином солі і висушували MgSO4). Розчинник випарювали при зниженому тиску. Залишок хроматографували (SiO2, гептан/етилацетат, 9:1) з одержанням 2.84г вказаної у При перемішуванні до розчину кислоти з презаголовку сполуки у вигляді безбарвного масла. паративного прикладу 4 (2.34г, 9.58ммоль) в дих1 H-ЯМР (400МГц, CDCI3): δ=2.28-7.21 (5Н, м), лорметані (30.5мл) при 0°С в атмосфері аргону 3.67 (3Н, с), 2.97 (1Н, д), 2.92 (1Н, д,), 2.65-2.60 додавали по краплям оксалілхлорид (0.92мл, 41 74879 42 10.5ммоль). Обережно додавали диметилформанатної температури і перемішували протягом 24 мід (0.3мл) і суміш залишали нагріватися до кімнагодин. Розчинник видаляли при зниженому тиску і тної температури, потім перемішували протягом залишок переносили в етилацетат (100мл), проподальших 4 годин. Розчинник видаляли у вакуумі мивали насиченим розчином гідрокарбонату наі залишок розбавляли дихлорметан (20мл). В аттрію (100мл), розбавленою соляною кислотою мосфері аргону обережно додавали до реакційної (100мл), насиченим розчином солі і висушували суміші 2-метилпропан-1-ол (10мл) в дихлорметані (МgSО4). Розчинник випарювали при зниженому (20мл), після додавали діізопропілетиламін (2.5мл, тиску з одержанням вказаної у заголовку сполуки у 14.4ммоль). Суміш перемішували протягом 17 вигляді масла жовтого кольору. -1 годин і потім переносили в етилацетат, промивали max(плівка)/см 1732. насиченим водним розчином гідрокарбонату наHPMC (XlAT): m/z [М-OтВu] 209. трію (2 200мл) і висушували (MgSO4). Розчинник Препаративний приклад 16 видаляли при зниженому тиску і залишок хромато[(1RS,5RS,6RS)-6-(2-Метокси-2графували (SiO2, гептан/етилацетат 95:5) з одероксоетил)біцикло[3.2.0]гепт-6-ил]оцтова кислота жанням вказаної у заголовку сполуки (2.40г) у вигляді масла жовтого кольору. -1 max(плівка)/см 1727. 1 Н-ЯМР (400МГц, CDCl3): δ=7.28-7.21 (5Η, м, Ph), 2.98 (1Н, д), 2.92 (1Н, д), 2.64-2.56 (2Н, м), Трифтороцтову кислоту (5мл) додавали по 2.16 (1Н, д), 2.09 (1Н, д), 2.04 (1Н, м), 1.80-1.50 краплям при перемішуванні до розчину естеру з (3Н, м), 1.48 (9Н, с), 1.47-1.20 (4Н, м). препаративного прикладу 15 (прибл. 6.63ммоль) в Препаративний приклад 14 дихлорметані (15мл) при 0°С. Суміш залишали [(1RS,5RS,6SR)-6-(2-трет-Бутокси-2нагріватися до кімнатної температури і перемішуоксоетил)біцикло[3.2.0]гепт-6-ил]оцтова кислота вали додатково протягом 17 годин. Суміш промивали насиченим водним розчином гідрокарбонату натрію поки не досягали нейтрального pH та екстрагували дихлорметаном (50мл). Потім повторно підкислювали до pH 4 розбавленою соляною кислотою. Суміш потім екстрагували дихлорметан Естер з препаративного прикладу 13 (2.4г, (2 50мл). Об'єднані органічні фракції промивали 7.99ммоль) та перйодат натрію (23.93г, насиченим розчином солі, висушували (MgSO4) і 111.8ммоль) перемішували разом в етилацетаті розчинник видаляли при зниженому тиску. Зали(24мл), ацетонітрилі (24мл) та воді (36мл) протяшок очищали хроматографією (SiO2, 8:2 до 6:4 гом 5 хвилин. Суміш охолоджували до 0°С і до гептан/етилацетат) з одержанням 0.63г вказаної у реакційної суміші додавали гідрат хлориду рутенію заголовку сполуки у вигляді безбарвного масла. (ІІІ) (0.033г, 0.16ммоль). Реакційну суміш залиша-1 max(плівка)/см 3200, 1738, 1705. ли нагріватися до кімнатної температури і перемі1 Н-ЯМР (400МГц, CDCl3): δ=68 (3Н, с), 2.84шували протягом 24 годин. Додавали діетиловий 2.73 (3Н, м), 2.61-2.48 (3Н, м), 2.03 (1Н, м), 1.80 етер (60мл) і суміш перемішували протягом 40 (1Н,м), 1.79-1.32 (6Н, м). хвилин. До суміші додавали розбавлену соляну НРМС (ΦlΑΤ): m/z [M-H] 225. кислоту (150мл) і потім екстрагували етилацетаПрепаративний приклад 17 том (3 100мл). Об'єднані органічні фракції промиМетил [(1RS,5RS,6RS)-6вали насиченим розчином солі, висушували (ізоціанатометил)біцикло[3.2.0]гепт-6-ил]ацетат (МgSО4) і розчинник випарювали при зниженому тиску з одержанням вказаної у заголовку сполуки (1.78г, 83%) у вигляді масла жовтого кольору. -1 max(плівка)/см 1728, 1714. 1 H-ЯМР (400МГц, CDCl3): δ=2.78 (1Η, д), 2.71 (1Н, д), 2.43 (1Н, д), 2.38 (1Н, д), 2.01 (1Н, м), 1.86Дифенілфосфорилазид (0.61мл, 2.82ммоль), 1.64 (3Н, м), 1.52-1.36 (6Н, м), 1.45 (9Н, с). триетиламін (0.40мл, 2.90ммоль), та кислоту з HPMC (ΦlΑΤ): m/z [M-H] 267. препаративного прикладу 16 (0.63г, 2.79ммоль) Препаративний приклад 15 кип'ятили в толуолі (15мл) протягом 6 годин. СуМетиловий естер [(1RS,5RS,6SR)-6-(2-третміш залишали охолоджуватися і розбавляли етибутокси-2-оксоетил)біцикло[3.2.0]гепт-6-іл]оцтової лацетатом (60мл). Одержаний розчин промивали кислоти насиченим водним розчином гідрокарбонату натрію (150мл), насиченим розчином солі і висушували (MgSO4). Розчинник видаляли при зниженому тиску з одержанням вказаної у заголовку сполуки у вигляді масла жовтого кольору. Rf(гептан-етилацетат, 9:1) 0.36. Триметилсилілдіазометан (4.3мл 2М розчину в -1 max(плівка)/см 2259, 2171, 1736. гексані, 8.6ммоль) додавали по краплям при пеПрепаративний приклад 18 ремішуванні до розчину кислоти з препаративного (1RS,6SR)-8.8-Дихлообіцикло[4.2.0]октан-7-он прикладу 14 (1.78г, 6.63ммоль) в суміші з толуолом (24мл) та метанолом (6мл) при 0°С у атмосфері аргону. Суміш залишали нагріватися до кім 43 74879 44 цетатом (200мл) і промивали 2Ν HCI (2 150мл). Органічну фазу збирали, висушували (MgSO4) і розчинник видаляли при зниженому тиску. Залишок очищали флеш-хроматографією (силікагель, Сульфат міді (ll) (2.0г, 8.0ммоль) розчиняли у ЕtOАс:гептан 3:20) з одержанням 5.49г вказаної у воді (75мл) і додавали до цинкового пилу (30г). заголовку сполуки у вигляді прозорого масла. -1 Суміш перемішували протягом 2 годин. Суміш фіmax(плівка)/см 2929, 1715, 1186. 1 льтрували, і тверду речовину збирали, промивали Н-ЯМР (400МГц, CDCl3): δ=5.63 та 5.58 (1Н двічі ацетоном і висушували у вакуумі при 100°С загальний - Ε/Ζ ізомери, 2 м), 4.15 (2Н, м), 3.38протягом 24 годин. Частину активованого цинку 2.98 (2Н, м), 2.79-2.35 (2Н, м), 2.13-1.05 (11Н, м). (8.0г) додавали до розчину циклогексану (10мл, НРМС (XlAT): m/z [MH+]195. 98.9ммоль) в діетиловому етері (180мл). Додавали Препаративний приклад 21 трихлорацетилхлорид (10.48мл, 93.96ммоль) в Етил [(1RS.6RS,7SR)-7діетиловому етері (20мл) з такою швидкістю, щоб (нітрометил)біцикло[4.2.0]окт-7-ил]ацетат підтримувати суміш при кипінні. Після того, як додавання закінчували, суміш нагрівали до кипіння протягом 4 годин. Суміш охолоджували до кімнатної температури, розбавляли діетиловим етером (50мл) і обережно виливали у водний насичений (2Z/Е)-(1RS,6RS)-Біцикло[4.2.0]окт-7розчин бікарбонату натрію. Суміш підкислювали за иліденетаноат (препаративний приклад 20) (5.47 г, допомогою 2N НСl і органічну фазу відокремлюва28.2ммоль) нагрівали до 60°С в тетрагідрофурані ли. Етерний екстракт промивали водою і потім (50мл) з нітрометаном (3.05мл, 56.4ммоль) та тетнасиченим водним розчином бікарбонату натрію. рабутиламонійфторидом (1М в ТГФ, 42мл, Органічну фазу збирали, висушували (MgSO4) і 42.0ммоль). Через 18 годин суміш охолоджували розчинник видаляли при зниженому тиску. Залидо кімнатної температури, розбавляли етилацеташок очищали флеш-хроматографією (силікагель, том (200мл) і промивали 2N НСl (2 100мл) і потім ЕtOАс:гептан 1:9) з одержанням 8.62г вказаної у насиченим розчином солі. Органічну фазу збиразаголовку сполуки у вигляді прозорого масла. ли, висушували (МgSО4) і розчинник видаляли у -1 max(плівка)/см 2939, 1802. вакуумі. Залишок очищали флеш-хроматографією 1 H-ЯМР (400МГц, CDCl3): δ=3.94 (1Η, м), 2.95 (силікагель, ЕtOАс:гептан 1:9) з одержанням 4.73г (1Н, м), 2.18-1.82 (2Н, м), 1.80-1.20 (6Н, м). вказаної у заголовку сполуки у вигляді прозорого Препаративний приклад 19 масла. -1 (1RS,6RS)-Біцикло[4.2.0]октан-7-он max(плівка)/см 1182, 1547, 1731, 2936. 1 Н-ЯМР (400МГц, CDCl3): δ=4.83 (2Н, м), 4.12 (2Н, кв), 2.66 (2Н, м), 2.57 (1Н, м), 2.22 (1Н, м), 2.05 (1Н, м), 1.86 (1Н, м), 1.76-1.31 (7Н, м), 1.26 (ЗН, т), (1RS,6SR)-8,8-дихлорбіцикло[4.2.0]октан-7-он 1.10 (1Н, м). НРМС (ΦίΑΤ): m/z [MH+] 256. (препаративний приклад 18) (8.60г, 44.6ммоль) Препаративний приклад 22 нагрівали до кипіння в оцтовій кислоті (100мл) з (1S,6S,7R)-Спіро[біцикло[4.2.0]октан-7.3'цинковим пилом (29.0г, 446ммоль). Через 4 години піролідин]-5'-он суміш охолоджували до кімнатної температури, розбавляли діетиловим етером (200мл) і промивали 2N NaOH (2 100мл) і потім насиченим водним розчином NaНСО3 (4 100мл). Етерну фазу збирали, висушували (MgSO4) і розчинник видаляЕтил [(1RS,6RS,7SR)-7ли при зниженому тиску з одержанням 4.79г вка(нітрометил)біцикло[4.2.0]окт-7-ил]ацетат (препазаної у заголовку сполуки у вигляді прозорого маративний приклад 21) (4.70г, 18.4ммоль) струшусла. вали у метанолі (150мл) при 30°С над каталізато-1 max(плівка)/см 2930, 1776. ром нікелем Ренея у атмосфері газоподібного 1 H-ЯМР (400МГц, CDCl3): δ=3.27 (1Н, м), 3.12 водню при 483кПа (70p.s.i.). Через 4 години каталі(1Н, м), 2.42 (2Н, м), 2.20-1. 02 (8Н, м). затор видаляли фільтруванням через целіт і розПрепаративний приклад 20 чинник видаляли при зниженому тиску з одержанЕтил (2Z/Έ)-(1RS,6RS)-біцикло[4.2.0]окт-7ням 3.23г вказаної у заголовку сполуки у вигляді илiденоктаноат прозорого масла: яке тверділо при стоянні. Гідрид натрію (60% дисперсія в маслі, 1.46г, 36.6ммоль) суспендували в сухому тетрагідрофурані (150мл) і охолоджували до 0°С. Додавали триетилфосфонацетат (7.65мл, 38.5ммоль) і суміш перемішували при 0°С протягом 15 хвилин. Потім додавали розчин (1RS,6RS)-біцикло[4.2.0]октан-7ону (препаративний приклад 19) (4.78 г, 38.5ммоль) в ТГФ (20мл) і суміш перемішували при 0°С. Через 1 годину суміш залишали нагріватися до кімнатної температури, розбавляли етила -1 max(плівка)/см 2919, 1712, 1677. H-ЯМР (400МГц, CDCI3): δ=5.61 (1Н, шс), 3.46 (2Н, м), 2.42 (2Н, м), 2.18-1.01(12Н, м). НРМС (ΦIΑΤ): m/z [MH+]180. Препаративний приклад 23 Етил (2E/Z)-(1RS,6RS)-біцикло[4.2.0]окт-7иліден(ціано)етаноат 1 Кетон з препаративного прикладу 19 (2.85г, 45 74879 46 1 23.0ммоль), етилціаноацетат (2.45мл, 23.0ммоль), Н-ЯМР (400МГц, CDCl3): δ=7.31-7.22 (5Н, м), ацетат амонію (1.77г, 23.0ммоль) та льодяну оцто3.08 (1Н, д), 3.00 (1Н, д), 2.56 (1Н, м), 2.44 (1Н, д), ву кислоту (1.32мл) кип'ятили в толуолі (40мл), 2.38 (1Н, д), 2.25 (1Н, м), 1.98 (1Н, м), 1.75 (1Н, т), використовуючи ловушку Діна-Старка. Через 6 1.71-1.30 (7Н, м), 1.10 (1Н, м). годин суміш залишали охолоджуватися і розбавHPMC (ES): m/z [M-H] 257. ляли етилацетатом (150мл), промивали водою Препаративний приклад 26 (50мл), насиченим розчином солі і висушували трет-Бутил [(1RS,6RS,7SR)-7(MgSO4). Розчинник випарювали при зниженому бензилбіцикло[4.2.0]окт-7-ил]ацетат тиску. Залишок хроматографували (SiO2, гептан/етилацетат, 4:1) з одержанням 2.76г суміші ціаноестерів у вигляді твердої речовини жовтого кольору. 1 Оксалілхлорид (0.67мл, 7.62ммоль) додавали Н-ЯМР (400МГц, CDCІ3): δ (основний ізомер); по краплям при перемішуванні до розчину кислоти 4.26 (2Н, к), 3.36 (1Н, м), 3.02 (2Н, м), 2.58 (1Н, м), з препаративного прикладу 25 (1.79г, 6.93ммоль) в 1.30-2.18 (8Н, м), 1.33 (3Н, т). дихлорметані (25мл) у атмосфері азоту при 0°С. δ (неосновний ізомер) =4.25 (2Н, к), 3.48 (1Н, Додавали обережно диметилформамід (0.25мл) і м), 3.23 (2Н, м), 2.58 (1Н, м), 1.30-2.18 (8Н, м), 1.32 суміш залишали нагріватися до кімнатної темпе(3Н, т). ратури, після чого перемішували протягом додатПрепаративний приклад 24 кових 4 годин. Розчинник видаляли у вакуумі і заЕтил [(1RS,6RS,7RS)-7лишок розбавляли дихлорметаном (20мл). До бензилбіцикло[4.2.0]окт-7-ил](ціано)аиетат реакційної суміші у атмосфері аргону обережно додавали 2-метилпропан-1-ол (9мл) в дихлорметані (20мл) з наступним додаванням діізопропілетиламіну (1.8мл, 10.4ммоль). Суміш перемішували протягом 18 годин і потім додавали насичений Ціаноестер з препаративного прикладу 23 водний розчин гідрокарбонату натрію (30мл). Су(2.75г, 12.5ммоль) в ТГФ (60мл) додавали близько міш екстрагували етилацетатом (3 50мл) і об'єд1 години при перемішуванні до розчину бензилманані органічні фракції промивали насиченим розгнійхлориду (20мл 1М розчину в етері, 20ммоль) в чином солі і висушували (MgSО4). Розчинник ТГФ (20мл) при -78°С у атмосфері аргону. Після видаляли при зниженому тиску і залишок хроматоперемішування протягом 2 годин при цій темпераграфували (SiО2, гептан/етилацетат 98:2) з одертурі суміш гасили додаванням насиченого розчину жанням естеру (2.42г). хлориду амонію (10мл). Суміш залишали нагріва1 Н-ЯМР (400МГц, CDCl3): δ=7.33-7.19 (5Н, м), тися до кімнатної температури, і додавали розба3.05 (1Н, д), 2.96 (1Н, д), 2.53 (1Н, м), 2.30-2.18 влену соляну кислоту (30мл). Водний шар екстра(3Н, м), 1.90 (1Н, м), 1.72 (1Н, т), 1.65-1.55 (2Н, м), гували етилацетатом (3 40мл). Об'єднані органічні 1.48 (9Н, с), 1.47-1.00 (6Н, м). шари промивали насиченим розчином солі, висуПрепаративний приклад 27 шували (МgSО4) і розчинник випарювали при зни[(1RS,6RS,7SR)-7-(2-трет-Бутокси-2женому тиску з одержанням суміші діастереомероксоетил)біцикло[4.2.0]окт-7-ил]оцтова кислота них ціаноестерів. Залишок хроматографували (SіО2, гептан/етилацетат, 4:1) з одержанням 3.53г суміші діастереомерних ціаноестерів у вигляді прозорого масла. Rf(гептан-етилацетат, 4:1)=0.30 -1 max(плівка)/см 2247, 1740. Препаративний приклад 25 [(1RS,6RS,7SR)-7-бензилбіцикло[4.2.0]окт-7ил]оцтова кислота Суміш діастереомерних ціаноестерів з препаративного прикладу 24 (3.52г, 11.3ммоль) та гідроксиду калію (3.8г, 67.9ммоль) нагрівали до 160°С в етиленгліколі (75мл) протягом 72 годин. Після цього суміш залишали охолоджуватися і обережно додавали розбавлену соляну кислоту поки розчин не ставав кислим: що визначалось pH папером. Суміш екстрагували етилацетатом (3 100мл) і об'єднані органічні фракції промивали насиченим розчином солі, висушували (МgSО4) і розчинник випарювали при зниженому тиску. Залишок хроматографували (SiO2, етилацетат:гептан 1:4) з одержанням 2.11г рацемічної діастереомерної кислоти у вигляді масла жовтого кольору. зЕстер з препаративного прикладу 26 (6.93ммоль) та перйодат натрію (20.75г, 97.02ммоль) перемішували разом в етилацетаті (20мл), ацетонітрилі (20мл) та воді (30мл) протягом 5 хвилин. Суміш охолоджували до 0°С і до реакційної суміші додавали гідрат хлориду рутенію (lll) (0.03г, 0.14ммоль). Реакційну суміш залишали нагріватися до кімнатної температури і перемішували протягом 24 годин. Додавали діетиловий етер (100мл) і суміш перемішували протягом 40 хвилин. Додавали до суміші розбавлену соляну кислоту (150мл) і потім екстрагували етилацетатом (3 100мл). Об'єднані органічні фракції промивали насиченим розчином солі, висушували (MgSO4) і розчинник випарювали при зниженому тиску з одержанням 0.64г кислоти. 1 Н-ЯМР (400МГц, CDCl3): δ=2.84 (1Н, д), 2.75 (1Н, д), 2.61-2.48 (3Н, м), 2.17 (1Н, м), 1.95-1.80 (3Н, м), 1.78-1.30 (7Н, м), 1.44 (9Н, с). Препаративний приклад 28 Метиловий естер [(1RS,6RS,7SR)-6-(2-третБутокси-2-оксоетил)біцикло[4.2.0]окт-7-ил]оцтова 47 74879 48 лад 1А) (25г, 226.9ммоль) в ТГФ (150мл), підтримуючи температуру в межах 5-15°С. Реакційну суміш перемішували при температурі навколишнього середовища протягом 30 хвилин і потім додавали воду (100мл). Фази розділяли і органічний Триметилсилілдіазометан (1.2мл 2М розчину в шар, який містив вказану в заголовку сполуку, вигексані, 2.4ммоль) додавали при перемішуванні по користовували безпосередньо в наступній стадії. 1 краплям до розчину кислоти з препаративного Н-ЯМР (400МГц, CDCl3): δ=5.55 (1Н, д), 4.15 прикладу 27 (0.64г, 2.28ммоль) в суміші толуолу (2Н, к), 3.40 (1Н, м), 3.20 (1Н, м), 2.90 (1Н, м), 2.55 (10мл) та метанолу (2.5мл) при 0°С у атмосфері (1Н, м), 1.8-1.5 (5Н, м), 1.30 (3Н, т). аргону. Суміш залишали нагріватися до кімнатної Препаративний приклад 31 температури і перемішували протягом 16 годин. Етил (1R,5R,6S)-[6Розчинник видаляли при зниженому тиску і зали(нітрометил)біцикло[3.2.0]гепт-6-ил]ацетат шок переносили в етилацетат (150мл), промивали насиченим розчином гідрокарбонату натрію (100мл), розбавленою соляною кислотою (100мл), насиченим розчином солі і висушували (MgSO4). Розчинник випарювали при зниженому тиску з одержанням 0.65г етсеру у вигляді масла жовтого Розчин в ТГФ сполуки з препаративного приккольору. ладу 30 (40.9г сполуки в загальному об'ємі 225мл) 1 Н-ЯМР (400МГц, CDСІ3): δ=3.66 (3Н, с), 2.83 розбавляли ТГФ (270мл). Додавали TBAF 3H2O (1Н, д), 2.74 (1Н, д), 2.57 (1Н, д), 2.49 (1Н, д), 2.15 (93.1г; 295.0ммоль) та MeNO2 (453.9ммоль) і роз(1Н, м), 1.94-1.78 (3Н, м), 1.72-1.06 (8Н, м), 1.43 чин нагрівали при кипіння протягом 4 годин. Реак(9Н, с). цій суміш охолоджували та концентрували при Препаративний приклад 29 зниженому тиску. Додавали толуол (330мл) і дво[(1RS,6RS,5SR)-7-(2-Метокси-2фазну суміш промивали водою (165мл), 2М водн. оксоетил)біцикло[4.2.0]окт-7-ил]оцтова кислота НСl (165мл+100мл) і потім додатковою кількістю води (165мл). Толуольний шар, який містив продукт, висушували над MgSO4 і концентрували при зниженому тиску з одержанням вказаної у заголовку сполуки у вигляді масла червоноТрифтороцтову кислоту (3мл) додавали по го/коричневого кольору (90% (за 2 стадії)). краплям при перемішуванні до розчину з препара1 Н-ЯМР (400МГц, CDCl3): δ=4.80 (2Н, м), 4.15 тивного прикладу 28 (0.65г, 2.19ммоль) в дихлор(2Н, м), 2.85 (1Н, м), 2.65 (1Н, м), 2.55 (2Н, м), 2.20 метані (9мл) при 0°С. Суміш залишали нагріватися (1Н, м), 1.9-1.4 (7Н, м), 1.25 (3Н, т). до кімнатної температури і перемішували протяПрепаративний приклад 32 гом додаткових 16 годин. Суміш промивали наси(1R,5R,6S)-[6-(нітpометил)біцикло[3.2.0]гепт-6ченим водним розчином гідрокарбонату натрію і ил]оцтова кислота потім екстрагували етилацетатом (50мл). Водний шар підкисляли до pH4 з допомогою розбавленої соляної кислоти і потім екстрагували етилацетатом (2 50мл). Об'єднані органічні фракції промивали насиченим розчином солі, висушували Розчин нітроестеру з препаративного прикла(MgSO4) і розчинник видаляли при зниженому тисду 31 (200г; 828.9ммоль) в ТГФ (1.0л) об'єднували ку. Залишок очищали хроматографією (SiC-2, 6:4 з 2М водн. NaOH (1.04л; 2.08моль) і перемішували гептан/етилацетат) з одержанням 0.47г кислоти у при температурі навколишнього середовища провигляді масла жовтого кольору. 1 тягом 18 годин. Двофазну суміш розбавляли толуH-ЯМР (400МГц, CDCl3): δ=3.67 (3Н, с), 2.84 олом (500мл) і шари розділяли. Доводили pH вод(1Н, д), 2.78 (1Н, д), 2.74 (1Н, д), 2.66 (1Н, д), 2.49 ного шару до значення 1-3 за допомогою (1Н, м), 2.14 (1Н, м), 1.95-1.81 (2Н, м), 1.70 (1Н, м), концентрованої водної НСl та екстрагували СН2О2 1.63 (1Н, м), 1.55-1.30 (5Н, м), 1.07 (1Н,м). (1.0л+600мл). Дихлорметанові шари, які містили Препаративний приклад 30 об'єднаний продукт, концентрували при зниженому Етил (2Е)-(1R,5R)-біцикло[3.2.0]гепт-6тиску з одержанням вказаної у заголовку сполуки у иліденаиетат/етил (2Ζ)-(1R,5R)-біцикло[3.2.0]гептвигляді масла оранжевого кольору, яке тверділо 6-иліденацетат при стоянні (163.4г). 1 Н-ЯМР (400МГц, CDCl3): δ=4.80 (2Н, м), 2.85 (1Н, м), 2.60 (3Н, м), 2.20 (1Н, м), 1.85 (1Н, м), 1.70 (2Н,м), 1.6-1.4(4Н, м). Препаративний приклад 33 Розчин триетилфосфонацетату (53.4г; (1RS,5RS,6SR)-Спіро[біцикло[3.2.0]гептан-6,3'238.3ммоль) в ТГФ (25мл) додавали до суспензії піролідин]-5'-он 60% дисперсії гідриду натрію (9.53г; 238.3ммоль) в ТГФ (75мл), підтримуючи температуру в межах 515°С. Додавали розчин (1R,5R)біцикло[3.2.0]гепатан-6-ону (препаративний приккислота 49 74879 50 Нітроестер з препаративного прикладу 31 Стеарат магнію 4 (13.0г, 53.9ммоль) струшували в метанолі (125мл) Попередньо желатинізований при 25°С над нікелевим пористим каталізатором у крохмаль NF15146 146 атмосфері газоподібного водню при 345кПа (50 400 p.s.i.). Через 24 години каталізатор видаляли фільтрацією через арбосел і розчинник видаляли Композиція Ε при зниженому тиску. Залишок потім хроматограмг/таблетка фували (SiO2, етилацетат) з одержанням лактаму Активний інгредієнт 250 (4.76г). Стеарат магнію 5 1 Н-ЯМР (400МГц, CDCI3): δ=5.86 (1Н, шс), 3.40 Лактоза 145 (2Н, с), 2.79-2.70 (1Н, м), 2.54-2.47 (1Н, м), 2.32 Авіцел 100 (1Н, д), 2.12 (1Н, т), 2.03 (1Н, д), 1.86-1.60 (3Н, м), 500 1.57-1.38 (4Н, м). Мікроаналіз: Одержано: С, 72.48; Н, 9.15; Ν, Композиція F (Композиція контрольованого 8.43. C10H15NO: розраховано С, 72.69; Н, 9.15; Ν, вивільнення) 8.48%. мг/таблетка [α]D-28.4° (25°С) (а) Активний інгредієнт 500 Приклади Фармацевтичних композицій (b) ГідроксипропілметилцелюВ наступних прикладах активною сполукою лоза (Метоцел К4М Преміум) 112 може бути будь-яка сполука формули ί-XXV та/або (с) Лактоза В.Р. 53 її фармацевтично прийнятна сіль, сольват або (d) Повідон В.P.C. 28 фізіологічно функціональна похідна. (e) Стеарат магнію 7 (і) Композиції таблеток 700 Наступні композиції А і В можуть бути одержані вологим гранулюванням інгредієнтів від (а) до Композиція може бути одержана вологим гра(с) та від (а) до (д) з розчином повідону, з наступнулюванням інгредієнтів (а)-(с) з розчином повідоним додаванням стеарату магнію та таблеткуну, з наступним додаванням стеарату магнію та ванням. пресуванням. Композиція А Композиція G (Таблетки з ентеральним покмг/табмг/табриттям) летка летка Таблетки з ентеральним покриттям можуть (а) Активний інгредієнт 250 250 одержані з композиції С шляхом покриття таблеток (b) Лактоза В.Р. 210 26 у кількості 25мг/табл. ентеральним полімером, (с) Натрієва сіль крохмальтаким як, ацетат-фталат целюлози, полівінілацегліколяту 20 12 татфталат, гідроксипропілметилцелюлозафталат (d) Повідон В.Р. 15 9 або аніонні полімери метакрилової кислоти та ме(e) Стеарат магнію 5 3 тилового естеру метакрилової кислоти (Eudraigit 500 300 L). За винятком Eudragit L, ці полімери можуть також включати до 10% (від ваги використовуваного Композиція В полімеру) пластифікатора для попередження руймг/табмг/табнування мембрани в процесі використання або летка летка зберігання. Придатними пластифікаторами є діе(а) Активний інгредієнт 250 250 тилфталат, і трибутилцитрат та триацетин. (b) Лактоза 150 150 Композиція Η (Таблетки контрольованого ви(с) Avicel PH 101 60 26 вільнення з ентеральним покриттям) (d) Натрієва сіль крохмальТаблетки з ентеральним покриттям з композигліколяту 20 12 ції F можуть одержані шляхом покриття таблеток (e) Повідон В.Р. 15 9 ентеральним полімером у кількості 50мг/табл., (f) Стеарат магнію 5 3 таким як, ацетат-фталат целюлози, полівінілаце500 300 татфталат, гідроксипропілметилцелюлозафталат, або аніонні полімери метакрилової кислоти та меКомпозиція С тилового естеру метакрилової кислоти (Eudgragit мг/таблетка L). За винятком Eudragit L, ці полімери можуть таАктивний інгредієнт 100 кож включати до 10% (від ваги використовуваного Лактоза 200 полімеру) пластифікатора для попередження руйКрохмаль 50 нування мембрани в процесі використання або Повідон 5 зберігання. Придатними пластифікаторами є діеСтеарат магнію 4 тилфталат, трибутилцитрат та триацетин. 359 (ii) Композиції капсул Наступні композиції D та Ε можуть бути одерКомпозиція А жані безпосереднім пресуванням змішаних інгреКапсули можуть бути одержані змішуванням дієнтів. Лактоза, використовувана в рецептурі Е, є інгредієнтів вищезгаданої композиції D та наповлактозою типу прямого пресування. ненням твердих желатинових капсул, що складаКомпозиція D ються з двох частин, одержаною сумішшю. Компомг/таблетка зицію В (наведену нижче) можна одержати Активний інгредієнт 250 аналогічним чином. Композиція В мг/капсула (а) Активний інгредієнт 250 (b) Лактоза В.Р. 143 (с) Натрієва сіль крохмаль-гліколяту 25 (d) Стеарат магнію 2 420 Композиція С мг/капсула 250 350 600 Капсули можуть бути одержані шляхом диспергування активного інгредієнту в лецетині та арахісовому маслі та наповнення м'яких, еластичних желатинових капсул дисперсією. (а) Активний інгредієнт (b) Макрогол 4000 ВР Композиція D мг/капсула Активний інгредієнт 250 Лецитин 100 Арахісове масло 100 450 Капсули можуть бути одержані шляхом розплавлення макроголу 4000 BP, наступного диспергування активного інгредієнту в розплаві та наповнення ним твердих желатинових капсул, що складаються з двох частин. Композиція Ε (Капсули контрольованого вивільнення) мг/капсула (а) Активний інгредієнт 250 (b) Мікрокристалічна целюлоза 125 (с) Лактоза В.Р. 125 (d) Етилцелюлоза 13 513 Рецептура капсул контрольованого вивільнення може бути одержана шляхом формування суміші інгредієнтів (а)-(с) з використанням шприцьмашини з наступною сферонізацією та висушуванням отриманого матеріалу. Висушені гранулки покривають мембраною контрольованого вивільнення (d) та наповнюють ними тверді желатинові капсули, що складаються з двох частин. Композиція F (Ентеральні капсули) мг/капсула 250 125 125 50 5 555 Композиція для ентеральних капсул може бути одержана шляхом формування суміші інгредієнтів (а)-(с) з використанням шприць-машини з наступною сферонізацією та висушуванням отриманого матеріалу. Висушені гранулки покривають ентера(а) Активний інгредієнт (b) Мікрокристалічна целюлоза (с) Лактоза В.Р. (d) Ацетат-фталат целюлози (e) Діетилфталат льною мембраною (d), яка містить пластифікатор (e), та наповнюють ними тверді желатинові капсули, що складаються з двох частин. Композиція G (Капсули контрольованого вивільнення з ентеральним покриттям) Ентеральні капсули з композиції Ε можуть бути одержанні покриттям гранулок контрольованого вивільнення ентеральним полімером у кількості 50мг/капсула, таким як, ацетат-фталат целюлози, полівінілацетатфталат, гідроксипропілметилцелюлозафталат або аніонні полімери метакрилової кислоти метилового естеру метакрилової кислоти (Eudragit L). За винятком Eudragit L, пластифікатора для попередження руйнування мембрани в процесі використання або зберігання. Придатними пластифікаторами є діетилфталат, трибутилцитрат та триацетин. (ііі) Композиції для внутрішньовенних ін'єкцій Активний інгредієнт 0,250г Стерильний апірогенний фосфатний буфер (pH9,0) до 10мл Активний інгредієнт розчиняють в більшій частині фосфатного буферу при 35-40°С, потім доводять до необхідного об'єму і фільтрують через стерильний мікропористий фільтр в стерильні скляні пляшечки об'ємом 10мл (Тип 1), які закривають герметично стерильними кришечками для пляшок та додатковим засобом, що попереджує несанкціоноване відкривання. iν) композиція для внутрішньом’язових ін'єкцій Активний інгредієнт 0,20г Бензиловий спирт 0,10г Глікофурол 751 1,45г Вода для ін'єкцій q.s. до 3,00мл Активний інгредієнт розчиняють в глікофуролі. Потім додають бензиловий спирт, розчиняють і додають воду до 3мл. Потім суміш фільтрують через стерильний мікропористий фільтр і запаюють в стерильні 3мл скляні ампули (Тип 1). (v) композиція сиропу Активний інгредієнт Розчин сорбіту Гліцерин Бензоат натрію Смаковий агент Очищена вода q.s. 0,25г 1,50г 1,00г 0,005г 0,0125мл до 5,0мл Бензоат натрію розчиняють в порції очищеної води і додають розчин сорбіту. Додають активний інгредієнт і розчиняють. Одержаний розчин змішують з гліцерином і потім доводять до необхідного об'єму очищеною водою. 53 (vi) композиція супозиторіїв Активний інгредієнт Твердий жир, BP (Witepsol H15 Dynamit NoBel) 74879 мг/супозиторій 250 1770 2020 Одну п'яту частину Witepsol H15 розплавляють в резервуарі з пароводяним кожухом при максимальній температурі 45°С. Активний інгредієнт просіюють через 200 tm сито і додають до розплавленої основи при перемішуванні, використовуючи Sitverson, оснащений різальною головкою, поки не отримують однорідну дисперсію. Підтримуючи суміш при 45°С, додають решту Witepsol H15 до суспензії, яку перемішують до одержання гомогенної суміші. Однорідну суспензію потім пропускають через 250 lm сито з нержавіючої сталі і при постійному перемішуванні залишають охолоджуватися до 40°С. При температурі 38-40°С аліквотами (2.02г) суміші наповнюють придатні пластмасові форми і залишають супозиторії охолоджуватися до кімнатної температури. Комп’ютерна верстка Т. Чепелева 54 (vii) Композиція пес арій мг/песарій 250 380 363 7 1000 Вищевказані інгредієнти повністю змішують, і песарії одержують пресуванням одержаної суміші. Активний інгредієнт (63 lm) Безводна декстроза Картопляний крохмаль Стеарат магнію (viii) Трансдермальна композиція Активний інгредієнт 200мг Спирт USP 0,1мл Гідроксиетилцелюлоза Активний інгредієнт та спирт USP желатинізують з гідроксиетилцелюлозою і компонують в трансдермальні засоби з площею поверхні 10см2. Біологічні дані Сполуку прикладів 1 та 4 випробували в тестах порадіолігандному зв'язуванню: описаному тут і визначили, що вони мають зв'язувальні афінності 46.8 та 600нМ, відповідно. Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюCondesated bicyclic or tricyclic amino acids
Автори англійськоюBryans Justin Stephen
Назва патенту російськоюКонденсированные бициклические или трициклические аминокислоты
Автори російськоюБраянс Джастин Стивен
МПК / Мітки
МПК: C07C 229/34, C07C 61/00, A61P 25/24, A61K 31/195, A61P 1/04, A61P 1/00, A61P 25/08, A61P 15/08, A61P 25/20, A61P 19/02, A61P 25/28, A61P 29/00, A61P 43/00, A61P 25/02, A61P 13/10, A61P 25/04, A61P 25/14, C07C 229/28, A61P 25/22, A61P 9/02, A61P 1/18
Мітки: конденсовані, біциклічні, амінокислоти, трициклічні
Код посилання
<a href="https://ua.patents.su/27-74879-kondensovani-biciklichni-abo-triciklichni-aminokisloti.html" target="_blank" rel="follow" title="База патентів України">Конденсовані біциклічні або трициклічні амінокислоти</a>
Біциклічні амінокислоти як фармацевтичні засоби

Номер патенту: 72931
Опубліковано: 16.05.2005
Автори: Ресевер Жан-Марі, Блейкмор Дейвід Клайв
МПК: A61P 9/00, A61P 5/24, A61P 25/22, A61P 25/02, A61P 25/14, A61P 25/08, A61P 25/28, A61P 21/04, A61P 9/02, A61P 15/00, A61P 17/02, A61P 19/02, C07C 229/28, A61P 7/02, A61P 25/16, A61P 25/00, A61P 25/04, A61P 43/00, A61P 25/24, A61P 9/10, A61P 21/02, A61P 11/00, A61P 3/10, A61P 25/20, A61K 31/195
Мітки: амінокислоти, біциклічні, фармацевтичні, засоби
Формула / Реферат:
1. (1,3,5) (3-амінометилдицикло[3,2,0]гепт-3-ил)оцтова кислота або її фармацевтично прийнятна сіль, або її пролікарська форма.2. Сполука за п.1 або її фармацевтично прийнятна сіль, або її пролікарська форма для використання як...
Фармацевтична композиція з антимікробною активністю та похідні дипептиду з a-амінокислоти або її похідного та циклопентан-b-амінокислоти або її похідного

Номер патенту: 46725
Опубліковано: 17.06.2002
Автори: Шмідт Аксель, Міттендорф Йоахім, Цігельбауер Карл, Куніш Франц, Мілітцер Ханс-Крістіан, Матцке Міхаель, Шьонфельд Вольфганг
МПК: A61K 31/195, C07K 5/023, A61K 38/05, C07C 237/04, C07C 231/00, A61P 31/04, A61P 31/10, A61K 31/27, C07C 229/48
Мітки: композиція, фармацевтична, похідні, антимікробною, похідного, циклопентан-b-амінокислоти, активністю, a-амінокислоти, дипептиду
Формула / Реферат:
1. Композиция с антимикробной активностью, содержащая минимум одну циклопентан-бета-аминокислоту и/или ее производное, отличающаяся тем, что композиция содержит дополнительно минимум одну альфа-аминокислоту и/или ее производное.2. Композиция по пункту 1, отличающаяся тем, что в качестве альфа-аминокислоты она содержит соединение общей формулы (Iа) , (Ia)в которойR3 означает циклоалкил с 3-8 атомами углерода или...
Похідні циклічної амінокислоти, фармацевтична композиція та спосіб лікування

Номер патенту: 49011
Опубліковано: 16.09.2002
Автори: Моррелл Ендрю І., Хартенштайн Йоханнес, Кнін Клер О., Реткліфф Джайлз С., Хоруелл Девід Крістофер, Брайанс Джастін С.
МПК: A61P 25/28, A61P 25/16, A61P 25/14, A61P 25/08, A61P 25/22, A61P 25/12, A61P 25/18, A61K 31/00, C07C 233/47, A61K 31/195, C07C 229/28, A61P 25/24
Мітки: амінокислоти, циклічної, похідні, фармацевтична, спосіб, лікування, композиція
Формула / Реферат:
1. Похідні циклічної амінокислоти загальної формули (І) , (I)деR1-R10 кожний незалежно вибраний з лінійного або розгалуженого алкілу з кількістю атомів вуглецю від 1 до 6, незаміщеного або заміщеного бензилу або фенілу, в яких замісники вибрані з галогену, алкілу, алкокси, гідрокси, карбокси, карбоалкокси, трифторметилу і нітро, ібудь-який R1-R10, якщо він не приймає значення, вказані вище, являє собою...
Конденсовані азепіни як агоністи вазопресину

Номер патенту: 73152
Опубліковано: 15.06.2005
Автори: Хадсон Пітер, Франклін Річард Джеремі, Йі Крістофер Мартін, Ешворт Дорін Мері, Пітт Гарі Роберт Уільям
МПК: A61P 13/00, A61P 5/10, C07D 471/04, C07D 403/12, A61K 31/553, A61K 31/5513, C07D 498/04, A61K 31/551, A61K 31/55, C07D 487/04, A61K 31/5517, C07D 413/12, A61P 7/04, C07D 519/00, A61P 43/00, C07D 495/04, A61P 7/12
Мітки: азепіни, вазопресину, агоністи, конденсовані
Формула / Реферат:
1. Сполука згідно із загальною формулою 1 або її фармацевтично прийнятна сіль, , 1у якій:А є біциклічною або трициклічною похідною азепіну, вибраною із загальних формул 2 - 7, 2 , 3
Біциклічні гетероцикли та лікувальний засіб на їх основі

Номер патенту: 73004
Опубліковано: 16.05.2005
Автори: Лангкопф Ельке, Хіммельсбах Франк, Золька Флавіо, Юнг Біргіт, Блех Штефан
МПК: C07D 239/94, A61P 37/08, A61P 11/08, A61K 31/517, A61P 43/00, C07D 413/14, A61P 1/04, A61P 1/16, A61P 11/00, A61P 11/06, A61P 35/04, A61P 17/00, C07D 413/12, A61P 13/12, C07D 405/12, A61P 27/16, A61P 35/00
Мітки: біциклічні, гетероцикли, засіб, основі, лікувальний
Формула / Реферат:
1. Біциклічні гетероцикли загальної формули,(І)у якійRa означає бензильну або 1-фенілетильну групу або заміщену залишками R1 та R2 фенільну групу, при цьомуR1 являє собою атом водню, фтору, хлору або брому, метильну, трифторметильну, ціано- або етинільну групу таR2 являє собою атом водню або фтору, таодин із замісників Rb або Rc означає R3-(СН2)m-О-групу, а інший з замісників Rbабо Rc означає...