Тетрагідропіролотіазинові сполуки
Номер патенту: 112941
Опубліковано: 10.11.2016
Автори: Уіннероскі мол., Леонард Ларрі, Грін Стівен Джеймс, Мерготт Дастін Джеймс, Уотсон Брайан Морган


























Формула / Реферат
1. Сполука формули:
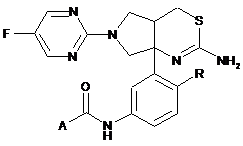 ,
,
де R являє собою Η або F; і А являє собою:
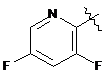 ,
, 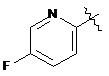 ,
,  або
або 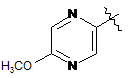 ;
;
або її фармацевтично прийнятна сіль.
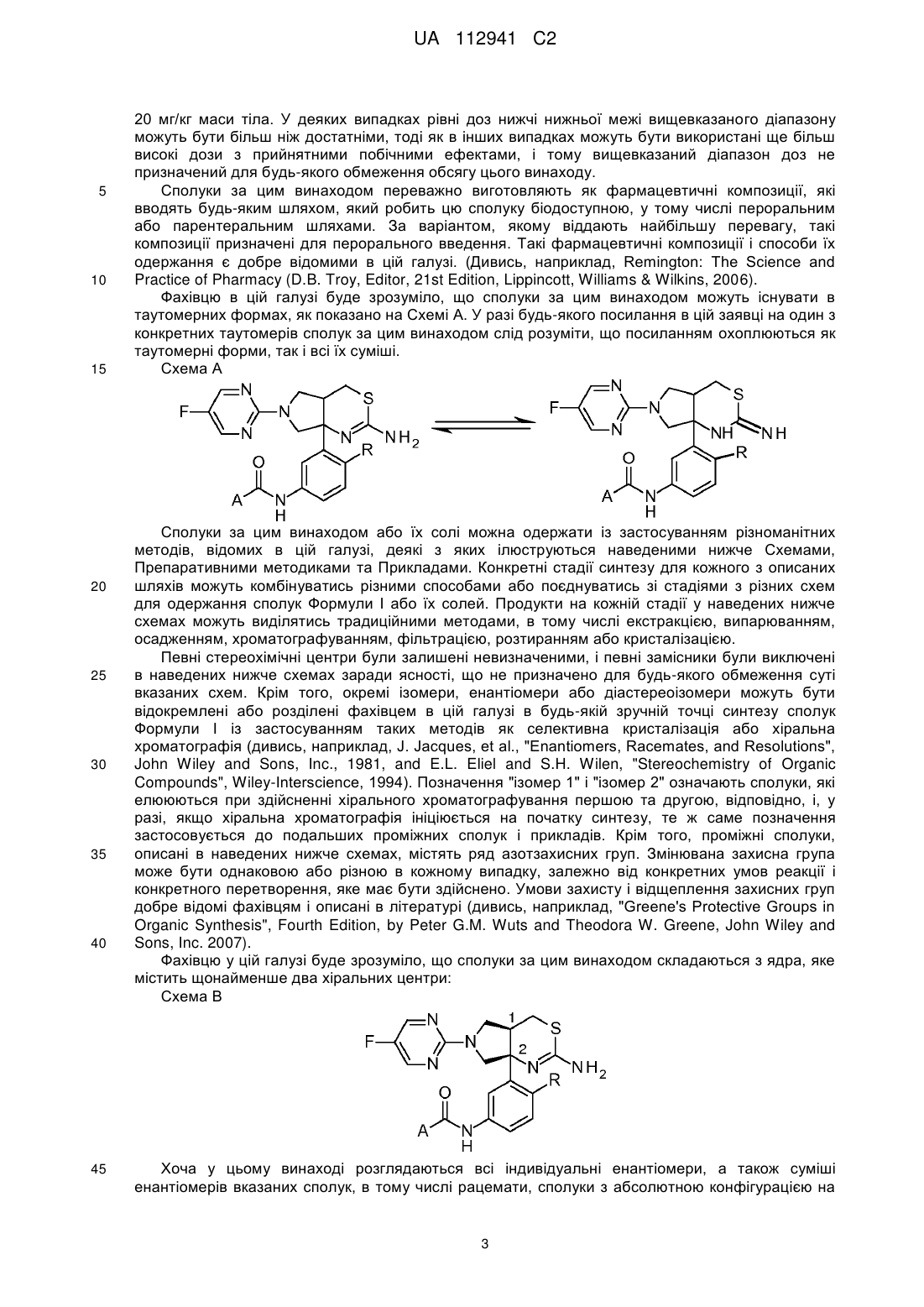
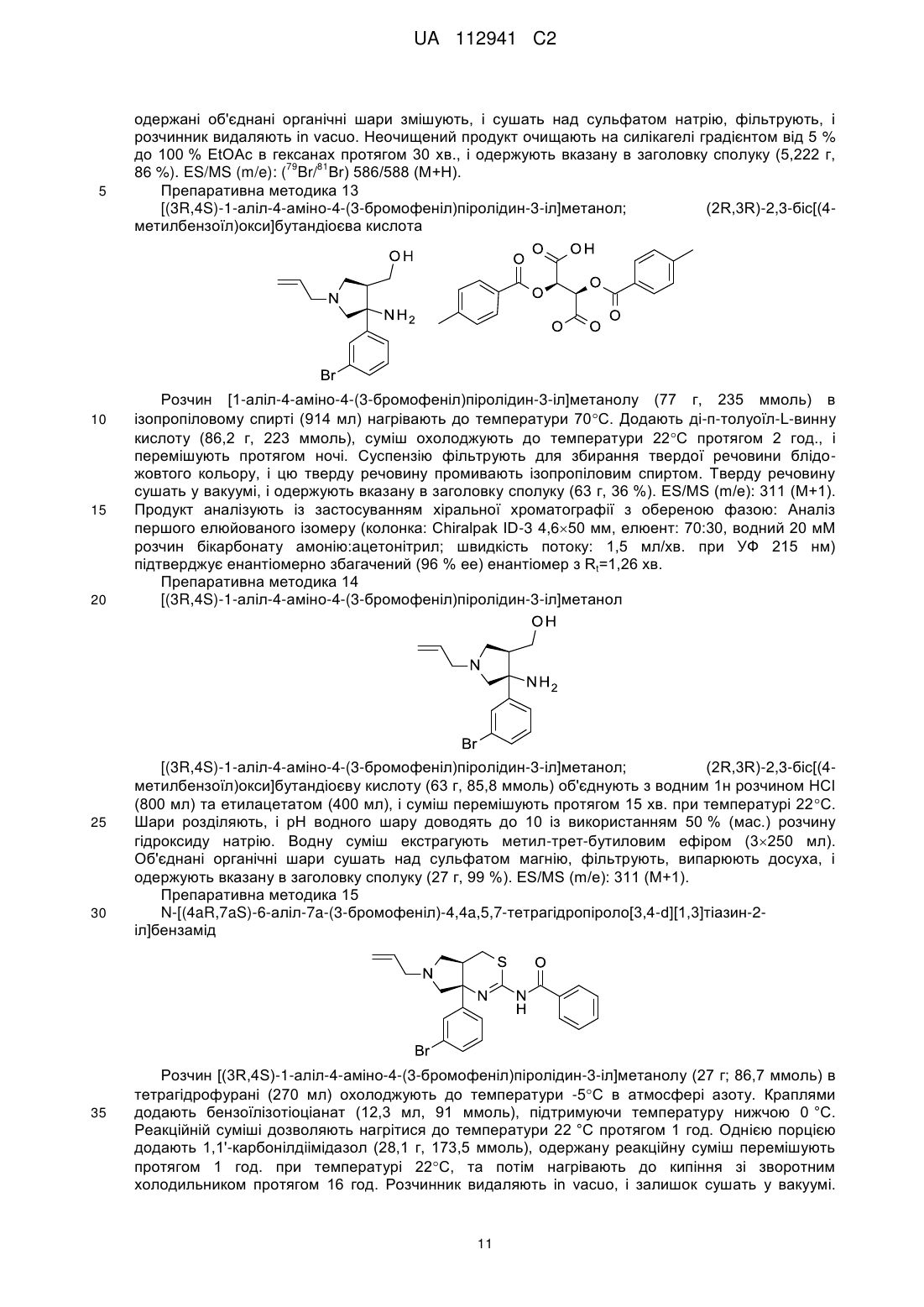
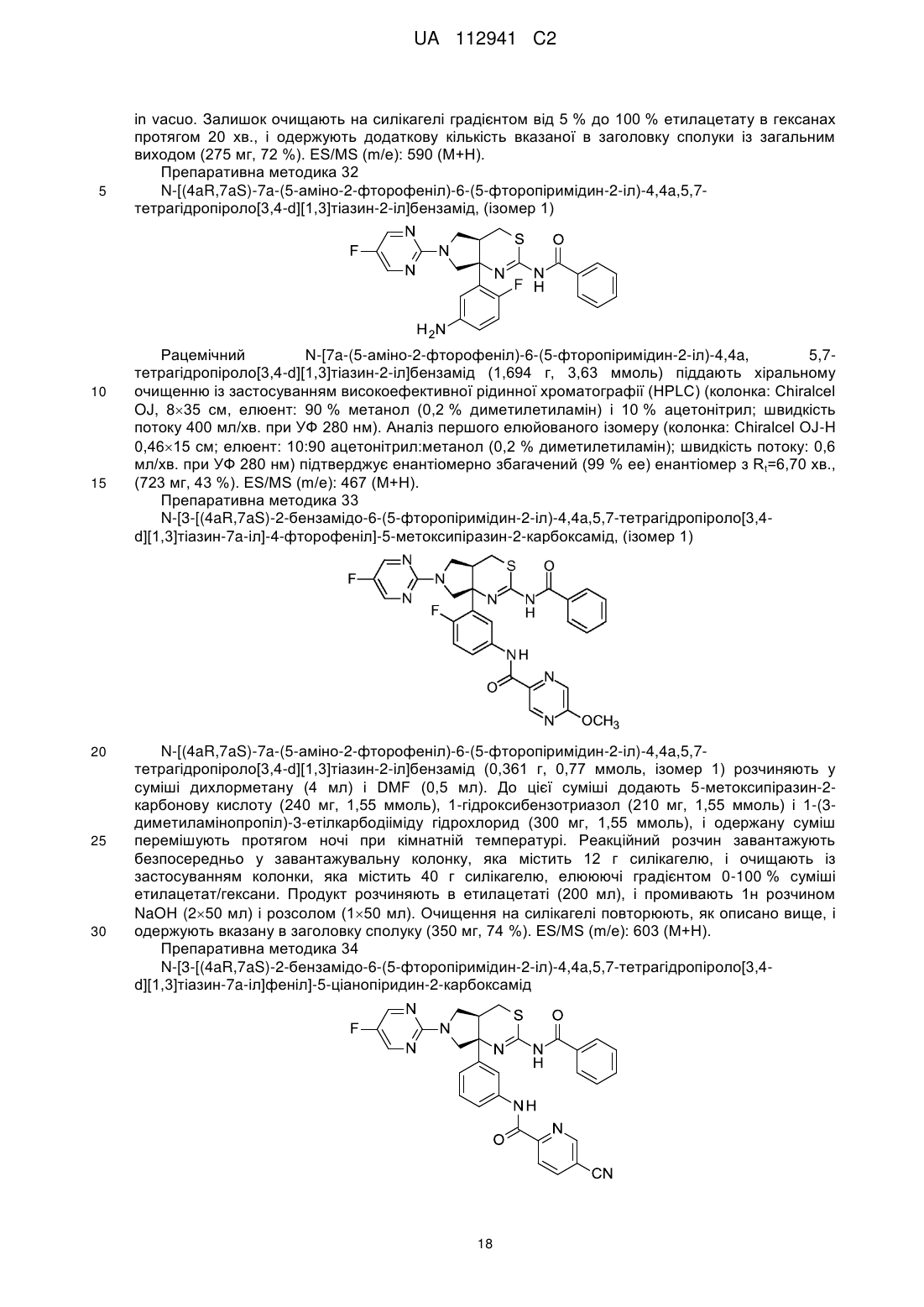
2. Сполука або сіль за п. 1, яка являє собою:
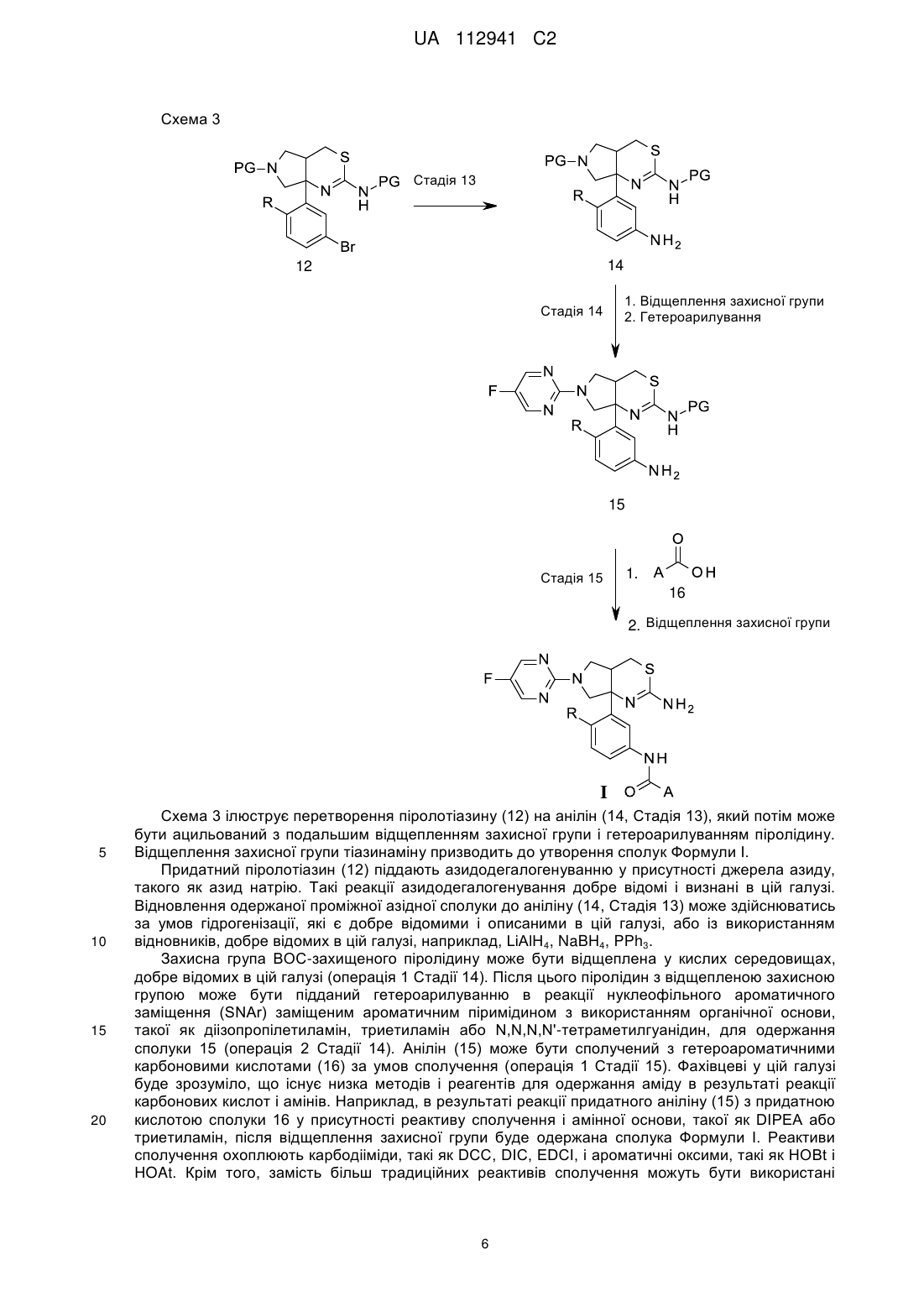
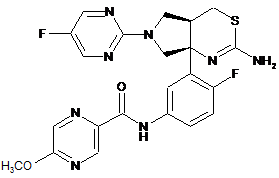 .
.
3. Сполука за п. 2, яка являє собою:
.
4. Спосіб лікування хвороби Альцгеймера у пацієнта, що включає введення пацієнту, який потребує такого лікування, ефективної кількості сполуки за будь-яким з пп. 1-3 або її фармацевтично прийнятної солі.
5. Спосіб запобігання розвитку хвороби Альцгеймера у пацієнта з підвищеним ризиком розвитку цієї хвороби, який включає введення пацієнту, який потребує такого лікування, ефективної кількості сполуки за будь-яким з пп. 1-3 або її фармацевтично прийнятної солі.
6. Сполука або її фармацевтично прийнятна сіль за будь-яким з пп. 1-3 для застосування в терапії.
7. Сполука або її фармацевтично прийнятна сіль за будь-яким з пп. 1-3 для застосування в лікуванні хвороби Альцгеймера.
8. Фармацевтична композиція, яка містить сполуку або її фармацевтично прийнятну сіль за будь-яким з пп. 1-3 з одним або декількома фармацевтично прийнятними носіями, розріджувачами або наповнювачами.
Текст
Реферат: Даний винахід пропонує сполуку Формули (І): N F S N N N O A R N H NH2 , де R являє собою Η або F; і А являє собою: (А), (В), (С) або (D) N F F , (A) F N N N , (B) NC , (C) H3CO N (D); UA 112941 C2 (12) UA 112941 C2 або її фармацевтично прийнятну сіль. UA 112941 C2 5 10 15 20 25 Цей винахід стосується нових тетрагідропіролотіазинових сполук, фармацевтичних композицій, які містять ці сполуки, способів застосування цих сполук для лікування фізіологічних розладів, та проміжних сполук і способів, придатних для синтезу згаданих сполук. Цей винахід стосується лікування хвороби Альцгеймера та інших захворювань і розладів, пов'язаних з -амілоїдним (A-бета) пептидом, нейротоксичним і високоагрегаційним пептидним сегментом білка-попередника амілоїду (АРР). Хвороба Альцгеймера являє собою руйнівний нейродегенеративний розлад, яким уражено мільйони пацієнтів у всьому світі. Приймаючи до уваги, що наявні на цей час у продажу схвалені лікарські засоби забезпечують лише тимчасовий симптоматичний вплив на пацієнта, існує значна незадовільнена потреба в лікуванні хвороби Альцгеймера. Хвороба Альцгеймера характеризується утворенням, агрегацією і осадженням бетаамілоїдного білка у головному мозку. Було показано, що повне або часткове пригнічування секретази (фермент, який розщеплює -сайт білка-попередника амілоїду; ВАСE) вчиняє істотний вплив на пов'язані з бляшками і залежні від бляшок патології у мишачих моделях, що й дозволяє висунути припущення про те, що навіть незначне зниження рівня -амілоїдного пептиду може призвести до довготривалого значного зменшення бляшкового навантаження і синаптичної недостатності із забезпеченням тим самим значного терапевтичного впливу, зокрема, при лікуванні хвороби Альцгеймера. У US 2009/0209755 розкриті сполучені амінодигідротіазинові похідні, які мають BACEпригнічувальну активність, і які розкриті як корисні терапевтичні засоби для нейродегенеративного захворювання, спричиненого -амілоїдним пептидом, такого як деменція альцгеймерівського типу. Крім того, у J. Neuroscience, 31(46), сторінки 16507-16516 (2011), розкритий (S)-4-(2,4-дифторо-5-піримідин-5-ілфеніл)-4-метил-5,6-дигідро-4Н-[1,3]тіазін-2-іламін, який є ЦНС-активним інгібітором BACE, який вводять перорально. Інгібітори BACE, які є сильнодіючими засобами з достатнім проникненням до ЦНС, є бажаними для здійснювання лікування опосередкованих -амілоїдним пептидом розладів, таких як хвороба Альцгеймера. Цей винахід пропонує певні нові сполуки, які є сильнодіючими інгібіторами BACE. Крім того, цей винахід пропонує певні нові сполуки з проникненням до ЦНС. Так, цим винаходом запропонована сполука Формули I Формула I , 30 де R являє собою H або F; і A являє собою: або 35 40 45 або її фармацевтично прийнятна сіль. Цим винаходом також запропонований спосіб лікування хвороби Альцгеймера у пацієнта, що включає введення пацієнту, який потребує такого лікування, ефективної кількості сполуки Формули I або її фармацевтично прийнятної солі. Крім того, цим винаходом запропонований спосіб запобігання розвитку помірних когнітивних порушень у хворобу Альцгеймера у пацієнта, який включає введення пацієнту, який потребує такого лікування, ефективної кількості сполуки Формули I або її фармацевтично прийнятної солі. Цим винаходом також запропонований спосіб пригнічування BACE у пацієнта, що включає введення пацієнту, який потребує такого лікування, ефективної кількості сполуки Формули I або її фармацевтично прийнятної солі. Цим винаходом також запропонований спосіб пригнічування BACE-опосередкованого розщеплення білка-попередника амілоїду, що включає введення пацієнту, який потребує такого лікування, ефективної кількості сполуки Формули I або її фармацевтично прийнятної солі. Цим винаходом також запропонований спосіб пригнічування продукування -амілоїдного пептиду, що включає введення пацієнту, який потребує такого лікування, ефективної кількості 1 UA 112941 C2 5 10 15 20 25 30 35 40 45 50 сполуки Формули I або її фармацевтично прийнятної солі. Крім того, цим винаходом запропонована сполука Формули I або її фармацевтично прийнятна сіль для застосування в терапії, зокрема, для лікування хвороби Альцгеймера або для запобігання розвитку помірних когнітивних порушень у хворобу Альцгеймера. На додаток до цього, цим винаходом запропоноване застосування сполуки Формули I або її фармацевтично прийнятної солі для виготовлення лікарського засобу для лікування хвороби Альцгеймера. Цим винаходом також запропоноване застосування сполуки Формули I або її фармацевтично прийнятної солі для виготовлення лікарського засобу для запобігання розвитку помірних когнітивних порушень у хворобу Альцгеймера. Цим винаходом також запропоноване застосування сполуки Формули I або її фармацевтично прийнятної солі для виготовлення лікарського засобу для пригнічування BACE. Крім того, цим винаходом також запропоноване застосування сполуки Формули I або її фармацевтично прийнятної солі для виготовлення лікарського засобу для пригнічування продукування -амілоїдного пептиду. Крім того, цим винаходом запропонована фармацевтична композиція, яка містить сполуку Формули I або її фармацевтично прийнятну сіль в комбінації з одним або декількома фармацевтично прийнятними носіями, розріджувачами або наповнювачами. У конкретному варіанті здійснення цього винаходу композиція додатково містить один або декілька інших терапевтичних засобів. Цей винахід також охоплює нові проміжні сполуки і способи синтезу сполук Формули I. Помірні когнітивні порушення визначаються як прихована продромальна фаза деменції, пов'язаної з хворобою Альцгеймера, на основі клінічних проявів і наростання у пацієнтів, у яких проявляються помірні когнітивні порушення, симптомів деменціі Альцгеймера з перебігом часу. (Morris, et al., Arch. Neurol., 58, 397-405 (2001); Petersen, et al., Arch. Neurol., 56, 303-308 (1999)). Термін "запобігання розвитку помірних когнітивних порушень у хворобу Альцгеймера" охоплює уповільнення, затримування або обертання напрямку розвитку помірних когнітивних порушень у хворобу Альцгеймера у пацієнта. Сполукою, якій віддається перевага, є: або її фармацевтично прийнятна сіль, при цьому особлива перевага віддається вільній основі. У значенні, вживаному у цьому описі, терміни "лікування" або "лікувати" охоплюють стримування, уповільнення, зупинення або обертання напрямку розвитку або тяжкості існуючого симптому або розладу. У значенні, вживаному у цьому описі, термін "пацієнт" означає людину. Термін "пригнічування продукування -амілоїдного пептиду" означає зниження рівнів амілоїдного пептиду у пацієнта in vivo. У значенні, вживаному у цьому описі, термін "ефективна кількість" означає кількість або дозу сполуки за цим винаходом або її фармацевтично прийнятної солі, яка, при введенні пацієнту разовою або кратною дозою, забезпечує бажаний ефект у пацієнта при діагностуванні або лікуванні. Ефективна кількість може бути легко визначена лікарем, який ставить діагноз, як фахівцем в цій галузі, із застосуванням відомих методик, та із дослідженням результатів, одержаних за аналогічних обставин. При визначенні ефективної кількості для пацієнта, лікарем, який ставить діагноз, до уваги приймається ряд факторів, в тому числі, але без обмеження ними: вигляд пацієнта; його розмір, вік і загальний стан здоров'я; конкретне захворювання або розлад, яким уражений пацієнт; ступінь ураження або тяжкість захворювання чи розладу; реакція окремого пацієнта; конкретна введена сполука; спосіб введення; характеристики біодоступності введеного препарату; вибрана схема приймання лікарського засобу; використання супутнього лікарського засобу; та інші відповідні обставини. Сполуки за цим винаходом є в більшості випадків ефективними в широкому діапазоні доз. Наприклад, добові дози звичайно знаходяться в діапазоні від приблизно 0,01 мг/кг до приблизно 2 UA 112941 C2 5 10 15 20 25 30 35 40 45 20 мг/кг маси тіла. У деяких випадках рівні доз нижчі нижньої межі вищевказаного діапазону можуть бути більш ніж достатніми, тоді як в інших випадках можуть бути використані ще більш високі дози з прийнятними побічними ефектами, і тому вищевказаний діапазон доз не призначений для будь-якого обмеження обсягу цього винаходу. Сполуки за цим винаходом переважно виготовляють як фармацевтичні композиції, які вводять будь-яким шляхом, який робить цю сполуку біодоступною, у тому числі пероральним або парентеральним шляхами. За варіантом, якому віддають найбільшу перевагу, такі композиції призначені для перорального введення. Такі фармацевтичні композиції і способи їх одержання є добре відомими в цій галузі. (Дивись, наприклад, Remington: The Science and Practice of Pharmacy (D.B. Troy, Editor, 21st Edition, Lippincott, Williams & Wilkins, 2006). Фахівцю в цій галузі буде зрозуміло, що сполуки за цим винаходом можуть існувати в таутомерних формах, як показано на Схемі А. У разі будь-якого посилання в цій заявці на один з конкретних таутомерів сполук за цим винаходом слід розуміти, що посиланням охоплюються як таутомерні форми, так і всі їх суміші. Схема А Сполуки за цим винаходом або їх солі можна одержати із застосуванням різноманітних методів, відомих в цій галузі, деякі з яких ілюструються наведеними нижче Схемами, Препаративними методиками та Прикладами. Конкретні стадії синтезу для кожного з описаних шляхів можуть комбінуватись різними способами або поєднуватись зі стадіями з різних схем для одержання сполук Формули I або їх солей. Продукти на кожній стадії у наведених нижче схемах можуть виділятись традиційними методами, в тому числі екстракцією, випарюванням, осадженням, хроматографуванням, фільтрацією, розтиранням або кристалізацією. Певні стереохімічні центри були залишені невизначеними, і певні замісники були виключені в наведених нижче схемах заради ясності, що не призначено для будь-якого обмеження суті вказаних схем. Крім того, окремі ізомери, енантіомери або діастереоізомери можуть бути відокремлені або розділені фахівцем в цій галузі в будь-якій зручній точці синтезу сполук Формули I із застосуванням таких методів як селективна кристалізація або хіральна хроматографія (дивись, наприклад, J. Jacques, et al., "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, Inc., 1981, and E.L. Eliel and S.H. Wilen, "Stereochemistry of Organic Compounds", Wiley-Interscience, 1994). Позначення "ізомер 1" і "ізомер 2" означають сполуки, які елююються при здійсненні хірального хроматографування першою та другою, відповідно, і, у разі, якщо хіральна хроматографія ініціюється на початку синтезу, те ж саме позначення застосовується до подальших проміжних сполук і прикладів. Крім того, проміжні сполуки, описані в наведених нижче схемах, містять ряд азотзахисних груп. Змінювана захисна група може бути однаковою або різною в кожному випадку, залежно від конкретних умов реакції і конкретного перетворення, яке має бути здійснено. Умови захисту і відщеплення захисних груп добре відомі фахівцям і описані в літературі (дивись, наприклад, "Greene's Protective Groups in Organic Synthesis", Fourth Edition, by Peter G.M. Wuts and Theodora W. Greene, John Wiley and Sons, Inc. 2007). Фахівцю у цій галузі буде зрозуміло, що сполуки за цим винаходом складаються з ядра, яке містить щонайменше два хіральних центри: Схема B Хоча у цьому винаході розглядаються всі індивідуальні енантіомери, а також суміші енантіомерів вказаних сполук, в тому числі рацемати, сполуки з абсолютною конфігурацією на 3 UA 112941 C2 5 10 15 20 25 атомах вуглецю, помічених 1 і 2, як показано на схемі B, є сполуками за цим винаходом, яким віддається перевага. Скорочення, які вжиті в цьому описі, визначені відповідно до Aldrichimica Acta, Vol. 17, No. 1, 1984. Інші скорочення визначені так: "APP" означає білок-попередник амілоїда; "BOC" означає трет-бутоксикарбоніл; "CSF" означає цереброспінальну рідину; "DCC" означає 1,3дициклогексилкарбодіімід; "DIC" означає діізопропілкарбодіімід; "DMEM" означає модифіковане за способом Дульбекко середовище Ігла; "DMSO" означає диметилсульфоксид; "EDCI" означає 1-(3-диметиламінопропіл)-3-етилкарбодііміду гідрохлорид; "ee" означає енантіомерний надлишок; "EtOAc" означає етилацетат; "Пр" означає приклад; "F12" означає середовище Ham F12; "FBS" означає ембріональну бичачу сироватку; "FRET" означає резонансний перенос енергії флуоресценції; "HEK" означає лінію клітин нирок людського зародку; "НОАc" означає оцтову кислоту; "HOAt" означає 1-гідрокси-7-азобензотриазол; "HOBt" означає 1гідроксибензотриазолу гідрат; "HPLC" означає високоефективну рідинну хроматографію; "год.” означає годину або години; "IC50" означає концентрацію засобу, яка спричинює 50 % максимальної реакції пригнічення, можливої для цього засобу; "хв" означає хвилину або хвилини; "MTBE" означає метил-трет-бутиловий ефір; "PDAPP" означає тромбоцитарний білокпопередник амілоїда; "Prep" означає препаративну методику; "RFU" означає відносну одиницю флуоресценції, "Rt" означає час утримування; "RT" означає кімнатну температуру; "SCX" означає сильну катіонобмінну смолу; "SFC" означає надкритичну рідинну хроматографію; і "THF" означає тетрагідрофуран. У наведених нижче схемах всі замісники, якщо не вказано інше, є такими, як визначено вище. Реагенти і вихідні матеріали зазвичай є легкодоступними для фахівця в цій галузі. Інші можуть бути виготовлені із застосуванням стандартних прийомів органічної хімії і хімії гетероциклічних сполук, які є аналогічними синтезу відомих структурно подібних сполук, і методик, описаних в наведених нижче Препаративних методиках та Прикладах, включаючи будь-які нові методики. Схема 1 Стадія 1 Step 1 Стадія 2 Step 2 2 1 4 3 Step 3 3 Стадія Х= галоген X = halogen PG= захисна група PG = protecting group 5 Step 7 7 Стадія Step 4 Стадія 4 6 30 35 Стадія 5 Step 5 7 Step 6 Стадія 6 8 9 Схема 1 ілюструє одержання оксимів (4) і (8). Кожен з оксимів може бути використаним для одержання біциклічного ізоксазола (5). Заміщена ароматична група може бути введена на різних стадіях синтезу, як показано на Схемі 1, Стадія 1 і Стадія 7. "PG" являє собою захисну групу, розроблену для аміногрупи, таку як карбаматні групи і алільна група. Такі групи добре відомі і визнані в цій галузі. У 2-стадійній реакції кетон з бета-галогеном (1) може бути алкілований (3, Стадія 1) захищеним аліламіном (2) із застосуванням неорганічної основи, такої як карбонат калію, і потім оброблений гідрохлоридом гідроксиламіну і органічною основою, такою як піридин, в полярному протонному розчиннику, такому як етанол, для одержання оксиму (4, Стадія 2). Потім цей оксим 4 UA 112941 C2 5 10 (4) може бути перетворений на біциклічний ізоксазол (5) шляхом 3+2 циклізації декількома методами, такими як прогрівання оксиму (4) в неполярному розчиннику, такому як толуол або ксилол, для утворення біциклічного ізоксазолу (5, Стадія 3). Альтернативно оксим може бути одержаний, виходячи з диметилацеталю (6), який обробляють кислотою, такою як мурашина кислота, для утворення альдегіду (7, Стадія 4). Потім, на Стадії 5, альдегід (7) може бути перетворений на оксим (8) гідрохлоридом гідроксиламіну і основою, такою як тригідрат ацетату натрію. Біциклічний ізоксазол (9) може бути одержаний з оксиму (8), як показано на Стадії 6, із застосуванням водного розчину гіпохлориту натрію. Потім, на Стадії 7, захищений біциклічний ізоксазол (9) вводять в реакцію з ароматичним літійорганічним реактивом або реактивом Гріньяра, і одержують захищений біциклічний ізоксазол (5). Схема 2 Стадія 8 Step 8 5 Стадія 9 Step 9 11 10 Стадія 10 Step 10 Стадія Step 1111 Стадія12 Step 12 12 13 15 20 25 Схема 2 ілюструє різні способи одержання захищеного піролотіазину (12). Захищений біциклічний ізоксазол (5) може бути оброблений порошковим Zn в оцтовій кислоті або нікелем Ренея в полярному розчиннику, такому як етанол, в умовах гідрогенізації під тиском, для одержання амінопіролідинметанолу (13, Стадія 11). Далі амінопіролідинметанол (13) вводять в реакцію з бензоїлізотіоціанатом в полярному розчиннику, такому як THF, після чого додають 1,1-карбонілдіімідазол (CDI), і одержують конденсований захищений піролідинтіазин (12, Стадія 12). Альтернативно, амін біциклічного ізоксазолу (5) може бути введений в реакцію з бензоїлізотіоціанатом для одержання тіосечовини (10, Стадія 8), і потім, на Стадії 9, ізоксазолове кільце може бути розкрите порошковим цинком в оцтовій кислоті для одержання гідроксилвмісної сполуки (11). Після цього згадана гідроксилвмісна сполука (11) може бути оброблена CDI в полярному апротонному розчиннику, такому як THF або 1-хлор-N,N,2триметилпропеніламін в DCM, для одержання конденсованого захищеного піролідинтіазину (12, Стадія 10). Конденсований піролідинтіазин (12) також можна одержати за допомогою реакції Міцунобу, такої, у якій використовують трифенілфосфін і діізопропілазодикарбоксилат (DIAD). 5 UA 112941 C2 Схема 3 Стадія 13 Step 13 14 12 1. Відщеплення захисної групи 1. Deprotect Стадія 14 Step 14 2. Гетероарилування 2. Heteroarylation 15 Стадія 15 Step 15 1. 16 Відщеплення захисної групи 2. Deprotect I 5 10 15 20 Схема 3 ілюструє перетворення піролотіазину (12) на анілін (14, Стадія 13), який потім може бути ацильований з подальшим відщепленням захисної групи і гетероарилуванням піролідину. Відщеплення захисної групи тіазинаміну призводить до утворення сполук Формули I. Придатний піролотіазин (12) піддають азидодегалогенуванню у присутності джерела азиду, такого як азид натрію. Такі реакції азидодегалогенування добре відомі і визнані в цій галузі. Відновлення одержаної проміжної азідної сполуки до аніліну (14, Стадія 13) може здійснюватись за умов гідрогенізації, які є добре відомими і описаними в цій галузі, або із використанням відновників, добре відомих в цій галузі, наприклад, LiAlH4, NaBH4, PPh3. Захисна група ВОС-захищеного піролідину може бути відщеплена у кислих середовищах, добре відомих в цій галузі (операція 1 Стадії 14). Після цього піролідин з відщепленою захисною групою може бути підданий гетероарилуванню в реакції нуклеофільного ароматичного заміщення (SNAr) заміщеним ароматичним піримідином з використанням органічної основи, такої як діізопропілетиламін, триетиламін або N,N,N,N'-тетраметилгуанідин, для одержання сполуки 15 (операція 2 Стадії 14). Анілін (15) може бути сполучений з гетероароматичними карбоновими кислотами (16) за умов сполучення (операція 1 Стадії 15). Фахівцеві у цій галузі буде зрозуміло, що існує низка методів і реагентів для одержання аміду в результаті реакції карбонових кислот і амінів. Наприклад, в результаті реакції придатного аніліну (15) з придатною кислотою сполуки 16 у присутності реактиву сполучення і амінної основи, такої як DIPEA або триетиламін, після відщеплення захисної групи буде одержана сполука Формули I. Реактиви сполучення охоплюють карбодііміди, такі як DCC, DIC, EDCI, і ароматичні оксими, такі як HOBt і HOAt. Крім того, замість більш традиційних реактивів сполучення можуть бути використані 6 UA 112941 C2 5 10 15 20 25 30 35 40 45 уронієві або фосфонієві солі ненуклеофільних аніонів, такі як HBTU, HATU, PyBOP та PyBrOP. Для прискорення реакції можуть бути використані домішки, такі як DMAP. Альтернативно, захищений анілінамін (15) може бути ацильований із використанням заміщених бензоїлхлоридів у присутності основи, такої як триетиламін або піридин. Потім для одержання сполук Формули I від захищеного тіазинаміну може бути відщеплена захисна група із використанням органічної основи, такої як піридин і метилгідроксиламіну гідрохлорид, в полярному апротонному розчиннику, такому як етанол, або із застосуванням неорганічної основи, такої як гідроксид літію, в метанолі. На факультативній стадії фармацевтично прийнятну сіль сполуки Формули I можна одержати шляхом реакції придатної вільної основи Формули I з придатною фармацевтично прийнятною кислотою у придатному розчиннику за стандартних умов. Крім того, утворення таких солей може відбуватись одночасно з відщепленням захисної групи азоту. Одержання таких солей є добре відомим і визнаним у цій галузі. Дивись, наприклад, Gould, P.L., "Salt selection for basic drugs," International Journal of Pharmaceutics, 33: 201-217 (1986); Bastin, R.J., et al., "Salt Selection and Optimization Procedures for Pharmaceutical New Chemical Entities," Organic Process Research and Development, 4: 427-435 (2000); та Berge, S.M., et al., "Pharmaceutical Salts," Journal of Pharmaceutical Sciences, 66: 1-19, (1977). Фахівцю в цій галузі буде зрозуміло, що сполука Формули I легко перетворюється на фармацевтично прийнятну сіль, і може бути виділена у вигляді фармацевтично прийнятної солі, такої як гідрохлоридна сіль. Препаративні методики та Приклади Наведені нижче препаративні методики та приклади пояснюють цей винахід. Якщо не вказано інше, сполуки, наведені у цьому описі, названі та пронумеровані із застосуванням програмного продукту Symyx Draw, версія 3.2 або версія 4.0 (компанія Symyx Solutions, Inc.) або IUPACNAME ACDLABS. Препаративна методика 1 1-(3-бромофеніл)-2-(діаліламіно)етанон Карбонат калію (38,8 г, 281 ммоль) додають до 3-бромфенацилброміду (60 г, 216 ммоль) в ацетонітрилі (430 мл), і суміш охолоджують в атмосфері азоту до температури 0С. Діаліламін (34,6 мл, 280,63 ммоль) додають краплями протягом 1 год., і дозволяють реакційній суміші протягом ночі нагрітися до температури 22С. Неочищену реакційну суміш концентрують, і залишок розподіляють у воді (300 мл) і MTBE (300 мл). Водний шар відкидають, а органічний шар промивають водою (100 мл, 2) і розсолом (100 мл). Цей органічний шар сушать над сульфатом натрію, фільтрують, і розчинник випарюють до постійної маси, і одержують вказану в заголовку сполуку (62 г, 98 %). ES/MS (m/e): 294 (М+1). Препаративна методика 2 Бензил-N-(2,2-диметоксиетил)карбамат Розчин аміноацетальдегіду диметилацеталю (25 мл, 229 ммоль) в толуолі (120 мл) обробляють при температурі 0С 4,85 М розчином гідроксиду натрію (70,8 мл, 343,5 ммоль). Суміш перемішують при температурі 0С протягом 10 хв., і додають бензілхлорформіат (33,8 мл, 229 ммоль), підтримуючи внутрішню температуру під час додавання нижчою 20С. Суміш нагрівають до кімнатної температури протягом 4 год. Органічний шар відокремлюють, промивають розсолом, сушать над сульфатом натрію, і концентрують досуха, і одержують вказану в заголовку сполуку (54 г, 98 %). ES/MS (m/e): 240 (М+Н). Препаративна методика 3 Бензил-N-аліл-N-(2,2-диметоксиетил)карбамат 7 UA 112941 C2 5 10 15 20 25 30 35 Розчин бензил-N-(2,2-диметоксиетил)карбамату (50 г, 208,9 ммоль) в толуолі (180 мл) обробляють твердим гідроксидом калію (51,6 г, 919,69 ммоль) в атмосфері азоту. Через 10 хв. додають бензилтриетиламонійхлорид (0,8 г, 3,1 ммоль). Ще через 10 хв. краплями протягом 10 хв. додають розчин алілброміду (33 г, 272,8 ммоль) в толуолі (50 мл). Одержану суміш перемішують при температурі 50С протягом 48 год. Суміш охолоджують до кімнатної температури, і гасять водою. Органічний шар відокремлюють, промивають розсолом, сушать над сульфатом магнію, і концентрують досуха, і одержують вказану в заголовку сполуку (44 г, 75 %). ES/MS (m/e): 280 (М+Н). Препаративна методика 4 Бензил-N-аліл-N-(2-оксоетил)карбамат Розчин бензил-N-аліл-N-(2,2-диметоксиетил)карбамату (30 г, 107 ммоль) в мурашиній кислоті (36,8 мл, 860 ммоль) і воді (4,84 мл) перемішують при кімнатній температурі протягом ночі. Суміш концентрують, і розбавляють сумішшю гексани/етилацетат (1:2) та вода. Органічний шар відокремлюють, промивають розсолом до pH=6, і сушать над сульфатом натрію. Розчинник випарюють, і одержують вказану в заголовку сполуку (25 г, 99 %). ES/MS (m/e): 234 (М+Н). Препаративна методика 5 1-(3-бромофеніл)-2-(діаліламіно)етаноноксим Розчин 1-(3-бромофеніл)-2-(діаліламіно)етанону (60 г, 204,7 ммоль) в етанолі (720 мл) і піридині (24,8 мл, 307 ммоль) перемішують 15 хв. при температурі 22С. До вказаного розчину порціями протягом 1 год. додають гідроксиламіну гідрохлорид (17 г, 246 ммоль). Реакційну суміш нагрівають до температури 50С протягом 2 год., після чого нагрівають до температури 70С протягом 16 год. Розчинник випарюють, і залишок розподіляють у воді (300 мл) і метилтрет-бутиловому ефірі (300 мл). Органічний шар відокремлюють, і промивають водою (100 мл, 2) і розсолом (100 мл). Цей органічний шар сушать над сульфатом натрію, фільтрують, і випарюють досуха, і одержують вказану в заголовку сполуку (75,5 г, 79 %). ES/MS (m/e): 309 (М+1). Препаративна методика 6 Бензил-N-аліл-N-[2-гідроксиіміноетил]карбамат Розчин бензил-N-аліл-N-(2-оксоетил)карбамату (25 г, 107 ммоль) в ацетонітрилі (150 мл) обробляють гідроксиламіну гідрохлоридом (9,68 г, 139 ммоль) і розчином тригідрату ацетату натрію (16 г, 117,9 ммоль) у воді (75 мл). Суміш перемішують при кімнатній температурі протягом ночі. Ацетонітрил випарюють, і водний розчин екстрагують етилацетатом. Органічний 8 UA 112941 C2 шар відокремлюють, сушать над сульфатом магнію, і концентрують у вакуумі, і одержують вказану в заголовку сполуку (24 г, 90 %). ES/MS (m/e): 249 (М+Н). Препаративна методика 7 5-аліл-6a-(3-бромофеніл)-3,3a,4,6-тетрагідро-1H-піроло[3,4-с]ізоксазол 5 10 15 20 25 30 35 40 Неочищений 1-(3-бромофеніл)-2-(діаліламіно)етаноноксим (75,5 г, 195,34 ммоль) розчиняють у толуолі (600 мл), і кип'ятять зі зворотним холодильником протягом 12 год. Розчинник випарюють in vacuo, і залишок розчиняють в суміші водного 1н розчину HCl (1 л) і метил-трет-бутилового ефіру (300 мл). Суміш перемішують протягом 15 хв., і додають діатомову землю (10 г). Цю суміш перемішують протягом додаткових 20 хв., і фільтрують через діатомову землю. Відфільтрований осад додатково промивають водним 1н розчином HCl (200 мл) і метил-трет-бутиловим ефіром (200 мл). Органічний шар відокремлюють, і промивають 1н розчином HCl (2100 мл). Водні шари об'єднують, і значення pH доводять до 9 із використанням NaOH (50 % мас.). Водну суміш екстрагують метил-трет-бутиловим ефіром (3250 мл). Органічні шари об'єднують, сушать над сульфатом натрію, і фільтрують. Фільтрат упарюють, сушать у вакуумі, і одержують тверду речовину червоного кольору (60 г). Цю тверду речовину червоного кольору розводять гептаном (600 мл), і суміш нагрівають до кипіння протягом 20 хв. Додають деревне вугілля (2 г), і суміш фільтрують через діатомову землю. Фільтрати концентрують при атмосферному тиску для доведення кінцевого об'єму до 300 мл. Розчин охолоджують до температури 22С, і перемішують протягом 3 год. Тверду речовину блідожовтого кольору відфільтровують, сушать у вакуумі до постійної маси, і одержують вказану в заголовку сполуку (40 г, 60 %). ES/MS (m/e): 309 (М+1). Препаративна методика 8 Бензил-3,3а, 4,6-тетрагідропіроло[3,4-с]ізоксазол-5-карбоксилат Розчин бензил-N-аліл-N-[2-гідроксиіміноетил]карбамату (24 г, 96,6 ммоль) в дихлорметані (338 мл) протягом 10 хв. обробляють краплями 5 % (мас.) водного розчину гіпохлориту натрію (106,08 ммоль, 143,06 мл). Одержану суміш перемішують при кімнатній температурі протягом ночі. Реакцію зупиняють 40 % водним розчином бісульфіту натрію (7 г). Органічний шар відокремлюють, сушать над сульфатом магнію, і концентрують у вакуумі. Неочищений продукт очищають на силікагелі, елююючі 5 % розчином етилацетату в гексанах, і одержують вказану в заголовку сполуку (18 г, 75 %). ES/MS (m/e): 247 (М+Н). Препаративна методика 9 Бензил-6a-(5-бромо-2-фторофеніл)-3,3a,4,6-тетрагідро-1H-піроло[3,4-с]ізоксазол-5карбоксилат 1,6 М розчин н-бутиллітію в гексанах (25,4 мл, 40,6 ммоль) додають краплями до розчину (78С) 4-бромо-1-фторо-2-йодбензолу (12,22 г, 40,6 ммоль) в тетрагідрофурані (60 мл), і одержують розчин жовтого кольору, який перемішують при температурі -78С протягом 15 хв. Ефірат трифториду бору (5,14 мл, 40,6 ммоль) додають до окремого розчину (-78C) бензил3,3a, 4,6-тетрагідропіроло[3,4-с]ізоксазол-5-карбоксилату (5 г, 20,3 ммоль) в тетрагідрофурані (60 мл), і суміш перемішують при температурі -78С протягом 5 хв. Цей розчин через канюлю 9 UA 112941 C2 5 10 15 20 25 30 35 додають до заздалегідь підготовленої (-78C) літійорганічної суміші. Об'єднану суміш перемішують протягом 30 хв. при температурі -78С. Суміш гасять насиченим водним розчином хлориду амонію, і нагрівають до кімнатної температури. Цю суміш екстрагують етилацетатом (3), і органічні екстракти об'єднують, сушать над сульфатом натрію, фільтрують, і розчинник видаляють in vacuo. Неочищений продукт очищають на силікагелі градієнтом від 5 % до 100 % етилацетату в гексанах протягом 35 хв., і одержують вказану в заголовку сполуку (2,27 г, 27 %). 79 81 ES/MS (m/e): ( Br/ Br) 421/423 (М+Н). Препаративна методика 10 Бензил-1-(бензоїлкарбамотіоїл)-6a-(5-бромо-2-фторофеніл)-3,3a,4,6-тетрагідропіроло[3,4c]ізоксазол-5-карбоксилат Бензоїлізотіоціанат (2,87 мл, 21,28 ммоль) додають краплями до розчину бензил-6а-(5бромо-2-фторофеніл)-3,3a,4,6-тетрагідро-1H-піроло[3,4-с]ізоксазол-5-карбоксилату (5,977 г, 14,2 ммоль) в тетрагідрофурані (95 мл), і перемішують протягом ночі в атмосфері азоту. Розчинник видаляють in vacuo. Неочищений продукт очищають на силікагелі градієнтом від 5 % до 100 % EtOAc в гексанах протягом 30 хв, і одержують вказану в заголовку сполуку (6,05 г, 73 %). ES/MS 79 81 (m/e): ( Br/ Br) 584/586 (М+Н). Препаративна методика 11 [1-аліл-4-аміно-4-(3-бромофеніл)піролідин-3-іл]метанол Розчин (22С) 5-аліл-6a-(3-бромофеніл)-3,3a,4,6-тетрагідро-1H-піроло[3,4-с]ізоксазолу (40 г, 129,4 ммоль) в оцтовій кислоті (400 мл) обробляють цинковим пилом (42,3 г, 646,8 ммоль), який додають однією порцією. Реакційну суміш енергійно перемішують при кімнатній температурі протягом 1 год. Додають етилацетат (400 мл), і суміш фільтрують через діатомову землю. Фільтрат випарюють, і залишок сушать у вакуумі. Залишок розподіляють у воді (300 мл) і MTBE (300 мл). Значення pH доводять до 8 із використанням 50 % (мас.) розчину гідроксиду натрію, і органічний шар відокремлюють, сушать над сульфатом натрію, і фільтрують. Фільтрат випарюють, і залишок сушать у вакуумі, і одержують вказану в заголовку сполуку (41 г, 97 %). ES/MS (m/e): 311 (М+1). Препаративна методика 12 Бензил-3-(бензоїлкарбамотіоїламіно)-3-(5-бромо-2-фторофеніл)-4-(гідроксиметил)піролідин1-карбоксилат Суміш бензил-1-(бензоїлкарбамотіоїл)-6a-(5-бромо-2-фторофеніл)-3,3a,4,6тетрагідропіроло[3,4-c]ізоксазол-5-карбоксилату (6,05 г, 10,4 ммоль) і цинку (пил, 10 мкм)(6,77 г, 103,5 ммоль) перемішують в оцтовій кислоті (52 мл) при кімнатній температурі протягом ночі в атмосфері азоту. Реакційну суміш розбавляють етилацетатом, і фільтрують через діатомову землю. Розчинник видаляють in vacuo, і залишок розбавляють етилацетатом, водою і насиченим водним розчином бікарбонату натрію. Суміш екстрагують етилацетатом (3), 10 UA 112941 C2 5 10 15 20 25 30 35 одержані об'єднані органічні шари змішують, і сушать над сульфатом натрію, фільтрують, і розчинник видаляють in vacuo. Неочищений продукт очищають на силікагелі градієнтом від 5 % до 100 % EtOAc в гексанах протягом 30 хв., і одержують вказану в заголовку сполуку (5,222 г, 79 81 86 %). ES/MS (m/e): ( Br/ Br) 586/588 (М+Н). Препаративна методика 13 [(3R,4S)-1-аліл-4-аміно-4-(3-бромофеніл)піролідин-3-іл]метанол; (2R,3R)-2,3-біс[(4метилбензоїл)окси]бутандіоєва кислота Розчин [1-аліл-4-аміно-4-(3-бромофеніл)піролідин-3-іл]метанолу (77 г, 235 ммоль) в ізопропіловому спирті (914 мл) нагрівають до температури 70С. Додають ді-п-толуоїл-L-винну кислоту (86,2 г, 223 ммоль), суміш охолоджують до температури 22С протягом 2 год., і перемішують протягом ночі. Суспензію фільтрують для збирання твердої речовини блідожовтого кольору, і цю тверду речовину промивають ізопропіловим спиртом. Тверду речовину сушать у вакуумі, і одержують вказану в заголовку сполуку (63 г, 36 %). ES/MS (m/e): 311 (М+1). Продукт аналізують із застосуванням хіральної хроматографії з обереною фазою: Аналіз першого елюйованого ізомеру (колонка: Chiralpak ID-3 4,650 мм, елюент: 70:30, водний 20 мМ розчин бікарбонату амонію:ацетонітрил; швидкість потоку: 1,5 мл/хв. при УФ 215 нм) підтверджує енантіомерно збагачений (96 % ee) енантіомер з Rt=1,26 хв. Препаративна методика 14 [(3R,4S)-1-аліл-4-аміно-4-(3-бромофеніл)піролідин-3-іл]метанол [(3R,4S)-1-аліл-4-аміно-4-(3-бромофеніл)піролідин-3-іл]метанол; (2R,3R)-2,3-біс[(4метилбензоїл)окси]бутандіоєву кислоту (63 г, 85,8 ммоль) об'єднують з водним 1н розчином HCl (800 мл) та етилацетатом (400 мл), і суміш перемішують протягом 15 хв. при температурі 22С. Шари розділяють, і pH водного шару доводять до 10 із використанням 50 % (мас.) розчину гідроксиду натрію. Водну суміш екстрагують метил-трет-бутиловим ефіром (3250 мл). Об'єднані органічні шари сушать над сульфатом магнію, фільтрують, випарюють досуха, і одержують вказану в заголовку сполуку (27 г, 99 %). ES/MS (m/e): 311 (М+1). Препаративна методика 15 N-[(4aR,7aS)-6-аліл-7a-(3-бромофеніл)-4,4a,5,7-тетрагідропіроло[3,4-d][1,3]тіазин-2іл]бензамід Розчин [(3R,4S)-1-аліл-4-аміно-4-(3-бромофеніл)піролідин-3-іл]метанолу (27 г; 86,7 ммоль) в тетрагідрофурані (270 мл) охолоджують до температури -5С в атмосфері азоту. Краплями додають бензоїлізотіоціанат (12,3 мл, 91 ммоль), підтримуючи температуру нижчою 0 °C. Реакційній суміші дозволяють нагрітися до температури 22 °C протягом 1 год. Однією порцією додають 1,1'-карбонілдіімідазол (28,1 г, 173,5 ммоль), одержану реакційну суміш перемішують протягом 1 год. при температурі 22С, та потім нагрівають до кипіння зі зворотним холодильником протягом 16 год. Розчинник видаляють in vacuo, і залишок сушать у вакуумі. 11 UA 112941 C2 5 10 15 20 25 30 35 40 Неочищений матеріал розподіляють в метил-трет-бутиловому ефірі (500 мл) і воді (250 мл). Органічний шар відокремлюють, сушать над сульфатом магнію, фільтрують, і випарюють досуха. Неочищений матеріал очищають на силікагелі градієнтом від 90/10 до 60/40 суміші метиленхлорид/етилацетат, і одержують вказану в заголовку сполуку (27 г, 68 %). ES/MS (m/e): 456 (М+1). Препаративна методика 16 Бензил-2-бензамідо-7a-(5-бромо-2-фторофеніл)-4,4a,5,7-тетрагідропіроло[3,4-d][1,3]тіазин-6карбоксилат 1,1'-карбонілдіімідазол (2,87 г, 17,7 ммоль) додають до розчину бензил-3(бензоїлкарбамотіоїламіно)-3-(5-бромо-2-фторофеніл)-4-(гідроксиметил)піролідин-1карбоксилату (5,198 г, 8,86 ммоль) в тетрагідрофурані (52 мл). Суміш перемішують протягом 1,5 год. при кімнатній температурі, після чого реакційну суміш гріють при кип'ятінні зі зворотним холодильником протягом ночі в атмосфері азоту. Реакційну суміш охолоджують, розбавляють водою, і екстрагують етилацетатом (3). Органічні шари об'єднують, сушать над сульфатом натрію, фільтрують, і розчинник видаляють in vacuo. Неочищений продукт очищають на силікагелі градієнтом від 5 % до 100 % EtOAc в гексанах протягом 30 хв., і одержують вказану в 79 81 заголовку сполуку (2,93 г, 58 %). ES/MS (m/e): ( Br/ Br) 568/570 (М+Н). Препаративна методика 17 N-[(4aR,7aS)-7a-(3-бромофеніл)-4a, 5,6,7-тетрагідро-4H-піроло[3,4-d][1,3]тіазін-2-іл]бензамід Суміш N-[(4aR,7aS)-6-аліл-7a-(3-бромофеніл)-4,4a, 5,7-тетрагідропіроло[3,4-d][1,3]тіазин-2іл]бензаміду (1 г, 2,19 ммоль) і N,N-диметилбарбітурової кислоти (0,868 г, 5,48 ммоль) в хлороформі (22 мл), яка має кімнатну температуру, знегажують шляхом барботування азоту через одержану суспензію при кімнатній температурі протягом 5 хв. Суміш обробляють тетракіс(трифенілфосфін)паладієм (0,261 г, 219 мкмоль), і перемішують протягом 1,5 год. в атмосфері азоту. В окремій колбі суміш N-[(4aR,7aS)-6-аліл-7a-(3-бромофеніл)-4,4a, 5,7-тетрагідропіроло[3,4d][1,3]тіазин-2-іл]бензаміду (22,2 г, 48,6 ммоль) і N,N-диметилбарбітурової кислоти (19,28 г, 121,6 ммоль) в хлороформі (486 мл) знегажують шляхом барботування азоту через одержану суспензію при кімнатній температурі протягом 5 хв. Суміш обробляють тетракіс(трифенілфосфін)паладієм (5,79 г, 4,86 ммоль), і перемішують протягом 2 год. в атмосфері азоту. Дві реакційні суміші об'єднують, розчинник видаляють in vacuo, і одержують неочищений продукт. Неочищений матеріал очищають на силікагелі градієнтом від 0,5 % до 10 % метанолу у дихлорметані протягом 30 хв., і одержують вказану в заголовку сполуку (22,4 г, 100 %). ES/MS 79 81 (m/e): ( Br/ Br) 416/418 (М+Н). Препаративна методика 18 N-[7a-(5-бромо-2-фторофеніл)-4a,5,6,7-тетрагідро-4H-піроло[3,4-d][1,3]тіазин-2-іл]бензамід Йодотриметилсилан (2,21 мл, 15,46 ммоль) краплями додають до розчину, що має кімнатну температуру, бензил-2-бензамідо-7a-(5-бромо-2-фторофеніл)-4,4a,5,7-тетрагідропіроло[3,4 12 UA 112941 C2 5 10 15 20 25 30 35 40 d][1,3]тіазин-6-карбоксилату (2,93 г, 5,15 ммоль) в ацетонітрилі (44 мл). Реакційну суміш перемішують при кімнатній температурі протягом двох годин, і розчинник видаляють in vacuo. Неочищений продукт очищають на колонці з SCX із застосуванням спочатку суміші дихлорметан:метанол (3:1), та потім суміші дихлорметан:7н розчин аміаку в метанолі (2:1), і 79 81 одержують вказану в заголовку сполуку (2,098 г, 94 %). ES/MS (m/e): ( Br/ Br) 434/436 (М+Н). Препаративна методика 19 трет-бутил-(4aR,7aS)-2-бензамідо-7a-(3-бромофеніл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-6-карбоксилат Розчин, що має кімнатну температуру, N-[(4aR,7aS)-7a-(3-бромофеніл)-4a,5,6,7-тетрагідро4H-піроло[3,4-d][1,3]тіазін-2-іл]бензаміду (22,4 г, 36,69 ммоль) в дихлорметані (367 мл) обробляють спочатку ди-трет-бутилдикарбонатом (8,81 г, 40,36 ммоль), потім - триетиламіном (7,67 мл, 55,04 ммоль), і одержану реакційну суміш перемішують при кімнатній температурі протягом 1 год. в атмосфері азоту. Розчинник видаляють in vacuo, і неочищений продукт очищають на силікагелі градієнтом від 5 % до 100 % етилацетату в гексанах протягом 25 хв., і 79 81 одержують вказану в заголовку сполуку (20,22 г, 100 %). ES/MS (m/e): ( Br/ Br) 516/518 (М+Н). Препаративна методика 20 трет-бутил-2-бензамідо-7a-(5-бромо-2-фторофеніл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-6-карбоксилат До розчину N-[7a-(5-бромо-2-фторофеніл)-4a,5,6,7-тетрагідро-4H-піроло[3,4-d][1,3]тіазин-2іл]бензаміду (2,098 г, 4,83 ммоль) в дихлорметані (48 мл) додають ди-трет-бутилдикарбонат (1,16 г, 5,31 ммоль) і триетиламін (1,01 мл, 7,25 ммоль). Реакційну суміш перемішують протягом 1 год. при кімнатній температурі в атмосфері азоту. Розчинник видаляють in vacuo, і неочищений продукт очищають на силікагелі градієнтом від 5 % до 100 % EtOAc в гексанах 79 81 протягом 30 хв., і одержують вказану в заголовку сполуку (2,556 г, 99 %). ES/MS (m/e): ( Br/ Br) 534/536 (М+Н). Препаративна методика 21 трет-бутил-(4aR,7aS)-7a-(3-амінофеніл)-2-бензамідо-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-6-карбоксилат Розчин трет-бутил-(4aR,7aS)-2-бензамідо-7a-(3-бромофеніл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-6-карбоксилату (5 г, 9,7 ммоль) і транс-N,N'-диметил-1,2-циклогександіаміну (220,3 мг, 1,5 ммоль) в етанолі (100 мл) обробляють азидом натрію (1,30 г, 19,4 ммоль). Додають водний розчин натрієвої солі L-аскорбінової кислоти (0,66 М, 3,2 мл, 2,1 ммоль) та воду (10 мл), і верхню частину колби продувають азотом. Суміш обробляють водним розчином пентагідрату сульфату міді (II) (0,33 М, 3,2 мл, 1,1 ммоль), і суміш негайно нагрівають на заздалегідь розігрітій нагрівальній плиті при температурі 80С протягом 1,5 год. в атмосфері азоту. При прогріванні одержують однорідну суміш. Реакційну суміш охолоджують, і додають талу воду. Суміш екстрагують етилацетатом (3). Органічні шари об'єднують, і сушать над сульфатом натрію, фільтрують, і розчинник видаляють in vacuo, і одержують неочищений азидний продукт. Неочищений азидний продукт об'єднують з 10 % паладієм на вугіллі (2 г) в холодному етанолі 13 UA 112941 C2 5 10 15 20 25 30 35 40 45 (150 мл), і цю суміш дезаерують із застосуванням комбінації вакуум/азот, і потім вакуум/водень. 2 Суміш перемішують при кімнатній температурі та при тиску водню 30 фунтів/дюйм (0,20684274 МПа) протягом 2 год. Реакційну суміш провітрюють, і фільтрують через діатомову землю з використанням дихлорметану для промивання відфільтрованого осаду. Розчинник видаляють з фільтрату in vacuo, і неочищений продукт очищають на силікагелі 50 % розчином етилацетату в дихлорметані, і одержують вказану в заголовку сполуку (4 г, 91 %). ES/MS (m/e): 453 (М+Н). Препаративна методика 22 трет-бутил-7a-(5-аміно-2-фторофеніл)-2-бензамідо-4,4а,5,7-тетрагідропіроло[3,4d][1,3]тіазин-6-карбоксилат Розчин трет-бутил-2-бензамідо-7a-(5-бромо-2-фторофеніл)-4,4а,5,7-тетрагідропіроло[3,4d][1,3]тіазин-6-карбоксилату (2,556 г, 4,8 ммоль) і транс-N,N'-диметил-1,2-циклогександіаміну (150 мг, 1,1 ммоль) в етанолі (50 мл) обробляють азидом натрію (933 мг, 14,3 ммоль). Додають водний розчин натрієвої солі L-аскорбінової кислоти (0,66 М, 3,2 мл, 2,1 ммоль) та воду (1 мл), і верхню частину колби продувають азотом. Суміш обробляють водним розчином пентагідрату сульфату міді (II) (0,33 М, 3,2 мл, 1,1 ммоль), і цю суміш прогрівають безпосередньо на заздалегідь розігрітій нагрівальній плиті при температурі 80С протягом 1,5 год. в атмосфері азоту. В результаті прогрівання одержують однорідну суміш. Реакційну суміш охолоджують, розбавляють талою водою, і цю суміш екстрагують етилацетатом (3). Органічні шари об'єднують, і сушать над сульфатом натрію, фільтрують, розчинник видаляють in vacuo, і одержують вказану в заголовку сполуку. Неочищений азидний продукт об'єднують з 10 % паладієм на вугіллі (2 г) в холодному етанолі (150 мл), і цю суміш деаерують із застосуванням комбінації вакуум/азот, і потім вакуум/водень. Суміш перемішують при кімнатній температурі та 2 при тиску водню 30 фунтів/дюйм (0,20684274 МПа) протягом 5 год. Реакційну суміш провітрюють, фільтрують через діатомову землю, і відфільтрований осад промивають дихлорметаном. Розчинник видаляють з фільтрату in vacuo, і неочищений продукт очищають на силікагелі 50 % розчином етилацетату в дихлорметані, і одержують вказану в заголовку сполуку (2,014 г, 89 %). ES/MS (m/e): 471 (М+Н). Препаративна методика 23 трет-бутил-(4aR,7aS)-2-бензамідо-7a-[3-[(5-фторопіридин-2-карбоніл)аміно]феніл]-4,4a,5,7тетрагідропіроло[3,4-d][1,3]тіазин-6-карбоксилат Суспензію трет-бутил-(4aR,7aS)-7a-(3-амінофеніл)-2-бензамідо-4,4a,5,7тетрагідропіроло[3,4-d][1,3]тіазин-6-карбоксилату (93 мг, 0,21 ммоль), 5-фторопіридин-2карбонової кислоти (31,9 мг, 0,23 ммоль), гідрату 1-гідроксибензотриазолу (56,7 мг, 0,41 ммоль) і 1-(2-диметиламінопропіл)-3-етилкарбодііміду гідрохлориду (40 мг, 0,21 ммоль) в дихлорметані (4 мл), який містить диметилформамід (1 мл), обробляють діізопропілетиламіном (179,2 мкл, 1,03 ммоль), і одержану суміш перемішують при кімнатній температурі протягом ночі. Реакційну суміш розбавляють дихлорметаном (5 мл) і насиченим водним розчином бікарбонату натрію (15 мл). Органічний шар відокремлюють, і промивають насиченим водним розчином хлориду натрію (10 мл), сушать над сульфатом натрію, фільтрують, розчинник видаляють in vacuo, і одержують вказану в заголовку сполуку (105 мг, 89 %). ES/MS (m/e): 576 (М+Н). Препаративна методика 24 N-[3-[(4aR,7aS)-2-бензамідо-4a,5,6,7-тетрагідро-4H-піроло[3,4-d][1,3]тіазин-7a-іл]феніл]-5фторопіридин-2-карбоксамід; 2,2,2-трифтороцтова кислота 14 UA 112941 C2 5 10 15 20 25 30 трет-бутил-(4aR,7aS)-2-бензамідо-7a-[3-[(5-фторопіридин-2-карбоніл)аміно]феніл]-4,4a,5,7тетрагідропіроло[3,4-d][1,3]тіазин-6-карбоксилат (105 мг, 0,18 ммоль) розчиняють у дихлорметані (2 мл), і обробляють трифтороцтовою кислотою (500 мкл, 6,6 ммоль). Одержаний розчин жовтого кольору перемішують протягом 4 год. при кімнатній температурі, розчинник видаляють in vacuo, і одержують неочищений вказаний в заголовку продукт (190 мг, 100 %). ES/MS (m/e): 476 (М+Н). Препаративна методика 25 N-[(4aR,7aS)-7a-(3-амінофеніл)-4a, 5,6,7-тетрагідро-4H-піроло[3,4-d][1,3]тіазин-2-іл]бензамід Трифтороцтову кислоту (25 мл) додають до розчину трет-бутил-(4aR,7aS)-7a-(3амінофеніл)-2-бензамідо-4,4a,5,7-тетрагідропіроло[3,4-d][1,3]тіазин-6-карбоксилату (4 г, 8,84 ммоль) в дихлорметані (100 мл), і суміш перемішують при кімнатній температурі в атмосфері азоту протягом 4 год. Розчинник видаляють in vacuo, і неочищений продукт очищають на колонці з SCX із застосуванням суміші дихлорметан:метанол (3:1), та потім суміші дихлорметан:7н розчин аміаку в метанолі (2:1), і одержують вказану в заголовку сполуку (2,49 г, 80 %). ES/MS (m/e): 353 (М+Н). Препаративна методика 26 N-[7a-(5-аміно-2-фторофеніл)-4a,5,6,7-тетрагідро-4H-піроло[3,4-d][1,3]тіазін-2-іл]бензамід Трифтороцтову кислоту (10 мл) додають до розчину трет-бутил-7a-(5-аміно-2-фторофеніл)2-бензамідо-4,4a,5,7-тетрагідропіроло[3,4-d][1,3]тіазин-6-карбоксилату (2,013 г, 4,28 ммоль) в дихлорметані (30 мл), і суміш перемішують при кімнатній температурі в атмосфері азоту протягом 4 год. Розчинник видаляють in vacuo, і неочищений продукт очищають на колонці з SCX із застосуванням суміші дихлорметан:метанол (3:1), а потім суміші дихлорметан:7н розчин аміаку в метанолі (2:1), і одержують вказану в заголовку сполуку (1,555 г, 98 %). ES/MS (m/e): 371 (М+Н). Препаративна методика 27 N-[(4aR,7aS)-7a-(3-амінофеніл)-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-2-іл]бензамід 15 UA 112941 C2 5 10 15 20 25 30 35 40 Розчин N-[(4aR,7aS)-7a-(3-амінофеніл)-4a,5,6,7-тетрагідро-4H-піроло[3,4-d][1,3]тіазин-2іл]бензаміду (2,49 г, 7,06 ммоль), 5-фторо-2-хлорпіримідину (3,74 г, 28,26 ммоль) та діізопропілетиламіну (6,16 мл, 35,32 ммоль) в 1,4-діоксані (60 мл) нагрівають до кипіння зі зворотним холодильником протягом 4 год. в атмосфері азоту. Реакційну суміш охолоджують, розбавляють водою, і екстрагують етилацетатом (3). Об'єднані органічні екстракти сушать над сульфатом натрію, фільтрують, і розчинник видаляють in vacuo, і одержують неочищений продукт. Неочищений продукт очищають на силікагелі градієнтом від 5 % до 100 % етилацетату в гексанах протягом 25 хв., і одержують вказану в заголовку сполуку (2,51 г, 79 %). ES/MS (m/e): 449 (М+Н). Препаративна методика 28 N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]феніл]-5-фторопіридин-2-карбоксамід Розчин N-[3-[(4aR,7aS)-2-бензамідо-4a,5,6,7-тетрагідро-4H-піроло[3,4-d][1,3]тіазин-7aіл]феніл]-5-фторопіридин-2-карбоксамід; 2,2,2-трифтороцтової кислоти (150 мг, 254 мкмоль), 5фторо-2-хлорпіримідину (68 мг, 51 мкмоль) та діізопропілетиламіну (98 мкл, 56 мкмоль) гріють в диметилсульфоксиді (5 мл) протягом ночі при температурі 40С. Додають додаткові 5-фторо-2хлорпіримідин (68 мг, 51 мкмоль) та діізопропілетиламін (98 мкл, 56 мкмоль), і цю суміш гріють протягом ночі при температурі 50С. Додають додаткові 5-фторо-2-хлорпіримідин (68 мг, 51 мкмоль) та діізопропілетиламін (98 мкл, 56 мкмоль), і одержану суміш гріють протягом третьої ночі при температурі 50С. Реакційну суміш охолоджують, розбавляють насиченим водним розчином карбонату натрію (50 мл) для одержання суспензії, яку фільтрують, і сушать у вакуумній сушильній шафі при температурі 50С протягом 4 год., і одержують вказану в заголовку сполуку (60 мг, 41 %). ES/MS (m/e): 449 (М+Н). Альтернативна препаративна методика 28 N-[(4aR,7aS)-7a-(3-амінофеніл)-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-2-іл]бензамід (282 мг, 628,73 мкмоль) і 5-фторопіридин-2-карбонову кислоту (106,46 мг, 754,47 мкмоль) змішують в дихлорметані (3 мл) і диметилформаміді (0,5 мл). Додають спочатку 1-гідроксибензотриазол (112,70 мг, 817,35 мкмоль), потім 1-(3-диметиламінопропіл)-3етилкарбодііміду гідрохлорид (159,07 мг, 817,35 мкмоль), і одержану суміш перемішують протягом 5 год. при кімнатній температурі в атмосфері азоту. Реакційну суміш розбавляють водою, і pH із застосуванням 1н розчину NaOH доводять до ~12. Суміш екстрагують етилацетатом (3). Органічні екстракти об'єднують, сушать над сульфатом натрію, фільтрують, розчинник видаляють in vacuo, і одержують неочищений продукт. Неочищений продукт очищають на силікагелі градієнтом від 5 % до 100 % етилацетату в гексанах протягом 20 хв., і одержують вказану в заголовку сполуку (327 мг, 91 %). ES/MS (m/e): 571 (М+Н). Препаративна методика 29 N-[7a-(5-аміно-2-фторофеніл)-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-2-іл]бензамід 16 UA 112941 C2 5 10 15 20 25 30 35 Розчин N-[7a-(5-аміно-2-фторофеніл)-4a,5,6,7-тетрагідро-4H-піроло[3,4-d][1,3]тіазин-2іл]бензаміду (705 мг, 1,90 ммоль), 5-фторо-2-хлорпіримідину (1,01 г, 7,61 ммоль) та діізопропілетиламіну (1,66 мл, 9,52 ммоль) нагрівають в 1,4-діоксані (20 мл) зі зворотним холодильником до кипіння протягом 4 год. в атмосфері азоту. Реакційну суміш охолоджують, розбавляють водою, і екстрагують етилацетатом (3). Органічні шари об'єднують, сушать над сульфатом натрію, фільтрують, і розчинник видаляють in vacuo, і одержують неочищений продукт. Неочищений продукт очищають на силікагелі градієнтом від 5 % до 100 % етилацетату в гексанах протягом 25 хв., і одержують вказану в заголовку сполуку (590 мг, 66 %). ES/MS (m/e): 467 (М+Н). Препаративна методика 30 N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]феніл]-5-метоксипіразин-2-карбоксамід N-[(4aR,7aS)-7a-(3-амінофеніл)-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-2-іл]бензамід (400 мг, 891,81 мкмоль) і 5-метоксипіразин-2-карбонову кислоту (165 мг, 1,07 ммоль) об'єднують в дихлорметані (4 мл) і диметилформаміді (0,5 мл). Додають спочатку 1-гідроксибензотриазол (160 мг, 1,16 ммоль), потім 1-(3-диметиламінопропіл)-3етилкарбодііміду гідрохлорид (226 мг, 1,16 ммоль), і одержану суміш перемішують протягом 5 год. при кімнатній температурі в атмосфері азоту. Реакційну суміш розбавляють водою, і pH із використанням 1н розчину NaOH доводять до ~12. Суміш екстрагують етилацетатом (3). Об'єднані органічні екстракти сушать над сульфатом натрію, фільтрують, і розчинник видаляють in vacuo. Неочищений продукт очищають на силікагелі градієнтом від 5 % до 100 % етилацетату в гексанах протягом 20 хв., і одержують вказану в заголовку сполуку (482 мг, 92 %). ES/MS (m/e): 585 (М+Н). Препаративна методика 31 N-[3-[2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4-d][1,3]тіазин-7a-іл]4-фторофеніл]-5-фторопіридин-2-карбоксамід N-[7a-(5-аміно-2-фторофеніл)-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-2-іл]бензамід (302 мг, 647 мкмоль) і 5-фторопіридин-2-карбонову кислоту (110 мг, 777 мкмоль) об'єднують в дихлорметані (3 мл) і диметилформаміді (0,5 мл). Додають спочатку 1-гідроксибензотриазол (116 мг, 842 ммоль), потім 1-(3-диметиламінопропіл)-3-етилкарбодііміду гідрохлорид (164 мг, 842 ммоль), і цю суміш перемішують протягом ночі при кімнатній температурі в атмосфері азоту. Реакційну суміш розбавляють водою, і pH із використанням 1н розчину NaOH доводять до ~12, після чого екстрагують етилацетатом (3). Органічні шари об'єднують, і фільтрують для збирання нерозчинного матеріалу. Тверді речовини промивають водою та етилацетатом, і сушать у вакуумі до одержання вказаної в заголовку сполуки. Органічний шар з фільтрату сушать над сульфатом натрію, фільтрують, і розчинник видаляють 17 UA 112941 C2 5 10 15 20 25 30 in vacuo. Залишок очищають на силікагелі градієнтом від 5 % до 100 % етилацетату в гексанах протягом 20 хв., і одержують додаткову кількість вказаної в заголовку сполуки із загальним виходом (275 мг, 72 %). ES/MS (m/e): 590 (М+Н). Препаративна методика 32 N-[(4aR,7aS)-7a-(5-аміно-2-фторофеніл)-6-(5-фторопіримідин-2-іл)-4,4a,5,7тетрагідропіроло[3,4-d][1,3]тіазин-2-іл]бензамід, (ізомер 1) Рацемічний N-[7a-(5-аміно-2-фторофеніл)-6-(5-фторопіримідин-2-іл)-4,4a, 5,7тетрагідропіроло[3,4-d][1,3]тіазин-2-іл]бензамід (1,694 г, 3,63 ммоль) піддають хіральному очищенню із застосуванням високоефективної рідинної хроматографії (HPLC) (колонка: Chiralcel OJ, 835 см, елюент: 90 % метанол (0,2 % диметилетиламін) і 10 % ацетонітрил; швидкість потоку 400 мл/хв. при УФ 280 нм). Аналіз першого елюйованого ізомеру (колонка: Chiralcel OJ-H 0,4615 см; елюент: 10:90 ацетонітрил:метанол (0,2 % диметилетиламін); швидкість потоку: 0,6 мл/хв. при УФ 280 нм) підтверджує енантіомерно збагачений (99 % ee) енантіомер з Rt=6,70 хв., (723 мг, 43 %). ES/MS (m/e): 467 (М+Н). Препаративна методика 33 N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]-4-фторофеніл]-5-метоксипіразин-2-карбоксамід, (ізомер 1) N-[(4aR,7aS)-7a-(5-аміно-2-фторофеніл)-6-(5-фторопіримідин-2-іл)-4,4a,5,7тетрагідропіроло[3,4-d][1,3]тіазин-2-іл]бензамід (0,361 г, 0,77 ммоль, ізомер 1) розчиняють у суміші дихлорметану (4 мл) і DMF (0,5 мл). До цієї суміші додають 5-метоксипіразин-2карбонову кислоту (240 мг, 1,55 ммоль), 1-гідроксибензотриазол (210 мг, 1,55 ммоль) і 1-(3диметиламінопропіл)-3-етілкарбодііміду гідрохлорид (300 мг, 1,55 ммоль), і одержану суміш перемішують протягом ночі при кімнатній температурі. Реакційний розчин завантажують безпосередньо у завантажувальну колонку, яка містить 12 г силікагелю, і очищають із застосуванням колонки, яка містить 40 г силікагелю, елююючі градієнтом 0-100 % суміші етилацетат/гексани. Продукт розчиняють в етилацетаті (200 мл), і промивають 1н розчином NaOH (250 мл) і розсолом (150 мл). Очищення на силікагелі повторюють, як описано вище, і одержують вказану в заголовку сполуку (350 мг, 74 %). ES/MS (m/e): 603 (М+Н). Препаративна методика 34 N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]феніл]-5-ціанопіридин-2-карбоксамід 18 UA 112941 C2 5 10 15 20 25 30 35 N-[(4aR,7aS)-7a-(3-амінофеніл)-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-2-іл]бензамід (0,30 г, 0,67 ммоль) розчиняють у дихлорметані (10 мл), і додають 5ціанопіридин-2-карбонову кислоту (129 мг, 0,87 ммоль), 1-гідроксибензотриазол (185 мг, 1,34 ммоль) та 1-(3-диметиламінопропіл)-3-етилкарбодііміду гідрохлорид (169 мг, 0,87 ммоль). Додають діізопропілетиламін (0,35 мл, 2 ммоль), і одержану реакційну суміш перемішують при кімнатній температурі протягом ночі. Матеріал очищають безпосередньо хроматографією на силікагелі, елююючі градієнтом 0-100 % суміші етилацетат/гексани, і одержують вказану в заголовку сполуку (360 мг, 88 %). ES/MS (m/e): 579 (М+Н). Препаративна методика 35 N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]феніл]-3,5-дифторопіридин-2-карбоксамід N-[(4aR,7aS)-7a-(3-амінофеніл)-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-2-іл]бензамід (0,30 г, 0,67 ммоль) розчиняють у дихлорметані (10 мл), і додають 3,5-дифторопіридин-2-карбонову кислоту (138 мг, 0,87 ммоль), 1-гідроксибензотриазол (185 мг, 1,34 ммоль) та 1-(3-диметиламінопропіл)-3-етилкарбодііміду гідрохлорид (169 мг, 0,87 ммоль). Додають діізопропілетиламін (0,35 мл, 2 ммоль), і одержану реакційну суміш перемішують при кімнатній температурі протягом ночі. Реакційну суміш очищають безпосередньо хроматографією на силікагелі, елююючі градієнтом 0-100 % суміші етилацетат/гексани, і одержують вказану в заголовку сполуку (330 мг, 84 %). ES/MS (m/e): 590 (М+Н). Препаративна методика 36 N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4а,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]-4-фторофеніл]-5-ціанопіридин-2-карбоксамід, (ізомер 1) N-[(4aR,7aS)-7a-(5-амін-2-фторофеніл)-6-(5-фторопіримідин-2-іл)-4,4а,5,7тетрагідропіроло[3,4-d][1,3]тіазин-2-іл]бензамід (0,180 г, 0,39 ммоль, ізомер 1) розчиняють у суміші дихлорметану (2 мл) та DMF (0,25 мл). Додають 5-ціанопіридин-2-карбонову кислоту (114 мг, 0,77 ммоль), 1-гідроксибензотриазол (106 мг, 0,77 ммоль) та 1-(3-диметиламінопропіл)-3етилкарбодііміду гідрохлорид (150 мг, 0,77 ммоль), і цю реакційну суміш перемішують при кімнатній температурі протягом ночі. Суміш розбавляють водою (10 мл), етилацетатом (10 мл), і додають до 1н розчину NaOH (100 мл). Суміш екстрагують EtOAc (2100 мл), органічні шари об'єднують, і промивають розсолом. Органічний шар сушать над MgSO 4, фільтрують, і концентрують. Залишок очищають хроматографією на силікагелі із застосуванням градієнта 0100 % суміші етилацетат/гексани, і одержують вказану в заголовку сполуку (133 мг, 57 %). ES/MS (m/e): 597 (М+Н). Препаративна методика 37 N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4а,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]-4-фторофеніл]-3,5-дифторопіридин-2-карбоксамід, (ізомер 1) 19 UA 112941 C2 5 10 15 20 25 30 35 40 N-[(4aR,7aS)-7a-(5-аміно-2-фторофеніл)-6-(5-фторопіримідин-2-іл)-4,4а,5,7тетрагідропіроло[3,4-d][1,3]тіазин-2-іл]бензамід (0,180 г, 0,39 ммоль, ізомер 1) розчиняють у суміші дихлорметану (2 мл) та DMF (0,25 мл). Додають 5-ціанопіридин-2-карбонову кислоту (114 мг, 0,77 ммоль), 1-гідроксибензотриазол (106 мг, 0,77 ммоль) та 1-(3-диметиламінопропіл)-3етилкарбодііміду гідрохлорид (150 мг, 0,77 ммоль), і цю реакційну суміш перемішують при кімнатній температурі протягом ночі. Суміш розбавляють водою (10 мл) та етилацетатом (10 мл), після чого виливають в 1н розчин NaOH (100 мл). Суміш екстрагують EtOAc (2100 мл), органічні екстракти об'єднують, і промивають розсолом. Органічний шар сушать над MgSO 4, фільтрують, і концентрують. Залишок очищають хроматографією на силікагелі із застосуванням градієнта 0-100 % суміші етилацетат/гексани, і одержують вказану в заголовку сполуку (190 мг, 80 %). ES/MS (m/e): 608 (М+Н). Приклад А N-[3-[2-аміно-6-(5-фторопіримідин-2-іл)-4,4а,5,7-тетрагідропіроло[3,4-d][1,3]тіазин-7a-іл]-4фторофеніл]-5-фторопіридин-2-карбоксамід Суміш N-[3-[2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4а,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]-4-фторофеніл]-5-фторопіридин-2-карбоксаміду (293 мг, 497 мкмоль), Ометилгідроксиламіну гідрохлориду (430 мг, 4,97 ммоль) і піридину (402 мкл, 4,97 ммоль) нагрівають в етанолі (13 мл) до температури 70С в закоркованій колбі протягом 2,5 год. Додають диметилсульфоксид (3 мл), і одержану суміш гріють при температурі 70С протягом ночі. Додають додаткову кількість диметилсульфоксиду (10 мл), і продовжують прогрівання при температурі 70 °C протягом 4 год. Додають додаткову кількість О-метилгідроксиламіну гідрохлориду (208 мг, 2,48 ммоль) та піридину (201 мкл, 2,48 ммоль), і цю суміш нагрівають до температури 60 °C протягом 3 год., та перемішують протягом 3 днів при кімнатній температурі. В окремій колбі суміш N-[3-[2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]-4-фторофеніл]-5-фторопіридин-2-карбоксаміду (276 мг, 468 мкмоль), Ометилгідроксиламіну гідрохлориду (405 мг, 4,68 ммоль) і піридину (478 мкл, 4,68 ммоль) нагрівають в етанолі (15 мл) та диметилсульфоксиді (4 мл) при температурі 70С в закоркованій колбі протягом ночі. Додають додаткову кількість диметилсульфоксиду (10 мл), і продовжують прогрівання при температурі 70С протягом 4 год. Додають додаткову кількість Ометилгідроксиламіну гідрохлориду (195 мг, 2,34 ммоль) та піридину (189 мкл, 2,34 ммоль), і прогрівання при температурі 70С продовжують протягом 3 год., після чого суміш перемішують протягом 3 днів при кімнатній температурі. Дві реакційні суміші об'єднують, і більшу частину розчинника видаляють in vacuo. Неочищений продукт очищають на колонці з SCX із застосуванням суміші дихлорметан:метанол (3:1), та потім суміші дихлорметан:7н розчин аміаку в метанолі (2:1). Неочищений продукт додатково очищають на силікагелі градієнтом 7н розчину аміаку в метанолі (від 0,5 % до 10 %) в дихлорметані, і одержують вказану в заголовку сполуку (451 мг, 96 %). ES/MS (m/e): 486 (М+Н). Приклад 1 N-{3-[(4aR,7aS)-2-аміно-6-(5-фторопіримідин-2-іл)-4а,5,6,7-тетрагідропіроло[3,4-d][1,3]тіазин7a(4H)-іл]феніл}-5-фторопіридин-2-карбоксаміду гідрохлорид 20 UA 112941 C2 5 10 15 20 25 30 35 Суміш N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]-5-фторопіридин-2-карбоксаміду (320 мг, 560 мкмоль), О-метилгідроксиламіну гідрохлориду (485 мг, 5,60 ммоль) і піридину (453 мкл, 5,60 ммоль) в етанолі (15 мл) гріють при температурі 65 °C в закоркованій посудині протягом п'яти годин. Реакційну суміш охолоджують, і розчинник видаляють in vacuo. Неочищений продукт очищають на силікагелі градієнтом 7н розчину аміаку в метанолі (від 0,5 % до 10 %) в дихлорметані, і одержують N-{3-[(4aR, 7aS)-2аміно-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4-d][1,3]тіазин-7a-іл]феніл}-5фторопіридин-2-карбоксаміду (219 мг, 84 %). Цей матеріал розчиняють у дихлорметані (1 мл), і додають метанол (0,5 мл) та 1М розчин хлориду водню в діетиловому ефірі (0,47 мл, 470 мкмоль). Розчинник видаляють in vacuo, і одержують вказану в заголовку сполуку (228 мг, 81 %). ES/MS (m/e): 468 (М+Н). Приклад 2 N-{3-[(4aR,7aS)-2-аміно-6-(5-фторопіримідин-2-іл)-4а,5,6,7-тетрагідропіроло[3,4-d][1,3]тіазин7a(4H)-іл]феніл}-5-метоксипіразин-2-карбоксаміду гідрохлорид Суміш N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]феніл]-5-метоксипіразин-2-карбоксаміду (479 мг, 819 мкмоль), Ометилгідроксиламіну гідрохлориду (709 мг, 8,19 ммоль) і піридину (663 мкл, 8,19 ммоль) в етанолі (20 мл) гріють при температурі 50 °C в закоркованій колбі протягом ночі. Додають диметилсульфоксид (4 мл), і цю суміш нагрівають до температури 70 °C протягом 4 год. для одержання розчину. Реакційну суміш охолоджують, і більшу частину розчинника видаляють in vacuo. Додають воду, і pH із використанням 1н розчину гідроксиду натрію доводять до ~12. Суміш екстрагують етилацетатом (5). Об'єднані органічні екстракти сушать над сульфатом натрію, фільтрують, і розчинник видаляють in vacuo. Неочищений продукт очищають на силікагелі градієнтом 7н розчину аміаку в метанолі (від 0,5 % до 10 %) в дихлорметані протягом 30 хв. Суміш знову очищають на колонці SCX із застосуванням суміші дихлорметан:метанол (3:1), а потім суміші дихлорметан:7н розчин аміаку в метанолі (2:1) для видалення залишкового диметилсульфоксиду. Суміш остаточно очищають на силікагелі градієнтом 7н розчину аміаку (від 0,5 % до 10 %) в метанолі в дихлорметані протягом 20 хв., і одержують N-[3-[(4aR,7aS)-2аміно-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4-d][1,3]тіазин-7a-іл]феніл]-5метоксипіразин-2-карбоксаміду. Цей матеріал розчиняють у дихлорметані (1 мл) і метанолі (0,5 мл), і додають 1М розчин хлориду водню в діетиловому ефірі (0,66 мл, 660 мкмоль). Розчинник видаляють in vacuo, і одержують вказану в заголовку сполуку (329 мг, 78 %). ES/MS (m/e): 481 (М+Н). Приклад 3 N-[3-[(4aR,7aS)-2-аміно-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4-d][1,3]тіазин7a-іл]-4- фторофеніл]-5- фторопіридин-2-карбоксаміду гідрохлорид 21 UA 112941 C2 5 10 15 20 25 30 35 Рацемічний N-[3-[2-аміно-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]-4-фторофеніл]-5-фторопіридин-2-карбоксамід (451 мг, 929 ммоль) піддають хіральному очищенню із застосуванням SFC (колонка: Chiralcel OD-H (5 мкм), 2,125 см, елюент: 40 % метанол (0,2 % ізопропіламін) в CO2; швидкість потоку 70 мл/хв. при УФ 225 нм). Хіральний аналіз першого елюйованого ізомеру (колонка: Chiralcel OD-H (5 мкм), 4,6150 мм, елюент: 40 % метанол (0,2 % ізопропіламін) в CO2; швидкість потоку 5 мл/хв. при УФ 225 нм) підтверджує енантіомерно збагачений (99 % ee) енантіомер з Rt=1,01 хв. (175 мг, 360 мкмоль). Цей матеріал (вільна основа, ізомер 1) розчиняють у дихлорметані (1 мл) і метанолі (0,5 мл), і додають 1М розчин хлориду водню в діетиловому ефірі (0,36 мл, 360 мкмоль). Розчинник видаляють in vacuo, і одержують вказану в заголовку сполуку (183 мг, 38 %). ES/MS (m/e): 486 (М+Н). Приклад 4 N-[3-[(4aR,7aS)-2-аміно-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4-d][1,3]тіазин7a-іл]-4-фторофеніл]-5-метоксипіразин-2-карбоксаміду гідрохлорид N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]-4-фторофеніл]-5-метоксипіразин-2-карбоксамід (0,350 г, 0,58 ммоль, ізомер 1) розчиняють у THF (2 мл), після чого додають метанол (4 мл) і етанол (4 мл). До цієї суміші додають О-метилгідроксиламіну гідрохлорид (495 мг, 5,81 ммоль) та піридин (470 мкл, 5,81 ммоль), і одержану реакційну суміш нагрівають до температури 50 °C, і перемішують протягом ночі. До реакційної суміші додають силікагель (~10 г), і суміш концентрують. Зразок, висушений на силікагелі, завантажують у порожній картридж, і очищають, елююючі градієнтом 7н розчину аміаку в метанолі (0-10 %) в дихлорметані. Цей продукт вдруге очищають на колонці SCX із застосуванням суміші дихлорметан:метанол (3:1), а потім суміші дихлорметан:7н розчин аміаку в метанолі (2:1). Продукт остаточно очищають на силікагелі градієнтом 7н розчину аміаку в метанолі (0-10 %) в дихлорметані з одержанням вільної основи вказаної в заголовку сполуки. Цей матеріал розчиняють у дихлорметані (5 мл), і додають 1М розчин хлориду водню в діетиловому ефірі (0,20 мл, 660 мкмоль). Розчинник видаляють in vacuo, і одержують вказану в заголовку сполуку (71 мг, 23 %). ES/MS (m/e): 498 (М+Н). Приклад 5 N-[3-[(4aR,7aS)-2-аміно-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4-d][1,3]тіазин7a-іл]феніл]-5-ціанопіридин-2-карбоксаміду гідрохлорид N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4 22 UA 112941 C2 5 10 d][1,3]тіазин-7a-іл]феніл]-5-ціанопіридин-2-карбоксамід (360 мг, 0,59 ммоль) розчиняють в етанолі (10 мл) і дихлорметані (2 мл). Додають О-метилгідроксиламіну гідрохлорид (504 мг, 5,91 ммоль) та піридин (478 мкл, 5,91 ммоль), і цю реакційну суміш перемішують при кімнатній температурі протягом уікенду (70 год.). Згадану реакційну суміш нагрівають до температури 60С, і перемішують протягом 24 год. Реакційну суміш концентрують з одержанням неочищеного продукту, і очищають із застосуванням хроматографії на силікагелі з використанням градієнту 7н розчину аміаку в метанолі (0-10 %) в дихлорметані, і одержують вільну основу вказаної в заголовку сполуки. Цей матеріал розчиняють у дихлорметані (5 мл), і додають 1М розчин хлориду водню в діетиловому ефірі (0,54 мл, 540 мкмоль). Розчинник видаляють in vacuo, і одержують вказану в заголовку сполуку (240 мг, 75 %). ES/MS (m/e): 475 (М+Н). Приклад 6 N-[3-[(4aR,7aS)-2-аміно-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4-d][1,3]тіазин7a-іл]феніл]-3,5-дифторопіридин-2-карбоксаміду гідрохлорид 15 20 25 N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]феніл]-3,5-дифторопіридин-2-карбоксамід (330 мг, 0,53 ммоль) розчиняють в THF (10 мл), і розбавляють етанолом (10 мл). Додають О-метилгідроксиламіну гідрохлорид (453 мг, 5,32 ммоль) та піридин (430 мкл, 5,91 ммоль), і одержану реакційну суміш перемішують при кімнатній температурі протягом уікенду (70 год.). Згадану реакційну суміш нагрівають до температури 60С, і перемішують протягом 24 год. Реакційну суміш концентрують на силікагелі (~10 г), і очищають із застосуванням хроматографії на силікагелі з використанням градієнту 7н розчину аміаку в метанолі (0-10 %) в дихлорметані, і одержують вільну основу вказаної в заголовку сполуки. Цей матеріал розчиняють у дихлорметані (5 мл), і додають 1М розчин хлориду водню в діетиловому ефірі (0,49 мл, 490 мкмоль). Розчинник видаляють in vacuo, і одержують вказану в заголовку сполуку (159 мг, 54 %). ES/MS (m/e): 486 (М+Н). Приклад 7 N-[3-[(4aR,7aS)-2-аміно-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4-d][1,3]тіазин7a-іл]-4-фторофеніл]-5-ціанопіридин-2-карбоксаміду гідрохлорид 30 35 40 N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]-4-фторофеніл]-5-ціанопіридин-2-карбоксамід (133 мг, 0,22 ммоль, ізомер 1) розчиняють в THF (1 мл), і розбавляють метанолом (3 мл) та етанолом (3 мл). Додають Ометилгідроксиламіну гідрохлорид (190 мг, 2,2 ммоль) та піридин (180 мкл, 2,2 ммоль). Реакційну суміш нагрівають до температури 50С, і перемішують протягом ночі. Суміш концентрують на силікагелі (~10 г), і очищають із застосуванням хроматографії на силікагелі, елююючі градієнтом 7н розчину аміаку в метанолі (0-10 %) в дихлорметані. Матеріал вдруге очищають на колонці SCX із застосуванням спочатку суміші дихлорметан:метанол (3:1), та потім суміші дихлорметан:7н розчин аміаку в метанолі (2:1). Одержану суміш остаточно очищають на силікагелі градієнтом 7н розчину аміаку в метанолі (0-10 %) в дихлорметані, і одержують вільну основу вказаної в заголовку сполуки. Цей матеріал розчиняють у дихлорметані (5 мл), і додають 1М розчин хлориду водню в діетиловому ефірі (0,27 мл, 270 мкмоль). Розчинник видаляють in vacuo, і одержують вказану в заголовку сполуку (114 мг, 97 %). ES/MS (m/e): 493 (М+Н). 23 UA 112941 C2 Приклад 8 N-[3-[(4aR,7aS)-2-аміно-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4-d][1,3]тіазин7a-іл]-4-фторофеніл]-3,5-дифторопіридин-2-карбоксаміду гідрохлорид 5 10 15 20 25 30 35 40 45 50 N-[3-[(4aR,7aS)-2-бензамідо-6-(5-фторопіримідин-2-іл)-4,4a,5,7-тетрагідропіроло[3,4d][1,3]тіазин-7a-іл]-4-фторофеніл]-3,5-дифторопіридин-2-карбоксамід (190 мг, 0,31 ммоль, ізомер 1) розчиняють в THF (1 мл), і розбавляють метанолом (3 мл) та етанолом (3 мл). Додають О-метилгідроксиламіну гідрохлорид (267 мг, 3,1 ммоль) та піридин (253 мкл, 3,1 ммоль), реакційну суміш нагрівають до температури 50 °C, і перемішують протягом ночі. Реакційну суміш очищають на колонці SCX із застосуванням спочатку суміші дихлорметан:метанол (3:1), а потім суміші дихлорметан:7н розчин аміаку в метанолі (2:1). Матеріал остаточно очищають на силікагелі градієнтом 7н розчину аміаку в метанолі (0-10 %) в дихлорметані, і одержують вільну основу вказаної в заголовку сполуки. Цей матеріал розчиняють у дихлорметані (5 мл), і додають 1М розчин хлориду водню в діетиловому ефірі (0,20 мл, 200 мкмоль). Розчинник видаляють in vacuo, і одержують вказану в заголовку сполуку (101 мг, 60 %). ES/MS (m/e): 504 (М+Н). Аналітичні методики in vitro Для ферментативних і клітинних аналізів експериментальні сполуки приготовляють в DMSO, та одержують 10 мМ маточний розчин. Перед проведенням ферментативних і цілоклітинних аналізів in vitro, цей маточний розчин послідовно розбавляють DMSO, так щоб одержати десятиточкову криву розведення з кінцевою концентрацією сполук від 10 мМ до 0,05 нМ в 96лунковому планшеті з круглим дном. Дослідження пригнічування протеаз in vitro: Експресія людського BACE1 Людський BACE1 (номер депонування AF190725) клонують з тотальної мозкової кДНК із застосуванням RT-PCR (полімеразної ланцюгової реакції). Нуклеотидні послідовності, які відповідають амінокислотним послідовностям від № 1 до № 460, вводять в кДНК, яка кодує поліпептид Fc-фрагменту людського IgG1 (Vassar et al., 1999). Цей гібридний білок з BACE1 (1460) і людського Fc, названий huBACE1:FC, вбудовують у вектор pJB02. Людський BACE1 (1460):FC (huBACE1:Fc) тимчасово експресується в клітинах HEK293. 250 мкг кДНК кожної генноінженерної конструкції змішують з Fugene 6, і додають до 1 л клітин НЕК293. Через чотири дні після трансфекції кондиціоновані середовища збирають для очищення. Очищення huBACE1: Fc. huBACE1: Fc очищають хроматографією з використанням сорбентів на основі імобілізованого протеїна А. Фермент зберігають при температурі -80С невеликими аліквотами. FRET-аналіз BACE1 Послідовно розведені розчини експериментальних сполук приготовляють так, як описано вище. Сполуки додатково розбавляють 20 в буфері КН2РО4. Десять мікролітрів кожного розведеного розчину додають в кожну лунку в рядку від А до Н відповідного чорного планшета з низьким зв'язуванням білка, яка вміщує реакційну суміш (25 мкл 50 мМ розчину КН 2РО4, pH 4,6, 1 мМ TRITON X-100, 1 мг/мл бичачого сироваткового альбуміну і 15 мкМ субстрату FRET) (дивись Yang, et al., J. Neurochemistry, 91(6) 1249-59 (2004)). Вміст добре перемішують на планшетному шейкері протягом 10 хв. Для ініціювання реакції на планшет, який вміщує субстрат і досліджувані сполуки, додають 15 мкл 200 пМ розчину людського BACE1 (1-460):Fc (дивись Vasser, et al., Science, 286, 735-741 (1999)) в буфері КН2РО4. Після нетривалого перемішування на планшетному шейкері, RFU суміші в момент часу 0 записують при довжині хвилі збудження 355 нм і довжині хвилі емісії 460 нм. Реакційний планшет вкривають алюмінієвою фольгою, і витримують в темному зволожуваному термостаті при кімнатній температурі протягом 16-24 год. RFU в кінці інкубації реєструють з тими ж параметрами збудження і емісії, які були використані на момент часу 0. Різниця RFU на момент часу 0 і в кінці інкубації є репрезентативною для активності BACE1 за умов вказаної обробки сполуками. Різниці RFU наносять на графік у зіставленні з інгібувальною концентрацією, і криву підганяють за 24 UA 112941 C2 допомогою чотирипараметричного логістичного рівняння для одержання значень ЕС50 та IC50. (Дивись Sinha, et al., Nature, 402, 537-540 (2000)). Сполуки за наведеними нижче прикладами були випробувані по суті так, як описано вище, і продемонстрували наведену нижче активність щодо BACE1: 5 Таблиця 1 Приклад № 1 2 3 4 5 25 30 35 0,358 (n=1/3) 8 20 0,739 (0,181, n=7) 7 15 0,450 (0,0911, n=4) 6 10 BACE1 IC50 (нМ) 0,610 (0,0948, n=8/9) 0,482 (0,0580, n=6/7) 0,554 (0,0674, n=3) 0,569 (0,0796, n=2) 0,730 (0,0951, n=3) Середнє SEM; SEM = середня квадратична помилка середнього Ці дані показують, що сполуки з Таблиці 1 ефективно пригнічують активність очищеного рекомбінантного фермента BACE1 in vitro. Цілоклітинні аналізи для визначення пригнічування бета-секретазної активності Цілоклітинний аналіз з використанням клітин HEK293Swe При проведенні традиційного цілоклітинного аналізу для визначення пригнічування бетасекретазної активності, використовують лінію ембріональних клітин нирки людини HEK293p (№ депонування АТСС: CRL-1573), які стабільно експресують кДНК людського APP751, що містить природну подвійну мутацію Lys651Met652 на Asn651Leu652, яку зазвичай називають шведською мутацією (позначена як HEK293Swe) і яка, як показано, забезпечує надвиробництво бета-амілоїдного пептиду (Citron, et al., Nature, 360, 672-674 (1992)). In vitro дослідження відновлення бета-амілоїдного пептиду були описані в літературі (дивись Dovey, et al., Journal of Neurochemistry, 76, 173-181 (2001); Seubert, et al., Nature, 361, 260 (1993); та Johnson-Wood, et al., Proc. Natl. Acad. Sci. USA, 94, 1550-1555 (1997)). 4 Клітини (HEK293Swe, 3,510 клітин/лунку, в яку вміщені 200 мкл культурального середовища, DMEM, до складу якого входить 10 % FBS) інкубують при температурі 37С протягом 4-24 год. у присутності/за відсутності інгібіторів (розведених в DMSO) в необхідній концентрації. Наприкінці інкубації кондиціоновані середовища аналізують на наявність активності бета-секретази, наприклад, за допомогою аналізу бета-амілоїдних пептидів. Загальну кількість бета-амілоїдних пептидів (Abeta 1-х) визначають сендвіч-ELISA, із використанням моноклонального антитіла 266 в ролі іммобілізованого антитіла, та біотинілованого антитіла 3D6 в ролі репортерного антитіла. Альтернативно бета-амілоїдні пептиди 1-40 та 1-42 визначають сендвіч-ELISA, із використанням моноклонального антитіла 2G3 в ролі іммобілізованого антитіла для бета амілоїдного пептиду 1-40, і моноклонального антитіла 21F12 в ролі іммобілізованого антитіла для бета-амілоїдного пептиду 1-42. При проведенні ELISA як для бета-амілоїдного пептиду 1-40, так і для бета-амілоїдного пептиду 142, біотиніловане антитіло 3D6 використовують як репортерне антитіло. Концентрація бетаамілоїдного пептиду, вивільненого в кондиціоноване середовище після обробки сполукою, відповідає активності BACE1 за таких умов. Накреслюють 10-точкову криву пригнічування, і підганяють її за допомогою чотирипараметричного логістичного рівняння для одержання значень ЕС50 та IC50 для впливу на зниження рівня бета-амілоїдного пептиду. Сполуки за наведеними нижче прикладами були випробувані по суті так, як описано вище, і продемонстрували наведену нижче активність щодо впливу на зниження рівня бета-амілоїдного пептиду. 40 25 UA 112941 C2 Таблиця 2 HEK 293 Swe A-beta (1HEK 293 Swe A-beta (140) ELISA 42) ELISA IC50 (нМ) IC50 (нМ) 1 0,619 0,437 2 0,324 0,289 3 1,26 0,299 5 0,0887 0,0785 6 0,220 0,211 Середнє SEM; SEM = середня квадратична помилка середнього Ці дані показують, що сполуки Таблиці 2 ефективно пригнічують продукування нативного бета-амілоїдного пептиду в цілих клітинах. Аналіз первинних PDAPP-нейронів Підтверджувальний цілоклітинний аналіз також здійснюють на первинних культурах нейронів, одержаних з ембріонів трансгенних мишей лінії PDAPP. Первинні кортикальні нейрони 4 приготовляють з 16-денних ембріонів PDAPP, і культивують в 96-лункових планшетах (1510 клітин/лунку в середовищі DMEM/F12 (1:1) плюс 10 % FBS). Після 2 діб in vitro, культуральне середовище замінюють на безсироваткове середовище DMEM/F12 (1:1), яке містить домішку B27 і 2 мкМ (кінцева концентрація) Ara-C (компанія Sigma, C1768). На 5-у добу in vitro, нейрони інкубують при температурі 37С протягом 24 год. у присутності/за відсутності інгібіторів (розведених в DMSO) в необхідній концентрації. Наприкінці інкубації кондиціоновані середовища піддають кількісному аналізу для підтвердження активності бета-секретази, наприклад, кількісним аналізом бета-амілоїдних пептидів. Загальну кількість бета-амілоїдних пептидів (Abeta 1-х) визначають сендвіч-ELISA, із використанням моноклонального антитіла 266 в ролі іммобілізованого антитіла, та біотинілованого антитіла 3D6 в ролі репортерного антитіла. Альтернативно бета-амілоїдні пептиди 1-40 та 1-42 визначають сендвіч-ELISA, із використанням моноклонального антитіла 2G3 в ролі іммобілізованого антитіла для бета амілоїдного пептиду 1-40, і моноклонального антитіла 21F12 в ролі іммобілізованого антитіла для бета-амілоїдного пептиду 1-42. При проведенні ELISA як для бета-амілоїдного пептиду 140, так і для бета-амілоїдного пептиду 1-42, біотиніловане антитіло 3D6 використовують як репортерне антитіло. Концентрація бета-амілоїдного пептиду, вивільненого в кондиціоноване середовище після обробки сполукою, відповідає активності BACE1 за таких умов. Накреслюють 10-точкову криву пригнічування, і підганяють її за допомогою чотирипараметричного логістичного рівняння для одержання значень ЕС50 та IC50 для впливу на зниження рівня бетаамілоїдного пептиду. Сполуки за наведеними нижче прикладами були випробувані по суті так, як описано вище, і продемонстрували наведену нижче активність щодо впливу на зниження рівня бета-амілоїдного пептиду: Приклад 5 10 15 20 25 30 Таблиця 3 Приклад 35 PDAPP Neuron A-beta (1- PDAPP Neuron A-beta (140) ELISA 42) ELISA IC50 (нМ) IC50 (нМ) 1 0,487 (0,0946, n=2) 0,591 (0,268, n=2) 2 0,244 (n=1/2) 1,22 (0,967, n=2) 3 0,309 (0,0478, n=2) 0,184 (0,0234, n=2) 4 0,134 0,131 5 0,0813 0,132 (0,0717, n=2) 6 0,279 (0,0607, n=2) 0,308 (0,115, n=2) 7 0,0873 0,0649 8 0,285 0,29 Середнє SEM; SEM = середня квадратична помилка середнього Ці дані показують, що сполуки Таблиці 3 ефективно пригнічують продукування нативного бета-амілоїдного пептиду в цілих клітинах. In vivo інгібування бета-секретази Декілька тваринних моделей, у тому числі моделей з використанням мишей, морських свинок, собак і мавп, можуть бути використані для скринінгу щодо пригнічування активності 26 UA 112941 C2 5 10 15 20 25 30 35 40 бета-секретази in vivo після обробки сполуками. Тварини, використані в цьому винаході, можуть бути тваринами дикого типу, трансгенні або з нокаутованим геном. Наприклад, моделі з мишами PDAPP, як описано у Games et al., Nature 373, 523-527 (1995), та іншими нетрансгенними тваринами або тваринами з нокаутованим геном, можуть бути корисними для дослідження in vivo пригнічування продукування бета-амілоїдного пептиду та sAPPbeta в присутності пригнічувальних сполук. Зазвичай мишам лінії PDAPP, яким є 2-12 місяців, мишам з нокаутованим геном або нетрансгенним тваринам вводять сполуку в суміші з носіями, такими як кукурудзяна олія, ціклодекстран, фосфатні буфери, PHARMASOLVE або інших придатних носіях. Через 1-24 год. після введення сполуки тварин умертвляють, і їх головний мозок, а також спинномозкову рідину і плазму, видаляють для аналізу бета-амілоїдних пептидів, C99 і sAPP фрагментів. (Дивись May, et al., Journal of Neuroscience, 31, 16507-16516 (2011)). Для здійснення стандартних in vivo фармакологічних досліджень тваринам вводять сполуки у різних концентраціях, і порівнюють з контрольною групою, обробленою носієм в той же самий час. Протягом деякого періоду досліджень, розпочинаючи з моменту часу 0, від обраних тварин одержують тканини мозку, плазму або спинномозкову рідину для встановлення базового рівня. Сполуку або придатний носій вводять тваринам інших груп, і умертвляють їх в різні моменти часу після введення дози. Мозкову тканину, плазму або спинномозкову рідину одержують з обраних тварин, і аналізують на наявність продуктів розщеплення АРР, у тому числі бетаамілоїдних пептидів, sAPPbeta та інших фрагментів АРР, наприклад, специфічними сендвічELISA аналізами. Наприкінці періоду дослідження тварин умертвляють, і тканини мозку, плазму або спинномозкову рідину піддають аналізу на присутність бета-амілоїдних пептидів, C99 і sAPPbeta, відповідно. Тканини мозку АРР-трансгенних тварин також можуть піддаватися аналізу на кількість бета-амілоїдних бляшок після обробки сполукою. Термін "пептид A-бета 1-х" у значенні, вживаному у цьому описі, означає суму різновидів А-бета, які починаються з залишку 1 і закінчуються С-кінцевим залишком, більшим за 28. Цей аналіз виявляє більшість різновидів А-бета, і його часто називають аналізом на "загальний A-бета". У тварин (PDAPP або інші APP-трансгенні або нетрансгенні миші), яким була введена пригнічувальна сполука, може спостерігатися зниження рівня бета-амілоїдного білка або sAPPbeta в тканинах мозку, плазмі або спинномозковій рідині і зниження кількості бетаамілоїдних бляшок в тканині головного мозку, у порівнянні з обробленими носієм контрольними тваринами або контрольними тваринами нульового моменту часу. Наприклад, через 3 год. після введення пероральним шляхом дози (1 мг/кг, 3 мг/кг або 10 мг/кг) сполуки Прикладу 1, у молодих PDAPP-мишей-самиць рівні пептиду А-бета 1-х зменшуються приблизно на 34 %, 48 % і 53 % в гіпокампі головного мозку, і приблизно на 43 %, 59 % і 66 % в корі головного мозку, відповідно, в порівнянні з мишами, обробленими носієм. Наприклад, через 3 год. після введення пероральним шляхом дози (1 мг/кг або 3 мг/кг) сполуки Прикладу 3, рівні пептиду А-бета 1-х зменшуються приблизно на 38 % і 50 % в гіпокампі головного мозку, і приблизно на 34 % і 53 % в корі головного мозку, відповідно, в порівнянні з мишами, обробленими носієм. Враховуючи активність сполук Прикладу 1 і Прикладу 3 проти ферменту BACE in vivo, ці впливи на зниження рівня бета-амілоїдного білка відповідають пригнічуванню BACE in vivo, і демонструють проникнення сполук Прикладу 1 і Прикладу 3 до ЦНС. Ці дослідження показують, що сполуки за цим винаходом пригнічують BACE і, отже, є корисними щодо зниження рівнів бета-амілоїдного білка. 45 ФОРМУЛА ВИНАХОДУ 1. Сполука формули: N F N N O A 50 S N R NH2 N H , де R являє собою Η або F; і А являє собою: 27 UA 112941 C2 N F F , F , NC або її фармацевтично прийнятна сіль. 2. Сполука або сіль за п. 1, яка являє собою: N F або H3CO N ; S N N N N N N F O NH2 N N H 5 H3CO N . 3. Сполука за п. 2, яка являє собою: N F S N N N O F NH2 N N H H3CO 10 15 20 N . 4. Спосіб лікування хвороби Альцгеймера у пацієнта, що включає введення пацієнту, який потребує такого лікування, ефективної кількості сполуки за будь-яким з пп. 1-3 або її фармацевтично прийнятної солі. 5. Спосіб запобігання розвитку хвороби Альцгеймера у пацієнта з підвищеним ризиком розвитку цієї хвороби, який включає введення пацієнту, який потребує такого лікування, ефективної кількості сполуки за будь-яким з пп. 1-3 або її фармацевтично прийнятної солі. 6. Сполука або її фармацевтично прийнятна сіль за будь-яким з пп. 1-3 для застосування в терапії. 7. Сполука або її фармацевтично прийнятна сіль за будь-яким з пп. 1-3 для застосування в лікуванні хвороби Альцгеймера. 8. Фармацевтична композиція, яка містить сполуку або її фармацевтично прийнятну сіль за будь-яким з пп. 1-3 з одним або декількома фармацевтично прийнятними носіями, розріджувачами або наповнювачами. Комп’ютерна верстка О. Гергіль Державна служба інтелектуальної власності України, вул. Василя Липківського, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 28
ДивитисяДодаткова інформація
Назва патенту англійськоюTetrahydropyrrolothiazine compounds
Автори англійськоюGreen, Steven James, Mergott, Dustin James, Watson, Brian Morgan, Winneroski Jr., Leonard Larry
Автори російськоюГрин Стивен Джэймс, Мерготт Дастин Джеймс, Уотсон Брайан Морган, Уиннероски мл., Леонард Ларри
МПК / Мітки
МПК: A61K 31/547, C07D 513/04, A61P 25/28
Мітки: сполуки, тетрагідропіролотіазинові
Код посилання
<a href="https://ua.patents.su/30-112941-tetragidropirolotiazinovi-spoluki.html" target="_blank" rel="follow" title="База патентів України">Тетрагідропіролотіазинові сполуки</a>
Сполуки бензимідазолу, фармацевтичні композиції, що містять ці сполуки, та спосіб лікування чутливих до позитивної модуляції гамк-рецепторних комплексів розладів цнс

Номер патенту: 54394
Опубліковано: 17.03.2003
Автори: ТЕУБЕР Лене, Ветьєн Франк, УСІРОДА Осаму, САСАКІ Тосіро, ФУКУДА Йосімаса
МПК: C07D 417/14, A61K 31/425, C07D 405/10, C07D 521/00, A61K 31/415, A61K 31/4433, C07D 417/10, A61K 31/505, A61K 31/4427, C07D 403/10, A61K 31/44, C07D 405/14, C07D 413/14, A61P 25/04, C07D 401/10, C07D 409/10, A61K 31/4164, A61P 43/00, C07D 235/06, A61K 31/506, A61K 31/443, A61P 25/00, A61K 31/4439, A61K 31/00, A61K 31/427, A61K 31/4184
Мітки: бензимідазолу, модуляції, сполуки, цнс, спосіб, фармацевтичні, лікування, чутливих, містять, комплексів, композиції, позитивної, розладів, гамк-рецепторних
Формула / Реферат:
1. Сполука формули:або її фармацевтично придатна сіль чи оксид, де R3 - ,де кожний А, В та D - СН, або один чи два з А, В та D - N, а інші - СН;R11 - феніл, бензимідазол або моноциклічний гетероарил, кожний з яких може бути заміщеним одним або більше замісниками,...
Спосіб одержання 3-n,n-дициклобутиламіно-8-фтор-3,4-дигідро-2н-1-бензопіран-5-карбоксаміду в вигляді рацемічної сполуки або r- чи s-енантіомерів та проміжні сполуки
Номер патенту: 61949
Опубліковано: 15.12.2003
Автори: Хансон Сверкер, Йоханссон Ларс, Сон Деніел Д.
МПК: C07D 311/58, A61K 31/352
Мітки: одержання, 3-n,n-дициклобутиламіно-8-фтор-3,4-дигідро-2н-1-бензопіран-5-карбоксаміду, спосіб, s)-енантіомерів, рацемічної, сполуки, вигляді, проміжні
Формула / Реферат:
Спосіб одержання рацемічної сполуки формули (І) , (І) її R-енантіомера (формула R-(І)) R-(І)та її S-енантіомера (формула S-(І)), S-(І)та їх фармацевтично...
Спіроазабіциклічні гетероциклічні сполуки, фармацевтична композиція на їх основі, спосіб лікування чи профілактики (варіанти), спосіб одержання сполуки (варіанти) та проміжні спіроазабіциклічні гетероциклічні с

Номер патенту: 61964
Опубліковано: 15.12.2003
Автори: Мек Роберт, Мейкор Джон, Семас Саймон, Філліпс Ейфіон
МПК: A61P 25/00, A61P 25/14, A61P 1/04, A61P 25/22, C07F 5/00, A61P 25/34, C07D 491/22, A61P 25/16, A61K 31/4738, A61P 25/18, C07F 7/18, A61P 25/04, A61K 31/444, A61K 31/439, A61P 25/28, A61P 43/00, A61K 31/695, A61K 31/496, A61K 31/5377, A61P 25/24
Мітки: основі, композиція, варіанти, лікування, профілактики, проміжні, одержання, спосіб, спіроазабіциклічні, гетероциклічні, фармацевтична, сполуки
Формула / Реферат:
1. Спіроазабіциклічні гетероциклічні сполуки формули І,де n дорівнює 0 чи 1,m дорівнює 0 чи 1,p дорівнює 0 чи 1,Х являє собою оксиген чи сульфур,Y являє собою CH, N чи NO,W являє собою оксиген, H2 чи F2,A являє собою N чи C(R2),G являє собою N чи C(R3),D являє собою N чи C(R4)за умови, що не більш ніж один з A, G та D являє собою нітроген, але принаймні один з Y, A,...
Спосіб одержання сполуки фенілпіразолу, проміжні сполуки та спосіб їх одержання

Номер патенту: 79295
Опубліковано: 11.06.2007
Автори: Відаль Жоель, Ансель Жан-Ерік
МПК: C07D 231/38, C07D 231/44, C07B 43/00, C07B 45/00, C07C 323/54, C07C 319/00, C07C 323/58
Мітки: сполуки, фенілпіразолу, спосіб, одержання, проміжні
Формула / Реферат:
1. Спосіб одержання сполуки (III), який відрізняється тим, що проводять реакцію між сполукою загальної формули (V) і диціаноацетиленом (IV) у присутності води, ,де R вибраний з CF3 або С1-С6-алкілу; таМ вибраний з лужного або лужноземельного металу або срібла.2. Спосіб за п. 1, який відрізняється тим, що R означає CF3, a M...
Триазолопіримідинові сполуки, спосіб їх одержання (варіанти) та проміжні сполуки (варіанти)

Номер патенту: 73182
Опубліковано: 15.06.2005
Автори: Мусіль Тібор, Магнуссон Маттіас, Ларссон Ульф, Пальмгрен Андреас
МПК: C07D 239/47, C07B 61/00, C07D 487/04, C07D 239/56, C07D 317/44, C07D 405/12
Мітки: одержання, триазолопіримідинові, спосіб, варіанти, сполуки, проміжні
Формула / Реферат:
1. Сполука формули (І): (I).2. Спосіб одержання сполуки формули (І), що включає взаємодію сполуки формули (II): (II)з сіллю сполуки формули (III): (III).3. Спосіб одержання сполуки формули...
Попередній патент: Пристрій і спосіб для бічного спрямування прокатного або відлитого виробу на транспортувальній лінії
Наступний патент: Спосіб виготовлення будівельних виробів із фосфогіпсу
Випадковий патент: Спосіб експрес-оцінки стану гідроекосистем за mn-тестом периферійної крові риб