Пероральна композиція, що запобігає зловживанню опіоїдними агоністами, що в ній містяться, при її псуванні








Формула / Реферат
1. Лікарська форма для перорального введення, яка включає (і) опіоїдний агоніст та (іі) біодоступний при пероральному введенні опіоїдний антагоніст у по суті невивільнюваній формі, коли лікарську форму вводять інтактною, яка відрізняється тим, що агоніст та антагоніст взаємно дисперговані.
2. Лікарська форма за п.1, де співвідношення кількості антагоніста, вивільненого з лікарської форми після її псування, і кількості вказаного антагоніста, вивільненого із вказаної інтактної лікарської форми, становить приблизно 4:1 або більше, на основі розчинення вказаної лікарської форми in vitro впродовж 1 години у 900 мл рідини, що імітує шлунковий сік, з використанням апарата USP типу II (лопатевого) на 75 об./хв. при 37 °С.
3. Лікарська форма за п.1, де співвідношення кількості антагоніста, вивільненого із вказаної лікарської форми після її псування, і кількості вказаного антагоніста, вивільненого із вказаної інтактної лікарської форми, становить приблизно 4:1 або більше, на основі розчинення вказаної лікарської форми in vitro впродовж 1 години у 900 мл рідини, що імітує шлунковий сік, з використанням апарата USP типу II (лопатевого) 75 об./хв. при 37 °С, яка відрізняється тим, що згаданий антагоніст знаходиться у формі множини частинок, окремо покритих секвеструвальним матеріалом, який по суті відвертає вивільнення антагоніста.
4. Лікарська форма за п.1, де співвідношення кількості антагоніста, вивільненого із вказаної лікарської форми після її псування, і кількості вказаного антагоніста, вивільненого із вказаної інтактної лікарської форми, становить приблизно 4:1 або більше, на основі розчинення вказаної лікарської форми in vitro впродовж 1 години у 900 мл рідини, що імітує шлунковий сік, з використанням апарата USP типу II (лопатевого) на 75 об./хв. при 37 °С, яка відрізняється тим, що згаданий антагоніст диспергований у матриксі, що включає секвеструвальний матеріал, який по суті відвертає вивільнення антагоніста.
5. Лікарська форма за п.1, де співвідношення кількості антагоніста, який міститься в указаній інтактній лікарській формі, і кількості вказаного антагоніста, вивільненого із вказаної інтактної лікарської форми через 1 годину, становить приблизно 4:1 або більше, на основі розчинення вказаної лікарської форми in vitro впродовж 1 години у 900 мл рідини, що імітує шлунковий сік, з використанням апарата USP типу II (лопатевого) на 75 об./хв. при 37 °С.
6. Лікарська форма за п.1, де кількість антагоніста, вивільненого із вказаної інтактної лікарської форми через 1 годину, менша за кількість, біоеквівалентну 0,25 мг налтрексону, а кількість вказаного антагоніста, вивільненого через 1 годину із вказаної лікарської форми після її псування, становить кількість, біоеквівалентну 0,25 мг налтрексону або більше, причому вказане співвідношення базується на розчиненні лікарської форми впродовж 1 години у 900 мл рідини, що імітує шлунковий сік, з використанням апарата USP типу II (лопатевого) на 75 об./хв. при 37°С.
7. Лікарська форма за п.1, де антагоністом є налтрексон та кількість налтрексону, вивільненого із вказаної інтактної лікарської форми через 1 годину, менша за 0,25 мг, а кількість вказаного налтрексону, вивільненого через 1 годину із вказаної лікарської форми після її псування, становить 0,25 мг або більше, причому вказане вивільнення базується на розчиненні лікарської форми впродовж 1 години у 900 мл рідини, що імітує шлунковий сік, з використанням апарата USP типу II (лопатевого) на 75 об./хв. при 37 °С.
8. Лікарська форма за п.1, яка відрізняється тим, що через 1 годину після перорального введення вказана лікарська форма вивільнює не більше 25% вказаного антагоніста, причому вказана лікарська форма забезпечує аналгезію, а вказаний антагоніст, що вивільнився, не впливає на анальгетичну ефективність.
9. Лікарська форма за п.1, яка відрізняється тим, що згаданий антагоніст перебуває у формі множини частинок, окремо покритих матеріалом, який по суті відвертає вивільнення антагоніста.
10. Лікарська форма за п.1, яка відрізняється тим, що згаданий антагоніст диспергований у матриксі, що включає матеріал, який по суті відвертає вивільнення антагоніста.
Текст
Опіоіди, відомі також як опіоідні агоністи, є групою лікарських засобів, які демонструють властивості, подібні до властивостей опію або морфіну. Опіоіди застосовують, головним чином, як анальгетики від помірної до сильної дії, але вони викликають також багато інших фармакологічних ефектів, включаючи сонливість, пригнічення дихання, зміну настрою та запаморочення свідомості, без втрати свідомості. Опіоіди здійснюють агоністичний вплив на стереоспецифічні та насичувані сайти зв'язування у головному мозку та інших тканинах. Ендогенні опіоідні пептиди присутні особливо у тих ділянках центральної нервової системи, які, як вважають, пов'язані зі сприйняттям болю, з рухом, настроєм і поведінкою, а також з регуляцією нейроендокринних функцій. Опій містить більше двадцяти різних алкалоїдів. Морфін, кодеїн і папаверин входять у цю групу. У середині дев'ятнадцятого століття в медицині почало отримувати розповсюдження використання чистих алкалоїдів, таких як морфін, а не препаратів опію-сирцю. Парентеральне застосування морфіну мало тенденцію до виникнення більш серйозної низки компульсивного застосування лікарських засобів, ніж препарати опію-сирцю. Проблема зловживання опіоідами стимулювала пошук ефективних анальгетиків, які б не мали потенціалу розвитку наркозалежності. До 1967р. дослідники дійшли висновку, що складні взаємодії між подібними до морфіну лікарськими засобами, антагоністами, які згодом називали "змішаними агоністамиантагоністами", можна найкращим чином пояснити, якщо прийняти постулат про те, що існують більш ніж один тип рецепторів для опіоідів і споріднених до них лікарських засобів. З появою нових, повністю синтетичних речовин з дією, подібною до морфіну, термін "опіоід" було в цілому збережено як родове позначення усіх ендогенних речовин, які у стереоспецифічний спосіб зв'язуються з будь-якими кількома підвидами опіоідних рецепторів і виявляють агоністичну дію. В той час, як це більш широке розуміння сприяло розвиткові фармакології як науки, воно не призвело до розробки анальгетичного опіоіду, який не має потенціалу виникнення зловживань. Потенціал розвитку толерантності та фізичної залежності з повторним застосуванням опіоідів є характерною рисою усіх опіоідних лікарських засобів, а можливість розвитку психологічної залежності (тобто наркоманії) становить одну з головних проблем використання лікування болю опіоідами, навіть незважаючи на те, що ятрогенна наркоманія є рідкою. Іншою основною проблемою, пов'язаною із застосуванням опіоідів, є перехід даних лікарських засобів від пацієнта, що страждає від болю, до іншого (не пацієнта) для незаконних цілей, наприклад, до наркомана. Загальний потенціал опіоіду відносно виникнення зловживань не визначається якимось одним фактором. Навпаки, існує комплекс факторів, включаючи здатність наркотика викликати такий вид фізичної залежності, при якому припинення приймання наркотика викликає дискомфорт, достатній для абстинентної поведінки; здатність пригнічування симптомів абстиненції, викликаних припиненням приймання наркотика, іншими агентами; ступінь, у якому він викликає ейфорію, схожу з ейфорією, що викликається морфіном та іншими опіоідами; профіль токсичності, яка спостерігається при перевищуванні дозою наркотика нормальних терапевтичних меж; і фізичні характеристики наркотиків, такі як розчинність у воді. Дані фізічні характеристики можуть визначати, чи існує ймовірність виникнення зловживань наркотиком при парентеральному способі введення. У Сполучених Штатах спроби контролювати компульсивних споживачів наркотиків включають спроби контролювати доступність наркотиків шляхом введення обмежень застосування опіоідів при лікуванні болю у компульсивних споживачів наркотиків. На практиці лікар часто постає перед вибором введення ефективних опіоідних анальгетиків навіть тим особам, які, як передбачається, схильні до розвитку психологічної залежності, тобто наркоманії, від таких лікарських засобів. З точки зору зазначеної проблеми рекомендується не призначати опіоід таким пацієнтам, якщо підходить інший лікарський засіб, що не має потенціалу виникнення зловживань; а також не давати таким пацієнтам таку лікарську форму, яку можна використовувати для парентеральних зловживань і у будь-який час давати запас лікарського засобу тільки на декілька днів. Ідентифіковано принаймні три види споживання опіоідів і залежності від них. Перший вид включає осіб, застосування наркотиків якими починається у контексті медичного лікування і які отримують свої перші запаси з легальних джерел, наприклад, від лікарів. Інший вид починається з експериментального або "рекреаційного" застосування наркотиків і прогресує до більш інтенсивного застосування. Третій вид включає споживачів, які починають з одного з попередніх видів, але потім переходять на пероральні опіоіди, такі як метадон, який одержують за допомогою ліцензованих програм лікування наркоманії. Толерантність відноситься до потреби збільшувати дозу опіоіду із плином часу, щоб досягати того самого рівня анальгезії або ейфорії, або до спостереження того, що повторне введення однієї й тієї самої дози викликає зниження анальгезії, ейфорії або інших ефектів опіоідів. Було встановлено, що виражений ступінь толерантності розвивається відносно пригнічування дихання, анальгезії, седації, блювотної дії та ейфоригенних ефектів опіоідів. Проте, ступінь, до якого може розвиватися ця толерантність, як у наркомана, так і у пацієнта, якому потрібне лікування болю, залежить від виду споживання. Якщо опіоід застосовується часто, може бути необхідним підвищення дози. Толерантність не розвивається у рівному або однаковому ступені відносно усіх ефектів опіоідів, і навіть у тих споживачів, у яких є висока толерантність до пригнічування дихання, продовжує спостерігатись міоз і запор. Толерантність до опіоідів здебільшого зникає, коли синдром абстиненції закінчується. Фізична залежність може розвинутись при повторних введеннях або тривалому застосуванні опіоідів. Фізична залежність проявляється поступово після припинення використання опіоідів або стрімко (наприклад, впродовж декількох хвилин) після введення антагоніста наркотиків (називається "стрімкою абстиненцією"). Залежно від наркотика, від якого виникла залежність, а також від тривалості вживання та дози, симптоми абстиненції варіюють за кількістю і видом, тривалістю і тяжкістю. Найбільш поширені симптоми абстинентного синдрому включають анорексію, втрату ваги, розширення зіниць, остуду, яка чергується з надмірним спітнінням, абдомінальні колики, нудоту, блювоту, спазми мускулатури, підвищену дратівливість, сльозоточивість, ринорею, гусячу шкіру та підвищення частоти серцевих скорочень. Справжні абстинентні синдроми звичайно починаються через 24-48 годин після останньої дози, сягають максимальної інтенсивності приблизно до третього дня і можуть не починати зменшуватись до третього тижня. Синдроми стрімкої абстиненції, викликані введенням опіоідного антагоніста, варіюють за інтенсивністю і тривалістю з дозою і конкретним антагоністом, але звичайно варіюють за тривалістю від декількох хвилин до декількох годин. Психологічна залежність (тобто наркоманія) від опіоідів характеризується намаганням одержати наркотик, спрямованим на досягнення ейфорії та вивільнення, наприклад, від психосоціоекономічного тиску. Наркоман продовжуватиме вводити собі опіоіди у немедичних цілях і всупереч шкоді, яку він собі завдає. Раніше спеціалісти робили спроби контролювати потенціал розвитку зловживань, пов'язаний з опіоідними анальгетиками. Наприклад, у таблетках, які є у продажу у Сполучених Штатах, використовується комбінація пентазоцину та налоксону під назвою Talwin®Nx від компанії Sanofi-Winthrop. Talwin®Nx містить пентазоцину гідрохлорид, еквівалентний 50мг основи, та налоксону гідрохлорид, еквівалентний 0,5мг основи. Talwin®Nx показаний для полегшення болю від помірного до сильного. Кількість налоксону, присутня у цій комбінації, має низьку активність при пероральному введенні і мінімально перешкоджає фамакологічній дії пентазоцину. Проте, ця кількість налоксону при парентеральному введенні виявляє виражену антагоністичну дію відносно наркотичних анальгетиків. Таким чином, включення налоксону призначене для стримування форми зловживання пероральним пентазоцином, яке спостерігається, якщо лікарську форму розчинити та ін'єкувати. Отже, для зазначеної лікарської форми існує більш низька ймовірність парентерального зловживання, ніж у попередніх пероральних композицій пентазоцину. Проте, вона все ще є об'єктом зловживань з боку пацієнта з використанням перорального способу введення, наприклад, якщо пацієнт приймає безліч доз одразу. Фіксований комбінований лікарський засіб, який включає тилідин (50мг) і налоксон (4мг), для лікування сильного болю є у продажу в Німеччині з 1978p. (Valoron®N, Goedecke). Обгрунтуванням для комбінування даних лікарських засобів є ефективне полегшення болю та запобігання зловживання тилідином завдяки індукованому налоксоном антагонізму відносно рецепторів морфіну. Фіксована комбінація бупренорфіну та налоксону є у продажу в Новій Зеландії з 1991p. (Temgesic®Nx, Reckitt & Colman) для лікування болю. Об'єктом цього винаходу є лікарська форма опіоідного агоніста для перорального введення, яка є придатною для зменшення ймовірності зловживань опіоідним агоністом, що в ній міститься. Об'єктом кращого варіанту здійснення цього винаходу є лікарська форма опіоідного агоніста для перорального введення, яка є придатною для зменшення ймовірності зловживань опіоідним агоністом, не впливаючи на анальгетичні ефекти опіоідного агоніста, або для зниження ризику розвитку стрімкої абстиненції. Об'єктом кращого варіанту здійснення цього винаходу є лікарська форма опіоідного агоніста для перорального введення, яка є стійкою відносно неправильного застосування, зловживань або відхилень; згадана стійкість не залежить від властивих окремому пацієнтові відмінностей у ефектах спільно введених сумішей опіоідного агоніста та антагоніста. Об'єктом кращого варіанту здійснення цього винаходу є лікарська форма для перорального введення, яка містить ефективну дозу опіоідного агоніста разом з такою дозою опіоідного антагоніста, яка не змінює анальгетичну ефективність опіоідного агоніста, коли лікарську дозу вводять інтактною перорально, але яка може запобігти зловживанням, перешкоджаючи ефекту опіоідного агоніста, якщо лікарську форму псують. Об'єктом кращого варіанту здійснення цього винаходу є спосіб запобігання зловживанням лікарською формою опіоіду для перорального введення, при якому лікарська форма включає також таку дозу опіоідного антагоніста, яка секвестрована, наприклад, не є біодоступною, коли лікарську форму псують (наприклад, у спробі зловживання дозою опіоідного анальгетика). Ще одним об'єктом кращого варіанту здійснення цього винаходу є лікарські форми для перорального введення, які призначені або придатні для застосування при лікуванні гострого або хронічного болю, коли необхідно уникати змін анальгетичних ефектів опіоідного агоніста, як у випадках толерантності, фізичної залежності або індивідуальних особливостей метаболізму або фізіології печінки. Ще одним об'єктом кращого варіанту здійснення цього винаходу є спосіб лікування болю у пацієнтівлюдей лікарською формою опіоідного агоніста для перорального введення з одночасним зниженням зловживання нею пероральним, парентеральним, інтраназальним та/або сублінгвальним шляхом. Деякі або усі згадані вище та інші об'єкти досягаються за допомогою варіантів здійснення цього винаходу, які спрямовані в одній своїй частині на лікарську форму для перорального введення, що включає опіоідний агоніст та опіоідний антагоінст, у якій опіоідний антагоніст присутній у практично невивільнюваній формі (тобто "секвестрований"). У кращих варіантах здійснення цього винаходу лікарська форма містить терапевтично ефективну при пероральному введенні кількість опіоідного агоніста; вказана лікарська форма здійснює бажану анальгетичну дію. Оскільки опіоідний антагоніст присутній у практично невивільнюваній формі, він практично не блокує анальгетичний ефект опіоідного агоніста, коли лікарську форму вводять перорально інтактною, і не становить ризику розвитку стрімкої абстиненції у толерантних до опіоідів або залежних пацієнтів. У кращих варіантах здійснення цього винаходу лікарська форма для перорального введення за цим винаходом спрямована на лікарську форму для перорального введення, яка включає (і) опіоідний агоніст у вивільнюваній формі та (іі) секвестрований опіоідний антагоніст, який практично не вивільнюється, коли лікарську форму вводять інтактною, таким чином, що співвідношення кількості антагоніста, вивільненого з лікарської форми після її псування, і кількості антагоніста, вивільненого з інтактної лікарської форми, становить приблизно 4:1 або більше, на основі розчинення лікарської форми in vitro впродовж 1 години у 900мл рідини, що імітує шлунковий сік, із використанням апарата USP типу II (лопасного) на 75об./хв. при 37°С, в якій агоніст та антагоніст взаємно дисперговані і не ізольовані один від одного у двох різних шарах. В інших варіантах здійснення даний винахід відноситься до лікарської форми для перорального введення, яка включає (і) опіоідний агоніст у вивільнюваній формі та (іі) секвестрований опіоідний антагоніст, який практично не вивільнюється, коли лікарську форму вводять інтактною, таким чином, що співвідношення кількості антагоінста, вивільненого з лікарської форми після її псування, і кількості антагоніста, вивільненого з інтактної лікарської форми, становить приблизно 4:1 або більше, на основі розчинення лікарської форми in vitro впродовж 1 години у 900мл рідини, що імітує шлунковий сік, із використанням апарата USP типу II (лопасного) на 75 об./хв. при 37°С, в якій антагоніст знаходиться у формі множини часток, окремо покритих секвеструвальним матеріалом, який практично відвертає вивільнення антагоніста. В інших варіантах здійснення даний винахід відноситься до лікарської форми для перорального введення, яка включає (і) опіоідний агоніст у вивільнюваній формі та (іі) секвестрований опіоідний антагоніст, який практично не вивільнюється, коли лікарську форму вводять інтактною, таким чином, що співвідношення кількості антагоніста, вивільненого з лікарської форми після її псування, і кількості антагоніста, вивільненого з інтактної лікарської форми, становить приблизно 4:1 або більше, на основі розчинення лікарської форми in vitro впродовж 1 години у 900мл рідини, що імітує шлунковий сік, із використанням апарата USP типу II (лопасного) на 75об./хв. при 37°С, в якій антагоніст диспергований у матриксі, що включає секвеструвальний матеріал, який практично відвертає вивільнення антагоніста. В інших варіантах здійснення даний винахід відноситься до лікарської форми для перорального введення, яка включає (і) опіоідний агоніст у вивільнюваній формі та (іі) секвестрований опіоідний антагоніст, який практично не вивільнюється, коли лікарську форму вводять інтактною, таким чином, що співвідношення кількості антагоніста, що міститься в інтактній лікарській формі, та кількості антагоніста, вивільненого з інтактної лікарської форми, становить приблизно 4:1 або більше, на основі розчинення лікарської форми in vitro впродовж 1 години у 900 мл рідини, що імітує шлунковий сік, із використанням апарата USP типу II (лопасного) на 75 об./хв. при 37°С, в якій агоніст та антагоніст взаємно дисперговані і не ізольовані один від одного у двох різних шарах. В інших варіантах здійснення даний винахід відноситься до лікарської форми для перорального введення, яка включає (і) опіоідний агоніст у вивільнюваній формі та (іі) секвестрований опіоідний антагоніст, який практично не вивільнюється, коли лікарську форму вводять інтактною, таким чином, що кількість антагоніста, вивільненого з інтактної лікарської форми через 1 годину, менше кількості, біоеквівапентної 0,25мг налтрексену, і кількість антагоніста, вивільненого через 1 годину з лікарської форми після її псування, становить кількість, біоеквівалентну 0,25мг налтрексрну або більше; вивільнення базується на розчиненні лікарської форми впродовж 1 години у 900мл рідини, що імітує шлунковий сік, із використанням апарата USP типу II (лопасного) на 75об./хв. при 37°С, в якій агоніст та антагоніст взаємно дисперговані і не ізольовані один від одного у двох різних шарах. Краще, якщо кількість антагоніста, вивільненого через 1 годину із зіпсованої лікарської форми, становить кількість, біоеквівалентну приблизно 0,5мг налтрексону або більше, та/або кількість антагоніста, вивільненого через 1 годину з інтактної лікарської форми, становить кількість, біоеквівалентну приблизно 0,125мг налтрексону або менше. В інших варіантах здійснення даний винахід відноситься до лікарської форми для перорального введення, яка включає (і) опіоідний агоніст у вивільнюваній формі та (іі) секвестрований налтрексон або його фармацевтично прийнятну сіль, який практично не вивільнюється, коли лікарську форму вводять інтактною, таким чином, що кількість налтрексону, вивільненого з інтактної лікарської форми через 1 годину, становить менше 0,25мг, і кількість налтрексону, вивільненого через 1 годину з лікарської форми після її псування, становить 0,25мг або більше; вивільнення базується на розчиненні лікарської форми впродовж 1 години у 900мл рідини, що імітує шлунковий сік, із використанням апарата USP типу II (лопасного) на 75об./хв. при 37°С, в якій агоніст та антагоніст взаємно дисперговані і не ізольовані один від одного у двох різних шарах. Альтернативно, у цьому варіанті здійснення даного винаходу кількість антагоніста, вивільненого через 1 годину із зіпсованої лікарської форми, становить приблизно 0,5мг налтрексону або більше, та/або кількість антагоніста, вивільненого через 1 годину з інтактної лікарської форми, становить приблизно 0,125мг налтрексону або менше. В інших варіантах здійснення даний винахід відноситься до лікарської форми для перорального введення, яка включає (і) терапевтично ефективну кількість опіоідного агоніста та (іі) секвестрований опіоідний антагоніст, таким чином, що через 1 годину після перорального введення інтактна лікарська форма вивільнює не більше приблизо 25% антагоніста, причому вказана лікарська форма забезпечує анальгезію, а вивільнений антагоніст не впливає на анальгетичну ефективність, в якій агоніст та антагоніст взаємно дисперговані і не ізольовані один від одного в двох різних шарах. Краще,-якщо інтактна лікарська форма вивільнює не більше приблизно 12,5% антагоніста. В інших варіантах здійснення даний винахід відноситься до лікарської форми для перорального введення, яка включає (і) опіоідний агоніст у вивільнюваній формі та (іі) опіоідний антагоніст у практично невивільнюваній формі, в якій антагоніст перебуває у формі множини часток, окремо покритих матеріалом, який практично відвертає вивільнення антагоніста. В інших варіантах здійснення даний винахід відноситься до лікарської форми для перорального введення, яка включає (і) опіоідний агоніст у вивільнюваній формі та (іі) опіоідний антагоніст у практично невивільнюваній формі, в якій антагоніст диспергований у матриксі, що включає матеріал, який практично відвертає вивільнення антагоніста. У певних варіантах здійснення даного винаходу інтактна лікарська форма за даним винаходом вивільнює деяку кількість опіоідного антагоніста, що міститься в ній, через 1 годину після перорального введення, наприклад, лікарська форма вивільнює принаймні 0,025мг налтрексону або біоеквівалентну дозу іншого антагоніста через 1 годину. У цих варіантах здійснення даного винаходу лікарська форма забезпечує анальгезію пацієнту, а вивільнений антагоніст не впливає на анальгетичну ефективність. У цих варіантах здійснення даного винаходу лікарська форма краще не вивільнює 0,25мг або більше налтрексону через 1 годину після введення. Вивільнення налтрексону з інтактної лікарської форми можна вимірити для цілей даних варіантів здійснення даного винаходу на основі розчинення in vitro лікарської форми впродовж 1 години у 900мл рідини, що імітує шлунковий сік, із використанням апарата USP типу II (лопасного) на 75об./хв. при 37°С. В інших варіантах здійснення даний винахід відноситься до лікарської форми дпд-перпряпьного вйєдєння, яка включає опіоідний агоніст і наптрексон або або його солі у практично невивільнюваній формі, в якій агоніст та налтрексон принаймні частково взаємно дисперговані. В інших варіантах здійснення даний винахід відноситься до лікарської форми для перорального введення, яка включає опіоідний агоніст і біодоступний при пероральному введенні опіоідний антагоніст у практично невивільнюваній формі, в якій агоніст та антагоніст принаймні частково взаємно дисперговані. У тих варіантах здійснення даного винаходу, в яких антагоніст перебуває у формі множини часток, покритих секвеструвальним матеріалом, множина часток може бути у формі інертних гранул, покритих антагоністом і зверху покритих матеріалом, або, альтернативно, у формі гранулята, що включає антагоніст і матеріал. Множина часток може бути диспергована у матриксі, який включає опіоідний агоніст, або може міститься у капсулі з опіоідним агоністом. У тих варіантах здійснення даного винаходу, в яких антагоніст диспергований у матриксі, що включає секвеструвальний матеріал, який практично відвертає вивільнення антагоніста, матрикс може бути у формі гранул. Гранули можуть бути дисперговані в іншому матриксі, що включає опіоідний агоніст, або можуть міститись у капсулі з опіоідним агоністом. В інших варіантах здійснення даного винаходу частина антагоніста перебуває в матриксі та/або частина антагоніста перебуває в гранулі з покриттям. У певних варіантах здійснення даного винаходу, в яких зберігається описане вище співвідношення приблизно 4:1 або більше, що стосується кількості антагоніста, вивільненого з лікарської форми після її псування, і кількості вказаного антагоніста, вивільненого з інтактної лікарської форми, на основі розчинення лікарської форми впродовж 1 години у 900мл рідини, що імітує шлунковий сік, із використанням апарата USP типу II (лопасного) на 75об./хв. при 37°С, інтактна лікарська форма вивільнює 22,5% або менше антагоніста через 1 годину, а зіпсована лікарська форма вивільнює 90% або більше антагоніста через 1 годину. В іншому варіанті здійснення даного винаходу інтактна лікарська форма вивільнює 20% або менше вказаного антагоніста через 1 годину, а зіпсована лікарська форма вивільнює 80% або більше антагоніста через 1 годину. В іншому варіанті здійснення даного винаходу інтактна лікарська форма вивільнює 10% або менше вказаного антагоніста через 1 годину, а > зіпсована лікарська форма вивільнює 40% або більше антагоніста через 1 годину. В , іншому варіанті здійснення даного винаходу інтактна лікарська форма вивільнює 5% або менше вказаного антагоніста через 1 годину, а зіпсована лікарська форма вивільнює 20% або більше антагоніста через 1 годину. У певних варіантах здійснення даного винаходу співвідношення кількості антагоніста, вивільненого з лікарської форми після її псування, і кількості вказаного антагоніста, вивільненого з інтактної лікарської форми, на основі розчинення лікарської форми впродовж 1 години у 900 мл рідини, що імітує шлунковий сік, із використанням апарата USP типу II (лопасного) на 75об./хв. при 37°С, становить 10:1 або більше, 50:1 або більше чи 100:1 або більше. У певних варіантах здійснення даного винаходу антагоністом є налтрексон або його фармацевтично прийнятна сіль. У зазначених варіантах інтактна лікарська форма переважно вивільнює менше 0,25мг, краще 0,125мг або менше налтрексону через 1 годину відповідно до описаних вище умов розчинення. Краще, якщо зіпсована лікарська форма вивільнює 0,25мг або більше налтрексону через 1 годину за тих самих умов. У певних варіантах здійснення даного винаходу співвідношення кількості антагоніста, вивільненої з лікарської форми після її псування, та кількості вказаного антагоніста, вивільненої з інтактної лікарської форми, на основі розчинення лікарської форми впродовж 1 години у 900мл рідини, що імітує шлунковий сік, із використанням апарата USP типу II (лопасного) на 75об./хв. при 37°С, становить 10:1 або більше, 50:1 або більше чи 100:1 або більше. У певних варіантах лікарської форми антагоніст у практично невивільнюваній формі адаптований для вивільнення менше 15% мас. in vivo через 36 годин. У певних варіантах лікарської форми антагоніст у практично невивільнюваній формі адаптований для вивільнення менше 8% мас. in vivo через 36 годин. У певних варіантах лікарської форми антагоніст у практично невивільнюваній формі адаптований для вивільнення менше 3% мас. in vivo через 36 годин. У певних варіантах лікарської форми антагоніст у практично невивільнюваній формі адаптований для вивільнення менше 1% мас. in vivo через 36 годин. У певних варіантах лікарської форми антагоніст у практично невивільнюваній формі адаптований для вивільнення менше 0,5% мас. in vivo через 36 годин. Даний винахід відноситься також до способів запобігання зловживань опіоідним агоністом із використанням лікарських форм, описаних у даному документі. Спосіб може включати забезпечення опіоідного агоніста у лікарській формі для перорального введення разом з опіоідним антагоністом, де опіоідний антагоніст присутній у формі, яка є практично невивільнюваною при травленні, коли цілість лікарської форми зберігається до самого початку травлення, але яка стає біодоступною, якщо зазнає псування (наприклад, роздавлювання, нарізання, які руйнують лікарську форму, тощо, дій розчинників або температур вище 45°С). Інший варіант здійснення даного винаходу відноситься до способу зменшення зловживань опіоідним агоністом у лікарській формі для перорального введення, який включає виготовлення лікарської форми для перорального введення, описаної у даному документі. Наприклад, спосіб може включати виготовлення лікарської форми, яка включає (і) терапевтично ефективну при пероральному введенні кількість опіоідного агоніста та (іі) опіоідний антагоніст у практично невивільнюваній формі, таким чином, що вказана лікарська форма забезпечує бажаний анальгетичний ефект, а вказаний антагоніст практично не блокує анальгетичний ефект опіоідного агоніста, коли вказану лікарську форму вводять інтактною. В альтернативних варіантах здійснення даного винаходу ефект опіоідного агоніста принаймні частково блокується, коли вказану лікарську форму псують, наприклад, розжовують, роздавлюють або розчиняють у розчиннику, і вводять перорально, інтраназально, парентерально або сублінгвально. Даний винахід відноситься також до способу лікування болю за допомогою лікарських форм, описаних у даному документі. Спосіб може включати виготовлення лікарської форми для перорального введення, яка містить опіоідний агоніст у вивільнюваній формі та опіоідний антагоніст у практично невивільнюваній формі, та пероральне введення інтактної лікарської форми для перорального введення. Інший варіант здійснення даного винаходу відноситься до способу лікування болю за допомогою лікарських форм, описаних у даному документі. У певних варіантах спосіб лікування болю у пацієнтів за допомогою лікарської форми, яка має менший потенціал виникнення зловживань, включає виготовлення лікарської форми для перорального введення, яка містить вивільнювану форму опіоідного агоніста і практично невивільнювану форму опіоідного антагоніста, та пероральне введення лікарської форми для перорального введення з метою забезпечення рівня агоніста в пламі крові, що перевищує мінімальну анальгетичну концентрацію опіоідного агоніста. Даний винахід відноситься також до способів виготовлення лікарських форм, описаних у цьому документі. У певних варіантах даний винахід включає спосіб виготовлення лікарської форми для перорального введення, яка включає попередню обробку опіоідного антагоніста для того, щоб зробити її практично невивільнюваною, та об'єднання попередньо обробленого антагоніста з вивільнюваною формою опіоідного агоніста, у такий спосіб, при якому зберігається цілісність невивільнюваної форми антагоніста. Певні варіанти здійснення даного винаходу відносяться до композицій, в яких агоніст та антагоніст взаємно дисперговані і не ізольовані один від одного в двох різних шарах. Проте, у певних варіантах агоніст та антагоніст частково взаємно дисперговані. Термін "анальгетична ефективність" визначений для цілей даного винаходу як задовільне зменшення або усунення болю при одночасному переносимому рівні побічних ефектів, що визначається пацієнтом-людиною. Фраза "практично не блокує анальгетичний ефект опіоідного агоніста" означає, що опіоідний антагоніст не блокує ефекти опіоідного агоніста до такого ступеня, який є достатнім для того, щоб зробити лікарську форму терапевтично менш ефективною для забезпечення анальгезії. Фраза "ризик розвитку стрімкої абстиненції" означає, що належна дія композиції не залежить від конкретного співвідношення агоніста та антагоніста або їх метаболізму, який відрізняється один від одного. Термін "опіоідний антагоніст у практично невивільнюваній формі" відноситься до опіоідного антагоніста, який не вивільнюється або практично не вивільнюється через одну годину після перорального введення інтактної лікарської форми, яка містить як опіоідний агоніст, так і опіоідний антагоніст (тобто незіпсованої). Для цілей даного винаходу кількість, вивільнена після перорального введення інтактної лікарської форми, можна вимірити in vitro за допомогою розчинення лікарської форми впродовж 1 години у 900 мл рідини, що імітує шлунковий сік, із використанням апарата USP типу II (лопасного) на 75 об./хв. при 37°С. Подібна лікарська форма також згадується як така, що включає "секвестрований антагоніст". Хоча кращі варіанти здійснення даного винаходу включають опіоідний антагоніст у такій формі, яка повністю запобігає вивільненню опіоідного антагоніста, даний винахід також включає антагоніст у практично невивільнюваній формі. Термін "практично не вивільнюється" відноситься до антагоніста, який може вивільнюватись у малій кількості до тих пір, поки вивільнена кількість не впливає або не впливає значним чином на анальгетичну ефективність, коли лікарську форму, як призначено, вводять перорально людям. У певних кращих варіантах здійснення даного винаходу практично невивільнювана форма антагоніста є стійкою до проносних засобів (наприклад, мінеральної олії), які використовуються для боротьби з уповільненим пасажем крізь товстий кишечник та з ахлоргідричними станами. У певних варіантах здійснення даного винаходу практично невивільнювана форма опіоідного антагоніста включає опіоідний антагоніст, який складений у композицію з одним або більше фармацевтично прийнятними гідрофобними матеріалами, таким чином, що антагоніст не вивільнюється або практично не вивільнюється під час його пасажу крізь шлунково-кишковий тракт при пероральному, як призначено, введенні, незіпсованої лікарської форми. У певних варіантах здійснення даного винаходу практично невивільнювана ' форма опіоідного антагоніста є чутливою до механічного, термічного та/або хімічного псування, наприклад, псування шляхом роздавлювання, нарізання, розмолування, розжовування та/або розчинення у розчиннику у поєднанні з нагріванням (наприклад, вище приблизно 45°С) лікарської форми для перорального введення. При руйнуванні такого роду цілісність практично невивільнюваної форми опіоідного антагоніста буде порушено, і опіоідний антагоніст стане доступним для вивільнення. У певних варіантах здійснення, коли лікарську форму розжовують, роздавлюють або розчиняють і нагрівають у розчиннику та вводять перорально, інтраназально, парентерально або сублінгвально, анальгетичний або ейфоричний ефект опіоіду знижується або усувається. У певних варіантах здійснення ефект опіоідного агоніста принаймні частково блокується опіоідним антагоністом. В інших певних варіантах здійснення ефект опірідного агоніста практично блокується опіоідним антагоністом. Термін "псування" означає будь-яку маніпуляцію За допомогою механічних, термічних та/або хімічних засобів, яка^змінює фізичні властивості лікарської форми, наприклад, для одержання опіоідного агоніста для негайного вивільнення, якщо він знаходиться у формі з уповільненим вивільненням, або для того, щоб зробити опіоідний агоніст доступним для неправильного застосування, такого як введення іншим шляхом, наприклад, парентерально. Псування можуть здійснювати, наприклад, шляхом роздавлювання, нарізання, розмолування, розжовування та/або розчинення у розчиннику у поєднанні з нагріванням (наприклад, вище приблизно 45°С) або будь-якого поєднання цих дій. Термін "принаймні частково блокує ефект опіоіду" для цілей даного винаходу означає, що опіоідний антагоніст принаймні значно блокує ейфоричний ефект опіоідного антагоніста, знижуючи, таким чином, потенціал виникнення зловживань опіоідним агоністом у лікарській формі. У певних кращих варіантах здійснення даного винаходу практично невивільнювана форма опіоідного антагоніста включає частки опіоідного антагоніста з покриттям, яке практично відвертає вивільнення антагоніста. У кращих варіантах здійснення покриття включає один або більше фармацевтично прийнятних гідрофобних матеріалів. Покриття переважно є непроникним для опіоідного антагоніста, що в ньому міститься, та нерозчинним у шлунково-кишковому тракті, практично відвертаючи, таким чином, вивільнення опіоідного антагоніста, коли лікарську форму вводять перорально, як призначено. Відповідно, коли лікарська форма для перорального введення не зіпсована з метою порушення цілісності покриття, опіоідний антагоніст, що в ній міститься, практично не вивільнятиметься впродовж першої години пасажу крізь шлунково-кишковий тракт і, таким чином, не буде доступним для усмоктування. У певних кращих варіантах здійснення даного винаходу гідрофобний матеріал включає целюлозний полімер або акриловий полімер, який є нерозчинним у рідинах шлунково-кишкового тракту і непроникним для опіоідного антагоніста. Термін "частки" опіоідного антагоніста, що використовується у даному документі, відноситься до гранул, сфероїдів, кульок або зерен, які включають опіоідний антагоніст. У певних кращих варіантах здійснення частки опіоідного антагоніста мають діаметр приблизно від 0,2 до 2мм, ще краще приблизно від 0,5 до 2мм. У певних варіантах здійснення даного винаходу лікарська форма для перорального введення додатково включає опіоідний антагоніст у вивільнюваній формі і, таким чином, здатний вивільнюватись з лікарської форми при пероральному введенні; співвідношення опіоідного агоніста та вивільнюваної форми опіоідного антагоніста є такою, що лікарська форма при пероральному введенні є анальгетично ефективною. Наприклад, коли опіоідний антагоніст має покриття, яке практично відвертає його вивільнення, а потім змішується з опіоідним агоністом і пресується у таблетки, деякі кількості покриття можуть потріскатись, призводячи, таким чином, до вивільнення опіоідного антагоніста при пероральному введенні. Краще, якщо опіоідний агоніст, придатний для даного винаходу, можна ~ вибрати з групи, яка складається з морфіну, гідроморфону, гідрокодону, оксикодону, кодеїну, леворфанолу, меперидину, метадону та їхніх сумішей. Кращі приклади опіоідного антагоніста, придатного для даного винаходу, включають налтрексон, налоксон, налмефен, циклазацин, леваллорфан, їх фармацевтично прийнятні солі та суміші. У певних варіантах здійснення даного винаходу співвідношення опіоідного агоніста та опіоідного антагоніста, присутнього у практично невивільнюваній формі, становить приблизно від 1:1 до 50:1 за масою, краще від 1:1 до 20:1 за масою або від 15:1 до 30:1. Масове співвідношення опіоідного агоніста та опіоідного антагоніста, Що використовується у даній заявці, відноситься до маси активних інгредієнтів. Так, наприклад, маса опіоідного антагоніста виключає масу покриття або матрикса, які роблять опіоідний антагоніст практично невивільнюваним, або інших можливих наповнювачів, пов'язаних із частками антагоніста. У певних кращих варіантах здійснення співвідношення становить приблизно від 1:1 до 10:1 за масою. Оскільки опіоідний антагоніст знаходиться у практично невивільнюваній формі, кількість вказаного антагоніста у лікарській формі можна варіювати більш широко, ніж у комбінованих лікарських формах опіоідного агоніста/опіоідного антагоніста, в яких обидва агенти є доступними для вивільнення після введення, оскільки належне фукціонування композиції не залежить від різного метаболізму або печінкового кліренсу. З метою безпеки кількість опіоідного антагоніста, присутнього у практично невивільнюваній формі, вибирають таким чином, щоб вона не була шкідливою для людей, навіть якщо вона повністю вивільниться внаслідок псування лікарської форми. У певних кращих варіантах здійснення даного винаходу опіоідний агоніст включає гідрокодон, оксикодон або їх фармацевтично прийнятні солі, а опіоідний антагоніст, присутній у практично невивільнюваній формі, включає налоксон, налтрексон або їх фармацевтично прийнятні солі. Лікарська форма для перорального введення, яка містить опіоідний агоніст у комбінації з практично невивільнюваною формою опіоідного антагоніста, включає, без обмеження, таблетки або капсули. Лікарські форми за даним винаходом можуть включати будь-які бажані фармацевтичні наповнювачі, відомі спеціалістам. Лікарські форми для перорального введення можуть додатково забезпечити негайне вивільнення опіоідного агоніста. У певних варіантах здійснення лікарські форми для перорального введення за даним винаходом забезпечують уповільнене вивільнення вміщуваного в них опіоідного агоніста. Лікарскі форми для перорального введення, що забезпечують уповільнене вивільнення опіоідного агоніста, можна виготовляти відповідно до композицій/способів виготовлення, відомих спеціалістам у галузі фармації, наприклад, шляхом інкорпорування носія з уповільненим вивільненням у матрикс, що містить практично невивільнювану форму опіоідного антагоніста; або шляхом нанесення покриття з уповільненим вивільненням на матрикс, що містить опіоідний агоніст і практично невивільнювану форму опіоідного антагоніста. Переваги лікарської форми, резистентної до зловживань, є особливо великими у зв'язку з лікарськими формами для перорального введення, що містять сильні опіоідні агоністи (наприклад, оксикодон або гідрокодон), які забезпечують цінні анальгетики, але піддаються зловживанням. Це стосується особливо препаратів опіоідних агоністів із уповільненим вивільненням, які містять у кожній дозованій одиниці більшу дозу бажаного опіоідного агоніста, призначеного для вивільнення впродовж певного проміжку часу. Наркомани придбають подібний препарат із уповільненим вивільненням і роздавлюють, розмолують, екстрагують або у будь-який інший спосіб руйнують препарат так, що весь вміст лікарської форми стає доступним для негайного усмоктування. Оскільки подібне псування лікарської форми за даним винаходом призводить до того, що опіоідний антагоніст також стає доступним для усмоктування, даний винахід забезпечує засіб для припинення зазначених зловживань. Крім того, даний винахід запобігає ризику передозування для звичайних пацієнтів, якого вони зазнають від ефекту "демпингу" повної дози опіоідного агоніста, якщо препарат був випадково розжований або роздавлений. Термін "уповільнене вивільнення" визначений для цілей даного винаходу як вивільнення опіоідного агоніста з лікарської форми для перорального введення з такою швидкістю, що концентрації (рівні) в крові (наприклад, у плазмі) підтримуються у межах терапевтичних значень (вище за мінімальну анальгетичну концентрацію або "МЕАС"), але нижче токсичних рівней, впродовж проміжку часу від 8 до 24 годин, краще, впродовж проміжку часу, показаного для прийняття композиції двічі на день або один раз на день. Даний винахід може забезпечити більш безпечний продукт (наприклад, з меншим пригніченням дихання), якщо його використовують неправильно, а також продукт, для якого існує менший ризик зловживань. У певних варіантах здійснення до композиції включають комбінацію двох опіоідних агоністів. В інших варіантах здійснення включають також один або більше опіоідних агоністів і додатковий неопіоідний лікарський засіб. Зазначені неопіоідні лікарські засоби переважно повинні забезпечувати додаткову анальгезію і включають, наприклад, аспірин, ацетамінофен, нестероідні протизапальні лікарські засоби ("НСПЗЗ"), антагоністи NMDATa інгібітори циклоксигенази-ІІ ("інгібітори СОХ-II"). У ще інших варіантах здійснення даного винаходу можна включати неопіоідний лікарський засіб, який забезпечує бажаний ефект, інший ніж анальгезія, наприклад, лікарські засоби проти кашлю, відхаркувальні, протинабрякові або антигістамінні лікарські засоби тощо. Для цілей даного винаходу термін "опіоідний агоніст" є взаємозамінним з терміном "опіоід" або "опіоідний анальгетик" і включатиме комбінації більше одного опіоідного агоніста, а також включати основу опіоіду, змішані агоністи-антагоністи, часткові агоністи, їх фармацевтично прийнятні солі, стереоізомери, прості і складні ефіри та суміші. Для цілей даного винаходу термін "опіоідний антагоніст" включатиме комбінації більше одного опіоідного антагоніста, а також включатиме основу, його фармацевтично прийнятні солі, стереоізомери, прості і складні ефіри та суміші. Описаний у даному документі винахід охоплює усі фармацевтично прийнятні солі описаних опіоідних агоністів та антагоністів. Фармацевтично прийнятні солі включають, без обмеження, солі металів, такі як натрієва сіль, калієва сіль, цезієва сіль тощо; солі лужноземельних металів, такі як кальцієва сіль, магнієва сіль тощо; солі органічних амінів, такі як солі триетиламіну, сіль піридину, сіль піколіну, сіль етаноламіну, сіль триетаноламіну, сіль дициклогексиламіну, сіль N,N'-дибензилетилендіаміну тощо; солі з неорганічними кислотами, такі як гідрохлорид, гідробромід, сульфат, фосфат тощо; солі з органічними кислотами, такі як форміат, ацетат, трифторацетат, малеат, тартрат тощо; сульфонати, такі як метансульфонат, бензолсульфонат, п-толуолсульфонат тощо; солі з амінокислотами, такі як аргінат, аспаргінат, глутамат тощо. Деякі з опіоідних агоністів та антагоністів, описані у даному документі, можуть містити один або більше асиметричних центрів і можуть, таким чином, утворювати енантіомери, діастереоізомери та інші стереоізомерні форми. Даний винахід також охоплює усі подібні можливі форми, а також рацемічні та розділені форми і їхні суміші. У випадку, коли сполуки, описані у даному документі, містять олефінові подвійні зв'язки або інші центри з геометричною асиметрією, і якщо не вказано інше, даний винахід включає як Е-, так і Z-геометричні ізомери. Даний винахід охоплює також усі таутомери. Використаний у даному документі термін "стереоізомери" є загальним терміном для усіх ізомерів окремих молекул, які можуть відрізнятись тільки орієнтацією своїх атомів у просторі. Він включає енантіомери та ізомери сполук, які мають більше одного хірального центру і не є дзеркальними відображеннями один одного (діастереоізомери). Термін "хіральний центр" відноситься до атома вуглецю, до якого приєднані чотири різні групи. Термін "енантіомер" або "енантіомерний" відноситься до молекули, яка не накладається на своє дзеркальне відображення і, відповідно, є оптично активною, коли енантіомер обертається у площині поляризованого світла в одному напрямку, а його дзеркальне відображення обертається у площині поляризованого світла у протилежному напрямку. Термін "рацемічний" відноситься до суміші рівних частин енантіомерів, яка є оптично неактивною. Термін "розділення" відноситься до відділення або концентрації чи вилучення однієї з двох енантіомірних форм молекули. Даний винахід відноситься також до способу зменшення можливості зловживань опіоідним агоністом у лікарській формі для перорального введення. Спосіб включає забезпечення опіоідного агоніста у лікарській формі для перорального введення, як описано у даному документі. Короткий опис креслень Фіг.1 являє собою графічне зображення результатів прикладу 20. Фіг.2 являє собою графічне зображення результатів прикладу 23. Фіг.3 являє собою графічне зображення результатів прикладу 24. Припускають, що існує принаймні три підвиди опіоідних рецепторів, позначуваних як мю, каппа та дельта. У цих межах мю-рецептор вважають учасником виникнення суперспинальної анальгезії, депресії дихання, ейфорії та фізичної залежності. Каппа-рецептор вважають учасником виникнення спинальної анальгезії, міозу та седації. Активація гамма-рецепторів викликає дисфорію та галюцінації, а також респіраторні та вазомоторні стимулюючі ефекти. Рецептор, що відрізняється від мю-рецептора та позначуваний гамма, було описано у [vas deferens миші, Lord, et al., Nature, 1077, 267, 495-99]. Опіоідні агоністи, як вважають, здійснюють свою агоністичну дію, перш за все, на мю-рецептор і, меншою мірою, на каппа-рецептор. Існує невелика кількість лікарських засобів, які, як виявилось, діють як часткові агоністи на той чи інший тип рецепторів. Зазначені лікарські засоби здійснюють перекривальну дію. Зазначені лікарські засоби включають налорфін, пропірам та бупренорфін. Ще одні лікарські засоби діють як конкурентні антагоністи на мю-рецептор і блокують ефекти морфіноподібних лікарських засобів, здійснюючи свій вплив на каппа- та омега-рецептори. Термін агоніст/антагоніст було введено для опису подібного механізму дії. Даний винахід відноситься до опіоідного анальгетика з контрольованим вивільненням, схожого за анальгетичним спектром з існуючими опіоідними анальгетиками з контрольованим вивільненням, з використанням якого створено композицію з метою зменшення або зведення до мінімуму неправильного застосування, зловживань та відхилень. У певних варіантах здійснення зазначені характеристики одержують за допомогою включення опіоідного антагоніста, такого як налтрексон НСІ, який, у свою чергу, входить до складу унікального матриксу з контрольованим вивільненням. Властивості вказаної композиції розробляли для вивільнення антагоніста в умовах неправильного застосування або псування, в той час як у приписаних умовах застосування повинна вивільнюватись зовсім мала кількість антагоніста (кількість, яка не впливає на анальгезію, викликану у пацієнта). У певних варіантах здійснення даного винаходу вивільнення антагоністичного компонента композиції виражають як співвідношення вивільнення, яке досягається псуванням, наприклад, роздавлюванням або жуванням, і кількості, вивільненої з інтактної композиції. Це співвідношення, таким чином, виражають як [зруйнований]: [цілий], і бажано, щоб це співвідношення мало чисельні межі принаймні 4:1 або більше (вивільнення при руйнуванні за 1 годину/інтактне вивільнення за 1 годину). Коли антагоністом є налтрексон, краще, щоб інтактна лікарська форма вивільнювала менше 0,25мг, краще 0,125мг або менше за 1 годину; і щоб 0,25мг або більше налтрексону вивільнювалось через 1 годину, коли лікарську форму роздавили або розжували. Походження цих величин описано у прикладах 17, 18 і 19. Даний винахід відноситься до лікарської форми опіоідного агоніста для перорального введення, придатної для зменшення ймовірності зловживань опіоідним агоністом, який у ній міститься. Даний винахід включає лікарську форму для перорального введення, яка включає терапевтично ефективну при пероральному введенні кількість опіоідного агоніста у комбінації з опіоідним антагоністом. Опіоідний антагоніст присутній у практично невивільнюваній формі. У певних кращих варіантах здійснення опіоідний антагоніст у практично невивільнюваній формі включає частки опіоідного антагоніста, на які нанесено покриття, яке практично відвертає його вивільнення. У кращих варіантах здійснення вказане покриття оточує частки антагоніста і є непроникним для лікарського засобу та нерозчинним у шлунково-кишковому тракті. Коли лікарську форму за даним винаходом вводять людям перорально, опіоідний антагоніст практично не вивільнюється з-під покриття і, отже, є недоступним для усмоктування в організм. Так, опіоідний антагоніст, хоча й присутній у лікарській формі, практично не блокує анальгетичну ефективність опіоідного агоніста. Проте, якщо лікарську форму для перорального введення за даним винаходом псують до порушення цілісності покриття, опіоідний антагоніст, що міститься в ній, стане доступним принаймні для часткового блокування ефекту опіоідного агоніста. Ця властивість зменшує ймовірність зловживань або неправильного використання опіоідного агоніста у лікарській формі для перорального введення. Нариклад, якщо хтось спробує зловжити лікарським засобом, що міститься у лікарській формі для перорального введення за даним винаходом, шляхом, наприклад, розжовування, роздавлювання, розмолування або розчинення її у розчині при нагріванні (наприклад, вище приблизно 45°С50°С), покриття буде пошкоджене і більше не запобігатиме вивільненню опіоідного антагоніста. Після введення опіоідний антагоніст вивільнятиметься і значно блокуватиме ейфоричну дію опіоідного агоніста. У певних варіантах здійснення даного винаходу співвідношення опіоідного агоніста та антагоніста з покриттям є таким, що, коли лікарську форму для перорального введення псують до порушення цілісності покриття, яке робить опіоідний антагоніт практично невивільнюваним, ейфоричний ефект агоніста повинен зводитись нанівець опіоідним антагоністом при неправильному використанні людиною перорально, парентерально, інтраназально або сублінгвально. У певних кращих варіантах здійснення даного винаходу ейфоричний ефект опіоідного агоніста зводитиметься нанівець опіоідним антагоністом при неправильному парентеральному або сублінгвальному застосуванні. Даний винахід відноситься також до лікарської форми для перорального введення, яка включає вивільнювану форму опіоідного антагоніста разом з опіоідним агоністом і частками антагоніста з покриттям; співвідношення опіоідного агоніста та антагоніста без покриття є таким, що при пероральному введенні, як призначено, лікарська форма для перорального введення є анальгетично ефективною. У певних варіантах здійснення даного винаходу опіоідний антагоніст у практично невивільнюваній формі включає опіоідний антагоніст, диспергований у матриксі, який робить антагоніст практично невивільнюваним; згаданий матрикс включає один або більше фармацевтично прийнятних гідрофобних матеріалів. Антагоніст практично не вивільнюється з матриксу, тому він є недоступним для усмоктування під час свого проходження крізь шлунково-кишковий тракт. У певних варіантах здійснення даного винаходу опіоідний антагоніст в матриксі, який робить антагоніст практично невивільнюваним, включає опіоідний антагоніст, диспергований у матриксі, одержаному екструзією розплаву; вказаний матрикс включає один або більше фармацевтично прийнятних гідрофобних матеріалів. У кращих варіантах здійснення даного винаходу опіоідні агоністи, придатні для даного винаходу, включають, без обмеження, альфентаніл, аллілпродин, алфапродин, анілеридин, бензилморфін, безитрамід, бупренорфін, буторфанол, клонітазен, кодеїн, дезоморфін, декстроморамід, дезоцин, діампромід, діаморфон, дигідрокодеїн, дигідроморфін, дименоксадол, димефептанол, диметилтіамбутен, діоксафетилу бутират, дипіпанон, ептазоцин, етогептазин, етилметилтіамбутен, етилморфін, етонітазен, еторфін, дигідроеторфін, фентаніл і похідні, героїн, гідрокодон, гідроморфон, гідроксипетидин, ізометадон, кетобемідон, леворфанол, левофенацилморфан, лофентаніл, меперидин, мептазинол, метазоцин, метадон, метопон, морфін, мірофін, нарцеін, нікоморфін, норлеворфанол, норметадон, налорфін, налбуфен, норморфін, норпіпанон, опій, оксикодон, оксиморфон, папаверетум, пентазоцин, фенадоксон, феноморфан, феназоцин, феноперидин, пімінодин, піритрамід, профептазин, промедол, проперидин, пропоксифен, суфентаніл, тилідин, трамадол, суміші будь-яких з перелічених агентів, солі будь-яких з перелічених агентів тощо. У певних варіантах здійснення кількість опіоідного агоніста у заявленій композиції опіоідів може становити приблизно від 75нг до 750мг. У певних кращих варіантах здійснення даного винаходу опіоідний агоніст вибирають з групи, яка складається з гідрокодону, морфіну, гідроморфону, оксикодону, кодеїну, леворфанолу, меперидину, метадону, оксиморфону, бупренорфіну, фентанілу та його похідних, дипіпанону, героїну, трамадолу, еторфіну, дигідроеторфін, буторфанолу, леворфанолу та їхніх солей або сумішей. У певних кращих варіантах здійснення даного винаходу опіоідним агоністом є оксикодон або гідрокодон. Еквіанальгетичні дози зазначених опіоідів порівняно до 15мг дози гідрокодону наведені у таблици нижче: Хоча гідрокодон та оксикодон є ефективними для лікування болю, спостерігається збільшення зловживань ними особами, які є психологічно залежними від опіоідів або які неправильно використовують опіоіди у нетерапевтичних цілях. Попередній досвід з іншими опіоідами показав знижену ймовірність зловживань, коли опіоіди вводять у комбінації з наркотичним антагоністом, особливо у пацієнтів, які є колишніми наркоманами. [Weinhold L.L., et al, Buprenorphine Alone and in Combination with Naltrexone in Non-Dependent Humans, Drug and Alcohol Dependence 1992; 30:263-274; Mendelson J., et al., Buprenorphine and Naloxone Interactions in Opiate-Dependent Volunteers, Clin. Pharm. Ther. 1996; 60:105-114]; обидва джерела включені до даного документа як посилання. Зазначені комбінації, проте, не містять опіоідного антагоніста, який знаходиться у практично невивільнюваній формі. Скоріше, опіоідний антагоніст вивільнюється у шлунково-кишковому тракті після перорального введення і стає доступним для усмоктування, базуючись на фізіології хазяїна, яка по-різному метаболізує агоніст та антагоніст і зводить нанівець ефекти агоніста. Гідрокодон являє собою напівсинтетичний наркотичний анальгетик і протикашлевий агент, що володіє різноманітною дією на центральну нервову систему та шлунково-кишковий тракт. З хімічної точки зору, гідрокодон є 4,5-епокси-3-метокси-17-метилморфінан-6-он і відомий також як дигідрокодеїнон. Як і інші опіоіди, гідрокодон може призводити до звикання та викликати лікарську залежність морфінового типу. У надмірних дозах гідрокодон, як і інші опіоіди, викликатиме депресію дихання. Гідрокодон для перорального введення також є доступним у Європі (Бельгії, Німеччині, Греції, Італії, Люксембурзі, Норвегії та Швейцарії) як протикашлевий агент. Парентеральна композиція також є доступною у Німеччині як протикашлевий агент. Для застосування як анальгетика гідрокодону бітартрат є комерційно доступним у Сполучених Штатах лише у вигляді фіксованої комбінації з неопіоідними лікарськими засобами (тобто ібупрофеном, ацетамінофеном, аспірином тощо) для полегшення помірного або помірно сильного болю. Поширена лікарська форма гідрокодону є комбінацією з ацетамінофеном і є комерційно доступною, наприклад, як Lortab® у США від компанії UCB Pharma, Inc., у формі таблеток по 2,5/500мг, 5/500мг, 7,5/500мг та 10/500мг гідрокодону/ацетамінофену. Таблетки є доступними також у співвідношенні 7,5мг гідрокодону бітартрата і 650мг ацетамінофену та 7,5мг гідрокодону бітартрата і 750мг ацетамінофену. Гідрокодон у комбінації з аспірином дають у вигляді лікарської форми для перорального введення дорослим звичайно по 12 таблетці кожні 4-6 годин, як це потрібно для полегшення болю. Таблеткова форма становить 5мг гідрокодону бітартрата та 224мг аспірину з 32мг кофеіну; або 5мг гідрокодону бітартрата та 500мг аспірину. Відносно нова композиція включає гідрокодону бітартрат та ібупрофен. Vicoprofen®, що є комерційно доступним у США від компанії Knoll Laboratories, є таблеткою, що містить 7,5мг гідрокодону бітартрата та 200мг ібупрофену. Даний винахід охоплює усі зазначені композиції із включенням часток опіоідного антагоніста з покриттям, яке робить антагоніст практично невивільнюваним. Оксикодон, хімічно відомий як 4,5-епокси-14-гідрокси-3-метокси-17-метилморфінан-6-он, є опіоідним агоністом, головною терапевтичною дією якого є анальгезія. Інші терапевтичні ефекти оксикодону включають заспокійливу дію, ейфорію та відчуття розслаблення. Точний механізм його анальгетичної дії не відомий, проте специфічні опіоідні рецептори у ЦНС для ендогенних сполук з опіоід-подібною активністю були виявлені по усьому головному та спинному мозку, і вони відіграють свою роль в анальгетичних ефектах зазначеного лікарського засобу. Оксикодон є комерційно доступним у Сполучених Штатах, наприклад, як Oxycontin® від компанії Purdue Pharma L.P., у вигляді таблеток з контрольованим вивільненням для перорального введення, які містять 10мг, 20мг, 40мг або 80мг оксикодону гідрохлориду, та як OxylR™, також від компанії Purdue Pharma L.P., у вигляді капсул з негайним вивільненням, які містять 5мг оксикодону гідрохлориду. Даний винахід охоплює усі зазначені композиції із включенням антагоніста у практично невивільнюваній формі. У кращих варіантах здійснення даного винаходу опіоідний антагоніст за даним винаходом включає налтрексон, налмефен, циклазацин, леваллорфан та їхні суміші. У певних кращих варіантах здійснення даного винаходу опіоідний антагоніст є налоксоном або налтрексоном. У певних варіантах здійснення кількість опіоідного антагоніста, присутня у практично невивільнюваній формі, може становити приблизно від 10нг до 275мг. Налоксон є опіоідним антагоністом, який майже вільний від агоністичних ефектів. Підшкірні дози до 12мг налоксону не виявляють помітної суб'єктивної дії, а 24мг налоксону викликають лише невелику сонливість. Малі дози (0,4-0,8мг) налоксону при внутрішньом'язовому або внутрішньовенному введенні людині відвертають або швидко реверсують ефекти морфіноподібного опіоідного агоніста. Одинмг налоксону при внутрішньовенному введенні, як сповіщають, повністю блокує ефект 25мг героїну. Ефекти налоксону виявляються майже негайно після внутрішньовенного введення. Зазначений лікарський засіб усмоктується після перорального введення, але, як сповіщають, метаболізується у неактивну форму швидко при першому його проходженні крізь печінку таким чином, що, як сповіщають, він має значно більш низьку ефективність, ніж при парентеральному введенні. Пероральна доза більше 1г, як сповіщають, майже повністю метаболізується менше ніж за 24 години. Сповіщають, що при сублінгвальному введенні усмоктується 25% налоксону. [Weinberg, et al., Sublingual Absorbtion of selected Opioid Analgesics, Clin. Pharmacol. Ther. (1988); 44-335-340]. Інші опіоідні антагоністи, наприклад, циклазоцин і налтрексон, у кожному з яких є циклопропілметильні заміщення по азоту, зберігають більшу частину своєї ефективності при пероральному введенні, а тривалість їхньої дії значно більша і сягає 24 годин після перорального введення. При лікуванні пацієнтів, які раніше були опіоідними наркоманами, налтрексон використовували у високих дозах перорально (понад 100мг) для запобігання ейфоригенних ефектів опіоідних агоністів. Налтрексон, як сповіщають, виявляє преференційну блокуючу дію проти мю порівняно до дельта-сайтів. Налтрексон відомий як синтетичний аналог оксиморфону, який не володіє властивостями опіоідного агоніста та відрізняється від оксиморфону за структурою заміною метильної групи, розташованої на атомі азоту оксиморфону, циклопропілметильною групою. Гідрохлорид налтрексону є розчинним у воді приблизно до 100мг/см3. Фармакологічні та фармакокінетичні властивості налтрексону було оцінено у численних дослідженнях на тваринах та людях. Див., наприклад, [статтю Gonzalez J.P., et al. Naltrexone: A review of its Pharmacodynamic and Pharmacokinetic Properties and Therapeutic Efficacy in the Management of Opioid Dependence. Drugs 1988; 35:192-213], включену у даний документ як посилання. Після перорального введення налтрексон швидко усмоктується (впродовж 1 години) та має пероральну біодоступність у межах 5-40%. Зв'язування налтрексону з білками становить приблизно 21%, а об'єм розподілу після введення одноразової дози становить 16,1л/кг. Налтрексон є комерційно доступним у формі таблеток (Revia®, DuPont) для лікування алкогольної залежності та для блокади екзогенно введених опіоідів. Див., наприклад, Revia (таблетки налтрексону гідрохлориду). [Physician's Desk Reference, 51-е вид., Montvale, N.J. "Medical Economics", 1997; 51:957-959]. Доза 50мг Revia® блокує фармакологічні ефекти 25мг в/в введеного героїну строком до 24 годин. Відомо, що при спільному введенні з морфіном, героїном або іншими опіоідами на постійній основі, налтрексон блокує розвиток фізичної залежності від опіоідів. Вважають, що спосіб, за допомогою якого налтрексон блокує ефекти героїну, являє собою конкурентне зв'язування з рецепторами опіоідів. Налтрексон використовували для лікування наркоманії шляхом повної блокади ефектів опіоідів. Було встановлено, що найбільш успішним застосуванням налтрексону для лікування наркоманії є застосування у наркоманів, які мають добрий прогноз, як частини багатосторонньої програми зайнятості та реабілітації, що включає контроль поведінки або інші способи поліпшення дотримання режиму та схеми лікування пацієнтами. Для лікування наркотичної залежності налтрексоном бажано, щоб пацієнт не приймав опіоідів принаймні впродовж 7-10 днів. Початкова доза налтрексону для таких цілей звичайно становила приблизно 25мг, та, якщо не спостерігаються симптоми абстиненції, дозу можна збільшувати до 50мг на день. Вважається, що добова доза 50мг викликає адекватну клінічну блокаду ефектів парентерально введених опіоідів. Налтрексон також використовувався для лікування алкоголізму як допоміжний засіб разом із соціальними та психотерапевтичними методами. У певних варіантах здійснення даного винаходу співвідношення опіоідного агоніста та практично невивільнюваної форми опіоідного антагоніста у лікарській формі для перорального введення таке, що ефект опіоідного агоніста принаймні частково блокується, коли лікарську форму розжовують, роздавлюють або розчиняють у розчиннику та нагрівають, а потім вводять перорально, інтраназально, парентерально або сублінгвально. Оскільки лікарська форма для перорального введення за даним винаходом, коли її вводять належним чином, практично не вивільнюватиме опіоідний антагоніст, кількість цього антагоніста може варіювати більш широко, ніж у випадку, якщо опіоідний антагоніст є доступним для вивільнення у шлунковокишковий тракт після перорального введення. Для цілей безпеки кількість антагоніста, присутнього у практично невивільнюваній формі, не повинна бути небезпечною для людей, навіть якщо вона вивільниться повністю. Співвідношення конкретного опіоідного агоніста та антагоніста спеціаліст може визначити без зайвого експериментування. У певних варіантах здійснення даного винаходу співвідношення опіоідного агоніста та опіоідного антагоніста, присутнього у практично невивільнюваній формі, становить приблизно від 1:1 до 50:1 за масою, краще приблизно від 1:1 до 20:1 за масою. У певних кращих варіантах здійснення даного винаходу зазначене співвідношення становить приблизно від 1:1 до 10:1 за масою. У кращому варіанті здійснення даного винаходу опіоідний агоніст включає оксикодон або гідрокодон і присутній у кількості приблизно 15-45мг, а опіоідний антагоніст включає налтрексон і присутній у кількості приблизно 0,5-5мг. Пероральна лікарська форма за даним винаходом може додатково включати, окрім опіоідного агоніста та антагоніста, один або більше лікарських засобів, які можуть або не можуть діяти з ними синергетично. Так, у певних варіантах здійснення даного винаходу до лікарської форми можна включати комбінацію двох опіоідних агоністів, окрім опіоідного антагоніста. Наприклад, лікарська форма може включати два опіоідні агоністи з різними властивостями, такими як напівперіод існування, розчинність, ефективність, і комбінацію будь-яких з вищезгаданих агентів. В інших варіантах здійснення даного винаходу включають один або більше опіоідних агоністів, а також додатковий неопіоідний лікарський засіб, окрім опіоідного антагоніста. Зазначені неопіоідні лікарські засоби, переважно, повинні забезпечувати додаткову анальгезію, і вони включають, наприклад, аспірин, ацетамінофен; нестероідні протизапальні лікарські засоби ("НСПЗЗ"), наприклад, ібупрофен, кетопрофен тощо; антагоністи N-метил-О-аспартатних (NMDA) рецепторів, наприклад, морфінан, такий як декстрометорфан чи декстрорфан, або кетамін; інгібітори циклооксигенази-ІІ ("інгібітори СОХ-ІГ); та/або антагоністи гліцинових рецепторів. У певних кращих варіантах здійснення даного винаходу даний винахід дозволяє використовувати більш низькі дози опіоідного анальгетика за допомогою включення додаткового неопіоідного агоніста, такого як НСПЗЗ або інгібітор СОХ-ІІ. Завдяки використанню більш малих кількостей одного або обох лікарських засобів зменшуються побічні ефекти, пов'язані з ефективним усуненням болю у людей. Придатні нестероідні протизапальні агенти включають ібупрофен, диклофенак, напроксен, беноксапрофен, флюрбіпрофен, фенопрофен, флюбуфен, кетопрофен, індопрофен, піропрофен, карпрофен, оксапрозин, прамопрофен, муропрофен, триоксапрофен, супрофен, амінопрофен, тіапрофенову кислоту, флюрпрофен, буклоксову кислоту, індометацин, суліндак, толметин, зомепірак, тіопінак, зидометацин, ацеметацин, фентіазак, кліданак, окспінак, мефенамінову кислоту, меклофенамінову кислоту, флюфенамінову кислоту, ніфлюмінову кислоту, толфенамінову кислоту, дифлюризал, флюфенізал, піроксикам, судоксикам або ізоксикам тощо. Придатні дозування вказаних лікарських засобів добре відомі спеціалістам. Антагоністи N-метил-О-аспартатних (NMDA) рецепторів добре відомі спеціалістам і включають, наприклад, морфінани, такі як декстрометорфан або декстрорфан, кетамін, d-метадон або їх фармацевтично прийнятні солі. Для цілей даного винаходу термін "антагоніст NMDA" також, як вважають, охоплює лікарські засоби, які блокують головний внутрішньоклітинний наслідок активації рецепторів NMDA, наприклад, гангліозид, такий як GM-i або GT1b, фенотіазин, такий як трифторперазин, або нафталінсульфонамід, такий як ІМ-(6-амінотексил)-5-хлор-1-нафталінсульфонамід. Згадані лікарські засоби інгібують розвиток толерантності та/або залежності від наркотиків, наприклад, наркотичних анальгетиків, таких як морфін, кодеїн тощо, як заявлено у [патентах США №№5321012 та 5556838 (обидва Mayer, et al.)], і лікують хронічний біль, як заявлено в [патенті США №5502058 (Mayer, et al.)]; усі зазначені патенти включені у даний документ як посилання. Антагоніст NMDA можна включати окремо або у комбінації з місцевим анестетиком, таким як лідокаїн, як описано в зазначених патентах Mayer, et al. Лікування хронічного болю шляхом використання антагоністів гліцинових рецепторів, а також ідентифікація вказаних лікарських засобів описані у [патенті США №5514680 (Weber, et al.)], включеному до даного документа як посилання. Інгібітори СОХ-2 описані в літературі, а також багато хімічних структур, які, як відомо, інгібують циклооксигеназу-2. Інгібітори СОХ-2 описані, наприклад, у [патентах США №№ 5616601, 5604260, 5593994, 5550142, 5536752, 5521213, 5475995, 5639780, 5604253, 5552422, 5510368, 5436265, 5409944 і 5130311], включених у даний документ як посилання. Деякі кращі інгібітори СОХ-2 включають целекоксиб (SC-58635), DUP-697, флозулід (CGP-28238), мелоксикам, 6-метокси-2-нафтилоцтову кислоту (6-MNA), МК-966 (відомий також як Vioxx), набуметон (пролікі 6-MNA), німезулід, NS-398, SC-5766, SC-58215, Т-614 або їхні комбінації. Рівні дози інгібітора СОХ-2 порядку приблизно від 0,005мг до 140мг на кілограм маси тіла в день є терапевтично ефективними у комбінації з опіоідним анальгетиком. Альтернативно, приблизно від 0,25мг до 7 г інгібітора СОХ-2 на пацієнта в день вводять у комбінації з опіоідним анальгетиком. В інших варіантах здійснення даного винаходу можна включати неопіоідний лікарський засіб, який виявляє бажану дію, іншу, ніж анальгезія, наприклад, лікарський засіб від кашлю, відхаркувальний лікарський засіб, протинабряковий лікарський засіб, антигістамінний лікарський засіб, місцеві анестетики тощо. Одержання опоідного антогоніста у практично невивільнюваній формі У певних варіантах здійснення цього винаходу опіоідний антагоніст у практично невивільнюваній формі можна одержувати шляхом об'єднання антагоніста з одним або більше фармацевтично прийнятним гідрофобним матеріалом. Наприклад, на частки опіоідного антагоніста можна наносити покриття, яке практично запобігає вивільненню антагоніста; зазначене покриття включає гідрофобний матеріал (матеріали). Іншим прикладом може слугувати опіоідний антагоніст, який диспергований у матриксі, який робить антагоніст практично невивільнюваним; зазначений матрикс включає гідрофобний матеріал (матеріали). У певних варіантах здійснення фармацевтично прийнятний гідрофобний матеріал включає целюлозний полімер, вибраний з групи, яка складається з етилцелюлози, ацетата целюлози, пропіоната целюлози (з малою, середньою або великою молекулярною масою), пропіоната ацетата целюлози, бутирата ацетата целюлози, фталата ацетата целюлози та триацетата целюлози. Прикладом етилцелюлози є етилцелюлоза із вмістом етокси від 44 до 55%. Етилцелюлозу можна використовувати у формі спиртового розчину. У деяких інших варіантах здійснення цього винаходу гідрофобний матеріал включає полімолочну кислоту, полігліколеву кислоту та сополімер полімолочної та полігліколевої кислоти. У певних варіантах здійснення цього винаходу гідрофобний матеріал може включати целюлозний полімер, вибраний з групи, яка складається з простого ефіру целюлози, складного ефіру целюлози, простого ефіру складного ефіру целюлози та целюлози. Целюлозні полімери мають ступінь заміщення, D.S., на ангідроглюкозній одиниці від більше ніж нуль і до 3 включно. Під ступенем заміщення мають на увазі середню кількість гідроксильних груп, присутніх на ангідроглюкозній одиниці, яка складає целюлозний полімер, які замінені на заміщувальну групу. Показові матеріали включають полімер, вибраний з групи, яка складається з ацилату целюлози, діацилату целюлози, триацилату целюлози, ацетату целюлози, діацетату целюлози, триацетату целюлози, алканілатів моно-, ди- та трицелюлози, ароілатів моно-, ди- та трицелюлози та алкенілатів моно-, ди- та трицелюлози. Показові приклади включають ацетат целюлози, що має D.S. і вміст ацетилу до 21%; ацетат целюлози, що має вміст ацетилу до 32-39,8%; ацетат целюлози, що має D.S. від 1 до 2 і вміст ацетилу до 21-35%; ацетат целюлози, що має D.S. від 2 до 3 і вміст ацетилу до 35-44,8%. Більш специфічні целюлозні полімери включають пропіонат целюлози, що має D.S. 1,8 і вміст пропілу від 39,2 до 45%, а вміст гідроксилу від 2,8% до 5,4%; бутират ацетата целюлози, що має D.S. 1,8, вміст ацетилу від 13 до 15% і вміст бутирилу від 34 до 39%% бутират ацетата целюлози, що має вміст ацетилу від 2 до 29%, вміст бутирилу від 17 до 53% та вміст гідроксилу від 0,5 до 4,7%; тріацилат целюлози, що має D.S. від 2,9 до 3, такий як триацетат целюлози, тривалеріат целюлози, трилаурат целюлози, трипальмітат целюлози, трисукцинат целюлози та триоктаноат целюлози; діацилати целюлози, що мають D.S. від 2,2 до 2,6, такі як дисукцинат целюлози, дипальмітат целюлози, діоктаноат целюлози, дипентаноат целюлози та коефіри целюлози, такі як бутират ацетата целюлози, бутират октаноата ацетата целюлози та пропіонат ацетата целюлози. Додаткові полімери целюлози, придатні для одержання опіоідного антагоніста у практично невивільнюваній формі, включають ацетат ацетальдегіддиметилцелюлози, етилкарбамат ацетата целюлози, метилкарбамат ацетата целюлози та ацетат целюлози-диметиламіноцелюлози. Акриловий полімер, придатний для одержання опіоідного антагоніста у формі, яка практично не вивільнюється, включає, без обмеження, акрилові смоли, що включають сополімери, синтезовані зі складних ефірів акрилової та метакрилової кислоти (наприклад, сополімер нижчого алкілового ефіру акрилової кислоти та нижчого алкілового ефіру метакрилової кислоти), які вміщують приблизно від 0,02 до 0,03 моль три(нижчий алкіл)амонієвої групи на моль використаних акрилових і метакрилових мономерів. Прикладом придатної акрилової смоли e полімер виробництва Rohm Pharma GmbH, що продається під товарним знаком Eudragit® RS. Кращим є Eudragit RS30D. Eudragit® RS є не розчинним у воді сополімером етилакрилату (ЕА), метилметакрилата (ММ) та хлориду триметиламонійметилметакрилата (ТАМ), в якому молярне співвідношення ТАМ та інших компонентів (ЕА та MM) становить 1:40. Акрилові смоли, такі як Eudragit® RS, можна використовувати у формі водної суспензії. У певних кращих варіантах здійснення цього винаходу акриловий полімер може бути вибраний з групи, що складається з сополімерів акрилової кислоти та метакрилової кислоти, метилметакрилатних сополімерів, етоксиетилметакрилатів, ціанетилметакрилатів, полі(акрилової кислоти), полі(метакрилової кислоти), алкіламідного сополімеру метакрилової кислоти, полі(метилметакрилата), поліметакрилата, сополімеру полі(метилметакрилата), поліакриламіду, аміноалкілового сополімеру метакрилата, полі(ангідриду метакрилової кислоти) та гліцидилових сополімерів метакрилата. У випадку, коли опіоідний антагоніст у формі, яка практично не вивільнюється, включає частки опіоідного антагоніста з покриттям, яке робить антагоніст практично невивільнюваним, і коли целюлозний полімер або акриловий полімер використовують для одержання композиції для покриття, з полімером можна також змішувати придатні підходящі пластифікатори, наприклад, ацетилтриетилцитрат та/або ацетилтрибутилцитрат. Покриття може також містити такі добавки, як барвники, тальк та/або стеарат магнію, які добре відомі спеціалістам у галузі виготовлення покриттів. Композицію покриття можна наносити на частки опіоідного антагоніста шляхом розпилювання її на частки з використанням відповідного обладнання для розпилювання, відомого спеціалістам. Наприклад, можна використовувати систему текучого шару Wurster, в якій повітряна форсунка, що діє знизу, забезпечує текучість матеріалу, що покривається, та сушіння, в той час як поверх розпилюється покриття з нерозчинного полімеру. Товщина покриття залежатиме від властивостей конкретної покривальної композиції, яка використовується. Проте, за допомогою звичайного експериментування спеціаліст цілком спроможний визначити оптимальну товщину конкретного покриття, яке потрібне для конкретної лікарської форми за цим винаходом. Фармацевтично прийнятний гідрофобний матеріал, придатний для одержання опіоідного антагоніста у практично невивільнюваній формі, включає полімер, який біорозкладається, включаючи полі(молочну/гліколеву кислоту) ("PLGA"), полілактид, полігліколід, поліангідрид, поліортоефір, полікапролактони, поліфосфазени, полісахариди, білкові полімери, поліефіри, полідіоксанон, поліглюконат, сополімери полімолочної кислоти-поліетиленоксиду, полі(гідроксибутират), поліфосфоефіри або суміші будьяких з перелічених агентів. У певних варіантах здійснення цього винаходу полімер, який біорозкладається, включає полі(молочну/гліколеву кислоту), сополімер молочної та гліколевої кислоти, що має молекулярну масу приблизно від 2000 до 500000 дальтон. Співвідношення молочної кислоти та гліколевої кислоти становить приблизно від 100:0 до 25:75, кращим є співвідношення молочної кислоти та гліколевої кислоти 65:35. Полі(молочну/гліколеву кислоту) можна одержати за допомогою процедури, описаної у патенті США № 4293539 (Ludwig, et al.), опис якого цілком включено до цього документа як посилання. Коротко, Ludwig одержує згаданий сополімер реакцією конденсації молочної кислоти та гліколевої кислоти у присутності каталізатора полімеризації, що легко видаляється (наприклад, іонообмінної з сильною кислотою смоли, такої як Dowex HCR-W2-H). Кількість каталізатора не є принциповою для полімеризації, але звичайно вона становить приблизно від 0,01 до 20 частин мас. від загальної маси об'єднаних молочної кислоти та гліколевої кислоти. Реакцію полімеризації можна здійснювати без розчинників при температурі приблизно від 100°С до 250°С впродовж приблизно від 48 до 96 годин, краще, при пониженому тиску для полегшення видалення води та побічних продуктів. Полі(молочну/гліколеву кислоту) потім збирають фільтруванням розплавленої реакціонної суміші в органічному розчиннику, такому як дихлорметан або ацетон, а потім фільтруванням для видалення каталізатора. Після того, як опіоідний антагоніст у формі, яка практично не вивільнюється, одержаний, його можна об'єднувати з опіоідним агоністом разом зі звичайними наповнювачами, відомими спеціалістам, для виготовлення лікарської форми для перорального введення за даним винаходом. У певних кращих варіантах здійснення цього винаходу лікарська форма для перорального введення є капсулою або таблеткою. При виготовленні таблетки опіоідний антагоніст та агоніст можна об'єднувати з одним або більше інертними нетоксичними фармацевтичними наповнювачами, які підходять для виготовлення таблеток. Зазначені наповнювачі включають, наприклад, інертний розріджувач, такий як лактоза; гранулюючий і розпушувальний агенти, такі як кукурудзяний крохмаль; зв'язуючі агенти, такі як крохмаль; та змащувальні агенти, такі як стеарат магнію. Лікарську форму для перорального введення за даним винаходом можна виготовляти для забезпечення негайного вивільнення опіоідного агоніста, що в ній міститься. В інших варіантах здійснення цього винаходу, проте, лікарська форма для перорального введення забезпечує уповільнене вивільненя опіоідного агоніста. У певних варіантах здійснення цього винаходу лікарську форму для перорального введення, яка забезпечує уповільнене вивільнення опіоідного агоніста, можна виготовляти шляхом змішування опіоідного антагоніста у практично невивільнюваній формі з агоністом і бажаними фармацевтичними наповнювачами, щоб виготовити таблетку, а потім - шляхом нанесення на таблетку покриття із уповільненим вивільненням. У певних варіантах здійснення цього винаходу таблетки опіоідного агоніста із уповільненим вивільненням можна виготовляти шляхом змішування опіоідного антагоніста у практично невивільнюваній формі з опіоідним антагоністом у матриксі, який надає таблеткам властивості уповільненого вивільнення. Докладний опис виготовлення лікарських форм із уповільненим вивільненням за даним винаходом наведено нижче. Виготовлення лікарських форм з контрольованим вивільненням, що містять опіоідний агоніст і практично невивільнювану форму опіоідного антагоніста Комбінацію опіоідного агоніста та форми опіоідного антагоніста, яка практично не вивільнюється, можна виготовляти у вигляді композиції для перорального введення із контрольованим або уповільненим вивільненням у будь-якій придатній таблетці, таблетці з покриттям або композиції з множини часток, відомих спеціалістам. Лікарська форма із уповільненим вивільненням може, необов'язково, включати носій із уповільненим вивільненням, який інкорпорований у матрикс разом з опіоідним агоністом і невивільнюваною формою опіоідного антагоніста, або може застосовуватись у вигляді покриття із уповільненим вивільненням. У тих варіантах здійснення, в яких опіоідний агоніст включає гідрокодон, лікарські форми з уповільненим вивільненням можуть включати анальгетичні дози приблизно від 8мг до 50мг гідрокодону на дозовану одиницю. У лікарських формах для перорального введення із уповільненим вивільненням, в яких терапевтично активним опіоідом є гідроморфон, його включають у кількості приблизно від 2мг до 64мг гідроморфону гідрохлориду. В іншому варіанті здійснення цього винаходу опіоідний агоніст включає морфій, а лікарські форми для перорального введення із уповільненим вивільненням за цим винаходом включають приблизно від 2,5мг до 800мг морфіну за масою. У ще одному варіанті здійснення цього винаходу опіоідний агоніст включає оксикодон, а лікарські форми для перорального введення із уповільненим вивільненням включають приблизно від 2,5мг до 800мг оксикодону. У певних кращих варіантах здійснення цього винаходу лікарські форми для перорального введення із уповільненим вивільненням включають приблизно від 20мг до 30мг оксикодону. Композиції оксикодону із контрольованим вивільненням відомі спеціалістам. Наступні документи описують різноманітні композиції оксикодону із контрольованим вивільненням, які підходять для використання в описаному винаході, а також способи їх виготовлення: [патенти США №№5266311, 5549912, 5508042 та 5656295]. Опіоідний агоніст може включати трамадол, а лікарські форми для перорального введення із уповільненим вивільненянм можуть включати приблизно від 25мг до 800мг трамадолу на дозовану одиницю. Лікарська форма може містити більше одного опіоідного агоніста, щоб забезпечувати практично еквівалентний терапевничний ефект. Альтернативно, лікарська форма може містити молярні еквівалентні кількості інших солей опіоідних агоністів, придатних для цього винаходу. В одному кращому варіанті здійснення цього винаходу лікарська форма із уповільненим вивільненням включає частки, які включають опіоідний агоніст; зазначені частки мають діаметр приблизно від 0,1мм до 2,5мм, краще приблизно від 0,5мм до 2мм. Частки опіоідного агоніста у кращому варіанті мають плівкове покриття з такого матеріалу, який дозволяє опіоідному агоністу у водному середовищі вивільнюватись із уповільненою швидкістю. Плівкове покриття вибране таким чином, щоб, у поєднанні з іншими заявленими властивостями досягати бажаної швидкості вивільнення in vitro. Композиції покриття із уповільненим вивільненням за цим винаходом повинні бутиздатними утворювати тривку безперервну плівку, яка є гладкою та елегантною, здатною утримувати пігменти та інші покривальні допоміжні агенти, нетоксичною, інертною та не липкою. Лікарські форми, які включають опіоідний агоніст та практично невивільнюваний опіоідний антагоніст, можуть бути, необов'язково, покриті одним або більше матеріалами, що підходять для регулювання вивільнення опіоідного агоніста або для захисту композиції. В одному варіанті здійснення цього винаходу створені покриття, які забезпечують як рН-залежне, так і рН-незалежне вивільнення, наприклад, під впливом шлунково-кишкової рідини. рН-залежне покриття слугує для вивільнення опіоідів у бажаних ділянках шлунково-кишкового (ШК) тракту, наприклад, у шлунку або тонкому кишечнику, таким чином, щоб отримати профіль усмоктування, який здатний забезпечити пацієнтові принаймні приблизно вісьмигодинну та, краще, приблизно від дванадцятигодинної до двадцятичотирьохгодинної альгезію. Якщо бажаним є рН-незалежне покриття, покриття виготовляють для досягнення оптимального вивільнення опіоіду незалежно від змін рН оточуючої рідини, наприклад, у ШК тракті. Можливим є також виготовлення композицій, які вивільнюють частину дози в одній потрібній ділянці ШК тракту, наприклад, у шлунку, а решту дози - в іншій ділянці ШК тракту, наприклад, у тонкому кишечнику. Композиції за даним винаходом, в яких використовуються рН-залежні покриття, можуть також набувати ефекту повторної дії, яка досягається, коли незахищений лікарський засіб наносять поверх ентеросолюбильного покриття, і воно вивільнюється у шлунку, в той час як решта його частина, захищена ентеросолюбильним покриттям, вивільнюється у віддаленіших ділянках шлунково-кишкового тракту. рНзалежні покриття, які можна використовувати відповідно до цього винаходу, включають шеллак, фталат ацетату целюлози (САР), фталат полівінілацетату (PVAP), фталат гідроксипропілметилцелюлози та сополімери складного ефіру метакрилової кислоти, цеїн тощо. У певних кращих варіантах здійснення цього винаходу субстрат (наприклад, ядро таблетки, частка матриксу), який містить опіоідний анальгетик (з інгібітором СОХ-2 або без нього), покривають гідрофобним матеріалом, вибраним з (і) алкіл целюлози, (іі) акрилового полімеру або (ііі) їхніх сумішей. Покриття можна наносити у формі органічного або водного розчину або дисперсії. Покриття можна наносити до збільшення маси субстратом приблизно від 2 до 25% з метою одержання бажаного профілю уповільненого вивільнення. Покриття, одержані з водних дисперсій, докладно описані, наприклад, у [патентах США №№5273760 та 5286493], які було переуступлено правонаступникові та які включені у цей документ як посилання. Інші приклади композицій із уповільненим вивільненням і покриттів, які можна використовувати відповідно до цього винаходу, включають переуступлені [патенти США №№5324351; 5356467 та 5472712], цілком включені у цей документ як посилання. Алкілцелюлозні полімери Целюлозні матеріали та полімери, включаючи алкілцелюлози, є гідрофобними матеріалами, які добре підходять як покриття для гранул за цим винаходом. Тільки як приклад один кращий алкілцелюлозний полімер є етилцелюлозою, хоча спеціаліст оцінить, що лего можна використовувати також інші целюлозні та/або алкілцелюлозні полімери, окремо або у будь-якій комбінації, які цілком становлять гідрофобне покриття за цим винаходом або є його частиною. Однією водною дисперсією етилцелюлози, яка є у продажу, є Aquacoat® (FMC Corp., Philadelphia, Pennsylvania, USA). Aquacoat® виготовляють шляхом розчинення етилцелюлози в органічному розчиннику, що не змішується з водою, а потім емульгування її у воді у присутності поверхово-активного агента та стабілізатора. Післа гомогенізації для одержання субмікронних крапель органічний розчинник випарюють в умовах вакууму з утворенням псевдолатексу. Під час стадії виробництва у псевдолатекс пластифікатор не вносять. Таким чином, перед використанням Aquacoat® як покриття необхідно ретельно змішати її з підходящим пластифікатором. Іншою водною дисперсією етилцелюлози, наявною у продажу, є Surelease® (Colorcon, Inc., West Point, Pennsylvania, USA). Зазначений продукт виготовляють шляхом включення пластифікатора до дисперсії під час процесу виробництва. Гарячий розплав полімеру, пластифікатора (дибутилсебакат) і стабілізатора (олеїнова кислота) виготовляють у вигляді гомогенної суміші, яку потім розріджують лужним розчином, щоб отримати водну дисперсію, яку можна безпосередньо наносити на субстрати. Акрилові полімери В інших кращих варіантах здійснення цього винаходу гідрофобний матеріал, який включає покриття із контрольованим вивільненням, є фармацевтично прийнятним акриловим полімером, включаючи, без обмеження, сополімери акрилової кислоти та метакрилової кислоти, метилметакрилатні сополімери, етоксиетилметакрилати, ціанетилматакрилати, полі(акрилову кислоту), полі(метакрилову кислоту), алкіламідний сополімер матакрилової кислоти, полі(метилметакрилат), поліметакрилат, сополімер полі(метилметакрилата), поліакриламід, аміноалкіловий сополімер метакрилата, полі(ангідрид метакрилової кислоти) та гліцидилові сополімери метакрилата. У певних кращих варіантах здійснення цього винаходу акриловий полімер складений з одного або більше амонієвих сополімерів метакрилата. Амонієві сополімери метакрилата добре відомі спеціалістам і описані у NF XVII як повністю полімеризовані сополімери складних ефірів акрилової та метакрилової кислот з низьким вмістом четвертинних амонієвих груп. З метою одержання бажаного профілю розчинення може бути необхідним включення двох або більше амонієвих сополімерів метакрилата, які мають відмінні фізичні властивості, такі як різні молярні співвідношення четвертинних амонієвих груп і нейтральних (мет)акрилових складних ефірів. Окремі полімери типу складних ефірів метакрилової кислоти є придатними для виготовлення рН-залежних покриттів, які можна використовувати відповідно до даного винаходу. Наприклад, існує родина сополімерів, синтезованих з діетиламіноетилметакрилата та інших нейтральних метакрилових складних ефірів, відомих також як сополімер метакрилової кислоти або полімерні метакрилати, які є у продажу як Eudragit® від Rohm Tech, Inc. Існує декілька різних типів Eudragit®. Наприклад, Eudragit® є прикладом сополімеру метакрилової кислоти, який набухає і розчиняється у кислих середовищах. Eudragit® L є сополімером метакрилової кислоти, який не набухає при рН приблизно 6. Eudragit® S не набухає при рН приблизно 7. Eudragit® RL та Eudragit® RS набрякають у воді, а кількість води, що адсорбується зазначеними полімерами, залежить від рН, проте, лікарські форми з покриттям від Eudragit® RL та RS є рН-незалежними. У певних кращих варіантах здійснення цього винаходу акрилове покриття включає суміш двох лаків з акрилової смоли, які є у продажу від Rohm Pharma під торговими найменуваннями Eudragit® RL30D та Eudragit® RS30D, відповідно. Eudragit® RL30D та Eudragit® RS30D є сополімерами акрилових і метакрилових складних ефірів з низьким вмістом четвертинних амонієвих груп, молярним співвідношенням амонієвих груп та решти нейтральних (мет)акрилових складних ефірів 1:20 для Eudragit® RL30D та 1:40 для та Eudragit® RS30D. Середня молекулярна маса складає приблизо 150000. Кодові позначення RL (висока проникність) та RS (низька проникність) відносяться до властивостей проникності зазначених агентів. Суміші Eudragit® RL/RS не розчиняються у воді та травних рідинах. Проте, покриття, утворені з них, у водних розчинах і травних рідинах набухають і є проникними. Дисперсії Eudragit® RL/RS за цим винаходом можна змішувати разом у будь-якому бажаному співвідношенні з метою одержання, в кінцевому підсумку, композиції із уповільненим вивільненням, яка має бажаний профіль розчинення. Бажані композиції із уповільненим вивільненням можна одержати, наприклад, із затримуючого покриття, одержаного з 100% Eudragit® RL, 50% Eudragit® RL та 50% Eudragit® RS та 10% Eudragit® RL : 90% Eudragit® RS. Звичайно, що спеціалісту буде зрозуміло, що можна використовувати також й інші акрилові полімери, такі як, наприклад, Eudragit® L. Пластифікатори У таких варіантах здійснення цього винаходу, в яких покриття включає водну дисперсію гідрофобного матеріалу, включення ефективної кількості пластифікатора у водну дисперсію гідрофобного матеріалу ще більше поліпшить фізичні властивості покриття із уповільненим вивільненням. Наприклад, оскільки етилцелюлоза має відносно високу температуру скловання і не утворює гнучких плівок за нормальних умов нанесення покриття, краще включати пластифікатор в етилцелюлозне покриття, яке містить покриття із уповільненим вивільненням, перед використанням його як покривального матеріалу. Звичайно кількість пластифікатора, включеного до покривального розчину, базується на концентрації плівкоутворювача, наприклад, найчастіше приблизно від 1 до 50% від маси плівкоутворювача. Концентрацію пластифікатора, проте, належним чином можна визначити тільки після ретельного експериментування з конкретним покривальним розчином і способом його нанесення. Приклади підходящих пластифікаторів для етилцелюлози включають не розчинні у воді пластифікатори, такі як дибутилсебакат, діетилфталат, триетилцитрат, трибутилцитрат та триацетин, хоча можливим є використання й інших не розчинних у воді пластифікаторів (таких як ацетильовані моногліцериди, фталатні складні ефіри, касторова олія тощо). Триетилцитрат є особливо кращим пластифікатором для водних дисперсій етилцелюлози за даним винаходом. Приклади придатних пластифікаторів для акрилових полімерів за цим винаходом включають, без обмеження, складні ефіри лимонної кислоти, такі як триетилцитрат NF XVI, трибутилцитрат, дибутилфталат та, можливо, 1,2-пропіленгліколь. Інші пластифікатори, які виявились придатними для підвищення еластичності плівок, утворених з акрилових плівок, таких як розчини лаків Eudragit® RL/RS, включають поліетиленгліколі, пропіленгліколь, діетилфталат, касторову олію та триацетин. Триетилцитрат є особливо кращим пластифікатором для водних дисперсій етилцелюлози за даним винаходом. Було встановлено також, що додавання малої кількості тальку зменшує тенденцію водної дисперсії до прилипання під час обробки і діє як полірувальний агент. Способи виготовлення гранул з покриттям У випадку, коли для нанесення покриття на інертні фармацевтичні гранули, такі як гранули нонпарель 18/20, на які вже нанесений опіоідний агоніст, використовується гідрофобний покривальний матеріал з контрольованим вивільненням, кількість одержаних твердих гранул з контрольованим вивільненням можна потім розміщувати у желатинову капсулу разом з опіоідним антагоністом у практично невивільнюваній формі. Зазначена лікарська форма забезпечує ефективну дозу опіоідного агоніста з контрольованим вивільненням після проточування та контактування з оточуючою рідиною, наприклад, шлунковим соком або розчинними середовищами. Гранульовані композиції з контрольованим вивільненням за даним винаходом повільно вивільнюють опіоідний агоніст, наприклад, після проточування та впливу на них шлункового соку, а потім - кишечних соків. Профіль контрольованого вивільнення композицій за даним винаходом можна змінювати, наприклад, шляхом варіювання кількості покривального гідрофобного матеріалу, зміни способу, за допомогою якого до гідрофобного матеріалу додають пластифікатор, варіювання кількості пластифікатора відносно гідрофобного матеріалу, включення додаткових інгредієнтів або наповнювачів, змінення способу виробництва тощо. Профіль розчинення кінцевого продукту також можна модифікувати, наприклад, шляхом збільшення або зменшення товщини затримуючого покриття. Сфероїди або гранули, покриті опіоідним агоністом, можна виготовити, наприклад, шляхом розчинення лікарського засобу у воді, а потім - розпилення розчину на субстрат, наприклад, гранули нонпарель 18/20, використовуючи вставку Wuster. He обов'язково додають також додаткові інгредієнти перед нанесенням покриття на гранули, щоб сприяти зв'язуванню опіоіду з гранулами та/або забарвленню розчину і т.п. Наприклад, продукт, який містить гідроксипропілметилцелюлозу і т.п., з барвником або без нього (наприклад, Opadry®, продається компанією Colorcon, Inc.), можна додавати до розчину і розчин перемішувати (наприклад, впродовж приблизно 1 години) перед нанесенням його на гранули. Одержаний субстрат з покриттям, у даному прикладі гранули, можна потім не обов'язково ще раз покрити бар'єрним агентом для відокремлення терапевтично активного агента від гідрофобного покриття з контрольованим вивільненням. Прикладом підходящого бар'єрного агента є такий агент, який включає гідроксипропілметилцелюлозу. Проте, можна використовувати будь-який плівкоутворювач, відомий спеціалістам. Краще, щоб бар'єрний агент не впливав на швидкість розчинення кінцевого продукту. Гранули потім можна ще раз покрити водною дисперсією гідрофобного матеріалу. Водна дисперсія гідрофобного матеріалу переважно додатково містить ефективну кількість пластифікатора, наприклад, триетилцитрата. Можна використовувати приготовані заздалегідь водні дисперсії етилцелюлози, такі як Aquacoat® або Surelease®. Якщо використовується Surelease®, то немає необхідності окремо додавати пластифікатор. Альтернативно, можна використовувати приготовані заздалегідь водні дисперсії акрилових полімерів, такі як Eudragit®. Покривальні розчини за даним винаходом переважно містять, крім плівкоутворювача, пластифікатора та системи розчинника (наприклад, води), ще і барвник, щоб забезпечити елегантність та можливість розрізнення продукту. Барвник можна додавати у розчин терапевтично активного агента замість або окрім водної дисперсії гідрофобного матеріалу. Наприклад, барвник можна додавати до Aquacoat® шляхом використання дисперсій барвника на основі спирту або пропіленгліколю, фарбувальних лаків на основі алюмінієвого порошку та агентів, які надають непрозорість, таких, як діоксид титану, шляхом додавання барвника зі зсувом до розчину водорозчинного полімеру, а потім застосування низького зсуву до пластифікованого Aquacoat®. Альтернативно, можна використовувати будь-який відповідний спосіб надання забарвлення композиціям за даним винаходом. Придатні інгредієнти для надання забарвлення композиції, коли використовується водна дисперсія акрилового полімеру, включають діоксид титану та фарбувальні пігменти, такі, як пігменти оксидів заліза. Однак, включення пігментів може збільшити затримувальний ефект покриття. Пластифікований гідрофобний матеріал можна наносити на субстрат, який містить терапевтично активний агент, шляхом розпилення, використовуючи будь-яке підходяще розпилювальне обладнання, відоме спеціалістам. У кращому способі використовують систему текучого шару Wurster, в якій повітряна форсунка, яка діє знизу, забезпечує текучість серцевинного матеріалу і сушіння, в той час як зверху розпилюється покриття із акрилового полімеру. Переважно наносять таку кількість гідрофобного матеріалу, яка є достатньою для одержання заздалегідь визначеного контрольованого вивільнення згаданого терапевтично активного агента, коли субстрат з покриттям піддається дії водних розчинів, наприклад, шлункового соку, з урахуванням фізичних властивостей терапевтично активного агента, способу внесення пластифікатора тощо. Після нанесення покриття із гідрофобного матеріалу на гранули, не обов'язково, поверх покриття наносять ще одне покриття, із плівкоутворювача, такого як Opadry®. Згадане додаткове покриття наносять, якщо наносять взагалі, для того, щоб значно зменшити агломерацію гранул. Вивільнення терапевтично активного агента із композиції з контрольованим вивільненням за даним винаходом можна також змінювати, тобто, доводити до бажаної швидкості, шляхом додавання одного або більше агентів, які модифікують вивільнення, або утворення одного або більше проходів через покриття. Співвідношення гідрофобного матеріалу і водорозчинного матеріалу визначається, серед інших факторів, необхідною швидкістю вивільнення та характеристиками розчинності вибраних матеріалів. Агенти, які модифікують вивільнення, що діють як пороутворювачі, можуть бути органічними або неорганічними, і включають матеріали, які можуть розчинятись, екстрагуватись або вилужуватись із покриття у середовищі застосування. Пороутворювачі можуть включати один або більше гідрофільних матеріалів, таких як гідроксипропілметилцелюлоза. Покриття з уповільненим вивільненням за даним винаходом можуть також включати агенти, які сприяють ерозії, такі як крохмаль та камеді. Покриття з уповільненим вивільненням за даним винаходом можуть також включати матеріали, придатні для утворення мікропористої пластини у середовищі застосування, такі як полікарбонати, які складаються з лінійних поліефірів карбонової кислоти, в яких карбонатні групи знову з'являються на полімерному ланцюгу. Агент, який модифікує вивільнення, може також включати напівпроникний полімер. У певних кращих варіантах здійснення даного винаходу агент, який модифікує вивільнення, вибирають із гідроксипропілметилцелюлози, лактози, стеаратів металів та сумішей будь-яких із перелічених агентів. Покриття з уповільненим вивільненням за даним винаходом можуть також включати засоби виходу, які включають принаймні один прохід, отвір або тому подібне. Прохід може бути утворений такими способами, які описані в [патентах США №№3845770, 3916889, 4063064 та 4088864] (всі вони включені у даний документ як посилання). Прохід може мати будь-яку форму, таку, як округла, трикутна, квадратна, еліптична, неправильна і т.п. Композиції матриксу В інших варіантах здійснення даного винаходу композицію з контрольованим вивільненням одержують за допомогою матриксу, який має покриття з контрольованим вивільненням, як описано вище. Даний винахід включає також таблетки з уповільненим вивільненням, які містять частки опіоідного агоніста і опіоідного антагоніста з покриттям, яке робить антагоніст таким, що він практично не вивільняється, в яких агоніст та антагоніст дисперговані у матриксі з контрольованим вивільненням, який забезпечує in vitro швидкості розчинення опіоідного агоніста у кращих межах і який вивільняє опіоідний агоніст рН-залежним або рНнезалежним чином. Матеріали, які підходять для включення у матрикс з контрольованим вивільненням, залежать від способу виготовлення матриксу. Наприклад, матрикс, крім опіоідного агоніста і такої форми опіоідного антагоніста з покриттям, яка практично не вивільняється, може містити: - гідрофільні і/або гідрофобні матеріали, такі, як камеді, ефіри целюлози, акрилові смоли, матеріали білкового походження; даний перелік не претендує на вичерпність, і будь-який фармацевтично прийнятний гідрофобний матеріал або гідрофільний матеріал, який здатний забезпечити контрольоване вивільнення опіоіду, можна використати за цим винаходом; - засвоювані довголанцюгові (С8-С5о, особливо Сі2-С4о), заміщені або незаміщені вуглеводні, такі як жирні кислоти, жирні спирти, складні ефіри гліцерину та жирних кислот, мінеральні та рослинні олії та воски, стеариловий спирт та поліалкіленгліколі. Із згаданих полімерів кращими є акрилові полімери, особливо Eudragit® RSPO, ефіри целюлози, особливо гідроксиал кіл целюлози та карбоксиалкілцелюлози. Лікарська форма для перорального введення може містити від 1% до 80% (за масою) принаймні одного гідрофільного або гідрофобного матеріалу. У тому випадку, коли гідрофобним матеріалом є вуглеводень, згаданий вуглеводень переважно має температуру плавлення від 25°С до 90°С. Серед довголанцюгових вуглеводневих матеріалів кращими є жирні (аліфатичні) спирти. Лікарська форма для перорального введення може містити до 60% (за масою) принаймні одного засвоюваного довголанцюгового вуглеводню. Переважно лікарська форма для перорального введення містить до 60% (за масою) принаймні одного поліалкіленгліколю. Гідрофобний матеріал переважно вибирають із групи, яка складається з алкілцелюлоз, полімерів і співполімерів акрилової та метакрилової кислот, шеллаку, цеїну, гідрогенізованої касторової олії та їх сумішей. У певних кращих варіантах здійснення даного винаходу гідрофобний матеріал являє собою фармацевтично прийнятний акриловий полімер, у тому числі, без обмеження, співполімери акрилової кислоти та метакрилової кислоти, метилметакрилат, метилметакрилатні співполімери, етоксиетилметакрилати, ціанетилметакрилати, аміноалкіловий співполімер метакрилата, полі(акрилову кислоту), полі(метакрилову) кислоту, алкіламіновий співполімер метакрилової кислоти, полі(метилметакрилат), полі(метакрилової кислоти) (ангідрид), поліметакрилат, поліакриламід, полі(ангідрид метакрилової кислоти) і гліцидилові співполімери метакрилата. В інших варіантах здійснення даного винаходу гідрофобний матеріал вибирають із таких матеріалів, як гідроксиалкілцелюлози, такі як гідроксипропілметилцелюлоза та суміші перелічених вище агентів. Кращими гідрофобними матеріалами є не розчинні у воді матеріали з більше або менше вираженими гідрофільними і/або гідрофобними тенденціями. Краще, щоб гідрофобні матеріали, придатні для даного винаходу, мали температуру плавлення приблизно від 45°С до 90°С. Конкретно, гідрофобний матеріал може включати натуральні або синтетичні воски, жирні спирти (такі, як лауриловий, міристиловий, стеариловий, цетиловий або краще цетостеариловий спирт), жирні кислоти, в тому числі, без обмеження, складні ефіри жирних кислот, гліцериди жирних кислот (моно-, ди- та тригліцериди), гідрогенізовані жири, вуглеводні, нормальні воски, стеариринову кислоту, стеариловий спирт і гідрофобні та гідрофільні матеріали, які мають вуглеводневі скелети. Підходящі воски включають, наприклад, бджолиний віск, гликовіск, касторовий віск та карнаубський віск. Для цілей даного винаходу воскоподібна речовина визначається як будь-який матеріал, який звичайно є твердою речовиною при кімнатній температурі і має температуру плавлення приблизно від 30°С до 100°С. Підходящі гідрофобні матеріали, які можна використовувати за даним винаходом, включають засвоювані довголанцюгові (С8-С50, особливо С12-С40), заміщені або незаміщені вуглеводні, такі як жирні кислоти, жирні спирти, складні ефіри гліцерину та жирних кислот, мінеральні та рослинні олії та натуральні і синтетичні воски. Вуглеводні, які мають температуру плавлення від 25°С до 90°С, є кращими. Із довголанцюгових вуглеводневих матеріалів у певних варіантах здійснення даного винаходу кращими є жирі (аліфатичні) спирти. Лікарська форма для перорального введення може містити до 60% (за масою) принаймні одного засвоюваного довголанцюгового вуглеводню. Переважно до складу матриці можна включати поєднання двох або більше гідрофобних матеріалів. Якщо включають додатковий гідрофобний матеріал, то краще вибирати його з натуральних і синтетичних восків, жирних кислот, жирних спиртів та їхніх сумішей. Приклади включають бджолиний віск, карнаубський віск, стеаринову кислоту і стеариловий спирт. Даний перелік не претендує на вичерпність. Приклад підходящого матриксу включає принаймні одну водорозчинну гідроксиалкілцелюлозу, принаймні один С-іг-Сзє, краще С14-С22 аліфатичний спирт і не обов'язково принаймні один поліалкіленгліколь. Принаймні одна гідроксиалкілцелюлоза переважно є гідрокси(Сі-С6)алкілцелюлозою, такою, як гідроксипропілцелюлоза, гідроксипропілметилцелюлоза та особливо гідроксиетилцелюлоза. Кількість принаймні однієї гідроксиалкілцелюлози у цій лікарській формі для перорального введення буде визначатись, серед іншого, точною необхідною швидкістю вивільнення опіоіду. Принаймні один аліфатичний спирт може, наприклад, бути лауриловим спиртом, міристиловим спиртом або стеариловим спиртом. В особливо кращих варіантах здійсненя даної лікарської форми для перорального введення, проте, принаймні один аліфатичний спирт є цетиловим спиртом або цетостеариловим спиртом. Кількість принаймні одного аліфатичного спирту у даній лікарській формі для перорального введення буде визначатись, як було сказано вище, точною необхідною швидкістю вивільнення опіоіду. Вона також залежатиме від того, чи присутній у лікарській формі для перорального введення принаймні один поліалкіленгліколь, чи ні. При відсутності принаймні одного поліалкіленгліколю лікарська форма для перорального введення переважно містить від 20% до 50% (за масою) принаймні одного аліфатичного спирту. У тому випадку, коли у лікарській формі для перорального введення присутній принаймні один поліалкіленгліколь, то об'єднана маса принаймні одного аліфатичного спирту і принаймні одного поліалкіленгліколю переважно становить від 20% до 50% (за масою) від загальної маси лікарської форми. В одному варіанті здійснення даного винаходу співвідношення, наприклад, принаймні однієї гідроксиалкілцелюлози або акрилової смоли і принаймні одного аліфатичного спирту/поліалкіленгліколю визначає значною мірою швидкість вивільнення опіоіду з композиції. Співвідношення принаймні однієї гідроксиалкілцелюлози і принаймні одного аліфатичного спирту/поліалкіленгліколю між 1:2 та 1:4 є кращим; співвідношення між 1:3 та 1:4 є особливо переважним. Принаймні один поліалкіленгліколь може бути, наприклад, поліпропіленгліколем або, що є кращим, поліетиленгліколем. Середньочислова молекулярна маса принаймні одного поліалкіленгліколю переважно становить від 1000 до 15000, особливо від 1500 до 12000. Інший придатний матрикс з контрольованим вивільненням включає алкілцелюлозу (особливо етилцелюлозу), С12-С36 аліфатичний спирт і, не обов'язково, поліалкіленгліколь. В іншому кращому варіанті здійснення даного винаходу матрикс включає фармацевтично прийнятне поєднання принаймні двох гідрофобних матеріалів. Крім перелічених вище інгредієнтів, матрикс з контрольованим вивільненням може також містити відповідні кількості інших матеріалів, наприклад, розріджувачів, змащувальних агентів, зв'язуючих агентів, агентів, які сприяють гранулюванню, барвників, коригентів та агентів, що забезпечують ковзання, які є звичайними в фармацевтиці. Способи виготовлення гранул на основі матриксу З метою полегшення виготовлення твердої лікарської форми для перорального введення з контрольованим вивільненням за даним винаходом можна використовувати будь-який спосіб виготовлення композиції матриксу, відомий спеціалістам. Наприклад, інкомпорування у матрикс можна здійснювати, наприклад, (а) формуванням гранул, які містять принаймні одну водорозчинну гідроксиалкілцелюлозу та опіоід чи сіль опіоіду; (Ь) змішуванням гранул, які містять гідроксиалкілцелюлозу, з принаймні одним С12-С36 аліфатичним спиртом; і (с) не обов'язково пресуванням та наданням форми гранулам. Краще, коли гранули формують за допомогою вологого гранулювання гідроксиалкілцелюлози/опіоіду з водою. В особливо кращому варіанті здійснення даного способу кількість води, доданої під час стадії вологого гранулювання, переважно у 1,5-5 разів, особливо у 1,75-3,5 разів більше, ніж суха маса опіоіду. В інших альтернативних варіантах можна використовувати сферонізувальний агент, який разом з активним інгредієнтом утворює сфероїди. Кращою є мікрокристалічна целюлоза. Придатною мікрокристалічною целюлозою є, наприклад, матеріал, який продається як Avicel PH 101 (товарний знак, FMC Corporation). У згаданих варіантах, крім активного інгредієнта і сферонізувального агента, сфероїди можуть містити також зв'язуючий агент. Придатні зв'язуючі агенти, такі як водорозчинні полімери з низькою в'язкістю, добре відомі спеціалістам у галузі фармацевтики. Однак, водорозчинна гідрокси (нижчий алкіл) целюлоза, така як гідроксипропілцелюлоза, є кращою. Додатково (або альтернативно) сфероїди можуть містити не розчинний у воді полімер, особливо акриловий полімер, акриловий співполімер, такий як співполімер метакрилової кислоти/етилакрилата, або етилцелюлоза. У цих варіантах здійснення покриття з уповільненим вивільннням звичайно містить гідрофобний матеріал, такий як (а) віск, як окремо, так і в суміші з жирним спиртом; або (b) шеллак чи цеін. Матрикс, одержаний екструзією розплаву Матрикси з уповільненим вивільненням можна також виготовляти за допомогою методики гранулювання розплаву або екструзії розплаву, оскільки згадані методики не порушують цілісність форми опіоідного антагоніста, яка практично не вивільняється, доданої під час виготовлення матриксу, до такого ступеню, щоб достатня кількість опіоідного антагоніста стала доступною для вивільнення у шлунково-кишковий тракт після перорального введення. Альтернативно, стадію екструдування розплаву можна здійснювати з опіоідним агоністом для одержання часток агоніста з уповільненим вивільненням, які потім можна поєднати з практично невивільнюваною формою опіоідного антагоніста. Звичайно методика гранулювання розплаву включає розплавлення звичайно твердого гідрофобного матеріалу, наприклад, воску, та інкорпорування у нього порошкоподібного лікарського засобу. Для одержання лікарської форми з уповільненим вивільненням може бути необхідним інкорпорування додаткової гідрофобної речовини, наприклад, етилцелюлози, або не розчинного у воді акрилового полімеру, у розплавлений восковий гідрофобний матеріал. Приклади композицій з уповільненим вивільненням, виготовлених за допомогою методики траулювання розплаву, можна знайти в [патенті США №4861598], переуступленому правонаступнику даного винаходу і повністю включеному у даний документ як посилання. Додатковий гідрофобний матеріал може містити одну або більше не розчинних у воді воскоподібних термопластичних речовин, можливо, змішаних з однією або більше воскоподібних термопластичних речовин, які є менш гідрофобними, ніж згадана одна або більше не розчинна у воді воскоподібна речовина. З метою досягнення постійного вивільнення окремі воскоподібні речовини в композиції мають бути практично такими, що не розкладаються і не розчиняються в шлунково-кишкових рідинах під час початкових фаз вивільнення. Придатними не розчинними у воді воскоподібними речовинам можуть бути такі речовини, у яких розчинність у воді нижче, ніж приблизно 1:5000 (мас/мас). Крім перелічених вище інгредієнтів, матрикс з уповільненим вивільненням може також містити підходящі кількості інших матеріалів, наприклад, розріджувачів, змащувальних агентів, зв'язуючих агентів, агентів, які сприяють гранулюванню, барвників, коригентів та агентів, які забезпечують ковзання, які є звичайними у фармацевтиці. Кількості згаданих додаткових матеріалів будуть достатніми для забезпечення бажаного ефекта для бажаної композиції. Крім перелічених вище інгредієнтів, матрикс з уповільненим вивільненням, в якому інкорпоровані множини часток, одержаних екструзією розплаву, може також містити підходящі кількості інших матеріалів, наприклад, розріджувачів, змащувальних агентів, зв'язуючих агентів, агентів, які сприяють гранулюванню, барвників, коригентів та агентів, що забезпечують ковзання, які є звичайними у фармацевтиці, у кількостях приблизно до 50% від маси часток, якщо це бажано. Конкретні приклади фармацевтично прийнятних носіїв і наповнювачів, які можна використовувати для виготовлення лікарських форм для перорального введення, описані у Handbook of Pharmaceutical Excipients, American Pharmaceutical Association (1986), включеному у даний документ як посилання. Множини часток, одержані екструзією розплаву Виготовлення підходящого матриксу способом екструзії розплаву за даним винаходом може, наприклад, включати стадії змішування опіоідного анальгетика разом з принаймні одним гідрофобним матеріалом і переважно додатковим гідрофобним матеріалом до одержання гомогенної суміші. Гомогенну суміш потім нагрівають до температури, достатньої принаймні для розм'якшення суміші, достатнього для її екструзії. Одержану суміш потім піддають екструзії з одержанням пасм. Екструдат переважно охолоджують і розрізають на множину часток за допомогою засобів, відомих спеціалістам. Пасма охолоджують і розрізають на множину часток. Множини часток потім змішують з частками опіоідного антагоніста з покриттям, яке робить антагоніст таким, що він практично не вивільняється, і розділяють на стандартні дози. Екструдат переважно має діаметр приблизно від 0,1 до 5мм і забезпечує уповільнене вивільнення опіоідного агоніста впродовж періоду часу приблизно від 8 до 24 годин. Оптимальний спосіб виготовлення екструзією розплаву за даним винаходом включає безпосереднє відмірювання в екструдер гідрофобного матеріалу, терапевтично активного агента та, не обов'язково, зв'язуючого агента; нагрівання гомогенної суміші; екструзію гомогенної суміші з формуванням паем; охолодження паем, які містять гомогенну суміш; розрізання паєм на частки розміром приблизно від 0,1 мм до 12 мм і поєднання часток з частками опіоідного антагоніста з покриттям і розділення їх на стандартні дози. У цьому аспекті даного винаходу реалізується відносно безперервний процес виробництва. Діаметр отвору або вихідного порту екструдера також можна підібрати, щоб варіювати товщину екструдованих паем. Крім того, вихідний порт екструдера не обов'язково має бути круглим; він може бути довгастим, прямокутним і т.п. Одержані пасма можна розрізати на частки, використовуючи різак із гарячого дроту, гільотину і т.п. Системи з множини часток, одержаних екструзією розплаву, можуть бути, наприклад, у формі гранул, сфероїдів або кульок, залежно від вихідного отвору екструдера. Для цілей даного винаходу терміни "множина (множини) часток, одержаних екструзією розплаву" і "система (системи) з множини часток, одержаних екструзією розплаву" і "частки, одержані екструзією розплаву", стосуватимуться множини одиниць, переважно у межах подібних розмірів та/або форми, і які містять один або більше активних агентів і один або більше наповнювачів, переважно у тому числі гідрофобний матеріал, як описано у даному документі. З цієї точки зору множини часток, одержаних екструзією розплаву, матимуть довжину у межах приблизно від 0,1 до 12мм і діаметр приблизно від 0,1 до 2мм. Крім того, треба розуміти, що множини часток, одержаних екструзією розплаву, можуть мати будь-яку геометричну форму у згаданих межах розмірів. Альтернативно, екструдат можна просто розрізати на необхідну довжину і розділяти на стандартні дози терапевтично активного агента, без необхідності етапу надання кулькоподібної форми. В одному кращому варіанті здійсненя даного винаходу лікарські форми для перорального введення включають ефективну кількість множини часток, одержаних екструзією розплаву, в капсулі. Наприклад, множину часток, одержаних екструзією розплаву, можна умістити в желатинову капсулу у кількості, достатній для забезпечення ефективної дози з уповільненим вивільненням після проковтування та контактування зі шлунковим соком. В іншому кращому варіанті здійснення даного винаходу відповідну кількість екструдату у вигляді множини часток поєднують з частками опіоідного антагоніста з покриттям і пресують у таблетку для перорального введення з використанням звичайного таблетувального обладнання і стандартних методик. Методики і композиції для виготовлення таблеток (пресованих і формованих), капсул (твердих і м'яких желатинових) і пілюль також описані в [Remington's Pharmaceutical Sciences (видавець Arthur Osol), 1553-1593 (1980)], включеній у даний документ як посилання. У ще одному кращому варіанті здійснення даного винаходу екструдату можна надавати форму таблеток, як описано в [патенті США №4957681 (Klimesch, et al.)], описаному докладно вище і включеному у даний документ як посилання. Не обов'язково на одержані екструзією розплаву системи із множини часток або таблетки з уповільненим вивільненням можна наносити покриття або на желатинову капсулу можна додатково наносити покриття, таке, як покриття з уповільненим вивільненням, описані вище. Згадані покриття переважно включають достатню кількість гідрофобного матеріалу, щоб приріст маси становив приблизно від 2 до 30%, хоча додаткове покриття може бути більшим, залежно від фізичних властивостей конкретної використаної опіоідної анальгетичної сполуки і бажаної швидкості вивільнення, серед інших факторів. Дозовані лікарські форми, одержані екструзією розплаву, за даним винаходом можуть додатково включати поєднання множин часток, одержаних екструзією розплаву, які містять один або більше терапевтично активних агентів, описаних вище, перед уміщенням у капсули. Крім того, дозовані лікарські форми можуть також містити кількість опіоідного агоніста з негайним вивільненням для досягнення швидкого терапевтичного ефекта. Опіоідний агоніст з негайним вивільненням можна інкорпорувати, наприклад, у вигляді окремих гранул, в желатинову капсулу, або можна наносити як покриття на поверхню множини часток після виготовлення лікарських форм (наприклад, покриття з контрольованим вивільненням або на основі матриксу). Дозовані лікарські форми за даним винаходом можуть також містити комбінацію гранул з контрольованим вивільненням і матричних множин часток для досягнення бажаного ефекту. Композиції з уповільненим вивільненням за даним винаходом переважно повільно вивільняють опіоідний агоніст, наприклад, після проковтування і під дією шлункового соку, а потім - кишкового соку. Профіль уповільненого вивільнення композицій, одержаних екструзією розплаву, за даним винаходом, можна змінювати, наприклад, варіюванням кількості уповільнювача, тобто, гідрофобного матеріалу, варіюванням кількості пластифікатора відносно гідрофобного матеріалу, включенням додаткових інгредієнтів або наповнювачів, зміною способу виробництва тощо. В інших варіантах здійснення даного винаходу матеріал, одержаний екструзією розплаву, виготовляють без включення опіоідного агоніста та/або покритих оболонкою часток опіоідного антагоніста, які додають до екструдату пізніше. У згаданих композиціях лікарські засоби звичайно будуть змішуватись з екструдованим матриксним матеріалом, а потім суміш буде таблетуватись з метою одержання опіоідного агоніста з уповільненим вивільненням. Згадані композиції можуть мати переваги, наприклад, коли терапевтично активний агент, включений до композиції, чутливий до температур, які необхідні для розм'якшення гідрофобного матеріалу та/або уповільнювача. Докладний опис кращих варіантів здійснення даного винаходу Наступні приклади ілюструють різні особливості даного винаходу. Вони не повинні тлумачитись як обмежуючі будь-яким чином формулу винаходу Приклад 1 У прикладі 1 практично невивільнювану форму опіоідного антагоніста (налтрексону НСІ) виготовляють шляхом нанесення на частки налтрексону покриття, яке робить антагоніст практично невивільнюваним. Процес: 1. Одержання розчину: Розчиняють налтрексон НСІ в очищеній воді. Після цього додають Opadry White і продовжують перемішування до одержання гомогенної дисперсії. 2. Навантаження: Наносять одержану дисперсію на цукрові кульки з використанням машини для нанесення покриття у текучому шарі. 3. Нанесення додаткового покриття: Одержують розчин додаткового покриття диспергуванням Opadry White в очищеній воді. Наносять одержану дисперсію на цукрові кульки, навантажені налтрексоном НСІ, з використанням машини для нанесення покриття у текучому шарі. 4. Нанесення затримуючого покриття: Одержують розчин для покриття, яке перешкоджає вивільненню, змішуванням Eudragit RS30D, триетилцитрата, тальку та очищеної води. Наносять одержану дисперсію на цукрові кульки, навантажені налтрексоном НСІ і які мають додаткове покриття, з використанням машини для нанесення покриття у текучому шарі. 5. Нанесення додаткового покриття: Одержують другий розчин додаткового покриття диспергуванням Opadry White в очищеній воді. Наносять одержану дисперсію на кульки з налтрексоном НСІ, які мають покриття, що перешкоджає вивільненню, з використанням машини для нанесення покриття у текучому шарі. 6. Висушування: Висушують кульки при 45°С впродовж приблизно 48 годин. Приклад 2 У прикладі 2 практично невивільнювану форму опіоідного антагоніста (налтрексону НСІ) виготовляють у вигляді гранулятів, які містять налтрексон НСІ. Грануляти складаються з налтрексону НСІ, диспергованого у матриксі, який робить антагоніст практично невивільнюваним. Процес: 1. Одержання розчину: Розчиняють PLGA в етилацетаті шляхом перемішування. 2. Гранулювання: Розміщують налтрексон НСІ і гідрофосфат кальцію в машину для нанесення покриття у текучому шарі та гранулюють розпиленням одержаного вище розчину. Приклад з У прикладі 3 практично невивільнювану форму опіоідного антагоніста (налтрексону НСІ) виготовляють у вигляді гранул налтрексону НСІ, одержаних екструзією. Процес: 1. Розмолування: Пропускають хлоп'я стеарилового спирту крізь молоткову дробарку. 2. Змішування: Змішують налтрексон НСІ, Eudragit і роздрібнений стеариловий спирт у мішалці з подвійним кожухом. 3. Екструзія: Безперервно подають змішаний матеріал в екструдер з двома гвинтами та збирають одержані пасмана транспортер. 4. Охолоджування: Дають пасмам остигнути на транспортері. 5. Гранулювання: Нарізають охолоджені пасма на гранули з використанням приладу для гранулювання. 6. Просіювання: Просіюють гранули та збирають потрібну просіяну порцію. Приклад 4 Таблетки гідрокодону бітартрата з контрольованим вивільненням з гранулами налтрексону НСІ Процес: 1. Розмолування: Пропускають хлоп'я стеарилового спирту крізь осцилюючу дробарку. 2. Змішування: Змішують гідрокодону бітартрат, роздрібнений стеариловий спирт, безводний гідрофосфат кальцію, мікрокристалічну целюлозу і ефір гліцерину та бегенової кислоти в мішалці з подвійним кожухом. 3. Екструзія: Безперервно подають змішаний матеріал в екструдер з двома гвинтами та збирають одержаний нагрітий матеріал на транспортер. 4. Охолоджування: Дають екструдату остигнути на транспортері. 5. Розмолування: Розмолюють охолоджений екструдат за допомогою осцилюючої дробарки. 6. Змішування: Змішують розмолотий екструдат, гранули налтрексону НСІ (з прикладу 1) та стеарат магнію. 7. Пресування: Пресують одержаний гранулят за допомогою таблеткового пресу. 8. Нанесення покриття: Виготовляють плівкоутворювальний розчин диспергуванням Opadry в очищеній воді та наносять його на ядра таблеток. Приклад 5 Таблетки гідрокодону бітартрату з контрольованим вивільненням з гранулятом налтрексону НСІ Процес: 1. Розмолування: Пропускають хлоп'я стеарилового спирту крізь осцилюючу дробарку. 2. Змішування: Змішують гідрокодону бітартрат, роздрібнений стеариловий спирт, безводний гідрофосфат кальцію, мікрокристалічну целюлозу і ефір гліцерину та бегенової кислоти в мішалці з подвійним кожухом. 3. Екструзія: Безперервно подають змішаний матеріал в екструдер з двома гвинтами та збирають одержаний нагрітий матеріал на транспортер. 4. Охолоджування: Дають екструдату остигнути на транспортері. 5. Розмолування: Розмолюють охолоджений екструдат за допомогою осцилюючої дробарки. 6. Змішування: Змішують розмолотий екструдат, гранулят налтрексону НСІ (з прикладу 2) та стеарат магнію. 7. Пресування: Пресують одержаний гранулят за допомогою таблеткового пресу. 8. Нанесення покриття- Виготовляють плівкоутворювальний розчин диспергуванням Opadry в очищеній воді та наносять його на ядра таблеток. Приклад 6 Таблетки оксикодону НСІ з контрольованим вивільненням з гранулами налтрексону НСІ Процес: 1. Виготовлення розчину: Пластифікують Eudragit з триацетином шляхом змішування. 2. Гранулювання: Розміщують оксикодон НСІ, висушену розпиленням лактозу та повідон у гранулятор у текучомі шарі та наносять одержаний вище розчин. 3. Розмолування: Пропускають гранулят крізь ротаційну турбинну дробарку. 4. Сушіння: Сушать гранулят, якщо вміст вологи надто високий. 5. Вощіння: Розплавлюють стеариловий спирт і вощать одержаний гранулят додаванням розплавленого стеарилового спирту до гранулята при перемішуванні. 6. Охолоджування: Охолоджують вощений гранулят у сушарці з текучім шаром. 7. Розмолування: Пропускають охолоджений вощений гранулят крізь ротаційну турбинну дробарку. 8. Змішування: Змішують розмолотий вощений гранулят, тальк, стеарат магнію та гранули налтрексону НСІ (з прикладу 1). 9. Пресування: Пресують одержаний гранулят з використанням таблеткового пресу. 10. Нанесення покриття: Виготовляють плівкоутворювальний розчин диспергуванням Opadry в очищеній воді та наносять його на ядра таблеток. Приклад 7 Таблетки оксикодону НСІ з контрольованим вивільненням з гранулятом налтрексону НСІ Процес: 1. Виготовлення розчину: Пластифікують Eudragit з триацетином шляхом змішування. 2. Гранулювання: Розміщують оксикодон НСІ, висушену розпиленням лактозу та повідон у гранулятор у текучомі шарі та наносять одержаний вище розчин. 3. Розмолування: Пропускають гранулят крізь ротаційну турбинну дробарку. 4. Сушіння: Сушать гранулят, якщо вміст вологи надто високий. 5. Вощіння: Розплавлюють стеариловий спирт і вощать одержаний гранулят додаванням розплавленого стеарилового спирту до гранулята при перемішуванні. 6. Охолоджування: Охолоджують вощений гранулят у сушарці з текучім шаром. 7. Розмолування: Пропускають охолоджений вощений гранулят крізь ротаційну турбинну дробарку. 8. Змішування: Змішують розмолотий вощений гранулят, тальк, стеарат магнію та гранулят налтрексону НСІ (з прикладу 2). 9. Пресування: Пресують одержаний гранулят з використанням таблеткового пресу. 10. Нанесення покриття: Виготовляють плівкоутворювальний розчин диспергуванням Opadry в очищеній воді та наносять його на ядра таблеток. Приклад 8 Капсули гідроморфону НСІ з контрольованим вивільненням з екструдованими гранулами налтрексону НСІ Процес: 1. Розмолування: Пропускають хлоп'я стеарилового спирту крізь молоткову дробарку. 2. Змішування: Змішують гідроморфон НСІ, Eudragit, етилцелюлозу та роздрібнений стеариловий спирт у мішалці з подвійним кожухом. 3. Екструзія: Безперервно подають змішаний матеріал в екструдер з двома гвинтами та збирають одержані пасма на транспортер. 4. Охолоджування: Дають пасмам остигнути на транспортері. 5. Гранулювання: Нарізають охолоджені пасма на гранули з використанням приладу для гранулювання. 6. Просіювання: Просіюють гранули та збирають потрібну просіяну порцію. 7. Інкапсуляція: Наповнюють тверді желатинові капсули екструдованими гранулами гідроморфону НСІ по 120мг та гранулами налтрексону НСІ (з прикладу 3) по 240мг. Приклад 9 Таблетки гідрокодону бітартрата з контрольованим вивільненням з гранулами налтрексону НСІ Процес: 1. Розмежування: Пропускають хлоп'я стеарилового спирту крізь осцилюючу дробарку. 2. Змішування: Змішують гідрокодону бітартрат, роздрібнений стеариловий спирт, безводний гідрофосфат кальцію, мікрокристалічну целюлозу і ефір гліцерину та бегенової кислоти в мішалці з подвійним кожухом. 3. Екструзія: Безперервно подають змішаний матеріал в екструдер з двома гвинтами та збирають одержаний нагрітий матеріал на транспортер. 4. Охолоджування: Дають екструдату остигнути на транспортері. 5. Розмолування: Розмолюють охолоджений екструдат за допомогою осцилюючої дробарки. 6. Змішування: Змішують розмолотий екструдат, гранули налтрексону НСІ (з прикладу 1) та стеарат магнію. 7. Пресування: Пресують одержаний гранулят за допомогою таблеткового пресу. 8. Нанесення покриття: Виготовляють плівкоутворювальний розчин диспергуванням Opadry в очищеній воді та наносять його на ядра таблеток. Приклад 10 Таблетки гідрокодону бітартрата з контрольованим вивільненням з гранулятом налтрексону НСІ Процес: 1. Розмолування: Пропускають хлоп'я стеарилового спирту крізь осцилюючу дробарку. 2. Змішування: Змішують гідрокодону бітартрат, роздрібнений стеариловий спирт, безводний гідрофосфат кальцію, мікрокристалічну целюлозу і ефір гліцерину та бегенової кислоти в мішалці з подвійним кожухом. 3. Екструзія: Безперервно подають змішаний матеріал в екструдер з двома гвинтами та збирають одержаний нагрітий матеріал на транспортер. 4. Охолоджування: Дають екструдату остигнути на транспортері. 5. Розмолування: Розмолюють охолоджений екструдат за допомогою осцилюючої дробарки. 6. Змішування: Змішують розмолотий екструдат, гранулят налтрексону НСІ (з прикладу 2) та стеарат магнію. 7. Пресування: Пресують одержаний гранулят за допомогою таблеткового пресу. 8. Нанесення покриття: Виготовляють плівкоутворювальний розчин диспергуванням Opadry в очищеній воді та наносять його на ядра таблеток. Приклад 11 Таблетки оксикодону НСІ з контрольованим вивільненням з гранулами налтрексону НСІ Процес: 1. Виготовлення розчину: Пластифікують Eudragit з триацетином шляхом змішування. 2. Гранулювання: Розміщують оксикодон НСІ, висушену розпиленням лактозу та повідон у гранулятор у текучомі шарі та наносять одержаний вище розчин. 3. Розмолування: Пропускають гранулят крізь ротаційну турбинну дробарку. 4. Сушіння: Сушать гранулят, якщо вміст вологи надто високий. 5. Вощіння: Розплавлюють стеариловий спирт і вощать одержаний гранулят додаванням розплавленого стеарилового спирту до гранулята при перемішуванні. 6. Охолоджування: Охолоджують вощений гранулят у сушарці з текучім шаром. 7. Розмолування: Пропускають охолоджений вощений гранулят крізь ротаційну турбинну дробарку. 8. Змішування: Змішують розмолотий вощений гранулят, тальк, стеарат магнію та гранулят налтрексону НСІ (з прикладу 1). 9. Пресування: Пресують одержаний гранулят з використанням таблеткового пресу. 10. Нанесення покриття: Виготовляють плівкоутворювальний розчин диспергуванням Opadry в очищеній воді та наносять його на ядра таблеток. Приклад 12 Таблетки оксикодону НСІ з контрольованим вивільненням з гранулятом налтрексону НСІ Процес: 1. Виготовлення розчину: Пластифікують Eudragit з триацетином шляхом змішування. 2. Гранулювання: Розміщують оксикодон НСІ, висушену розпиленням лактозу та повідон у гранулятор у текучомі шарі та наносять одержаний вище розчин. 3. Розмолування: Пропускають гранулят крізь ротаційну турбин ну дробарку. 4. Сушіння: Сушать гранулят, якщо вміст вологи надто високий. 5. Вощіння: Розплавлюють стеариловий спирт і вощать одержаний гранулят додаванням розплавленого стеарилового спирту до гранулята при перемішуванні. 6. Охолоджування: Охолоджують вощений гранулят у сушарці з текучім шаром. 7. Розмолування: Пропускають охолоджений вощений гранулят крізь ротаційну турбинну дробарку. 8. Змішування: Змішують розмолотий вощений гранулят, тальк, стеарат магнію та гранулят налтрексону НСІ (з прикладу 2). 9. Пресування: Пресують одержаний гранулят з використанням таблеткового пресу. 10. Нанесення покриття: Виготовляють плівкоутворювальний розчин диспергуванням Opadry в очищеній воді та наносять його на ядра таблеток. Приклад 13 Капсули гідроморфону НСІ з контрольованим вивільненням з екструдованими гранулами налтрексону НСІ Процес: 1. Розмолування: Пропускають хлоп'я стеарилового спирту крізь молоткову дробарку. 2. Змішування: Змішують гідроморфон НСІ, Eudragit, етилцелюлозу та роздрібнений стеариловий спирт у мішалці з подвійним кожухом. 3. Екструзія: Безперервно подають змішаний матеріал в екструдер з двома гвинтами та збирають одержані пасма на транспортер. 4. Охолоджування: Дають пасмам остигнути на транспортері. 5. Гранулювання: Нарізають охолоджені пасма на гранули з використанням приладу для гранулювання. 6. Просіювання: Просіюють гранули та збирають потрібну просіяну порцію. 7. Інкапсуляція: Наповнюють тверді желатинові капсули екструдованими гранулами гідроморфону НСІ по 120мг та гранулами налтрексону НСІ (з прикладу 3) по 240мг. Приклад 14 Таблетки оксикодону гідрохлориду з контрольованим вивільненням по 10мг-Органічне виробництво Оксикодону гідрохлорид (10мг/на таблетку) та висушену розпиленням лактозу (71,25мг/на таблетку) переносять у мішалку відповідного розміру та змішують впродовж приблизно б хвилин. Порошок Eudragit® RS РМ (6мг/на таблетку) диспергують в етанолі. В той час як порошки перемішуються, вони гранулюються, і диспергування та перемешування тривають до утворення вологої гранулярної маси. Додають додаткову кількість етанолу для досягнення кінцевої точки гранулювання. Гранулят переносять у сушарку з текучим шаром і сушать при 30°С, а потім пропускають крізь сито меш 12. Eudragit® RS РМ (9мг/на таблетку), що залишився, диспергують у розчиннику з 90 частин етанолу та 10 частин очищеної води та розпилюють на гранули в грануляторі/сушарці з текучим шаром при 30°С. Потім гранулят пропускають крізь сито меш 12. Розплавляють стеариловий спирт (25мг/на таблетку) при температурі близько 60-70°С. Теплі гранули повертають у мішалку. При перемішуванні додають розплавлений стеариловий спирт. Гранули з покриттям видаляють з мішалки та дозволяють їм остуднути. Потім їх пропускають крізь сито меш 12. Після цього гранулят змішують з частками налоксону (приблизно 1-5мг на таблетку) з покриттям, яке робить налоксон практично невивільнюваним, і фармацевтично бажаними наповнювачами для таблетування, наприклад, тальком і стеаратом магнія, у підходящому змішувачі та пресують у таблетки. Частки налоксону мають діаметр приблизно від 0,5 до 2мм. Частки налоксону з покриттям, яке робить налоксон практично невивільнюваним, можна виготовити розпиленням на частки композиції покриття, що включає целюлозний полімер або акриловий полімер, який не розчиняється у воді та є непроникним для налоксону. Підходящі частки включають гранули, зерна, сфероїди або кульки, що включають налоксон. У випадку, коли частки є гранулами або зернами, їх можна виготовляти розчиненням налоксону в розчині та розпиленням його на інертні гранули або зерна. Краще, якщо композиція покриття включає Eudragit® RS, який можна використовувати у формі водної суспензії та у комбінації з пластифікатором, таким як, наприклад, ацетилтриетилцитрат та/або ацетилтрибутилцитрат. Приклад 15 Спосіб лікування болю Лікарську форму для перорального введення за даним винаходом можна вводити пацієнтові для полегшення болю. Лікарська форма для перорального введення може включати ефективну при переральному введенні кількість опіоідного агоніста та опіоідного антагоніста, який знаходиться у практично невивільнюваній формі. Коли лікарська форма для перорального введення введена перорально та доставлена у шлунковокишковий тракт пацієнта, який потребує лікування болю, опіоідний агоніст вивільнюється з лікарської форми у перебігу нормального травлення, забезпечуючи пацієнтові анальгезію. Але опіоідний антагоніст, оскільки він знаходиться у практично невивільнюваній формі, практично не вивільнюється під час свого проходження крізь шлунково-кишковий тракт. Краще, якщо практично невивільнювана форма антагоніста є резистентною до проносних засобів (мінеральної олії), які використовуються проти уповільненого пасажу крізь товстий кишечник або проти ахлоргідричних станів. У пацієнтів, які приймають лікарську форму для перорального введення належним чином, без її псування (наприклад, шляхом механічного пошкодження, нагрівання або розчинення в розчиннику), опіоідний антагоніст не буде усмоктуватись у достатній кількості впродовж будьякого інтервалу часу дозування композиції таким чином, щоб антагоніст зменьшував або усував анальгетичну ефективність опіоідного антагоніста. Інакше кажучи, кількість опіоідного антагоніста, яка вивільнилась з лікарської форми (при пероральному введенні в інтактному вигляді) та яке усмокталось у шлунковокишковому тракті та акумулювалось в організмі пацієнта, не підвищується до того рівня, який значно перешкоджає або змінює анальгетичну ефективність дози опіоідного агоніста, що включена у лікарську форму. Приклад 16 Спосіб запобігання зловживанням опіоідним агоністом Лікарську форму для перорального введення за даним винаходом можна використовувати для зменшення ймовірності виникнення зловживань опіоідним агоністом, що в ній міститься. Лікарська форма для перорального введення включає опіоідний агоніст у комбінації з опіоідним антагоністом. Опіоідний антагоніст присутній у формі, яка практично не вивільнюється під час травлення. Таким чином, коли лікарська форма для перорального введення доставляється до шлунково-кишкового тракту перорально, як призначено, без псування, вивільнення антагоніста до шлунково-кишкового тракту практично не відбувається. Але якщо лікарська форма зазнала псування, наприклад, шляхом механічного пошкодження (наприклад, роздавлювання, нарізання, розмолування), нагрівання (наприклад, до температур вище 45°С, краще, від 45 до 50°С) або розчинення лікарської форми у розчиннику (з нагріванням або без нього), опіоідний антагоніст псує лікарську форму, оскільки він притупляє ефекти опіоідів. Таким чином, коли лікарську форму розжовують, роздавлюють, нагрівають або розчиняють у розчиннику, а потім вводять перорально, інтраназально, парентерально або сублінгвально, ефект опіоідного агоніста, принаймні частково, блокується опіоідним антагоністом. Приклад 17 У даному дослідженні на людях 12 морфінонезалежних осіб оцінювали на предмет стрімкої абстиненції після введення таблеток гідрокодону з негайним вивільненням, які давали одночасно з дозою налтрексону від 0,25 до 8мг. Дослідження було одинарним сліпим, з використанням одноразової дози та контролю плацебо при зростаючій дозі налтрексону. Після введення досліджуваних лікарських засобів здійснювали суб'єктивні та психологічні оцінки схильності до зловживання та абстиненції у 32-кратних межах доз налтрексону. Дані передбачають, що при дозі налтрексону 1мг опіоід-залежним особам агоніст подобається менше порівняно із комбінацією з плацебо, і досягається концентрація у плазмі крові, яка призводить до 50% від максимального оцінного балу симптомів абстиненції. Приклад 18 Дане випробування становило рандомізоване, подвійне сліпе, з контролем плацебо дослідження, що стосувалося порога абстиненції, індукованої налтрексоном з негайним вивільненням, у 12 метадон-залежних осіб. Під час проведення цього дослідження проміжний аналіз показав, що у даній популяції 0,5мг налтрексону здатні викликати ознаки та симптоми абстиненції. Зазначені дослідження передбачають, що доза налтрексону, яка потрібна для того, щоб викликати симптоми абстиненції у опіоід-залежних осіб, становить від 0,25 до 1мг. Приклад 19 Дане випробування становило рандомізоване, одинарне сліпе, з однократною дозою та контролем плацебо, перехресне, з 10 підрозділами дослідження, яке стосувалося впливу налтрексону на суб'єктивний і психологічний ефект 15мг гідрокодону у 16 нормальних осіб. Дози налтрексону варіювали від 0,4 до 12,8мг. У цьому дослідженні 0,4мг налтрексону були здатні протистояти декільком центральним опіоідним ефектам гідрокодону, включаючи звуження зіниць. На підставі цих даних, значно більш низькі дози нижче 0,25мг налтрексону демонструватимуть незначний антагонізм відносно одночасно діючого агоніста. Це підтверджується відсутністю ознак абстиненції у осіб з прикладу 17, які отримували 0,25мг. Клінічні дані прикладів 17, 18 та 19 передбачають, що біодоступні, з негайним вивільненням дози 0,125мг налтрексону (або еквівалентні, швидко вивільнювані з лікарської форми з контрольованим вивільненням) не впливатимуть на анальгезію до будь-якого значного ступеня, в той час як більш високі дози біодоступного лікарського засобу (0,25мг або більше) при швидкому вивільненні робитимуть це. Зазначені клінічні дані показують, що навантаження налтрексоном опіоідного матриксу, для даного прикладу, становить співвідношення від 1:15 до 1:30мг налтрексону/мг гідрокодону, і що співвідношення кількості антагоніста, вивільненого з лікарської форми після її псування, та кількості антагоніста, вивільненого з інтактної лікарської форми, становить принаймні 4:1 та, краще, вище. Або, альтернативно, можна визначити, що менше 0,25мг налтрексону вивільнюється з інтактної лікарської форми та 0,25мг або більше налтрексону вивільнюється зі зруйнованої лікарської форми. Приклад 20 Гранули налтрексону НСІ Процедура виготовлення гранул 1. Розчиняють налтрексон НСІ та Opadry Clear у воді. Розпилюють розчин лікарського засобу на гранули нонпарель в апараті для нанесення покриття у текучому шарі зі вставкою Wurster. 2. Диспергують Eudragit L30D, трибутилцитрат і тальк у воді. Розпилюють дисперсію на гранули, навантажені лікарським засобом, в апараті для нанесення покриття у текучому шарі. 3. Диспергують Eudragit RS30D, трибутилцитрат і тальк у воді. Розпилюють дисперсію на гранули в апараті для нанесення покриття у текучому шарі. 4. Розчиняють Opadry Clear у воді. Розпилюють розчин на гранули в апараті для нанесення покриття у текучому шарі. 5. Сушать гранули при 60°С впродовж 24 годин. Спосіб розчинення 1. Апарат: USP типу II (лопасний), 75 об./хв. при 37°С 2. Час відбирання зразків: 1, 2, 4, 8, 12, 24 та 36 годин. 3. Середовища: SGF впродовж однієї години/потім SIF. 4. Аналітичний метод: Рідинна хроматографія високого дозволу. Результати та обговорення: Як було встановлено, гранули (108мг) показали наступні результати розчинення: Результати розчинення показують, що лише близько 10% налтрексону НСІ (0,06мг) вивільнилось через 36 годин на бані для розчинення. Зазначені гранули не будуть біодоступними, якщо їх вживатимуть перорально незруйнованими. Налтрексон НСІ дуже добре розчиняється у воді. У нього спостерігається тенденція мігрувати крізь плівку з уповільненим вивільненням під час водного процесу нанесення покриття (стадія 3). Якщо під час даної стадії нанесення покриття спостерігається міграція, плівка стане пористою під час розчинення, і швидкість вивільнення лікарського засобу має бути відносно високою. Аніонне покриття (стадія 2) утворює не розчинний у воді комплексний шар із протонованою сіллю налтрексону НСІ і перешкоджатиме міграції лікарського засобу крізь покриття з уповільненим вивільненням, яке з'являється згодом. Розчинення зруйнованих гранул Імітація процесу псування Приблизно 108мг гранул налтрексону розмолували у ступці з товкачем для отримання порошку, що використовується для вивчення розчинення. Спосіб розчинення: як описано вище. Результати та обговорення: Як було встановлено, зруйновані гранули (108мг) показали наступні результати розчинення: Можна бачити, таким чином, що через 1 годину з інтактних гранул не було виявлено вивільнення NTX, в той час як зі зруйнованих гранул вивільнився весь NTX, 0,6мг. Це графічно подано показано на Фіг.1. Таким чином, співвідношення зруйнований/цілий через 1 годину становило 100:0, і це більше, ніж критерій >4:1, як можна зробити висновок з прикладів 17,18 і 19. Приклад 21 Капсули оксикодону НСІ з НВ з гранулами налтрексону НСІ Процедура виготовлення 1. Розчиняють оксикодон НСІ і НРМС у воді. Розпилюють розчин лікарського засобу на гранули нонпарель в апараті для нанесення покриття у текучому шарі зі вставкою Wurster. 2. Розчиняють забарвлений Opadry у воді. Наносять плівкове покриття на гранули, навантажені лікарським засобом, в апараті для нанесення покриття у текучому шарі. 3. Змішують рівні кількості гранул Оху з НВ і гранул налтрексону. Інкапсулюють у тверді желатинові капсули. Приклад 22 Капсули морфіна сульфата з контрольованим вивільненням з гранулами налтрексону НСІ Процедура виготовлення. 1. Диспергують повідон та Eudragit RS30D воді. Змішують морфіна сульфат і лактозу. 2. Навантажують гранули в роторному процесорі. Розпилюють порошкоподібну суміш лікарських засобів і розчин зв'язуючого агента на гранули. 3. Наносять плівкове покриття на одержані вище гранули у роторному процесорі. 4. Диспергують Eudragit RS30D, RL30D, триетилцитрат і тальк у воді. Наносять покриття на одержані вище гранули в апараті для нанесення покриття у текучому шарі зі вставкою Wurster. 5. Сушать гранули (гранули MSCR). 6. Змішують рівні кількості гранул MSCR і гранул налтрексону. Інкапсулюють у тверді желатинові капсули. Приклад 23 Екструдовані гранули налтрексону НСІ Процес: 1. Розмолування: Пропускають хлоп'я стеарилового спирту крізь дробарку. 2. Змішування: Змішують налтрексон НСІ, Eudragit, роздрібнений стеариловий спирт, стеаринову кислоту та ВНТ у мішалці з подвійним кожухом. 3. Екструзія: Безперервно подають змішаний матеріал в екструдер з двома гвинтами та збирають одержані пасма на транспортер. 4. Охолоджування: Дають пасмам остигнути на транспортері. 5. Гранулювання: Нарізають охолоджені пасма на 1 мм гранули з використанням приладу для гранулювання. 6. Просіювання: Просіюють гранули та збирають потрібну просіяну порцію. СПОСІБ РОЗЧИНЕННЯ 1. Апарат: LJSP типу II (лопасний), 75 об./хв. при 37°С. 2. Час відбирання зразків: 1, 2, 4, 8, 12, 24 та 36 годин. 3. Середовища: SGF впродовж однієї години/потім SIF. 4. Аналітичний метод: Рідинна хроматографія високого дозволу. Імітація процесу псування Гранули налтрексону розмолували в ступці з товкачем для отримання порошку, що використовується для вивчення розчинення. Спосіб розчинення: як описано вище. Результати: Таким чином, вивільнення через 1 годину з інтактних гранул становило 0,026мг, в той час як зі зруйнованих гранул за 1 годину вивільнилось 0,67мг. Дане співвідношення зруйнованого та цілого також було більше 4:1. Це графічно показано на Фіг.2. Приклад 24 Екструдовані гранули налтрексону НСІ Процес: 1. Розмолування: Пропускають хлоп'я стеарилового спирту крізь дробарку. 2. Змішування: Змішують налтрексон НСІ, Eudragit, роздрібнений стеариловий спирт, двоосновний фосфат кальцію та ВНТ у мішалці з подвійним кожухом. 3. Екструзія: Безперервно подають змішаний матеріал в екструдер з двома гвинтами та збирають одержані пасма на транспортер. 4. Охолоджування: Дають пасмам остигнути на транспортері. 5. Гранулювання: Нарізають охолоджені пасма на гранули з використанням приладу для гранулювання. 6. Просіювання: Просіюють гранули та збирають потрібну просіяну порцію. Спосіб розчинення 1. Апарат: USP типу II (лопасний), 75об./хв. при 37°С. 2. Час відбирання зразків: 1, 2, 4, 8, 12, 24 та 36 годин. 3. Середовища: SGF впродовж однієї години/потім SIF. 4. Аналітичний метод: Рідинна хроматографія високого дозволу. Результати: Імітація процесу псування Гранули налтрексону розмолували в ступці з товкачем для отримання порошку, що використовується для вивчення розчинення. Спосіб розчинення: як описано вище. Таким чином, вивільнення через 1 годину з інтактних гранул становило 0,062мг, в той час як зі зруйнованих гранул за 1 годину вивільнилось 0,728мг. Дане співвідношення зруйнованого та цілого також було більше 4:1. Це графічно показано на Фіг.2. Приклад 25 Можливі капсули гідроморфону НСІ з KB з екструдованими гранулами налтрексону НСІ Процес: 1. Розмолування: Пропускають хлоп'я стеарилового спирту крізь молоткову дробарку. 2. Змішування: Змішують гідроморфон НСІ, Eudragit, етилцелюлозу та роздрібнений стеариловий спирту мішалці з подвійним кожухом. 3. Екструзія: Безперервно подають змішаний матеріал в екструдер з двома гвинтами та збирають одержані пасма на транспортер. 4. Охолоджування: Дають пасмам остигнути на транспортері. 5. Гранулювання: Нарізають охолоджені пасма на гранули з використанням приладу для гранулювання. 6. Просіювання: Просіюють гранули та збирають потрібну просіяну порцію. 7. Інкапсуляція: Наповнюють тверді желатинові капсули екструдованими гранулами гідроморфону НСІ по 120мг та гранулами налтрексону НСІ (з прикладу 23) по 121мг. Приклад 26 Можливі капсули гідроморфону НСІ з KB з екструдованими гранулами налтрексону НСІ Процес: 1. Розмолування: Пропускають хлоп'я стеарилового спирту крізь молоткову дробарку. 2. Змішування: Змішують гідроморфон НСІ, Eudragit, етилцелюлозу та роздрібнений стеариловий спирт у мішалці з подвійним кожухом. 3. Екструзія: Безперервно подають змішаний матеріал в екструдер з двома гвинтами та збирають одержані пасма на транспортер. 4. Охолоджування: Дають пасмам остигнути на транспортері. 5. Гранулювання: Нарізають охолоджені пасма на гранули з використанням приладу для гранулювання. 6. Просіювання: Просіюють гранули та збирають потрібну просіяну порцію. 7. Інкапсуляція: Наповнюють тверді желатинові капсули екструдованими гранулами гідроморфону НСІ по 120мг та твердими желатиновими капсулами налтрексону НСІ (з прикладу 24) по 127мг. Приклад 27а Гранули налтрексону з KB Розроблені гранули налтрексону з контрольованим вивільненням, які можна інкорпорувати у гранулят опіоідів з контрольованим вивільненням, а суміш потім пресувати у таблетки. Як приклад з гранулами налтрексону використовується гранулят оксикодону НСІ з контрольованим вивільненням. Процедура виготовлення гранул 1. Розчиняють налтрексон НСІ та НРМС у воді. Розпилюють розчин лікарського засобу на гранули нонпарель в апараті для нанесення покриття у текучому шарі зі вставкою Wurster. 2. Диспергують Eudragit L, трибутилцитрат і тальк у воді. Розпилюють дисперсію на гранули, навантажені лікарським засобом, в апараті для нанесення покриття у текучому шарі. 3. Диспергують Eudragit RS, трибутилцитрат і тальк у воді. Розпилююто дисперсію на гранули в апараті для нанесення покриття у текучому шарі. 4. Розчиняють НРМС у воді. Розпилюють розчин на гранули в апараті для нанесення покриття у текучому шарі. 5. Сушать гранули при 60°С впродовж 24 годин. Спосіб розчинення 1. Апарат: USP типу II (лопасний), 75об./хв. при 37°С. 2. Час відбирання зразків: 1, 2, 4, 8, 12, 24 та 36 годин. 3. Середовища: SGF впродовж однієї години/потім SIF. 4. Аналітичний метод: Рідинна хроматографія високого дозволу. Результати та обговорення: Розчинення налтрексону з інтактних гранул Процедура виготовлення гранул 1. Розчиняють дисперсію Eudragit/триацетин на оксикодон НСІ, висушену розпиленням лактозу та повідон з використанням гранулятора з текучим шаром. 2. Вивантажують гранули та пропускають крізь дробарку. 3. Розплавляють стеариловий спирт і додають до розмолотого гранулята за допомогою дробарки. Дають остуднути. 4. Пропускають охолоджений гранулят крізь дробарку.
ДивитисяДодаткова інформація
Назва патенту англійськоюPeroral composition preventing from abuse of opioid agonists contained therein
Автори англійськоюWright Curtis
Назва патенту російськоюПероральная композиция, которая предотвращает злоупотребление опиоидными агонистами, которые в ней содержатся, при ее повреждении
Автори російськоюРайт Куртис
МПК / Мітки
МПК: A61P 25/36, A61K 31/445, A61K 31/485
Мітки: ній, зловживанню, опіоїдними, псуванні, пероральна, композиція, агоністами, містяться, запобігає
Код посилання
<a href="https://ua.patents.su/30-79069-peroralna-kompoziciya-shho-zapobigaeh-zlovzhivannyu-opiodnimi-agonistami-shho-v-nijj-mistyatsya-pri-psuvanni.html" target="_blank" rel="follow" title="База патентів України">Пероральна композиція, що запобігає зловживанню опіоїдними агоністами, що в ній містяться, при її псуванні</a>
Пероральна фармацевтична композиція на основі сполуки, що має антигрибкову активність, і спосіб її одержання

Номер патенту: 64009
Опубліковано: 16.02.2004
Автор: Пікорнелль Дардер Карлос
МПК: A61K 9/16
Мітки: активність, одержання, пероральна, основі, спосіб, має, антигрибкову, композиція, сполуки, фармацевтична
Формула / Реферат:
1. Пероральна фармацевтична композиція в формі гранул, що включає як активний інгредієнт сполуку, що має антигрибкову активність, вибрану з ітраконазолу і саперконазолу, інертне ядро і покриття, що включає вказаний активний інгредієнт, яка відрізняється тим, що інертне ядро має розмір частинок між 50 і 600 мкм, і в якій вказане покриття включає єдиний шар, отриманий розпиленням на вказане інертне ядро розчину, що включає вказану сполуку, яка...
Пероральна композиція контрольованого вивільнення левозимендану (варіанти)

Номер патенту: 68376
Опубліковано: 16.08.2004
Автори: Бякман Мааріт, Лехтонен Лассе, Антіла Сайла, Ларма Ількка
МПК: A61K 9/20, A61K 31/50, A61K 9/00, A61P 9/04
Мітки: пероральна, композиція, вивільнення, левозимендану, варіанти, контрольованого
Формула / Реферат:
1. Композиція контрольованого вивільнення для перорального введення, що включає а) терапевтично ефективну кількість левозимендану і б) компонент, що контролює вивільнення лікарського засобу, для забезпечення вивільнення левозимендану протягом тривалого періоду часу і постійного вмісту метаболіту (II) в плазмі, меншого за 20 нг/мл, переважно, меншого за 10 нг/мл.2. Композиція за п. 1, в якій вказаний компонент, що контролює вивільнення...
Похідне триазолу, його фармацевтично прийнятні солі або проліки, що є агоністами 5-нt1-подібних рецепторів, спосіб його отримання та фармацевтична композиція
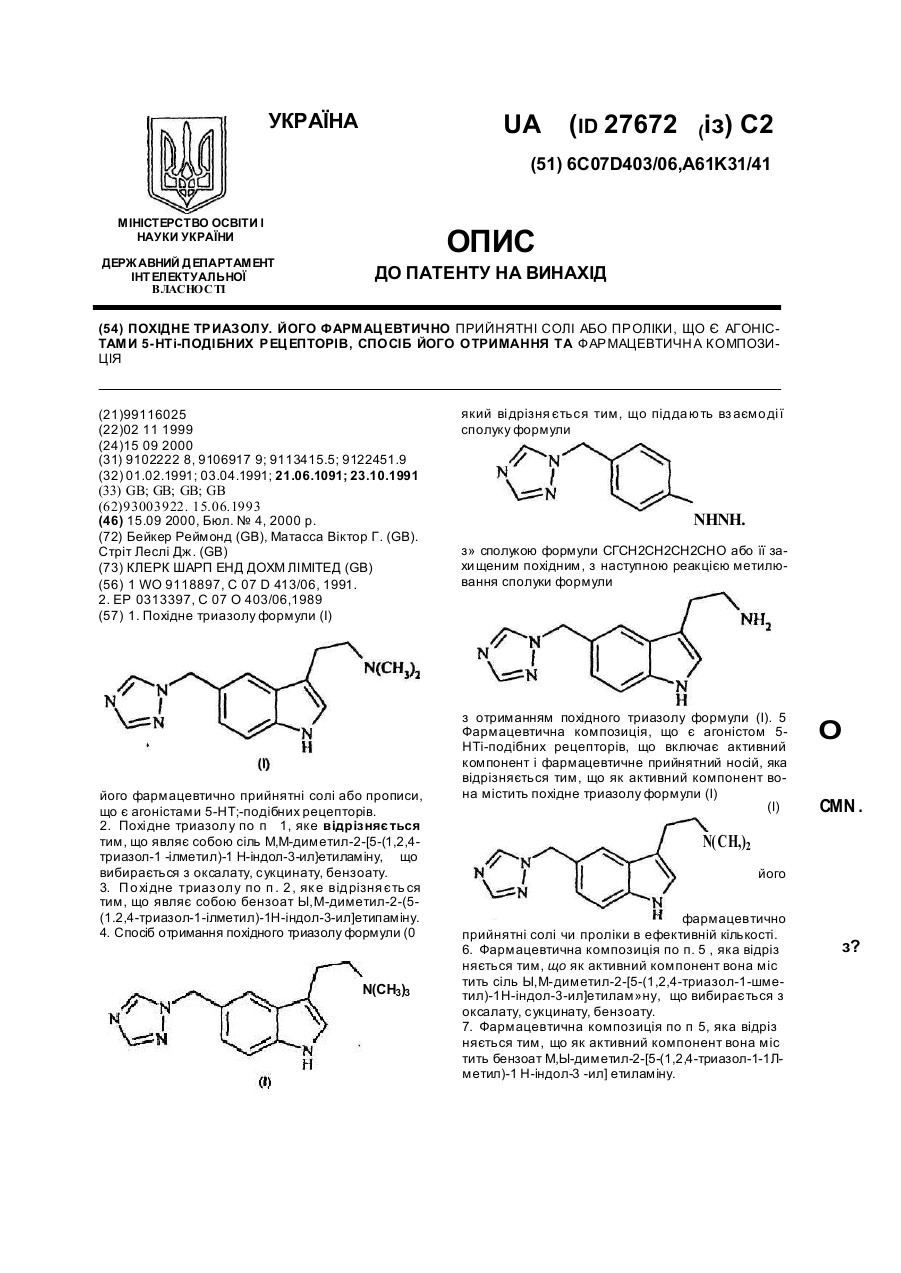
Номер патенту: 27672
Опубліковано: 15.09.2000
Автори: Матасса Віктор Г., Стріт Леслі Дж., Бейкер Реймонд
МПК: C07D 409/04, C07D 403/06, C07D 401/14
Мітки: солі, прийнятні, фармацевтично, триазолу, проліки, 5-нt1-подібних, похідне, отримання, фармацевтична, спосіб, рецепторів, агоністами, композиція
Текст:
...призначення або призначення шля хом ін'єкцій , включають водні розчини , ароматизо вані сиропи, во дні або масляні емульсії, ароматизовані емульсі ї, з такими маслами, як бавовняне масло, сезамове масло, кокосове масло, а також еліксири та подібні засоби . Відпо відні диспер гуючі та суспензуючі аген ти для водни х суспензій включають синте ти чні і натуральні смоли, такі як трагант, акація, альгінат, декстран, натрієва сіль...
Похідні азолів, їх фармацевтично прийнятні солі або проліки, що є агоністами 5-нt1-подібних рецепторів, спосіб їх отримання, фармацевтична композиція
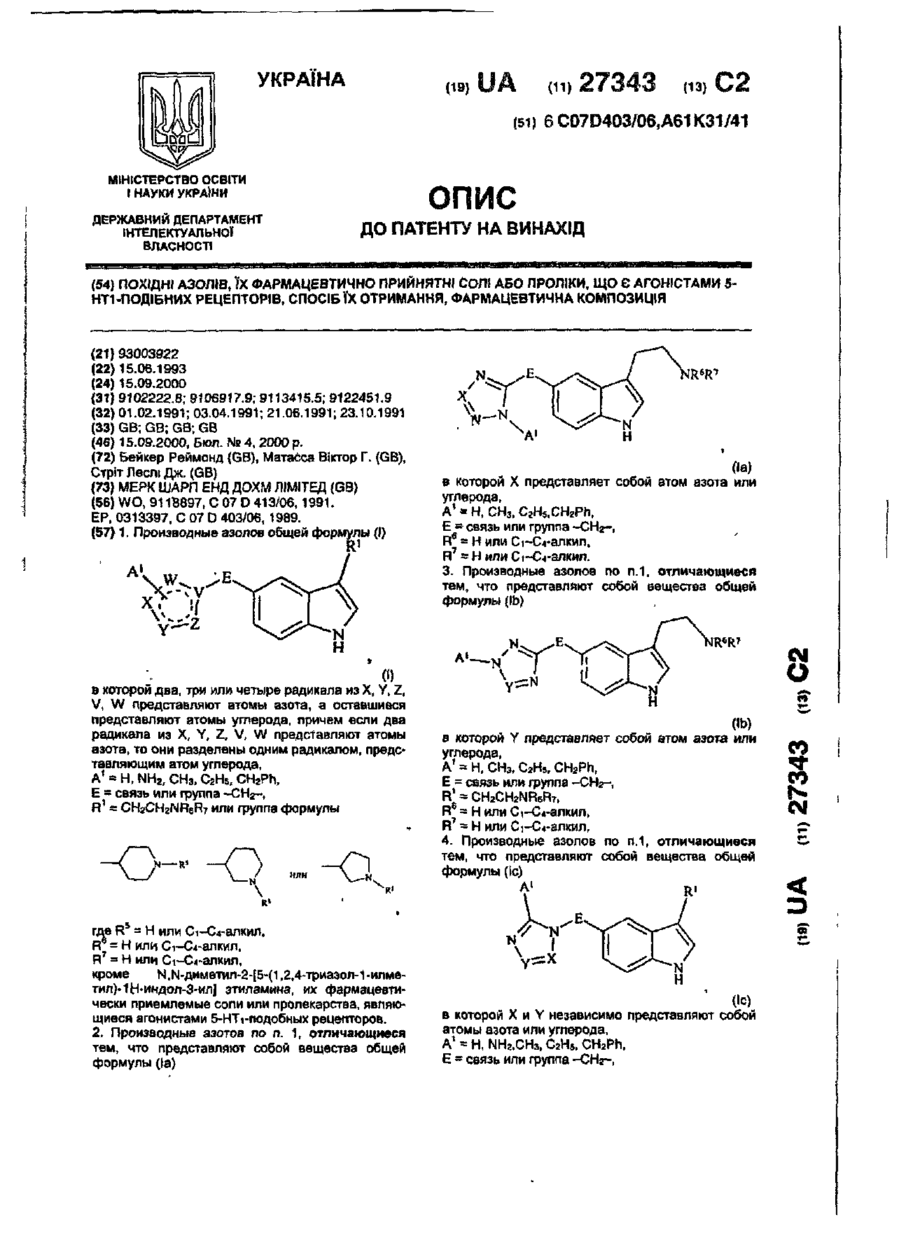
Номер патенту: 27343
Опубліковано: 15.09.2000
Автори: Матасса Віктор Г., Стріт Леслі Дж., Бейкер Реймонд
МПК: A61K 31/425, A61K 31/415, A61K 31/445, A61K 31/41, C07D 401/14, C07D 521/00, C07D 417/06, C07D 403/06, C07D 403/04, C07D 409/06, C07D 413/14, A61K 31/42, C07D 403/14, C07D 417/14, A61K 31/4433, C07D 413/06, C07D 409/14, C07D 409/04, A61P 25/04, A61K 31/4427
Мітки: рецепторів, спосіб, отримання, похідні, проліки, фармацевтична, агоністами, солі, азолів, композиція, фармацевтично, 5-нt1-подібних, прийнятні
Текст:
...2-15-(1-бензилтетразол-5-илметил)-1Н-индол-3ил]этиламин М,М-диметил-2-{5-(2-метилтетразол-5-илметил)1 Н-индол-3-ил]этиламин М,М-диметил-2-[5-(1-метилтетразол-5-илметил)1 Н-индол-3-ил]этиламин М,М-диметил-2-[5-(тетрээол-2-ил метил )• 1 Н-индол3-ил]этиламин N,N-димeтил-2-[5-(тeтpaзoл-1-илмeтил)-1 Н-индол3-ил]этиламин N,N-flHMeTHn-2-tS-{1 -метил-1,2,4-триазол-5-и л метил) 1Н-индол-3-ил]этиламин...
Пероральна тверда фармацевтична композиція та спосіб її одержання

Номер патенту: 27525
Опубліковано: 15.09.2000
Автори: Клебович Імре, Драбант Сандор, Жлаві Жужа, Марошелі Біборка, Беззег Денес, Жанто Марта, Сандорфалві Андреа, Юджфалуссі Джорджі, Гора Магдолна, Фекете Паль, Манді Аттіла, Феллнер Ержебет
МПК: A61K 31/40, A61K 31/185
Мітки: тверда, пероральна, композиція, одержання, фармацевтична, спосіб
Текст:
...активный ингредиент в сухом виде гом огенизируют с наполнителем (наприм ер, целлюлозой, лактозой, м аннитом , крахм алом , м икрокристаллической целлюлозой, фос фатом кальция или т.п.), взятым в количестве 0-40 % м асс, по отношению к содержимом у капсулы или сердцевины непокрытой таблетки. По ж еланию, сухой гом огенизат м ож ет быть гранулирован известным и способам и. Гранулирование м ож ет быть осуществлено с использованием процедур...
Попередній патент: Пристрій для захисту від заливання водою приміщення, групи приміщень або будівлі
Наступний патент: Нові lнrн-антагоністи, спосіб їх одержання та застосування як лікарського засобу
Випадковий патент: Гідропривід вертикальних шламових насосів