Способи одержання аналогів камптотецину та проміжних сполук
Номер патенту: 76449
Опубліковано: 15.08.2006
Автори: Ушіда Міюкі, Савада Сейго, Огава Таканорі, Нішіяма Хіроюкі





















Формула / Реферат
1. Спосіб одержання 2'-аміно-5'-гідроксипропіофенону для синтезу аналогів камптотецину, в якому з сполуки (а):
 (а)
(а)
одержують сполуку (b):
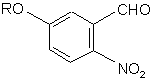 (b)
(b)
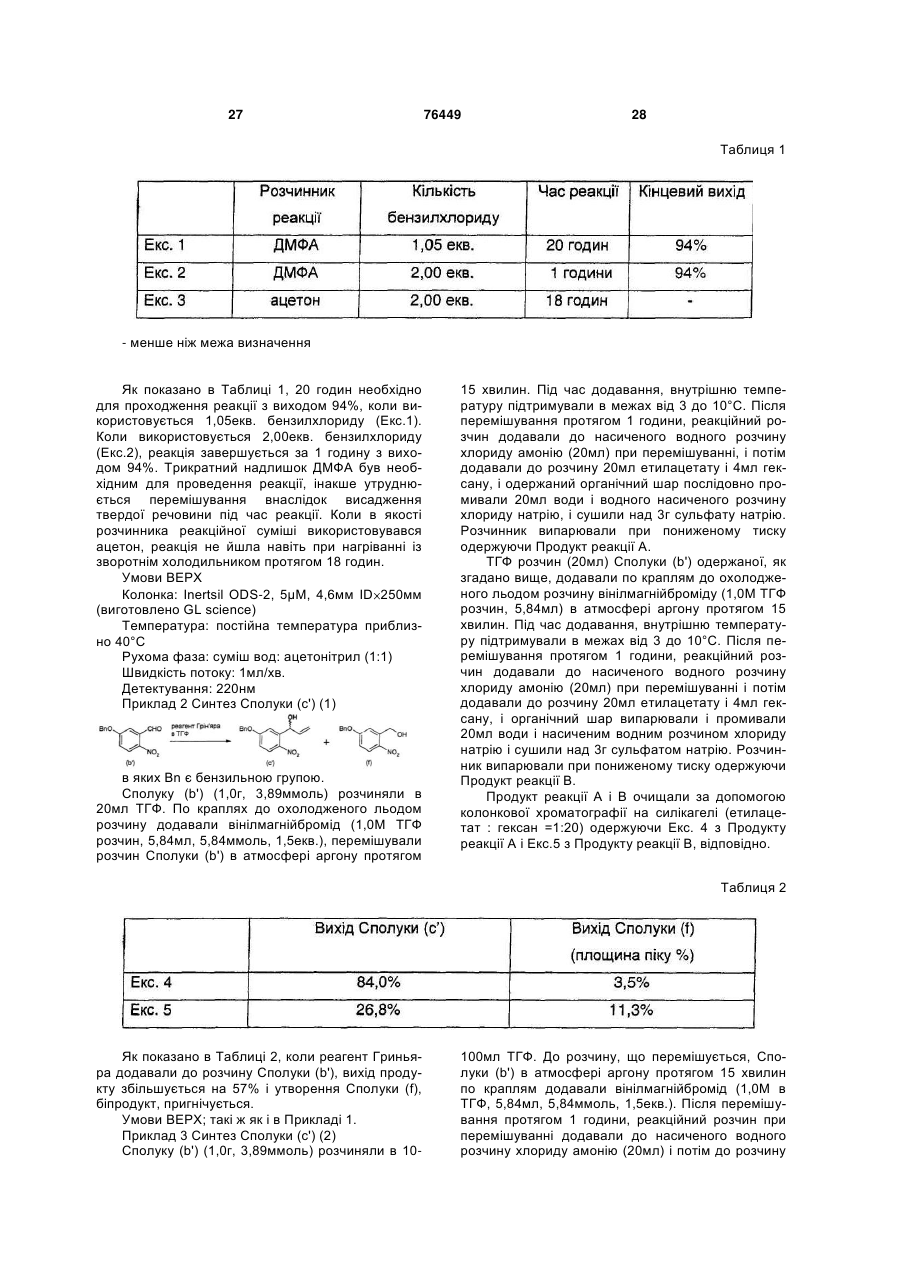
і з сполуки (b) одержують сполуку (с):
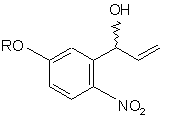 , (c)
, (c)
і з сполуки (с) одержують сполуку (d):
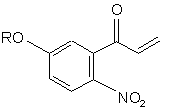 , (d)
, (d)
і з сполуки (d) одержують сполуку (е):
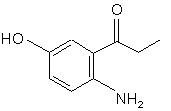 , (e)
, (e)
де R є захисною групою, яка може бути видалена шляхом каталітичного відновлення.
2. Спосіб згідно з пунктом 1, в якому захисною групою, яка може бути видалена каталітичним відновленням, є бензильна група.
3. Спосіб згідно з пунктом 1 або 2, де він включає одну або більшу кількість стадій, що вибирають з групи, яка містить
(1) стадію одержання сполуки (b) шляхом змішування сполуки (а), бензилюючого реагенту і основи і перемішування згаданої суміші в розчиннику при кип'ятінні із зворотним холодильником;
(2) стадію одержання сполуки (с) шляхом додавання краплинами реагенту Гриньяра до сполуки (b) в атмосфері інертного газу;
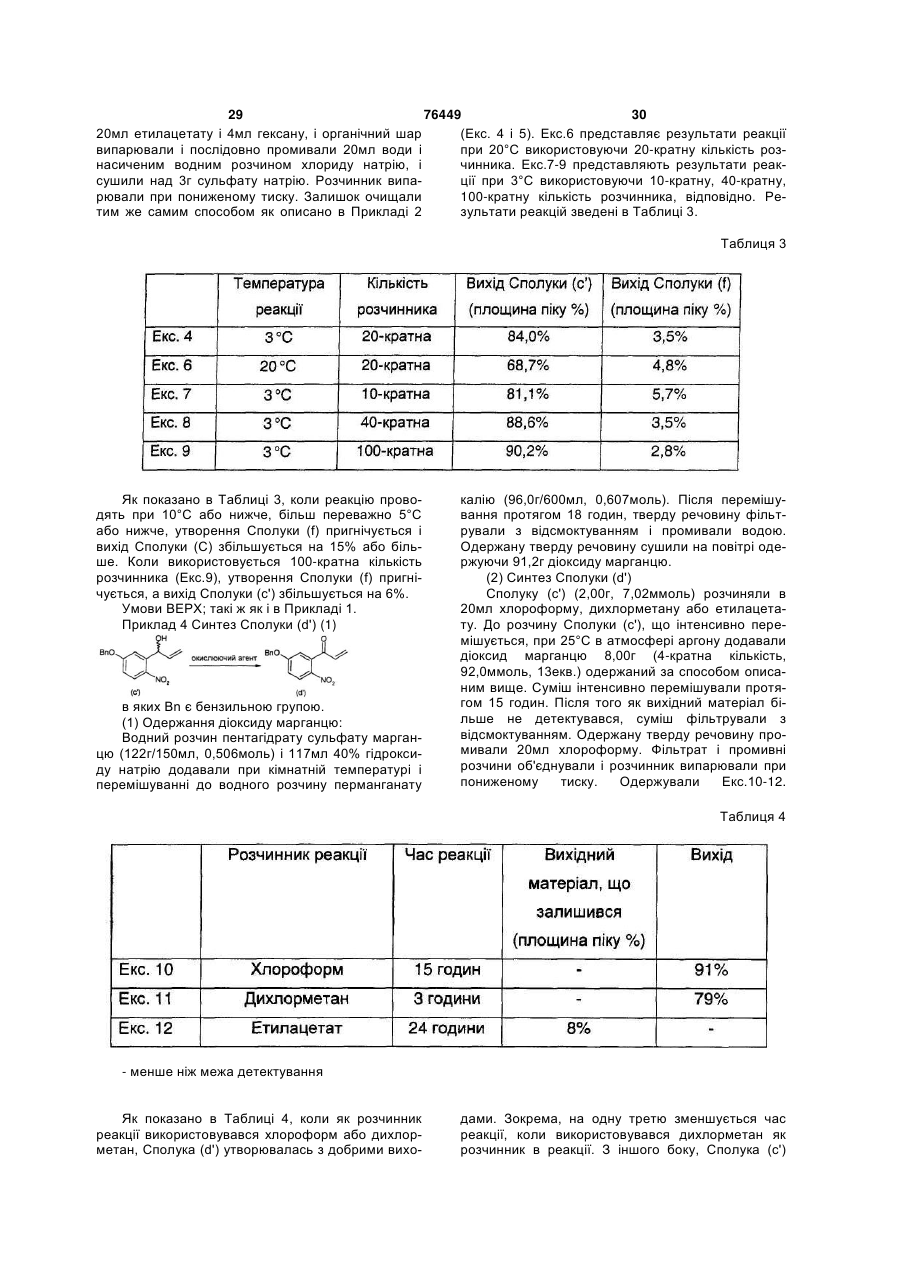
(3) стадію одержання сполуки (d) шляхом змішування сполуки (с) і окислюючого агента і перемішування суміші; і
(4) стадію одержання сполуки (е) шляхом каталітичного відновлення сполуки (d).
4. Спосіб згідно з пунктом 3, в якому на стадії (1) розчинником є диметилформамід.
5. Спосіб згідно з пунктом 3, в якому на стадії (2) реагентом Гриньяра є вінілмагнійбромід.
6. Спосіб згідно з пунктом 3, в якому на стадії (3) окислюючим агентом є реагент Джоунса, діоксид марганцю або (2,2,6,6-тетраметилпіперидин-1-оксил)-гіпохлорит натрію (ТЕМРО-гіпохлорит натрію).
7. Сполука формули (с'):
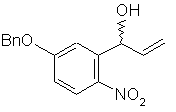 , (с')
, (с')
в якій Вn є бензильною групою.
8. Сполука формули (d'):
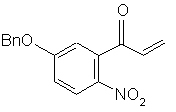 , (d')
, (d')
в якій Вn є бензильною групою.
9. Спосіб одержання 2'-аміно-5'-гідроксипропіофенону для синтезу аналогів камптотецину, в якому з cполуки (а):
 (а)
(а)
одержують cполуку (с"):
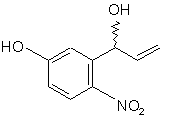 (c'')
(c'')
і з cполуки (с") одержують cполуку (d''):
 , (d")
, (d")
і з cполуки (d") одержують cполуку (е):
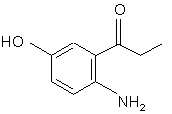 (e).
(e).
10. Спосіб згідно з пунктом 9, де він включає одну або більшу кількість стадій, що вибирають з групи, яка містить
(1) стадію одержання cполуки (с") шляхом додавання краплинами реагенту Гриньяра до cполуки (а) в атмосфері інертного газу;
(2) стадію одержання cполуки (d") шляхом змішування cполуки (с") і окислюючого агента і перемішування суміші; і
(3) стадію одержання полуки (е) шляхом каталітичного відновлення cполуки (d").
11. Спосіб згідно з пунктом 10, в якому на стадії (1) реагентом Гриньяра є вінілмагнійбромід.
12. Спосіб згідно з пунктом 11, в якому на стадії (2) окислюючим агентом є реагент Джоунса, діоксид марганцю або ТЕМРО-гіпохлорит натрію.
13. Застосування 2'-аміно-5'-гідроксипропіофенону, який одержують за способом згідно з будь-яким з пунктів 1-6, для одержання аналогів камптотецину.
14. Застосування 2'-аміно-5'-гідроксипропіофенону, який одержують за способом згідно з будь-яким з пунктів 9-12, для одержання аналогів камптотецину.
15. Спосіб одержання аналогів камптотецину, що включає реакцію 2'-аміно-5'-гідроксипропіофенону, одержаного за способом згідно з будь-яким з пунктів 1-6, і трициклічного кетону.
16. Спосіб одержання аналогів камптотецину, що включає реакцію 2'-аміно-5'-гідроксипропіофенону, одержаного за способом згідно з будь-яким з пунктів 9-12, і трициклічного кетону.
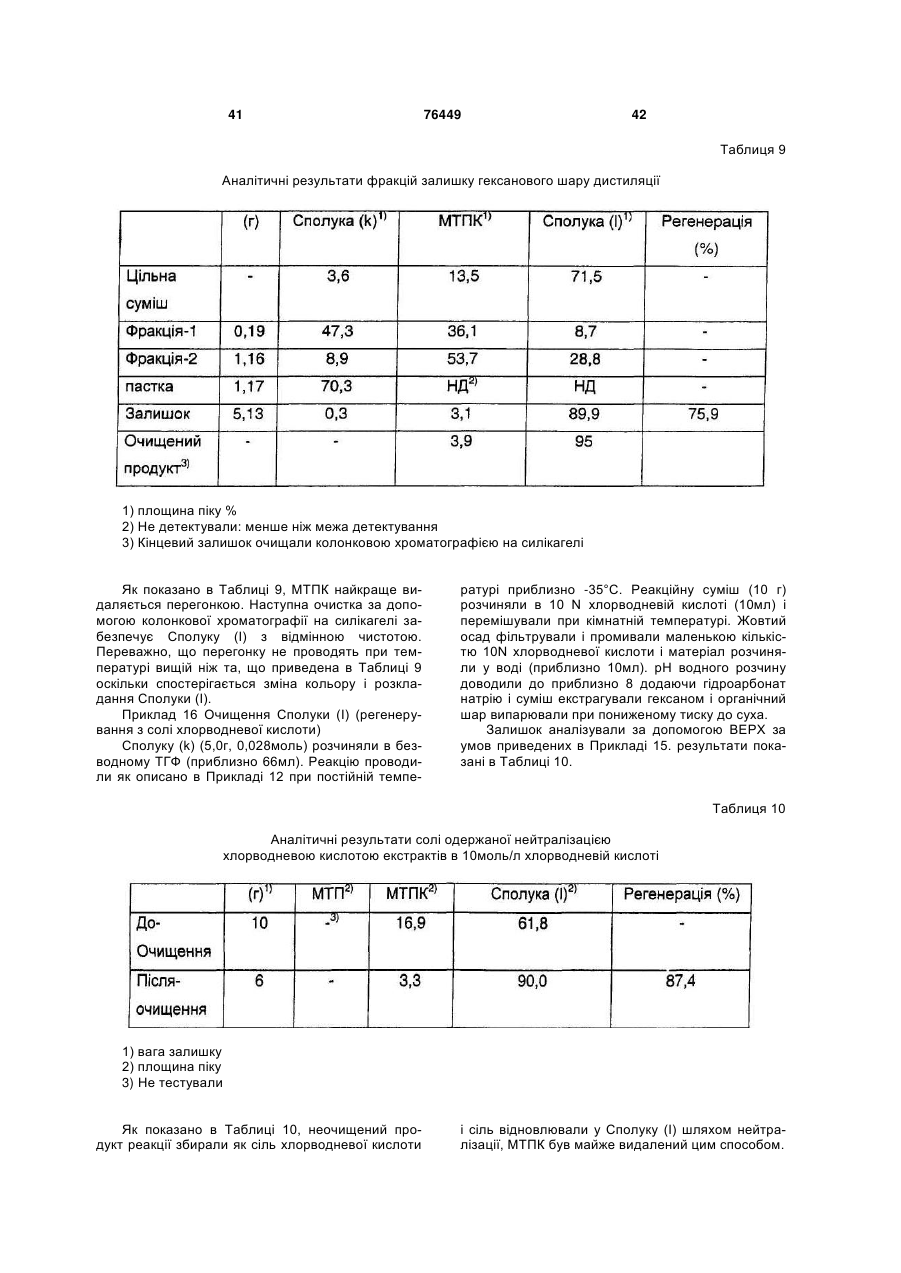
17. Спосіб одержання трициклічного кетону для синтезу аналогів камптотецину, в якому з cполуки (k):
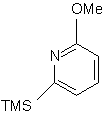 , (k)
, (k)
в якій TMS є триметилсилільною групою і Me є метильною групою,
або cполуки (v):
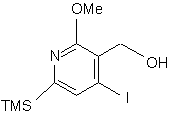 , (v)
, (v)
в якій TMS є триметилсилільною групою і Me є метильною групою,
одержують cполуку (l):
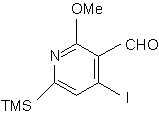 , (l)
, (l)
в якій TMS є триметилсилільною групою і Me є метильною групою,
і з cполуки (І) одержують cполуку (m):
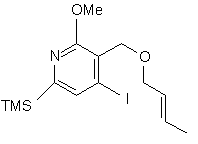 , (m)
, (m)
в якій TMS є триметилсилільною групою і Me є метильною групою,
і з cполуки (m) одержують cполуку (n):
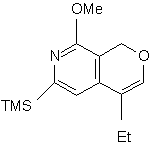 , (n)
, (n)
в якій TMS є триметилсилільною групою, Me є метильною групою і Et є етильною групою,
і з cполуки (n) одержують cполуку (о):
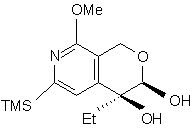 , (o)
, (o)
в якій TMS є триметилсилільною групою, Me є метильною групою і Et є етильною групою,
і з cполуки (о) одержують cполуку (р):
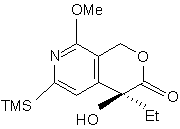 , (p)
, (p)
в якій TMS є триметилсилільною групою, Me є метильною групою і Et є етильною групою,
і з cполуки (р) одержують cполуку (q):
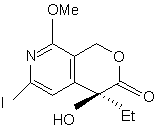 , (q)
, (q)
в якій Me є метильною групою і Et є етильною групою,
і з cполуки (q) одержують cполуку (r):
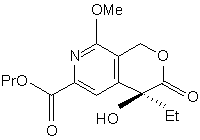 , (r)
, (r)
в якій Me є метильною групою, Et є етильною групою і Рr є пропільною групою,
і з cполуки (r) одержують cполуку (s):
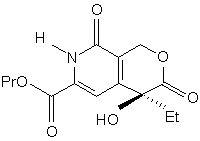 , (s)
, (s)
в якій Et є етильною групою і Рr є пропільною групою,
і з cполуки (s) одержують cполуку (t):
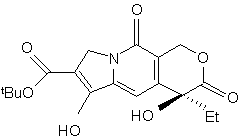 , (t)
, (t)
в якій Et є етильною групою і tВu є т-бутильною групою,
і з cполуки (t) одержують cполуку (h):
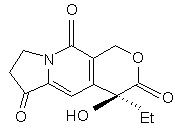 , (h)
, (h)
в якій Et є етильною групою,
де спосіб включає одну або більшу кількість стадій, що вибирають з групи, яка містить:
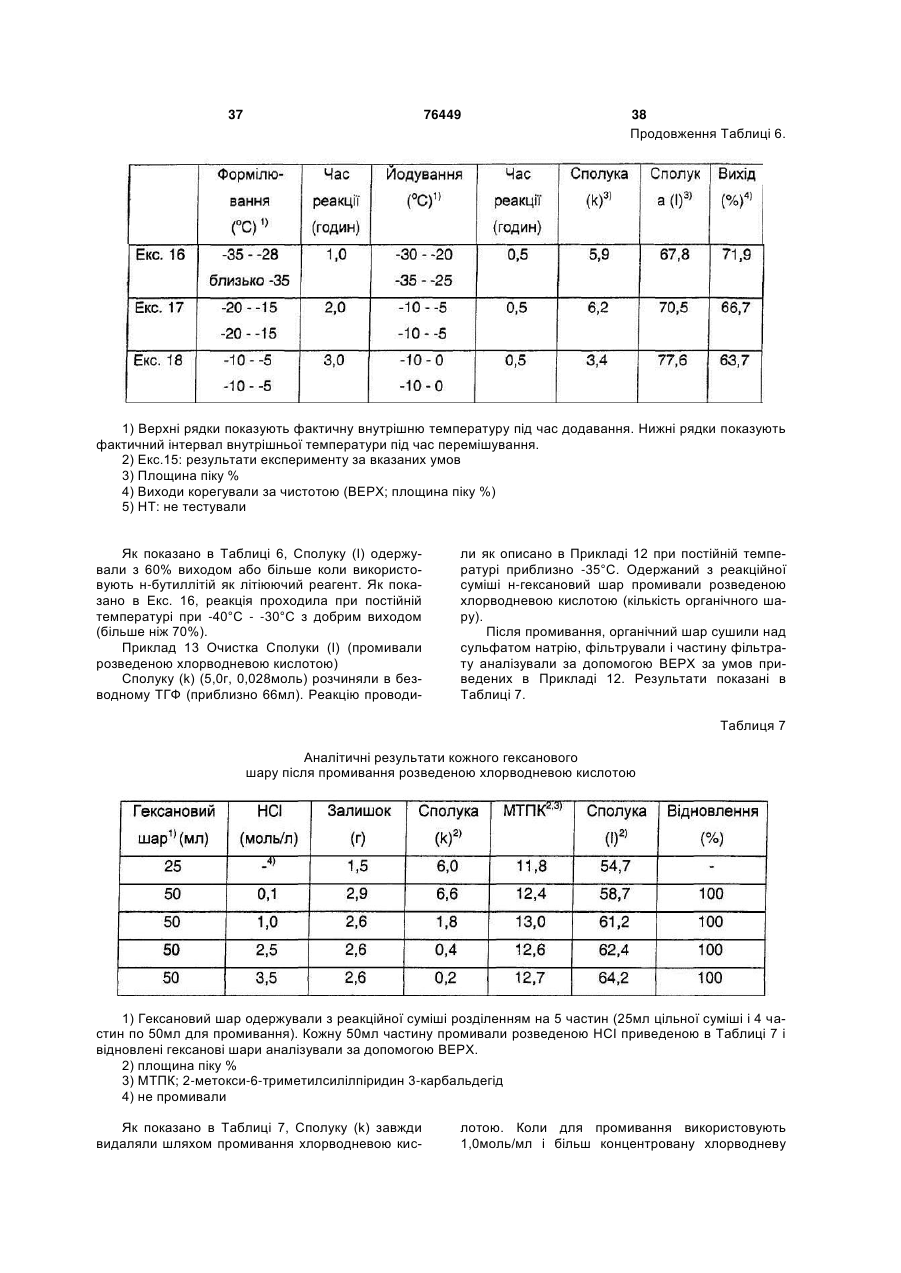
(1) стадію одержання cполуки (І) шляхом змішування cполуки (k), літіюючого агента, формілюючого реагенту і йодуючого реагенту;
(2) стадію одержання cполуки (m) шляхом змішування cполуки (І), кротилового спирту, триетилсилану і кислоти і взаємодію згаданої суміші без використання розчинника;
(3) стадію одержання cполуки (I) шляхом змішування cполуки (v), біпродукту стадії (2) з окислюючим агентом і основою;
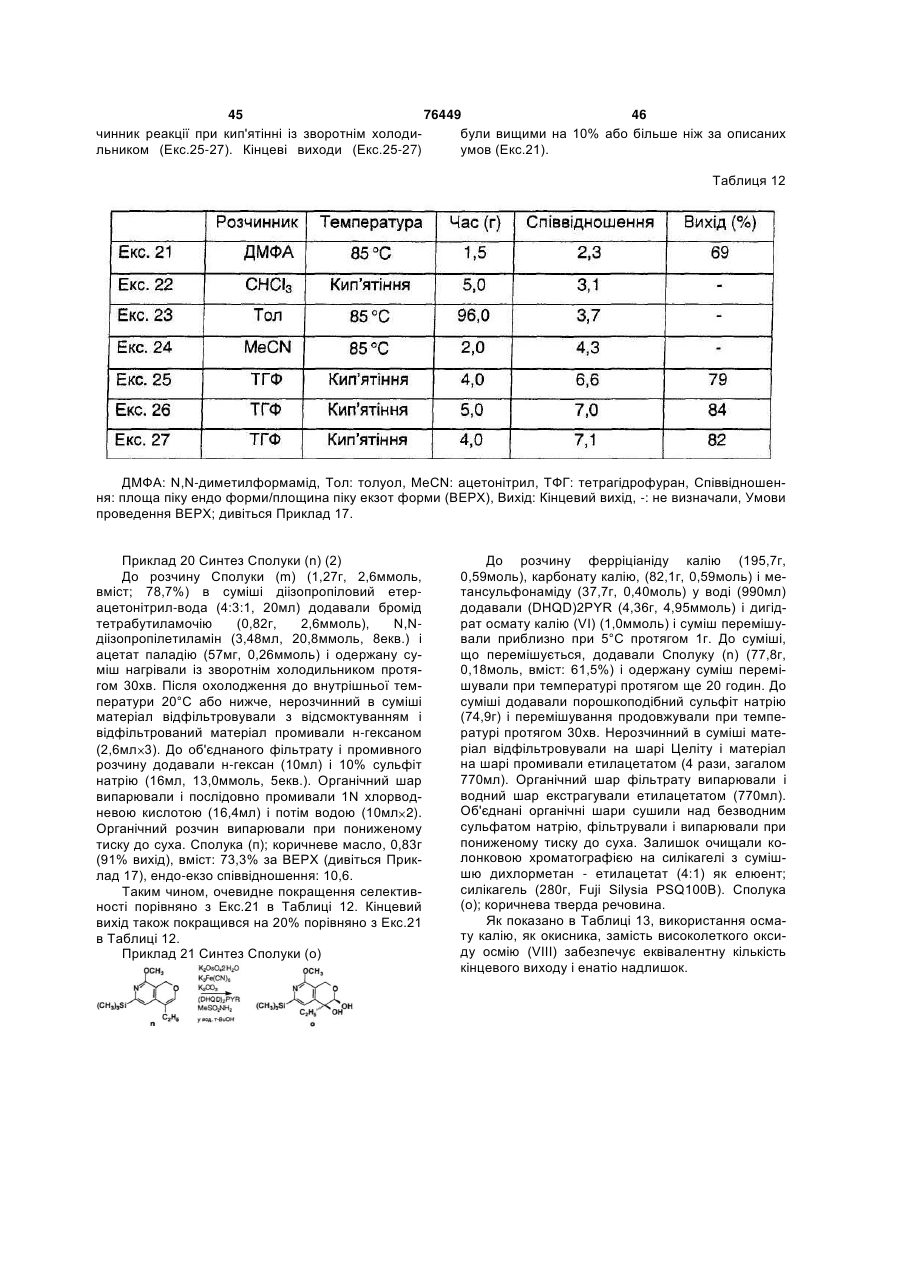
(4) стадію одержання cполуки (n) шляхом змішування cполуки (m), паладієвого каталізатора, основи і каталізатора переносу фаз і кип'ятіння згаданої суміші в розчиннику;
(5) стадію одержання cполуки (о) шляхом змішування cполуки (n), осмієвого каталізатора, співокислюючого агента, основи і асиметричного реагенту;
(6) стадію одержання cполуки (р) шляхом змішування cполуки (о), основи і йоду і кип'ятіння згаданої суміші в рідкій суміші спирт-вода;
(7) стадію одержання cполуки (q) шляхом змішування cполуки (р) і десилілюючого-йодуючого реагенту;
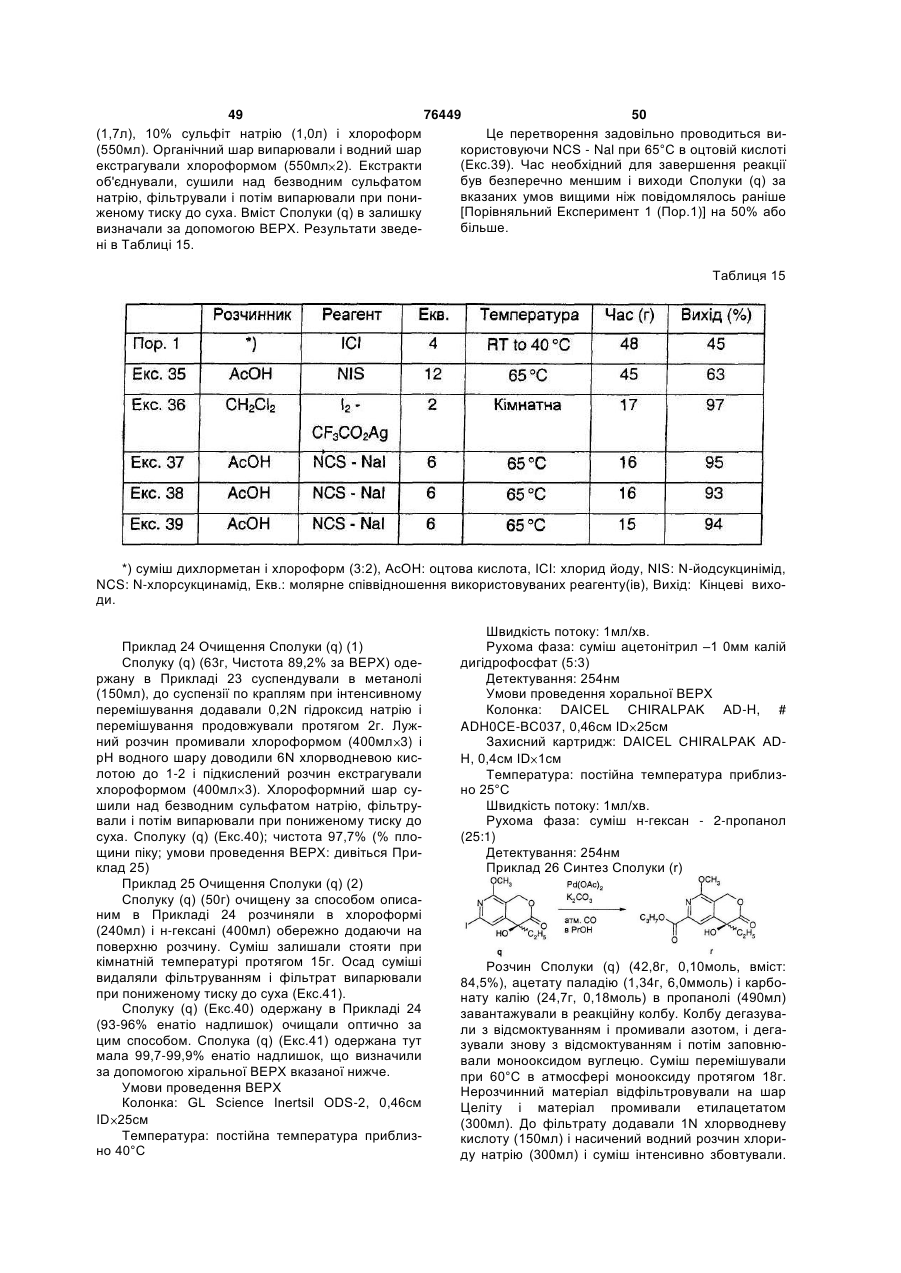
(8) стадію одержання cполуки (r) шляхом змішування cполуки (q), паладієвого каталізатора і основи і взаємодію згаданої суміші в 1-пропанолі в атмосфері монооксиду вуглецю;
(9) стадію одержання cполуки (s) шляхом змішування cполуки (r) і деметилюючого реагенту і взаємодію згаданої суміші при кімнатній температурі;
(10) стадію одержання cполуки (t) шляхом взаємодії cполуки (s) з т-бутилакрилатом в присутності основи.
18. Спосіб згідно з пунктом 17, в якому на стадії (1) літіюючим агентом є н-бутиллітій.
19. Спосіб згідно з пунктом 17 або 18, в якому на стадії (1) температура реакції є постійною температурою в межах -30 - -40°С.
20. Спосіб згідно з пунктом 17, в якому на стадії (3) окислюючим агентом є ТЕМРО-гіпохлорит натрію.
21. Спосіб згідно з пунктом 17, в якому на стадії (4) основою є карбонат калію або діізопропілетиламін.
22. Спосіб згідно з пунктом 17 або 21, в якому на стадії (4) розчинником є тетрагідрофуран або рідка суміш діізопропіловий етер-ацетонітрил-вода.
23. Спосіб згідно з пунктом 17, в якому на стадії (5) осмієвим каталізатором є осмат калію (VI).
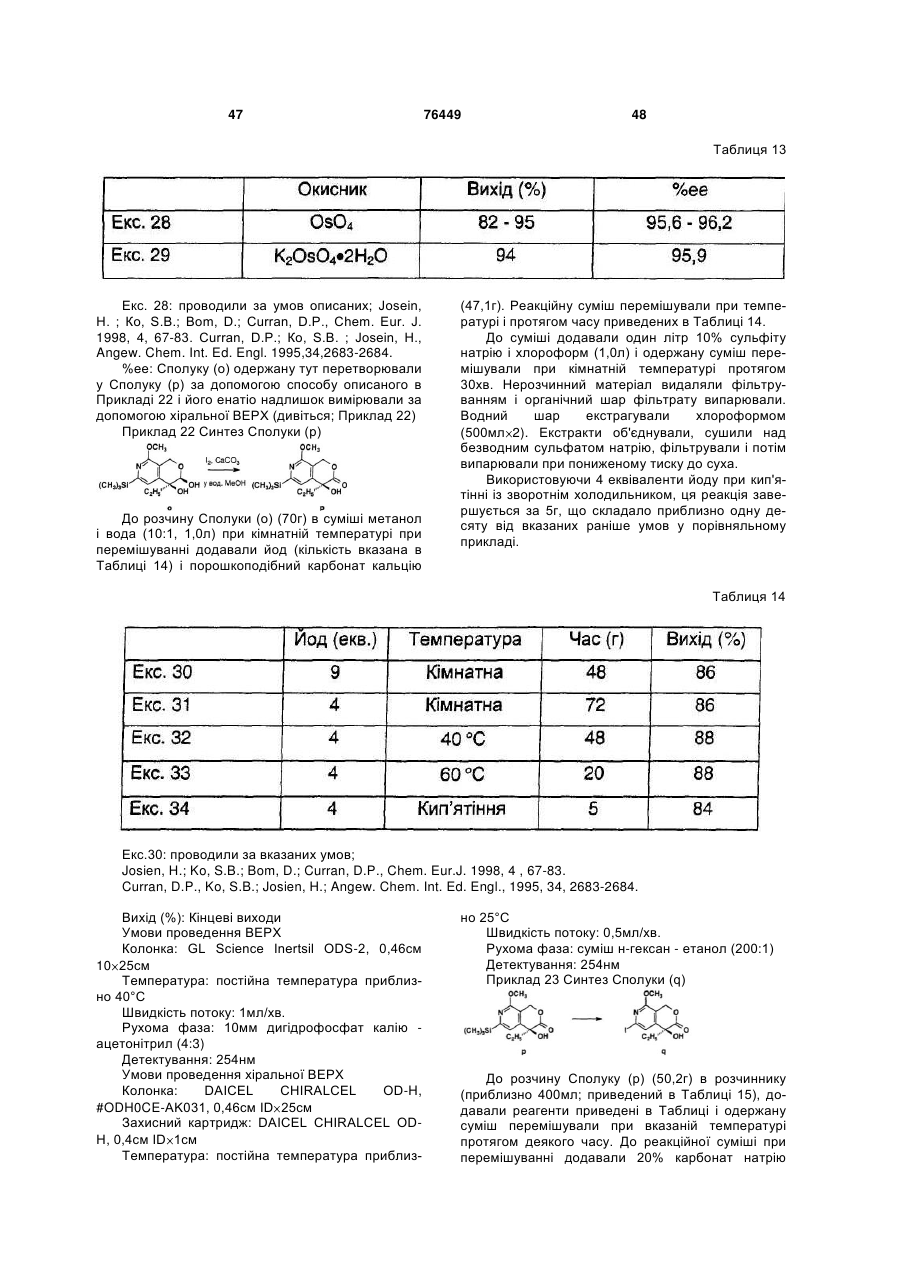
24. Спосіб згідно з пунктом 17, в якому на стадії (6) використовують 4 еквіваленти йоду по відношенню до сполуки (о).
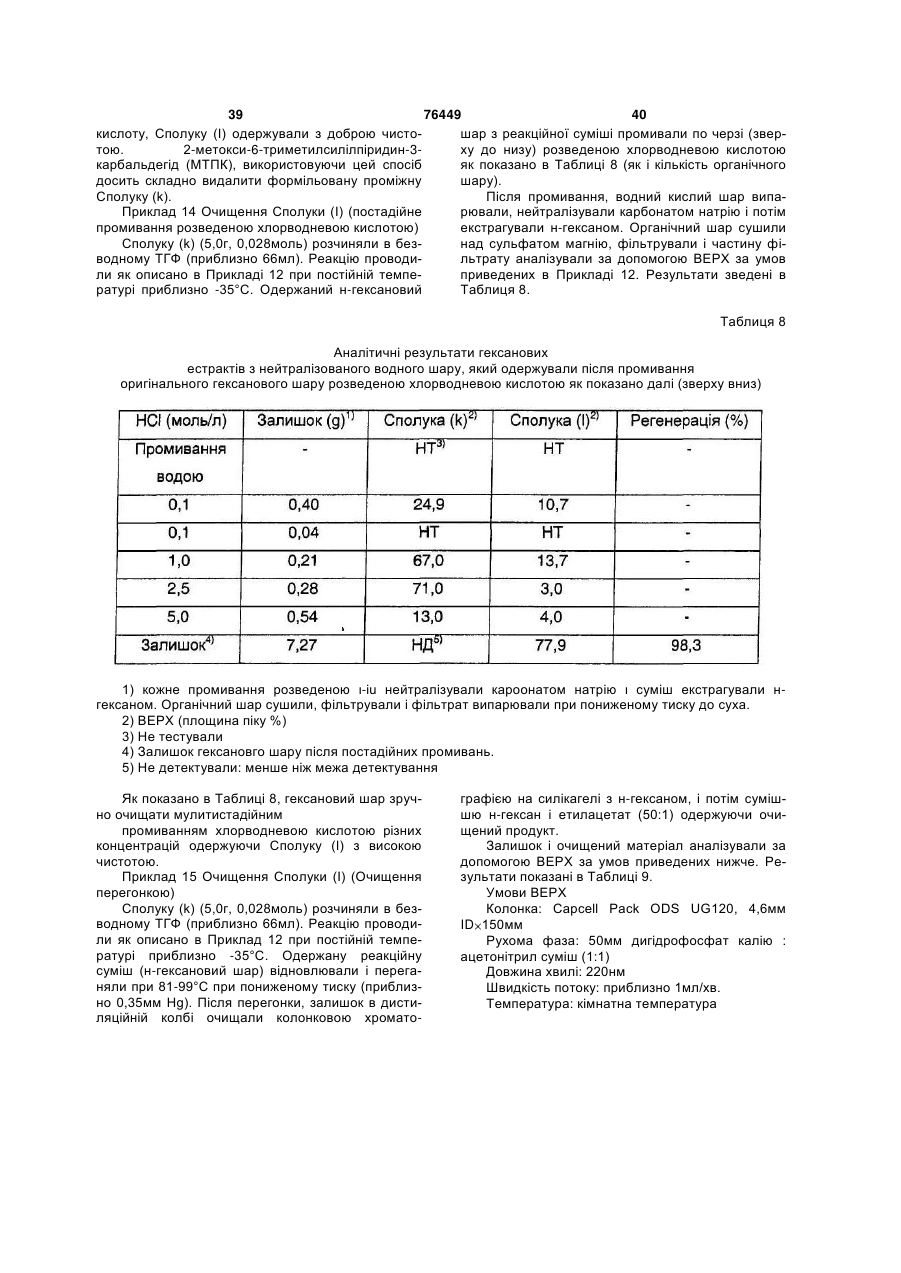
25. Спосіб згідно з пунктом 17, в якому на стадії (7) десилілюючим-йодуючим реагентом є йод-трифторацетат срібла або N-хлорсукцинімід-йодид натрію.
26. Спосіб згідно з пунктом 17 або 25, в якому cполуку (q) очищають хімічно за допомогою стадій очистки, що включають:
стадію додавання продукту реакції, одержаного на стадії одержання cполуки (q) з cполуки (р), до водного лужного розчину і перемішування;
стадію додавання органічного розчинника і перемішування з наступним видаленням органічного шару; і
стадію підкислення водного шару і екстрагування органічним розчинником.
27. Спосіб згідно з пунктом 26, в якому водним лужним розчином є водний розчин гідроксиду натрію.
28. Спосіб згідно з пунктом 26, в якому органічним розчинником є хлороформ.
29. Спосіб згідно з пунктом 17 або 25, в якому cполуку (q) оптично очищають за допомогою стадій очистки, що включає:
стадію розчинення продукту реакції, одержаного на стадії одержання cполуки (q) з cполуки (р), у високополярному розчиннику, з наступним розшаровуванням низькополярним розчинником і
стадію фільтрування осаду з наступним концентруванням фільтрату досуха при зниженому тиску.
30. Спосіб згідно з пунктом 29, в якому високополярним розчинником є хлороформ.
31. Спосіб згідно з пунктом 29, в якому низькополярним розчинником є н-гексан.
32. Спосіб згідно з пунктом 17, в якому на стадії (10) основою є карбонат калію.
33. Застосування трициклічного кетону, одержаного за способом згідно з будь-яким з пунктів 17-32, для одержання аналогів камптотецину.
34. Спосіб одержання аналогів камптотецину, в якому трициклічний кетон, який одержують за способом згідно з будь-яким з пунктів 17-32, реагує з 2'-аміно-5'-гідроксипропіофеноном.
35. Спосіб згідно з пунктом 34, в якому 2'-аміно-5'-гідроксипропіофеноном є 2'-аміно-5'-гідроксипропіофенон, одержаний за способом згідно з будь-яким з пунктів 1-6.
36. Спосіб згідно з пунктом 34, в якому 2'-аміно-5'-гідроксипропіофеноном є 2'-аміно-5'-гідроксипропіофенон, одержаний за способом згідно з будь-яким з пунктів 9-12.
37. Спосіб згідно з пунктом 15, в якому трициклічний кетон і 2'-аміно-5'-гідроксипропіофенон змішують і згадана суміш реагує в атмосфері інертного газу.
38. Спосіб згідно з пунктом 16, в якому трициклічний кетон і 2'-аміно-5'-гідроксипропіофенон змішують, і згадана суміш реагує в атмосфері інертного газу.
39. Спосіб згідно з будь-яким з пунктів 34-36, в якому трициклічний кетон і 2'-аміно-5'-гідроксипропіофенон, змішують і згадана суміш реагує в атмосфері інертного газу.
Текст
1. Спосіб одержання 2'-аміно-5'-гідроксипропіофенону для синтезу аналогів камптотецину, в якому з сполуки (а): 2 3 76449 4 OMe O BnO N NO2 , (d') в якій Вn є бензильною групою. 9. Спосіб одержання 2'-аміно-5'-гідроксипропіофенону для синтезу аналогів камптотецину, в якому з сполуки (а): HO TMS , (k) в якій TMS є триметилсилільною групою і Me є метильною групою, або сполуки (v): OMe CHO N NO2 , (а) одержують сполуку (с"): OH HO OH I TMS , (v) в якій TMS є триметилсилільною групою і Me є метильною групою, одержують сполуку (l): OMe NO2 , (c'') і з сполуки (с") одержують сполуку (d''): O HO NO 2 , (d") і з сполуки (d") одержують сполуку (е): CHO N I TMS , (l) в якій TMS є триметилсилільною групою і Me є метильною групою, і з сполуки (І) одержують сполуку (m): OMe O N O HO I TMS NH2 , (e). 10. Спосіб згідно з пунктом 9, де він включає одну або більшу кількість стадій, що вибирають з групи, яка містить (1) стадію одержання сполуки (с") шляхом додавання краплинами реагенту Гриньяра до сполуки (а) в атмосфері інертного газу; (2) стадію одержання сполуки (d") шляхом змішування сполуки (с") і окислюючого агента і перемішування суміші; і (3) стадію одержання сполуки (е) шляхом каталітичного відновлення сполуки (d"). 11. Спосіб згідно з пунктом 10, в якому на стадії (1) реагентом Гриньяра є вінілмагнійбромід. 12. Спосіб згідно з пунктом 11, в якому на стадії (2) окислюючим агентом є реагент Джоунса, діоксид марганцю або ТЕМРО-гіпохлорит натрію. 13. Застосування 2'-аміно-5'-гідроксипропіофенону, який одержують за способом згідно з будьяким з пунктів 1-6, для одержання аналогів камптотецину. 14. Застосування 2'-аміно-5'-гідроксипропіофенону, який одержують за способом згідно з будьяким з пунктів 9-12, для одержання аналогів камптотецину. 15. Спосіб одержання аналогів камптотецину, що включає реакцію 2'-аміно-5'-гідроксипропіофенону, одержаного за способом згідно з будь-яким з пунктів 1-6, і трициклічного кетону. 16. Спосіб одержання аналогів камптотецину, що включає реакцію 2'-аміно-5'-гідроксипропіофенону, одержаного за способом згідно з будь-яким з пунктів 9-12, і трициклічного кетону. 17. Спосіб одержання трициклічного кетону для синтезу аналогів камптотецину, в якому з сполуки (k): , (m) в якій TMS є триметилсилільною групою і Me є метильною групою, і з сполуки (m) одержують сполуку (n): OMe N O TMS Et , (n) в якій TMS є триметилсилільною групою, Me є метильною групою і Et є етильною групою, і з сполуки (n) одержують сполуку (о): OMe N O OH TMS OH Et , (o) в якій TMS є триметилсилільною групою, Me є метильною групою і Et є етильною групою, і з сполуки (о) одержують сполуку (р): OMe N O O TMS Et HO , (p) в якій TMS є триметилсилільною групою, Me є метильною групою і Et є етильною групою, і з сполуки (р) одержують сполуку (q): OMe N I O O HO Et , (q) в якій Me є метильною групою і Et є етильною групою, 5 і з сполуки (q) одержують сполуку (r): 76449 6 (9) стадію одержання сполуки (s) шляхом змішуOMe вання сполуки (r) і деметилюючого реагенту і взаємодію згаданої суміші при кімнатній температурі; N O (10) стадію одержання сполуки (t) шляхом взаємоPrO дії сполуки (s) з т-бутилакрилатом в присутності O основи. Et HO O , (r) 18. Спосіб згідно з пунктом 17, в якому на стадії (1) в якій Me є метильною групою, Et є етильною грулітіюючим агентом є н-бутиллітій. пою і Рr є пропільною групою, 19. Спосіб згідно з пунктом 17 або 18, в якому на і з сполуки (r) одержують сполуку (s): стадії (1) температура реакції є постійною темпеO ратурою в межах –30 - -40°С. H 20. Спосіб згідно з пунктом 17, в якому на стадії (3) O N окислюючим агентом є ТЕМРО-гіпохлорит натрію. PrO 21. Спосіб згідно з пунктом 17, в якому на стадії (4) O основою є карбонат калію або діізопропілетиламін. Et HO O 22. Спосіб згідно з пунктом 17 або 21, в якому на , (s) стадії (4) розчинником є тетрагідрофуран або рідка в якій Et є етильною групою і Рr є пропільною грусуміш діізопропіловий етер-ацетонітрил-вода. пою, 23. Спосіб згідно з пунктом 17, в якому на стадії (5) і з сполуки (s) одержують сполуку (t): O осмієвим каталізатором є осмат калію (VI). 24. Спосіб згідно з пунктом 17, в якому на стадії (6) O O N використовують 4 еквіваленти йоду по відношенню tBuO до сполуки (о). O HO 25. Спосіб згідно з пунктом 17, в якому на стадії (7) Et HO , (t) десилілюючим-йодуючим реагентом є йодt в якій Et є етильною групою і Вu є т-бутильною трифторацетат срібла або N-хлорсукцинімід-йодид групою, натрію. і з сполуки (t) одержують сполуку (h): 26. Спосіб згідно з пунктом 17 або 25, в якому споO луку (q) очищають хімічно за допомогою стадій очистки, що включають: O N стадію додавання продукту реакції, одержаного на стадії одержання сполуки (q) з сполуки (р), до водO HO O Et , (h) ного лужного розчину і перемішування; стадію додавання органічного розчинника і перев якій Et є етильною групою, мішування з наступним видаленням органічного де спосіб включає одну або більшу кількість сташару; і дій, що вибирають з групи, яка містить: стадію підкислення водного шару і екстрагування (1) стадію одержання сполуки (І) шляхом змішуорганічним розчинником. вання сполуки (k), літіюючого агента, формілюючо27. Спосіб згідно з пунктом 26, в якому водним го реагенту і йодуючого реагенту; лужним розчином є водний розчин гідроксиду на(2) стадію одержання сполуки (m) шляхом змішутрію. вання сполуки (І), кротилового спирту, триетилси28. Спосіб згідно з пунктом 26, в якому органічним лану і кислоти і взаємодію згаданої суміші без вирозчинником є хлороформ. користання розчинника; 29. Спосіб згідно з пунктом 17 або 25, в якому спо(3) стадію одержання сполуки (I) шляхом змішулуку (q) оптично очищають за допомогою стадій вання сполуки (v), біпродукту стадії (2) з окислююочистки, що включає: чим агентом і основою; стадію розчинення продукту реакції, одержаного (4) стадію одержання сполуки (n) шляхом змішуна стадії одержання сполуки (q) з сполуки (р), у вання сполуки (m), паладієвого каталізатора, освисокополярному розчиннику, з наступним розшанови і каталізатора переносу фаз і кип'ятіння згаровуванням низькополярним розчинником і даної суміші в розчиннику; стадію фільтрування осаду з наступним концент(5) стадію одержання сполуки (о) шляхом змішуруванням фільтрату досуха при зниженому тиску. вання сполуки (n), осмієвого каталізатора, співоки30. Спосіб згідно з пунктом 29, в якому високопослюючого агента, основи і асиметричного реагенлярним розчинником є хлороформ. ту; 31. Спосіб згідно з пунктом 29, в якому низькопо(6) стадію одержання сполуки (р) шляхом змішулярним розчинником є н-гексан. вання сполуки (о), основи і йоду і кип'ятіння згада32. Спосіб згідно з пунктом 17, в якому на стадії ної суміші в рідкій суміші спирт-вода; (10) основою є карбонат калію. (7) стадію одержання сполуки (q) шляхом змішу33. Застосування трициклічного кетону, одержановання сполуки (р) і десилілюючого-йодуючого реаго за способом згідно з будь-яким з пунктів 17-32, генту; для одержання аналогів камптотецину. (8) стадію одержання сполуки (r) шляхом змішу34. Спосіб одержання аналогів камптотецину, в вання сполуки (q), паладієвого каталізатора і осякому трициклічний кетон, який одержують за спонови і взаємодію згаданої суміші в 1-пропанолі в собом згідно з будь-яким з пунктів 17-32, реагує з атмосфері монооксиду вуглецю; 2'-аміно-5'-гідроксипропіофеноном. 7 76449 8 35. Спосіб згідно з пунктом 34, в якому 2'-аміно-5'ють і згадана суміш реагує в атмосфері інертного гідроксипропіофеноном є 2'-аміно-5'-гідроксипрогазу. піофенон, одержаний за способом згідно з будь38. Спосіб згідно з пунктом 16, в якому трициклічяким з пунктів 1-6. ний кетон і 2'-аміно-5'-гідроксипропіофенон змішу36. Спосіб згідно з пунктом 34, в якому 2'-аміно-5'ють, і згадана суміш реагує в атмосфері інертного гідроксипропіофеноном є 2'-аміно-5'-гідроксипрогазу. піофенон, одержаний за способом згідно з будь39. Спосіб згідно з будь-яким з пунктів 34-36, в яким з пунктів 9-12. якому трициклічний кетон і 2'-аміно-5'-гідроксипро37. Спосіб згідно з пунктом 15, в якому трициклічпіофенон, змішують і згадана суміш реагує в атмоний кетон і 2'-аміно-5'-гідроксипропіофенон змішусфері інертного газу. Представлений винахід стосується способу синтезу камптотецинподібних сполук(и). Більш особливо, винахід стосується способу одержання проміжних сполук для синтезу аналогів камптотецину, що мають антиракову активність і використовуються як згадані проміжні сполуки, і стосується загального синтезу аналогів камптотецину. Камптотецин (тут далі згадується як СРТ) виділений з кори, кореня, плодів, листя і подібного Camtotheca acuminata китайського походження є пентациклічним алкалоїдом і відомий як такий, що проявляє антиракову активність шляхом інгібування синтезу нуклеїнової кислоти. Між тим, повідомлялось, що похідне камптотецину викликає діарею і подібні сторонні ефекти (Gann to Kagaku Ryohou 17, p.115-120, 1990), і, відповідно, існує потреба у вирішенні проблеми, що є причиною розладу гастроінтестинального тракту, визначенні різновидів похідних з меншою токсичністю, підвищені ефективністю і так далі. Таким чином, винахідники вже описали 7-етил10-[4-(1-піперидино)-1піперидино]карбонілоксикамптотецинтідрохлоридтригідрат (тут далі згадується як СРТ-11), водорозчинне напівсинтетичне похідне СРТ, як сполуку, яка має меншу токсичність порівняно з СРТ, і на сьогодні широко використовується як протираковий агент (загальна назва; іринотекану гідрохлорид). Аналоги камптотецину, такі як СРТ-11 можна модифікувати за допомогою хімічних модифікацій СРТ одержаного з природних джерел. Однак, з природних джерел, таких як Camtotheca acuminata, одержується доволі низька кількість СРТ, який є вихідним матеріалом, і очікується, що внаслідок все зростаючої потреби у СРТ-11, який є корисним похідним і таке інше, достатнє забезпечення СРТ стає досить складним не дивлячись на те, що необхідна кількість необхідного вихідного матеріалу забезпечується насадженням плантацій. Хоча також розроблений загальний спосіб синтезу, на сьогоднішній день ситуація в цьому питанні ще є далекою до практичного використання. Як загальний спосіб синтезу відомий спосіб Шен, В. та ін. (Shen, W. et al.) показаний нижче за допомогою реакційної схеми через реакцію Фріендландера (Friedlander) амінопропіофенону і трициклічного кетону (J. Org. Chem. 1993, 58, 611-617 "Concise Total Syntheses of dl-Camptothecin і Related Anticancer Drugs.", через це існує проблема, оскільки стадії є досить складними, виходи не є достатніми і утворюється тільки рацемат. В той же час, не дивлячись на те, що Курран, Д та ін. (Curran, D. P. et al.) провели загальний синтез за допомогою способу, в якому використовується серія радикальних циклізацій арилізонітрилу і йодпіридону, як показано на реакційній схемі нижче (Chem. Eur. J. 1998, 4, 67-83 "A General Synthetic Approach to the (20S)-Camptothecin Family of Antitumor Agents by a Regiocontrolled Cascade Radical Cyclization of Aryl Isonitriles."), проблеми полягають у виході реакції циклізації, що не є достатніми і необхідності видалення захисної групи після циклізації. Крім того, хоча вище Куран, Д.П. та ін. (Curran, D.P. et al.) синтезували 4-йод-2-метокси-6триметилсилілпіридин-3-карбальдегід, проміжну сполуку у синтезі трициклічного кетону частини аналога СРТ, згідно приведеної нижче схеми, Josien, Η; Ко, S.-B.; Bom, D.; Ourran, D.P. Chem.Eur.J. 1998, 4, No.1, 67. цей спосіб є високонебезпечним внаслідок необхідності використання т-BuLi, що легко спалахує при використанні у великих кількостях в промислових масштабах, і для проведення реакції необхідна температура -78°С, що робіть неможливим збільшення розмірів завантаження. Крім того, внаслідок складностей необхідного температурного контролю в системі загальної реакції відсутня промислово придатна реакційна система. Об'єкт винаходу раціонально забезпечує СРТ, який є вихідним матеріалом для іринотекану гідро 9 76449 10 хлориду і різних видів похідних камптотецину, і аналогів камптотецину, таких як 7-етил-10гідроксикамптотецин, який є ключовою проміжною сполукою для синтезу іринотекану гідрохлориду, за допомогою практичного загального синтезу. Особливо, об'єктом винаходу є синтез проміжної сполуки, що відповідає АВ-кільцевій частині скелету камптотецину і проміжної сполуки, що відповідає CDE-кільцевій частині, відповідно, і, крім того, (де TMS є триметилсилільної групою, Me є метисинтезу аналогів камптотецину використовуючи ці льною групою, Et є етильною групою, Рr є пропільпроміжні сполуки. ною групою, і 'Вu є т-бутильною групою), що ґрунЧерез ці обставини винахідники провели інтетується на методиці Curran (Josien, Η,; Ко, S. В.; нсивний пошук, і в результаті цього, як АВ-кільцеву Bom, D.: Curran, D. P. Chem. Eur. J. 1998, 4 67-83) і частину, одержали Сполуку (а) (5-гідрокси-2методиці Pharmacia & Upjohn (тут і далі згадується нітробензальдегід): як методика P&U; Heneger, Κ. Ε.; Ashford, c, W.; (a) вихідний матеріал, і розробили спосіб одержання СРТ і його стабільних похідних за допомогою раціонального одержання 2'-аміно-5'гідроксипропіофенону, що відповідає АВ-кільцевій частині СРТ скелету, і як CDE-кільцеву частину виходять з Сполуки (к) (2-метокси-6триметилсилілпіридину (МТР)): (k) (де TMS представляє триметилсилільну групу, і Me представляє метильну групу.) розробили спосіб одержання СРТ і його стійких похідних за допомогою раціонального одержання трициклічного кетону, що відповідає CDE-кільцевій частині СРТ скелету, і встановили загальну методику синтезу аналогів СРТ використовуючи прийнятну комбінацію цих способів без використання природних матеріалів, для завершення винаходу. Тобто, винахід стосується способу одержання 2'-аміно-5'-гідроксипропіофенону, що відповідає АВ-кільцевій частині скелету СРТ згідно з послідовністю; Baughman, Τ. Α.; Sih, J. С; Gu, R. L J. Org. Chem. 1997, 62, 6588-6597.) які є синтетичними методиками відомими на сьогоднішній день. Крім того, оскільки Сполука (ν) є біпродуктом, що виникає в процесі синтезу 3-(2-бутенілоксиметил)-4-йод-2метокси-6-триметилсилілпіридину (Сполука (m)), в згаданій вище синтетичній методиці Сполука (І) є описаною нижче. Зокрема, винахід стосується способу одержання 2'-аміно-5'-гідроксипропіофенону для синтезу аналогів камптотецину, де з Сполуки (а): (a) одержують Сполуку (b): (b) і з Сполуки (b) одержують Сполуку (с): (c) і з Сполуки (c) одержують Сполуку (d): (d) і з Сполуки (d) одержують Сполуку (e): (e) (де R означає захисну групу), і стосується загального способу синтезу аналогів СРТ за допомогою прийнятної комбінації процесу для трициклічного кетону, що відповідає CDE-кільцевій частині скелету СРТ, що включає, зокрема, синтез 3-форміл-4-йод-2-метокси-6триметилсилілпіридину (Сполука (І)) з 2-метокси-бтриметилсилілпіридину (Сполука (k)) або Згідроксиметил-4-йод-2-метокси-6триметилсилілпіридину (Сполука (ν)) шляхом удосконалення і оптимізації процесу згідно з синтетичною послідовністю: де R є захисною групою, яка може бути видалена шляхом каталітичного відновлення. Також, винахід стосується описаного вище способу, в якому захисною групою, яка може бути видалена за допомогою каталітичного відновлення, є бензильна група. Крім того, винахід стосується описаного вище способу, де він включає одну або більшу кількість стадій, що вибирають з групи, яка містить (1) стадію одержання Сполуки (b) шляхом змішування Сполуки (а), бензилуючого реагенту і основи, і перемішування згаданої суміші в розчиннику при температурі кипіння; (2) стадію одержання Сполуки (с) шляхом прикапування реагенту Гриньяра до Сполуки (b) в атмосфері інертного газу; (3) стадію одержання Сполуки (d) шляхом змішування Сполуки (с) і окислюючого агенту і 11 76449 12 перемішування суміші; аміно-5'-гідроксипропіофенону, який одержують (4) стадію одержання Сполуки (e) шляхом каописаним вище способом, для одержання аналогів талітичного відновлення Сполуки (d). камптотецину. Крім того, винахід стосується згаданого вище Крім того, винахід стосується способу одерспособу, в якому на стадії (1) розчинником є димежання аналогів камптотецину, що включає реакцію тилформамід. 2'-аміно-5'-гідроксипропїофенону одержаного з Винахід також стосується згаданого вище сповикористанням описаного вище способу і трициксобу, в якому на стадії (2) реагентом Гриньяра є лічного кетону. вінілмагнійбромід. Винаходом також є спосіб одержання трицикКрім того, винахід стосується згаданого вище лічного кетону для синтезу аналогів камптотецину, способу, в якому на стадії (3) окислюючим агентом в якому з Сполуки (k): є реагент Джоунса, діоксид марганцю або ТЕМРО(2,2,6,6-тетраметилпіперидин-1-оксил)-гіпохлорит (k) натрію. Також, винахід стосується сполуки представ(в якій TMS є триметилсилільною групою, і Me леної формулою (с'): є метильною групою), або Сполуки (ν): (c') (v) (в якій Вn є бензильною групою). Крім того, винахід стосується сполуки представленої формулою (d'): (в якій TMS є триметилсилільною групою, і Me є метильною групою), одержують Сполуку (І): (d') (l) (в якій Вn є бензильною групою). Також, винаходом є спосіб одержання 2'аміно-5'-гідроксипропіофенону для синтезу аналогів камптотецину, в якому з Сполуки (а): (в якій TMS є триметилсилільною групою, і Me є метильною групою), і з Сполуки (І) одержують Сполуку (m): (a) (m) (c'') (в якій TMS є триметилсилільною групою, і Me є метильною групою), і з Сполуки (m) одержують Сполуку (n): одержують Сполуку (с"): і з Сполуки (с") одержують Сполуку (d"): (n) (d'') (в якій TMS є триметилсилільною групою, Me є метильною групою, і Et є етильною групою), і з Сполуки (п) одержують Сполуку (о): (e) (o) Крім того, винахід стосується згаданого вище способу, який включає одну або більшу кількість стадій, які вибирають з групи, що містить (1) стадію одержання Сполуки (с") шляхом прикапування реагенту Гриньяра до Сполуки (а) в атмосфері інертного газу; (2) стадію одержання Сполуки (d") шляхом змішування Сполуки (с") і окислюючого агенту і перемішування суміші; і (3) стадію одержання Сполуки (e) шляхом каталітичного відновлення Сполуки (d"). Винахід також стосується згаданого вище способу, в якому на стадії (1) реагентом Гриньяра є вінілмагнійбромід. Крім того, винахід стосується згаданого вище способу, в якому на стадії (2) окислюючим агентом є реагент Джоунса, діоксид марганцю або ТЕМРОгіпохлорит натрію. Винахід також стосується використання 2' (в якій TMS є триметилсилільною групою, Me є метильною групою, і Et є етильною групою), і з Сполуки (о) одержують Сполуку (р): і з Сполуки (d") одержують Сполуку (e): (p) (в якій TMS є триметилсилільною групою, Me є метильною групою, і Et є етильною групою), і з Сполуки (р) одержують Сполуку (q): (q) (в якій Me є метильною групою, і Et є етильною групою), і з Сполуки (q) одержують Сполуку (r): 13 76449 (r) (в якій Me є метильною групою, Et є етильною групою, і Рr є пропільною групою), і з Сполуки (r) одержують Сполуку (s): (s) (в якій Et є етильною групою, і Рr є пропільною групою), і з Сполуки (s) одержують Сполуку (t): (t) (в якій Et є етильною групою, і 'Вu є тбутильною групою), і з Сполуки (t) одержують Сполуку (h): (h) (в якій Et є етильною групою), і де він включає одну або більшу кількість стадій, що вибирають з групи, що містить: (1) стадію одержання Сполуки (І) шляхом змішування Сполуки (k), літіюючого агенту, формілюючого реагенту і йодуючого реагенту; (2) стадію одержання Сполуки (m) шляхом змішування Сполуки (І), кротилового спирту, триетилсилану і кислоти і реакції згаданої суміші без використання розчинника; (3) стадію одержання Сполуки (І) шляхом змішування Сполуки (ν), біпродукту стадії (2), з окислюючим агентом і основою; (4) стадію одержання Сполуки (n) шляхом змішування Сполуки (m), паладієвого каталізатора, основи і каталізатору переносу фаз і кип'ятіння згаданої суміші в розчиннику; (5) стадію одержання Сполуки (о) шляхом змішування Сполуки (n), осмієвого каталізатора, співокислюючого агенту, основи і асиметричного реагенту; (6) стадію одержання Сполуки (р) шляхом змішування Сполуки (о), основи і йоду, і кип'ятіння згаданої суміші в рідкій спиртово-водній суміші; (7) стадію одержання Сполуки (q) шляхом змішування Сполуки (р) і десиліліючого-йодуючого реагенту; (8) стадію одержання Сполуки (r) шляхом змішування Сполуки (q), паладієвого каталізатора і основи і взаємодію згаданої суміші в 1-пропанолі в атмосфері монооксиду вуглецю; (9) стадію одержання Сполуки (s) шляхом змішування Сполуки (r) і деметилюючого реагенту і взаємодію згаданої суміші при кімнатній температурі; (10) стадію одержання Сполуки (t) з Сполуки (s) в присутності т-бутил акрилату і основи. Крім того, винахід стосується згаданого вище способу, в якому на стадії (1) літіюючим агентом є н-бутиллітій. 14 Винахід також стосується згаданого вище способу, в якому на стадії (1) температура реакції є постійною температурою в межах - 30-40°С. Крім того, винахід стосується згаданого вище способу, в якому на стадії (3) окислюючим агентом є ТЕМРО-гіпохлорит натрію. Винахід також стосується згаданого вище способу, в якому на стадії (4) основою є карбонат калію або Ν,Ν-діізопропілетиламін. Крім того, винахід стосується згаданого вище способу, в якому на стадії (4) розчинником є тетрагідрофуран або рідка суміш діізопропіловий етерацетонітрил-вода. Винахід також стосується згаданого вище способу, в якому на стадії (5) осмієвим каталізатором є осмат калію (VI). Крім того, винахід стосується згаданого вище способу, в якому на стадії (6) йод по відношенню до Сполуки (о) використовують у 4 еквівалентному надлишку. Винахід також стосується згаданого вище способу, в якому на стадії (7) десилілюючимйодуючим реагентом є йод-трифторацетат срібла або N-хлорсукцинімід-йодид натрію. Крім того, винахід стосується згаданого вище способу, в якому Сполуку (q) очищають хімічно за допомогою стадій очистки, що включають стадію додавання до реакційної суміші продукту одержаного на стадії одержання Сполуки (q) з Сполуки (р) до водного лужного розчину і перемішування; стадію додавання органічного розчинника і перемішування, з наступним видаленням органічного шару; і стадію підкислення водного шару і екстрагування органічним розчинником. Винахід також стосується згаданого вище способу, в якому водним лужним розчином є водний розчин гідроксиду натрію. Крім того, винахід стосується згаданого вище способу, в якому органічним розчинником є хлороформ. Винахід також стосується згаданого вище способу, в якому Сполуку (q) оптично очищають за допомогою стадій очистки, що включає стадію розчинення продукту реакційної суміші одержаного на стадії одержання Сполуки (q) з Сполуки (р) в високополярному розчиннику, з наступним розшаровуванням низькокополярним розчинником; і стадію фільтрування осаду і подальше концентрування фільтрату до суха при пониженому тиску. Крім того, винахід стосується згаданого вище способу, в якому високополярним розчинником є хлороформ. Винахід також стосується згаданого вище способу, в якому низькополярним розчинником є нгексан. Крім того, винахід стосується згаданого вище способу, в якому на стадії (10) основою є карбонат калію. Винахід також стосується використання трициклічного кетону, одержаного за допомогою описаного вище способу, для одержання аналогів камптотецину. Крім того, винахід стосується способу одержання аналогів камптотецину, де трициклічний кетон, одержаний за допомогою описаного вище способу, реагує з 2'-аміно-6'-гідроксипропіофено 15 76449 16 ном. ли з різних видів природних матеріалів, і природВинахід також стосується згаданого вище споних матеріалів, що містять Сполуку (а). Також мособу, в якому 2'-аміно-5'-гідроксипропіофеноном є же бути використаний комерційно доступний реа2'-аміно-5'-гідроксипропіофенон, одержаний за гент. допомогою описаного вище способу. Далі більш особливо обговорюються згадані Крім того, винахід стосується згаданого вище вище стадії (1)-(4). способу, в якому трициклічний кетон і 2'-аміно-5'На стадії (1), Сполуку (а) розчиняють або сугідроксипропіофенон змішують і згадана суміш спендують в розчиннику, після чого додають бенреагує у інертній атмосфері. зилюючий реагент і основу і завдяки нагріванню Винахід робить можливим раціональне одерпри перемішуванні одержують Сполуку (b). жання 2'-аміно-5'-гідроксипропіофенону, що відпоЯк розчинник можуть бути використані Ν,Νвідає АВ-кільцевій частині СРТ скелету шляхом диметилформамід (ДМФА), диметилсульфоксид, адаптування цих компонентів і робить можливим хлороформ, ацетонітрил, етанол, вода і їм подібні, пропонування загального синтезу СРТ для практиі ДМФА є особливо переважним завдяки його розчного використання. Крім того, як проміжні сполуки чиненню і реактивності. Сполука (с') і Сполука (d') в способі винаходу ще Використовувана кількість ДМФА може в три не були описані для використання в цьому синтезі, або більше разів бути більшою щодо Сполуки (а), і тому вони є корисними новими сполуками. переважно в інтервалі від 3 до 20 разів. Винахід також робить можливим проведення Як бензилюючий реагент може бути викориспрактично асиметричного синтезу сполук(и) шлятаний будь-який придатний реагент. Специфічнихом адаптування цих компонентів, в наслідок чого ми прикладами є бензилхлорид, бензилбромід, сполука(и) має скелет, що відповідає CDEбензилйодид, фенілдіазометан, дибензилкарбонат кільцевій частині (частина трициклічного кетону) в і їм подібні, і, зокрема, переважно може бути викоСРТ скелеті. ристаний бензилхлорид. Для синтезу 2'-аміно-5'-гідроксипропіофенону, Використовувана кількість бензилюючого реащо є АВ-кільцем в СРТ скелеті, спосіб одержання генту може бути прийнятною для використовува2'-аміно-5'-гідроксипропіофенону включає одну ного реагенту, хоча у випадку коли використовуабо більшу кількість стадії приведених далі: ється, наприклад, бензилхлорид, використову(1) стадію синтезу 5-бензилокси-2-нітробенється 1-5 еквівалентів виходячи з Сполуки (а), пезальдегіду (Сполука (b')) з 5-гідрокси-2реважно 1-2 еквіваленти. нітробензальдегіду (Сполука (а)); Як основа може бути використаний будь-який (2) стадію синтезу 1-(5-бензилокси-2-нітропридатний агент. Специфічними прикладами є фенол)-2-пропен-1-олу (Сполука (с')) з Сполуки карбонат калію, карбонат натрію, карбонат цезію, (b'); гідроксид натрію, гідроксид калію і їм подібні, і, (3) стадію синтезу 1-(5-бензилокси-2-нітрозокрема, карбонат калію є особливо переважним. фенол)-2-пропен-1-ону (Сполука (d')) з СполуВикористовувана кількість основи може бути ки (с'); і прийнятною для використовуваного реагенту, хоча (4) стадію синтезу 2'-аміно-5'-гідроксиу випадку коли використовується, наприклад, карпропіофенону (Сполука (e)) з Сполуки (d'). Як тибонат калію, вона використовується у 1-10 еквіваповий шлях синтезу, приводиться наступний синлентному надлишку до Сполуки (а), переважно 1-4 тетичний шлях: еквівалент. Як температура нагрівання є інтервал в межах від 60 до 100°С, переважно 60-80°С. Крім того, час реакції знаходиться в інтервалі від 0,5 до 24 годин, переважно від 1 до 20 годин. На стадії (2), Сполуку (с) одержують шляхом прикапування реагенту Гриньяра до Сполуки (b) в атмосфері інертного газу. Як інертний газ може бути використаний будьякий газ, у нашому випадку це інертний газ, такий як аргон, гелій, неон, криптон, ксенон, радон або (де R є захисною групою, яка може бути видаїм подібні, або газ з низькою реакційною здатністю, лена шляхом каталітичного відновлення). такий як азот, і аргон і азот є особливо переважУ винаході, у випадку коли R є захисною груними з огляду на їх вартість. пою, яка може бути видалена шляхом каталітичноЯк реагент Гриньяра може бути використаний го відновлення, це не повинно бути як обмеження, будь-який придатний реагент. Специфічними приале типовими прикладами є захисні групи типу кладами є вінілмагнійбромід, вінілмагнійхлорид, бензиловий етер, такі як бензил, метоксибензил, вінілмагніййодид і їм подібні, і вінілмагнійбромід є 2,5-диметилбензил або 4-нітробензил, і захисні особливо переважним для використання. групи типу бензилкарбонат, такі як бензилоксикарВикористовувана кількість реагенту Гриньяра бонільна група, хоча бензильна група є більш підможе бути прийнятною для використовуваного ходящою для використання завдяки ціні реагенту. реагенту, хоча у випадку, наприклад, вінілмагнійбКрім того, може бути використана Сполуки (а), роміду, використовується 1-2 еквіваленти виходяяка є вихідним матеріалом, що синтезується за чи з Сполуки (b), переважно 1-1,5 еквіваленти. У допомогою відомого способу, що хімічно перетвовипадку коли реагент Гриньяра прикапують до рюється з подібної сполуки, що виділяли і очищарозчину Сполуки (b) або навпаки розчин Сполуки 17 76449 18 (b) прикапують до реагенту Гриньяра, можливий чинності. синтез Сполуки (с), хоча для того щоб зменшити Використовувана кількість розчинника станоутворення біпродукту (тут далі описана як Сполука вить 5-50 частин, переважно 10-20 частин. (f)) Крім того, час реакції становить 0,1-24 годин і, зокрема, переважно 1-3 години. (f) Крім того, замість синтезу Сполуки (e) через описані вище стадії (1)-(4) її можна одержати на(в якій R є захисною групою яка може бути виступним чином: далена каталітичним відновленням), переважно з Сполуки (а): реагент Гриньяра прикапують до розчину Сполуки (b). (a) Використовувана кількість розчинника в реакції, наприклад, тетрагідрофурану (тут і далі згадуодержують Сполуку (с"): ється як ТГФ) може бути кількість 10-100 частин, і для зменшення утворення спирту переважною є (c") кількість 50-100 частин. Також, температура реакції переважно не пеі з Сполуки (с") одержують Сполуку (d"): ревищує 10°С, і для зменшення продукування спирту переважною є температура –78 - -40°С. (d") Крім того, час реакції становить 0,1-3 годин, і, зокрема, переважно, він становить 0,5-1 годин. і з Сполуки (d") одержують Сполуку (e): На стадії (3), Сполуку (d) можна одержати шляхом змішування Сполуки (с) з окислюючим (e) агентом і перемішування суміші. Як окислюючий агент може бути використаний В цій синтетичній методиці, Сполуку (с") можна будь-який придатний агент. Прикладами таких одержати шляхом прикапування реагенту Гриньяокислюючих агентів є, наприклад, діоксид марганра до Сполуки (а) у інертній атмосфері. Крім того, цю, перйодат Десса-Мартіна, реагент Джоунса Сполуку (d") можна одержати шляхом змішування (Nа2Сr2O7/Н2SO4), РСС, PDC, ДМСО/оксалілСполуки (с") і окислюючого агенту і перемішування хлорид/триетиламін (окислення Сверна), ТЕМРОсуміші, і Сполуку (e) можна одержати шляхом кагіпохлорит натрію і їм подібні, і, зокрема, переважталітичного відновлення Сполуки (d"). Тут, реагенно можуть бути використані діоксид марганцю, том Гриньяра і окислюючим агентом, який може перйодат Десса-Мартіна, реагент Джоунса і ТЕМбути використаний є тими ж самими як і ті що були РО-гіпохлорит натрію. згадані на стадіях (2) і (3). В цій синтетичній метоЩодо цих оксилюючих агентів, агент одержудиці, оскільки не використовується захисної групи, ють безпосередньо перед використанням, і у висинтез АВ-кільцевої частини може бути проведепадку, наприклад, діоксиду марганцю, агент одерний простіше. жують перед використанням з перманганату калію Крім того, аналоги камптотецину можна одері сульфату марганцю. жати за допомогою реакції Сполуки (є) одержаної Використовувана кількість окислюючого агенту на стадії (4) або синтетичної методики тільки що може бути прийнятною для використовуваного описаної вище і трициклічного кетону, хоча може реагенту, хоча у випадку, наприклад, діоксиду мабути використаний трициклічний кетон подібний, рганцю, використовують 2 - 50-кратний надлишок наприклад, Сполуці (h): щодо Сполуки (с), переважно 4-10-кратний надлишок. Як розчинник може бути доцільно використо(h) вувати, наприклад, хлороформ, метиленхлорид, етилацетат, бензол, толуол і їм подібні, і, зокрема, Стосовно синтезу CDE-кільцевої частини (часпереважно хлороформ і метиленхлорид. тина трициклічного кетону) СРТ скелету, одержанВикористовувана кількість розчинника станоня трициклічного кетону проводять за допомогою вить 5-50 частин, переважно 10-20 частин. наступної методику синтезу. Крім того, час реакції становить 1-48 годин і, зокрема, переважно 1-18 годин. На стадії (4), Сполуку (e) можна одержати шляхом каталітичного відновлення Сполуки (d). Як каталізатор для відновлення доцільно використовувати паладій-вугілля, гідроксид паладіювугілля, родій-оксид алюмінію і їм подібні, і, зокрема, переважними є паладій-вугілля і гідроксид паладію-вугілля. Використовувана кількість каталізатору для відновлення становить 0,01-0,5 еквівалентів стосовно Сполуки (d), переважно 0,05-0,2 еквіваленти. Як розчинник може бути доцільно використо(в якій TMS є триметилсилільною групою, Me є вувати будь-який прийнятний розчинник, хоча етиметильною групою, Et є етильною групою, Рr є лацетат є переважним завдяки його добрій роз 19 76449 20 пропільною групою, і 'Вu є т-бутильною групою.) лука (r)) з Сполуки (q), використовують ацетат паЯк вихідна Сполука (k) у описаній вище метоладію як паладієвий каталізатор; диці синтезу, може бути використана сполука, що (11) на стадії синтезу пропіл (S)-4-етилсинтезується за допомогою описаного вище мето3,417,8-тетрагідро-4-гідрокси-3,8-діоксо-1Нду Куррана (Josien, H.; Ко, S. В.; Bom, D.: Curran, пірано[3,4-с]піридин-6-карбоксилату (далі познаD. P. Chem. Eur. J. 1998, 4 67-83), що хімічно перечена як Сполука (s)) з Сполуки (r), реакцію провотворюється у подібну сполуку, яку виділяли і очидять при кімнатній температурі; щали з різних видів природних матеріалів або (12) на стадії синтезу 1,1-диметилетил (3)-4природного матеріалу, що містить Сполуку (k). етил-3,4,8,10-тетрагідро-4,6-дигідрокси-3,10Переважна методика синтезу для синтезу діоксо-1Н-пірано[3,4-f]індолідин-7-карбоксилату трициклічного кетону за допомогою описаної вище (далі позначена як Сполука (t)) з Сполуки (s), пропослідовності включає одну або більшу кількість водять конденсування Мікаеля використовуючи стадій з 12 стадій приведених далі; карбонат калію. (1) на стадії синтезу 4-йод-2-метокси-6Крім того, (13) на стадії одержують SN-38 з триметилсиліл-3-піридинкарбальдегіду (тут і далі (S)-4-етил-7,8-дигідро-4-гідрокси-1Н-іірано[3,4позначена як Сполука (І)) з 2-метокси-6f]індолідин-3,6,10(4'Н)-триону (далі позначена як триметилсилілпіридину (далі позначена як СполуСполука (n)) і Сполуки (e), SN-38 можна одержати ка (k)), н-бутиллітій використовують як основу і проводячи реакцію в атмосфері інертного газу реакцію проводять при постійній температурі одержуючи SN-38. -30 - -40°C; Надалі, згадані вище 13 стадій розкривають (2) на стадії синтезу 3-(2-бутенілоксиметил)-4більш детально. йод-2-метокси-6-триметилсилілпіридину (далі позНа стадії (1), Сполуку (k) розчиняють у розначена як Сполука (m)) з Сполуки (І), розчинник в чиннику, після чого додають літіюючий, Ізормілюреакції не використовується; ючий і йодуючий реагенти і перемішують одержу(3) на стадії синтезу Сполуки (І) з 3ючи Сполуку (І). гідроксиметил-4-йод-2-метокси-6-триметилсилілЯк розчинник може бути використаний тетрагіпіридину (далі позначена як Сполука (ν)), ТЕМРОдрофуран (ТГФ), діетиловий етер, ексан, гептан і гіпохлорит натрію використовують як окислюючий їм подібні, і ТГФ є особливо переважним завдяки агент; добрій розчинюваності і інертності. (4) на стадії синтезу 4-етил-8-метокси-6Як літіюючий реагент може бути використаний триметилсиліл-1Н-пірано[3,4-с]піридину (далі позбудь-який доцільний реагент, якщо ін є традиційно начена як Сполука (n)) з Сполуки (m), як розчинник використовуваним. Специфічними прикладами є нреакційної суміші використовують суміш діізопробутиллітій, в-бутиллітій, т-бутиллітій, діізопропілапілового етеру, ацетонітрилу і води, і Ν,Νмід літію (LDA), біс(триметилсиліл)амід літію діізопропілетиламін використовують як основу; (LiHMDS) і їм подібні, і н-бутиллітій є особливо (5) на стадії синтезу (S)-4-етил-3,4-дигідро-3,4переважним для використання завдяки добрій обдигідрокси-8-метокси-6-триметилсиліл-1Нробці і реакційності. пірано[3,4-с]піридину (далі позначена як Сполука Використовувана кількість літіюючого реагенту (о)) з Сполуки (n), осмат калію (VI) використовують може бути підходящою відповідно до реагенту, як осмієвий каталізатор; хоча у випадку використання, наприклад, н(6) на стадії синтезу (S)-4-етил-3,4-дигідро-4бутиллітію, використовують 2-10 еквівалентів стогідрокси-8-метокси-6-триметилсиліл-3-оксо-1Нсовно Сполуки (k), переважно 2-5 еквівалентів. пірано[3,4-с]піридину (далі позначена як Сполука Специфічними прикладами формілюючого ре(р)) з Сполуки (о), реакційну суміш кип'ятять з йоагенту є Ν-форміл-Ν,Ν',Ν'-триметилетилендіамін, дом (4 еквіваленти); диметилформамід (ДМФА) і їм подібні, і Ν-форміл(7) на стадії синтезу (S)-4-етил-3,4-дигідро-4Ν,Ν',Ν'-триметилетилендіамін доцільно використогідрокси-6-йод-8-метокси-3-оксо-1Н-пірано[3,4вувати враховуючи наступне йодування. с]піридину (далі позначена як Сполука (q)) з СпоВикористовувана кількість формілюючого реалуки (р), N-хлорсукцинімід-йодид натрію викорисгенту, наприклад, у випадку використання Nтовують в оцтовій кислоті; форміл-N,N',N'-триметилетилендіаміну використо(8) на стадії хімічної очистки Сполуки (q), сувується 1-10 еквівалентів стосовно Сполуки (k), міш розводять розчином лугу, таким як водний переважно 1-3 еквіваленти. розчин гідроксиду натрію роблячи його лужним, Як йодуючий реагент можуть бути використані промивають органічним розчинником, таким як йод, N-йодсукцинімід (NIS) і їм подібні, і йод є осохлороформ, і потім водний шар після підкислюбливо переважним з огляду на вартість і реакційну вання екстрагують органічним розчинником, таким здатність. як хлороформ; Використовувана кількість йодуючого реагенту (9) на стадії оптичної очистки Сполуки (q), становить 1-10 еквівалентів стосовно Сполуки (k), Сполуку (q) розчиняють в високополярному розпереважно 1-5 еквіваленти. чиннику, такому як хлороформ і розшаровують Температура реакції знаходиться в інтервалі низькополярним розчинником, таким як н-гексан від 0 до -78°С, переважно використовується посодержуючи осад, який видаляють фільтруванням, тійна температура -30 - -40°С. після чого концентрують фільтрат; На стадії (2), Сполуку (І) додають до кротило(10) на стадії синтезу пропіл (3)-4-етил-3,4вого спирту, триетилсилану і кислоти і перемішудигідро-4-гідрокси-8-метокси-3-оксо-1Н-пірано[3,4ють без розчинника одержуючи Сполуку (m). с]піридин-6-карбоксилату (далі позначена як СпоВикористовувана кількість кротилового спирту 21 76449 22 становить 1-10 еквівалентів стосовно Сполуки (k), товувати ацетат паладію, тетрапереважно 2-5 еквівалентів. кіс(трифенілфосфін)паладій, дихлорВикористовувана кількість триетилсилану стабіс(трифенілфосфін)паладій, хлорид паладію і їм новить 1-10 еквівалентів стосовно Сполуки (k), подібні, і ацетат паладію є особливо переважним переважно 1-4 еквіваленти. завдяки його реакційній здатності. Як кислота можуть бути використані трифтоВикористовувана кількість паладієвого каталіроцтова кислота (TFA), сірчана кислота, метансузатора становить 0,01-1 еквівалент стосовно Спольфонова кислота, хлорводнева кислота і їм поділуки (m), переважно 0,05-0,2 еквіваленти. бні, і TFA є особливо переважною завдяки Як основа може бути використана будь-яка реакційній здатності. доцільна основа, якщо вона є загальноприйнятною Використовувана кількість кислоти, наприклад, до використання. Прикладами таких основ є, нау випадку TFA становить 1-15 еквівалентів стосовприклад, карбонат натрію, карбонату калію, карбоно Сполуки (І), переважно 5-10 еквівалентів. нат кальцію, карбонат цезію, триетиламін (TEA), На стадії (3), Сполуку (І) можна одержати Ν,Ν-діізопропілетиламін (DIPEA), гідроксид натрію, шляхом розчинення Сполуки (ν), біпродукту стадії гідроксид калію і їм подібні, і карбонат калію і (2), в розчиннику, з наступним додаванням окисDIPEA є особливо переважними. люючого агенту і основи, і перемішування. Використовувана кількість основи, наприклад, Як розчинник може бути використаний будьу випадку DIPEA становить 1-20 еквівалентів стоякий доцільний розчинник, якщо він є загальнопсовно Сполуки (m), переважно 5-10 еквівалентів. рийнятним для використання. Прикладами таких Як каталізатору переносу фаз може бути вирозчинників є дихлорметан, хлороформ, ацетоніткористаний будь-який доцільний каталізатор, якщо рил, толуол, н-гексан і їм подібні, і толуол і нвін є четвертинною амонієвою сіллю або краунгексан є особливо переважними завдяки їх реакетером, які зазвичай використовуються, і бромід ційній здатності. тетрабутиламонію є особливо переважним. Прикладами окислюючих агентів є діоксид маВикористовувана кількість каталізатору перерганцю, перйодинан Десса-Мартіна, реагент Джоносу фаз, наприклад, у випадку броміду тетрабуунса (Nа2Сr2O7/Н2SО4), РСС, PDC, ДМСОтиламонію становить 0,1-3 еквіваленти стосовно оксалілхлорид-триетиламін (окислення Сверна), Сполуки (m), переважно 0,5-1,5 еквіваленти. ТЕМРО-гіпохлорит і їм подібні, і, зокрема, ТЕМРОКрім того, час реакції у випадку використання гіпохлорит є переважним, більш переважним є ТГФ знаходиться в інтервалі від 1 до 20 годин, ТЕМРО-гіпохлорит натрію. переважно 4-10 годин. У випадку використання Використовувана кількість окислюючого агенрідкої суміші ацетонітрил-ІРЕ-вода час реакції знату, наприклад, у випадку ТЕМРО-гіпохлорит находиться в інтервалі від 0,5 до 10 годин, переважтрію, становить 0,001-0,1 еквівалентів TEMPO стоно 1-5 годин. совно Сполуки (ν), переважно 0,005-0,02 На стадії (5), Сполуку (n) розчиняють у рідкій еквіваленти. Крім того, використовується 1-5 еквісуміші спирт-вода, додають осмієвий каталізатор, валентів гіпохлориту натрію, переважно 1-2 еквіспівокислюючий агент, асиметричний каталізатор, валенти. основу і метансульфонамід і перемішують одерЯк основа може бути використана будь-яка жуючи Сполуку (о). доцільна основа, якщо вона є загальноприйнятною Прикладами спиртів є метанол, етанол, 1до використання. Прикладами таких основ є бікарпропанол, ізопропанол (ІРА), 1-бутанол, 2бонат натрію, карбонат натрію, карбонат калію, бутанол, т-бутиловий спирт і їм подібні, і ткарбонат кальцію, гідроксид натрію, гідроксид кабутиловий спирт є особливо переважним завдяки льцію, триетиламін і їм подібні, де бікарбонат найого реакційній здатності. трію є особливо переважним. Як осмієвий каталізатор доцільно використоВикористовувана кількість основи, наприклад, вувати тетраоксид осмію, осмат калію (VI) і їм поу випадку бікарбонату натрію, становить 1-10 еквідібні, і осмат калію (VI) є особливо переважним валентів стосовно Сполуки (ν), переважно 2-4 екзавдяки легкості використання. віваленти. Використовувана кількість осмієвого каталізаТемпература реакції знаходиться в інтервалі тора становить 0,001-0,1 еквівалентів стосовно від -10 до 30°С, особливо переважно -10 - 10°С, Сполуки (n), переважно 0,002-0,01 еквіваленти. для запобігання сторонніх реакцій. Як співокислюючий агент доцільно використоКрім того, час реакції знаходиться в інтервалі вувати гексаціаноферрат (III) калію, Nвід 0,5 до 10 годин, переважно 0,5-5 годин. метилморфолін N-оксид (NMO) і їм подібні, і гекНа стадії (4), Сполуку (m) розчиняють у розсаціаноферрат (III) калію є особливо переважним чиннику, додають паладієвий каталізатор, основу і завдяки його реакційній здатності. каталізатор переносу фаз і нагрівають із зворотнім Використовувана кількість співокислюючого холодильником одержуючи Сполуку (n). агенту, наприклад, у випадку гексаціаноферрату Як розчинник можуть бути використані ацето(III) калію, становить 1-10 еквівалентів стосовно нітрил, тетрагідрофуран (ТГФ), діізопропіловий Сполуки (n), переважно 2-5 еквівалентів. етер (ІРЕ), діетиловий етер, толуол, вода і їм подіПрикладами асиметричних каталізаторів є бні, і ацетонітрил, ТГФ, ІРЕ і вода є особливо пе(DHQD)2PYR, (DHQD)2PHAL, (DHQD)2AQN і їм пореважними завдяки їх реакційній здатності, більш дібні, і (DHQD)2PYR є особливо переважним запереважними є ТГФ або рідка суміш ацетонітрилвдяки оптичному виходу. ІРЕ-вода. Використовувана кількість асиметричного каЯк паладієвий каталізатор доцільно використалізатору, наприклад, у випадку (DHQD)2PYR, 23 76449 24 становить 0,005-0,1 еквіваленти стосовно Сполуки На стадії (8), Сполуку (q) додають до основно(n), переважно 0,01-0,05 еквіваленти. го розчинника, наприклад, такого як водний 0,2N Як основу можна використати карбонат нагідроксид натрію, і перемішують одержуючи розктрію, карбонат калію, карбонат кальцію, карбонат риття лактонового циклу сполуки (Сполука (u)): цезію, гідроксид натрію, гідроксид калію і їм подібні, і карбонат калію є особливо переважним завдя(u) ки його реакційній здатності. Використовувана кількість основи, наприклад, (в якій Me є метильною групою, і Et є етильною у випадку карбонату калію, становить 1-20 еквівагрупою.), який розчиняють у водному основному лентів стосовно Сполуки (n), переважно 4-10 еквірозчині. Після промивання розчину органічним валенти. розчинником, виділяють нейтрально-основну реВикористовувана кількість метансульфонаміду човину в органічний шар. Органічний шар відокрестановить 0,1-5 еквівалентів стосовно Сполуки (n), млюють, після чого підкислюють водний шар киспереважно 0,5-2 еквіваленти. лотою і екстрагують органічним розчинником Температура реакції знаходиться в інтервалі одержуючи Сполуку (q) з добрим виходом. від -10 до 30°С, переважно -10 - 10°С. На стадії Основність розчинника знаходиться в інтерва(6), Сполуку (о) розчиняють у розчиннику, додають лі від 0,01-5N, переважно 0,1-1N, більш переважно основу і йод і кип'ятять одержуючи Сполуку (р). 0,2-0,5N. Прикладами розчинника є метанол, етанол, 1Прикладами використовуваних основ є гідрокпропанол, ізопропанол (ІРА), вода і їм подібні, і сид калію, гідроксид кальцію, гідроксид натрію, рідка суміш метанол-вода є особливо переважною карбонат калію, карбонат натрію і їм подібні, і гідзавдяки її реакційній здатності. роксид натрію є особливо переважним. Як основа може бути використана будь-яка Як органічний розчинник може бути викорисдоцільна основа, якщо вона є загальноприйнятною таний будь-який доцільний розчинник, якщо він є до використання. Прикладами таких основ є карзагальноприйнятним до використання. Прикладабонат натрію, карбонат калію, карбонат кальцію, ми такого розчинника є дихлорметан, хлороформ, карбонат цезію, гідроксид натрію, гідроксид калію і етилацетат, толуол, діетиловий етер, діізопропілоїм подібні, і карбонат кальцію є особливо перевавий етер і їм подібні, і, зокрема, і дихлорметан і жним. хлороформ є особливо переважним. Використовувана кількість основи, наприклад, Прикладами використовуваних кислот є хлору випадку карбонату кальцію, становить 1 -10 еквіводнева кислота, сірчана кислота, азотна кислота, валентів стосовно Сполуки (о), переважно 2-5 екоцтова кислота, фосфорна кислота, трифтороцтовівалентів. ва кислота і їм подібні, і хлорводнева кислота є Використовувана кількість йоду становить 1-10 особливо переважною. еквівалентів стосовно Сполуки (о), переважно 3-5 На стадії (9), Сполуку (q) розчиняють в висоеквівалентів. Крім того, час реакції знаходиться в кополярному розчиннику, і розшаровують низькоінтервалі від 0,5 до 20 годин, переважно 1-5 годин. полярним розчинником одержуючи кристали які На стадії (7), Сполуку (р) розчиняють у розфільтрують. Фільтрат концентрують при понижечиннику і обробляють йод-трифторацетат срібла ному тиску до суха. Одержані кристали є рацеміч(далі позначений як l2-CF3COOAg) або Nними, і більш оптично чисту Сполуку (q) одержухлорсукцинімід-йодид натрію (далі позначений як ють як залишок. NCS-Nal) одержуючи Сполуку (q). Як високополярний розчинник може бути виЯк розчинник, у випадку l2-CF3COOAg доцількористаний хлороформ, дихлорметан, етилацетат, но використовувати дихлорметан, тетрахлорид метанол, етанол, пропанол і їм подібні, і, зокрема, вуглецю, хлороформ і їм подібні, і, зокрема, диххлороформ є переважним. Використовувана кільлорметан є переважним. Крім того, у випадку NCSкість високополярного розчиннику, наприклад, у Nal можна використовувати оцтову кислоту, ацевипадку хлороформу, становить 1-10мл, переважтонітрил і їм подібні, і оцтова кислота є особливо но 3-6мл, до 1г Сполуки (q). переважною завдяки її реакційній здатності. Прикладами низькополярного розчинника є нВикористовувана кількість l2-CF3COOAg стагексан, н-гептан, діетиловий етер і їм подібні, і нновить 1-10 еквівалентів І2 стосовно Сполуки (р), гексан є особливо переважним. переважно 2-4 еквіваленти. Крім того, використоСпіввідношення високополярний розчинвується 1-10 еквівалентів CF3COOAg, переважно ник:низькополярний розчинник, наприклад, у ви2-4 еквіваленти. падку хлороформ:н-гексан, знаходиться в інтерваВикористовувана кількість NCS-Nal становить лі від 10:1-1:20, переважно 2:1-1:2. 1-20 еквівалентів NCS стосовно Сполуки (р), переТемпература кристалізації не є вищою за кімважно 5-8 еквівалентів. Крім того, використовуєтьнатну температуру, переважно не вище ніж 5°С. ся 1-20 еквівалентів Nal стосовно Сполуки (р), пеНа стадії (10), Сполуку (q) розчиняють в 1реважно 5-8 еквівалентів. пропанолі, додають паладієвий каталізатор і осноТемпература реакції у випадку використання ву, і проводять взаємодію у атмосфері монооксиду l2-CF3COOAg становить 10-60°С, переважно 20вуглецю одержуючи Сполуку (r). 40°С. Крім того, у випадку використання NCS-Nal Як паладієвий каталізатор доцільно викорисвона становить 20°С - температура кипіння, перетовувати, ацетат паладію, тетракіс(трифенілважно 50-80°С. фосфін)паладій, дихлорбіс(трифеніл-фосфін)паКрім того, час реакції знаходиться в інтервалі ладій, хлорид паладію і їм подібні, і ацетат палавід 5 до 48 годин, переважно 15-24 години. 25 76449 26 дію є особливо переважним завдяки його реакційльше ніж 24 годин для того щоб уникнути розкланій здатності. дання одержаної Сполуки (t). Використовувана кількість паладієвого каталіНа стадії (13), Сполуку (h) і Сполуку (e) розчизатора становить 0,005-0,5 еквівалентів стосовно няють у розчиннику, додають кислоту і нагрівають Сполуки (q), переважно 0,01-0,1 еквіваленти. в атмосфері інертного газу і перемішують одержуЯк основа може бути використана будь-яка ючи SN-38. доцільна основа, якщо вона є загальноприйнятною Як розчинник доцільно використовувати толудо використання. Прикладами таких основ є, наол, оцтову кислоту і їм подібні, і, зокрема, рідка приклад, карбонат натрію, карбонат калію, карбосуміш толуол-оцтова кислота є переважною. нат кальцію, карбонат цезію, триетиламін (TEA), Як інертний газ може бути використаний будьΝ,Ν-діізопропілетиламін (DIPEA), гідроксид натрію, який інертний газ, в нашому випадку це є інертний гідроксид калію і їм подібні, і карбонат калію є осогаз такий як аргон, гелій, неон, криптон, ксенон, бливо переважним. радон або їм подібні, або газ з низькою чутливістю, Використовувана кількість основи, наприклад, такий як азот, і аргон і азот є особливо переважу випадку карбонату калію, становить 1-20 еквіваними завдяки їх вартості. лентів стосовно Сполуки (q), переважно 4-10 еквіЯк кислота може бути використана толуолсувалентів. льфонова кислота, метансульфонова кислота, Температура реакції знаходиться в інтервалі трифтороцтова кислота і їм подібні, і толуолсульвід 20°С до температури кипіння, переважно не фонова кислота є особливо переважною завдяки її вище ніж 50°С до температури кипіння. реакційній здатності. На стадії (11), Сполуку (r) розчиняють у розВикористовувана кількість кислоти, наприклад, чиннику, додають деметилюючий реагент, і провоу випадку толуолсульфонової кислоти, становить дять реакцію при кімнатній температурі одержую1-100мг стосовно 1г Сполуки (h), переважно чи Сполуку (s). 10-30мг. Як розчинник може бути використаний ацетоВикористовувана кількість Сполуку (e) станонітрил, хлороформ, дихлорметан, толуол і їм подівить 1-3 еквіваленти стосовно Сполуки (h), перебні, і, зокрема, ацетонітрил є переважним. важно 1-1,5 еквівалентів. Прикладами деметилюючих реагентів є хлортТемпература реакції знаходиться в інтервалі риметилсилан-йодид натрію, йодтриметилсилан, від 50°С до температури кипіння, переважно 80°С йодводнева кислота, бромводнева кислота і їм - температура кипіння. подібні, і хлортриметилсилан-йодид натрію є осоНадалі, винахід ілюструється більш детально бливо переважним. наступними прикладами, але винахід не обмежуВикористовувана кількість деметилюючого реється ними. агенту, наприклад, у випадку хлортриметилсиланПриклад 1 Синтез Сполуки (b') йодид натрію, становить 1-10 еквівалентів стосовно Сполуки (r), переважно 2-5 еквівалентів. На стадії (12), Сполуку (s) розчиняють у розчиннику, додають основу і перемішують в атмосде Вn є бензильною групою. фері інертного газу. До одержаної суміші по крапСполуку (а) (38,5г, 0,230моль) розчиняли в лям додають т-бутил акрилат, і перемішують в 116мл ДМФА або ацетону. До розчину, що переатмосфері інертного газу одержуючи Сполуку (t). мішується, Сполуки (а) при кімнатній температурі в Як розчинник доцільно використати диметилатмосфері аргону додавали карбонат калію (33,4г, сульфоксид (ДМСО), Ν,Ν-диметилформамід 0,242моль, 2,1екв.) і 27,8мл (0,242моль, 1,05екв.) (ДМФА) і їм подібні, і ДМСО є особливо переважабо 59,95мл (0,461моль, 2екв.) бензилхлориду. ним завдяки його реакційній здатності. Після додавання, суміш нагрівали при 60°С і інтеЯк основу можна використати карбонат калію, нсивно перемішували протягом 20 годин періодичкарбонат натрію, гідроксид натрію, гідроксид калію но перевіряючи наявність Сполуки (а). Після того, і їм подібні, і, зокрема, карбонат калію є переяк Сполука (а) не детектувалась, суміш фільтруважним. вали з відсмоктуванням. Використовувана кількість основи, наприклад, Тверду речовину промивали тим же самим роу випадку карбонату калію, становить 1-20 еквівазчинником, що і використовувався для реакції. лентів стосовно Сполуки (s), переважно 2-5 еквіФільтрат і промивні розчини об'єднували і розчинвалентів. ник випарювали при пониженому тиску. До залишЯк інертний газ може бути використаний будьку додавали воду (300мл). Суміш перемішували і який інертний газ, в нашому випадку це є інертний осади фільтрували з відсмоктуванням і сушили на газ такий як аргон, гелій, неон, криптон, ксенон, повітрі. Після висушування на повітрі, відфільтрорадон або їм подібні, або газ з низькою чутливістю, ваний матеріал розчиняли в 170мл етилацетату. такий як азот, і аргон і азот є особливо переважЦей розчин додавали до 1л гексану при перемішуними завдяки їх вартості. ванні. Осаджену тверду речовину фільтрували з Використовувана кількість т-бутил акрилату відсмоктуванням, промивали 300мл суміші етиластановить 1-20 еквівалентів стосовно Сполуки (s), цетат і гексан (1:10) і сушили при пониженому переважно 8-12 еквівалентів. тиску. Температура реакції знаходиться в інтервалі Експеримент (Екс.) 1 і 2, в яких відрізняються від 20 до 80°С, переважно 40-60°С. кількості бензилхлориду і в Екс. 3, ацетон викорисКрім того, час реакції знаходиться в інтервалі товували як розчинник реакції. від 5 до 48 годин, і, зокрема, він становить не бі 27 76449 28 Таблиця 1 - менше ніж межа визначення Як показано в Таблиці 1, 20 годин необхідно для проходження реакції з виходом 94%, коли використовується 1,05екв. бензилхлориду (Екс.1). Коли використовується 2,00екв. бензилхлориду (Екс.2), реакція завершується за 1 годину з виходом 94%. Трикратний надлишок ДМФА був необхідним для проведення реакції, інакше утруднюється перемішування внаслідок висадження твердої речовини під час реакції. Коли в якості розчинника реакційної суміші використовувався ацетон, реакція не йшла навіть при нагріванні із зворотнім холодильником протягом 18 годин. Умови ВЕРХ Колонка: Inertsil ODS-2, 5μΜ, 4,6мм ID 250мм (виготовлено GL science) Температура: постійна температура приблизно 40°С Рухома фаза: суміш вод: ацетонітрил (1:1) Швидкість потоку: 1мл/хв. Детектування: 220нм Приклад 2 Синтез Сполуки (с') (1) в яких Вn є бензильною групою. Сполуку (b') (1,0г, 3,89ммоль) розчиняли в 20мл ТГФ. По краплях до охолодженого льодом розчину додавали вінілмагнійбромід (1,0Μ ТГФ розчин, 5,84мл, 5,84ммоль, 1,5екв.), перемішували розчин Сполуки (b') в атмосфері аргону протягом 15 хвилин. Під час додавання, внутрішню температуру підтримували в межах від 3 до 10°С. Після перемішування протягом 1 години, реакційний розчин додавали до насиченого водного розчину хлориду амонію (20мл) при перемішуванні, і потім додавали до розчину 20мл етилацетату і 4мл гексану, і одержаний органічний шар послідовно промивали 20мл води і водного насиченого розчину хлориду натрію, і сушили над 3г сульфату натрію. Розчинник випарювали при пониженому тиску одержуючи Продукт реакції А. ТГФ розчин (20мл) Сполуки (b') одержаної, як згадано вище, додавали по краплям до охолодженого льодом розчину вінілмагнійброміду (1,0Μ ТГФ розчин, 5,84мл) в атмосфері аргону протягом 15 хвилин. Під час додавання, внутрішню температуру підтримували в межах від 3 до 10°С. Після перемішування протягом 1 години, реакційний розчин додавали до насиченого водного розчину хлориду амонію (20мл) при перемішуванні і потім додавали до розчину 20мл етилацетату і 4мл гексану, і органічний шар випарювали і промивали 20мл води і насиченим водним розчином хлориду натрію і сушили над 3г сульфатом натрію. Розчинник випарювали при пониженому тиску одержуючи Продукт реакції В. Продукт реакції А і В очищали за допомогою колонкової хроматографії на силікагелі (етилацетат : гексан =1:20) одержуючи Екс. 4 з Продукту реакції А і Екс.5 з Продукту реакції В, відповідно. Таблиця 2 Як показано в Таблиці 2, коли реагент Гриньяра додавали до розчину Сполуки (b'), вихід продукту збільшується на 57% і утворення Сполуки (f), біпродукт, пригнічується. Умови ВЕРХ; такі ж як і в Прикладі 1. Приклад 3 Синтез Сполуки (с') (2) Сполуку (b') (1,0г, 3,89ммоль) розчиняли в 10 100мл ТГФ. До розчину, що перемішується, Сполуки (b') в атмосфері аргону протягом 15 хвилин по краплям додавали вінілмагнійбромід (1,0Μ в ТГФ, 5,84мл, 5,84ммоль, 1,5екв.). Після перемішування протягом 1 години, реакційний розчин при перемішуванні додавали до насиченого водного розчину хлориду амонію (20мл) і потім до розчину 29 76449 30 20мл етилацетату і 4мл гексану, і органічний шар (Екс. 4 і 5). Екс.6 представляє результати реакції випарювали і послідовно промивали 20мл води і при 20°С використовуючи 20-кратну кількість рознасиченим водним розчином хлориду натрію, і чинника. Екс.7-9 представляють результати реаксушили над 3г сульфату натрію. Розчинник випації при 3°С використовуючи 10-кратну, 40-кратну, рювали при пониженому тиску. Залишок очищали 100-кратну кількість розчинника, відповідно. Ретим же самим способом як описано в Прикладі 2 зультати реакцій зведені в Таблиці 3. Таблиця 3 Як показано в Таблиці 3, коли реакцію проводять при 10°С або нижче, більш переважно 5°С або нижче, утворення Сполуки (f) пригнічується і вихід Сполуки (С) збільшується на 15% або більше. Коли використовується 100-кратна кількість розчинника (Екс.9), утворення Сполуки (f) пригнічується, а вихід Сполуки (с') збільшується на 6%. Умови ВЕРХ; такі ж як і в Прикладі 1. Приклад 4 Синтез Сполуки (d') (1) в яких Вn є бензильною групою. (1) Одержання діоксиду марганцю: Водний розчин пентагідрату сульфату марганцю (122г/150мл, 0,506моль) і 117мл 40% гідроксиду натрію додавали при кімнатній температурі і перемішуванні до водного розчину перманганату калію (96,0г/600мл, 0,607моль). Після перемішування протягом 18 годин, тверду речовину фільтрували з відсмоктуванням і промивали водою. Одержану тверду речовину сушили на повітрі одержуючи 91,2г діоксиду марганцю. (2) Синтез Сполуки (d') Сполуку (с') (2,00г, 7,02ммоль) розчиняли в 20мл хлороформу, дихлорметану або етилацетату. До розчину Сполуки (с'), що інтенсивно перемішується, при 25°С в атмосфері аргону додавали діоксид марганцю 8,00г (4-кратна кількість, 92,0ммоль, 13екв.) одержаний за способом описаним вище. Суміш інтенсивно перемішували протягом 15 годин. Після того як вихідний матеріал більше не детектувався, суміш фільтрували з відсмоктуванням. Одержану тверду речовину промивали 20мл хлороформу. Фільтрат і промивні розчини об'єднували і розчинник випарювали при пониженому тиску. Одержували Екс.10-12. Таблиця 4 - менше ніж межа детектування Як показано в Таблиці 4, коли як розчинник реакції використовувався хлороформ або дихлорметан, Сполука (d') утворювалась з добрими вихо дами. Зокрема, на одну третю зменшується час реакції, коли використовувався дихлорметан як розчинник в реакції. З іншого боку, Сполука (с') 31 76449 32 залишається навіть через 24 години, коли викорищеної Сполуки (d') (виходили з 3,0г; очищеної стовувався етилацетат. Сполуки (d'), 2,3г, одержали з: 77%, чистота: 95,2% Приклад 5 Синтез Сполуки (d') (2) за ВЕРХ). Водний розчин гіпохлориту натрію (доступний Умови ВЕРХ; такі ж як і в Прикладі 1. хлор мін. 5,0%; 42мл) і водний розчин гідрокарбоПриклад 6 Синтез Сполуки (e) нату натрію (7,1г в 60мл води) додавали до суміші 7,0г (3,5ммоль) Сполуки (с'), толуолу (70мл), етилацетату (70мл), води (10мл) і 38,3мг (1моль%) TEMPO, що інтенсивно перемішується і охолоджув яких Вn є бензильною групою. ється льодом (при 2-6°С, 55хв.). Через 5 хвилин, До охолодженого льодом розчину 1,84г детектувалось 0,4% (ВЕРХ, площина піку %) вихі(6,50ммоль) Сполуки (d'), що перемішується, в дного матеріалу. Суміш залишали і відокремлений 37мл етилацетату, в атмосфері аргону додавали органічний шар і послідовно промивали сумішшю 0,69г (0,65ммоль, 10моль%) 10% паладію на вуйодиду калію і гідросульфату калію (жовтий гіллі. Суміш інтенсивно перемішували при 25°С в червоно-коричневий), насиченим водним розчиатмосфері водню і періодично відбирали частину ном тіосульфату натрію і потім водою. Розчинник суміші на зразок для ВЕРХ. Реакційну суміш фільвипарювали при пониженому тиску одержуючи трували і фільтрат випарювали. Одержували 6,4г Сполуки (d') (вихід 91%, чистота 92,6% за Екс.13-14. ВЕРХ), яку очищали шляхом перекристалізації з суміші метанол і вода (25:1) одержуючи 2,3г очиТаблиця 5 Як показано в Таблиці 5, реакція триває більше ніж 13 годин, вихід Сполуки (e) збільшується на 10% і утворення біпродукту, Сполуки (g), пригнічується. Умови ВЕРХ Колонка: Inertsil ODS-2,5μм, 4,6мм ID 250мм (виготовлена GL science) Температура: постійна температура приблизно 40°С Рухома фаза: суміш вод : ацетонітрил (1:1) Швидкість потоку: 1мл/хв. Детектування: 254нм Приклад 7 Повністю синтетичний спосіб одержання 2'-аміно-5'-гідроксипропіофенону Повністю синтетичний спосіб одержання 2'аміно-5'-гідроксипропіофенону є наступним. (1) Синтез Сполуки (b') Сполуку (а) (1,00г, 5,98ммоль) розчиняли в 3мл ДМФА. До розчину Сполуки (а), що перемішується, при кімнатній температурі в атмосфері аргону додавали карбонат калію (0,87г, 6,28ммоль, 2,1екв.) і 0,72мл (6,28ммоль, 1,05екв.) бензилхлориду. Після додавання, суміш нагрівали при 60°С і інтенсивно перемішували протягом 20 годин з періодичною перевіркою вмісту Сполуки (а) за допомогою ВЕРХ. Після того як Сполука (а) більше не детектувалась, суміш фільтрували з відсмоктуванням. Тверду речовину промивали 3мл ДМФА. Фільтрат і промивні розчини об'єднували, і розчинник випарювали при пониженому тиску. Після випарювання, залишок додавали до 100мл води. Після цього суміш перемішували, нерозчинний матеріал фільтрували з відсмоктуванням і сушили на повітрі. Після висушування на повітрі, матеріал сушили при пониженому тиску (1мм Hg, при 20°С) одержуючи 1,45г (вихід 95%) Сполуки (b') як блідожовту тверду речовину. Фізичні властивості Сполуки (b'), включаючи ЯМР спектр, є наступними. Сполука (b'); Тпл. 71-73°С. 1 Н-ЯМР (400МГц, CDCI3): δ 5,21 (2Н, с, PhCH2O), 7,21 (1H, дд, J=2,8, 9,3Гц), 7,35-7,44 (6Н, м), 8,16 (1Н, д, J=9,3Гц), 10,48 (1Н, с, СHO). ІЧ (KВr): 1250, 1333, 1514, 1589, 1697 см-1. EI-MC: m/z 257 (М+). (2) Синтез Сполуки (с') Сполуку (b') (1,0г, 3,89ммоль) розчиняли в 20мл ТГФ. По краплям протягом 15 хвилин до охолодженого льодом розчину Сполуки (b'), що перемішується, в атмосфері аргону додавали розчин вінілмагнійброміду (1,0Μ розчин в ТГФ, 5,84мл, 5,84ммоль, 1,5екв.). Під час додавання, внутрішню температуру підтримували в межах від 3 до 10°С. Після перемішування протягом 1 години, реакційний розчин додавали до насиченого водного розчину хлориду амонію, що перемішується. Потім додавали 20мл етилацетату і 4мл гексану, і органічний шар випарювали і промивали 20мл води і насиченим водним розчином хлориду натрію і сушили над 3г сульфату натрію. Розчинник випарювали при пониженому тиску. Залишок (1,19г) очищали колонковою хроматографією на силікагелі (етилацетат:гексан =1:20) одержуючи 0,93г Сполуки (с') (вихід 84%) як оранжева тверда речовина. Фізичні властивості Сполуки (с'), включаючи 33 76449 34 ЯМР спектр, є наступними. Сполуку (а) (500мг, 2,99ммоль) розчиняли в Сполука (с'); Тпл. 60-63°С. 15мл ТГФ. До охолодженого льодом розчину Спо1Н-ЯМР (400МГц, CDCI3) δ; 5,15 (2Н, с, луки (а), що перемішується, по краплям протягом PhCH2O), 5,22-5,26 (1H, м), 5,39-5,44 (1Н, м), 5,90 приблизно 5 хвилин в атмосфері аргону додавали (1Н, д, J=5,1Гц), 6,06 (1Н, ддд, J=5,1, 10,5, 15,6Гц), вінілмагнійбромід (1,0Μ в ТГФ, 7,5мл, 7,5ммоль, 6,94 (1Н, дд, J=2,9, 9,0Гц), 7,34 (1Н, д, J=2,9Гц), 2,5екв.). Після перемішування протягом 1 години, 7,35-7,44 (5Н, м), 8,04 (1Н, д, J=9,0Гц). реакційну суміш додавали до охолодженої льодом ІЧ (KВr): 3298, 1614, 1582, 1506, 1292, 1229сім1 моль/л хлорводневої кислоти (30мл). Потім до1. давали 30мл етилацетату і 5мл гексану і органічEI-MC: m/z 285 (М+). ний шар випарювали і промивали 50мл води і на(3) Синтез Сполуки (d') сиченим водним розчином хлориду натрію і Сполуку (с') (2,00г, 7,02ммоль) розчиняли в сушили над 3г сульфату натрію. Розчинник випа20мл хлороформу. До розчину Сполуки (с'), що рювали при пониженому тиску. Залишок очищали інтенсивно перемішується, при 25°С в атмосфері колонковою хроматографією на силікагелі (етилааргону додавали діоксид марганцю (8,00г, 4цетат:гексан =1:10 1:3) одержуючи 541мг Сполукратна кількість, 92,0моль, 13екв.). Суміш інтенсики (с") (вихід 93%) як жовто-коричневу тверду ревно перемішували протягом 15 годин. Після того човину. як вихідний матеріал більш не детектувався, суміш Сполука (с"); 1 фільтрували з відсмоктуванням. Одержану тверду Н-ЯМР (400МГц, CDCI3) δ: 5,22-5,26 (1Н, м), речовину промивали 20мл хлороформу. Фільтрат і 5,35-5,40 (1Н, м), 5,90-5,92 (1Н, м), 6,06 (1Н, ддд, промивні розчини об'єднували, і розчинник випаJ=5,2, 10,5, 15,6Гц), 6,83 (1 Η, дд, J=2,7, 9,0Гц), рювали при пониженому тиску. Залишок очищали 7,19 (1Н, д, J=2,7Гц), 8,00 (1Н, д, J=9,0Гц). колонковою хроматографією на силікагелі (етилаСинтез Сполуку (d") з Сполуки (с") цетат:гексан =1:20) одержуючи 1,88г Сполуки (d') (вихід 95%) як білу тверду речовину. Фізичні властивості Сполуки (d'), включаючи ЯМР спектр, є наступними. Сполуку (d'); Тпл. 84-85°С. 1 Сполуку (с") (1,00г, 5,13ммоль) розчиняли в Н-ЯМР (400МГц, CDCI3) δ: 5,17 (2Н, с, 8мл ацетону. Де охолодженого льодом розчину PhCH2O), 5,83 (1H, д, J=17,7Гц), 6,01 (1Н, д, Сполуки (с"), при перемішуванні додавали реагент J=10,6Гц), 6,62 (1Н, дд, J=10,6, 17,7Гц), 6,91 (1Н, Джоунса (3,0мл, 5ммоль, 1,5екв.). Після перемішуд, J=2,7Гц), 7,10 (1Н, дд, J=2,7, 9,0Гц), 7,37-7,43 вання протягом 0,5 годин до реакційної суміші до(5Н, м), 8,17 (1Н, д, J=9,0Гц). давали три частини льоду і насичений водний розІЧ (KВr): 1686, 1578, 1506, 1342, 1244см 1. чин гідросульфату натрію (5мл). Потім додавали EI-MC: m/z 283 (М+). 50мл етилацетату і 5мл гексану і шари розділяли і (4) Синтез Сполуки (e) послідовно промивали 50мл води і насиченим воДо охолодженого льодом розчину 1,84г дним розчином хлориду натрію і сушили над 5г (6,50ммоль) Сполуки (d'), що перемішується, в сульфату натрію. Розчинник випарювали при по37мл етилацетату, в атмосфері аргону додавали ниженому тиску одержуючи 0,82г Сполуки (d") (ви0,69г (0,65ммоль, 10моль%) 10% паладію на вухід 83%). Сполука (d"); гіллі. Суміш інтенсивно перемішували при 25°C в 1 H-ЯМР (400МГц, ДMCO-d6) δ; 5,84 (1Н, д, атмосфері водню. Після перемішування протягом J=17,6Гц), 6,11 (1Н, д, J=10,7Гц), 6,60 (1Н, ад, 13 годин, каталізатор з реакційної суміші видаляли J=10,7, 17,7Гц), 6,75 (1Н, д, 2,7Гц), 7,03 (1Н, дд, фільтруванням. Фільтрат випарювали при пони9,1Гц), 8,13 (1Н, д, J=9,1Гц), 11,41 1Н,с). женому тиску одержуючи 0,87г (вихід 81%, чистота 3) Синтез Сполуку (е) з Сполуки (d") 91,14% за ВЕРХ) неочищеного продукту, як оранжеву тверду речовину. 500мг одержаного продукту реакції очищали колонковою хроматографією на силікагелі (етилацетат:гексан =1:10 1:4) одержуючи 421мг Сполуки (e) (вихід 84%, чистота 95,59% Сполуку (d") (100мг, 0,513ммоль) розчиняли в за ВЕРХ) як жовта тверда речовина. Фізичні влас1мл етилацетату. До охолодженого льодом розчитивості Сполуки (e), включаючи ЯМР спектр, є нану при перемішуванні в атмосфері аргону додаваступними. ли 55мг (0,0513ммоль, 10моль%) 10% паладію на Сполука (e); Тпл. 131-140°C 1 вугіллі. Суміш перемішували при кімнатній темпеН-ЯМР (400МГц, CDCI3) δ: 1,20 (3Н, т, ратурі в атмосфері водню протягом 18 годин. КаJ=7,2Гц), 2,93 (2H, к, J=7,2Гц), 6,59 (1Н, д, талізатор фільтрували і фільтрат випарювали при J=8,8Гц), 6,88 (1Н, дд, J=2,9, 8,8Гц), 7,23 (1Н, д, пониженому тиску одержуючи 64мг Сполуки (е) J=2,9Гц). (вихід 76%), як жовта тверда речовина. ІЧ (KВr): 3379, 3296, 1670, 1447, 1194см-1. + Приклад 9 Синтез 7-етил-10EI-MC:m/z165(M ). гідроксикамптотецину (SN-38) Приклад 8 Синтетичний спосіб одержання Сполуки (e) без захисної групи R (1) Синтез Сполуки (с") з Сполуки (а) Сполуку (е) (0,36г, 2,14ммоль) одержали в Прикладі 7 і Сполуку (n) (0,50г, 1,82ммоль) су 35 76449 36 спендували в суміші оцтова кислота і толуол Рухома фаза: суміш диметила(АсОН-толуол; 1:1, 10мл). До суспензії при кімнатмін:гексан:етанол (1:250:250) ній температурі додавали моногідрат пДетектування: 254нм Приклад 11 Синтез 7-етил-10-[4-(1толуолсульфонової кислоти (п-TsOH H2O; 10мг) і піперидино)-1-піперидино]карбонілоксикамптотесуміш перемішували при 100°С протягом 18 годин. цину (СРТ-11) Реакційну суміш конденсували при пониженому тиску, до залишку додавали толуол (10мл), і суміш знову конденсували при пониженому тиску. До залишку додавали ацетон (9мл) і суміш перемішували при кімнатній температурі 2 години, нерозчинний матеріал фільтрували і промивали ацетоSN-38B-11 (1,00г, 1,7ммоль) одержаний в Прином (2мл, двічі). Відфільтрований матеріал кладі 10 суспендували в 1/10 Ν хлорводневій киссушили при пониженому тиску одержуючи SN-38 лоті (20мл, 2,0ммоль) і суспензію нагрівали приб(0,63г, чистота 97,7% за ВЕРХ, вихід 89%) як чорлизно при 80°С до розчинення. До розчину додана тверда речовина. вали ацетонітрил (100мл) і суміш перемішували Умови ВЕРХ при кімнатній температурі протягом ночі. Осади Колонка: Inertsil ODS-2, 5μм, 4,6мм ID 250мм фільтрували, сушили і зволожували до 75% RH (виготовлено GL science) одержуючи СРТ-11 (0,95мг, вихід 89,8%) як блідоТемпература: постійна температура приблизжовтий кристалічний порошок. но 40°С Приклад 12 Синтез Сполуки (І) (1) Швидкість потоку: 1мл/хв. Сполуку (І) одержували шляхом формілюванРухома фаза: метанол - ацетонітрил -10мМ ня Сполуки (k) приблизно при -30°С - -20°С ндигідрофосфат калію (1:1:3) бутиллітієм і N-форміл-N,N',N'-триметилетиленДетектування: 254нм діаміном з наступним йодуванням приблизно при SN-38; 30°С - -20°С н-бутиллітієм і йодом. 1 Н-ЯМР (400МГц, CDCI3) δ: 0,98 (3Н, т, J=7Гц, Сполуку (k) (5,0г; 0,028моль) розчиняли в безCH3), 1,38 (3Н, т, J=7Гц, СН3), 1,90 (2Н, к, J=7Гц, водному ТГФ (приблизно 66мл) в атмосфері азоту. СН2), 3,08 (2Н, к, J=7Гц, СН2), 5,17 (2Н, с, СН2О), Суміш охолоджували до приблизно -30°С - -20°С. 5,23 (1Н, д, J=16Гц), 5,54 (1Н, д, J=16Гц), 6,83 (1Н, До розчину по краплям додавали н-бутиллітій д, J=9Гц), 7,34-7,39 (3Н, м). (1,6моль/л в гексані; 21,2мл, 0,034 моль, 1,2екв.) і Приклад 10 Синтез 7-етил-10-[4-(1-піперисуміш перемішували при охолодженні. Потім до дино)-1-піперидино]карбонілоксикамптотецину реакційної суміші додавали N-формт-Ν,Ν',Ν'(SN-38B-11) триметилетилендіамін (4,4г, 0,0034моль, 1,2екв.), SN-38B-11 (1,22г, 2,08ммоль, вихід 89%, енатіочистота 99,8% ее) одержували з SN-38 (0,91г, 2,32ммоль) синтезовано в Прикладі 9 за допомогою описаної методики (Sawada, S.; Okajima, S.; Aiyama, R.; Nokata, K.; Furuta, Т.; Yokokura, Т.; Sugino, E.; Yamaguchi, K.; Miyasaka, T. Chem. Pharm. Bull. 1991, 39,1446.). Умови хіральної ВЕРХ Колонка: DAICEL CHIRALCEL OD-H, 0,46см ID 25см (#ODHOCE-AK031) Захисний картридж: DAICEL CHIRALCEL ODH, 0,4см ID 1см Введена кількість: 10μг/10μл Температура: постійна температура приблизно 40°С Швидкість потоку: 1мл/хв. як формілюючий реагент, і суміш перемішували при охолодженні. По краплях додавали н-бутиллітій (1,6моль/л в гексані; 35мл, 0,05моль, 1,2екв.) додавали і перемішували при температурі показаній в Таблиці 6. Потім по краплям до суміші, що перемішується, додавали йод (18,4г) в безводному ТГФ (19мл). До суміші додавали водний розчин гідросульфату натрію (12г в 200мл). Після перемішування відновлений органічний шар (гексан) аналізували за допомогою ВЕРХ. Результати показані в Таблиці 6. Умови ВЕРХ Колонка: Capcell Pack ODS UG120, 4,6мм ID 150мм Рухома фаза: 50мм суміш дигідрофосфат калію:ацетонітрил (9:11) Детектування: 220нм Швидкість потоку: приблизно 1мл/хв. Температура: кімнатна температура. Таблиця 6 37 76449 38 Продовження Таблиці 6. 1) Верхні рядки показують фактичну внутрішню температуру під час додавання. Нижні рядки показують фактичний інтервал внутрішньої температури під час перемішування. 2) Екс.15: результати експерименту за вказаних умов 3) Площина піку % 4) Виходи корегували за чистотою (ВЕРХ; площина піку %) 5) НТ: не тестували Як показано в Таблиці 6, Сполуку (І) одержували з 60% виходом або більше коли використовують н-бутиллітій як літіюючий реагент. Як показано в Екс. 16, реакція проходила при постійній температурі при -40°С - -30°С з добрим виходом (більше ніж 70%). Приклад 13 Очистка Сполуки (І) (промивали розведеною хлорводневою кислотою) Сполуку (k) (5,0г, 0,028моль) розчиняли в безводному ТГФ (приблизно 66мл). Реакцію проводи ли як описано в Прикладі 12 при постійній температурі приблизно -35°С. Одержаний з реакційної суміші н-гексановий шар промивали розведеною хлорводневою кислотою (кількість органічного шару). Після промивання, органічний шар сушили над сульфатом натрію, фільтрували і частину фільтрату аналізували за допомогою ВЕРХ за умов приведених в Прикладі 12. Результати показані в Таблиці 7. Таблиця 7 Аналітичні результати кожного гексанового шару після промивання розведеною хлорводневою кислотою 1) Гексановий шар одержували з реакційної суміші розділенням на 5 частин (25мл цільної суміші і 4 частин по 50мл для промивання). Кожну 50мл частину промивали розведеною НСІ приведеною в Таблиці 7 і відновлені гексанові шари аналізували за допомогою ВЕРХ. 2) площина піку % 3) МТПК; 2-метокси-6-триметилсилілпіридин 3-карбальдегід 4) не промивали Як показано в Таблиці 7, Сполуку (k) завжди видаляли шляхом промивання хлорводневою кис лотою. Коли для промивання використовують 1,0моль/мл і більш концентровану хлорводневу 39 76449 40 кислоту, Сполуку (І) одержували з доброю чистошар з реакційної суміші промивали по черзі (звертою. 2-метокси-6-триметилсилілпіридин-3ху до низу) розведеною хлорводневою кислотою карбальдегід (МТПК), використовуючи цей спосіб як показано в Таблиці 8 (як і кількість органічного досить складно видалити формільовану проміжну шару). Сполуку (k). Після промивання, водний кислий шар випаПриклад 14 Очищення Сполуки (І) (постадійне рювали, нейтралізували карбонатом натрію і потім промивання розведеною хлорводневою кислотою) екстрагували н-гексаном. Органічний шар сушили Сполуку (k) (5,0г, 0,028моль) розчиняли в безнад сульфатом магнію, фільтрували і частину фіводному ТГФ (приблизно 66мл). Реакцію проводильтрату аналізували за допомогою ВЕРХ за умов ли як описано в Прикладі 12 при постійній темпеприведених в Прикладі 12. Результати зведені в ратурі приблизно -35°С. Одержаний н-гексановий Таблиця 8. Таблиця 8 Аналітичні результати гексанових естрактів з нейтралізованого водного шару, який одержували після промивання оригінального гексанового шару розведеною хлорводневою кислотою як показано далі (зверху вниз) 1) кожне промивання розведеною ι-iu нейтралізували кароонатом натрію ι суміш екстрагували нгексаном. Органічний шар сушили, фільтрували і фільтрат випарювали при пониженому тиску до суха. 2) ВЕРХ (площина піку %) 3) Не тестували 4) Залишок гексановго шару після постадійних промивань. 5) Не детектували: менше ніж межа детектування Як показано в Таблиці 8, гексановий шар зручно очищати мулитистадійним промиванням хлорводневою кислотою різних концентрацій одержуючи Сполуку (І) з високою чистотою. Приклад 15 Очищення Сполуки (І) (Очищення перегонкою) Сполуку (k) (5,0г, 0,028моль) розчиняли в безводному ТГФ (приблизно 66мл). Реакцію проводили як описано в Приклад 12 при постійній температурі приблизно -35°С. Одержану реакційну суміш (н-гексановий шар) відновлювали і переганяли при 81-99°С при пониженому тиску (приблизно 0,35мм Hg). Після перегонки, залишок в дистиляційній колбі очищали колонковою хромато графією на силікагелі з н-гексаном, і потім сумішшю н-гексан і етилацетат (50:1) одержуючи очищений продукт. Залишок і очищений матеріал аналізували за допомогою ВЕРХ за умов приведених нижче. Результати показані в Таблиці 9. Умови ВЕРХ Колонка: Capcell Pack ODS UG120, 4,6мм ID 150мм Рухома фаза: 50мм дигідрофосфат калію : ацетонітрил суміш (1:1) Довжина хвилі: 220нм Швидкість потоку: приблизно 1мл/хв. Температура: кімнатна температура 41 76449 42 Таблиця 9 Аналітичні результати фракцій залишку гексанового шару дистиляції 1) площина піку % 2) Не детектували: менше ніж межа детектування 3) Кінцевий залишок очищали колонковою хроматографією на силікагелі Як показано в Таблиці 9, МТПК найкраще видаляється перегонкою. Наступна очистка за допомогою колонкової хроматографії на силікагелі забезпечує Сполуку (І) з відмінною чистотою. Переважно, що перегонку не проводять при температурі вищій ніж та, що приведена в Таблиці 9 оскільки спостерігається зміна кольору і розкладання Сполуки (І). Приклад 16 Очищення Сполуки (І) (регенерування з солі хлорводневої кислоти) Сполуку (k) (5,0г, 0,028моль) розчиняли в безводному ТГФ (приблизно 66мл). Реакцію проводили як описано в Прикладі 12 при постійній темпе ратурі приблизно -35°С. Реакційну суміш (10 г) розчиняли в 10 N хлорводневій кислоті (10мл) і перемішували при кімнатній температурі. Жовтий осад фільтрували і промивали маленькою кількістю 10N хлорводневої кислоти і матеріал розчиняли у воді (приблизно 10мл). рН водного розчину доводили до приблизно 8 додаючи гідроарбонат натрію і суміш екстрагували гексаном і органічний шар випарювали при пониженому тиску до суха. Залишок аналізували за допомогою ВЕРХ за умов приведених в Прикладі 15. результати показані в Таблиці 10. Таблиця 10 Аналітичні результати солі одержаної нейтралізацією хлорводневою кислотою екстрактів в 10моль/л хлорводневій кислоті 1) вага залишку 2) площина піку 3) Не тестували Як показано в Таблиці 10, неочищений продукт реакції збирали як сіль хлорводневої кислоти і сіль відновлювали у Сполуку (І) шляхом нейтралізації, МТПК був майже видалений цим способом. 43 76449 44 Сполука (І): жовте масло. гексан (56мл) і органічний шар випарювали і вод1 Н-ЯМР(499мгц.СОСІ3) 6: 0,30 (9Н, с) 4,05 (3Н, ний шар екстрагували н-гексаном (56мл). Об'єднас), 7,67 (1Н, с), 10,19(1 Н, с), ні органічні шари випарювали при пониженому ЕІ:МС: m/z 335 (М+). тиску до суха. Залишок очищали колонковою хроПриклад 17 Синтез Сполуки (m) матографією на силікагелі; силікагель (80г, Fuji Silysia PSQ100B) з сумішшю н-гексан-етилацетат (73:3) як елюент. Результати цього Прикладу зведені в Таблиці 11 (Екс.20). Екс. 9 в Таблиці показує результати повтореного експерименту за умов приведених; До суміші Сполуки (І) (20,0г, 56,0ммоль, вміст: Josein, Η.; Ко, S. В.; Bom, D.; Curran, D. P., Chem. 93,9% за ВЕРХ), триетилсилану, (17,9мл, Eur. J. 1998, 4, 67-83. Curran, D. P.; Ко, S. B.; 112,0ммоль, 2екв.) і кротилового спирту (15,7мл, Josein, H., Angew. Chem. Int. Ed. Engl. 1995, 34, 184,8ммоль, 3,3екв.) по краплям при перемішу2683-2684. В приведеному способі, дихлорметан ванні при 0-5°С в атмосфері азоту додавали тривикористовували як розчинник реакції. Одержувафтороцтову кислоту (28,5мл, 375,3ммоль, 6,7екв.). ли за якістю і виходом еквівалентний рівень проПісля перемішування при температурі протягом дукту (m) без дихлорметану. 30хв., суміш перемішували при кімнатній температурі протягом 20г. До суміші додавали водний розчин карбонату натрію (20,8г в 277мл води) і нТаблиця 11 Умови проведення ВЕРХ Колонка: GL Science Inertsil ODS-2, 0,46см ID 25см Температура: Постійна температура приблизно 40°С Швидкість потоку: 1мл/хв. Рухома фаза: ацетонітрил - 10мм дигідрофосфат калію (5:1) Детектування: 254нм Приклад 18 Синтез Сполуки (І) (2) До суміші Сполуки (ν) (1,00г, вміст; 98,43%, 2,9ммоль), TEMPO (2,3мг, 0,015ммоль, 0,005екв.) і 7% (ваг/об) гідрокарбонату натрію (7,0мл) в толуолі (8,7мл) при 0-5°С додавали водний розчин гіпохлориту натрію (доступний хлор; мінімально 5%, 4,5г, 3,0ммоль, 1,05екв.) і потім суміш перемішували при 0-5°С протягом 2г. До суміші додавали 10% сульфіт натрію (3,7мл, 2,9ммоль) і одержану суміш перемішували при 0-5°С протягом 30хв. Нерозчинний матеріал в суміші видаляли фільтруванням і матеріал на фільтрувальному папері промивали толуолом (1мл 3). Органічний шар фільтрату випарювали і промивали водою (10мл), сушили над сульфатом натрію (2г), фільтрували і десикант промивали толуолом. Фільтрат і промивні розчини об'єднували і потім випарювали при пониженому тиску до суха. Сполуку (І): жовте масло, 0,93г (87% вихід), вміст: 90,60% за ВЕРХ (ди віться Приклад 17). Приклад 19 Синтез Сполуки (n) (1) Сполуку (m) (1,60г) розчиняли в розчинниках приведених в Таблиці 12, до розчину додавали бромід тетрабутиламонію (0,83г), карбонат калію (0,71г) і ацетат паладію (57мг). Кожну реакцію проводили за умов приведених в Таблиці 12. Реакційну суміш при перемішуванні виливали в охолоджений льодом н-гексан (18мл). Нерозчинний матеріал фільтрували з відсмоктуванням і матеріал промивали н-гексаном (6мл 3). Фільтрат і промивні розчини об'єднували і промивали водою (9мл 2), сушили над безводним сульфатом натрію і потім випарювали при пониженому тиску до суха. Залишок очищали колонковою хроматографією на силікагелі з н-гексан-етилацетат (95:5) як елюент. Екс.21 в Таблиці 12 розкриває результати експерименту проведеного за умов описаних; Josien, Η.; Ко, S. В.; Bom, D.; Curran, D. P., Chem. Eur.J. 1998, 4, 67-83. Curran, D. P.; Ко, S. В.; Josien, H., Angew. Chem. Int. Ed. Engl. 1995, 34, 2683-2684. Вимірювали Співвідношення ендо і ексзо форм кожного очищеного продукту за допомогою ВЕРХ. Як показано в Таблиці 12, одержували достатню селективність (ендо -екзо співвідношення) і кінцевий вихід, коли використовувся ТГФ як роз 45 76449 46 чинник реакції при кип'ятінні із зворотнім холодибули вищими на 10% або більше ніж за описаних льником (Екс.25-27). Кінцеві виходи (Екс.25-27) умов (Екс.21). Таблиця 12 ДМФА: N,N-диметилформамід, Тол: толуол, MeCN: ацетонітрил, ТФГ: тетрагідрофуран, Співвідношення: площа піку ендо форми/площина піку екзот форми (ВЕРХ), Вихід: Кінцевий вихід, -: не визначали, Умови проведення ВЕРХ; дивіться Приклад 17. Приклад 20 Синтез Сполуки (n) (2) До розчину Сполуки (m) (1,27г, 2,6ммоль, вміст; 78,7%) в суміші діізопропіловий етерацетонітрил-вода (4:3:1, 20мл) додавали бромід тетрабутиламочію (0,82г, 2,6ммоль), Ν,Νдіізопропілетиламін (3,48мл, 20,8ммоль, 8екв.) і ацетат паладію (57мг, 0,26ммоль) і одержану суміш нагрівали із зворотнім холодильником протягом 30хв. Після охолодження до внутрішньої температури 20°С або нижче, нерозчинний в суміші матеріал відфільтровували з відсмоктуванням і відфільтрований матеріал промивали н-гексаном (2,6мл 3). До об'єднаного фільтрату і промивного розчину додавали н-гексан (10мл) і 10% сульфіт натрію (16мл, 13,0ммоль, 5екв.). Органічний шар випарювали і послідовно промивали 1N хлорводневою кислотою (16,4мл) і потім водою (10мл 2). Органічний розчин випарювали при пониженому тиску до суха. Сполука (п); коричневе масло, 0,83г (91% вихід), вміст: 73,3% за ВЕРХ (дивіться Приклад 17), ендо-екзо співвідношення: 10,6. Таким чином, очевидне покращення селективності порівняно з Екс.21 в Таблиці 12. Кінцевий вихід також покращився на 20% порівняно з Екс.21 в Таблиці 12. Приклад 21 Синтез Сполуки (о) До розчину ферріціаніду калію (195,7г, 0,59моль), карбонату калію, (82,1г, 0,59моль) і метансульфонаміду (37,7г, 0,40моль) у воді (990мл) додавали (DHQD)2PYR (4,36г, 4,95ммоль) і дигідрат осмату калію (VI) (1,0ммоль) і суміш перемішували приблизно при 5°С протягом 1г. До суміші, що перемішується, додавали Сполуку (n) (77,8г, 0,18моль, вміст: 61,5%) і одержану суміш перемішували при температурі протягом ще 20 годин. До суміші додавали порошкоподібний сульфіт натрію (74,9г) і перемішування продовжували при температурі протягом 30хв. Нерозчинний в суміші матеріал відфільтровували на шарі Целіту і матеріал на шарі промивали етилацетатом (4 рази, загалом 770мл). Органічний шар фільтрату випарювали і водний шар екстрагували етилацетатом (770мл). Об'єднані органічні шари сушили над безводним сульфатом натрію, фільтрували і випарювали при пониженому тиску до суха. Залишок очищали колонковою хроматографією на силікагелі з сумішшю дихлорметан - етилацетат (4:1) як елюент; силікагель (280г, Fuji Silysia PSQ100B). Сполука (о); коричнева тверда речовина. Як показано в Таблиці 13, використання осмату калію, як окисника, замість високолеткого оксиду осмію (VIII) забезпечує еквівалентну кількість кінцевого виходу і енатіо надлишок. 47 76449 48 Таблиця 13 Екс. 28: проводили за умов описаних; Josein, Η. ; Ко, S.B.; Bom, D.; Curran, D.P., Chem. Eur. J. 1998, 4, 67-83. Curran, D.P.; Ко, S.B. ; Josein, H., Angew. Chem. Int. Ed. Engl. 1995,34,2683-2684. %ее: Сполуку (о) одержану тут перетворювали у Сполуку (р) за допомогою способу описаного в Прикладі 22 і його енатіо надлишок вимірювали за допомогою хіральної ВЕРХ (дивіться; Приклад 22) Приклад 22 Синтез Сполуки (р) До розчину Сполуки (о) (70г) в суміші метанол і вода (10:1, 1,0л) при кімнатній температурі при перемішуванні додавали йод (кількість вказана в Таблиці 14) і порошкоподібний карбонат кальцію (47,1г). Реакційну суміш перемішували при температурі і протягом часу приведених в Таблиці 14. До суміші додавали один літр 10% сульфіту натрію і хлороформ (1,0л) і одержану суміш перемішували при кімнатній температурі протягом 30хв. Нерозчинний матеріал видаляли фільтруванням і органічний шар фільтрату випарювали. Водний шар екстрагували хлороформом (500мл 2). Екстракти об'єднували, сушили над безводним сульфатом натрію, фільтрували і потім випарювали при пониженому тиску до суха. Використовуючи 4 еквіваленти йоду при кип'ятінні із зворотнім холодильником, ця реакція завершується за 5г, що складало приблизно одну десяту від вказаних раніше умов у порівняльному прикладі. Таблиця 14 Екс.30: проводили за вказаних умов; Josien, H.; Ko, S.B.; Bom, D.; Curran, D.P., Chem. Eur.J. 1998, 4 , 67-83. Curran, D.P., Ko, S.B.; Josien, H.; Angew. Chem. Int. Ed. Engl., 1995, 34, 2683-2684. Вихід (%): Кінцеві виходи Умови проведення ВЕРХ Колонка: GL Science Inertsil ODS-2, 0,46см 10 25см Температура: постійна температура приблизно 40°С Швидкість потоку: 1мл/хв. Рухома фаза: 10мм дигідрофосфат калію ацетонітрил (4:3) Детектування: 254нм Умови проведення хіральної ВЕРХ Колонка: DAICEL CHIRALCEL OD-H, #ODH0CE-AK031, 0,46см ID 25см Захисний картридж: DAICEL CHIRALCEL ODH, 0,4см ID 1см Температура: постійна температура приблиз но 25°С Швидкість потоку: 0,5мл/хв. Рухома фаза: суміш н-гексан - етанол (200:1) Детектування: 254нм Приклад 23 Синтез Сполуки (q) До розчину Сполуку (p) (50,2г) в розчиннику (приблизно 400мл; приведений в Таблиці 15), додавали реагенти приведені в Таблиці і одержану суміш перемішували при вказаній температурі протягом деякого часу. До реакційної суміші при перемішуванні додавали 20% карбонат натрію 49 76449 50 (1,7л), 10% сульфіт натрію (1,0л) і хлороформ Це перетворення задовільно проводиться ви(550мл). Органічний шар випарювали і водний шар користовуючи NCS - Nal при 65°С в оцтовій кислоті (Екс.39). Час необхідний для завершення реакції екстрагували хлороформом (550мл 2). Екстракти був безперечно меншим і виходи Сполуки (q) за об'єднували, сушили над безводним сульфатом вказаних умов вищими ніж повідомлялось раніше натрію, фільтрували і потім випарювали при пони[Порівняльний Експеримент 1 (Пор.1)] на 50% або женому тиску до суха. Вміст Сполуки (q) в залишку більше. визначали за допомогою ВЕРХ. Результати зведені в Таблиці 15. Таблиця 15 *) суміш дихлорметан і хлороформ (3:2), АсОН: оцтова кислота, ІСІ: хлорид йоду, NIS: N-йодсукцинімід, NCS: N-хлорсукцинамід, Екв.: молярне співвідношення використовуваних реагенту(ів), Вихід: Кінцеві виходи. Приклад 24 Очищення Сполуки (q) (1) Сполуку (q) (63г, Чистота 89,2% за ВЕРХ) одержану в Прикладі 23 суспендували в метанолі (150мл), до суспензії по краплям при інтенсивному перемішування додавали 0,2N гідроксид натрію і перемішування продовжували протягом 2г. Лужний розчин промивали хлороформом (400мл 3) і рН водного шару доводили 6N хлорводневою кислотою до 1-2 і підкислений розчин екстрагували хлороформом (400мл 3). Хлороформний шар сушили над безводним сульфатом натрію, фільтрували і потім випарювали при пониженому тиску до суха. Сполуку (q) (Екс.40); чистота 97,7% (% площини піку; умови проведення ВЕРХ: дивіться Приклад 25) Приклад 25 Очищення Сполуки (q) (2) Сполуку (q) (50г) очищену за способом описаним в Прикладі 24 розчиняли в хлороформі (240мл) і н-гексані (400мл) обережно додаючи на поверхню розчину. Суміш залишали стояти при кімнатній температурі протягом 15г. Осад суміші видаляли фільтруванням і фільтрат випарювали при пониженому тиску до суха (Екс.41). Сполуку (q) (Екс.40) одержану в Прикладі 24 (93-96% енатіо надлишок) очищали оптично за цим способом. Сполука (q) (Екс.41) одержана тут мала 99,7-99,9% енатіо надлишок, що визначили за допомогою хіральної ВЕРХ вказаної нижче. Умови проведення ВЕРХ Колонка: GL Science Inertsil ODS-2, 0,46см ID 25см Температура: постійна температура приблизно 40°С Швидкість потоку: 1мл/хв. Рухома фаза: суміш ацетонітрил –1 0мм калій дигідрофосфат (5:3) Детектування: 254нм Умови проведення хоральної ВЕРХ Колонка: DAICEL CHIRALPAK AD-H, # ADH0CE-BC037, 0,46см ID 25см Захисний картридж: DAICEL CHIRALPAK ADH, 0,4см ID 1см Температура: постійна температура приблизно 25°С Швидкість потоку: 1мл/хв. Рухома фаза: суміш н-гексан - 2-пропанол (25:1) Детектування: 254нм Приклад 26 Синтез Сполуки (r) Розчин Сполуки (q) (42,8г, 0,10моль, вміст: 84,5%), ацетату паладію (1,34г, 6,0ммоль) і карбонату калію (24,7г, 0,18моль) в пропанолі (490мл) завантажували в реакційну колбу. Колбу дегазували з відсмоктуванням і промивали азотом, і дегазували знову з відсмоктуванням і потім заповнювали монооксидом вуглецю. Суміш перемішували при 60°С в атмосфері монооксиду протягом 18г. Нерозчинний матеріал відфільтровували на шар Целіту і матеріал промивали етилацетатом (300мл). До фільтрату додавали 1N хлорводневу кислоту (150мл) і насичений водний розчин хлориду натрію (300мл) і суміш інтенсивно збовтували. 51 76449 52 Органічний шар випарювали і водний шар екстраперемішуваної суміші по краплям додавали третгували етилацетатом (300мл). Об'єднані органічні бутил акрилат (1,8г) і одержану суміш перемішушари сушили над безводним сульфатом натрію, вали при 50°С в атмосфері аргону протягом 24г. фільтрували і потім випарювали при пониженому До охолодженої льодом суміші додавали воду тиску до суха. Залишок очищали колонковою хро(10мл) і концентрували хлорводневою кислотою матографією на силікагелі; силікагель (100г, Fuji (1мл) і суміш екстрагували сумішшю толуол і етиSilysia PSQ100B) з сумішшю хлороформ і метанол лацетат (4:1, 7мл 4). Об'єднані екстракти сушили (99:1), як елюент. Сполуку (r): коричневе масло, над безводним сульфатом натрію, фільтрували і 30,3г (70% вихід), вміст: 73,4% визначали за доповипарювали при пониженому тиску до суха могою ВЕРХ. (Екс.42-43). Залишок досліджували за допомогою Умови проведення ВЕРХ ВЕРХ (як показано далі). Колонка: GL Science Inertsil ODS-2, 0,46см Як показано в Таблиці 16, Сполуку (t) одержуID 25см вали з 72% виходом використовуючи карбонат Температура: постійна температура приблизцезію як основу (Екс.42), з іншого боку, викорисно 40°C тання недорогого карбонату калію як основи заШвидкість потоку: 1мл/хв. безпечувало еквівалентний вихід Екс. 42. Рухома фаза: 10мм дигідрофосфат калію ацетонітрил (4:3) Таблиця 16. Детектування: 254нм Приклад 27 Синтез Сполуки (s) До розчину, що перемішується, Сполуки (r) (28,7г, 68,2ммоль, вміст: 73,4%) і йодиду натрію (27,6г, 0,18моль) в абсолютному ацетонітрилі (141мл), по краплям при кімнатній температурі в атмосфері азоту додавали хлортриметилсилан (23,3мл, 0,18ммоль). Суміш перемішували при кімнатній температурі протягом 3г. До суміші додавали 1N хлорводневу кислоту (8мл) і 10% сульфіт натрію (232мл) і одержану суміш перемішували протягом 30хв. Суміш екстрагували етилацетатом і органічний шар випарювали і випарювали при пониженому тиску до суха. Сполуку (s): 22,3г (95% вихід), вміст: 85,6% визначали за допомогою ВЕРХ (як вказано далі). Умови проведення ВЕРХ Колонка: GL Science Inertsil ODS-2, 0,46см ID 25см Температура: постійна температура приблизно 40°C Швидкість потоку: 1мл/хв. Рухома фаза: 10мм калій дигідрофосфат ацетонітрил (5:2). Детектування: 254нм Приклад 28 Синтез Сполуки (t) Розчин Сполуки (s) (0,50г) в диметилсульфоксиді (ДМСО, 7мл) в присутності неорганічної основи (карбонат калію або цезію, 0,4г) перемішували при 50°С в атмосфері аргону протягом 20хв. До Умови проведення ВЕРХ Колонка: GL Science Inertsil ODS-2 , 0,4 см ID 25см Температура: постійна температура приблизно 40°С Швидкість потоку: 1мл/хв. Рухома фаза: 10мм дигідрофосфат калію ацетонітрил (5:2) Детектування: 254нм Приклад 29 Синтез SN-38 Суміш Сполуки (п) (0,50г, 1,82ммоль, вміст: 96,6%) і Сполуки (e) (0,36г, 2,18ммоль) в суміші толуол - оцтова кислота (1:1, 10мл) в присутності n-TsOH H2O (10мг) нагрівали при 90°С при перемішуванні в атмосфері азоту протягом 7г. Після охолодження, суміш випарювали при пониженому тиску до суха. Після 2 азеотропних видалень оцтової кислоти за допомогою толуолу (10мл), до залишку додавали ацетон (9мл) і суспензію перемішували в атмосфері азоту протягом 30хв. Тверду речовину збирали фільтруванням, промивали ацетоном (2мл 2) і потім сушили при пониженому тиску. SN-38 (Екс.45): тверда речовина охрового кольору, 0,63г (89,1% вихід), чистота; 99,6% за ВЕРХ (дивіться Приклад 9). Екс.44 в Таблиця 17 показані результати експерименту за умов описаних; Henegar, Κ. Ε.; Ashford, S. W.; Baughman, Τ. Α.; Sih, J. C; Gu, R. L, J. Org. Chem. 1997, 62, 6588-6597. В атмосфері азоту, чистота і вихід SN-38 покращуються, як показано в Таблиці 17. Таблиця 17 53 76449 54 Приклад 30 Синтез трициклічного кетону дібний сульфіт натрію (74,9г). Суспензію переміПовний синтетичний спосіб трициклічного кешували приблизно при 5°С протягом 30хв. і нерозтону (h) виходячи з Сполуки (І) приведений нижче; чинний матеріал фільтрували крізь шар Целіту. (1) Синтез Сполуки (m) Матеріал шару промивали етилацетатом (4 рази, До перемішуваної суміші Сполуки (І) (20,0г, загалом 770мл). Органічний шар фільтрату випа56,0ммоль, 2екв., вміст: 93,9%), триетилсилану рювали і водний шар надалі екстрагували етила(17,9мл, 112ммоль, 2екв.) і кротилового спирту цетатом (770мл). Об'єднані органічні шари сушили (15,7мл, 184,8ммоль, 3,3екв.) по краплям при 0над безводним сульфатом натрію і випарювали 5°С в атмосфері азоту додавали трифтороцтову при пониженому тиску до суха. Залишок очищали кислоту (28,5мл, 375,2ммоль, 6,7екв.) і суміш пеколонковою хроматографією на силікагелі; силікаремішували при температурі протягом 30хв. Суміш гель (700г, Fuji Silysia PSQ100B), елюент: дихлорперемішували при кімнатній температурі протягом метан - етилацетат (4:1). Сполука (о): коричнева 20г і потім до суміші додавали водний розчин картверда речовина. бонату натрію (20,8г в 277мл води) і н-гексан (4) Синтез Сполуки (р) (56мл). Органічний шар випарювали і водний шар Суміш Сполуки (о) (70,2г), йоду (183,7г, екстрагували н-гексаном (57мл). Об'єднані органі0,72моль) і карбонату кальцію (36,23г, 0,36моль) в чні шари випарювали при пониженому тиску до метанол - вода (10:1, 1,0л) нагрівали із зворотнім суха. Залишок очищали за допомогою колонкової холодильником протягом 5г. Після охолодження хроматографії на силікагелі; силікагель (80г, Fuji до суміші додавали 10% сульфіт натрію (1,0л) і Silysia PSQ100B), елюент; н-гексан-етилацетат хлороформ (1,0л) і одержану суміш перемішували (73:3) видаляючи біпродукт, Сполука (ν) (4,95г, при кімнатній температурі протягом 15хв. Нероз14,68ммоль, чистота 98,43%, вихід 26%). Сполука чинний матеріал фільтрували з відсмоктуванням і (m); 17,8г (64% вихід), вміст: 80,0% за ВЕРХ (диматеріал промивали хлороформом (0,5л). Об'єдвіться Приклад 17). нані органічні шари фільтрату і промивні розчини 1 Н-ЯМР (400МГц, CDCI3) δ: 0,24 (9Н, с, TMS), розділяли і водний шар надалі екстрагували хло1,69 (3Н, дд, J=1,0, 6,1Гц, =СНСН3), 3,85-4,05 (2Н, роформом (0,5л). Об'єднані органічні шари сушили м, ОСН2СН=), 3,93 (3Н, с, СН3О), 4,55 (2Н, с, над безводним сульфатом натрію, фільтрували і ОСН2), 5,55-5,83 (2Н, м, СН=СН),7,4 7(1Н, с). випарювали при пониженому тиску до суха. СпоСполука (ν); 5,0г (26% вихід), вміст: 98,4% луку (р): коричневе масло, 53,6г (повний 81% вихід (ВЕРХ). 1Н-ЯМР (400МГц, CDCI3) δ: 0,27 (9Н, с, з Сполуки (m)), вміст: 80,4% за ВЕРХ (дивіться TMS), 2,45 (1Н, т, J=6,8Гц, ОН), 3,99 (3Н, с, СН3О), Приклад 22), 96,2%ее за допомогою хіральної 4,79 (2Н, д, J=6,8Гц, СН2ОН), 7,49 (1Н, с). ВЕРХ (дивіться Приклад 22). 1 (2) Синтез Сполуки (n) Н-ЯМР (400МГц, CDCI3) δ: 0,28 (9Н, с, TMS), Суміш Сполуки (m) (1,27г, 2,56ммоль, вміст: 0,94 (3H, т, J=7,4Γц СН2СH3), 1.76 (2Η, κ, J=7,4Гц, 78,73%), бромід тетрабутиламонію (0,82г, СH2СН3), 3,61 (1Н, с, ОН), 3,98 (3Н, с, ОСН3), 5,23 2,56ммоль) і ацетат паладію (57мг, 0,26ммоль) в (1Н, д, J=15,6Гц), 5,54 (1Н, д, J=15,6Гц), 7,33 (1Н, суміш діізопропіловий етер - ацетонітрил - вода с, ароматика-Н). (4:3:1, 20мл) нагрівали із зворотнім холодильником (5) Синтез Сполуки (q) протягом 30хв. Після охолодження до внутрішньої Суміш Сполуки (p) (50,2г, 0,14моль, вміст: температури 20°С або нижче, нерозчинний мате80,4%, 96,2%ее), N-хлорсукциніміду ріал суміші видаляли фільтруванням і матеріал (107,36г, 0,80моль) і йодиду натрію (120,52г, промивали н-гексаном (10мл). Фільтрат і промивні 0,80моль) в оцтовій кислоті (411мл) нагрівали прирозчини об'єднували і до розчину додавали нблизно при 65°С при перемішуванні протягом 16г. гексан (10мл) і 10% сульфіт натрію (16мл, Після охолодження до суміші послідовно додавали 113ммоль, 5екв.). Органічний шар суміші випарю20% карбонат натрію (1,7л), 10% сульфіт натрію вали і послідовно промивали 1 N хлорводневою (1,0л) і хлороформ (0,6л). Органічний шар суміші випарювали і водний шар екстрагували хлорофокислотою (16,4мл) і водою (10мл 2). Органічний рмом (0,6л 2). Об'єднані органічні шари сушили шар випарювали при пониженому тиску до суха. Сполука (n): коричневе масло, 0,83г (91% вихід), над безводним сульфатом натрію, фільтрували і вміст: 73,34% за ВЕРХ, ендо-екзо співвідношення: потім випарювали при пониженому тиску до суха 10,6 за ВЕРХ (дивіться Приклад 17). (неочищений q). 1 Н-ЯМР (400МГц, CDCI3) δ: 0,26 (9Н, с, TMS), (6) Очищення Сполуки (q) (1) 1,12 (3Н, т, J=7,3Γц, СН2СH3, 2,31 (2Η, дк, J=1,0, Суспензію неочищеної Сполуки (q) (залишок 7,3Гц, СН2СНз), 3,94 (3Н, с, ОСН3), 5,00 (2Н, с, попередньої стадії, чистота: 89,2% за ВЕРХ) в меОСН2), 6,51 (1Н, т, J=1,0Γц, ОСН=), 6,83 (1Н, с, танолі (150мл) додавали по краплям до 0,2N гідароматика-Н роксиду натрію (0,40моль) при перемішуванні. Су3) Синтез Сполуки (о) міш перемішували при кімнатній температурі До розчину ферроціаніду калію (195,7г, протягом 2г. Лужну суміш промивали хлорофор0,59моль), карбонату калію (82,1г, 0,59моль) і мемом (400мл 3) і водний шар випарювали і довотансульфонаміду (37,7г, 0,40моль) у воді (990мл) дили рН до 1-2 використовуючи 6N хлорводневу додавали (DHQD)2PYR (4,36г, 4,95ммоль) і дигідкислоту. Кислий розчин екстрагували хлорофоррат осмату калію (VI) (0,99ммоль) і суміш перемімом (400мл 3). Органічний шар випарювали і сушували приблизно при 5°С протягом 1г. До суміші шили над безводним сульфатом натрію, фільтрудодавали Сполуку (n) (77,8г, 0,18моль, вміст: вали і потім випарювали при пониженому тиску до 61,5%) і одержану суміш перемішували приблизно суха. Напівочищений (q), чистота: 97,7% за ВЕРХ при 5°С протягом 20г і потім додавали порошкопо(дивіться Приклад 25). 55 76449 56 (7) Очищення Сполуки (q) (2) ної суміші по краплям в атмосфері аргону додаваНапівочищену (q) розчиняли в хлороформі ли трет-бутил акрилат (2,1мл, 14,6ммоль) і пере(280мл) і н-гексані (400мл) додаючи на поверхню мішування продовжували протягом 20г. До суміші, розчину і одержану суміш залишали при кімнатній що охолоджується на бані з льодом і перемішутемпературі на 15г. Осад видаляли фільтруванням ється, по краплям додавали воду (10мл) і концені фільтрат випарювали при пониженому тиску до тровану хлорводневу кислоту (1мл). Суміш екстрасуха. Сполуку (q); желеподібне масло, 47,4г (86% гували толуол-етилацетат (4:1, 7мл 4). Об'єднані вихід), вміст: 84,5% за ВЕРХ (дивіться Приклад екстракти промивали водою (5мл 3), сушили над 25), 99,7%ее за хіральною ВЕРХ (дивіться Прикбезводним сульфатом натрію, фільтрували і потім лад 25). випарювали при пониженому тиску до суха. Спо1 Н-ЯМР (400МГц, CDCI3) δ: 0,94 (3Н, т, лука (t): 0,55г (77% вихід), вміст: 75,0% за ВЕРХ J=7,3Гц, СН2СН3). 1-75 (2H, к, а=7,3Гц, СН2СН3), (дивіться Приклад 28). 1 3,58 (1H, с, ОН), 3,96 (3Н, с, ОСН3), 5,16 (1Н, д, Н-ЯМР (400МГц, CDCI3) δ: 0,99 (3Н, т, J=15,6Γ4), 5,47 (1Н, д, J=15,6R4), 7,59 (1 Η, с, ароJ=7,4Гц, СН2СН3), 1.58 (9Н, с, т-Вu), 1,83 (2Н, м, матика-Н). [ ]D20 =+51,3 (с=0,981, СНСl3) СH2СН3), 4,68 (2Н, с, СН2), 5,25 (1Н, д, J=17,8Гц), (8) Синтез Сполуки (r) 5,69 (1Н, д, J=17,8Гц), 7,01 (1Н, с, ароматика-Н). Розчин Сполуки (q) (42,8г, 0,10моль, вміст: (11) Синтез Сполуки (h) 84,5%), ацетату паладію (1,34г, 5,95ммоль) і карДо розчину Сполуки (t) (1,02г, 1,84ммоль, бонату калію (24,67г, 0,179моль) в пропанолі вміст: 66,0%) в толуолі (17мл), при перемішуванні (490мл) дегазували з відсмоктуванням і промивав атмосфері аргону додавали трифтороцтову кисли аргоном і дегазували з відсмоктуванням і потім лоту (1,7мл). Суміш перемішували при 110°С в наповнювали монооксидом вуглецю. Суміш переатмосфері аргону протягом 100хв. Після охоломішували при 60°С протягом 4г. Після охолоджендження, суміш випарювали при пониженому тиску ня, нерозчинний матеріал видаляли на шар Целіту до суха. Залишок суспендували в дихлорметані і матеріал на шарі промивали етилацетатом (50мл) і нерозчинний матеріал фільтрували на (300мл). Фільтрат промивали 1N хлорводневою шар Целіту. Фільтрат промивали водою (10мл) і кислотою (150мл) і насиченим водним розчином органічний шар випарювали і водний шар екстрахлориду натрію (300мл) і водний шар випарювали і гували дихлорметаном (20мл 3). Об'єднані екстекстрагували етилацетатом (300мл). Об'єднані ракти сушили над безводним сульфатом натрію, органічні шари сушили над безводним сульфатом фільтрували і випарювали при пониженому тиску натрію, фільтрували і потім випарювали при понидо суха. Сполука (h); (3)-4-етил-7,8-дигідро-4женому тиску до суха. Залишок очищали колонкогідрокси-1Н-пірано-[3,4-f]індолідин-3,6,10(4Н)вою хроматографією на силікагелі; силікагель трион; 0,46г (77% вихід), вміст: 80,7% за ВЕРХ (як (200г), елюент: хлороформ - метанол (99:1). Спопоказано далі). 1 лука (r): коричневе масло, 30,3г (70% вихід), вміст: Н-ЯМР (400МГц, CDCI3) δ: 0,98 (3Н, т, 73,4% за ВЕРХ (дивіться Приклад 26). J=7,3Гц, СН2СН3, 1.81 (2H, м, СН2СН3), 2,97 (2H, т, 1 Н-ЯМР (400МГц, CDCI3) δ: 0,88 (3Н, т, J=6,3Гц, CН2CH2), 3,64 (1H, с, ОН), 4,34 (2H, м, J=7,3Гц, CH3), 1,04 (3Н, т, J=7,3Гц, CH3), 1,82 (4H, CH2CH2), 5,25 (1Н, д, J=17,1Γц), 5,68 (1Н, д, м, СН2 2), 3,69 (1Н, с, ОН), 4,09 (3Н, с, ОСН3), 4,34 J=17,1Гц), 7,22 (1Н, с, ароматика-Н). (2Н, т, J=6,8Гц, СН2), 5,31 (1Н, д, J=16,3Гц), 5,61 Умови проведення ВЕРХ (1Н, д, J=16,3Гц), 7,94 (1Н, с, ароматика-Н) Колонка: Inertsil ODS-2, 0,46см ID 25см (9) Синтез Сполуки (s) Температура: постійна температура приблизДо розчину Сполуки (r), що перемішується, но 40°С (28,7г, 68,2ммоль, вміст: 73,4%) і йодиду натрію Швидкість потоку: 1мл/хв. (27,6г, 0,18моль) в абсолютному ацетонітрилі Рухома фаза: 10мм дигідрофосфат калію - ме(141мл) по краплям при кімнатній температурі в танол (4:1) атмосфері азоту додавали хлортриетилсилан Детектування: 254нм (23,3мл, 0,18ммоль). Суміш перемішували при Приклад 31Синтез 7-етил-10кімнатній температурі протягом 3г і потім гасили гідроксикамптотецину (SN-38) 1N хлорводневою кислотою (8мл) і 10% сульфітом натрію (232мл). Одержану суміш перемішували при кімнатній температурі протягом 30хв. Суміш екстрагували етилацетатом і органічний шар випарювали і випарювали при пониженому тиску до Суспензію Сполуки (h) (0,50г, 1,82ммоль, суха. Сполука (s): 22,3г (95% вихід), вміст: 85,6% вміст: 96,6%), одержували як описано в Прикладі за ВЕРХ (дивіться Приклад 27). 1 30 (11), і Сполуки (е) (0,36г, 2,14ммоль) в присутН-ЯМР (400МГц, CDCI3) δ: 1,00 (3Н, т, ності моногідрату п-толуолсульфонової кислоти J=7,3Γ4, СН3), 1,02 (3Н, т, J=7,3Гц, СН3), 1,83 (4Н, (10мг) в оцтова кислота - толуол (1:1, 10мл) перем, СН2 2), 3,75 (1Н, с, ОН), 4,35 (2Н, т, J=6,8Гц, мішували при 100°С в атмосфері азоту протягом СН2), 5,21 (1Н, д, J=17,1Гц), 5,61 (1H, д, J=17,1Γц), 18г. Суміш випарювали при пониженому тиску і до 7,28 (1Н, с, ароматика-Н), 9,59 (1Н, шс, ОН). залишку додавали толуол (10мл) і потім випарю(10) Синтез Сполуки (t) вали при пониженому тиску до суха. Залишок суРозчин Сполуки (s) (0,50г, 1,46ммоль, вміст: спендували в ацетоні (9мл) і суспензію перемішу86,6%) і карбонату калію (0,40г, 2,92ммоль) в дивали при кімнатній температурі протягом 2г. метилсульфоксиді (7мл) перемішували при 50°С в Суспензію фільтрували з відсмоктуванням і зібраатмосфері аргону протягом 20хв. До перемішува 57 76449 58 піперидино]карбонілоксикамптотецину (СРТ-11) ну тверду речовину промивали ацетоном (2мл 2) і потім сушили при пониженому тиску. SN-38: коричнева тверда речовина, 0,63г (89% вихід), вміст: 97,7% за ВЕРХ (дивіться Приклад 9). 1 Н-ЯМР (400МГц, CDCI3) δ: 0,98 (3Н, т, J=7Гц, CH3), 1,38 (3Н, т, J=7Гц, СН3), 1,90 (2Н, к, J=7Гц, СН2), 3,08 (2Н, к, J=7Гц, СН2), 5,17 (2Н, с, СН2О), SN-38B-11 (1,00г, 1,7ммоль), одержували як 5,23 (1Н, д, J=16Гц), 5,54 (1Н, д, J=16Гц), 7,34-7,39 описано в Прикладі 32, розчиняли в 0,1N хлорвод(3Н, м), 6,83 (1Н, д, J=9Гц). невій кислоті (20мл) шляхом нагрівання приблизно Приклад 32 Синтез 7-етил-10-[4-(1при 80°С. До розчину додавали ацетонітрил піперидино)-1-піперидино]карбонілоксикамптоте(100мл) і суміш обережно перемішували при кімцину (SN-38B-11) натній температурі протягом 15г. Осади фільтрували з відсмоктуванням і сушили при пониженому тиску і потім зволожували. СРТ-11: блідо-жовтий кристалічний порошок, 0,95мг (89,9% вихід). SN-38 (0,91г, 2,32ммоль), одержували як опиПромислова застосовуваність сано в Приклад 31, перетворювали у SN-38B-11 Використовуючи синтетичний спосіб винаходу (1,22г, 89% вихід, 99,8%ее за хіральною ВЕРХ; можна в короткий час з високим виходом одержадивіться Приклад 10) за допомогою описаного ти високочистий 2'-аміно-5'-гідроксипропіофенон і способу (S. Sawada, etal., Chem. Pharm. Bull., 1991, трициклічний кетон, і використовуючи ці проміжні 39, 1446). сполуки можна раціонально провести загальний Приклад 33] Синтез гідрохлориду тригідрату 7синтез аналогів СРТ. етил-10-[4-(1-піперидино)-1 Комп’ютерна верстка Т. Чепелева Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethods for producing camptothecin analogs and intermediary compounds
Автори англійськоюSawada Seigo
Назва патенту російськоюСпособы получения аналогов камптотецина и промежуточных соединений
Автори російськоюСавада Сейго
МПК / Мітки
МПК: C07D 491/00, C07C 205/00, C07D 213/00, C07C 225/00
Мітки: сполук, аналогів, камптотецину, одержання, способи, проміжних
Код посилання
<a href="https://ua.patents.su/29-76449-sposobi-oderzhannya-analogiv-kamptotecinu-ta-promizhnikh-spoluk.html" target="_blank" rel="follow" title="База патентів України">Способи одержання аналогів камптотецину та проміжних сполук</a>
Спосіб одержання [(1s)-[1a,2b,3b,4a(s)]]-4-[7-[[1-(3-хлор-2-тієніл)метил] пропіл]аміно]-3н-імідазо[4,5-b]піридин-3-іл]-n-етил-2,3-дигідроксициклопентанкарбоксаміду, способи одержання проміжних сполук для його одержання та проміжні сполуки

Номер патенту: 54479
Опубліковано: 17.03.2003
Автори: Леон Патрік, Паунер Торі Х., О'Брайєн Майкл К., Цуей Чінг Т., Томпсон Майкл Д., Гарсіа Ерве, Ванасс Бенуа Дж., Шах Харшавадан К., Рейллі Лоренс, Вальтер Френсіс Л.
МПК: C07D 409/14, C07D 409/12, C07D 213/69, A61P 3/06, C07D 471/04, A61P 9/00, A61P 9/12
Мітки: проміжні, способи, пропіл]аміно]-3н-імідазо[4,5-b]піридин-3-іл]-n-етил-2,3-дигідроксициклопентанкарбоксаміду, одержання, проміжних, спосіб, сполук, сполуки, 1s)-[1a,2b,3b,4a(s)]]-4-[7-[[1-(3-хлор-2-тієніл)метил
Формула / Реферат:
1. Спосіб одержання [(1S)-[1a,2b,3b,4a(S*)]]-4-[7-[[1-(3-хлор-2-тієніл)метил]пропіл]аміно]-3H-імідазо[4,5-b]піридин-3-іл]-N-етил-2,3-дигідроксициклопентанкарбоксаміду, який включає взаємодію [(1S)-[1a,2b,3b,4a(S*)]]-4-[[3-аміно-4-[[1-(3-хлор-2-тієніл)метил]пропіл]аміно]-2-піридиніл]аміно]-N-етил-2,3-дигідроксициклопентанкарбоксаміду зі складним ефіром ортоформіату, ацетатом формамідину або диметилацеталем диметилформаміду.2. Спосіб за...
Спосіб одержання похідних 4-трифтрометилсульфінілпіразолу та способи одержання проміжних сполук

Номер патенту: 73752
Опубліковано: 15.09.2005
Автори: Пельта Ізабелль, Клавель Жан-Луї, Шарро Філіпп, Ле Бар Сільві
МПК: C07D 231/44, C07D 401/04
Мітки: 4-трифтрометилсульфінілпіразолу, спосіб, похідних, проміжних, сполук, одержання, способи
Формула / Реферат:
1. Спосіб (А) одержання сполуки формули (І):, (I)в якій W означає азот або -CR3; R1 означає галоген, галоалкіл, галоалкокси, R4S(O)n- або -SF5; R2 означає водень або галоген; R3 означає галоген; R4 означає алкіл або галоалкіл і n означає 0, 1 або 2, що включає окислення сполуки формули (II):
Способи одержання діарилпіридинів, корисних як інгібітори cox-2, та проміжних сполук

Номер патенту: 61119
Опубліковано: 17.11.2003
Автори: Пай Філіп Дж., Девіс Ян В., Россен Кай, Журне Мішель, Джерена Лінда, Ларсен Роберт Д.
МПК: C07F 3/00, C07D 213/50, A61P 29/00, C07C 317/24, C07D 213/61, C07D 213/53, C07C 315/00, A61K 31/44, C07D 213/85, A61P 43/00, A61P 37/08, A61K 31/444, C07D 213/82
Мітки: діарилпіридинів, корисних, cox-2, способи, сполук, інгібітори, проміжних, одержання
Формула / Реферат:
1. Спосіб одержання сполук формули 5 , 5у якій R1 вибраний з групи, що включає(a) СН3,(b) NH2,(c) NНС(О)СF3, (d) NНСН3, іАr представляє моно-, ди- або тризаміщений феніл чи піридиніл (або його N-оксид), у яких замісники обрані з групи, що включає(a) гідроген,(b) галоген,(c) С1-4-алкокси,(d) С1-4-алкілтіо,(e) CN,(f)...
Способи одержання проміжних сполук b-метилкарбапенему, проміжні cполуки

Номер патенту: 50705
Опубліковано: 15.11.2002
Автори: Воланте Ральф П., Чой Ву-Баєг, Томпсон Ендрю С., Шінкай Ічіро, Рейдер Пол Дж., Хамфрі Гай Р.
МПК: C07D 205/00, C07D 405/04
Мітки: способи, сполук, cполуки, b-метилкарбапенему, одержання, проміжних, проміжні
Формула / Реферат:
1. Способ получения промежуточного соединения b-метилкарбапенема формулы (VI):, (VI)где R представляет собой:(a) водород,(b) метил, или(c) гидроксизащитную группу, иР' представляет собой азотзащитную группу,Nu представляет собой нуклеофильную группу, выбранную из –СН2СО2-т-Вu и -SR2,где R2 представляет собой водород, прямой или разветвленный C1-C10aлкил, прямой или разветвленный...
Способи одержання проміжних продуктів пестицидів

Номер патенту: 69451
Опубліковано: 15.09.2004
Автори: Верспрумі П'єр, Ансель Жан-Ерік, Ванжелісті Манюель, Перрен-Жане Жилль
МПК: C07D 231/44, C07C 209/42, C07D 213/73, C07B 61/00, C07C 211/45, C07C 211/52
Мітки: одержання, продуктів, проміжних, способи, пестицидів
Формула / Реферат:
1. Спосіб одержання сполуки формули (І) , (I)в якій R1 являє собою галогеналкіл, галогеналкокси або -SF5; W являє собою N або CR3; і R2 і R3 кожний незалежно являє собою водень або хлор; або її кислотно-адитивної солі; який передбачає гідрогеноліз сполуки формули (II) (II)або...
Попередній патент: Компресуюча пластинка для остеосинтезу
Наступний патент: Декоративний папір-основа з покращеною непрозорістю
Випадковий патент: Спосіб комбінованої фармакотерапії хронічної серцевої недостатності