Заміщені 8-[6-аміно-3-піридил]ксантини
Номер патенту: 105164
Опубліковано: 25.04.2014
Автори: Ріґер Джейсон М., Томпсон Роберт Д., Вонґ Ґуок'юан


















Формула / Реферат
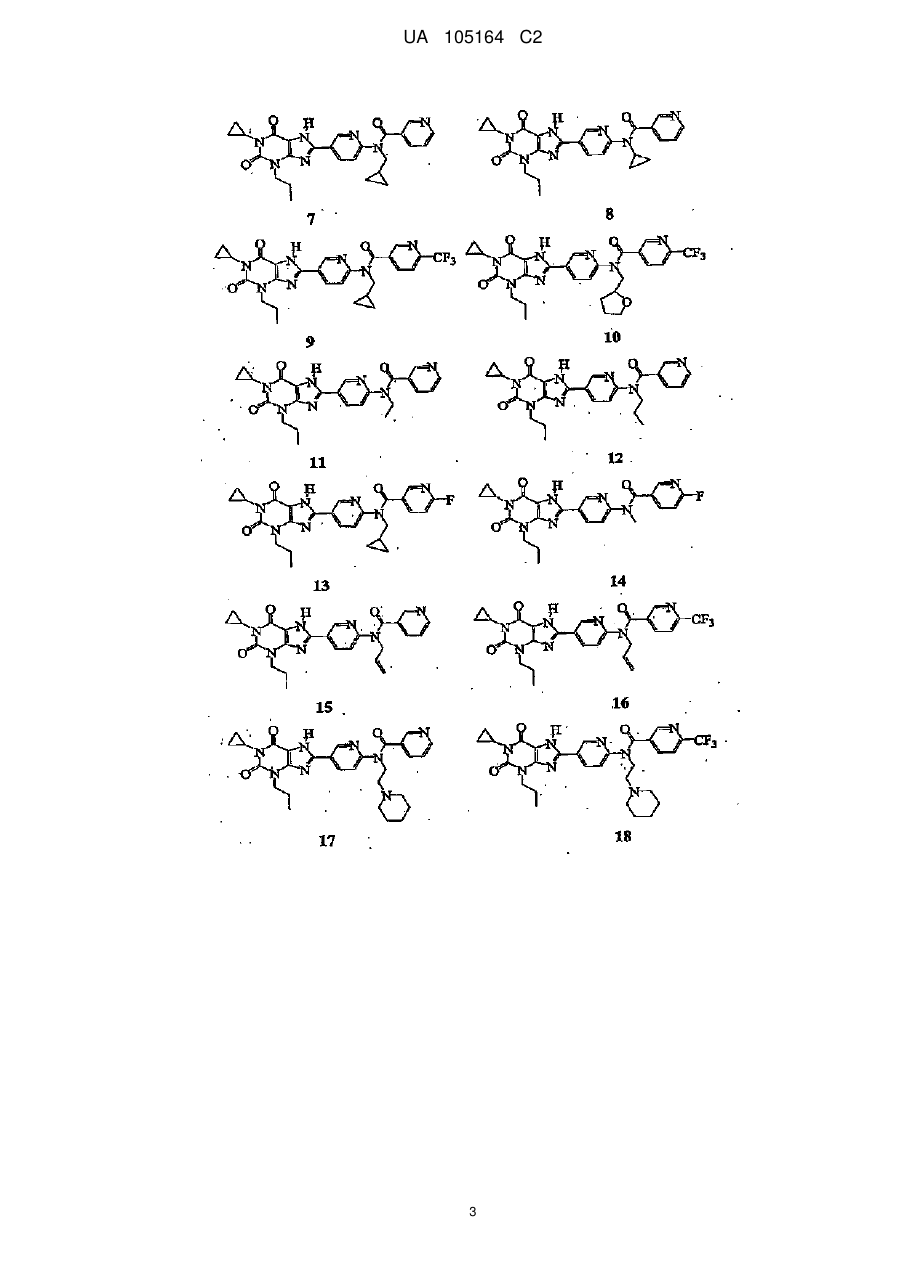
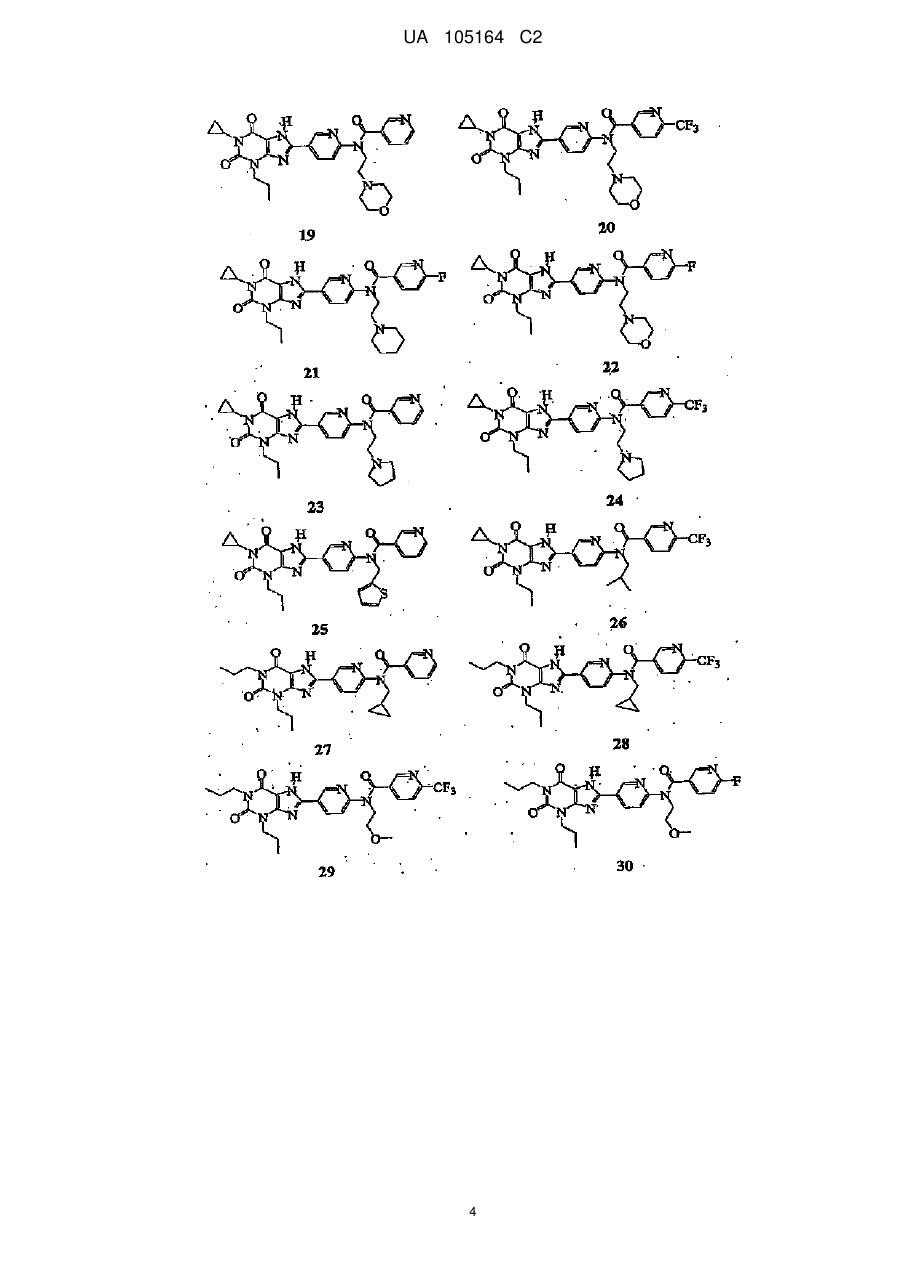
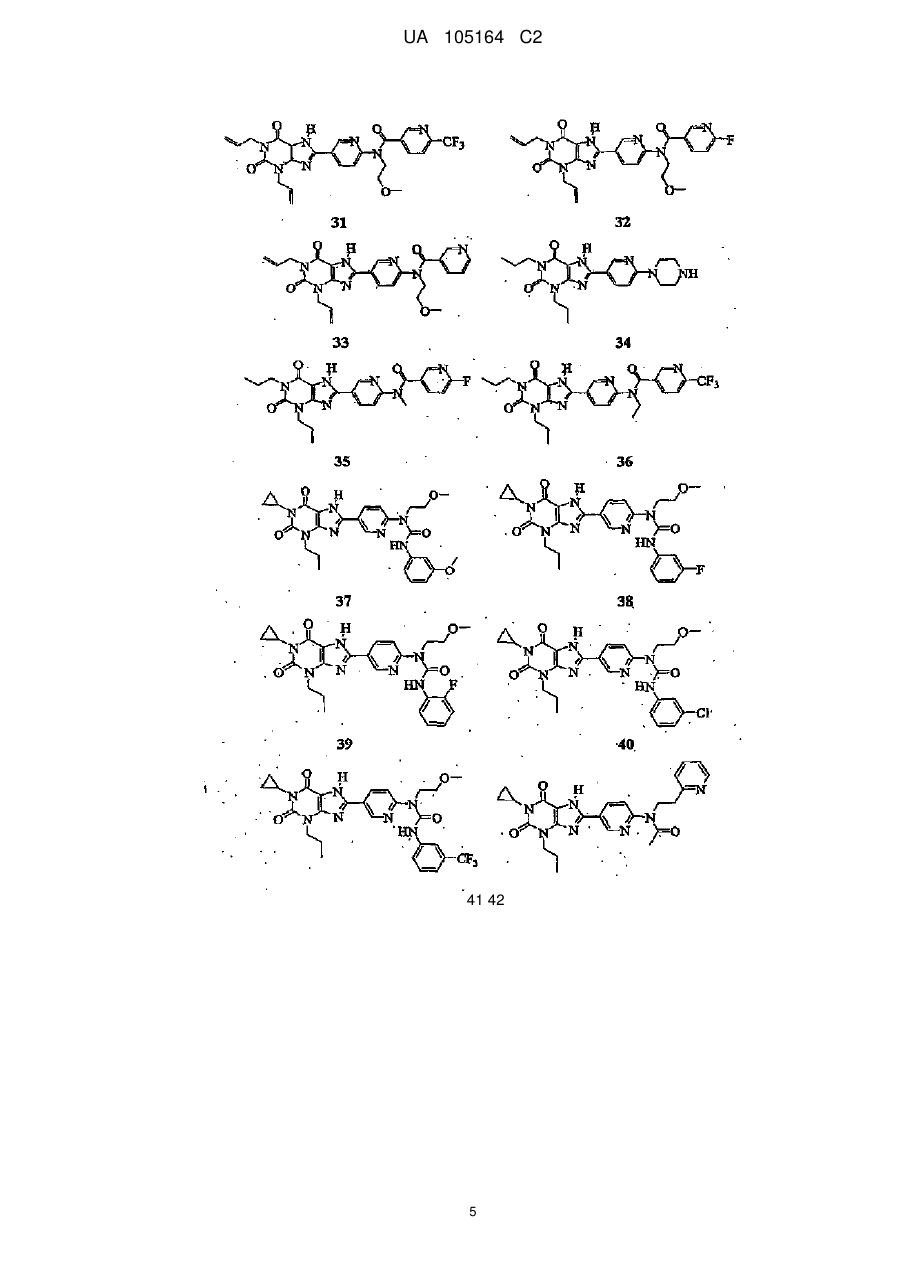
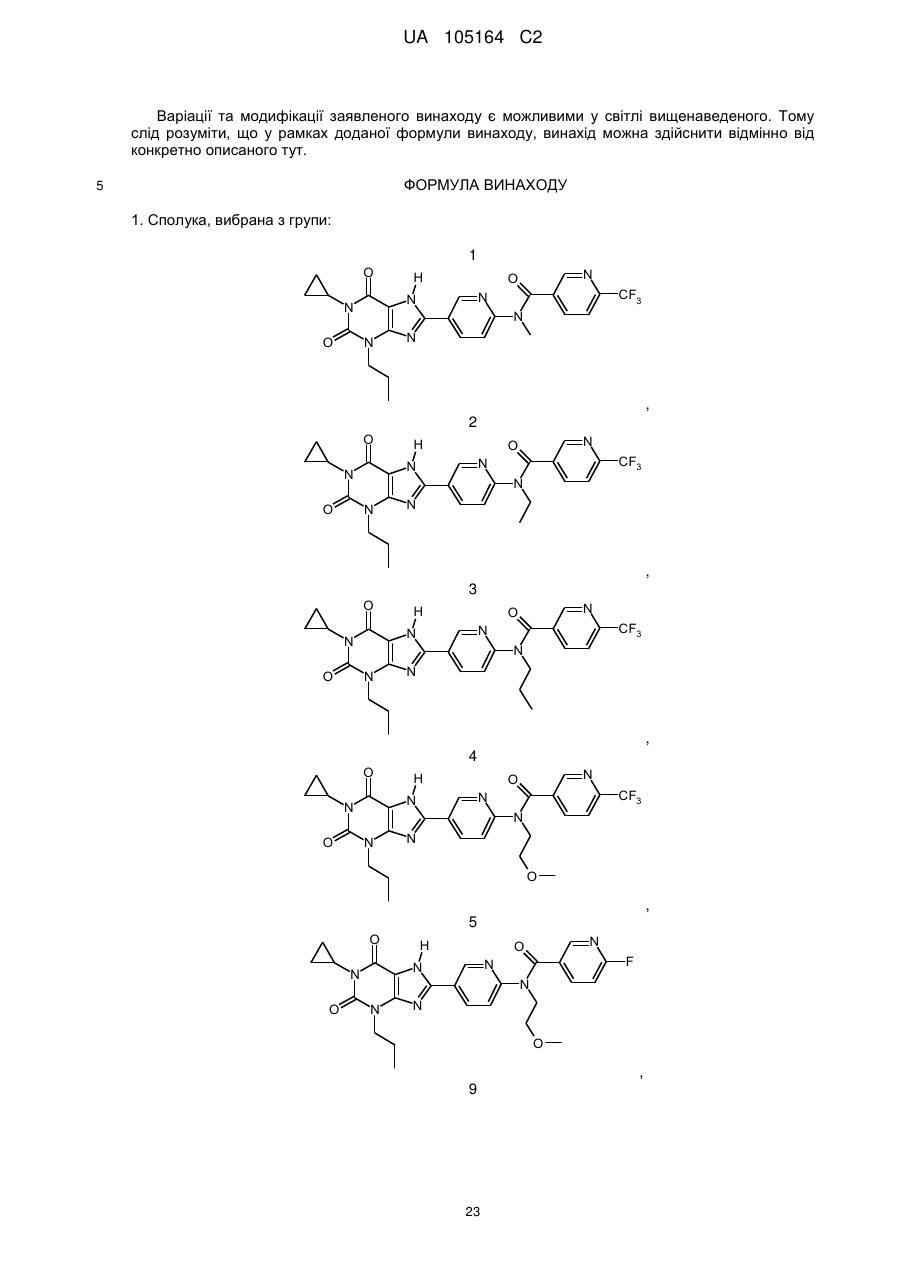
1. Сполука, вибрана з групи:
1
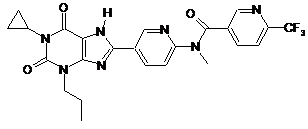 ,
,
2
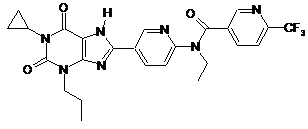 ,
,
3
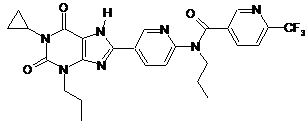 ,
,
4
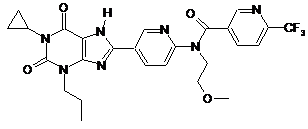 ,
,
5
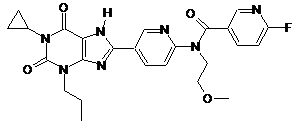 ,
,
9
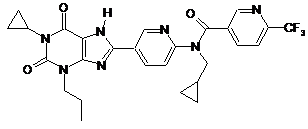 ,
,
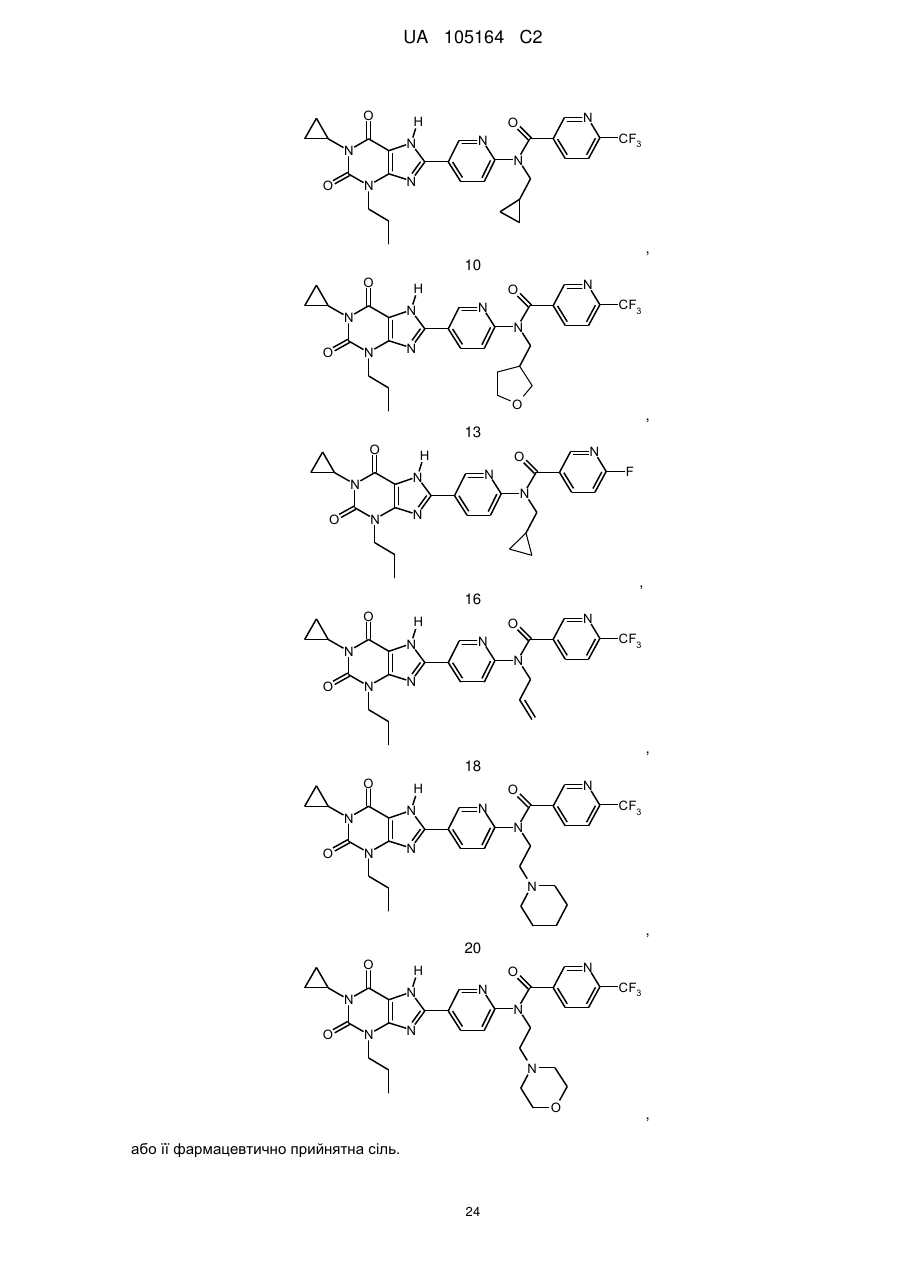
10
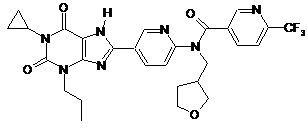 ,
,
13
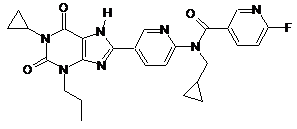 ,
,
16
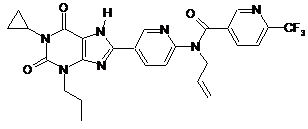 ,
,
18
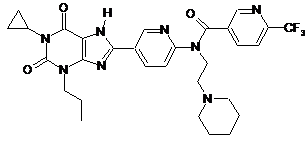 ,
,
20
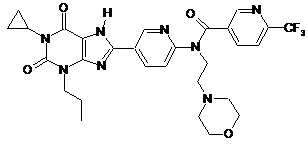 ,
,
або її фармацевтично прийнятна сіль.
2. Фармацевтична композиція, що містить:
(a) терапевтично ефективну кількість сполуки за п. 1; та
(b) фармацевтично прийнятний наповнювач.
3. Спосіб лікування астми, в якому вводять ефективну кількість сполуки за п. 1 ссавцю при необхідності такого лікування.
4. Спосіб лікування діабетичної ретинопатії, в якому вводять ефективну кількість сполуки за п. 1 або її фармацевтично прийнятної солі ссавцю при необхідності такого лікування.
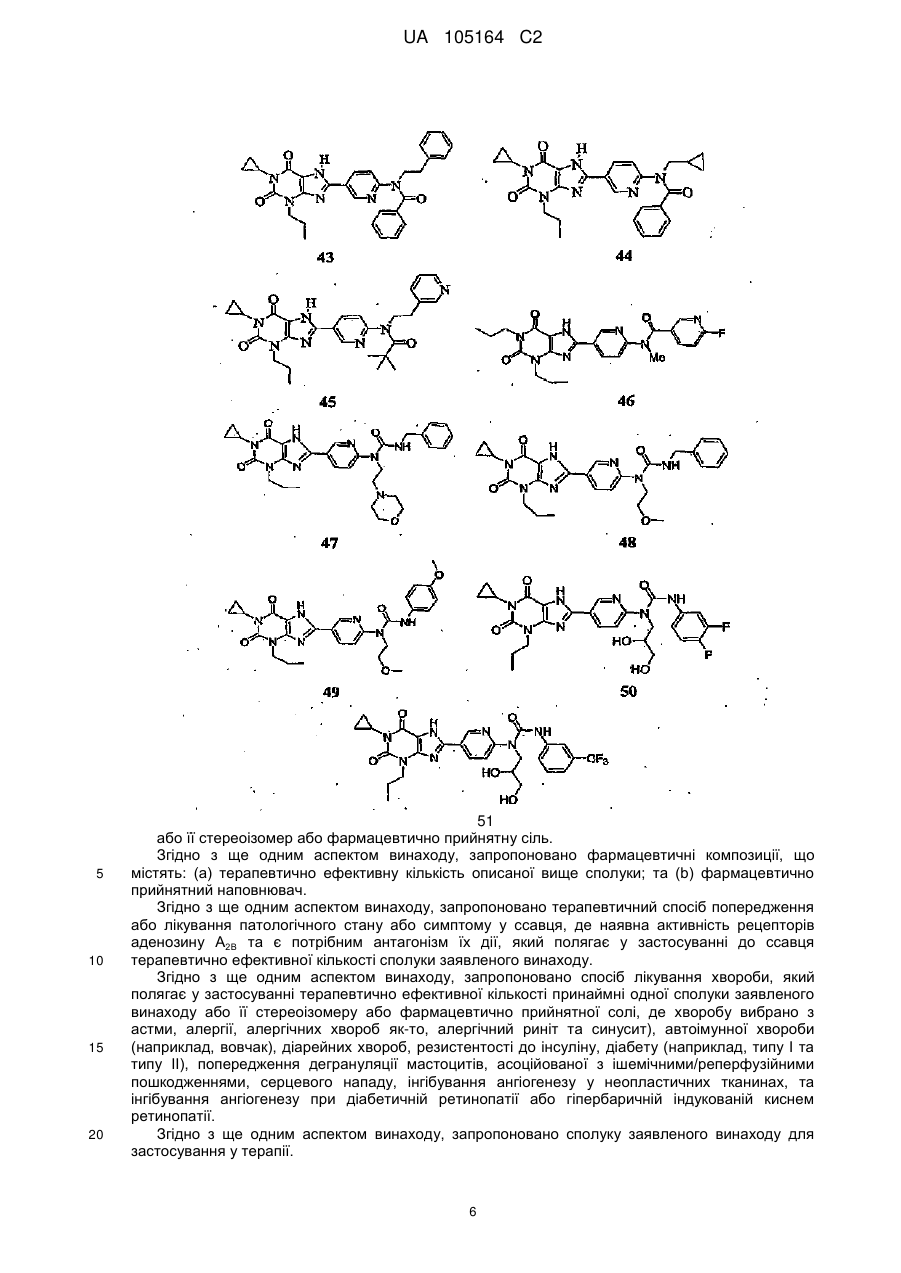
5. Сполука за п. 1, якою є
5
,
або її фармацевтично прийнятна сіль.
6. Спосіб лікування астми, в якому вводять ефективну кількість сполуки за п. 5 ссавцю при необхідності такого лікування.
7. Спосіб лікування діабетичної ретинопатії, в якому вводять ефективну кількість сполуки за п. 5 ссавцю при необхідності такого лікування.
8. Сполука, вибрана з групи:
50
 ,
,
51
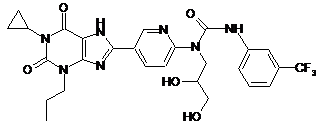 ,
,
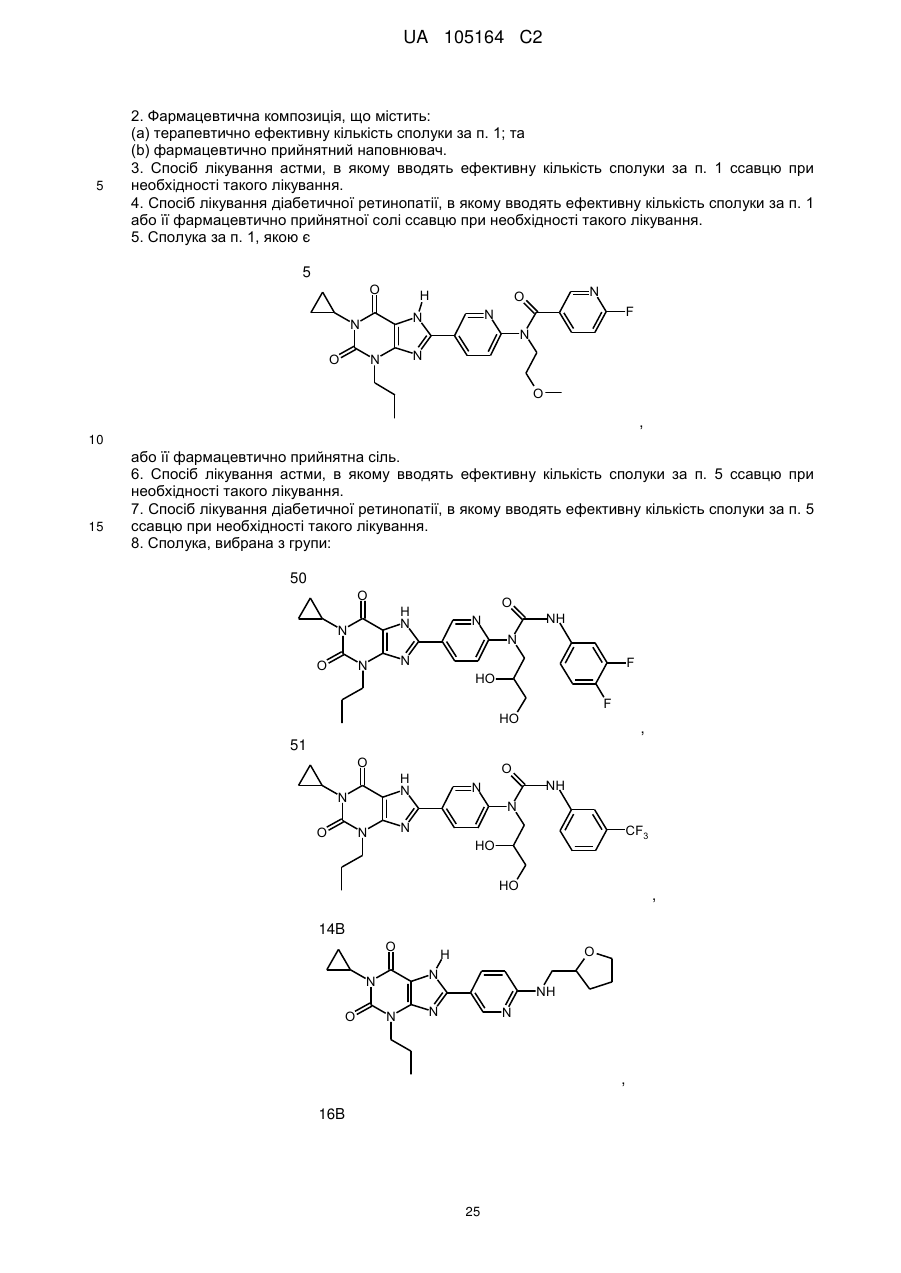
14B
 ,
,
16B
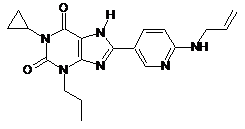 ,
,
або її фармацевтично прийнятна сіль.
9. Фармацевтична композиція, що містить:
(a) терапевтично ефективну кількість сполуки 50 або 51 за п. 8; та
(b) фармацевтично прийнятний наповнювач.
10. Спосіб інгібування активності А2B-рецепторів аденозину у ссавця, при якому ссавцю вводять ефективну кількість сполуки 50 або 51 за п. 8.
11. Спосіб лікування астми, в якому вводять ефективну кількість сполуки 50 або 51 за п. 8 ссавцю при необхідності такого лікування.
12. Спосіб лікування діабетичної ретинопатії, в якому вводять ефективну кількість сполуки 50 або 51 за п. 8 або її фармацевтично прийнятної солі ссавцю при необхідності такого лікування.
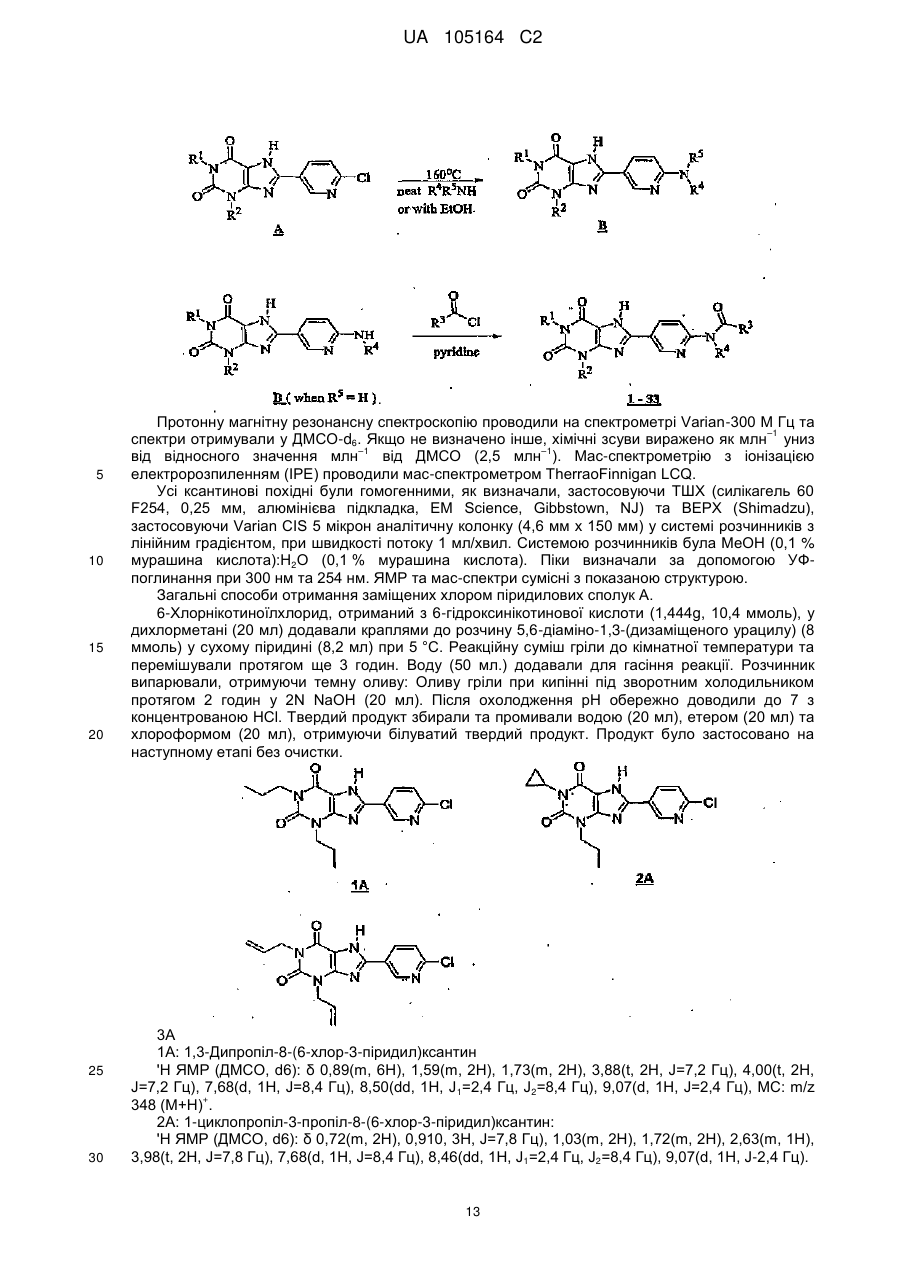
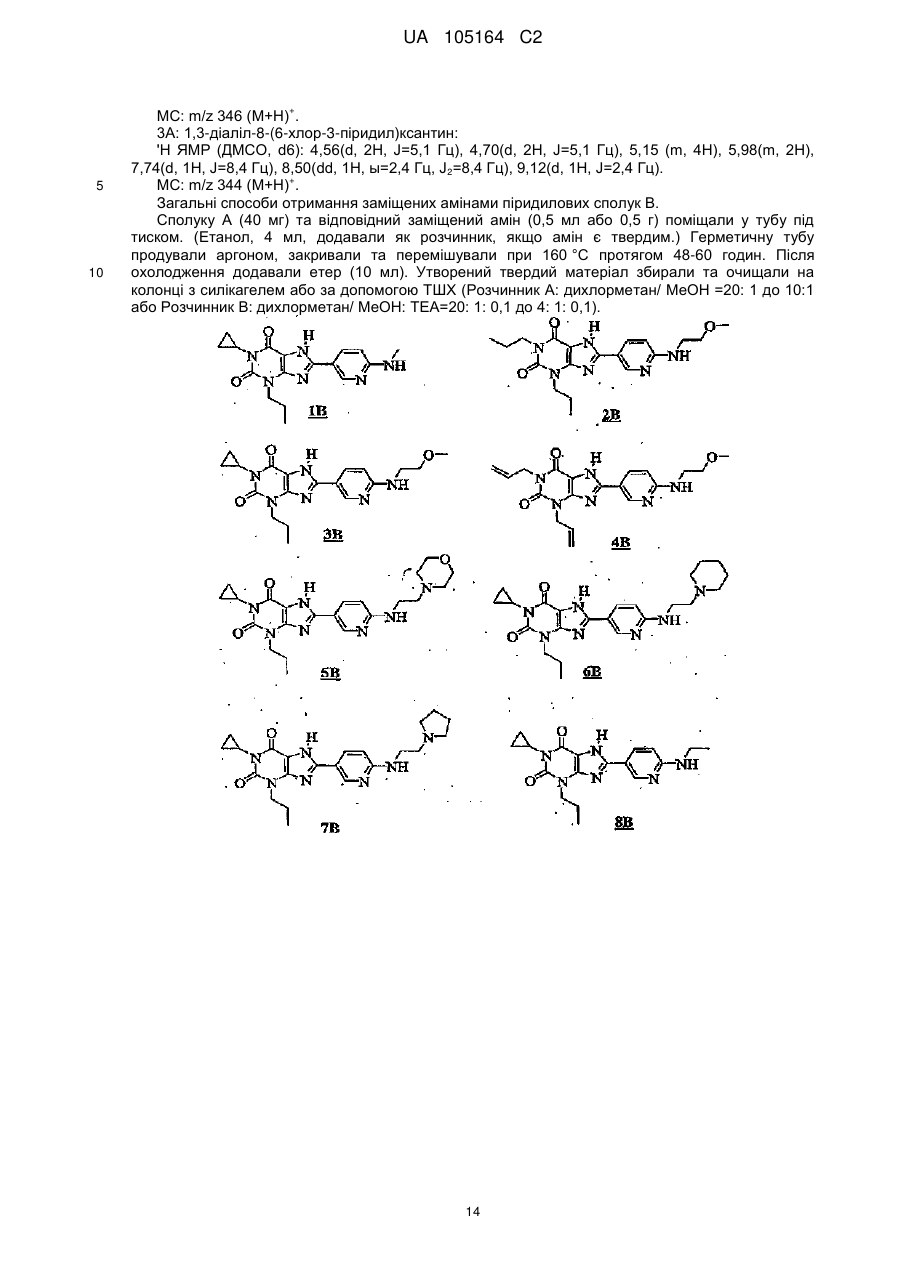
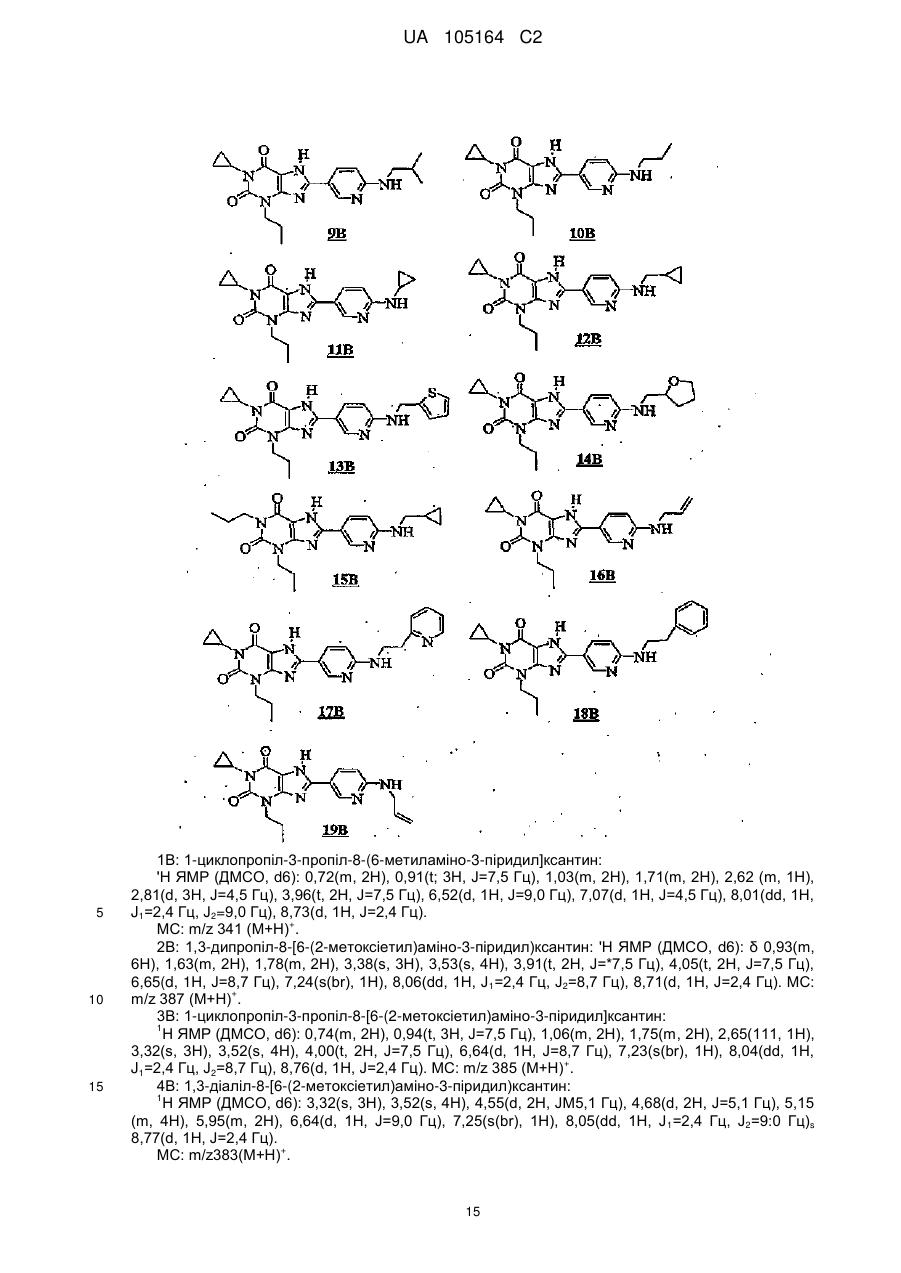
Текст
Реферат: Заявлений винахід стосується заміщених 8-[6-аміно-3-піридил]ксантинів та фармацевтичних композицій, що є селективними антагоністами рецепторів А2в аденозину (AR). Ці сполуки та композиції є корисними як фармацевтичні агенти. UA 105164 C2 (12) UA 105164 C2 UA 105164 C2 5 10 15 20 25 30 35 40 45 50 Ця заявка заявляє на права попередньої патентної заявки США № 60/805,030, зареєстрованої 16.06.2006, та попередньої патентної заявки США № 60/805,864, зареєстрованої 22.06.2006, зміст котрих уведено тут як посилання. Заявлений винахід стосується заміщених 8-[6-аміно-3-піридил]ксантинів та фармацевтичних композицій, що є селективними антагоністами рецепторів А2В аденозину (АR). Ці сполуки та композиції є корисними як фармацевтичні агенти. Алкілксантин теофілін (нижче), слабкий неселективний антагоніст аденозину (Дивись Lіndеn, J., еt аl, Саrdіоvаsсulаr Віоlоgу оf Рurіnеs, еds. G. Вurnstосk, еt аl, 1998, рр 120), є корисним терапевтично для лікування астми. Однак, його застосування асоційоване з неприємною побічною дією, як-то безсоння та діурез. В останні роки застосування теофіліну як бронходилататору для полегшення астми, витиснено ліками інших класів, наприклад, селективними піадренергічними агоністами, кортикостероїдами, та нещодавно, антагоністами лейкотриєнів. Ці сполуки також мають обмеження. Тому, розробка ліків типу теофіліну зі зменшеною побічною дією є бажаною. Виявлено, що теофілін та його близький аналог кофеїн блокують ендогенну дію аденозину, діючи як локальні модулятори рецепторів аденозину у мозку та інших органах у терапевтично корисних дозах. Аденозин активує чотири підтипи сполучених з G-білком рецепторів аденозину (АR), А1/А2а/А2В/А3. Енпрофілін (нижче) є ще одним прикладом ксантину, який, як повідомлено, блокує рецептори аденозину А2В та є застосовуваним для лікування астми. Також показано LаNоuе еt аl (патент США № 6,060,481), що селективні антагоністи аденозину А2В є корисними для поліпшення чутливості до інсуліну у пацієнта. Повідомлено, що терапевтичні концентрації теофіліну або енпрофіліну блокують А 2Врецептори людини, та припущено, що антагоністи, селективні для цього підтипу, можуть мати потенційне застосування як антіастматичні агенти. (Дивись Fеоktіstоv, І., еt аl; Рhаrmасоl Rеv. 1997, 49, 381-402; та Rоbеvа, А.S., еt аl, Drug Dеv. Rеs. 1996, 39, 243-252). Енпрофілін має повідомлене значення Кі 7 мкМ та є почасти селективним у зв'язуванні до АR людини А 2В. (Дивись Rоbеvа, А.S., еt аl. Drug Dеv. Rеs. 1996, 39, 243-252 та Lіndеn, J., еt аl., Моl. Рhаrmасоl. 1999, 56, 705-713). А2В-АR експресуються у деяких мастоцитах, як-то лінія ВR мастоцитоми 2+ клітин собак, котрі виявлені як відповідні за запуск гострої мобілізації та дегрануляції Са . (Дивись Аuсhаmрасh, J.А., еt аl, Моl. Рhаrmасоl 1997, 52, 846-860 та Fоrsуth, Р., еt аl, Іnflаmm. 2+ Rеs. 1999, '48, 30,1-307). А2В-АR також запускають мобілізацію Са , та приймають участь в уповільненому DL8 вивільненні з мастоцитів людини НМС-1. Іншими функціями, асоційованими з А2В-АR, є регуляція росту клітин та експресія гена, (Дивись Nеаrу, J:, еt аl., Тrеnds Nеurоsсі. 1996; 19, 13-18) залежна від ендотелію вазодилатація (Дивись Маrtіn, РХ., еt аl, J. Рhаrmасоl Ехр. Тhеr. 1993, 265, 248-253), та секреція рідини з кишкового епітелію. (Дивись Strоhmеіеr, G.R., еt аl, J. Віоl. Сhеm. 1995, 270, 2387-2394). Аденозин, діючи через А2В АR, як також повідомлено, стимулює проникність хлориду у клітини, що експресують регулятор транспорту кістозного фіброзу. (Дивись Сlаnсу, J.Р., еt аl, Аm. J. Рhуsіоl 1999, 276, С361-С369.). Нещодавно Lіndеn еt аl (патент США № 6,545,002) описали нову групу сполук та фармацевтичних композицій, що є селективними антагоністами А2В-рецепторів аденозину (АR). Хоча селективні стосовно підтипів рецепторів аденозину зонди є тільки для АR А 1, А2А, та А3, кілька селективних антагоністів є відомими для рецептору А2В. Тому, існує постійна необхідність у сполуках, що є селективними антагоністами рецептору А2В. Заявлений винахід стосується заміщених 8-[6-аміно-3-піридил]ксантинів або стереоізомерів чи фармацевтично прийнятних солей, що діють як антагоністи рецепторів аденозину А2В. Винахід також стосується фармацевтичних композицій, що містять сполуку заявленого винаходу або її стереоізомер чи фармацевтично прийнятну сіль у комбінації із фармацевтично прийнятним наповнювачем. Крім того, винахід стосується терапевтичного способу лікування патологічного стану або симптому у ссавця, як-то людини, де активність, наприклад, 1 UA 105164 C2 5 10 15 20 25 надактивність, А2В-рецепторів аденозину призводить до одного або більше симптомів патології, а антагонізм (тобто, блокування) є потрібним для полегшення таких симптомів. Тому, заявлений винахід стосується способу лікування хвороб, який полягає у застосуванні терапевтично ефективної кількості принаймні одної сполуки заявленого винаходу або її стереоізомеру або фармацевтично прийнятної солі, де хворобу вибрано з астми, алергії, алергічних хвороб (як-то, алергічний риніт та синусит), автоімунної хвороби (наприклад, вовчак), діарейних хвороб, резистентості до інсуліну, діабету (наприклад, типу І та типу ІІ), попередження дегрануляції мастоцитів, асоційованої з ішемічними/реперфузійними пошкодженнями, серцевого нападу, інгібування ангіогенезу у неопластичних тканинах, та інгібування ангіогенезу при діабетичній ретинопатії або гіпербаричній індукованій киснем ретинопатії. Винахід стосується нової сполуки заявленого винаходу для застосування у терапії. Винахід також стосується застосування нової сполуки заявленого винаходу для виробництва медикаменту для лікування патологічного стану або симптому у ссавця, що асоційовано зі шкідливою активацією або активністю А2В-рецепторів. Винахід також охоплює спосіб, який полягає у контакті сполуки заявленого винаходу, що необов'язково має радіоактивний ізотоп (нуклід), як-то, наприклад, тритій, радіоактивний йод 125 (наприклад, І для аналізів зв'язування або для спектрального відображення) тощо, з цільовими ділянками А2В-рецепторів аденозину, що містять вказані рецептори, іn vіvо або іn vіtrо, для зв'язування з вказаними рецепторами. Мембрани клітин, що містять зв'язані ділянки А2В-рецепторів аденозину можна застосовувати для виміру селективності тест-сполук стосовно підтипів рецепторів аденозину або їх можна застосовувати як засоби для ідентифікації потенційних терапевтичних агентів для лікування хвороб або станів, асоційованих із посередництвом А2В -рецептору, контактуванням вказаних агентів із вказаними радіолігандами та рецепторами, та виміром ступеню заміщення радіоліганду та/або зв'язування агенту. Заявники виявили, що показані нижче заміщені 8-[6-аміно-3-піридил]ксантини можуть бути корисними для лікування хвороб або станів, асоційованих зі шкідливою активацією або активністю А2В-рецепторів. Згідно з аспектом винаходу, запропоновано сполуку вибрану з групи: 2 UA 105164 C2 3 UA 105164 C2 4 UA 105164 C2 41 42 5 UA 105164 C2 5 10 15 20 51 або її стереоізомер або фармацевтично прийнятну сіль. Згідно з ще одним аспектом винаходу, запропоновано фармацевтичні композиції, що містять: (а) терапевтично ефективну кількість описаної вище сполуки; та (b) фармацевтично прийнятний наповнювач. Згідно з ще одним аспектом винаходу, запропоновано терапевтичний спосіб попередження або лікування патологічного стану або симптому у ссавця, де наявна активність рецепторів аденозину А2В та є потрібним антагонізм їх дії, який полягає у застосуванні до ссавця терапевтично ефективної кількості сполуки заявленого винаходу. Згідно з ще одним аспектом винаходу, запропоновано спосіб лікування хвороби, який полягає у застосуванні терапевтично ефективної кількості принаймні одної сполуки заявленого винаходу або її стереоізомеру або фармацевтично прийнятної солі, де хворобу вибрано з астми, алергії, алергічних хвороб як-то, алергічний риніт та синусит), автоімунної хвороби (наприклад, вовчак), діарейних хвороб, резистентості до інсуліну, діабету (наприклад, типу І та типу ІІ), попередження дегрануляції мастоцитів, асоційованої з ішемічними/реперфузійними пошкодженнями, серцевого нападу, інгібування ангіогенезу у неопластичних тканинах, та інгібування ангіогенезу при діабетичній ретинопатії або гіпербаричній індукованій киснем ретинопатії. Згідно з ще одним аспектом винаходу, запропоновано сполуку заявленого винаходу для застосування у терапії. 6 UA 105164 C2 5 10 15 20 25 30 35 40 45 50 Згідно з ще одним аспектом, запропоновано застосування сполуки винаходу для виробництва медикаменту, корисного для лікування хвороби у ссавця. Слід розуміти, що будь-який аспект або особливість заявленого винаходу у всякому разі, що характеризуються як кращі або не характеризуються як кращі, можуть бути поєднаним з будьяким іншим аспектом або особливістю винаходу, у всякому разі така інша особливість характеризується як краща або не характеризується як краща. Як ясно спеціалісту, імідазольне кільце сполук заявленого винаходу може існувати у таутомерних формах або як таутомери, та тому це також охоплено рамками винаходу. Таутомерні ізомери є представленими як структури (lа) та (lb): 1 2 де R, R , R , Х, та Z визначені тут. Визначення одної сполуки, як слід розуміти, означає також її відповідний таутомер. Спеціалістам ясно, що сполуки винаходу, що мають хіральний центр, можуть існувати та бути виділеними в оптично активних та рацемічних формах. Деякі сполуки можуть виявляти поліморфізм. Слід розуміти, що заявлений винахід стосується будь-яких рацемічних, оптичноактивних, поліморфних або стереоізомерних форм, або їх суміші, стосовно сполук винаходу, котрі виявляють корисні властивості, описані тут; добре відомо у рівні техніки, як отримати оптично активні форми (наприклад, розділенням рацемічної форми способами перекристалізації, синтезом з оптично-активних вихідних матеріалів, хіральним синтезом, або хроматографічним розділенням, застосовуючи хіральну стаціонарну фаз) та як визначити терапевтичну активність, застосовуючи стандартні описані тут тести або застосовуючи інші подібні тести, котрі є добре відомими у рівні техніки. Ссавці та пацієнти охоплюють тепло-кровних ссавців, що звичайно перебувають під медичним наглядом (наприклад, людей та домашніх тварин). Приклади ссавців охоплюють (а) кішок, собак, коней, корів, та людини та (b) людину. "Лікування" охоплює лікування стану хвороби у ссавця, а зокрема: (а) попередження стану хвороби ссавця, зокрема, коли так ссавець є схильним до стану хвороби, але не має ще діагнозу; (b) інгібування стану хвороби, наприклад, затримка його розвинення; (с) полегшення стану хвороби, наприклад, регрес стану хвороби до досягнення потрібного результату; та/або (d) усунення стану хвороби, наприклад, припинення стану хвороби та/або її дії. Лікування також охоплює полегшення симптому хвороби (наприклад, зменшення болю або нездужання), де таке полегшення може або ні безпосередньо діяти на хворобу (наприклад, причину, хід, виразність, тощо). "Фармацевтично прийнятні солі" стосуються похідних розкритих сполук, де вихідну сполуку модифікують до солей її кислоти або основи. Приклади фармацевтично прийнятних солей охоплюють, але без обмеження, солі основних залишків мінеральних або органічних кислот, якто аміни; лужні або органічні солі кислотних залишків, як-то карбонових кислот; тощо. Фармацевтично прийнятні солі охоплюють звичайні нетоксичні солі або солі четвертинного амонію вихідної сполуки, утворені, наприклад, з нетоксичних неорганічних або органічних кислот. Наприклад, такі звичайні нетоксичні солі охоплюють, але без обмеження, похідні від неорганічних та органічних кислот, вибраних з групи: 1,2-етандисульфонова, 2ацетоксибензойна, 2-гідроксіетансульфонова, оцтова, аскорбінова, бензенсульфонова, бензойна, карбонатна, лимонна, едетова, етандисульфонова, етансульфонова, фумарова, глюкогептонова, глюконова, глутамінова, гліколева, гліколліарсанілова, гексилрезорцинова, гідрабамінова, бромідна; хлоридна, йодидна, гідроксималеїнова, гідроксинафтойна, ізетіонова, молочна, лактобіонова, лаурилсульфонова, малеїнова, яблучна, мигдальна, метансульфонова, напсилова, нітратна, щавлева, памова, пантотенова, фенілоцтова, фосфатна, полігалактуронова, пропіонова, саліцилова, стеаринова, субоцтова, бурштинова, сульфамінова, сульфанілова, сульфатна, дубильна, винна, та толуєнсульфонова. Фармацевтично прийнятні солі заявленого винаходу можна синтезувати з вихідної сполуки, що містить основну або кислотну частину звичайними хімічними способами. Загалом, так солі можна отримувати реакцією вільної кислоти або основи цих сполук зі стехіометричною кількістю прийнятної основи або кислоти у воді або органічному розчиннику, або у їх суміші; загалом, неводні середовища, як-то етер, етилацетат, етанол, ізопропанол, або ацетонітрил є кращими. 7 UA 105164 C2 5 10 15 20 25 Перелік придатних солей є виявленим у Rеmіngtоn's Рhаrmасеutісаl Sсіеnсеs, 18th еd., Масk Рublіshіng Соmраnу, Еаstоn, РА, 1990, р 1445, розкриття якої уведене як посилання. "Терапевтично ефективна кількість" охоплює кількість сполуки заявленого винаходу, що є ефективною при застосуванні поодинці або у комбінації для лікування перерахованих тут показань. "Терапевтично ефективна кількість" також охоплює кількість комбінації заявленої сполуки, що є ефективною для лікування потрібного показання. Комбінація сполук є переважно синергічною комбінацією. Синергія, що описано, наприклад, Сhоu та Таlаlау, Аdv; Еnzуmе Rеgul. 1984, 22:27-55, відбувається, коли дія сполуки при застосуванні у комбінації є більше адитивної дії сполуки при застосуванні як одиничного агенту. Загалом, синергічна дія є краще показана при субоптимальній концентрації сполуки. Синергія може бути виражена, як нижча цитотоксичність, збільшена дія або деяка інша корисна дія комбінації порівняно з індивідуальними компонентами. Конкретні та кращі значення, перераховані для радикалів, замісників та меж, є тільки для ілюстрації; вони не обмежують інших визначених значень або інших значень у визначених межах для радикалів та замісників. Синтез Сполуки заявленого винаходу можна отримувати способами, описаними у US2005/0065341, вміст якого уведено тут як посилання. Сполуки заявленого винаходу можна також отримувати способами, описаними у Р. J. Sсаmmеlls, еt аl, J. Меd. Сhеm, 37, 2704-2712 (1994). Діаміно-1,3-дизаміщений урацил ацилують 6-хлорнікотиноїлхлоридом у піридині при 5 °C, отримуючи сполуки формули (5а). Утворений амід (5а) циклізують при кипінні під зворотним холодильником у водному розчині натрій гідроксиду, отримуючи сполуку А. 6-Хлорнікотиноїлхлорид отримують при кипінні під зворотним холодильником 6-гідроксинікотинової кислоти у тіонілхлориді, застосовуючи ДМФ як каталізатор як показано у схемі реакцій 1. Сполуку А можна алкілувати алкілбромідом або йодидом, 1 1 отримуючи сполуки формули А . Сполуки А або А реагують із заміщеним аміном при 1501 1 4 160 °C у тубі під тиском з утворенням сполуки формули В або В . Сполуки формули В , де R – 4 6 гідроген, можуть реагувати з ацилхлоридом, даючи сполуки, де R – -С(О)R (С). схема реакцій 1, руrіdіnе – піридин, DМF – ДМФ, оr – або, whеrе – де, hуdrоgеn – гідроген 30 8 UA 105164 C2 Скорочення: 125 І]АВА І-АВОРХ АR СGS СРХ DМЕМ ДМФ ДМСО ЕДТА НЕК-клітини Кі NЕСА R-VІА ТЕА ТШХ. [ 125 ZМ 241385 5 10 15 20 25 30 35 40 125 6 І]N -4-амінобензил)-аденозин І-3-(4-аміно-3-йодбензил)-8-оксиацетат-1-пропіл-ксантин рецептор аденозину 216802-[4-[(2-карбоксіетил)феніл]етил-аміно]-5N7іV'-етилкарбамоїл аденозин 8-циклопентил-1,3-дипропілксантин модифіковане Дульбекко середовище Ігла N, N-диметилформамід диметилсульфоксид етилендіамінтетраацетат клітини нирок ембріона людини константа рівноваги інгібування 5'-(N-етилкарбамоїл)аденозин 6 R-N - -фенілізопропіладенозин триетиламін тонко-шарова хроматографія 4-(2-[7-аміно-2-{фурил}{1,2,4}триазоло{2,3-а}{l, 3,5}триазин-5іламіноетил)фенол [ 125 Сполуки заявленого винаходу можна формувати як фармацевтичні композиції та застосовувати до ссавця, як-то людини, у різних формах, адаптованих до вибраного шляху застосування, наприклад, перорально або парентерально, внутрішньовенно, внутрішньом'язово, місцево, інгаляційно або підшкірно. Приклади фармацевтичних композицій є у "Rеmіngtоn: Тhе Sсіеnсе та Рrасtісе оf Рhаrmасу", А. Gеnnаrо, еd., 20th еdіtіоn, Lірріnсоtt, Wіllіаms & Wіlkіhs, Рhіlаdеlрhіа, РА. Тому заявлені сполуки можна застосовувати системно, наприклад, перорально, у комбінації із фармацевтично прийнятним наповнювачем, як-то інертний розріджувач або засвоюваний їстівний носій. Вони можуть бути інкапсульованими у твердих чи м'яких желатинових капсулах, можуть бути спресованими у таблетки або можуть бути уведеними безпосередньо з їжею. Для перорального терапевтичного застосування, активна сполука може бути поєднаною з одним або більше наповнювачами та застосовуваною у формі таблеток для ковтання, букальних таблеток, пастилок, капсул; еліксирів, суспензій, сиропів, облаток, тощо. Такі композицій та препаратів містять принаймні 0,1 % активної сполуки. Вміст композицій та препаратів може, безумовно, варіювати і бути приблизно між 2 – 60 мас. % даної форми одиничної дози. Кількість активної сполуки у такій терапевтично корисній композиції є такою, щоб отримати ефективну дозу. Таблетки, пастилки, пілюлі, капсули, тощо можуть також містити нижченаведене: зв'язуючі, як-то камедь трагаканту, гуміарабік, кукурудзяний крохмаль або желатин; наповнювачі, як-то дикальцій фосфат; дезинтегратор, як-то кукурудзяний крохмаль, картопляний крохмаль, альгінова кислота тощо; лубрикант, як-то магній стеарат; та. підсолоджувач, як-то сахароза, фруктоза, лактоза або аспартам або ароматизатор, як-то м'ята, олія гаультерії або вишні. Коли формою одиничної дози є капсула, вона може містити, на додаток до вищенаведених матеріалів, рідкий носій, як-то рослинна олія або поліетиленгліколь. Різні інші матеріали можуть бути для покриття, або іншої модифікації фізичної форми твердої одиничної дози. Наприклад, таблетки, пілюлі або капсули можуть бути покритими желатином, воском чи цукром тощо. Сироп або еліксир можуть містити активну сполуку, сахарозу або фруктозу як підсолоджувач, метилта пропілпарабени як консерванти, барвник та ароматизатор, як-то вишневий або апельсиновий. Безумовно, будь-який матеріал, застосовуваний у отриманні будь-якої форми одиничної дози повинен бути фармацевтично прийнятним та по суті нетоксичним у застосовуваній кількості. На додаток, активна сполука може бути уведеною у препарат з безперервним вивільненням та пристрої. Активну сполук можна також застосовувати внутрішньовенно або інтраперитонеально вливанням або ін'єкціями. Розчини активної сполуки або її солі можна отримувати у воді, необов'язково змішаною із нетоксичною ПАР Дисперсії можна також отримувати у гліцерині, рідкому поліетиленгліколі, триацетині та їх суміші та у олії. У звичайних умовах зберігання та застосування, ці препарати містять консервант для попередження росту мікроорганізмів. Фармацевтичні придатні для ін'єкції чи вливання форми дозування охоплюють стерильні водні розчини або дисперсії, або стерильні порошки, що містять активний інгредієнт, котрі є адаптованими для негайного отримання стерильних розчинів або дисперсій для вливання або 9 UA 105164 C2 5 10 15 20 25 30 35 40 45 50 55 60 ін'єкцій, необов'язково інкапсульованих у ліпосомах. В усіх випадках форма дозування повинна бути стерильною, текучою та стабільною в умовах виробництва та зберігання. Рідким носієм або середовищем може бути розчинник або рідке дисперсійне середовище, що містить, наприклад, воду, етанол, поліол (наприклад, гліцерин, пропіленгліколь, рідкий поліетиленгліколі, тощо), рослинні олії, нетоксичні гліцерил-естери та їх придатні суміші. Текучість препарату можна підтримувати, наприклад, утворенням ліпосом, підтримкою потрібного розміру частинок у випадку дисперсій або застосуванням ПАР. Попередження дії мікроорганізмів може бути за допомогою різних антибактеріальних та антигрибкових засобів, якто, парабени, хлорбутанол, фенол, сорбінова кислота, тимеросал, тощо. У багатьох випадках це містить ізотонічні засоби, наприклад, цукри, буфери або натрій хлорид. Подовженого поглинання композицій для ін'єкцій можна досягти застосуванням у композиціях засобів затримуваного поглинання, як-то, алюміній моностеарат та желатин. Стерильні розчини для ін'єкцій отримують уведенням активної сполуки у потрібній кількості у прийнятний розчинник з різними іншими інгредієнтам, переліченими вище, якщо потрібно, а потім фільтр-стерилізацією. У випадку стерильних порошків для отримання стерильних розчинів для ін'єкцій, кращими способами отримання є вакуумна сушка та сушка сублімацією, котрі дають порошок активного інгредієнта з будь-яким додатковим потрібним інгредієнтом у раніше стерильно-фільтрованих розчинах. Для місцевого застосування заявлені сполуки можуть бути у чистій формі, тобто коли вони є рідинами. Однак, загалом бажано застосовувати їх на шкіру як композиції, у комбінації із дерматологічно прийнятним носієм, котрий може бути твердим або рідким. Корисні тверді носії охоплюють мілко подрібнені тверді матеріали, як-то тальк, глина, мікрокристалічна целюлоза, силіцій діоксид, алюміній оксид тощо. Корисні рідкі носії охоплюють воду, спирти або гліколі або водно-спиртові/гліколеві суміші, у котрих заявлені сполуки можна розчиняти або диспергувати на ефективному рівні, необов'язково за допомогою нетоксичної ПАР. Ад'юванти, як-то ароматизатори та додаткові антимікробні агенти можна додавати для оптимізації препарату для застосування. Утворені рідкі композиції можна наносити тампонами, застосовувати як просочені бандажі та інші матеріали для перев'язування або розпилювати на вражену зону, застосовуючи розпилювачі. Загусники, як-то синтетичні полімери, жирні кислоти, солі та естери жирних кислот, жирні спирти, модифікована целюлоза або модифіковані мінеральні матеріали можна також застосовувати з рідкими носіями з утворенням паст, гелів, мазі, мила, тощо, для нанесення безпосередньо на шкіру. Приклади корисних дерматологічних композиції, котрі можна застосовувати для нанесення сполуки заявленого винаходу на шкіру, є відомими; наприклад, дивись Jасquеt еt. аl. (патент США № 4,608,392), Gеrіа (патент США № 4,992,478), Smіth еt аl. (патент США № 4,559,157) та Wоrtzmаn (патент США № 4,820,508). Корисні дози сполук заявленого винаходу можна визначати порівнянням їх активності іn vіtrо та активності іn vіvо у тваринних моделях. Способи екстраполяції ефективного дозування у мишей та інших тварин до людей є відомими; наприклад, дивись патент США № 4,938,949. Загалом, концентрація сполук заявленого винаходу у рідких композиціях, як-то лосьйон, повинна бути (а) приблизно 0,1-25 мас. % та (b) приблизно 0,5-10 мас. %. Концентрація у напівтвердій або твердій композиції, як-то гель або порошок, повинна бути (а) приблизно 0,1-5 мас. % та (b) приблизно 0,5-2,5 мас. %. Кількість сполуки або її активної солі або похідного, потрібна для застосування у лікуванні залежить не тільки ввід конкретної вибраною сполуки або солі, але також шляху застосування, природи лікованого стану, віку та стану пацієнта, та повинна бути вибраною на розсуд лікаря або клініциста. Загалом, однак, придатна доза повинна бути у межах (а) приблизно 1,0-100 мг/кг маси тіла на добу, (b) приблизно 10-75 мг/кг маси тіла на добу, та (с) приблизно 5-20 мг/кг маси тіла на добу. Сполуку можна звичайно застосовувати у формі одиничної дози; наприклад, таблетки, що містить (а) приблизно 4-400 мг, (b) приблизно 10-200 мг, та (с) приблизно 20-100 мг активного інгредієнта на форму одиничної дози. В ідеалі, активний інгредієнт слід застосовувати до досягнення пікової концентрації у плазмі активної сполуки (а) приблизно 0,02-20 рМ, (b) приблизно 0,1-10 рМ, та (с) приблизно 0,5-5 мкМ. Цих концентрацій можна досягти, наприклад, внутрішньовенною ін'єкцією 0,005-0,5 % розчину активного інгредієнту, або перорально застосовувати як болюс, що містить приблизно 4-400 мг активного інгредієнта. Сполуки винаходу можна також застосовувати інгаляціями з інгалятора, пристрою для вдування, розпилювачу або упаковки під тиском або іншими засобами розподілу аерозолю спрею. Упаковки під тиском можуть містити придатний пропелент, як-то карбон діоксид або інший придатний газ. У випадку аерозолю під тиском, одиничну дозу можна 10 UA 105164 C2 5 10 15 20 25 30 35 40 45 50 55 60 визначати підбором умов для випуску виміряної кількості. Інгалятори, пристрої для вдування, розпилювачі описані у фармацевтичних довідниках, як-то Rеmіngtоn's Рhаrmасеutісаl Sсіеnсеs Vоlumеs 16 (1980) або 18 (1990) Масk Рublіshіng Со. Потрібна доза може звичайно бути одиничною дозою або як поділеними дозами, застосовуваними з прийнятними інтервалами, наприклад, два, три, чотири або більше піддоз на добу. Піддоза сама може бути поділеною, наприклад, на ряд дискретних неточних застосувань; як-то багатократними інгаляціями з пристрою для вдування або застосуванням крапель в очі. Усі патенти, патентні заявки та Джерела інформації:, наведені в описі є уведеними як посилання. У випадку будь-якого протиріччя представлене розкриття, у тому числі будь-яке визначення тут, повинне превалювати. Винахід описано різними конкретними та кращими втіленнями та способами. Однак, слід розуміти, що варіації та модифікації можна отримати без виходу за рамки винаходу. Приклади Фармакологія Здатність сполук винаходу діяти як антагоністи А2В-рецептору аденозину можна визначати, застосовуючи фармакологічні моделі, котрі є добре відомими, або способами тестування, описаними нижче. кДНК А2В-рецептору щурів субклонували у плазміду експресії рDоublеТrоublе способами, описаними у Rоbеvа, А. еt аl, Віосhеm. Рhаrmасоl., 51. 545-555 (1996). Плазміду ампліфікували у компетентних клітинах JМ109 та плазмідну ДНК виділяли, застосовуючи колонки Wіzаrd Меgарrер (Рrоmеgа Соrроrаtіоn, Маdіsоn, WІ). А2В-рецептори аденозину уводили у клітини НЕК-293 за допомогою ліпофектину як описано Fеіgnеr, Р. L. еt аl, Рrос. Nаtl. Асаd. Sсі. USА. 84, 7413-7417 (1987). Культура клітин Трансфектовані НЕК-клітини вирощували у зволоженій атмосфері 5 % СО2/95 % О2 при температурі 37 °C. Колонії були вибраними за ростом клітин у 0,6 мг/мл G418. Трансфектовані клітини тримали у DМЕМ, доповненому живильною сумішшю Наms F12 (1/1), 10 % сироватки новонародженого теляти, 2 мМ глутаміну та 50 ІU/мл пеніциліну, 50 мг/мл стрептоміцину, та 0,2 мг/мл генетицину (G418, Воеhrіngеr Маnnhеіm). Клітини культивували у круглих планшетах діаметром 10 см та субкультивували при зростанні конфлюентності (приблизно через 72 години). Дослідження зв'язування радіоліганду Стосовно А2В-рецепторів: Конфлюєнтні моношари клітин НЕКL-А2В далі промивали РВS, а потім охолодженим льодом буфером А (10 мМ ГЕПЕС, 10 мМ ЕДТА, рН 7,4) з інгібіторами протеази (10 мкг/мл бензамідину, 100 мкМ фенілметансульфонілфлуориду, та 2 мкг/мл, кожного, апротиніну, пепстатину та лейпептину). Клітини гомогенізували у Роlуtrоn (Вrіhkmаnn) протягом 20 с, центрифугували при 30000 х g, та пелети промивали двічі буфером НЕ (10 мМ ГЕПЕС, 1 мМ ЕДТА, рН 7,4 з інгібіторами протеази). Кінцеву пелету ресуспендували у буфері НЕ, доповненому 10 % сахарози та заморожували в аліквотах при -80 °C Для аналізів зв'язування мембрани розморожували та розбавляли 5-10-кратно НЕ до кінцевої концентрації білку приблизно 1 мг/мл. Для визначення концентрації білку мембрани та альбумін сироватки корови розчиняли у 0,2 % NаОН/0,01 % SDS та білок визначали, застосовуючи флуоресценцію флуорескаміну. Stоwеll, С. Р. еt аl., Аnаl. Віосhеm. 85, 572-580 (1978). Аналізи насичення зв'язування для А2В-рецепторів аденозину щура проводили із 3 [ Н]ZМ214,385 (17 Кі/ммоль, Тосrіs Сооksоn, Вrіstоl UК) (Jі, Х. еt аl, Drug Dеsіgn Ріsсоv. 16, 216125 12S 226 (1999)) або І-АВОРХ (2200 Кі/ммоль). Для отримання І-АВОРХ, 10 мкл 1 мМ АВОРХ у метанолі/1 М NаОН (20:1) додавали до 50 мл 100 мМ фосфатного буферу, рН 7,3. Один або 2 125 мКі Nа І додавали, а потім 10 мкл 1 мг/мл хлораміну-Т у вод. Після інкубування 20 хвилин при кімнатній температурі 50 мкл 10 мг/мл Nа-метабісульфіту у воді додавали для гасіння реакції. Реакційну суміш переносили на ВЕРХ-колонку С18, елюючи сумішшю метанолу та 5 мМ фосфату, рН 6,0. Через 5 хвил при 35 % метанолу концентрацію метанолу доводили до 100 % 125 протягом 15 хвил. Непрореагований АВОРХ елювали протягом 11-12 хвилин; І-АВОРХ 125 елювали при 18-19 хвил, отримуючи 50-60 % відносно початкового І. 127 125 В аналізах урівноваженого зв'язування співвідношення І/ І-АВОРХ було 10-20/1. Експерименти зв'язування радіоліганду проводили у потроєнні з 20-25 мкг білку мембран у загальному об'ємі 0,1 мл буферу НЕ, доповненого 1 U/мл аденозиндеамінази та 5 мМ МgСl2. Час інкубування був 3 годин при 21 °C. Неспецифічне зв'язування вимірювали у присутності 100 125 мкМ NЕСА. Конкурентні експерименти проводили з 0,6 нМ І-АВОРХ. Мембрани фільтрували на фільтрах Whаtmаn GF/С збирачем клітин Вrаndеll (Gаіthеrsburg, МD) та промивали 3 рази 11 UA 105164 C2 5 10 15 20 25 30 35 40 45 50 55 протягом 15-20 с охолодженим льодом буфером (10 мМ Трис, 1 мМ МgСl2, рН 7,4). Значення Вмакс та Кd розраховували інтерполяцією методом найменших квадратів для моделі одиничної ділянки зв'язування. Маrquаrdt, D. М, J. Sос. Іndust. АddІ. Маth., Н, 431-441,21 (1963). Значення Кі для відмінних сполук були похідними від значень ІК50, як описано Lіndеn, J., J. Сусl. Nuсl. Rеs., г, 163-172 (1982). Дані повторних експериментів виражали як значення ± СВ. 3 Стосовно інших рецепторів аденозину: [ Н]СРХ. Вrаns, R. F. еt аl„ Nаunvn-Sсhmіеdеbеrе's 125 125 Аrсh. Рhаrmасоl. 335. 59-63 (1987). І-ZМ241385 та І-АВА застосовували в аналізах зв'язування радіолігандів з мембранами, похідними від клітин НЕК-293, що експресують 3 6 рекомбінантні АR щура А1, А2А та А3, відповідно. Зв'язування [Н]R-N -фенілізопропіладенозину 3 Sсhwаbе, U. еt аl, Nаunvn-Sсhmіеdеbеrg's Аrсh. Рhаrmасоl. 313, 179-187 (1980). ([ Н]А-РІА, Аmеrshаm, Сhісаgо, ІL) з А1-рецепторами з кортикальними церебральними мембранами щурів 3 та [ Н]СGS 21680. Jаrvіs, М.F. еt аl, J. Рhаrmасоl. Ехn. Тhеrар. 251. 888-893 (1989). (Duроnt NЕN, Воstоn, МА) з А2А-рецепторами від стріарних мембран щурів проводили як описано. Аденозиндеаміназу (3 од/мл) було представлено при отриманні мембран мозку при попередньому інкубуванні 30 хвил при 30 °C, та при інкубуванні з радіолігандами. Усі нерадіоактивні сполуки спочатку розчиняли у ДМСО, та розбавляли буфером до кінцевої концентрації, де кількість ДМСО не перевищувала 2 %. Інкубування припиняли швидким фільтруванням через фільтри Whаtmаn GF/В, збирачем клітин Вrаndеll (Вrаndеll, Gаіthеrsburg, МD). Туби промивали три рази 3 мл буферу, кожну. Принаймні 6 відмінних концентрацій конкуренту, що перекривають на 3 порядки величину, доведену прийнятно для ІК50 кожної сполуки були застосовуваними. Значення ІК50, розраховували способом нелінійної регресії (Grарh-Раd Рrіsm, Sаn Dіеgо, СА)5 перетворювали у справжні значення Кі як описано. Lіndеn, J., J. Сусl. Nuсl. Rеs. 8:163-172 (1982). Коефіцієнти Хілла тестованих сполук були у межах 0,8 – 1,1. Функціональний аналіз. НЕК-А2В-клітини з одної конфлюєнтної колби Т75 промивали буферованим фосфатом 2+ 2+ 2+ фізіологічним розчином Дульбекко (РВS) без Са та Мg та тоді інкубували у НВSS без Са та 2+ Мg з 0,05 % трипсину та 0,53 мМ ЕДТА до відокремлення клітин. Клітини промивали двічі центрифугуванням при 250 х g у РВS та ресуспендували у 10 мл НВSS з 137 мМ NаСl, 5 мМ КСl, 0,9 мМ МgSО4, 1,4 мМ СаСl2, 3 мМ NаНСО3, 0,6 мМ Nа2НРО4,0,4 мМ КН3РО4,5,6 мМ 2+ глюкози, та 10 мМ ГЕПЕС, рН 7,4 та Са -чутливим флуоресцентним індикатором індо-1-АМ (5 мкМ) 37 °C протягом 60 хвил. Клітини промивали та ресуспендували у 25 мл НВSS без індикатору без доповнення 1 од/мл аденозин-деаміназою та тримали при кімнатній температурі. Антагоністи рецептору аденозину. отримані як 100Х вихідні розчини у ДМСО або середовищі, додавали та клітини переносили у баню 37 °C на 2 хвилини. Тоді клітини (1 мільйон у 2 мл) переносили при перемішуванні у кювету і тримали при 37 °C у Аmіnсо SLМ 8000 спектрофлуорометрі (SМL іnstrumеnts, Urbаnа ІL). Співвідношення флуоресценції індо-1, отримані при 400 та 485 нм (збудження, 332 нм) реєстрували, застосовуючи ширину щілини 4 нм. NЕСА додавали через 100 с урівноваження. Акумуляція циклічного АМФ. Створення циклічного АМФ проводили у буфері DМЕМ/ГЕПЕС (DМЕМ, що містить 50 мМ ГЕПЕС, рН 7,4, 37 °C). Кожну лунку клітин далі промивали двічі буфером DМЕМ/ГЕПЕС, та тоді 100 мкл аденозин-деамінази (кінцева концентрація 10 ІU/мл) та додавали 100 мкл розчинів роліпраму та цилостаміду (кожний при кінцевій концентрації 10 мкМ), а потім 50 мкл тестсполуки (прийнятна концентрація) або буферу. Через 15 хвилин інкубування при 37 °C зупиняли видаленням середовища та додаванням 200 мкл 0,1 М НСl. Кислотні екстракти зберігали при 20 °C до аналізу. Кількості циклічного АМФ були визначені за протоколом, котрий застосовував білок зв'язування цАМФ (РКА) [vаn dеr Wеndеn еt аl, 1995], з невеликими модифікаціями. Аналізаційний буфер містив 150 мМ К2НРО4/10 мМ ЕДТА/0,2 % альбуміну сироватки корів FV при рН. 7,5. Зразки (20 мл) інкубували протягом 90 хвилин при 0 °C. Інкубати фільтрували через скляні фільтри GF/С у збирачі клітин Вrаndеll М-24: Фільтри додатково промивали 4 рази 2 мл 150 мМ К2НРО4/10 мМ ЕДТА (рН 7,5, 4 °C). Перфоровані фільтри підраховували у сцинтиляційній рідині Расkаrd Еmulsіfіеr Sаfе через 2 години екстракції. [0,071] Зразкові сполуки заявленого винаходу показані як активні у вищенаведеному тестуванні афінності. Синтез та охарактеризування nеаt – чистий, оr wіth – або з, whеn – коли 12 UA 105164 C2 5 10 15 20 25 30 Протонну магнітну резонансну спектроскопію проводили на спектрометрі Vаrіаn-300 М Гц та –1 спектри отримували у ДМСО-d6. Якщо не визначено інше, хімічні зсуви виражено як млн униз –1 –1 від відносного значення млн від ДМСО (2,5 млн ). Мас-спектрометрію з іонізацією електророзпиленням (ІРЕ) проводили мас-спектрометром ТhеrrаоFіnnіgаn LСQ. Усі ксантинові похідні були гомогенними, як визначали, застосовуючи ТШХ (силікагель 60 F254, 0,25 мм, алюмінієва підкладка, ЕМ Sсіеnсе, Gіbbstоwn, NJ) та ВЕРХ (Shіmаdzu), застосовуючи Vаrіаn СІS 5 мікрон аналітичну колонку (4,6 мм х 150 мм) у системі розчинників з лінійним градієнтом, при швидкості потоку 1 мл/хвил. Системою розчинників була МеОН (0,1 % мурашина кислота):Н2О (0,1 % мурашина кислота). Піки визначали за допомогою УФпоглинання при 300 нм та 254 нм. ЯМР та мас-спектри сумісні з показаною структурою. Загальні способи отримання заміщених хлором піридилових сполук А. 6-Хлорнікотиноїлхлорид, отриманий з 6-гідроксинікотинової кислоти (1,444g, 10,4 ммоль), у дихлорметані (20 мл) додавали краплями до розчину 5,6-діаміно-1,3-(дизаміщеного урацилу) (8 ммоль) у сухому піридині (8,2 мл) при 5 °C. Реакційну суміш гріли до кімнатної температури та перемішували протягом ще 3 годин. Воду (50 мл.) додавали для гасіння реакції. Розчинник випарювали, отримуючи темну оливу: Оливу гріли при кипінні під зворотним холодильником протягом 2 годин у 2N NаОН (20 мл). Після охолодження рН обережно доводили до 7 з концентрованою НСl. Твердий продукт збирали та промивали водою (20 мл), етером (20 мл) та хлороформом (20 мл), отримуючи білуватий твердий продукт. Продукт було застосовано на наступному етапі без очистки. 3А 1А: 1,3-Дипропіл-8-(6-хлор-3-піридил)ксантин 'Н ЯМР (ДМСО, d6): δ 0,89(m, 6Н), 1,59(m, 2Н), 1,73(m, 2Н), 3,88(t, 2Н, J=7,2 Гц), 4,00(t, 2Н, J=7,2 Гц), 7,68(d, 1Н, J=8,4 Гц), 8,50(dd, 1Н, J1=2,4 Гц, J2=8,4 Гц), 9,07(d, 1Н, J=2,4 Гц), МС: m/z + 348 (М+Н) . 2А: 1-циклопропіл-3-пропіл-8-(6-хлор-3-піридил)ксантин: 'Н ЯМР (ДМСО, d6): δ 0,72(m, 2Н), 0,910, 3Н, J=7,8 Гц), 1,03(m, 2Н), 1,72(m, 2Н), 2,63(m, 1Н), 3,98(t, 2Н, J=7,8 Гц), 7,68(d, 1Н, J=8,4 Гц), 8,46(dd, 1Н, J1=2,4 Гц, J2=8,4 Гц), 9,07(d, 1Н, J-2,4 Гц). 13 UA 105164 C2 + 5 10 МС: m/z 346 (М+Н) . 3А: 1,3-діаліл-8-(6-хлор-3-піридил)ксантин: 'Н ЯМР (ДМСО, d6): 4,56(d, 2Н, J=5,1 Гц), 4,70(d, 2Н, J=5,1 Гц), 5,15 (m, 4Н), 5,98(m, 2Н), 7,74(d, 1Н, J=8,4 Гц), 8,50(dd, 1Н, ы=2,4 Гц, J2=8,4 Гц), 9,12(d, 1Н, J=2,4 Гц). + МС: m/z 344 (М+Н) . Загальні способи отримання заміщених амінами піридилових сполук В. Сполуку А (40 мг) та відповідний заміщений амін (0,5 мл або 0,5 г) поміщали у тубу під тиском. (Етанол, 4 мл, додавали як розчинник, якщо амін є твердим.) Герметичну тубу продували аргоном, закривали та перемішували при 160 °C протягом 48-60 годин. Після охолодження додавали етер (10 мл). Утворений твердий матеріал збирали та очищали на колонці з силікагелем або за допомогою ТШХ (Розчинник А: дихлорметан/ МеОН =20: 1 до 10:1 або Розчинник В: дихлорметан/ МеОН: ТЕА=20: 1: 0,1 до 4: 1: 0,1). 14 UA 105164 C2 5 10 15 1В: 1-циклопропіл-3-пропіл-8-(6-метиламіно-3-піридил]ксантин: 'Н ЯМР (ДМСО, d6): 0,72(m, 2Н), 0,91(t; 3Н, J=7,5 Гц), 1,03(m, 2Н), 1,71(m, 2Н), 2,62 (m, 1Н), 2,81(d, 3Н, J=4,5 Гц), 3,96(t, 2Н, J=7,5 Гц), 6,52(d, 1Н, J=9,0 Гц), 7,07(d, 1Н, J=4,5 Гц), 8,01(dd, 1Н, J1=2,4 Гц, J2=9,0 Гц), 8,73(d, 1Н, J=2,4 Гц). + МС: m/z 341 (М+Н) . 2В: 1,3-дипропіл-8-[6-(2-метоксіетил)аміно-3-піридил)ксантин: 'Н ЯМР (ДМСО, d6): δ 0,93(m, 6Н), 1,63(m, 2Н), 1,78(m, 2Н), 3,38(s, 3Н), 3,53(s, 4Н), 3,91(t, 2Н, J=*7,5 Гц), 4,05(t, 2Н, J=7,5 Гц), 6,65(d, 1Н, J=8,7 Гц), 7,24(s(br), 1Н), 8,06(dd, 1Н, J 1=2,4 Гц, J2=8,7 Гц), 8,71(d, 1Н, J=2,4 Гц). МС: + m/z 387 (М+Н) . 3В: 1-циклопропіл-3-пропіл-8-[6-(2-метоксіетил)аміно-3-піридил]ксантин: 1 Н ЯМР (ДМСО, d6): 0,74(m, 2Н), 0,94(t, 3Н, J=7,5 Гц), 1,06(m, 2Н), 1,75(m, 2Н), 2,65(111, 1Н), 3,32(s, 3Н), 3,52(s, 4Н), 4,00(t, 2Н, J=7,5 Гц), 6,64(d, 1Н, J=8,7 Гц), 7,23(s(br), 1Н), 8,04(dd, 1Н, + J1=2,4 Гц, J2=8,7 Гц), 8,76(d, 1Н, J=2,4 Гц). МС: m/z 385 (М+Н) . 4В: 1,3-діаліл-8-[6-(2-метоксіетил)аміно-3-піридил)ксантин: 1 Н ЯМР (ДМСО, d6): 3,32(s, 3Н), 3,52(s, 4Н), 4,55(d, 2Н, JМ5,1 Гц), 4,68(d, 2Н, J=5,1 Гц), 5,15 (m, 4Н), 5,95(m, 2Н), 6,64(d, 1Н, J=9,0 Гц), 7,25(s(br), 1Н), 8,05(dd, 1Н, J 1=2,4 Гц, J2=9:0 Гц)s 8,77(d, 1Н, J=2,4 Гц). + МС: m/z383(М+Н) . 15 UA 105164 C2 5 10 15 20 25 30 35 40 45 50 55 60 5В: 1-циклопропіл-3-пропіл-8-[6-(2-морфоліноетил)аміно-3-піридил]ксантин: 1 Н ЯМР (ДМСО, d6): 0,74(m, 2Н), 0,94(t, 3Н, J=7,5 Гц), 1,06(m, 2Н), 1,75(m, 2Н), 2,46(t, 4Н, J=4,5 Гц), 2,52(m, 2Н), 2,65(m, 1Н), 3,46(m, 2Н), 3,63(t, 4Н. J=4,5 Гц), 4,00(t, 2Н, J=7,2 Гц), 6,62(d, 1Н, J=8,7 Гц), 7,23(t, 1Н, J=5,4 Гц), 8,04(dd, 1Н, Jі-2,4 Гц, J2=8,7 Гц), 8,75(d, 1Н, J=2,4 Гц). МС: + m/z 440 (М+Н) . 1 6В: 1-циклопропіл-3-пропіл-8-[6-(2-(піперидин-1-іл)етиламіно)-3-піридил]ксантин: Н ЯМР (ДМСО, d6): 0,74(m, 2Н), 0,94(t, 3Н, J=7,5 Гц), 1,07(m, 2Н), 1,44(m, 2Н), 1,57(m, 4Н), 1,75(m, 2Н), 2,51(m, 6Н), 2,65(m, 1Н), 3,48(m, 2Н), 4,00(t, 2Н, J=7,2 Гц), 6,63(d, 1Н, J=9,0 Гц), 7,05(t, 1Н), 8,05(dd, 1Н, J1=2,4 Гц, J2=9,0 Гц), 8,76(d, 1Н, J=2,4 Гц). + МС: m/z438(М+Н) . 7В: 1-циклопропіл-3-пропіл-8-[6-(2-(піролідин-1-іл)етиламіно)-3-піридил]ксантин: + МС: m/z 424 (М+Н) . Загальні способи отримання амідних сполук (1-33). Аміно-заміщену піридильну сполуку В (50 мг) розчиняли у піридині (25 мг) при 80-100 °C. Після охолодження до кімнатної температури, потрібний хлорангідрид кислоти (4-6 еквіваленти) додавали при кімнатній температурі. Суміш перемішували при кімнатній температурі протягом 24-60 годин. Реакцію гасили льодом та розчинник видаляли та залишок очищали на колонці з силікагелем (дихлорметан: МеОН =96: 4) з утворенням сполук 1-36 та 46-51 при 60-80 % виходу. 1: 1-циклопропіл-3-пропіл-8-[6-(N-[6-(трифлуорметил)нікотиноїл]-N-метиламіно)-3піридил)ксантин: Умови ВЕРХ: МеОН 40 %-95 % градієнт протягом 10 хвилин, тоді МеОН 95 %. Час утримування=9,77 хвил. ! Н ЯМР (ДМСО, d6): 0;72(m, 2Н), 0,89(t, 3Н, J=7.S Гц), 1,01(а, 2Н), 1,71 (m, 2Н), 2,62(m, 1Н), 3,53(s, 3Н), 3,96 (t, 2Н, J=7,5 Гц), 7,53(d, 1Н, J=*8,4 Гц, ), 7,88(d, 1Н, J=8,4 Гц, ), 8,00(dd, 1Н, ' J1=1,8 Гц, J2=7,8 Гц), 8,38(dd, 1Н, J1=2,4 Гц, J2=8,4 Гц), '8,70(s, 1Н), 8,94(d, 1Н, J=2,4 Гц). МС: m/z 514(М+Н)t 2: 1-циклопропіл-3-пропіл-8-[6-(N-[6-(трифлуорметил)нікотиноїлї-N-етиламіно)-3піридил]ксантин; Умови ВЕРХ: МеОН 40 %-95 % градієнт протягом 10 хвилин, тоді МеОН 95 %. Час утримування=10,13 хвил. ! Н ЯМР (ДМСО, d6): 0,70(m, 2Н), 0,88(t, 3Н, J=7,5 Гц), 1,02(m, 2Н), 1,19(3Н, J=7,2 Гц), 1,69(m, 2Н), 2,61(m, 1Н), 3,95(t, 2Н, J=7,2 Гц), 4,08(q, 2Н, J=7,5 Гц), 7,46(d, 1Н, J=S, 7 Гц), 7,85(d, 1Н, J=8,1 Гц), 7,96(dd, 1Н, J1=8,1 Гц, J2=2,1 Гц), 8,36(dd, 1Н, J1=8,7 Гц, J2=2,1 Гц), 8,66(s, 1Н), 8,96(d, 1Н, J=2.l Гц). + МС: m/z 528 (М+Н) . 3: 1-циклопропіл-3-пропіл-8-[6-(N-(6-(трифлуорметил)нікотиноїл]-N-пропіламіно)-3піридил]ксантин: Умови ВЕРХ: МеОН 40 %-95 % градієнт протягом 10 хвилин, тоді МеОН 95 %. Час утримування=10,80 хвил. 1 'Н ЯМР (ДМСО, d6): Н ЯМР (ДМСО, d6): 0 0,71(10, 2Н), 0.£l(m, 6Н), 1,03(m, 2Н), 1,57-1,73(m, 4Н), 2,61 (m, 1Н), 3,92-4,04(m, 4Н), 7,47(d, 1Н, J-8,7 Гц), 7,85(0 % 1Н, J=8,4 Гц), 7,95(d, 1Н, J=8,4 Гц), 8,36(dd, 1Н, J1=8,7 Гц, J2=2,4 Гц), 8,66(s, 1Н), 8,95(d, 1Н, J=2,4 Гц). + МС: m/z 542 (М+Н) . 4: 1-циклопропіл-3-пропіл-8-[6-(N-(6-(трифлуорметил)нікотиноїл]-N-(2-метоксіетил)аміно)-3піридил]ксантин: Умови ВЕРХ: МеОН 40 %-95 % градієнт протягом 10 хвилин, тоді МеОН 95 %. Час 1 утримування=10,08 хвил-. Н ЯМР (ДМСО, d6):] Н ЯМР (ДМСО, d6): 0,71(m, 2Н), 0,88(t, 3Н, J-7,5 Гц), 1,05(m, 2Н), 1,70(m, 2Н), 2,62(m, 1Н), 3,19(s, 3Н), 3,62(t 2Н, J=5,4 Гц), 3,96(t, 2Н, J=7.S Гц), 4,21 (t, 2Н, J=5,4 Гц), 7,47(d, 1Н, J=8,7 Гц), 7,87(d, 1Н, J=8,1 Гц), 7.&6(d, 1Н, J=8,1ttz),-8;34(dd, + 1Н, J1=8,7 Гц, J2=4 Гц), 8,66(s, 1Н), 8,95(d, 1Н, J=2,4 Гц). МС: m/z 558 (М+Н) . 5: 1-циклопропіл-3-пропіл-8-[6-(6-(N-(6-флуорнікотиноїл]-N-(2-метоксіетил)аміно)-3пірпіридил]ксантин: Умови ВЕРХ: МеОН 40 %-95 % градієнт протягом 10 хвилин, тоді МеОН 95 %. Час утримування=9,32 хвил. 1 Н ЯМР (ДМСО, d6): 0,74(m, 2Н), 0,92(t, 3Н, J=7,5 Гц), 1,06(m, 2Н), 1,73(m, 2Н), 2,65(m, 1Н), 3,19(s, 3Н), 3,64(t, 2Н, J=5,7 Гц), 3,99(t, 2Н, J=7,5 Гц), 4,22(t, 2Н, J=5,7 Гц), 7,18(dd, 1Н, J1=8,4 Гц, J2=2-4 Гц), 7,41(d, 1Н, J=8,4 Гц), 7,91(td, 1Н, J1=8,4 Гц, J2=2,4 Гц), 8,18(d, 1Н, J=2,4 Гц), 8,36(dd, 1Н, J1=8,4 Гц, J2=2,1 Гц), 8,76(d, 1Н, J=2,1 Гц). + МС: m/z 508 (М+Н) . 16 UA 105164 C2 5 10 15 20 25 30 35 40 45 50 55 60 6: 1-циклопропіл-3-пропіл-8-[6-(N-нікотиноїл-N-(2-метоксіетил)аміно)-3-піридил]ксантин: Умови ВЕРХ: МеОН 40 %-95 % градієнт протягом 10 хвилин, тоді МеОН 95 %. Час утримування=8,53 хвил. + МС: m/z 490 (М+Н) . 7: 1-циклопропіл-3-пропіл-8-[6-(N-нікотиноїл-N-(циклопропілметил)аміно)-3-піридил]ксантин: Умови ВЕРХ: МеОН 40 %-95 % градієнт протягом 10 хвилин, тоді МеОН 95 %. Час утримування=9,77 хвил. + МС: m/z 486 (М+Н) . 8: 1-циклопропіл-3-пропіл-8-[6-(N-нікотиноїл-N-(циклопропіл)аміно)-3-піридил]ксантин: Умови ВЕРХ: МеОН 40 %-95 %-градієнт протягом 10 хвилин, тоді МеОН 95 %. Час утримування =8,67 хвил. + МС: m/z 472 СМ+Н) .* 9: 1-циклопропіл-3-пропіл-8-[6-(N-[6-(трифлуорметилнікотиноїл]-N(циклопропілметил)аміно)-3-піридил]ксантин: Умови ВЕРХ: МеОН 40 %-95 % градієнт протягом 10 хвилин, тоді МеОН 95 %. Час утримування=10,71 хвил. : Н ЯМР (ДМСО, d6): 0,19(m, 2Н), 0,41(m, 2Н), 0,72(m, 2Н), 0,91(t, 3Н, J=7,2 Гц), 1,00-U6(m, 3Н), 1,70 (m, 2Н), 2,62(m, 1Н), 3,96 (m, 4Н), 7,47(d, 1Н, J=8,4 Гц), 7,86(d, 1Н, J=8,1 Гц, ), 7,97.(dd, 1Н, Jr=2,1 Гц, J2
ДивитисяДодаткова інформація
Автори російськоюThompson, Robert, D.
МПК / Мітки
МПК: A61K 31/497
Мітки: 8-[6-аміно-3-піридил]ксантини, заміщені
Код посилання
<a href="https://ua.patents.su/28-105164-zamishheni-8-6-amino-3-piridilksantini.html" target="_blank" rel="follow" title="База патентів України">Заміщені 8-[6-аміно-3-піридил]ксантини</a>
Мезилат 4-аміно-6,7-диметокси-2-(5-метансульфонамідо-1,2,3,4-тетрагідроізохінол-2-іл)-5-(2-піридил)хіназоліну та поліморфні форми

Номер патенту: 72311
Опубліковано: 15.02.2005
Автори: Ходжсон Пол Блейз, Басфорд Патриція Ен
МПК: A61P 13/08, C07D 401/14, A61K 31/517
Мітки: форми, мезилат, поліморфні, 4-аміно-6,7-диметокси-2-(5-метансульфонамідо-1,2,3,4-тетрагідроізохінол-2-іл)-5-(2-піридил)хіназоліну
Формула / Реферат:
1. 4-Аміно-6,7-диметокси-2-(5-метансульфонамідо-1,2,3,4-тетрагідроізохінол-2-іл)-5-(2-піридил)хіназоліну мезилат формули .2. Мезилатна сіль за п. 1, яка відрізняється тим, що в процесі диференціальної сканувальної калориметрії вона проявляє ендотермічний пік при приблизно 279°C .3. Мезилатна сіль за п. 1 або п. 2, яка характеризується порошковою рентгенограмою, отриманою при опроміненні рентгенівськими променями K-альфа1...
Заміщені 3-аміно-5-карбамоїл-6-метилселенофено[2,3-b]піридини

Номер патенту: 104154
Опубліковано: 10.01.2014
Автор: Кривоколиско Сергій Геннадійович
МПК: C07D 421/00
Мітки: 3-аміно-5-карбамоїл-6-метилселенофено[2,3-b]піридини, заміщені
Формула / Реферат:
Заміщені 3-аміно-5-карбамоїл-6-метилселенофено[2,3-b]піридини загальної формули (І), (І) в якій R означає алкіл (С1-С6 або i-Pr); R1 означає С6Н5арил, який може бути заміщений Hal (Cl, F, Br, I), OMe, OEt, Me, Et; R2 означає Н, C(O)Alk (де Alk означає С1-С6алкіл або i-Pr), СООAlk (де Alk...
Натрієва сіль 5-[4-[2-(n-метил-n-(2-піридил)аміно)етокси]бензил]тіазолідин-2,4-діону та фармацевтична композиція (варіанти)

Номер патенту: 74842
Опубліковано: 15.02.2006
Автори: Міллан Майкл Джон, Крейг Ендрю Саймон
МПК: A61K 31/427, C07D 417/12, A61P 3/10
Мітки: фармацевтична, сіль, натрієва, варіанти, 5-[4-[2-(n-метил-n-(2-піридил)аміно)етокси]бензил]тіазолідин-2,4-діону, композиція
Формула / Реферат:
1. Сполука, що являє собою натрієву сіль 5-[4-[2-(N-метил-N-(2-піридил) аміно)етокси]бензил]тіазолідин-2,4-діону або її фармацевтично прийнятний сольват, яка відрізняється тим, що натрієва сіль є негігроскопічною.2. Сполука за п. 1, яка відрізняється тим, що має одну або декілька характеристик:(і) інфрачервоний спектр по суті відповідно до фігури 1; (іі) спектр комбінаційного розсіювання по суті відповідно до фігури...
Поліморф 5-[4-[2- (n-метил-n-(2-піридил)аміно)етокси]бензил]тіазолідин-2,4-діону солі малеїнової кислоти

Номер патенту: 67845
Опубліковано: 15.07.2004
Автори: Сейсс Майкл Джон, Мур Стефен, Гілес Роберт Гордон, Блеклер Пол Девід Джеймс
МПК: A61P 3/10, A61K 31/4439, C07D 417/12
Мітки: n-метил-n-(2-піридил)аміно)етокси]бензил]тіазолідин-2,4-діону, солі, 5-[4-[2, малеїнової, кислоти, поліморф
Формула / Реферат:
1. Поліморфна форма 5-[4-[2-(N-метил-N-(2-піридил)аміно)етокси]бензил]тіазолідин-2,4-діону солі малеїнової кислоти (“поліморф”), яка відрізняється тим, що вона має:(і) інфрачервоний спектр, що містить піки в області 1752, 1546, 1154, 621 і 602 см-1; та/або(іі) спектр комбінаційного розсіювання, що містить піки в області 1751, 1243 і 602 см-1; та/або(ііі) спектр ядерного магнітного резонансу твердого тіла, що містить піки...
Сіль гідрохлориду 5-[4-[2-(n-метил-n-(2-піридил)аміно)етокси]бензил]тіазолідин-2,4-діону, спосіб її одержання та фармацевтична композиція

Номер патенту: 74003
Опубліковано: 17.10.2005
Автори: Крейг Ендрю Саймон, Міллан Джон Майкл
МПК: A61P 9/10, A61P 1/14, A61P 13/12, A61P 3/00, C07D 417/12, A61P 3/04, A61K 31/4439, A61P 9/12, A61P 5/50, A61P 3/10, A61P 25/00
Мітки: сіль, композиція, 5-[4-[2-(n-метил-n-(2-піридил)аміно)етокси]бензил]тіазолідин-2,4-діону, одержання, спосіб, фармацевтична, гідрохлориду
Формула / Реферат:
1. Гідрохлоридна сіль 5-[4-[2-(N-метил-N-(2-піридил)аміно)етокси]бензил]тіазолідин-2,4-діону, що характеризується:(і) спектром поглинання інфрачервоного випромінювання, в основному, відповідно до фіг. 1; і/або(іі) спектром комбінаційного розсіювання, в основному, відповідно до фіг. 2; і/або(ііі) порошковою рентгенограмою (XRPD), в основному, відповідно до фіг. 3; і(iv) спектром 13С ЯМР твердого стану, в основному,...