Спосіб покращення фармакокінетики інгібіторів інтегрази віл

























Формула / Реферат
1. Застосування (2S,3S,5S)-5-(N-(N-((N-метил-N-((2-ізопропіл-4-тіазоліл)метил)аміно)карбоніл)-L-валініл)аміно)-2-(N-((5-тіазоліл)метоксикарбоніл)аміно)-1,6-дифеніл-3-гідроксигексану (ритонавіру) або його фармацевтично прийнятної солі у терапії, що включає введення пацієнту, який цього потребує, терапевтично ефективної кількості інгібітора інтегрази ВІЛ, де інгібітор інтегрази являє собою сполуку формули Іа
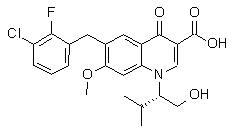 Iа
Iа
або її фармацевтично прийнятну сіль, та терапевтично ефективної кількості ритонавіру.
2. Застосування за п. 1, у якому ритонавір або його фармацевтично прийнятну сіль вводять перорально один раз на добу у дозі від 20 мг до 200 мг та сполуку формули Іа вводять перорально один раз на добу у дозі від 20 мг до 500 мг.
3. Застосування за п. 1 або п. 2, де пацієнту додатково вводять один або більше агентів, вибраних з групи, що складається з ставудину, емтрицитабіну, тенофовіру, абакавіру, ламівудину, зидовудину, диданозину, залцитабіну, фосфазиду, ефавіренцу, невірапіну, делавірдину, типранавіру, саквінавіру, індинавіру, атазанавіру, нелфінавіру, ампренавіру, сампренавіру, фосампренавіру, лопінавіру, енфувіртиду, фозивудину тидоксилу, аловудину, декселвуцитабіну, априцитабіну, амдоксовіру, елвуцитабіну (АСН126443), рацивіру (рацемічного FTC, PSI-5004), MIV-210, KР-1461, фосалвудину тидоксилу (HDP 99.0003), AVX756, діоксалану тиміну (DOT), TMC-254072, INK-20, 4'-Ed4T, TMC-125 (етравірину), каправірину, ТМС-278 (рилпівірину), GW-695634, каланоліду A, BILR 355 BS та VRX 840773 або їх фармацевтично прийнятних солей.
4. Спосіб лікування або профілактики ретровірусної інфекції у пацієнта, за яким пацієнту вводять терапевтично ефективну кількість сполуки формули Іа: 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метоксі-4-оксо-1,4-дигідрохінолін-3-карбонової кислоти, або її фармацевтично прийнятної солі, та прийом їжі пацієнтом проводять протягом періоду між від приблизно 1 години до введення сполуки формули Іа до приблизно 2 годин після введення сполуки формули Іа; або
за яким пацієнту вводять терапевтично ефективну кількість сполуки формули Іа або її фармацевтично прийнятної солі протягом періоду між від приблизно 1 години до прийому їжі до приблизно 2 годин після прийому їжі.
5. Спосіб за п. 4, у якому пацієнту додатково вводять терапевтично ефективну кількість ритонавіру або його фармацевтично прийнятної солі.
6. Спосіб за п. 5, де сполуку формули Іа або її фармацевтично прийнятну сіль вводять перорально один раз на добу у дозі від 20 мг до 500 мг та ритонавір вводять перорально один раз на добу у дозі від 20 мг до 200 мг.
7. Спосіб підвищення біодоступності сполуки формули Іа: 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метоксі-4-оксо-1,4-дигідрохінолін-3-карбонової кислоти, або її фармацевтично прийнятної солі у пацієнта при терапії, за яким пацієнту вводять терапевтично ефективну кількість сполуки формули Іа або її фармацевтично прийнятної солі, та прийом їжі пацієнтом проводять протягом періоду між від приблизно 1 години до введення сполуки формули Іа до приблизно 2 годин після введення сполуки формули Іа; або
за яким пацієнту вводять терапевтично ефективну кількість сполуки формули Іа або її фармацевтично прийнятної солі протягом періоду між від приблизно 1 години до прийому їжі до приблизно 2 годин після прийому їжі.
8. Спосіб за п. 7, у якому пацієнту додатково вводять терапевтично ефективну кількість ритонавіру або його фармацевтично прийнятної солі.
9. Спосіб за п. 8, де сполуку формули Іа або її фармацевтично прийнятну сіль вводять перорально один раз на добу у дозі від 20 мг до 500 мг та ритонавір вводять перорально один раз на добу у дозі від 20 мг до 200 мг.
10. Спосіб підвищення абсорбції сполуки формули Іа: 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метоксі-4-оксо-1,4-дигідрохінолін-3-карбонової кислоти, або її фармацевтично прийнятної солі у пацієнта при терапії, за яким пацієнту вводять терапевтично ефективну кількість сполуки формули Іа або її фармацевтично прийнятної солі, та прийом їжі пацієнтом проводять протягом періоду між від приблизно 1 години до введення сполуки формули Іа до приблизно 2 годин після введення сполуки формули Іа; або
за яким пацієнту вводять терапевтично ефективну кількість сполуки формули Іа або її фармацевтично прийнятної солі протягом періоду між від приблизно 1 години до прийому їжі до приблизно 2 годин після прийому їжі.
11. Спосіб за п. 10, у якому пацієнту додатково вводять терапевтично ефективну кількість ритонавіру або його фармацевтично прийнятної солі.
12. Спосіб за п. 11, де сполуку формули Іа або її фармацевтично прийнятну сіль вводять перорально один раз на добу у дозі від 20 мг до 500 мг та ритонавір вводять перорально один раз на добу у дозі від 20 мг до 200 мг.
13. Спосіб інгібування активності інтегрази ретровірусу у пацієнта, який приймає сполуку формули Іа: 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метоксі-4-оксо-1,4-дигідрохінолін-3-карбонову кислоту, або її фармацевтично прийнятну сіль, у терапії, за яким пацієнту вводять терапевтично ефективну кількість сполуки формули Іа або її фармацевтично прийнятної солі, та прийом їжі пацієнтом проводять протягом періоду між від приблизно 1 години до введення сполуки формули Іа до приблизно 2 годин після введення сполуки формули Іа; або
за яким пацієнту вводять терапевтично ефективну кількість сполуки формули Іа або її фармацевтично прийнятної солі протягом періоду між від приблизно 1 години до прийому їжі до приблизно 2 годин після прийому їжі.
14. Спосіб за п. 13, у якому пацієнту додатково вводять терапевтично ефективну кількість ритонавіру або його фармацевтично прийнятної солі.
15. Спосіб за п. 14, де сполуку формули Іа або її фармацевтично прийнятну сіль вводять перорально один раз на добу у дозі від 20 мг до 500 мг та ритонавір вводять перорально один раз на добу у дозі від 20 мг до 200 мг.
16. Набір, що містить:
(1) фармацевтичну композицію у формі одиничної дози, що містить 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метоксі-4-оксо-1,4-дигідрохінолін-3-карбонову кислоту або її фармацевтично прийнятну сіль у дозі від 20 мг до 500 мг; та фармацевтично прийнятний носій;
(2) ритонавір або його фармацевтично прийнятну сіль у дозі від 20 мг до 200 мг;
(3) інформацію, що стосується призначень, та
(4) контейнер,
причому інформація, що стосується призначень, включає рекомендацію щодо введення 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метоксі-4-оксо-1,4-дигідрохінолін-3-карбонової кислоти або її фармацевтично прийнятної солі з їжею.
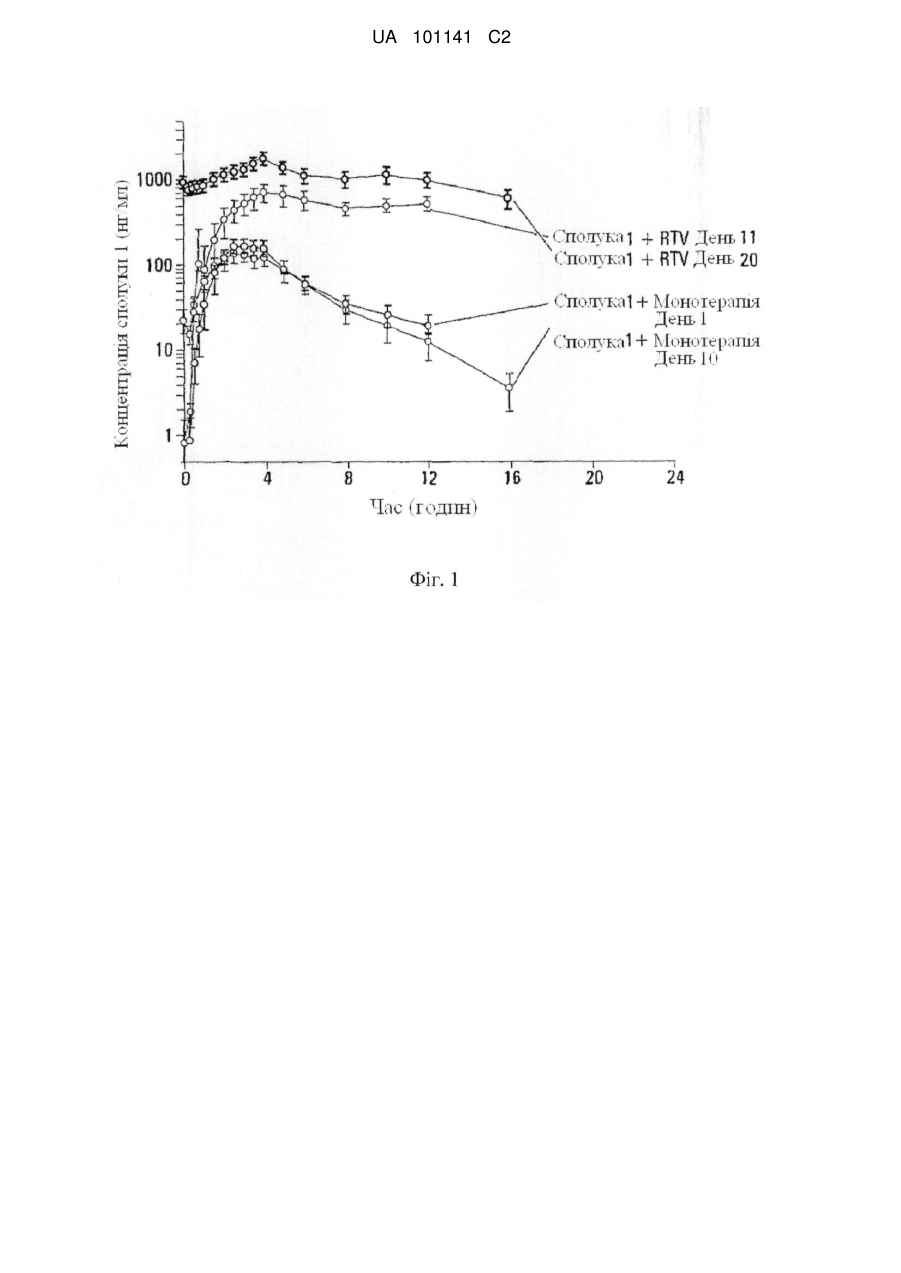
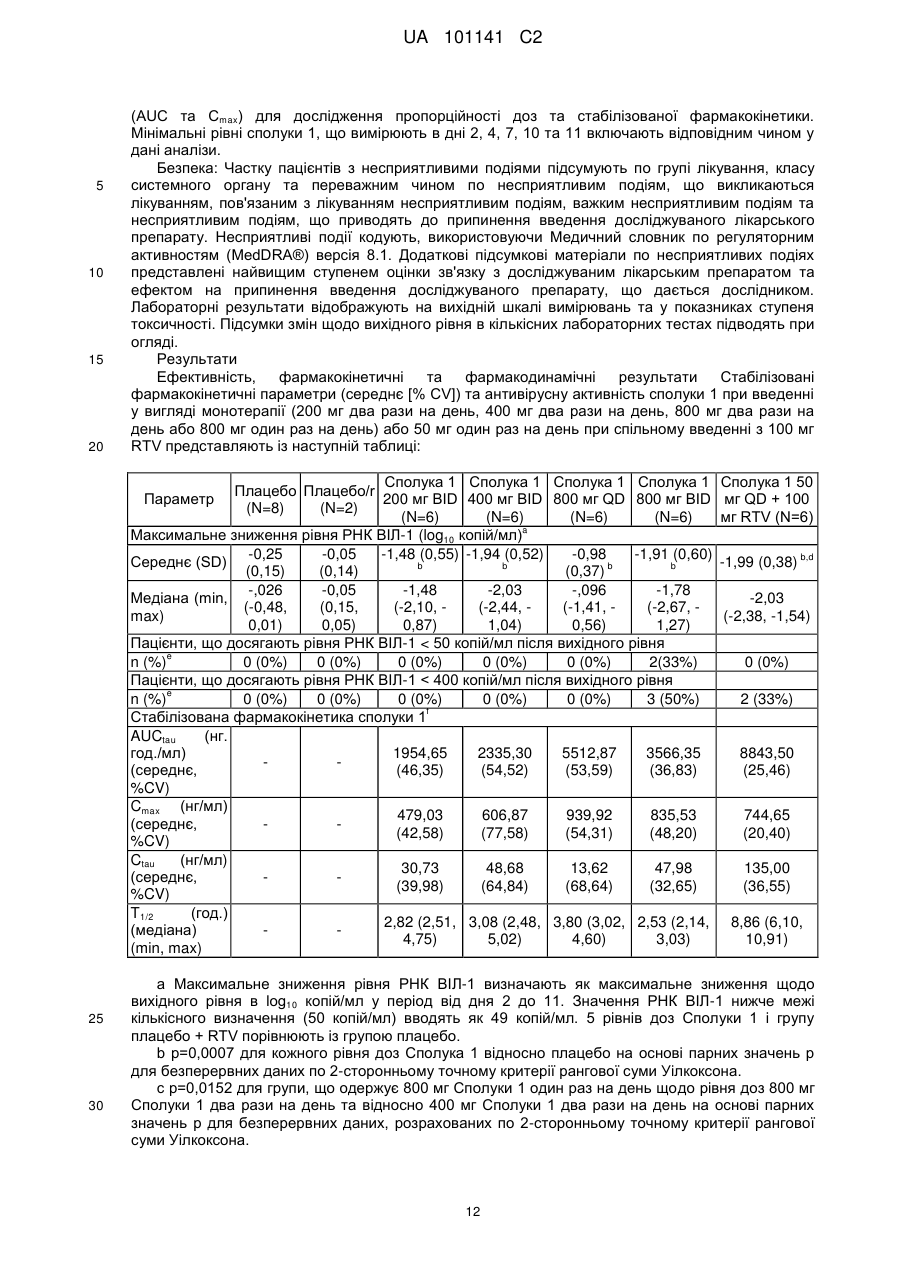
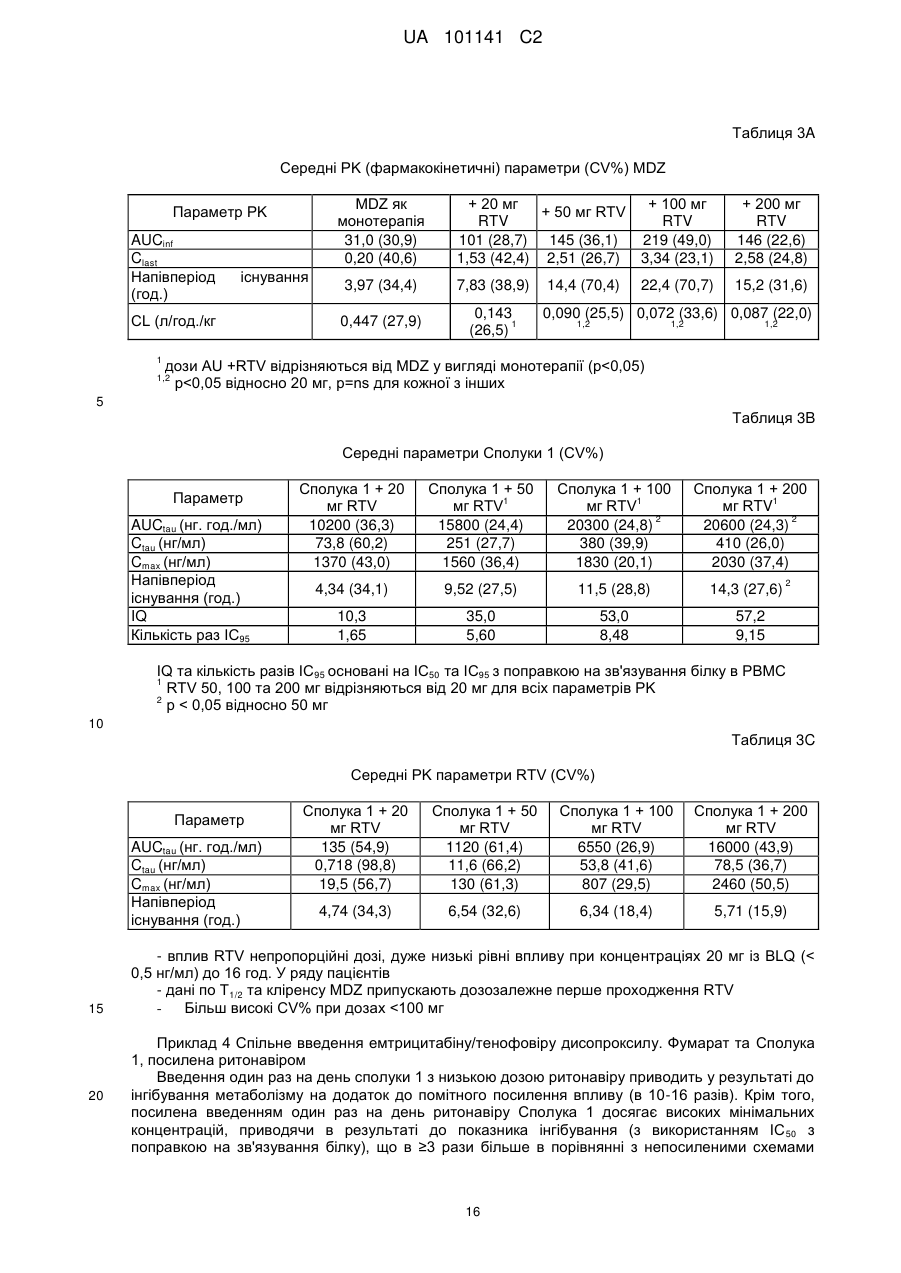
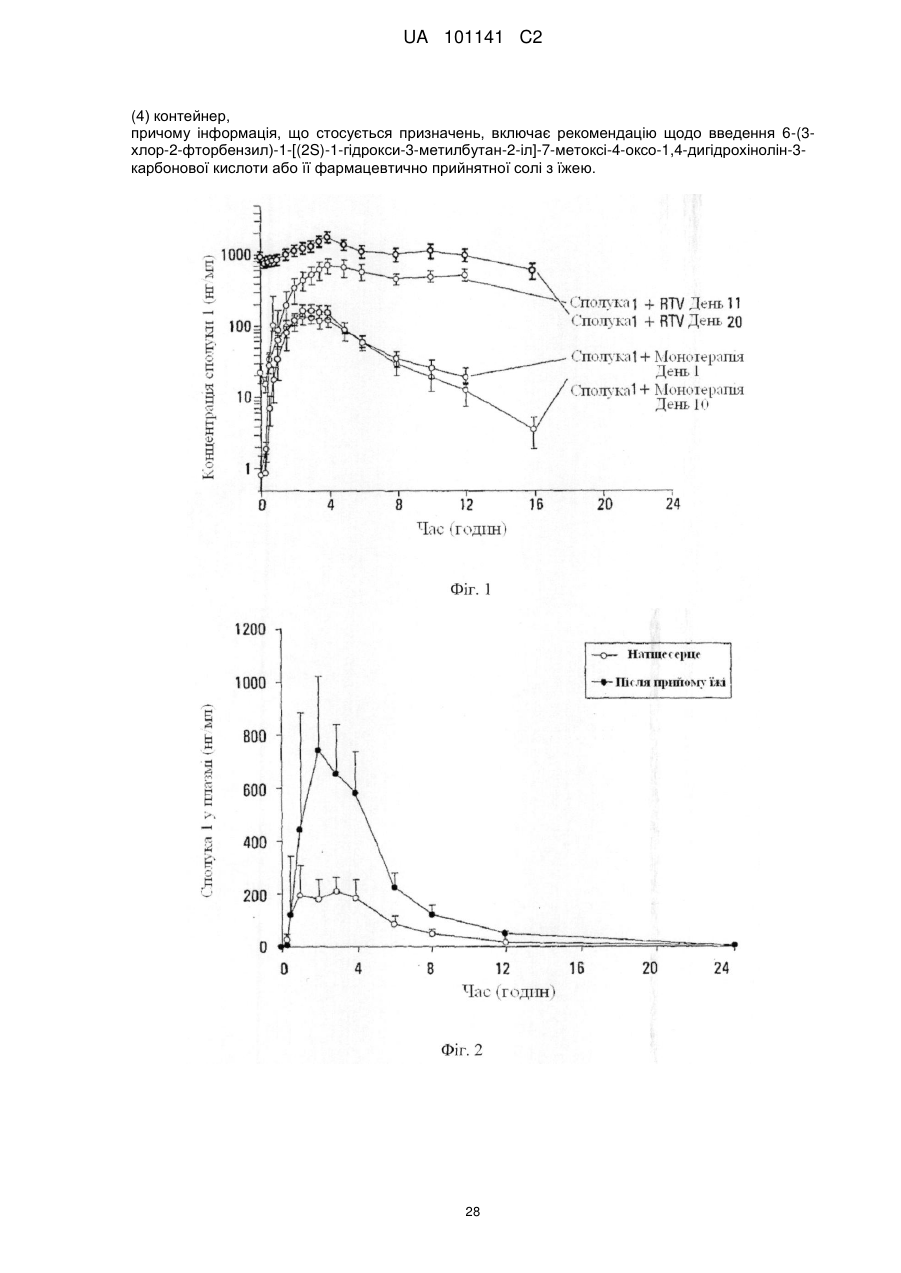
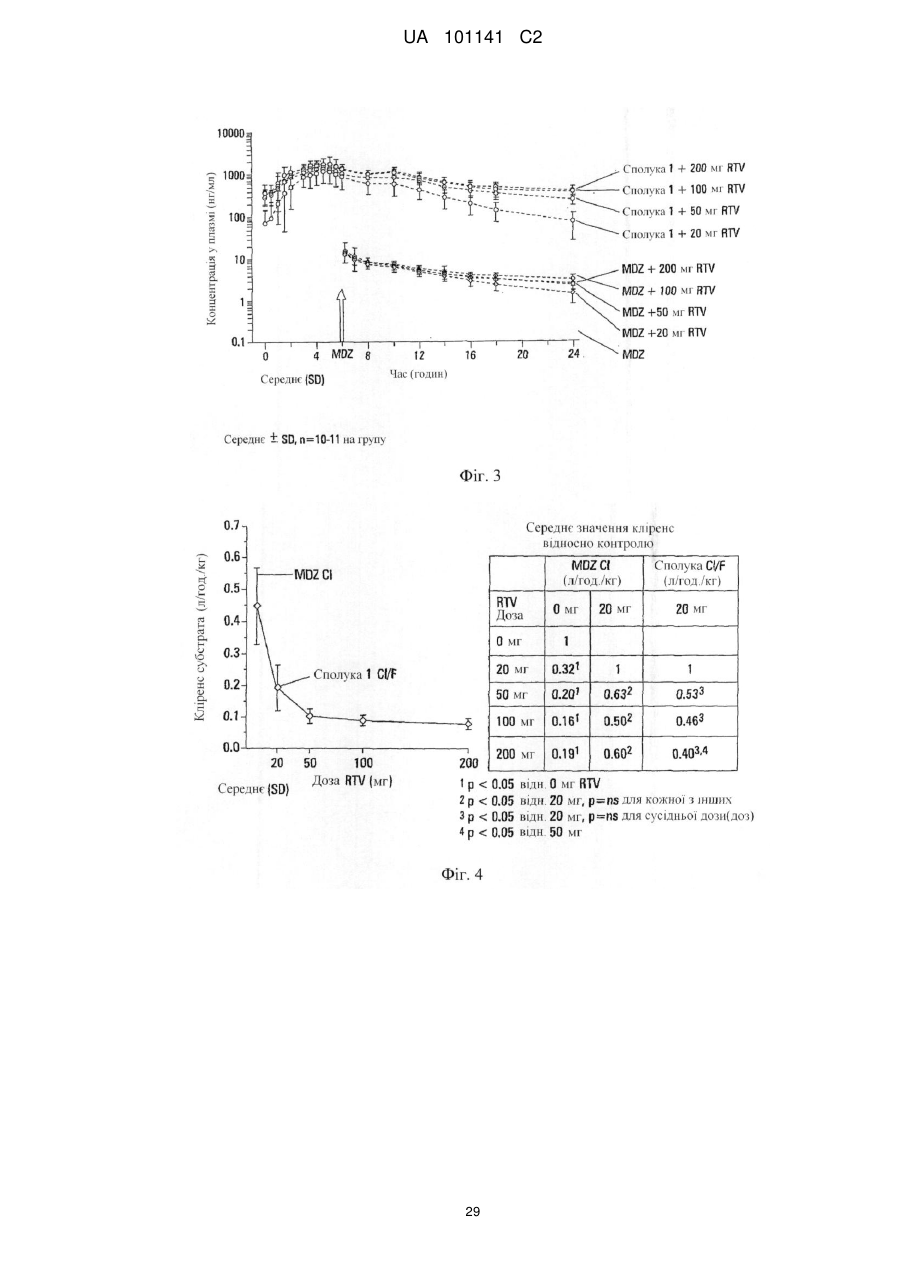
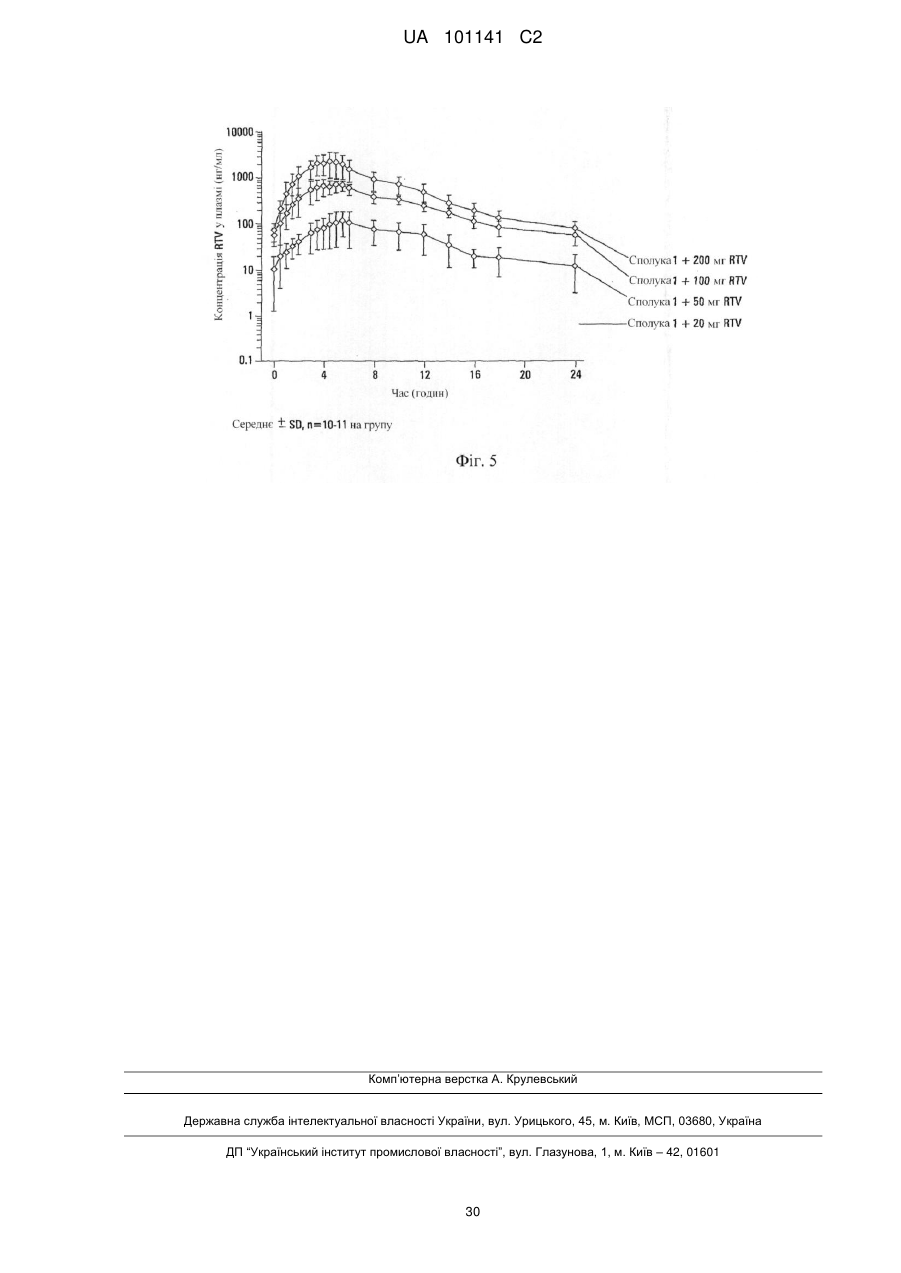
Текст
Реферат: Винахід стосується застосування ритонавіру або його фармацевтично прийнятної солі в комбінованій терапії з 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метоксі4-оксо-1,4-дигідрохінолін-3-карбоновою кислотою або її фармацевтично прийнятною сіллю сполукою формули Іа: , Iа а також способу лікування, за яким сполуку формулу Іа приймають з їжею. UA 101141 C2 (12) UA 101141 C2 UA 101141 C2 5 10 15 20 25 30 35 40 45 50 55 Пріоритет винаходу Дана заявка затверджує пріоритет попередніх патентних заявок США 60/755039, поданої 30 грудня 2005 р., 60/756631, поданої 6 січня 2006 р. та 60/763901, поданої 1 лютого 2006 р. Попередній рівень техніки Інфекція ретровірусу, відомого як вірус імунодефіциту людини (ВІЛ), продовжує залишатися серйозною проблемою охорони здоров'я. Способи лікування ВІЛ-інфекцій включають введення агентів, які інгібують активність вірусних ферментів, які є основними для життєвого циклу вірусу. Ритонавір (2S,3S,5S)-5-(N-(N-((N-Метил-N-((2-ізопропіл-4-тіазоліл)метил)аміно)-карбоніл)-Lвалініл)аміно)-2-(N-((5-тіазоліл)метоксикарбоніл)аміно)-1,6-дифеніл-3-гідроксигексан), являє собою інгібітор протеази ВІЛ, який можна синтезувати способами, описаними в публікації міжнародної патентної заявки № WO 1994/14436 та патенті США № 5567823. Як інгібітор протеази, ритонавір може бути ефективний у плані інгібування ВІЛ-інфекції у людини. Показано також, що ритонавір як інгібітор ферменту обміну речовин цитохром P450 монооксигенази, особливо ізоформи 3A4 (CYP 3A4), включений у шлях метаболізму багатьох лікарських препаратів. Див. патенти США №№. 5541206, 5635523, 5648497, 5674882, 5846987 та 5886036. Інгібітори протеази метаболізуються цитохром P450 монооксигеназою, що приводить до несприятливої фармакокінетики та необхідності використання більш частих та більш високих доз, ніж це бажано. Введення даних лікарських препаратів з агентом, що інгібує метаболізм, опосередкований цитохром P450 монооксигеназою, може поліпшити фармакокінетику (наприклад, збільшити напівперіод існування, час до максимальної концентрації в плазмі та рівні в крові) лікарського препарату. Ритонавір можна використовувати для покращення фармакокінетики ряду інгібіторів протеази ВІЛ, які метаболізуються цитохром P450 монооксигеназою. Див. патенти США №№ 6037157 та 6703403. Спільне введення ритонавіру з лікарським препаратом, що метаболізується цитохром P450 монооксигеназою, особливо ізоформою P450 3A4 (ізоферментом), може привести до покращення фармакокінетики даного лікарського препарату. Більш докладно, спільне введення ритонавіру з іншим інгібітором протеази ВІЛ, що метаболізується цитохром P450 монооксигеназою, може привести в результаті до покращення фармакокінетики інгібітору протеази ВІЛ. У комбінованій терапії даного типу ритонавір можна використати в субтерапевтичних дозах, тобто дозах, які менші ніж використовувані для значимої супресії реплікації вірусу, але досить високі для інгібування цитохром P450 монооксигенази та підтримання фармакокінетики іншого інгібітору протеази ВІЛ. Ряд 4-оксохінолінів, включаючи сполуку 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3-карбонова кислота, ідентифікований як агенти проти вірусу імунодефіциту людини (ВІЛ). Див. патентну заявку США № 10/492833, подану 20 листопада 2003 р., що опублікована як публікація патентної заявки США № 2005/0239819. Зокрема, 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7метокси-4-оксо-1,4-дигідрохінолін-3-карбонова кислота описана як така, що має інгібуючу здатність відносно білку інтегрази ВІЛ. Див. там же. ВІЛ належить до сімейства ретровірусів та є етіологічним фактором синдрому набутого імундефіциту (СНІДу). Відповідно фармацевтичний агент, що знижує вірусне навантаження, кількість вірусного геному або реплікацію ВІЛ в організмі, може бути ефективним для лікування або профілактики СНІДу. У цей час існує потреба в агентах та способах, які ефективні в плані підвищення біодоступності або всмоктування інгібіторів інтегрази, таких як 6-(3-хлор-2-фторбензил)-1-[(2S)1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3-карбонова кислота, з метою підвищення їхнього терапевтичного ефекту у пацієнта. Зокрема, існує потреба в агентах та способах, які поліпшують фармакокінетику даного інгібітора інтегрази, таким чином, що прийнятний терапевтичний ефект може досягатися введенням один раз на день. Крім того, у цілому є потреба в покращенні фармакокінетики лікарських препаратів (наприклад, інгібіторів інтегрази), які використають для лікування ВІЛ-інфекції. Розкриття винаходу Винахід відноситься до способу покращення фармакокінетики 4-оксохінолінових сполук. Крім того, винахід відноситься до способу інгібування ретровірусних інтеграз, зокрема, інгібування інтегрази вірусу імунодефіциту людини (ВІЛ), та способу інгібування ретровірусної інфекції, особливо ВІЛ-інфекції. В одному варіанті здійснення винахід передбачає агенти та способи, які використовують для підвищення біодоступності або всмоктування інгібіторів інтегрази, таких як 6-(3-хлор-2фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3карбонова кислота 60 1 UA 101141 C2 5 10 (Сполука 1), з метою підвищення їхнього терапевтичного ефекту у пацієнта. В одному варіанті здійснення винахід передбачає спосіб покращення фармакокінетики інгібітора інтегрази, такої як сполука 1, шляхом введення інгібітора інтегрази пацієнтові з ритонавіром або його фармацевтично прийнятною сіллю. В одному варіанті здійснення винахід передбачає спосіб покращення фармакокінетики інгібітора інтегрази ВІЛ, що полягає у введенні пацієнтові, що потребує інгібітору, ефективної посилюючої дію кількості ритонавіру або його фармацевтично прийнятної солі, таким чином, що інгібітор має більше ефективний фармакокінетичний профіль, ніж був би без додавання ритонавіру. В іншому варіанті здійснення винахід передбачає спосіб покращення фармакокінетики 4оксохінолінової сполуки відповідно до формули (I): 15 20 25 30 35 40 де цикл Cy являє собою C3-10 вуглецеву циклічну групу або гетероциклічну групу, причому кожна група є необов'язково заміщеною 1-5 замісниками, що вибирають із групи A; гетероциклічна група являє собою насичений або ненасичений цикл, що включає щонайменше один гетероатом, що вибирають із групи, що включає азот, кисень та сірку; група A являє собою ціаногрупу, феніл, нітрогрупу, галоген, C 1-4алкіл, галоген C1-4алкіл, a1 a1 a1 a2 a1 a2 a1 a2 a3 галоген C1-4алкілоксигрупу, -OR , -SR , -NR R , -CONR R , - SO2NR R , -COR , a1 a3 a3 a1 a3 a1 a2 a3 NR COR , -SO2R , -NR SO2R , -COOR або -NR COOR ; a1 a2 R та R однакові або різні та кожний являє собою H, C1-4алкіл або бензил; a3 R являє собою С1-4алкіл; 1 R вибрано із групи B або являє собою C 1-10алкіл, необов'язково заміщений 1-3 замісниками, вибраними з галогену або групи B; група B являє собою: C3-10 вуглецевий цикл, необов'язково заміщений 1-5 замісниками, вибраними з групи A, гетероциклічну групу, необов'язково заміщену 1-5 замісниками, вибраними з групи A, a4 -OR , a4 -SR , a4 a5 -NR R , a4 a5 -CONR R , a4 a5 -SO2NR R , a6 -COR , a4 a6 -NR COR , a6 -SO2R , a4 a6 -NR SO2R , a4 -COOR або a5 a6 -NR COOR ; a4 a5 R та R є однаковими або різними та кожний являє собою: 2 UA 101141 C2 5 10 15 20 25 30 35 40 45 50 55 60 H, C1-4алкіл, C3-10 вуглецеву циклічну групу, необов'язково заміщену 1-5 замісниками, вибраними з групи A, або гетероциклічну групу, необов'язково заміщену 1-5 замісниками, вибраними з групи A; a6 R являє собою C1-4алкіл, C3-10вуглецеву циклічну групу, необов'язково заміщену 1-5 замісниками, вибраними з групи A, або гетероциклічну групу, необов'язково заміщену 1 - 5 замісниками, вибраними з групи A; 2 R являє собою H або С1-4алкіл; 31 R являє собою H, ціано-, гідрокси-, аміно-, нітрогрупу, галоген, C1-4алкіл, C1-4алкоксигрупу, C1-4алкілсульфаніл, галоген C1-4алкіл або галоген C1-4алкоксигрупу; 32 X являє собою C-R або N; 33 Y являє собою C-R або N; 32 33 R та R є однаковими або різними та кожний являє собою: H, ціано-, нітрогрупу, галоген, C3-10 вуглецеву циклічну групу, необов'язково заміщену 1-5 замісниками, вибраними з групи A, гетероциклічну групу, необов'язково заміщену 1-5 замісниками, вибраними з групи A, C110алкілу, необов'язково заміщеного 1-3 замісниками, вибраними з галогену або групи B, a7 -OR , a7 -SR , a7 a8 -NR R , a7 a9 -NR COR , a10 -COOR або a10 a11 -N=CH-NR R ; a7 a8 R та R є однаковими або різними та кожний вибраний з H, групи B або C 1-10алкілу, необов'язково заміщеного 1-3 замісниками, вибраними з галогену та групи B; a9 R являє собою C1-4алкіл; та a10 a11 R та R є однаковими або різними та кожний являє собою H або C1-4алкіл, що полягає у введенні сполуки формули (I) або її фармацевтично прийнятної солі та ритонавіру або його фармацевтично прийнятної солі пацієнтові, який цього потребує. В одному варіанті здійснення винахід передбачає спосіб покращення фармакокінетики сполуки формули (I), що полягає у введенні пацієнтові, що потребує сполуки або її фармацевтично прийнятної солі, ефективної кількості ритонавіру або його фармацевтично прийнятної солі. В одному варіанті здійснення винахід передбачає спосіб підвищення в крові рівня сполуки формули (I) у пацієнта, якого лікують сполукою або її фармацевтично прийнятною сіллю, що полягає у введенні пацієнтові, що потребує сполуки або її фармацевтично прийнятної солі, ефективної кількості ритонавіру або його фармацевтично прийнятної солі. В одному варіанті здійснення винахід передбачає спосіб інгібування інтегрази ВІЛ у пацієнта, що потребує даного лікування, що полягає у введенні сполуки формули (I) або її фармацевтично прийнятної солі та ефективної кількості ритонавіру або його фармацевтично прийнятної солі. В одному варіанті здійснення винахід передбачає спосіб підвищення біодоступності сполуки 1 у пацієнта. Спосіб полягає у введенні пацієнтові терапевтично ефективної кількості сполуки 1 з їжею. Підвищення біодоступності сполуки можна спостерігати за підвищенням максимальної концентрації в плазмі або збільшенням площі під кривою залежності концентрації плазми від часу (AUC) у порівнянні із площею, що була б, якби сполуку вводили без їжі. В одному варіанті здійснення винахід передбачає спосіб підвищення рівня всмоктування сполуки 1 у пацієнта, що полягає у введенні пацієнтові терапевтично ефективної кількості сполуки 1 із їжею. Всмоктування сполуки можна виміряти за концентрацією, що досягається в кровотоці після введення сполуки. Підвищення рівня всмоктування можна спостерігати за підвищенням максимальної концентрації в плазмі або за підвищенням площі під кривою залежності концентрації плазми від часу (AUC) у порівнянні із площею, що була б, якби сполуку вводили без їжі. В одному варіанті здійснення винахід передбачає спосіб інгібування активності інтегрази 3 UA 101141 C2 5 10 15 20 25 30 35 40 45 50 55 60 ретровірусу у пацієнта, що полягає у введенні пацієнтові терапевтично ефективної кількості сполуки 1 з їжею. В одному варіанті здійснення винахід передбачає спосіб лікування або профілактики ретровірусної інфекції у пацієнта, що полягає у введенні пацієнтові терапевтично ефективної кількості сполуки 1 з їжею. В одному варіанті здійснення винахід передбачає набір, що включає: (1) фармацевтичну композицію, що містить сполуку 1 або її фармацевтично прийнятну сіль та фармацевтично прийнятний носій; (2) інформацію, що стосується приписань, та (3) контейнер. Інформація, що стосується приписань, включає попередження або інструкцію для пацієнта, що відноситься до застосування сполуки 1 з їжею. В іншому варіанті здійснення винахід передбачає набір, що включає інгібітор інтегрази, такий як сполука 1, інформацію, що стосується приписань, та контейнер, причому інформація, що стосується приписань, включає інформацію, що відноситься до введення сполуки з метою покращення її біодоступності. В одному варіанті здійснення винахід передбачає спосіб покращення фармакокінетики інгібітора інтегрази, такого як сполука 1, шляхом введення пацієнтові інгібітора інтегрази з ритонавіром або його фармацевтично прийнятної солі та з їжею. В одному варіанті здійснення винахід передбачає спосіб підвищення біодоступності сполуки 1 у пацієнта, що полягає у введенні пацієнтові терапевтично ефективної кількості сполуки 1 з ритонавіром та з їжею. В одному варіанті здійснення винахід передбачає спосіб підвищення рівня всмоктування сполуки 1 у пацієнта, що полягає у введенні пацієнтові терапевтично ефективної кількості сполуки 1 з ритонавіром та з їжею. В одному варіанті здійснення винахід передбачає спосіб інгібування активності ретровірусної інтегрази у пацієнта, що полягає у введенні пацієнтові терапевтично ефективної кількості сполуки 1 з ритонавіром та з їжею. В одному варіанті здійснення винахід передбачає спосіб лікування або профілактики ретровірусної інфекції у пацієнта, що полягає у введенні пацієнтові терапевтично ефективної кількості сполуки 1 з ритонавіром та з їжею. В одному варіанті здійснення винахід передбачає застосування ритонавіру або його фармацевтично прийнятної солі для виготовлення лікарського препарату для покращення фармакокінетики інгібітора інтегрази ВІЛ (наприклад, сполука формули (I)) або його фармацевтично прийнятної солі у пацієнта. В одному варіанті здійснення винахід передбачає застосування ритонавіру або його фармацевтично прийнятної солі та сполуки формули (I) або її фармацевтично прийнятної солі для виготовлення лікарського препарату для інгібування інтегрази ВІЛ у пацієнта. В одному варіанті здійснення винахід передбачає застосування сполуки 6-(3-хлор-2фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3карбонової кислоти або її фармацевтично прийнятної солі для виготовлення лікарського препарату для підвищення біодоступності сполуки, що полягає у введенні пацієнтові терапевтично ефективної кількості сполуки або її фармацевтично прийнятної солі, що призначено для введення з їжею. В одному варіанті здійснення винахід передбачає застосування сполуки 6-(3-хлор-2фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3карбонової кислоти або її фармацевтично прийнятної солі для виготовлення лікарського препарату для підвищення всмоктування сполуки у пацієнта, що полягає у введенні пацієнтові терапевтично ефективної кількості сполуки або її фармацевтично прийнятної солі, що призначено для введення з їжею. В одному варіанті здійснення винахід передбачає застосування сполуки 6-(3-хлор-2фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3карбонової кислоти або її фармацевтично прийнятної солі для виготовлення лікарського препарату для інгібування активності ретровірусної інтегрази у пацієнта, що полягає у введенні пацієнтові терапевтично ефективної кількості сполуки або її фармацевтично прийнятної солі, що призначено для введення з їжею. В одному варіанті здійснення винахід передбачає застосування сполуки 6-(3-хлор-2фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3карбонової кислоти або її фармацевтично прийнятної солі для виготовлення лікарського препарату для лікування або профілактики ретровірусної інфекції у пацієнта, що полягає у введенні пацієнтові терапевтично ефективної кількості сполуки або її фармацевтично прийнятної солі, що призначено для введення з їжею. 4 UA 101141 C2 5 10 15 20 25 30 35 40 45 50 55 60 В одному варіанті здійснення винахід передбачає набір, що включає: (1) фармацевтичну композицію, що містить 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7метокси-4-оксо-1,4-дигідрохінолін-3-карбонову кислоту або її фармацевтично прийнятну сіль та фармацевтично прийнятний носій; (2) інформацію, що стосується приписань, та (3) контейнер, причому інформація, що стосується приписань, включає рекомендацію щодо введення 6-(3хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3карбонової кислоти або її фармацевтично прийнятної солі з їжею. В одному варіанті здійснення набір може необов'язково включати ритонавір або його фармацевтично прийнятну сіль. В одному варіанті здійснення винахід передбачає застосування сполуки 6-(3-хлор-2фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3карбонової кислоти або її фармацевтично прийнятної солі для виготовлення лікарського препарату для підвищення біодоступності сполуки 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3-карбонової кислоти у пацієнта при введенні лікарського препарату з їжею, призначеного для введення з ритонавіром або його фармацевтично прийнятною сіллю. В одному варіанті здійснення винахід передбачає застосування сполуки 6-(3-хлор-2фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3карбонової кислоти або її фармацевтично прийнятної солі для виготовлення лікарського препарату для підвищення всмоктування сполуки 6-(3-хлор-2-фторбензил)-1-[(2,S)-1-гідрокси-3метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3-карбонової кислоти у пацієнта при введенні лікарського препарату з їжею, призначеного для введення з ритонавіром або його фармацевтично прийнятною сіллю. В одному варіанті здійснення винахід передбачає застосування сполуки 6-(3-хлор-2фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3карбонової кислоти або її фармацевтично прийнятної солі для виготовлення лікарського препарату для інгібування активності ретровірусної інтегрази у пацієнта при введенні лікарського препарату з їжею, призначеного для введення з ритонавіром або його фармацевтично прийнятною сіллю. В одному варіанті здійснення винахід передбачає застосування сполуки 6-(3-хлор-2фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3карбонової кислоти або її фармацевтично прийнятної солі для виготовлення лікарського препарату для лікування або профілактики ретровірусної інфекції у пацієнта при введенні лікарського препарату з їжею, призначеного для введення з ритонавіром або його фармацевтично прийнятною сіллю. В одному варіанті здійснення винахід передбачає фармацевтичну композицію, що включає ритонавір або його фармацевтично прийнятну сіль, для покращення фармакокінетики інгібітора інтегрази ВІЛ у пацієнта. В одному варіанті здійснення винахід передбачає фармацевтичну композицію, що включає 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4дигідрохінолін-3-карбонову кислоту або її фармацевтично прийнятну сіль, для підвищення біодоступності сполуки, що призначено для введення з їжею. В одному варіанті здійснення винахід передбачає фармацевтичну композицію, що включає 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4дигідрохінолін-3-карбонову кислоту або її фармацевтично прийнятну сіль, для підвищення всмоктування сполуки в пацієнта, що призначено для введення з їжею. В одному варіанті здійснення винахід передбачає фармацевтичну композицію, що включає 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4дигідрохінолін-3-карбонову кислоту або її фармацевтично прийнятну сіль для інгібування активності ретровірусної інтегрази у пацієнта, що призначено для введення з їжею. В одному варіанті здійснення винахід передбачає фармацевтичну композицію, що включає 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4дигідрохінолін-3-карбонову кислоту або її фармацевтично прийнятну сіль, для лікування або профілактики ретровірусної інфекції у пацієнта, що призначено для введення з їжею. В одному варіанті здійснення винахід передбачає антиретровірусний агент, що включає ритонавір або його фармацевтично прийнятну сіль, для покращення фармакокінетики інгібітора інтегрази ВІЛ у пацієнта. В одному варіанті здійснення винахід передбачає композицію антиретровірусних агентів, що включає 6-(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4дигідрохінолін-3-карбонову кислоту або її фармацевтично прийнятну сіль, для підвищення біодоступності сполуки, що призначено для введення з їжею. 5 UA 101141 C2 5 10 15 20 25 30 35 40 45 50 55 60 В одному варіанті здійснення винахід передбачає антиретровірусний агент, що включає 6(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4дигідрохінолін-3-карбонову кислоту або її фармацевтично прийнятну сіль, для підвищення всмоктування сполуки у пацієнта, що призначено для введення з їжею. В одному варіанті здійснення винахід передбачає антиретровірусний агент, що включає 6(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4дигідрохінолін-3-карбонову кислоту або її фармацевтично прийнятну сіль, для інгібування активності ретровірусної інтегрази у пацієнта, що призначено для введення з їжею. В одному варіанті здійснення винахід передбачає антиретровірусний агент, що включає 6(3-хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4дигідрохінолін-3-карбонову кислоту або її фармацевтично прийнятну сіль, для лікування або профілактики ретровірусної інфекції у пацієнта, що призначено для введення з їжею. В одному варіанті здійснення винахід передбачає застосування інгібітора інтегрази або його фармацевтично прийнятної солі для виготовлення лікарського препарату для перорального введення з їжею для досягнення підвищеної біодоступності інгібітора інтегрази або його фармацевтично прийнятної солі при лікуванні чутливого до інтегрази стану (наприклад, ретровірусної інфекції, такої як ВІЛ-інфекція або СНІД). В одному варіанті здійснення винахід передбачає застосування інгібітора інтегрази (наприклад, сполуки формули (I), такої як Сполука 1) або його фармацевтично прийнятної солі для виготовлення лікарського препарату для перорального введення з їжею з метою досягнення підвищеного всмоктування інгібітора інтегрази або його фармацевтично прийнятної солі при лікуванні стану, чутливого до інтегрази (наприклад, ретровірусної інфекції, такої як ВІЛінфекція або СНІД). В одному варіанті здійснення винахід передбачає застосування інгібітора інтегрази (наприклад сполуки Формули (I), такої як Сполука 1) або його фармацевтично прийнятної солі для виготовлення лікарського препарату для введення з ритонавіром або його фармацевтично прийнятною сіллю для лікування стану, чутливого до інтегрази (наприклад, ретровірусної інфекції, такої як ВІЛ-інфекція або СНІД). В одному варіанті здійснення винахід передбачає застосування ритонавіру або його фармацевтично прийнятної солі для виготовлення лікарського препарату для введення з інгібітором інтегрази (наприклад, сполукою Формули (I), такою як Сполука 1) або його фармацевтично прийнятною сіллю для лікування стану, чутливого до інтегрази (наприклад, ретровірусної інфекції, такої як ВІЛ-інфекція або СНІД). В одному варіанті здійснення винахід передбачає застосування інгібітора інтегрази (наприклад сполуки Формули (I), такої як Сполука 1) або його фармацевтично прийнятної солі та ритонавіру або його фармацевтично прийнятної солі для виготовлення лікарського препарату для лікування стану, чутливого до інтегрази (наприклад, ретровірусної інфекції, такої як ВІЛ-інфекція або СНІД). В одному варіанті здійснення винахід передбачає застосування ритонавіру або його фармацевтично прийнятної солі для виготовлення лікарського препарату для введення з інгібітором інтегрази (наприклад, сполукою Формули (I), такою як Сполука 1) або його фармацевтично прийнятною сіллю для лікування стану, чутливого до інтегрази (наприклад, ретровірусної інфекції, такої як ВІЛ-інфекція або СНІД). В одному варіанті здійснення винахід передбачає застосування інгібітора інтегрази (наприклад, сполуки Формули (I), такої як Сполука 1) або його фармацевтично прийнятної солі для виготовлення лікарського препарату для введення з ритонавіром або його фармацевтично прийнятною сіллю та для введення з їжею для лікування стану, чутливого до інтегрази (наприклад, ретровірусної інфекції, такої як ВІЛ-інфекція або СНІД). В одному варіанті здійснення винахід передбачає застосування ритонавіру або його фармацевтично прийнятної солі для виготовлення лікарського препарату для введення з інгібітором інтегрази (наприклад, сполукою Формули (I), такою як Сполука 1) або його фармацевтично прийнятною сіллю та для застосування з їжею для лікування стану, чутливого до інтегрази (наприклад, ретровірусної інфекції, такої як ВІЛ-інфекція або СНІД). Дані та інші переваги винаходу, а також додаткові винахідницькі ознаки будуть очевидні з опису винаходу, представленого в даному контексті. Короткий опис креслень На фіг. 1 представлений графік залежності концентрації в плазмі від часу для Сполуки 1 у вигляді монокомпоненту та у комбінації з ритонавіром. На фіг. 2 представлений графік, побудований на лінійній шкалі концентрації в плазмі 6-(3хлор-2-фторбензил)-1-[(2S)-1-гідрокси-3-метилбутан-2-іл]-7-метокси-4-оксо-1,4-дигідрохінолін-3 6 UA 101141 C2 5 карбонової кислоти (Сполука 1) після її введення в умовах натще та після прийому їжі. Фіг. 3 ілюструє дані, отримані в Прикладі 3. Фіг. 4 ілюструє дані, отримані в Прикладі 3. Фіг. 5 ілюструє дані, отримані в Прикладі 3. Детальний опис винаходу Ефекти ритонавіру Сполуки формули (I) являють собою інгібітори інтегрази ВІЛ. Спеціальна група сполук формули (I) являє собою сполуки формули (II): 10 15 20 25 30 35 40 де, 4 6 R та R є однаковими або різними та кожен вибраний із групи A; 5 R вибрано з H або групи A; 4 5 або R та R спільно формують цикл, що зливається з бензольним циклом, з яким вони зв'язані; m означає 0-3; та 1 31 32 33 кожен з R , R , R та R визначає те ж саме, що у формулі (I); 6 за умови, коли m означає 2 або 3, R кожного m необов'язково є однаковими або різними. Краща сполука формули (I) являє собою Сполуку 1: Сполуки формули (I) описані в публікації патентної заявки США № 2005/0239819, що у такий спосіб включена в даному контексті у всій своїй повноті. Сполуку 1 можна знайти на стор. 76 у прикладах 4-32 даного документу. Способи одержання та застосування даної та інших сполук формул (I) та (II) також описані в даному документі. Відповідно до одного варіанту здійснення винахід розкриває спосіб покращення фармакокінетики лікарського препарату (або його фармацевтично прийнятної солі) шляхом введення ритонавіру або його фармацевтично прийнятної солі разом з лікарським препаратом. Переважно, коли лікарський препарат являє собою інгібітор інтегрази ВІЛ. Крім того, переважно, коли лікарський препарат метаболізується цитохром P450 монооксигеназою. При введенні два агенти можуть бути отримані у вигляді роздільних композицій, які вводять у той самий або різний час (наприклад, одночасно, безперервно або послідовно), або вони можуть бути отримані та введені у вигляді однієї композиції. Деякі інгібітори протеази ВІЛ, які метаболізуються цитохром P450 монооксигеназою та які було б сприятливо вводити з ритонавіром, описані в патентах США №№ 6037157 та 6703403, які в такий спосіб включені в даному контексті у вигляді посилання у всій своїй повноті. Дані патенти описують одержання та схеми дозування, які можна використати з ритонавіром. Одержання та схеми дозування інгібіторів інтегрази ВІЛ можна знайти в публікації патентної заявки США № 2004/0167124, що у такий спосіб включена в даному контексті у вигляді 7 UA 101141 C2 5 10 15 20 25 30 35 40 45 50 55 60 посилання у всій своїй повноті. Вищевказані схеми можна використати у винаході, описаному в даному контексті. У випадку комбінованої терапії, наприклад, приблизно від 20 мг до приблизно 500 мг сполуки формули (I) або (II), такої як Сполука 1, можна ввести із приблизно від 10 мг до приблизно 1200 мг ритонавіру/день. В одному спеціальному варіанті здійснення винаходу приблизно від 20 мг до приблизно 500 мг сполуки формули (I) або (II), такої як Сполука 1, можна ввести із приблизно від 10 мг до приблизно 600 мг ритонавіру/день. У варіанті здійснення винаходу прийнятна доза сполуки 1 становить приблизно 20 мг, 50 мг, 75 мг, 85 мг, 100 мг, 125 мг, 150 мг, 175 мг або 200 мг, більш спеціально приблизно 85 мг, приблизно 125 мг або приблизно 150 мг. У варіанті здійснення винаходу прийнятна доза ритонавіру становить від приблизно 20 мг до приблизно 200 мг, зокрема, від приблизно 50 мг до приблизно 125 мг, більш докладно приблизно 100 мг. Однак можуть бути ефективними більш високі та більш низькі дози кожного агенту. В одному варіанті здійснення добового введення кращі кількості сполуки 1 та ритонавіру являють собою дози, які можуть досягати концентрації сполуки 1 у крові, що підтримують IC 95 (наприклад, з поправкою на зв'язування білку in vitro IC95 100 нМ) протягом 24 годин. В одному варіанті здійснення добового введення кращі кількості сполуки 1 та ритонавіру являють собою дози, які можуть досягати концентрації сполуки 1 у крові, що підтримують більше EC90 (наприклад приблизно 170 нг/мл на моделі Emax) протягом 24 годин. В одному варіанті здійснення кращі кількості сполуки 1 являють собою дози, які одержують пацієнти, які можуть досягати такого рівня, що середнє зниження рівнів РНК ВІЛ як активність у відношенні ВІЛ становить більше ніж 1,5 log10 копій/мл, переважно 2,0 log10. У спеціальному варіанті здійснення винахід передбачає спосіб покращення фармакокінетики інгібітора інтегрази ВІЛ (або його фармацевтично прийнятної солі), що, зокрема, метаболізується цитохром P450 монооксигеназою, більш докладно, ізоформою CYP 3 A4, у пацієнта, який потребує даного лікування, за допомогою введення або спільного введення ритонавіру або його фармацевтично прийнятної солі з інгібітором інтегрази. Дану комбінацію ритонавіру або його фармацевтично прийнятної солі та інгібітора інтегрази ВІЛ або його фармацевтично прийнятної солі, що метаболізується цитохром P450 монооксигеназою, використовують для пригнічення, лікування або профілактики ВІЛ-інфекції або СНІДу (синдрому набутого імундефіциту) у пацієнта. Пацієнти включають будь-яких живих істот, зокрема, ссавців (наприклад, людину). Один аспект винаходу представляє застосування ефективної кількості ритонавіру для покращення фармакокінетики інгібітора інтегрази ВІЛ. Ефективна кількість ритонавіру, тобто кількість, що вимагається для посилення дії інгібітора інтегрази ВІЛ, являє собою кількість, необхідну для покращення фармакокінетичного профілю інгібітора інтегрази ВІЛ у порівнянні з його профілем при використанні у вигляді монотерапії. Інгібітор має більш ефективний фармакокінетичний профіль, ніж був би без додавання ритонавіру. Кількість ритонавіру, що використовують для посилення дії інгібітора інтегрази, може бути субтерапевтичною (наприклад, дози нижче кількості ритонавіру, звичайно використовувані для терапевтичного лікування ВІЛ-інфекції у пацієнта). Посилююча доза ритонавіру є субтерапевтичною для лікування ВІЛ-інфекції, але досить високою для того, щоб викликати модуляцію метаболізму сполук формул (I) та (II), таким чином, що їхній вплив на пацієнта підсилюється за допомогою підвищеної біодоступності, підвищених рівнів у крові, збільшеного напівперіоду існування, збільшеного періоду часу до досягнення максимальної концентрації в плазмі, підвищеного/прискореного інгібування інтегрази ВІЛ та/або зниженого системного кліренсу. Сполука 1 являє собою інгібітор інтегрази ВІЛ, що метаболізується цитохром P450 монооксигеназою, особливо ізоформою CYP 3A. У цей час виявлено, що ритонавір можна використати для покращення фармакокінетики сполук формули (I), а також інших інгібіторів інтегрази ВІЛ. Ритонавір особливо ефективний у плані посилення ефектів інгібіторів інтегрази, які метаболізуються цитохром P450 монооксигеназою (наприклад, ізоформою CYP 3A). Ступінь даного посилення виявляється неочікувано істотним. Ритонавір може обмежувати ефект першого проходження даних сполук. Ритонавір може також обмежувати ефекти вторинного проходження (системний або печіночний метаболізм/кліренс) даних сполук. Відповідно до способів, що відповідають винаходу, інгібітор інтегрази (або сполука формули (I)) або його фармацевтично прийнятну сіль можна також вводити з одним або двома іншими агентами, які використовують для лікування вірусних інфекцій, такими як ставудин, емтрицитабін, тенофовір, абакавір, ламівудин, зидовудин, діданозин, залцитабін, фосфазид, ефавіренц, невирапін, делавірдин, типранавір, саквинавір, індинавір, атазанавір, нелфінавір, ампренавір, сампренавір, фосампренавір, лопінавір, ритонавір, енфувіртид, фозивудин тидоксил, аловудин, декселвуцитабін, априцитабін, амдоксовір, елвуцитабін (ACH126443), 8 UA 101141 C2 5 10 15 20 25 30 35 40 45 50 55 60 рацивір (рацемічний FTC, PSI-5004), MIV-210, KP-1461, фосалвудин тидоксил (HDP 99.0003), AVX756, діоксалан тимін (DOT), TMC-254072, INK-20, 4'-Ed4T, TMC-125 (етравірин), каправірин, TMC-278 (рилпівірин), GW-695634, каланолід A, BILR 355 BS та VRX 840773 або їх фармацевтично прийнятні солі. Хоча вищенаведений перелік включає торговельні назви ряду сполук, слід мати на увазі, що посилання на торговельну назву включає також лежачий в основі активний хімічний агент незалежно від джерела. Набори, що відповідають винаходу, можуть також необов'язково далі включати один або більше агентів, вибраних з вищенаведеного переліку. В одному варіанті здійснення винаходу способи, що відповідають винаходу, далі включають введення одного або більше інших агентів, вибраних з тенофовіру DF (TDF), емтрицитабіну (FTC), зидовудину (AZT), діданозину (ddl), ставудину (d4T), абакавіру (ABC), атазанавіру (ATV), лопінавіру (LPV), сакцинавіру (SQV), типранавіру (TPV), фосампренавіру (FosAPV) та ефавіренцу (EFV). Набори, що відповідають винаходу, можуть також далі включати один або більше інших агентів з вищенаведеного переліку. Даний винахід також представляє набір, що включає (i) інгібітор інтегрази (наприклад, Сполуку 1) або його фармацевтично прийнятну сіль, (ii) ритонавір або його фармацевтично прийнятну сіль, (iii) інформацію, що відноситься до приписань, та (iii) один або більше контейнерів. Інформація, що відноситься до приписань, може представляти інформацію, що відноситься до приписань, що відповідає способам, представленим у винаході, та/або як іншим способом обговорюється в даному контексті. У варіанті здійснення винаходу інформація, що відноситься до приписань, включає введення інгібітора інтегрази або його фармацевтично прийнятної солі з ритонавіром для покращення фармакокінетики інгібітора інтегрази. Ефекти ритонавіру на біодоступність репрезентативного інгібітора інтегрази тепер будуть проілюстровані наступними необмежуючими прикладами. Приклад 1. Підтримуючі ефекти ритонавіру на фармакокінетику сполуки 1 Визначають ефекти спільного введення 100 мг ритонавіру (RTV) на стабілізовану фармакокінетику сполуки 1. Визначають також фармакокінетику сполуки 1 при одноразовій дозі та багаторазовій дозі. Крім того, визначають безпека багаторазової дози сполуки 1 при введенні у вигляді монотерапії та з RTV. Способи Дослідження являє собою фармакокінетичне дослідження на 12 пацієнтах з відкритими етикетками, фіксованою послідовністю, перекриванням. Пацієнти являють собою здорових чоловіків та невагітних нелактуючих жінок у віці від 18 до 45 років включно. Тривалість дослідження становить 20 днів з періодом 1 від 1 до 10 дня та періодом 2 від 11 до 20 дня. Наступний контакт здійснюють у день 27. Сполуку 1 (100 мг) та RTV (100 мг) вводять два рази на день перорально відразу після прийняття їжі. Сполуку 1 (100 мг) вводять два рази на день перорально відразу після прийняття їжі. Критерії оцінки Фармакокінетика: Для Сполуки 1 (і метаболітів при можливості) у плазмі розраховують наступні параметри: Cmax, Tmax, Clast, Tlast, Ctau, λz, AUC0-last, AUCinf, %AUCexp, AUCtau, T1/2, VZ/F та CL/F. Безпека: Безпеку визначають за оцінкою клінічних лабораторних тестів на вихідному рівні та у різних точках часу протягом дослідження, по періодичних фізичних дослідженнях та документації несприятливих подій протягом дослідження. Статистичні методи Фармакокінетика: Підсумки дослідження фармакокінетики сполуки 1 та RTV підводять, використовуючи описову статистику. Крім того, параметричний аналіз (нормальна теорія) варіанси (ANOVA) з використанням моделі змішаних ефектів, що підходить для типу, що перекривається, відповідає натуральному логарифмічному перетворенню фармакокінетичних параметрів сполуки 1 (AUC та Cmax). Порівняння фармакокінетики сполуки 1 при одноразовій дозі та багаторазовій дозі та фармакокінетики сполуки 1 при багаторазовій дозі із введенням і без введення RTV проводять, використовуючи 90% довірчі інтервали для співвідношення геометричних середніх для кожної пари варіантів лікування. Безпека: У даному фармакокінетичному дослідженні не роблять ніякого статистичного висновку відносно даних по безпеці. Результати Фармакокінетичні результати: Середні (%CV) фармакокінетичні параметри в плазмі сполуки 1 та RTV після одноразового перорального введення дози 1 (100 мг, день 1) та багаторазового перорального введення дози сполуки 1 (100 мг два рази в день, день 10) під час відсутності 9 UA 101141 C2 RTV або в присутності RTV, що вводять перорально у вигляді одноразової дози (100 мг, день 11) або багаторазових доз (100 мг два рази в день, день 20) є наступними. Параметри Сполука 1 a AUC (нг. год./мл) Cmax (нг/мл) Ctau (нг/мл) с T1/2 (год.) Ритонавір d AUC (нг. год./мл) Cmax (нг/мл) Ctau (нг/мл) с T1/2 (год.) Сполука 1 як монотерапія (N=12) День1 День 10 Сполука 1 + RTV (N=12) День 11 День 20 908,1 (28,3) 200,1 (30,4) 19,2 (52,5) 3,1 (2,2, 4,8) 719,3 (26,2) 164,1 (28,8) 12,4 (63,7) 3,5 (2,2 4,1) 6167,3 (29,1) 795,3 (38,4) 543,3 (30,4) 18,2 (9,0, 42,6) 14302,1 (23,7) 1826,4 (26,4) 1035,6 (32,0) 9,5 (5,9, 78,2) Незастосовно Незастосовно Незастосовно Незастосовно Незастосовно Незастосовно Незастосовно Незастосовно 4979,4 (57,8) 616,3 (53,5) 219,8 (61,8) 5,1 (2,2 8,3) 9402,5 (46,9) 1686,5 (46,5) 544,8 (44,3) 4,8 (4,3 6,9) 5 a Для Сполуки 1, AUC являє собою AUCinf у день 1 та AUCtau у дні 10, 11 та 20. b Ctau являє собою концентрацію наприкінці інтервалу дозування для днів 1, 10, 11 та 20. c Середнє (min, max) d Для ритонавіру, AUC являє собою AUCinf у день 11 та AUCtau у день 20. 10 Під час введення сполуки 1 у вигляді монотерапії середній стабілізований системний вплив (AUCtau) Сполуки 1 (День 10) становить приблизно на 20% нижче в порівнянні із середнім впливом (AUCinf) після одноразового введення (День 1), вказуючи на автоіндукцію метаболізму сполуки 1. Спільне введення з RTV приводить у результаті до мережного інгібування метаболізму Сполуки 1, що доводять як по стабілізованих впливах, що перевищують прогнозовані, так і по відносно довгому середньому напівперіоді існування до елімінації (9,5 відносно 3,5 годин у стабілізованому стані). Підвищення ступеня впливу Сполуки 1 після спільного введення RTV, імовірно, обумовлено комбінацією підвищеної пероральної біодоступності внаслідок зниженого метаболізму при першому проходженні з компонентом зниженого системного кліренсу, як показано шляхом зміни спостережуваного T 1/2. У цілому дані результати підтримують застосування RTV (наприклад, RTV у низькій дозі) як фармакокінетичного підсилювача сполуки 1, щоб зробити можливим, наприклад, досягнення більш високих мінімальних концентрацій та менш частих інтервалів дозування. Результати по безпеці: Виникаючі при лікуванні несприятливі події (AEs) описані в 4 з 12 пацієнтів (33%, п'ять подій) під час застосування сполуки 1 у вигляді монотерапії та в 7 з 12 пацієнтів (58%, 44 події) при застосуванні сполуки 1+RTV. Ніякі окремі AE не описані в більше ніж одного пацієнта при застосуванні сполуки 1 у вигляді монотерапії. При застосуванні сполуки 1+RTV найбільш частим описуваним AE, що виникає при лікуванні, є нудота, що мають 4 пацієнти (33%). Більшість AEs, що виникають при лікуванні, відносяться до подій низької важкості (Ступінь 1) та усуваються без лікування. Виникаючі при лікуванні AEs, розглянуті автором як пов'язані з досліджуваними лікарськими препаратами, описані у 2 з 12 пацієнтів (17%, дві події) після введення сполуки 1 у вигляді монотерапії та у 5 з 12 пацієнтів (42%, 21 подія) після введення сполуки 1+RTV. Ніяких пов'язаних з лікуванням AE не описане в більш ніж одного пацієнта після введення сполуки 1 у вигляді монотерапії. Пов'язані з лікуванням AEs, описані в більш ніж одного пацієнта після введення сполуки 1+RTV, являють собою нудоту (три пацієнти), блювоту (два пацієнти), головний біль (два пацієнти) та сверблячку (два пацієнти). Ніяких серйозних несприятливих подій не відбулося, жоден пацієнт не виключений з випробування внаслідок несприятливої події та ні однієї вагітності не трапилося під час даного дослідження. Жоден з пацієнтів не припинив приймати досліджувані лікарські препарати внаслідок клінічної лабораторної патології та жодна клінічна лабораторна патологія не описана як AE. Висновки Спільне введення з RTV приводить у результаті до мережного інгібування метаболізму сполуки 1 та в істотній мірі підвищеним системним впливам, зокрема, мінімальним концентраціям. Дані підтримують застосування RTV у низькій дозі як фармакокінетичного підсилювача сполуки 1. Пероральне введення сполуки 1 (100 мг два рази в день) у період до 20 добре переноситься пацієнтами, що беруть участь у дослідженні. Після введення сполуки 1 у вигляді монотерапії всі несприятливі події є слабкими та тимчасовими. Одночасне пероральне введення сполуки 1 (100 мг два рази на день) та RTV (100 мг два рази на день) у період до 10 15 20 25 30 35 40 45 10 UA 101141 C2 5 10 15 20 25 30 35 40 45 50 55 60 днів в основному добре переноситься пацієнтами, що беруть участь у дослідженні. Після введення сполуки 1+RTV більшість описаних несприятливих подій є слабкими та тимчасовими та в основному відповідними описаним для RTV. Приклад 2. Безпека, фармакокінетика та антивірусна активність сполуки 1 після перорального введення пацієнтам, інфікованим ВІЛ-1 Досліджують безпеку, фармакокінетику та антивірусну активність сполуки 1, що вводять перорально у вигляді 10 послідовних добових доз (два рази на день для груп 1, 2 та 4; один раз на день для груп 3 та 5) пацієнтам, хронічно інфікованим ВІЛ-1, що у цей час не одержують антиретровірусного лікування. Досліджують також фармакокінетику та фармакодинаміку сполуки 1. Способи Дослідження являють собою проведені подвійним сліпим методом, рандомізовані, з використанням контрольної групи із плацебо, з послідовними групами, ранжирувані за дозами дослідження фази 1/2 лікування сполукою 1 нелікованих або лікованих антиретровірусними препаратами ВІЛ-інфікованих дорослих, які в цей час не проходять антиретровірусне лікування. При відборі пацієнти повинні мати навантаження РНК ВІЛ-1 у плазмі від > 10000 до 200 клітин/мм . П'ять послідовних груп з 8 специфічних пацієнтів (6 пацієнтів - на активному препараті та 2 на плацебо) лікують протягом 10 послідовних днів, починаючи із дня 1 досліджуваним лікарським препаратом або плацебо в умовах після прийому їжі. Безпеку, переносимість, фармакокінетику та ефективність контролюють до дня 21 після введення дози. Активними варіантами лікування в 5 групах є наступні: Група 1, 400 мг сполуки 1 два рази на день; Група 2, 800 мг сполуки 1 два рази на день; Група 3, 800 мг сполуки 1 один раз на день; Група 4, 200 мг сполуки 1 два рази на день; Група 5, 50 мг Сполуки 1+100 мг RTV (RTV), кожен препарат один раз на день. Група плацебо в Групі 5 також одержує 100 мг RTV. У групу включають вісім пацієнтів, 2 - що одержують плацебо та 6 - активний препарат (рандомізовані: 48, вводять дозу 40). Безпеку оцінюють для 8 пацієнтів/групу (2, що одержують плацебо, 6 - активний препарат), усього 40 пацієнтів. Фармакокінетику оцінюють у 28 пацієнтів у день 1, у 30 пацієнтів у день 10. Фармакокінетику оцінюють в 30 пацієнтів та РНК ВІЛ-1 визначають в 40 пацієнтів. Ефективність визначають в 8 пацієнтів/групу (2, що одержують плацебо, 6 - активний препарат), усього в 40 пацієнтів. Пацієнти являють собою чоловіків та жінок у віці від 18 до 60 років включно, які хронічно інфіковані ВІЛ-1 та у цей час не проходять антиретровірусне лікування з обумовленим при скринінгу рівнем РНК ВІЛ-1 у плазмі від > 10,000 до 200 клітин/мм . Пацієнти можуть бути такими, що пройшли або не пройшли антиретровірусного лікування, але не одержують антиретровірусні препарати протягом 90 днів до початку експерименту (День 0). Тривалість дослідження становить 21 день, причому 10 днів лікування та 11 днів вивчення віддалених результатів. Таблетки по 50 мг або 200 мг Сполуки 1 вводять перорально в умовах після прийому їжі. Придатне плацебо вводять перорально в умовах після прийому їжі. Тільки в Групі 5 капсули по 100 мг RTV перорально вводять спільно в умовах після прийому їжі. Критерії оцінки Ефективність: Первинна кінцева точка ефективності являє собою максимальне зниження рівня РНК ВІЛ-1 (log10 копій/мл) щодо вихідного рівня до дня 11. Максимальне зниження для окремого пацієнта становить максимальне падіння серед змін щодо вихідного рівня в дні 2-11. Фармакокінетика: Для Сполуки 1 розраховують наступні фармакокінетичні параметри: C max, Tmax, Clast, Tlast, Ctau, λz, AUC0-last, AUCinf, %AUCexp, AUCtau, VZ/F та CL/F. Визначають концентрації сполуки 1 у мононуклеарних клітинах периферичної крові (PBMC) та використовують фармакокінетику Сполуки 1 в PBMC. Безпека: Несприятливі події, життєві показники, електрокардіограму, дані офтальмологічного дослідження та клінічних лабораторних аналізів оцінюють на безпеку. Статистичні методи Ефективність: Попарні порівняння проводять із використанням точного критерію рангової суми Уілкоксона для порівняння групи плацебо (2 пацієнта, кожний з об'єднаних Груп 1-4) та кожного з 5 рівнів доз Сполуки 1 (6 пацієнтів для кожного рівня дози) і між кожною парою серед 5 рівнів доз Сполуки 1.2 пацієнтів, які одержують плацебо + 100 мг RTV, лікують як окрему групу. Значення РНК ВІЛ-1 нижче межі визначення (50 копій/мл) вводять як 49 копій/мл. Фармакокінетика: Фармакокінетичні параметри сполуки 1 у плазмі та мононуклеарних клітинах периферичної крові підсумують, використовуючи описову статистику. Крім того, для кожного рівня дози Сполуки 1 проводять дисперсійний аналіз фармакокінетичних параметрів 11 UA 101141 C2 5 10 15 20 (AUC та Сmax) для дослідження пропорційності доз та стабілізованої фармакокінетики. Мінімальні рівні сполуки 1, що вимірюють в дні 2, 4, 7, 10 та 11 включають відповідним чином у дані аналізи. Безпека: Частку пацієнтів з несприятливими подіями підсумують по групі лікування, класу системного органу та переважним чином по несприятливим подіям, що викликаються лікуванням, пов'язаним з лікуванням несприятливим подіям, важким несприятливим подіям та несприятливим подіям, що приводять до припинення введення досліджуваного лікарського препарату. Несприятливі події кодують, використовуючи Медичний словник по регуляторним активностям (MedDRA®) версія 8.1. Додаткові підсумкові матеріали по несприятливих подіях представлені найвищим ступенем оцінки зв'язку з досліджуваним лікарським препаратом та ефектом на припинення введення досліджуваного препарату, що дається дослідником. Лабораторні результати відображують на вихідній шкалі вимірювань та у показниках ступеня токсичності. Підсумки змін щодо вихідного рівня в кількісних лабораторних тестах підводять при огляді. Результати Ефективність, фармакокінетичні та фармакодинамічні результати Стабілізовані фармакокінетичні параметри (середнє [% CV]) та антивірусну активність сполуки 1 при введенні у вигляді монотерапії (200 мг два рази на день, 400 мг два рази на день, 800 мг два рази на день або 800 мг один раз на день) або 50 мг один раз на день при спільному введенні з 100 мг RTV представляють із наступній таблиці: Сполука 1 Сполука 1 Сполука 1 Сполука 1 Плацебо Плацебо/r 200 мг BID 400 мг BID 800 мг QD 800 мг BID (N=8) (N=2) (N=6) (N=6) (N=6) (N=6) a Максимальне зниження рівня РНК ВІЛ-1 (log10 копій/мл) -0,25 -0,05 -1,48 (0,55) -1,94 (0,52) -0,98 -1,91 (0,60) Середнє (SD) b b b b (0,15) (0,14) (0,37) -,026 -0,05 -1,48 -2,03 -,096 -1,78 Медіана (min, (-0,48, (0,15, (-2,10, (-2,44, (-1,41, (-2,67, max) 0,01) 0,05) 0,87) 1,04) 0,56) 1,27) Пацієнти, що досягають рівня РНК ВІЛ-1 < 50 копій/мл після вихідного рівня e n (%) 0 (0%) 0 (0%) 0 (0%) 0 (0%) 0 (0%) 2(33%) Пацієнти, що досягають рівня РНК ВІЛ-1 < 400 копій/мл після вихідного рівня e n (%) 0 (0%) 0 (0%) 0 (0%) 0 (0%) 0 (0%) 3 (50%) f Стабілізована фармакокінетика сполуки 1 AUCtau (нг. год./мл) 1954,65 2335,30 5512,87 3566,35 (середнє, (46,35) (54,52) (53,59) (36,83) %CV) Cmax (нг/мл) 479,03 606,87 939,92 835,53 (середнє, (42,58) (77,58) (54,31) (48,20) %CV) Ctau (нг/мл) 30,73 48,68 13,62 47,98 (середнє, (39,98) (64,84) (68,64) (32,65) %CV) T1/2 (год.) 2,82 (2,51, 3,08 (2,48, 3,80 (3,02, 2,53 (2,14, (медіана) 4,75) 5,02) 4,60) 3,03) (min, max) Параметр 25 30 Сполука 1 50 мг QD + 100 мг RTV (N=6) -1,99 (0,38) b,d -2,03 (-2,38, -1,54) 0 (0%) 2 (33%) 8843,50 (25,46) 744,65 (20,40) 135,00 (36,55) 8,86 (6,10, 10,91) а Максимальне зниження рівня РНК ВІЛ-1 визначають як максимальне зниження щодо вихідного рівня в log10 копій/мл у період від дня 2 до 11. Значення РНК ВІЛ-1 нижче межі кількісного визначення (50 копій/мл) вводять як 49 копій/мл. 5 рівнів доз Сполуки 1 і групу плацебо + RTV порівнюють із групою плацебо. b р=0,0007 для кожного рівня доз Сполука 1 відносно плацебо на основі парних значень р для безперервних даних по 2-сторонньому точному критерії рангової суми Уілкоксона. с р=0,0152 для групи, що одержує 800 мг Сполуки 1 один раз на день щодо рівня доз 800 мг Сполуки 1 два рази на день та відносно 400 мг Сполуки 1 два рази на день на основі парних значень р для безперервних даних, розрахованих по 2-сторонньому точному критерії рангової суми Уілкоксона. 12 UA 101141 C2 5 10 15 20 25 30 35 40 45 50 55 60 d Рівень доз 50 мг Сполуки 1 + RTV один раз на день відносно плацебо + RTV, p=0,0714; відносно 200 мг два рази на день р=0,1797; відносно 400 мг два рази на день р=0,9372; відносно 800 мг один раз на день р=0,0022; відносно 800 мг два рази на день р=0,8182. e Відсоток базується на всіх рандомізованих та лікованих пацієнтах f 12- та 24-годинні фармакокінетичні зразки, що відповідають закінченню інтервалу введення доз (tau), зібрані в День 10 (введення доз два рази на день) або 11 (введення доз один раз на день) встановлюють номінальний час 12 та 24 години, відповідно. Монотерапія Сполукою 1 істотно знижує рівні РНК ВІЛ-1 при всіх рівнях доз у порівнянні із плацебо (p < 0,0007). Протягом 10 наступних один за одним днів введення дози Сполука 1 400 мг два рази на день, 800 мг два рази на день або 50 мг при введенні спільно з 100 мг RTV один раз на день приводять у результаті до максимальних знижень рівнів РНК ВІЛ-1 щодо вихідного рівня -1,94 ± 0,52, -1,91 ± 0,60 та -1,99 ± 0,38 log10 копій/мл (середнє ± SD(стандартне відхилення)), відповідно. Через 10 днів монотерапії Сполукою 1, у жодного пацієнта не з'являються мутації інтегрази ВІЛ-1, які відповідають мутаціям стійкості до Сполуки 1, що спостерігаються в експериментах по селекції in vitro, або мутаціям, які з'являються при селекції на тлі інших експериментальних інгібіторів інтегрази. Сполука 1 не проявляє пропорційних дозі збільшень у фармакокінетиці з дозами, що підвищуються (200, 400 та 800 мг два рази на день) і не демонструє дозозалежної аутоіндукції метаболізму. Спільне введення сполуки 1 у дозі 50 мг + 100 мг RTV один раз на день приводить у результаті до мережного інгібування CYP3A-опосередкованого метаболізму та високим системним впливам, зокрема, мінімальних концентрацій. Фармакокінетичні результати в PBMC погоджуються з фармакокінетичними даними в плазмі. Зв'язок впливу Сполуки 1 - відповіді ідентифікують із використанням значень Ctau Сполуки 1, що цілком відповідає простій моделі Emax з EC50 14,4 нг/мл і зниженню Emax 2,32 log10 копій/мл щодо вихідного рівня. Передбачуваний інгібуючий коефіцієнт сполуки 1, розрахований як отримане середнє C tau, поділене на скоректоване на зв'язування білку IC50 in vitro 7,17 нг/мл становить 5,9, 6,7 та 18,8 при рівнях доз 400 мг два рази на день, 800 мг два рази на день та 50 мг + RTV один раз на день, відповідно. Мінімальні концентрації Сполуки 1 при даних дозах також перевищують скоректоване на зв'язування білку IC95 in vitro 44,9 нг/мл (100 нМ) для всього інтервалу дозування. Результати по безпеці Викликані лікуванням несприятливі події мають більшість пацієнтів, але головний біль та діарея є єдиними несприятливими подіями, описаними більше ніж в 1 пацієнта в групі. Імовірність викликаних лікуванням несприятливих подій у пацієнтів, які одержують Сполуку 1, близька або нижче, ніж імовірність у групах, що одержують плацебо, і класи несприятливих подій подібні. Ніякі несприятливі події, які автор вважає пов'язаними з досліджуваним лікарським препаратом, не мають більше ніж 2 пацієнти в групі, і тільки 3 викликані лікуванням несприятливі події переважним чином мають місце у 2 пацієнтів у групі: діарея (плацебо), нудота, (плацебо та 200 мг сполуки 1 два рази на день) та втома (200 мг сполуки 1 два рази на день). Всі 40 пацієнтів, які одержують досліджуваний лікарський препарат завершують дослідження. Відсутні порушення доз, припинення або серйозні несприятливі події. П'ять пацієнтів мають викликані лікуванням лабораторні патології ступеня 3 або 4: 2 пацієнти в групі плацебо та по 1 пацієнтові в кожній з наступних груп - плацебо + RTV один раз у день, 400 мг сполуки 1 два рази в день та 50 мг сполуки 1 + RTV один раз на день. Одну з лабораторних патологій ступеня 3 у групі плацебо, високий рівень тригліцеридів, що усувають без лікування, автор розглядає як несприятливу подію, що не пов'язана з досліджуваним лікарським препаратом. 2 пацієнта, що лікували Сполукою 1, які мають лабораторні патології ступеня 3 (підвищений не на голодний шлунок рівень тригліцеридів або підвищений рівень сироваткової амілази), також мають патологічні значення на вихідному рівні. У процесі дослідження не відбувається ніяких клінічно значимих змін у результатах гематологічного аналізу та аналізу сечі, життєвих показниках, масі тіла або електрокардіограмі та очних дослідженнях. Висновки Ефективність: Введення сполуки 1 у дозах 200, 400 або 800 мг два рази на день; 800 мг один раз на день або 50 мг + 100 мг RTV один раз на день не підданих або підданих впливу антивірусних препаратів пацієнтів, інфікованих ВІЛ-1, протягом 10 наступних один за одним днів у вигляді монотерапії істотно знижує рівень РНК ВІЛ-1 у порівнянні із плацебо на всіх рівнях доз. Дози Сполуки 1 400 мг два рази на день, 800 мг два рази на день та 50 мг + 100 мг RTV один раз на день приводять у результаті до середнього зниження щодо вихідного рівня -1,94, -1,91 та -1,99 log10 копій/мл, відповідно. У гені інтегрази якого-небудь пацієнта не визначають ніякі мутації стійкості до Сполуки 1 до дня 21. 13 UA 101141 C2 5 10 15 20 25 30 35 40 45 Фармакокінетика/фармакодинаміка: Сполука 1 проявляє фармакокінетичні властивості, що підтримують дозування два рази в день у вигляді монотерапії або дозування один раз у день у поєднанні з низькою дозою (100 мг) RTV. Зв'язок впливу Сполуки 1 - відповіді ідентифікують із використанням значень Ctau Сполуки 1, що цілком відповідає простій моделі Emax. Передбачуваний інгібуючий коефіцієнт сполуки 1, розрахований з використанням скоректованого на зв'язування білку IC50 in vitro 7,17 нг/мл становить 5,9, 6,7 та 18,8 при рівнях доз 400 мг два рази на день, 800 мг два рази на день та 50 мг + RTV один раз на день, відповідно. Мінімальні концентрації Сполуки 1 при даних дозах також перевищують скоректоване на зв'язування білку IC95 in vitro 44,9 нг/мл (100 нМ) для всього інтервалу дозування. Безпека Сполука 1, як правило, добре переноситься при всіх рівнях дози без порушень доз, припинень або серйозних несприятливих подій. Більшість несприятливих подій є слабкими та усуваються за допомогою лікування. Найпоширенішою викликуваною лікуванням несприятливою подією є головний біль, та найпоширенішою пов'язаною з лікуванням несприятливою подією є нудота. Класи, частота та важкість несприятливих подій та лабораторних патологій близькі між групами, що одержують активний препарат та плацебо. Приклад 3. Ефекти доз ритонавіру на фармакокінетику Сполуки 1 Оцінюють ефекти інтервалу доз ритонавіру (RTV) (20, 50, 100 та 200 мг один раз на день) на фармакокінетику сполуки 1. Оцінюють також ефекти інтервалу доз RTV (20, 50, 100 та 200 мг один раз на день) на активність печіночного цитохрому P450 3A (CYP3A), використовуючи субстрат CYP3 A. Крім того, оцінюють безпеку та переносимість інтервалу доз RTV у комбінації із Сполукою 1. Способи Проводять рандомізоване, з відкритими етикетками, одноцентрове, з використанням багатьох доз дослідження фази 1 на двох групах (24 пацієнта (16 піддаються оцінці) у двох групах; 12 (8 піддаються оцінці) у кожній групі) із приблизно рівним розподілом пацієнтів здорових чоловіків і невагітних нелактуючих жінок у віці від 18 до 45 років включно. Підходящими пацієнтами є чоловіки та невагітні, нелактуючі жінки з коефіцієнтом маси тіла (BMI) 19 < BMI 80 мл/хв. Використовують наступні досліджувані варіанти лікування Сполукою 1/р: Варіант лікування A: Сполука 1, 1 таблетка x 125 мг та RTV 20 мг (80 мг/мл розчину, розведеного для одержання дози 20 мг) QD (обидва лікарських препарати в AM). Варіант лікування B: Сполука 1, 1 таблетка x 125 мг та RTV 50 мг (80 мг/мл розчину, розведеного для одержання дози 50 мг) QD (обидва лікарських препарати в AM). Варіант лікування C: Сполука 1, 1 таблетка x 125 мг та RTV 100 мг (80 мг/мл розчину, розведеного для одержання дози 100 мг) QD (обидва лікарських препарати в AM). Варіант лікування D: Сполука 1, 1 таблетка x 125 мг та RTV 200 мг (80 мг/мл розчину, розведеного для одержання дози 200 мг) QD (обидва лікарських препарати в AM). У кожному варіанті лікування спочатку вводять RTV, з наступним негайним введенням Сполуки 1. Повільне внутрішньовенне введення мідазоламу (MDZ; 1 мг протягом 1 хвилини) як зонд активності CYP3A проводять кожному пацієнтові в 14:00 у день 1 та через 6 годин після введення дози Сполуки 1/р у Дні 11 та 21. Після процедур відбору та оцінок вихідного рівня підходящих пацієнтів рандомізовано розподіляють в одну із двох груп лікування, як описано нижче: 50 14 UA 101141 C2 День 1 2-10 11 12-20 21 Група 2 1 2-10 11 12-20 21 5 10 15 20 25 30 35 Група лікування та/або зонд MDZ Група 1 MDZ A: в АМ A: в АМ MDZ: через 6 год. після дозування С: в АМ С: в АМ MDZ: через 6 год. після дозування MDZ B: в АМ B: в АМ MDZ: через 6 год. після дозування D: в АМ D: в АМ MDZ: через 6 год. після дозування Проведена оцінка PK MDZ Немає Сполука 1, RTV та MDZ Немає Сполука 1, RTV та MDZ MDZ Немає Сполука 1, RTV та MDZ Немає Сполука 1, RTV та MDZ Всі дози Сполуки 1/р вводять відразу після завершення прийому їжі до полудня з 240 мл води. Серійні зразки крові для аналізу концентрацій сполуки 1 та RTV у плазмі збирають у дні 11 та 21 у наступні точки часу після введення дози Сполуки 1/р: 0 (перед введенням дози), 0,5, 1, 1,5, 2, 3, 3,5, 4, 4,5, 5, 5,5, 6, 8, 10, 12, 14, 16, 18 та 24 години після введення дози. Серійні зразки крові для аналізу концентрацій MDZ у плазмі збирають у Дні 1, 11 та 21 у наступні точки часу після введення дози MDZ: 0 (перед введенням дози), 5 хвилин, 10 хвилин, 15 хвилин, 30 хвилин та 1, 2, 4, 6, 8, 10, 12 та 18 годин після введення MDZ. Оцінюють фармакокінетичні параметри сполуки 1, RTV та MDZ. Оцінку ЕКГ (електрокардіограми) проводять при відборі. Клінічні лабораторні тести, психічні дослідження та життєві показники (температуру, кров'яний тиск, частоту серцевих скорочень та частоту дихання) проводять при відборі, на вихідному рівні та у дні 10 та 20. Життєві показники (температуру, кров'яний тиск, частоту серцевих скорочень та частоту дихання) визначають також у дні 1, 11 та 21 в 0 годин (перед введенням дози мідазоламу) та через 0,5, 1,0, 1,5 та 2,0 години після введення дози мідазоламу. Продукт, що тестують, доза та спосіб введення: 1. Сполука 1, 1 таблетка x 125 мг та RTV 20 мг QD (в AM) 2. Сполука 1, 1 таблетка x 125 мг та RTV 50 мг QD (в AM) 3. Сполука 1, 1 таблетка x 125 мг та RTV 200 мг QD (в AM) Всі дози Сполука 1/р вводять відразу після завершення прийому стандартної їжі. Еталонне лікування, доза та спосіб введення Сполука 1, 1 таблетка x 125 мг та RTV 100 мг QD (в AM) Всі дози Сполуки 1/р вводять відразу після завершення прийому стандартної їжі. Критерії оцінки: Безпеку визначають на підставі оцінки клінічних лабораторних тестів, ЕКГ, періодичних психічних досліджень, включаючи життєві показники в різних точках часу під час дослідження та по документації несприятливих подій. Розраховують наступні фармакокінетичні параметри в плазмі: для Сполуки 1 та RTV: Cmax, Tmax, Clast, Tlast, Ctau, λz, , AUCtau, T1/2, CL/F та Vz/F. Для MDZ: AUC0-last, AUCinf, %AUCexp, Clast, Tlast, λz, T1/2, CL та Vz. Статистичні методи: Дані по безпеці та фармакокінетичним параметрам для Сполуки 1, RTV та MDZ підсумують по пацієнтах та рівню дози RTV, використовуючи описову статистику. Підбирають моделі активності та/або моделі ANOVA (відповідно), використовуючи фармакокінетичні параметри Сполуки 1 (AUC, Cmax та Ctau), отримані у всіх варіантах лікування. Нахил кривої математичного очікування визначають разом з відповідним 90% CI. Дане дослідження проводять відповідно до настанови по Сучасній клінічній практиці (GCP). Результати: Результати, отримані при дослідженні, приводять у наступних трьох таблицях 3A-3C та на Фігурах 3-5. 40 15 UA 101141 C2 Таблиця 3A Середні PK (фармакокінетичні) параметри (CV%) MDZ MDZ як монотерапія 31,0 (30,9) 0,20 (40,6) AUCinf Clast Напівперіод (год.) існування CL (л/год./кг 1 + 20 мг + 50 мг RTV RTV 101 (28,7) 145 (36,1) 1,53 (42,4) 2,51 (26,7) + 100 мг RTV 219 (49,0) 3,34 (23,1) + 200 мг RTV 146 (22,6) 2,58 (24,8) 3,97 (34,4) 7,83 (38,9) 22,4 (70,7) 15,2 (31,6) 0,447 (27,9) Параметр PK 0,143 1 (26,5) 14,4 (70,4) 0,090 (25,5) 0,072 (33,6) 0,087 (22,0) 1,2 1,2 1,2 дози AU +RTV відрізняються від MDZ у вигляді монотерапії (p
ДивитисяДодаткова інформація
Назва патенту англійськоюMethods for improving the pharmacokinetics of hiv integrase inhibitors
Автори англійськоюKearney, Brian, P., Kakee, Atsuyuki, Kawaguchi, Isao
Назва патенту російськоюСпособ улучшения фармакокинетики ингибиторов интегразы вич
Автори російськоюКарни Брайан П., Каки Атцуюки, Кавагучи Исао
МПК / Мітки
МПК: A61P 31/18, A61K 31/47, A61K 31/426
Мітки: інтегрази, віл, спосіб, інгібіторів, покращення, фармакокінетики
Код посилання
<a href="https://ua.patents.su/32-101141-sposib-pokrashhennya-farmakokinetiki-ingibitoriv-integrazi-vil.html" target="_blank" rel="follow" title="База патентів України">Спосіб покращення фармакокінетики інгібіторів інтегрази віл</a>
Біциклічні гетеросполуки як інгібітори віл-інтегрази

Номер патенту: 85097
Опубліковано: 25.12.2008
Автори: Крістал Марк Р., Реміллард Роджер, Пламондон Серж, Больйо Франсіс, Матіскелла Джон Д., Конноллі Тімоті П., Наіду Б. Нарасімхулу, Волкер Майкл А., Банвілле Жак, Соренсон Маргарет Е., Еллет Карл, Уеда Ясутсугу
МПК: A61K 31/519, C07D 498/04, A61P 31/18
Мітки: віл-інтегрази, гетеросполуки, біциклічні, інгібітори
Формула / Реферат:
1. Сполука за формулою І, Iде:R1 є С1-6(Аr1)алкіл, С1-6(Аr1)(СОN(R8)(R9))алкіл, С1-6(Аr1)(СО2R14)алкіл, С1-6(Аr1)гiдpoкciaлкiл або C1-6(Ar1)oкciaлкiл;R2 є водень, С1-6алкіл або OR14;R3 є водень, галоїд, гідрокси, ціано, С1-6алкіл, С3-7циклоалкіл, С5-7циклоалкеніл, С1-6галоїдалкіл, С1-6алкокси, С1-6алкілтіо, С1-6галоїдалкокси, N(R8)(R9),...
N-заміщені гідроксипіримідинон карбоксамідні інгібітори віл інтегрази

Номер патенту: 77454
Опубліковано: 15.12.2006
Автори: Ніці Емануела, Пескаторе Джованна, Пома Марко, Паче Паола, Гарделлі Крістіна, Крешенци Бенедетта, Муралья Естер, Сумма Вінченцо, Орвьєто Федеріка, Скарпеллі Ріта, Роулі Майкл, Петроккі Алєссія
МПК: C07D 417/04, C07D 403/14, A61K 31/513, A61P 43/00, C07D 513/04, C07D 401/04, C07D 401/12, C07D 413/04, C07D 401/06, A61P 31/18, C07D 239/557, C07D 403/12, C07D 239/46, C07D 403/10, C07D 401/14, C07D 413/12, C07D 403/04, A61K 31/5377
Мітки: n-заміщені, інтегрази, інгібітори, гідроксипіримідинон, карбоксамідні, віл
Формула / Реферат:
1. Сполука формули (І): (I),де R1 являє собою(1) -Н,(2) -С1-6алкіл, який необов'язково заміщений одним або декількома замісниками, кожний з яких незалежно являє собою галоген, -ОН, -CN, -O-С1-6алкіл, -O-С1-6галогеналкіл, -C(=O)Ra, -CO2Ra, -SRa, -S(=O)Ra, -N(RaRb), -C(=O)-C0-6алкіл-N(RaRb), N(Ra)-C(=O)-C0-6алкіл-N(RbRc), -SO2Ra, -N(Ra)SO2Rb,...
Комбінація тіазолідиндіону і сульфонілсечовини, фармацевтична композиція та спосіб лікування діабету

Номер патенту: 70302
Опубліковано: 15.10.2004
Автори: Бекінгем Робін Едвін, Сміт Стефен Елістер
МПК: A61P 3/10, A61K 31/44, A61K 31/64
Мітки: фармацевтична, діабету, тіазолідиндіону, лікування, спосіб, комбінація, композиція, сульфонілсечовини
Формула / Реферат:
1. Комбінація, яка включає 2-8 мг 5-[4-[2-(N-метил-N-(2-піридил)аміно) етокси]бензил]тіазолідин-2,4-діону (Сполуки І) або його таутомерної форми або фармацевтично прийнятної похідної та субмаксимальну кількість сульфонілсечовини.2. Комбінація за п. 1, яка відрізняється тим, що сульфонілсечовина являє собою глібенкламід, гліпізид, гліклазид, глімепірид, толазамід, толбутамід, ацетогексамід, карбутамід, хлорпропамід, гліборнурид,...
Мультичастинкова композиція контрольованого вивільнення флувоксаміну та спосіб лікування станів, які лікуються за допомогою селективних інгібіторів зворотного захоплення серотоніну

Номер патенту: 74335
Опубліковано: 15.12.2005
Автори: Морріссей Кетрін Енн, Старк Пол, Джері Тереза Енн
МПК: A61K 9/52, A61P 25/24, A61K 31/137
Мітки: композиція, серотоніну, лікуються, спосіб, контрольованого, флувоксаміну, зворотного, захоплення, допомогою, селективних, станів, лікування, мультичастинкова, інгібіторів, вивільнення
Формула / Реферат:
1. Мультичастинкова композиція контрольованого вивільнення селективного інгібітора зворотного захоплення серотоніну (SSRI) для перорального введення, яка включає частинки, що складаються з ядра, яке містить флувоксамін або його фармацевтично прийнятну сіль, і покриття, що забезпечує контрольоване вивільнення вказаного SSRI протягом періоду часу не менше ніж приблизно 12 годин після перорального введення, при цьому вказане покриття є...
Спосіб одержання інгібіторів віл-протеази
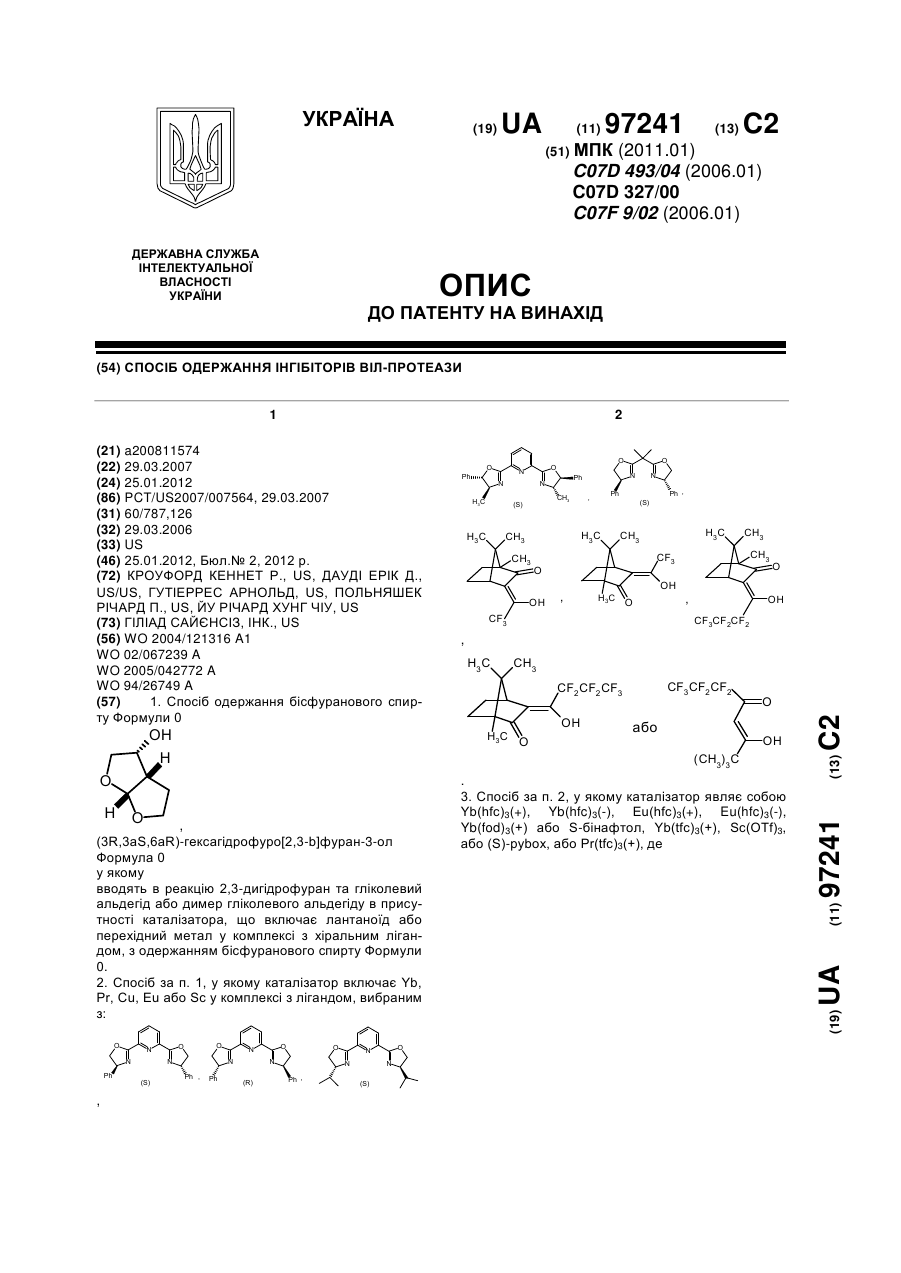
Номер патенту: 97241
Опубліковано: 25.01.2012
Автори: Гутіеррес Арнольд, Йу Річард Хунг Чіу, Польняшек Річард П., Кроуфорд Кеннет Р., Дауді Ерік Д.
МПК: C07D 327/00, C07D 493/04, C07F 9/02
Мітки: одержання, інгібіторів, віл-протеази, спосіб
Формула / Реферат:
1. Спосіб одержання бісфуранового спирту Формули 0,(3R,3aS,6аR)-гексагідрофуро[2,3-b]фуран-3-олФормула 0 у якомувводять в реакцію 2,3-дигідрофуран та гліколевий альдегід або димер гліколевого альдегіду в присутності каталізатора, що включає лантаноїд або перехідний метал у комплексі з хіральним лігандом, з одержанням...
Попередній патент: Комбінація рекомбінантної мікобактерії і біологічно активного агента як вакцини
Наступний патент: Спосіб розробки антитіла
Випадковий патент: Спосіб обробки продуктів тваринного походження перед збереженням