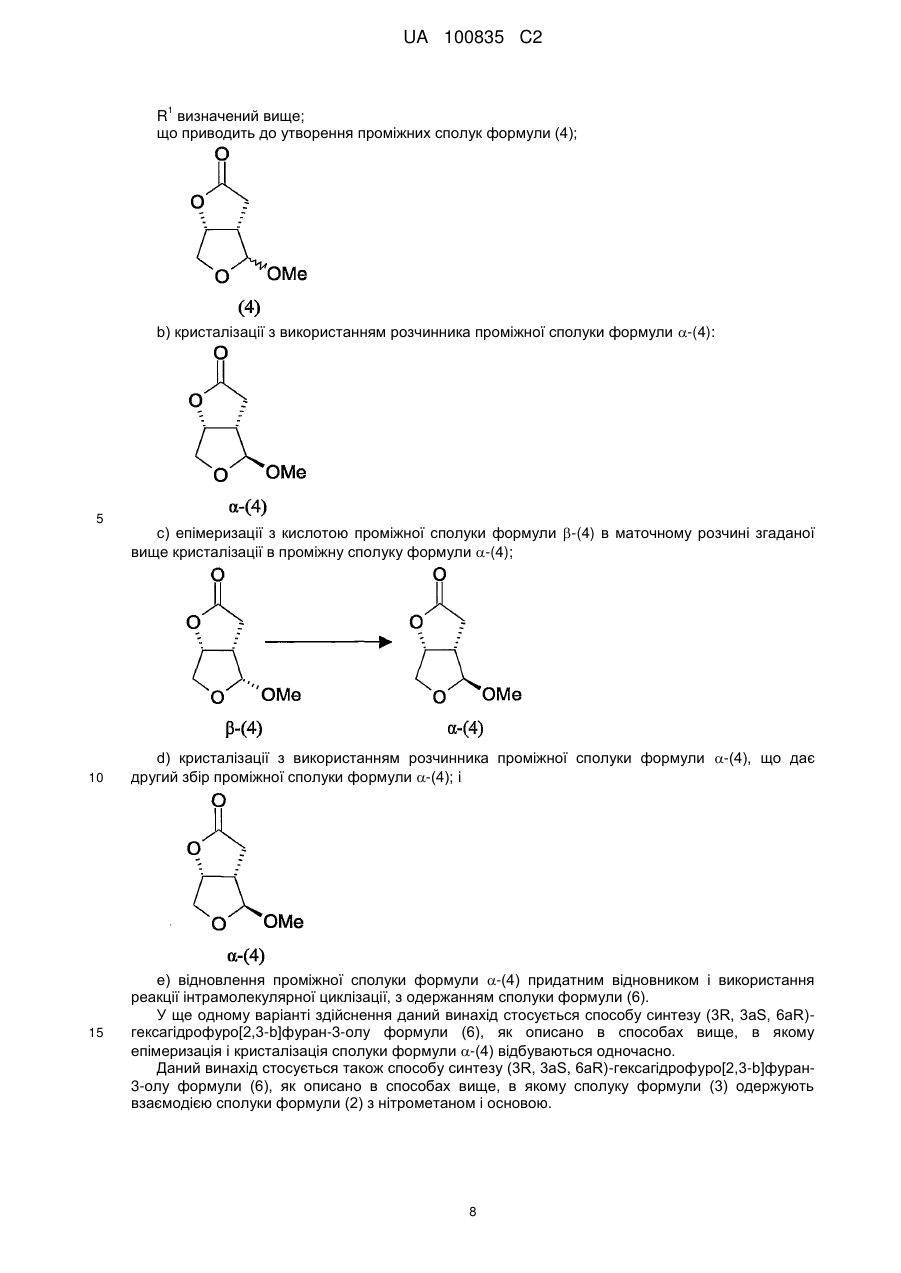
Спосіб синтезу (3r,3as,6ar)-гексагідрофуро[2,3-b]фуран-3-олу, проміжна сполука та спосіб її одержання
Номер патенту: 100835
Опубліковано: 11.02.2013
Автори: Кваєдфлієг Пітер Ян Леонар, Ломмен Франціскус Альфонс Марі, Кестелін Барт Рудольф Романі, Віджн Роберт Ян, Ліебрегтс Константінус Сімон Марія, Кооістра Якоб Херманус Матеус Херо




























Формула / Реферат
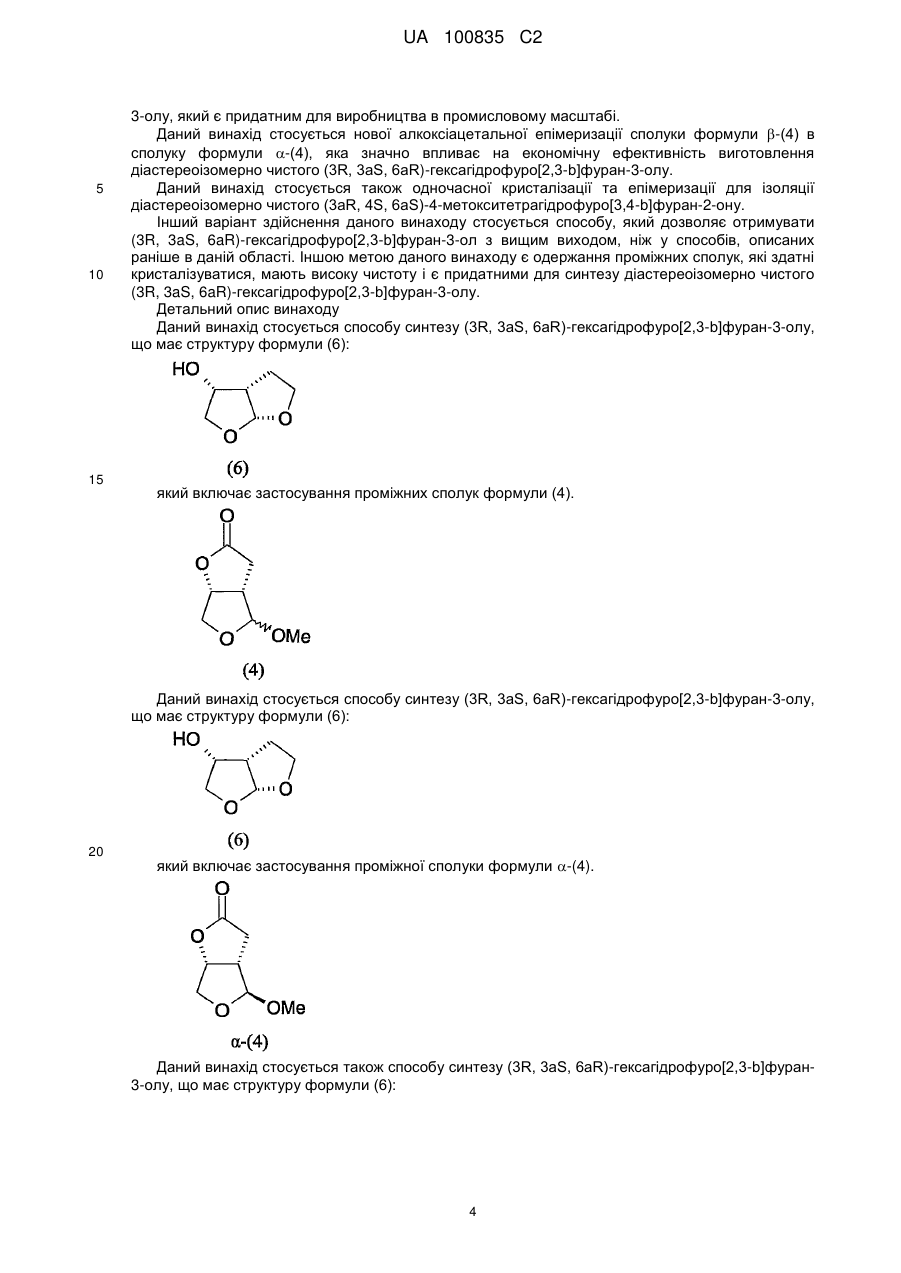
1. Спосіб синтезу (3R,3аS,6аR)-гексагідрофуро[2,3-b]фуран-3-олу, що має структуру формули (6):
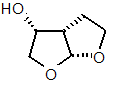 , (6)
, (6)
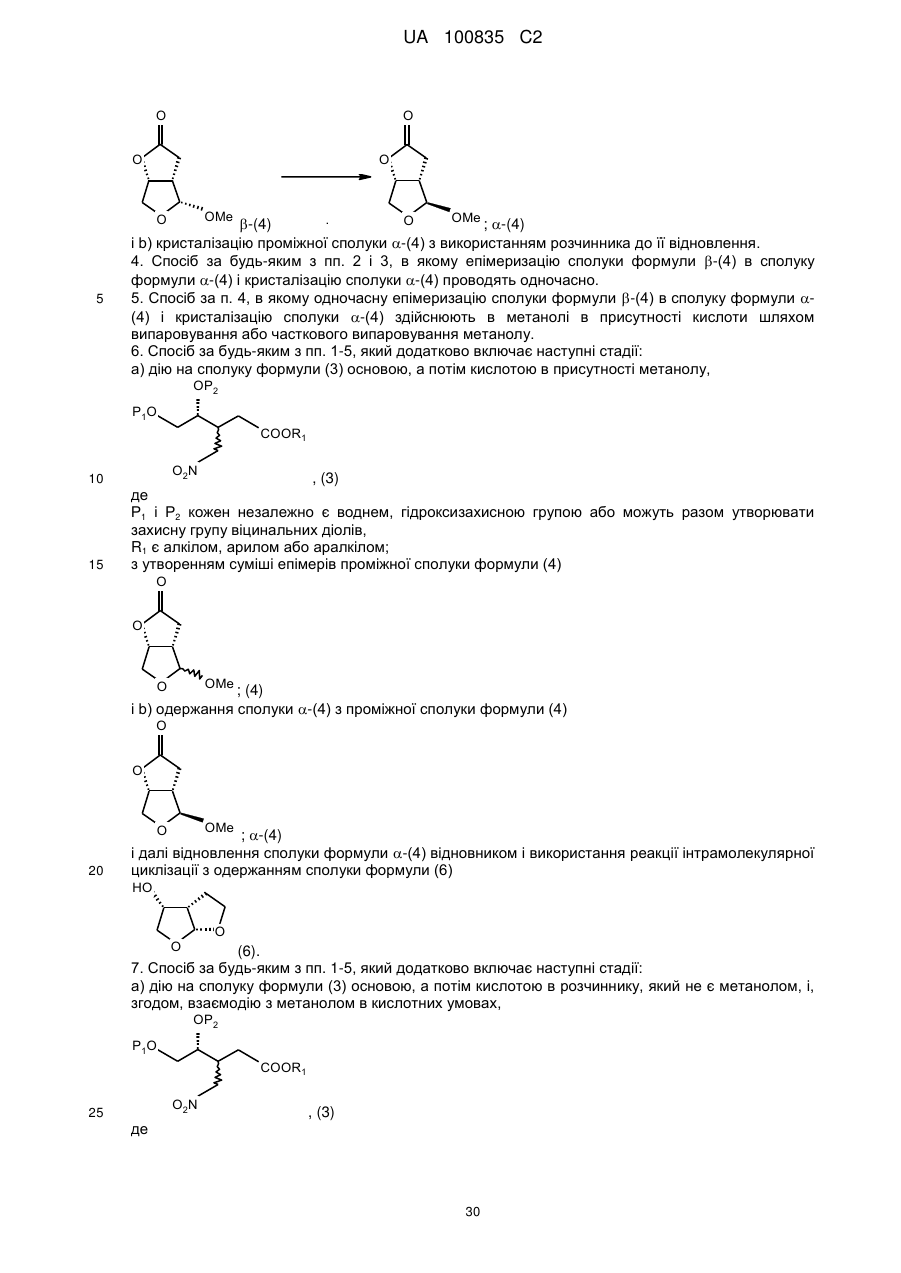
який включає стадію відновлення проміжної сполуки формули a-(4)
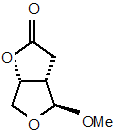 a-(4)
a-(4)
та який додатково включає кристалізацію проміжної сполуки формули a-(4) з використанням розчинника до її відновлення.
2. Спосіб за п. 1, який додатково включає:
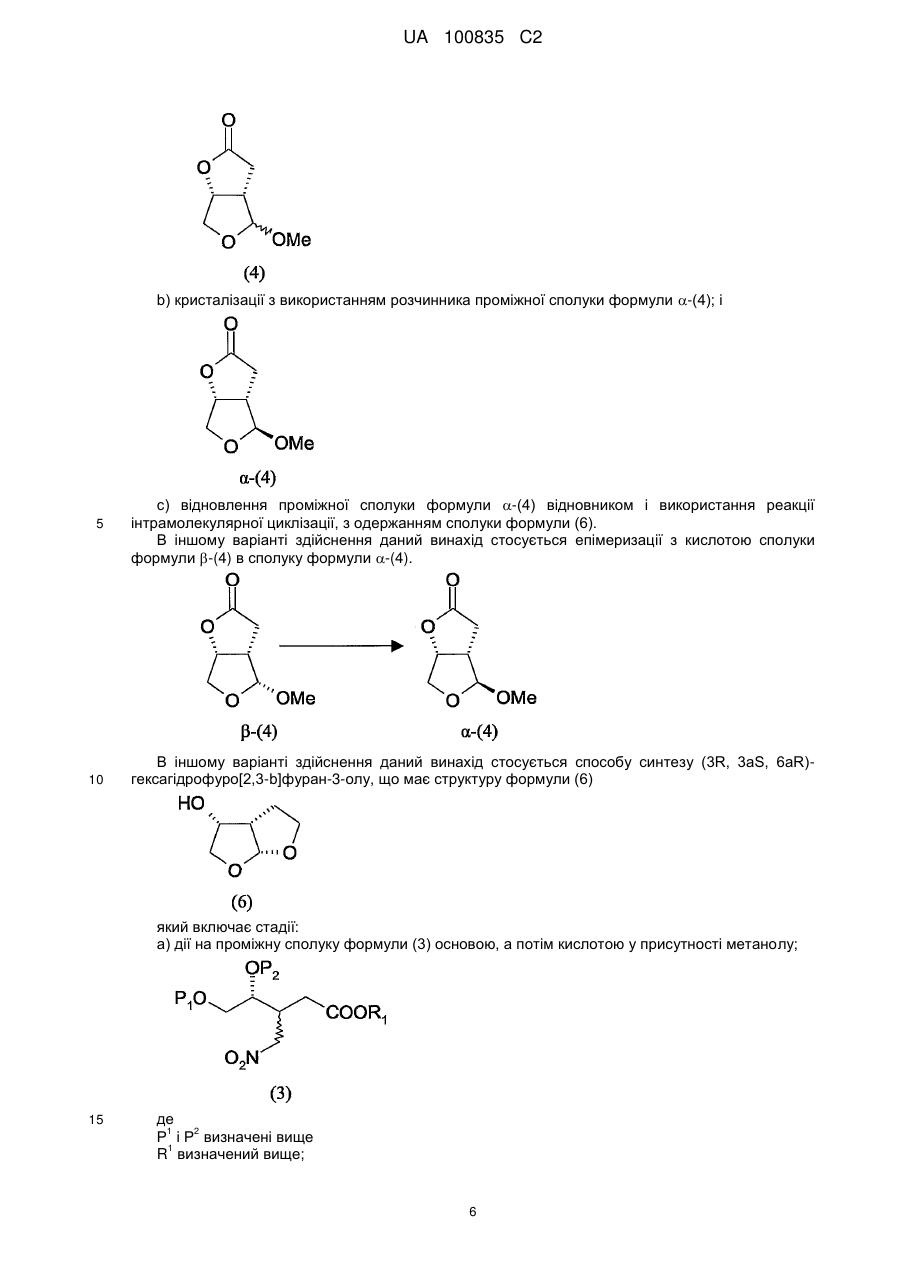
а) епімеризацію з кислотою проміжної сполуки формули b-(4) в проміжну сполуку формули a-(4); і
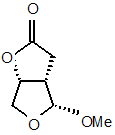 b-(4)
b-(4)  a-(4)
a-(4)
b) кристалізацію проміжної сполуки a-(4) з використанням розчинника до її відновлення.
3. Спосіб за п. 2, який додатково включає після кристалізації проміжної сполуки a-(4):
a) епімеризацію з кислотою проміжної сполуки формули b-(4) в проміжну сполуку формули a-(4)
b-(4) ; a-(4)
і b) кристалізацію проміжної сполуки a-(4) з використанням розчинника до її відновлення.
4. Спосіб за будь-яким з пп. 2 і 3, в якому епімеризацію сполуки формули b-(4) в сполуку формули a-(4) і кристалізацію сполуки a-(4) проводять одночасно.
5. Спосіб за п. 4, в якому одночасну епімеризацію сполуки формули b-(4) в сполуку формули a-(4) і кристалізацію сполуки a-(4) здійснюють в метанолі в присутності кислоти шляхом випаровування або часткового випаровування метанолу.
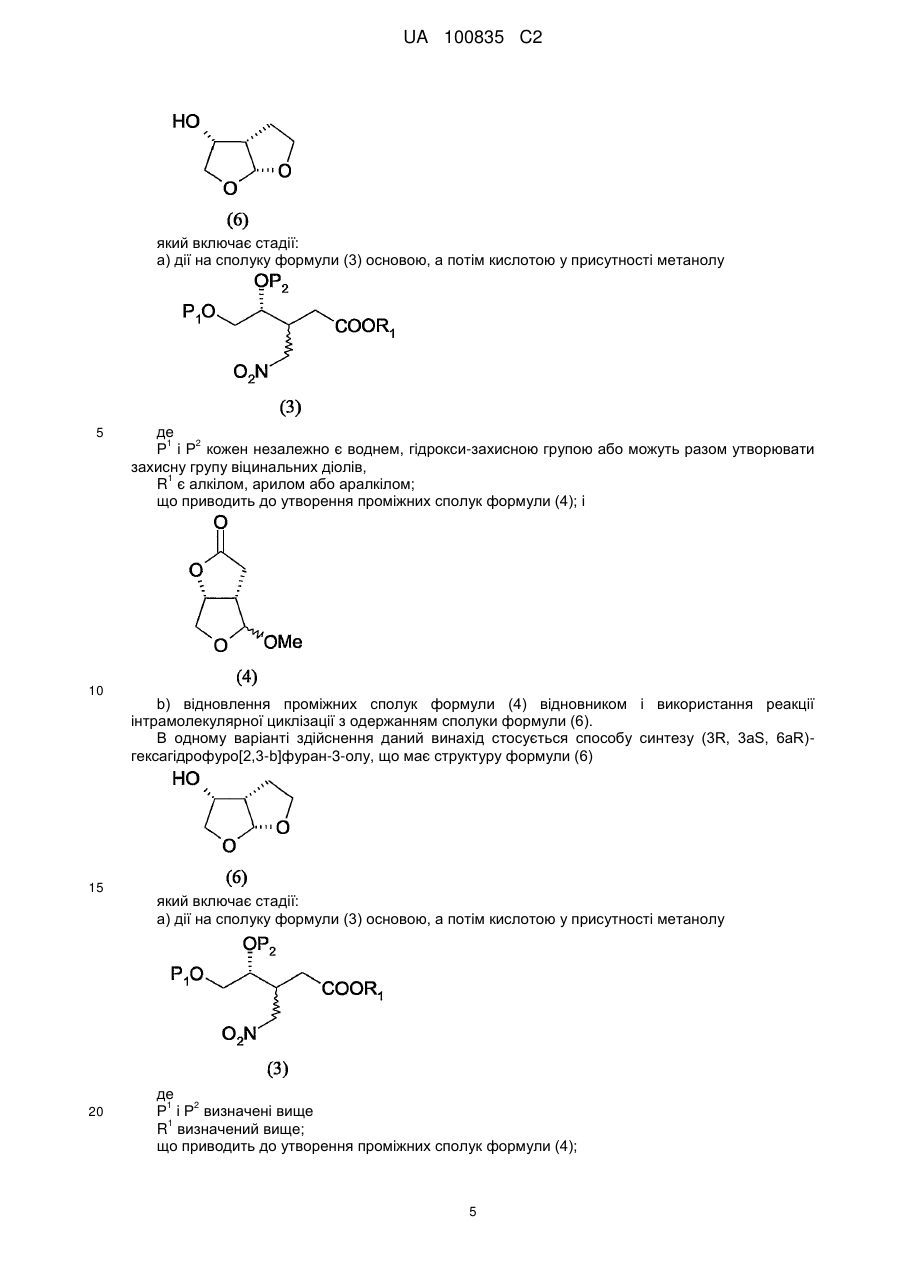
6. Спосіб за будь-яким з пп. 1-5, який додатково включає наступні стадії:
а) дію на сполуку формули (3) основою, а потім кислотою в присутності метанолу,
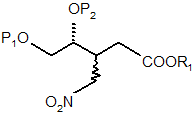 , (3)
, (3)
де
Р1 і Р2 кожен незалежно є воднем, гідроксизахисною групою або можуть разом утворювати захисну групу віцинальних діолів,
R1 є алкілом, арилом або аралкілом;
з утворенням суміші епімерів проміжної сполуки формули (4)
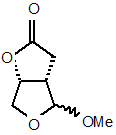 ; (4)
; (4)
і b) одержання сполуки a-(4) з проміжної сполуки формули (4)
; a-(4)
і далі відновлення сполуки формули a-(4) відновником і використання реакції інтрамолекулярної циклізації з одержанням сполуки формули (6)
 (6).
(6).
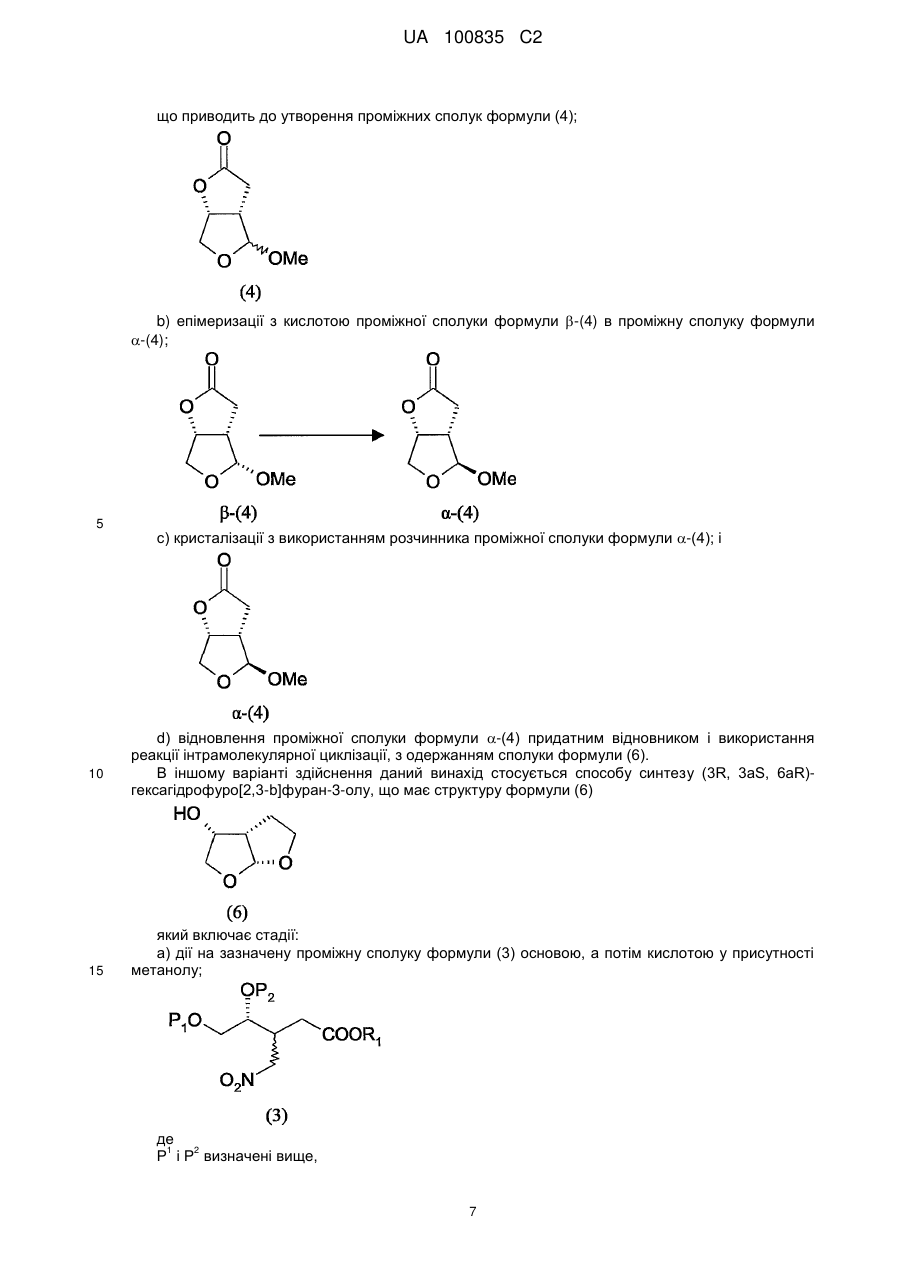
7. Спосіб за будь-яким з пп. 1-5, який додатково включає наступні стадії:
а) дію на сполуку формули (3) основою, а потім кислотою в розчиннику, який не є метанолом, і, згодом, взаємодію з метанолом в кислотних умовах,
, (3)
де
P1 і Р2 кожен незалежно є воднем, гідроксизахисною групою або можуть разом утворювати захисну групу віцинальних діолів,
R1 є алкілом, арилом або аралкілом;
з утворенням проміжної сполуки формули (4)
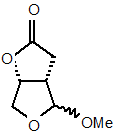 ; (4)
; (4)
і b) одержання сполуки a-(4) з проміжної сполуки формули (4)
; a-(4)
і далі відновлення сполуки формули a-(4) відновником і використання реакції інтрамолекулярної циклізації з одержанням сполуки формули (6)
(6).
8. Спосіб за будь-яким з пп. 6 і 7, в якому додатково одержують сполуку формули (3) взаємодією сполуки формули (2) з нітрометаном і основою
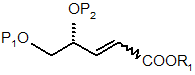 (2).
(2).
9. Спосіб за п. 8, в якому додатково одержують сполуку формули (2) конденсацією проміжної сполуки формули (1) або її гідрату, напівгідрату чи їхньої суміші з фосфонатами формули (R6O)2P(=O)-CH2-C(=O)OR1, де
Р1 і Р2 визначені вище,
R1 визначений вище,
R6 є алкілом, арилом або аралкілом,
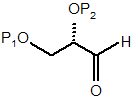 (1).
(1).
10. Спосіб за будь-яким з пп. 6-9, в якому Р1 і Р2 разом утворюють радикал діалкілметилен.
11. Спосіб за п. 8, в якому основа, використовувана для конденсації сполуки формули (2) в сполуку формули (3), є 1,8-діазабіцикло[5.4.0]ундец-7-еном або 1,1,3,3-тетраметилгуанідином або їхніми похідними.
12. Спосіб за п. 9, в якому фосфонат формули (R6O)2P(=O)-CH2-C(=O)OR1 є триетилфосфоноацетатом.
13. Спосіб за будь-яким з пп. 6 і 7, в якому конверсію сполуки формули (3) в сполуку формули (4) здійснюють з використанням основи, вибраної з групи метоксиду натрію, метоксиду літію, 1,8-діазабіцикло[5.4.0]ундец-7-ену чи 1,1,3,3-тетраметилгуанідину або їхніх сумішей.
14. Спосіб за будь-яким з пп. 8 і 11, в якому конверсію сполуки формули (2) в сполуку формули (4) здійснюють з використанням 1,8-діазабіцикло[5.4.0]ундец-7-ену або 1,1,3,3-тетраметилгуанідину як основи для конверсії сполуки формули (2) в сполуку формули (3), не ізолюючи сполуку формули (3) і використовуючи метоксид натрію або літію як додаткову основу для конверсії сполук формул (3) в сполуку формули (4).
15. Спосіб за будь-яким з пп. 6, 7, 13 і 14, в якому кислота, використовувана для конверсії сполуки формули (3) в сполуку формули (4), є концентрованою сірчаною кислотою, в кількості від 2,5 до 5 еквівалентів в розрахунку на сполуку формули (2), у вигляді 20-80 % мас. розчину в метанолі.
16. Спосіб за будь-яким з пп. 1-15, в якому кристалізацію сполуки формули a-(4) здійснюють в спирті.
17. Спосіб за п. 16, в якому спирт є ізопропанолом, трет-аміловим спиртом або трет-бутанолом.
18. Спосіб конверсії сполуки формули b-(4) в сполуку формули a-(4), який включає епімеризацію з використанням кислоти
b-(4) a-(4)
та додатково включає кристалізацію проміжної сполуки формули a-(4) з використанням розчинника.
19. Спосіб за будь-яким з пп. 4-7, в якому епімеризацію сполуки формули b-(4) в сполуку формули a-(4) здійснюють з використанням 0,05-1,5 еквівалентів МеSO3Н в метанолі.
20. Спосіб за п. 19, в якому епімеризацію сполуки формули b-(4) в сполуку формули a-(4) здійснюють з використанням 0,05-1,5 еквівалентів МеSO3Н в метанолі.
21. Спосіб за будь-яким з пп. 4-7, в якому епімеризацію здійснюють при температурі від 40 °С і до температури кипіння із зворотним холодильником.
22. Спосіб за будь-яким з пп. 19-20, в якому епімеризацію здійснюють при температурі від 40 °С і до температури кипіння із зворотним холодильником.
23. Проміжна сполука, що має формулу a-(4), причому вказана проміжна сполука знаходиться в кристалічній формі.
Текст
Реферат: Даний винахід стосується способів одержання діастереоізомерно чистого (3R,3аS,6аR)гексагідрофуро[2,3-b]фуран-3-олу (6), а також нової проміжної речовини (3аR,4S,6аS)-4метокситетрагідрофуро[3,4-b]фуран-2-ону (4), для застосування у зазначених способах. Конкретніше, винахід стосується стереоселективного способу одержання діастереомерно чистого (3R,3aS,6aR)-гексагідрофуро[2,3-b]фуран-3-олу, а також способів кристалізації (3aR,4S,6aS)-4-метокситетрагідрофуро[3,4-b]фуран-2-ону і епімеризації (3aR,4R,6aS)-4метокситетрагідрофуро[3,4-b]фуран-2-ону до (3aR,4S,6aS)-4-метокситетрагідрофуро[3,4b]фуран-2-ону. UA 100835 C2 (12) UA 100835 C2 UA 100835 C2 5 10 15 20 25 30 35 40 45 50 55 Даний винахід стосується способів одержання (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3олу, а також нової проміжної речовини (3aR, 4S, 6aS)-4-метокситетрагідрофуро[3,4-b]фуран-2ону, для застосування у зазначених способах. Конкретніше, винахід стосується стереоселективного способу одержання (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-олу і способу, придатного для виробництва в промислових масштабах. Гексагідрофуро[2,3-b]фуран-3-ол є важливою фармакологічною частиною, присутньою в структурі інгібіторів ретровірусної протеїнази, таких як ті, що описані Ghosh et al. у J. Med. Chem. 1996, 39(17), 3278-3290, EP 0715618, WO 99/67417 і WO 99/65870. Вказані публікації включені в цей документ як посилання. Відомо декілька способів одержання гексагідрофуро[2,3-b]фуран-3-олу. Ghosh et al. у J. Med. Chem. 1996, 39(17), 3278-3290 описують енантіоселективний синтез для одержання як (3R, 3aS, 6aR), так і (3S, 3aR, 6aS)-гексагідрофуро[2,3-b]фуран-3-олу в оптично чистій формі, починаючи з 3(R)-діетилмалату і 3(S)-діетилмалату, відповідно. Ghosh et al. також описують синтез рацемічної суміші (3R, 3aS, 6aR) і (3S, 3aR, 6aS)-енантіомерів гексагідрофуро[2,3-b]фуран-3-олу, починаючи з 2,3-дигідрофурану, з подальшим ферментативним розділенням кінцевого продукту. Pezeck et al. у Tetrahedron Lett. 1986, 27, 3715-3718 також описують шлях синтезу гексагідрофуро[2,3-b]фуран-3-олу з використанням озонолізу. Гексагідрофуро[2,3-b]фуран-3-ол також описаний як проміжна речовина в синтезі оптично активних похідних пергідрофуро[2,3-b]фурану в публікації Uchiyama et al., у Tetrahedron Lett. 2001, 42, 4653-4656. WO 03/022853 стосується альтернативного способу, який включає синтез (3R, 3aS, 6aR)гексагідрофуро[2,3-b]фуран-3-олу; вказаний спосіб починається з 2,3-двозахищеного-2,3дигідроксипропіональдегіду, який трансформується в похідне, яке включає нітрометил і одну або дві карбоксилатні частини. Зазначене похідне згодом трансформується реакцією Нефа (Nef) в тетрагідрофуранову сполуку, яку відновлюють і піддають реакції інтрамолекулярної циклізації, з одержанням (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-олу. Для того, щоб трансформувати вихідний матеріал, тобто, 2,3-двозахищений-2,3дигідроксипропіональдегід, в похідне, яке включає одну або дві карбоксилатні частини, WO03/022853 описує різні шляхи, що включають реакцію Віттіга (Wittig) з використанням фосфорних ілідів; реакцію Хорнера-Еммонса (Horner-Emmons) з використанням фосфонатів у присутності основи; реакцію конденсації типу Knoevenagel з використанням похідних малонату; або, альтернативно, із застосуванням реагентів Реформатского (Reformatsky), тобто, попередників частин -С(=О)-О-, таких як ціанид. Зокрема, наведені тут приклади фокусуються на двох шляхах: шляхах Knoevenagel і Віттіга. Шлях Knoevenagel, як показано у WO 03/022853, полягає в додаванні диметилмалонату до сухого розчину вихідного матеріалу 2,3-О-ізопропіліденгліцеральдегіду, з одержанням диметилового ефіру 2-(2,2-диметил-[1,3]діоксолан-4-ілметилен)малонової кислоти з 2 інкорпорованими карбоксилатами. Оскільки вихідний матеріал одержують у водному розчині, необхідно здійснити складну процедуру ізоляції, яка включає екстракцію тетрагідрофураном і видалення води. Зазначена екстракція і видалення води потребують великих кількостей тетрагідрофурану і витрат часу. Крім того, вихід реакції Knoevenagel з 2,3-Оізопропіліденгліцеральдегіду до диметилового ефіру 2-(2,2-диметил-[1,3]діоксолан-4ілметилен)малонової кислоти має граничне значення приблизно 77 %, оскільки спостерігаються неминучі побічні реакції, навіть після оптимізації умов. Завдяки тому факту, що одержана дикарбоксильована проміжна сполука, тобто, диметиловий ефір 2-(2,2-диметил-[1,3]діоксолан-4-ілметилен)малонової кислоти, є в'язкою маслянистою рідиною, її необхідно вводити в подальшу реакцію приєднання Michael (Міхаеля) у вигляді розчину в метанолі. Дистиляція метанолу після гасіння водним розчином NaHCO 3, після кислотних реакцій Нефа і циклізації, але до екстракції органічним розчинником, таким як етилацетат, має недоліки. Оскільки проміжна сполука, яка утворюється в результаті кислотних реакцій Нефа і циклізації, тобто, метиловий ефір 4-метокси-2-оксогексагідрофуро[3,4-b]фуран-3карбонової кислоти, є лабільною у воді сполукою, а дистиляція метанолу вимагає відносно високих температур (до 30-40 °C), спостерігається розклад проміжної сполуки до полярних сполук. Зазначені полярні сполуки залишаються у водній фазі і надалі втрачаються, оскільки 1 UA 100835 C2 5 10 15 20 25 30 35 40 45 50 вони не екстрагуються в органічну фазу. Оскільки метанол не можна видалити до екстракції, потрібний значний об'єм для обробки метилового ефіру 4-метокси-2-оксогексагідрофуро[3,4b]фуран-3-карбонової кислоти. Під час декарбоксилювання метилового ефіру 4-метокси-2-оксогексагідрофуро[3,4-b]фуран3-карбонової кислоти спостерігається значне утворення побічного продукту, тобто, (4-гідрокси-2метокситетрагідрофуран-3-іл)оцтової кислоти. Крім того, кристалізація 4метокситетрагідрофуро[3,4-b]фуран-2-ону дає тверду речовину коричневого кольору із-за супутніх полімеризацій. Крім цього, для очищення 4-метокситетрагідрофуро[3,4-b]фуран-2-ону потрібні щонайменше два кислотно-основних екстрактивних каскади для видалення кислоти для циклізації, внаслідок чого загальний вихід 4-метокситетрагідрофуро[3,4-b]фуран-2-ону складає 52 % від метилового ефіру 4-метокси-2-оксогексагідрофуро[3,4-b]фуран-3-карбонової кислоти і розцінюється як субоптимальний. Всі фактори, згадані вище, перешкоджають використанню шляху Knoevenagel. Фактично, стадія декарбоксилювання в даному шляху є недоліком у порівнянні зі шляхом Віттіга, оскільки подібна стадія декарбоксилювання при останньому шляху є непотрібною. WO03/022853 в прикладі I описує шлях Віттіга, в якому використовується триетилфосфоноацетат (ТЕРА) для одержання етилового ефіру 3-(2,2-диметил-[1,3]діоксолан4-іл)акрилової кислоти. Подальше приєднання Міхаеля до етилового ефіру 3-(2,2-диметил[1,3]діоксолан-4-іл)акрилової кислоти має обмеження в тому, що воно утворює нітрометановий аддукт, тобто, етиловий ефір 3-(2,2-диметил-[1,3]діоксолан-4-іл)-4-нітромасляної кислоти, із співвідношенням син:анти приблизно 8:2. Подальше відновлення, за яким йдуть реакції Нефа/циклізації, дає (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-ол, серйозно контамінований його екзо-діастереоізомером, тобто, (3R, 3aR, 6aS)-гексагідрофуро[2,3-b]фуран-3-олом, із співвідношенням ендо:екзо приблизно 8:2. Не дивлячись на те, що даний процес не має декількох недоліків, властивих процесу Knoevenagel, він не дає чистого ендо-діастереоізомеру, оскільки немає доступної стадії очищення, такої як кристалізація, для видалення небажаних екзо-діастереоізомерів, що утворилися в результаті реакції приєднання Міхаеля в антиконфігурації. При альтернативному шляху Віттіга, як описано в прикладі II WO 03/022853, продукт реакції приєднання Міхаеля має таке ж невигідне співвідношення син:анти (8:2), як і в прикладі I. Проміжні етоксисполуки (3aR, 4S, 6aS)-4-етокситетрагідрофуро[3,4-b]фуран-2-он і (3aR, 4R, 6aS)-4-етокситетрагідро-фуро[3,4-b]фуран-2-он, одержані після реакції Нефа/циклізації, присутні в співвідношенні (3aR, 4S, 6aS)/(3aR, 4R, 6aS) приблизно 2,5/1, разом із значною кількістю анти-ізомерів, тобто, із співвідношенням син:анти приблизно 8:2. Очищення проміжних сполук (3aR, 4S, 6aS)-4-етокситетрагідрофуро[3,4-b]фуран-2-ону і (3aR, 4R, 6aS)-4етокситетрагідрофуро[3,4-b]фуран-2-ону шляхом видалення небажаних анти-діастереоізомерів кристалізацією в даний час представляється неможливим. Відновлення суміші та циклізація давали (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-ол, серйозно контамінований його екзодіастереоізомером, тобто, (3R, 3aR, 6aS)-гексагідрофуро[2,3-b]фуран-3-олом, із співвідношенням ендо:екзо приблизно 8:2. Подібно до процесу Віттіга прикладу I вище, даний процес не має декількох недоліків, пов'язаних з процесом Knoevenagel, але в своїй існуючій формі все ж таки не дає чистого (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-олу з високими промисловими виходами. Крім того, об'єми реакторів, використовуваних у відомих фахівцям процедурах, є надто великими, а кількість операцій надто високою; зазначені фактори погіршують показники економічної ефективності процесу і, таким чином, роблять дані процеси не оптимальними в промисловому масштабі. Таким чином, існує потреба в оптимізованих процесах для промислового виготовлення діастереоізомерно чистого (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-олу. Несподівано було встановлено, що у разі, коли використовується шлях Віттіга і утворюються ізомери проміжної сполуки формули (4) і (4") WO 03/022853 у формі метилацеталю (тобто, R’’’ є метилом, а R’’ є воднем), вихід сирої проміжної сполуки формули (4), в перерахунку на проміжну сполуку формули (2), є набагато вищим порівняно, наприклад, з етокси- або ізопропоксіацеталями (де R’’’ є етилом або ізопропилом, відповідно, а R’’ є воднем). 2 UA 100835 C2 5 10 15 20 25 30 35 Більш того, несподівано було встановлено, що зазначену метилацетальну форму проміжної сполуки формули (4) в ізомерній формі (3aR, 4S, 6aS) можна кристалізувати з суміші (3aR, 4S, 6aS) і (3aR, 4R, 6aS)-ізомерів сполук формули (4) і відносно великої кількості ізомерів (4"). Підвищений вихід і можливість кристалізації ізомерної форми (3aR, 4S, 6aS) дозволяють виробляти (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-ол в діастереоізомерно чистій формі і з підвищеним виходом. Сполука формули (4) (3aR, 4S, 6aS)-4-метокситетрагідрофуро[3,4-b]фуран-2-он, далі в цьому документі називатиметься сполукою формули -(4) або альфа-епімером або -ізомером. Аналогічно, (3aR, 4R, 6aS)-4-метокситетрагідрофуро[3,4-b]фуран-2-он далі в цьому документі називатиметься сполукою формули -(4) або бета-епімером або -ізомером. сполуки формули (4) Дивно не лише те, що метоксіацеталь формули -(4) може бути кристалізований, але ще дивніше, що зазначена кристалізація є успішною, не зважаючи на низьке співвідношення альфа/бета - менше 4/1 у необробленій проміжній сполуці формули (4), з якої починають кристалізацію. Слід розуміти, що в процесі Knoevenagel було потрібне співвідношення альфа/бета щонайменше 6:1, щоб отримати здатну до кристалізації проміжну сполуку формули -(4). Таким чином, даний винахід стосується удосконаленого способу Віттіга і застосування 4альфа-ізомеру 4-метокситетрагідрофуро[3,4-b]фуран-2-ону, зокрема, (3aR, 4S, 6aS), який вносить значний внесок до можливості промислового виготовлення (3R, 3aS, 6aR)гексагідрофуро[2,3-b]фуран-3-олу в діастереоізомерно чистій формі. Крім того, несподівано було встановлено, що суміш з будь-яким співвідношенням альфа- і бета-епімерів формули (4) може бути трансформована в суміш з переважним змістом альфаепімеру, який може бути згодом ізольований в чистій формі шляхом кристалізації. По суті, даний винахід стосується нової алкоксіацетальної епімеризації сполуки формули (4), яка значно впливає на економічну ефективність процесу виготовлення (3R, 3aS, 6aR)-гексагідрофуро[2,3b]фуран-3-олу. Крім того, також несподівано було встановлено, що суміш майже з будь-яким співвідношенням альфа- і бета-епімерів формули (4) може бути трансформована в ході єдиної стадії в кристалічний альфа-епімер шляхом одночасної кристалізації і епімеризації, також відомої як індукована кристалізацією асиметрична трансформація. По суті, даний винахід стосується також одночасної кристалізації та епімеризації для ізоляції чистого (3aR, 4S, 6aS)-4метокситетрагідрофуро[3,4-b]фуран-2-ону. Суть винаходу Даний винахід стосується удосконаленого способу Віттіга і застосування (3aR, 4S, 6aS)-4метокситетрагідрофуро[3,4-b]фуран-2-ону як проміжної сполуки, конкретніше, в кристалічній формі, для виготовлення діастереоізомерно чистого (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран 3 UA 100835 C2 5 10 15 3-олу, який є придатним для виробництва в промисловому масштабі. Даний винахід стосується нової алкоксіацетальної епімеризації сполуки формули -(4) в сполуку формули -(4), яка значно впливає на економічну ефективність виготовлення діастереоізомерно чистого (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-олу. Даний винахід стосується також одночасної кристалізації та епімеризації для ізоляції діастереоізомерно чистого (3aR, 4S, 6aS)-4-метокситетрагідрофуро[3,4-b]фуран-2-ону. Інший варіант здійснення даного винаходу стосується способу, який дозволяє отримувати (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-ол з вищим виходом, ніж у способів, описаних раніше в даній області. Іншою метою даного винаходу є одержання проміжних сполук, які здатні кристалізуватися, мають високу чистоту і є придатними для синтезу діастереоізомерно чистого (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-олу. Детальний опис винаходу Даний винахід стосується способу синтезу (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-олу, що має структуру формули (6): який включає застосування проміжних сполук формули (4). Даний винахід стосується способу синтезу (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-олу, що має структуру формули (6): 20 який включає застосування проміжної сполуки формули -(4). Даний винахід стосується також способу синтезу (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран3-олу, що має структуру формули (6): 4 UA 100835 C2 який включає стадії: а) дії на сполуку формули (3) основою, а потім кислотою у присутності метанолу 5 10 15 20 де 1 2 Р і Р кожен незалежно є воднем, гідрокси-захисною групою або можуть разом утворювати захисну групу віцинальних діолів, 1 R є алкілом, арилом або аралкілом; що приводить до утворення проміжних сполук формули (4); і b) відновлення проміжних сполук формули (4) відновником і використання реакції інтрамолекулярної циклізації з одержанням сполуки формули (6). В одному варіанті здійснення даний винахід стосується способу синтезу (3R, 3aS, 6aR)гексагідрофуро[2,3-b]фуран-3-олу, що має структуру формули (6) який включає стадії: а) дії на сполуку формули (3) основою, а потім кислотою у присутності метанолу де 1 2 Р і Р визначені вище 1 R визначений вище; що приводить до утворення проміжних сполук формули (4); 5 UA 100835 C2 b) кристалізації з використанням розчинника проміжної сполуки формули -(4); і 5 10 с) відновлення проміжної сполуки формули -(4) відновником і використання реакції інтрамолекулярної циклізації, з одержанням сполуки формули (6). В іншому варіанті здійснення даний винахід стосується епімеризації з кислотою сполуки формули -(4) в сполуку формули -(4). В іншому варіанті здійснення даний винахід стосується способу синтезу (3R, 3aS, 6aR)гексагідрофуро[2,3-b]фуран-3-олу, що має структуру формули (6) який включає стадії: а) дії на проміжну сполуку формули (3) основою, а потім кислотою у присутності метанолу; 15 де 1 2 Р і Р визначені вище 1 R визначений вище; 6 UA 100835 C2 що приводить до утворення проміжних сполук формули (4); b) епімеризації з кислотою проміжної сполуки формули -(4) в проміжну сполуку формули -(4); 5 10 15 с) кристалізації з використанням розчинника проміжної сполуки формули -(4); і d) відновлення проміжної сполуки формули -(4) придатним відновником і використання реакції інтрамолекулярної циклізації, з одержанням сполуки формули (6). В іншому варіанті здійснення даний винахід стосується способу синтезу (3R, 3aS, 6aR)гексагідрофуро[2,3-b]фуран-3-олу, що має структуру формули (6) який включає стадії: а) дії на зазначену проміжну сполуку формули (3) основою, а потім кислотою у присутності метанолу; де 1 2 Р і Р визначені вище, 7 UA 100835 C2 1 R визначений вище; що приводить до утворення проміжних сполук формули (4); b) кристалізації з використанням розчинника проміжної сполуки формули -(4): 5 10 15 с) епімеризації з кислотою проміжної сполуки формули -(4) в маточному розчині згаданої вище кристалізації в проміжну сполуку формули -(4); d) кристалізації з використанням розчинника проміжної сполуки формули -(4), що дає другий збір проміжної сполуки формули -(4); і е) відновлення проміжної сполуки формули -(4) придатним відновником і використання реакції інтрамолекулярної циклізації, з одержанням сполуки формули (6). У ще одному варіанті здійснення даний винахід стосується способу синтезу (3R, 3aS, 6aR)гексагідрофуро[2,3-b]фуран-3-олу формули (6), як описано в способах вище, в якому епімеризація і кристалізація сполуки формули -(4) відбуваються одночасно. Даний винахід стосується також способу синтезу (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран3-олу формули (6), як описано в способах вище, в якому сполуку формули (3) одержують взаємодією сполуки формули (2) з нітрометаном і основою. 8 UA 100835 C2 5 10 15 20 25 30 35 40 45 У ще одному варіанті здійснення даний винахід стосується способу синтезу (3R, 3aS, 6aR)гексагідрофуро[2,3-b]фуран-3-олу формули (6), як описано в способах вище, в якому сполуку формули (2) одержують конденсацією проміжної сполуки формули (1) або її гідрату, 6 1 напівгідрату або їхньої суміші з фосфонатами формули (R O)2P(=O)-CH2-C(=O)OR , де 1 2 Р і Р визначені вище, 1 R визначений вище, 6 R є алкілом, арилом або аралкілом. Термін "гідрокси-захисна група", використовуваний в цьому документі, стосується замісника, який захищає гідроксильні групи від небажаних реакцій під час процедур синтезу, такого як Озахисні групи, описані в Greene and Muts, "Protective Groups In Organic Synthesis" (John Wiley & Sons, New York, 3-е видання, 1999). Гідрокси-захисні групи включають заміщені метилові ефіри, наприклад, метоксиметил, бензилоксиметил, 2-метоксіетоксиметил, 2(триметилсиліл)етоксиметил, т-бутил, бензил і трифенілметил; тетрагідропіранільні ефіри; заміщені етилові ефіри, наприклад, 2,2,2-трихлоретил; силілові ефіри, наприклад, триметилсиліл, т-бутилдиметилсиліл і т-бутилдифенілсиліл; і складні ефіри, наприклад, ацетат, пропіонат, бензоат і т.п. Термін "захисна група віцинальних діолів", використовуваний в цьому документі, стосується захисних груп у формі ацеталю або кеталю і в ортоефірній формі. Конкретні приклади захисної групи у формі ацетального або кетального радикала включають метилен, дифенілметилен, етиліден, 1-т-бутилетиліден, 1-фенілетиліден, (4-метоксифеніл)етиліден, 2,2,2-трихлоретиліден, ізопропіліден, циклопентиліден, циклогексиліден, циклогептиліден, бензиліден, пметоксибензиліден, 2,4-диметоксибензиліден, 3,4-диметоксибензиліден, 2-нітробензиліден і т.п., а конкретні приклади захисної групи в ортоефірній формі включають метоксиметилен, етоксиметилен, 1-метоксіетиліден, 1-етоксіетиліден, 1,2-диметоксіетиліден, альфаметоксибензиліден, 1-(N, N-диметиламін)етиліден, альфа-(N, N-диметиламін)бензиліден, 2оксациклопентиліден і т.п. У кращому варіанті здійснення даного винаходу захисною групою віцинальних діолів є ізопропіліден. Термін "алкіл", використовуваний в цьому документі окремо або як частина групи, стосується насичених одновалентних вуглеводневих радикалів, які мають прямі або розгалужені вуглеводневі ланцюги, або, у разі присутності щонайменше 3 атомів вуглецю, циклічних вуглеводнів або їхніх комбінацій, і містять від 1 до 20 атомів вуглецю (С 1-20-алкіл), бажано, від 1 до 10 атомів вуглецю (С1-10-алкіл), краще, від 1 до 8 атомів вуглецю (С 1-8-алкіл), ще краще, від 1 до 6 атомів вуглецю (С1-6-алкіл), і найкраще, від 1 до 4 атомів вуглецю (С 1-4алкіл). Приклади алкільних радикалів включають метил, етил, пропіл, ізопропіл, н-бутил, ізобутил, втор-бутил, трет-бутил, пентил, ізоаміл, гексил, циклопропіл, циклобутил, циклопентил, циклогексил і т.п. Термін "алкеніл", використовуваний в цьому документі, окремо або як частина групи, стосується одновалентних вуглеводневих радикалів, які мають прямі або розгалужені вуглеводневі ланцюги, що мають один чи більше подвійних зв'язків, і містять від 2 до 18 атомів вуглецю, краще, від 2 до 8 атомів вуглецю, ще краще, від 2 до 5 атомів вуглецю. Приклади придатних алкенільних радикалів включають етеніл, пропеніл, алкіл, 1,4-бутадієніл і т.п. Термін "алкініл", використовуваний в цьому документі, окремо або як частина групи, стосується одновалентних вуглеводневих радикалів, які мають прямі або розгалужені вуглеводневі ланцюги, що мають один чи кілька потрійних зв'язків, і містять від 2 до 10 атомів вуглецю, ще краще, від 2 до 5 атомів вуглецю. Приклади придатних алкінільних радикалів включають етиніл, пропініл, (пропаргіл), бутиніл і т.п. 9 UA 100835 C2 5 10 15 20 25 30 35 40 45 50 55 60 Термін "арил", використовуваний в цьому документі, окремо або як частина групи, стосується органічного радикала, одержуваного з ароматичного вуглеводня видаленням одного атома водню, і включає моноциклічні та поліциклічні радикали, такі як феніл, дифеніл, нафтил. Термін "алкокси", використовуваний в цьому документі, окремо або як частина групи, стосується алкільного ефірного радикала, де термін "алкіл" визначений вище. Приклади алкільного ефірного радикала включають метокси, етокси, н-пропокси, ізопропокси, н-бутокси, ізобутокси, втор-бутокси, трет-бутокси і т.п. Терміни "аралкіл" і "аралкокси", використовувані в цьому документі, окремо або в комбінації, означають алкільні або алкоксильні радикали, визначені вище, в яких щонайменше один атом водню замінений на арильний радикал, визначений вище, такий як бензил, бензилокси, 2фенілетил, дибензилметил, гідроксифенілметил, метилфенілметил і т.п. Термін "аралкоксикарбоніл", використовуваний в цьому документі, окремо або в комбінації, означає радикал формули аралкіл-О-С(О)-, в якому термін "аралкіл" визначений вище. Прикладами аралкоксикарбонільного радикала є бензилоксикарбоніл і 4метоксифенілметоксикарбоніл. Термін "циклоалкіл", використовуваний в цьому документі окремо або в комбінації, означає насичений або частково насичений моноциклічний, біциклічний або трициклічний алкільний радикал, в якому кожна циклічна частина містить приблизно від 3 до 8 атомів вуглецю, ще краще, приблизно від 3 до 6 атомів вуглецю. Приклади зазначених циклоалкільних радикалів включають циклопропіл, циклобутил, циклопентил, циклогексил і т.п. Термін "циклоалкілалкіл", використовуваний в цьому документі, окремо або в комбінації, означає алкільний радикал, визначений вище, заміщений циклоалкільним радикалом, визначеним вище. Приклади зазначених циклоалкілалкільних радикалів включають циклопропілметил, циклобутилметил, циклопентилметил, циклогексилметил, 1циклопентилетил, 1-циклогексилетил, 2-циклопентилетил, 2-циклогексилетил, циклобутилпропіл, циклопентилпропіл, циклогексилбутил і т.п. Термін "гетероциклоалкіл", використовуваний в цьому документі, окремо або в комбінації, стосується насиченого або частково ненасиченого, моноциклічного, біциклічного або трициклічного гетероциклу, який має, краще, від 3 до 12 членів кільця, ще краще, від 5 до 10 членів кільця і, найкраще, від 5 до 6 членів кільця, і включає один або більше гетероатомних членів кільця, вибраних з азоту, кисню та сірки, і який, необов'язково, є заміщеним на одному або більше атомах вуглецю галогеном, алкілом, алкокси, гідрокси, оксо, арилом, аралкілом і т.п., та/або на вторинному атомі азоту (тобто, –NH-) - гідроксилом, алкілом, аралкоксикарбонілом, алканоїлом, фенілом або фенілалкілом, та/або на третинному атомі азоту (тобто, =NH-) - оксидогрупою. Гетероциклоалкіл також включає конденсовані з бензолом моноциклічні циклоалкільні групи, які містять щонайменше один зазначений гетероатом. Гетероциклоалкіл, крім сірки та азоту, також включає сульфони, сульфоксиди і N-оксиди третинного азоту, що містять гетероциклоалкільні групи. Термін "гетероарил", використовуваний в цьому документі, окремо або в комбінації, стосується ароматичного моноциклічного, біциклічного або трициклічного гетероциклоалкільного радикала, визначеного вище, і, необов'язково, заміщеного, як визначено вище для арилу і гетероциклоалкілу. Прикладами зазначених гетероциклоалкільних і гетероарильних груп є піролідиніл, піперидиніл, піперазиніл, морфолініл, тіаморфолініл, піроліл, імідазол-4-іл, 1бензилоксикарбонілімідазол-4-іл, піразоліл, піридил, 2-(1-піперидиніл)піридил, 2-(4бензилпіперазин-1-іл-1-піридиніл), піразиніл, піримідиніл, фурил, тетрагідрофурил, тієніл, триазоліл, оксазоліл, тіазоліл, 2-індоліл, 2-хінолініл, 3-хінолініл, 1-оксидо-2-хінолініл, ізохінолініл, 1-ізохінолініл, 3-ізохінолініл, тетрагідрохінолініл, 1,2,3,4-тетрагідро-2-хіноліл, 1,2,3,4тетрагідроізохінолініл, 1,2,3,4-тетрагідро-1-оксоізохінолініл, хіноксалініл, 2-бензофуранкарбоніл, 1-, 2-, 4- або 5-бензімідазоліл і т.п. Термін "силіл", використовуваний в цьому документі, стосується атома кремнію, необов'язково, заміщеного однією чи більше алкільною, арильною або аралкільною групами. Терміни "ізомер", "ізомерна форма", "стереохімічно ізомерні форми" або "стереоізомерні форми", використовувані в цьому документі, визначають всі можливі ізомерні, а також конформаційні форми, що складаються з одних і тих же атомів, зв'язаних однією і тією ж послідовністю зв'язків, але мають відмінні просторові структури, що не є взаємозамінними, які можуть мати сполуки або проміжні сполуки, одержані під час зазначеного процесу. Якщо не згадано або не зазначено інше, хімічне позначення сполуки охоплює суміш всіх можливих стереохімічно ізомерних форм, які може мати зазначена сполука. Зазначена суміш може містити всі діастереоізомери, епімери, енантіомери та/або конформери базової молекулярної 10 UA 100835 C2 5 10 15 20 25 30 35 40 структури зазначеної сполуки. Конкретніше, стереогенні центри можуть мати R- або Sконфігурацію, діастереоізомери можуть мати син- або анти-конфігурацію, замісники на двовалентних циклічних насичених радикалах можуть мати цис- або транс-конфігурацію, а алкенільні радикали можуть мати Е- або Z-конфігурацію. Все стереохімічно ізомерні форми зазначеної сполуки, як в чистій формі, так і в суміші одна з одною, входять в об'єм даного винаходу. Термін "діастереоізомер" або "діастереоізомерна форма" стосується молекул з ідентичною хімічною будовою, що містять більш ніж один стереоцентр, які відрізняються одна від одної за конфігурацією по відношенню до одного або більше зазначених стереоцентрів. Термін "епімер" в даному винаході стосується молекул з ідентичною хімічною будовою, що містять більш ніж один стереоцентр, але які відрізняються одна від одної за конфігурацією по відношенню до тільки одного із зазначених стереоцентрів. Зокрема, термін "епімер" включає сполуки формули (4), які відрізняються за орієнтацією зв'язку між атомом вуглецю 4 (С-4) і метоксизамісником, тобто, сполуки формули -(4) і -(4), відповідно, де С-4 є 4S і 4R, відповідно. Чисті стереоізомерні форми проміжної сполуки формули (1), (4), (6) і вихідного матеріалу, згадані в цьому документі, визначаються як ізомери, що практично не містять інших енантіомерних або діастереоізомерних форм однієї і тієї ж базової молекулярної структури зазначених сполук або вихідного матеріалу. Відповідно, термін "стереоізомерно чиста" сполука або вихідний матеріал стосується сполуки або вихідного матеріалу, що має стереоізомерний надлишок від щонайменше 50 % (тобто, мінімум 75 % одного ізомеру і максимум 25 % інших можливих ізомерів) і аж до стереоізомерного надлишку 100 % (тобто, 100 % одного ізомеру і ніяких інших), краще, сполуки або вихідного матеріалу, що має стереоізомерний надлишок від 75 % до 100 %, ще краще, сполук, вихідного матеріалу або реагентів, що мають стереоізомерний надлишок від 90 % до 100 %, ще краще, сполук або проміжних сполук, що мають стереоізомерний надлишок від 94 % до 100 % і, найкраще, таких, що мають стереоізомерний надлишок від 97 % до 100 %. Терміни "енантіомерно чисті" і "діастереоізомерно чисті" слід розуміти аналогічно, але по відношенню до енантіомерного надлишку, і, відповідно, діастереоізомерного надлишку в даній суміші. По суті, в кращому варіанті здійснення даного винаходу використовують S-2,3-Oізопропіліденгліцеральдегід як вихідний матеріал в енантіомерному надлишку більше 95 %, ще краще, в енантіомерному надлишку більше 97 %, найкраще, в енантіомерному надлишку більше 99 %. Сполуки формули (1) Сполуку формули (1) можна отримати з комерційно доступних джерел. Синтез сполуки формули (1), як в енантіомерно чистій формі, так і в рацемічній формі, описаний в літературі. Наприклад, одержання 2,3-О-ізопропіліден-S-гліцеральдегіду описано в C. Hubschwerlen, Synthesis 1986, 962; одержання 2,3-О-ізопропіліден-R-гліцеральдегіду описано в C.R. Schmid et al., J. Org. Chem. 1991, 56, 4056-4058; і одержання 2,3-О-ізопропіліден-(R, S)-гліцеральдегіду описано в A. Krief et al., Tetrahedron Lett. 1998, 39, 1437-1440. Таким чином, зазначену проміжну сполуку формули (1) можна придбати, одержати до проведення реакції або утворити in situ. В кращому варіанті здійснення даного винаходу зазначену сполуку утворюють in situ, наприклад, 11 UA 100835 C2 5 10 15 20 25 30 35 40 45 50 55 окисненням у водному або частково водному розчині. У разі, коли зазначена сполука знаходиться у водному або частково водному розчині, вона звичайно частково присутня у формі свого гідрату або напівгідрату. 1 2 Відповідно, винахід стосується способу, в якому Р і Р разом утворюють захисну групу віцинальних діолів, яка, особливо, є кислотно-лабільною захисною групою, що залишається без змін під час стадії обробки основою подальшої реакції Нефа. Краще, зазначену захисну групу віцинальних діолів вибирають з групи, що складається з метилену, дифенілметилену, етилідену, 1-т-бутилетилідену, 1-фенілетилідену, (4-метоксифеніл)етилідену, 2,2,2трихлоретилідену, ізопропілідену, циклопентилідену, циклогексілідену, циклогептилідену, бензилідену, п-метоксибензилідену, 2,4-диметоксибензилідену, 3,4-диметоксибензилідену і 21 2 нітробензилідену. У ще кращому варіанті здійснення даного винаходу Р і Р разом утворюють діалкілметилен, такий як ізопропіліденовий або 3-пентиліденовий радикал. У найкращому 1 2 варіанті здійснення даного винаходу Р і Р разом утворюють ізопропіліденовий радикал. Особливою перевагою використання ізопропілідену порівняно з іншими захисними групами є те, що реагенти, потрібні для захисту діолу, тобто, диметоксипропан, 2-метоксипропен або ацетон, є комерційно доступними і недорогими. Цікавими захисними групами віцинальних діолів є такі захисні групи, що утворюють один чи більше додаткових стереогенних центрів в проміжних сполуках формул (1), (2) і (3). Згадані вище гідрокси-захисні групи і захисні групи віцинальних діолів легко відщеплюються способами, відомими фахівцям, такими як гідроліз, відновлення і т.п., які вибирають належним чином залежно від використовуваної захисної групи. Згідно із кращим варіантом здійснення даного винаходу, захисна група віцинальних діолів є кислотно-лабільною захисною групою, де термін "кислотно-лабільний", використовуваний в цьому документі, відносяться до захисних груп віцинальних діолів, які легко відщеплюються при використанні кислотних умов. Сполука формули (2) Сполуки формули (1) або їхні гідрати, напівгідрати чи суміші згодом трансформуються в сполуку формули (2) з використанням фосфонатів у присутності основи. В реакції 6 1 використовуються фосфонати формули (R O)2P(=O)-CH2-C(=O)OR , де 1 R є алкілом, арилом або аралкілом, 6 R є алкілом, арилом або аралкілом. 1 Відповідно, R позначає С1-6-алкіл, арил або арил-С1-6-алкіл, зокрема, С1-6-алкіл, 1 конкретніше, R позначає метил, етил, пропіл, ізопропіл, н-бутил, ізобутил, втор-бутил, трет1 1 бутил і пентил, краще, R є метилом, етилом або трет-бутилом і, найкраще, R є етилом. Приклади фосфонатів включають, поміж інших, етил-2-(діетилфосфоно)-пропіонат, етил-2(диметилфосфоно)пропіонат, триетилфосфоноацетат (ТЕРА). Краще, сполука формули (1) і фосфонат присутні в реакційній суміші в межах молярного співвідношення приблизно від 0,9:1 до 1,1:0,9, найкраще, в молярному співвідношенні приблизно 1:1. Коли сполуку формули (1) одержують in situ, її вміст в реакційній суміші слід визначати і основувати приблизно на 1 еквіваленті фосфонату, що додається. Відповідні температури для реакції конденсації знаходяться в межах приблизно від -5 °C до 50 °C, краще, приблизно від -2 °C до 35 °C, ще краще, приблизно від 0 °C до 35 °C. Приклади придатних основ, які можуть використовуватися для конверсії сполуки формули (1) в сполуку формули (2), включають, без обмеження, алкіламіни, карбонати натрію, калію, літію або цезію, гідроксиди або алкоксиди натрію, калію, літію або цезію, та їхні суміші. Краще, основа є карбонатом калію, найкраще, основу додають у вигляді твердої речовини, а не розчину у воді. Також, краще, кількість карбонату калію у вигляді твердої речовини складає щонайменше приблизно 2,5 еквіваленти, у розрахунку на сполуку формули (1). Краще, рН реакційної суміші підтримують в межах приблизно від 7 до 13, ще краще, в межах від 8 до 12, найкраще, рН підтримують в межах приблизно від 9 до 11. Відповідними розчинниками для даної реакції є вода, будь-який вуглеводень, ефір, галогенований вуглеводень або ароматичні розчинники, відомі фахівцям для реакцій конденсації. Останні включатимуть, без обмеження, пентан, гексан, гептан, толуол, ксилол (ксилоли), бензол, мезитилен (мезитилени), т-бутилметиловий ефір, діалкілові ефіри (етилові, бутилові), дифеніловий ефір, хлорбензол, метиленхлорид, хлороформ, чотирихлористий 12 UA 100835 C2 5 10 15 20 25 30 35 40 вуглець, ацетонітрил, дихлорбензол, дихлоретан, трихлоретан, циклогексан, етилацетат, ізопропілацетат, тетрагідрофуран, діоксан, метанол, етанол та ізопропанол. Краще, як розчинник використовується вода, будь то як єдиний розчинник або в суміші з іншим розчинником, наприклад, тетрагідрофураном. В одному варіанті здійснення даного винаходу до реакційної суміші, що містить сполуку формули (2), може бути застосована процедура обробки за допомогою розділення органічної та водної фаз і подальшої екстракції з водної фази додаткової порції сполуки формули (2) з використанням органічного розчинника, відмінного від органічної фази. По суті, тетрагідрофуранову фазу можна відокремити від водної фази, а останню можна екстрагувати, наприклад, двома порціями толуолу. Кращими розчинниками для екстракції є етилацетат, толуол, тетрагідрофуран. Найкращим розчинником є толуол. Сполуки формули (2), краще, не очищають на силікагелі. Хоча в результаті одержують менш чисті сполуки формули (2), ніж очищений на силікагелі продукт, якість є достатньою для одержання сполуки формули (4) із задовільною якістю та виходом. Відсутність стадії очищення в остаточному рахунку спрощує промисловий процес за даним винаходом. Сполуки формули (2) можна одержати в двох ізомерних формах, E- і Z-ізомерів; при цьому Е-ізомер є кращим ізомером. Сполука формули (3) Сполуки формули (2) можна згодом піддавати реакції приєднання Міхаеля, при якому нітрометан додають як попередник формільної групи до ,-ненасиченого ефіру проміжних сполук формули (2), разом з основою. Нітрометан є комерційно доступним у вигляді розчину в метанолі, і є кращим у зазначеній композиції. Прикладами основ, які є придатними для каталізу реакцій приєднання Міхаеля, є гідроксиди або алкоксиди натрію, калію, літію, цезію, TBAF (фторид тетра-н-бутиламонію), DBU (1,8діазабіцикло[5.4.0.]ундец-7-ен), TMG (1,1,3,3-тетраметилгуанидин), краще, гідроксид натрію, гідроксид калію, гідроксид літію, метоксид натрію, метоксид літію, TBAF, DBU, TMG та їхні суміші, ще краще, DBU і ТMG і, найкраще, DBU. Коли DBU використовується як основа в конверсії сполук формули (2) в сполуки формули (3), кількість доданої основи, краще, вище приблизно 0,5 еквівалента, в розрахунку на сполуки формули (2), ще краще, вище приблизно 0,8 еквівалента, найкраще, приблизно від 0,8 до 1,2 еквівалента, найкраще, приблизно від 0,9 до 1,1 еквівалента. У кращому варіанті здійснення даного винаходу DBU присутній в кількості приблизного 1 еквівалента. Можна використовувати будь-який розчинник, придатний для здійснення реакції приєднання Міхаеля. Прикладами придатних розчинників є метанол, етанол і ацетонітрил. Краще, розчинником є метанол, який дозволяє здійснити процедуру в одній посудині, з подальшими трансформаціями одержаних сполук формули (3) в сполуки формули (4). Присутня переважно син-форма приєднання сполуки формули (3). Співвідношення син/анти складає приблизно 8/2. Сполуки формули (4) 13 UA 100835 C2 5 10 15 20 25 30 35 40 Сполуки формули (4), тобто, -(4) і -(4), одержують за допомогою ряду трансформацій, починаючи зі сполук формули (3), з використанням реакції Нефа, у відповідне формільне похідне, одночасного зняття захисту діолу з кислотним каталізом і двох реакцій циклізації. Зазначені трансформації здійснюють дією на проміжні сполуки формули (3) основою, з подальшою дією на реакційну суміш кислотою в присутності метанолу, краще, додаванням або виливанням реакційної суміші в кислоту у присутності метанолу, внаслідок чого одержують сполуки формули (4). Згадані вище реакції також дають сполуки формули (4"). В реакції Нефа первинний або вторинні нітроалкан конвертують у відповідну карбонільну сполуку (N. Kornblum, Organic reactions 1962, 12, 101 і H.W. Pinnick, Organic Rеactions 1990, 38, 655). У класичній процедурі нітроалкан депротонують основою в -позиції до нітрофункції з подальшим кислотним гідролізом проміжної "нітронатної" соли шляхом додавання до сильної кислоти, присутньої в надлишку, з одержанням карбонільного похідного. Придатні основи може вибрати фахівець в області органічного синтезу. Придатні основи включають, без обмеження, неорганічні основи, такі як гідроксиди та алкоксиди лужних металів, лужноземельних металів і амонію. Прикладами придатних основ є діізопропіламід літію, метоксид натрію, метоксид калію, метоксид літію, т-бутоксид калію, гідроксид кальцію, гідроксид барію і гідроксиди четвертинного алкіламонію, DBN (1,3-діазабіцикло[3.4.0]нон-5-ен), DBU, DABCO (1,4-діазабіцикло[2.2.2]октан), TBAF, TMG, карбонат калію та карбонат натрію або їхні суміші. Кращими основами є метоксид натрію, метоксид калію, метоксид літію, TBAF, DBU, TMG або їхні суміші, ще кращими основами є метоксид натрію, метоксид літію, DBU або TMG або їхні суміші, і найкращим є метоксид натрію. Що стосується кислоти, можна використовувати будь-яку кислоту, краще, сильну кислоту, ще краще, мінеральну кислоту, таку як концентрована сірчана кислота, концентрована хлористоводнева кислота і, найкраще, концентрована сірчана кислота. Шляхом використання безводих умов або майже безводих умов, а також метанолу як розчинника в реакції Нефа, одержують циклічний метилацеталь формільної групи. Метильні замісники в проміжних сполуках формули (4) і (4") походять з метанольного розчинника. Альтернативно, якщо реакцію Нефа і попередню реакцію приєднання Міхаеля здійснюють в неметанольному розчиннику, наприклад, в ацетонітрилі, замість них утворюватимуться інші ацеталі, відмінні від сполук формули (4) і (4"), звичайно, суміш напівацеталей та алкілацеталей, 1 що відповідають заміснику R в сполуках формули (3). Зазначені напівацетальні та ацетальні споріднені сполуки можуть бути трансформовані в бажані метилацеталі формули (4) і (4") шляхом нової обробки останніх метанолом в кислотних умовах. Альтернативно, коли попередню реакцію приєднання Міхаеля здійснюють з використанням DBU або TMG і сполуки формули (3) не ізолюють, а подальшу реакцію Нефа здійснюють з використанням сильної основи, зокрема, метоксиду натрію або метоксиду літію, несподівано спостерігається значне підвищення виходу сполук формули (4). По суті, присутність DBU або TMG під час реакції Нефа з сильною основою є кращим варіантом здійснення даного винаходу. Наприклад, коли реакцію приєднання Міхаеля з нітрометаном здійснюють в метанолі з гідроксидами, алкоксидами або TBAF в різних кількостях, вихід сполук формули (3), в розрахунку на сполуки формули (2), складає приблизно 80 %. Коли подальші реакції Нефа і циклізації здійснюють з неізольованими сполуками формули (3), з використанням метоксиду 14 UA 100835 C2 5 10 15 20 25 30 35 40 45 50 55 60 натрію як додаткової основи і сірчаної кислоти в метанолі як кислотного розчину, можна одержати вихід сполуки формули (4) 43 %, в розрахунку на сполуки формули (2), із співвідношенням (4)/(4) щонайменше приблизно 3/1. Коли реакцію приєднання Міхаеля здійснюють з використанням приблизно 1 еквівалента DBU або TMG, в розрахунку на сполуки формули (2), вихід сполуки формули (3), в розрахунку на сполуки формули (2), також складає приблизно 80 %. Проте, коли згодом реакції Нефа і циклізації здійснюють з неізольованими сполуками формули (3), об'єднаними з 1,0 еквівалентом метоксиду натрію або літію, в розрахунку на сполуки формули (2), сполуку формули (4) можна одержати з 53-58 % виходом в розрахунку на сполуки формули (2), із співвідношенням (4)/(4) щонайменше приблизно 3/1. Біциклічні проміжні сполуки формули (4) є очікуваними продуктами циклізації, що походять від проміжних сполук формули (3) в син-конфігурації. Проміжні сполуки формули (4") є очікуваними продуктами реакції, що походять від проміжної сполуки формули (3) в антиконфігурації, яка не циклізується, а також очікуваними є продукти реакції, що походять від проміжної сполуки формули (3) в син-конфігурації, оскільки циклізація син-ізомерів зазвичай не завершується повністю. Транс-конфігурація замісників по атому вуглецю номер 3 (С-3) і атому вуглецю номер 4 (С-4) на тетрагідрофурановому кільці проміжної сполуки формули (4") запобігає утворенню лактонного кільця, як це спостерігається з проміжними сполуками формули (4). Краще, гасіння кислотою реакцій Нефа і циклізації здійснюють з використанням надлишку концентрованої сірчаної кислоти, краще, від 2 до 10 еквівалентів в розрахунку на сполуки формули (2), ще краще, від 2,5 до 5 еквівалентів, найкраще, від 3 до 4 еквівалентів і, найкраще, приблизно 3,5 еквівалентів, у вигляді розчину в метанолі від 20 % мас. до 80 % мас., краще, у вигляді розчину в метанолі від 40 % мас. до 60 % мас. Більший надлишок сірчаної кислоти дає вище співвідношення альфа/бета для сполуки формули (4), але також потребує більшої кількості основи для подальшої нейтралізації при лужному гасінні. Наприклад, коли 3,5 еквівалента сірчаної кислоти, в розрахунку на сполуки формули (2), використовують для кислотного гасіння у вигляді 50 % мас. розчину в метанолі, може досягатися співвідношення (4)/(4) до 4/1. Гасіння кислотою реакцій Нефа і циклізації можна здійснювати при температурі в межах приблизно від -40 °C до 70 °C, краще, при температурі в межах приблизно від -25 °C до 15 °C, ще краще, при температурі в межах приблизно від -20 °C до 5 °C, найкраще, при температурі в межах приблизно від -15 °C до 0 °C. Час реакції може варіювати аж до приблизно 24 годин, відповідно, в межах приблизно від 15 хвилин до 12 годин, ще краще, в межах приблизно від 20 хвилин до 6 годин. Для виділення сполук формули (4) може знадобитися обробка водою для видалення солей і частини проміжних сполук формули (4"). Основа нейтралізує раніше використану кислоту, оскільки кислотні водні умови викликають гідроліз метилацеталю сполуки формули (4) до напівацетальної спорідненої сполуки, що приводить до втрат продукту. По суті, виділення сполуки формули (4) оптимально здійснюється лужною реакцією гасіння, краще, водною лужною реакцією гасіння, з подальшою екстракцією сполуки формули (4) органічним розчинником, незмішуваним з водою. Краще, кислотну суміш, що утворюється в результаті реакцій Нефа і циклізації, додають до лужного водного розчину. Оскільки під час лужної водної реакції гасіння потрібний великий об'єм реактора, краще мінімізувати зазначений об'єм настільки, наскільки це можливо. Це можна здійснити різними способами, наприклад, шляхом застосування основ з високою розчинністю, або з використанням основ у формі суспензії. По суті, придатними основами для обробки сполук формули (4) є бікарбонат або карбонат, краще, бікарбонат натрію, калію, літію або цезію, найкраще, бікарбонат натрію або калію, найкраще, бікарбонат калію, як повністю в розчині, так і у формі суспензії. По суті, застосування насиченого розчину гідрокарбонату калію для лужного гасіння замість насиченого розчину гідрокарбонату натрію має, завдяки його вищій розчинності, ту перевагу, що об'єм водної фази може бути додатково зменшений, і несподівано виявилося, що утворюваний сульфат калію набагато краще фільтрується, ніж сульфат натрію. Краще, під час лужного гасіння рН підтримують в межах приблизно від 2 до 9, краще, приблизно від 3 до 8, ще краще, приблизно від 3,5 до 7,5. Також краще, в кінці лужного гасіння рН встановлюється приблизно від 3,5 до 6, краще, приблизно від 3,5 до 5, найкраще, приблизно від 3,8 до 4,5. Зазначені потрібні рівні рН можна забезпечити шляхом використання карбонатів і бікарбонатів, як зазначено вище. Необов'язково, можна використовувати додаткову основу або кислоту для підтримання рН на певному рівні до кінця реакції гасіння. У кращих межах рН метанол можна випаровувати з реакційної суміші після лужного гасіння і до екстракції 15 UA 100835 C2 5 10 15 20 25 30 35 40 45 50 55 60 органічним розчинником, при температурі в межах приблизно від 0 °C до 65 °C, краще, приблизно від 20 °C до 45 °C. За зазначених умов сполуки формули (4) не розкладаються, навіть при великій тривалості реакції. Видалення метанолу випаровуванням до екстракцій органічним розчинником має ту перевагу, що ефективність екстракції значно зростає, таким чином, витрачається менше органічного розчинника, і продуктивність ще більш зростає. Відповідними органічними розчинниками, що не змішуються з водою, є будь-який складний ефір, вуглеводень, простій ефір, галогенований вуглеводень або ароматичні розчинники. Зазначені розчинники включають, без обмеження, пентан, гексан, гептан, толуол, ксилол (ксилоли), бензол, мезитилен (мезитилени), т-бутилметиловий ефір, діалкілові ефіри (етиловий, бутиловий), дифеніловий ефір, хлорбензол, дихлорметан, хлороформ, чотирихлористий вуглець, ацетонітрил, дихлорбензол, 1,2-дихлоретан, 1,1,1-трихлоретан, етилацетат, ізопропілацетат, краще, етилацетат. Для того, щоб поліпшити вихід після екстракції сполук формули (4), можна додавати до суміші до екстракції водорозчинні солі. Краща сіль включає NaCl. Одна з переваг способу, описаного в даному винаході, в порівнянні з раніше відомим шляхом Knoevenagel, полягає в тому, що під час лужного водного гасіння не є необхідним одночасно екстрагувати сполуки формули (4) органічним розчинником. Відсутність органічного розчинника під час лужного гасіння додатково сприяє зменшенню об'єму реактора, а фільтрація неорганічних солей, що утворилися, відбувається набагато легше. При використанні шляху Knoevenagel потрібна присутність органічного розчинника під час лужного гасіння, щоб уникнути втрат продукту. Для того, щоб надалі ізолювати сполуку формули -(4), можна використати кристалізацію зазначеної сполуки. Кристалізація Сполуку формули -(4) можна кристалізувати з розчинника, такого як органічні, неорганічні розчинники або вода та їхні суміші. Придатні розчинники для кристалізації включають ізопропанол, т-аміловий спирт, т-бутанол, етилацетат, етанол і метилізобутилкетон. Особливо кращими є ізопропанол, т-аміловий спирт і т-бутанол, оскільки вони дають високий вихід кристалізації та продукт високого ступеня чистоти. Ще краще використовувати ізопропанол або т-аміловий спирт, найкраще, ізопропіловий спирт. У випадку, якщо розчинником, використовуваним для кристалізації, є ізопропіловий спирт, краща концентрація перед кристалізацією сполуки формули -(4) складає приблизно від 5 до 30 % мас., ще краще, приблизно від 10 до 25 % мас., найкраще, приблизно від 15 до 20 % мас. Кристалізація дає сполуку -(4) високої чистоти, хоча можуть бути присутніми малі кількості сполуки формули -(4), тобто, менш ніж приблизно 5 %, зокрема, в кількостях менше приблизно 3 %. Епімерізація Сполука формули (4) в її бета-ізомерній формі може бути епімеризована в сполуку формули -(4) з використанням кислоти, наприклад, органічної або неорганічної кислоти, краще, у відсутності води і в присутності метанолу. Епімерізацію краще здійснюють з використанням MeSO3H в метанолі або з використанням будь-якої порівнянної кислоти схожої сили, оскільки це запобігає утворенню побічних продуктів. Краще, застосовувана кількість MeSO3H в метанолі знаходиться в межах приблизно від 0,05 до 1,5 еквівалента в розрахунку на сполуки формули (4), ще краще, приблизно від 0,1 до 0,3 еквівалента. Температура для здійснення епімеризації складає приблизно від 0 °C і приблизно до температури кипіння із зворотним холодильником, краще, приблизно від 20 °C і приблизно до температури кипіння із зворотним холодильником, найкраще, приблизно при температурі кипіння із зворотним холодильником. Для деяких зі способів, описаних вище, існують декілька альтернативних способів. Наприклад, в одному варіанті здійснення даного винаходу, після одержання суміші сполуки формули -(4) і сполуки формули -(4), сполуку формули -(4) кристалізують, і процедуру синтезу продовжують для одержання сполуки формули (6). В іншому варіанті здійснення даного винаходу фахівець може вибрати кристалізацію сполуки формули -(4), здійснення епімеризації маточного розчину, що залишився, який містить відносно велику кількість небажаного епімеру -(4), з одержанням суміші з відносно великою кількістю епімеру -(4), і здійснення другої кристалізації епімеру -(4). Наприклад, коли кристалізують необроблену суміш сполуки формули (4), що має співвідношення -(4)/-(4) в межах приблизно від 3,5/1 до 4/1, виділяють перший збір -(4), а маточний розчин, що залишився, має співвідношення -(4)/-(4) в межах приблизно від 0,3/1 до 1,5/1. Після епімеризації епімеру -(4) співвідношення -(4)/-(4) в 16 UA 100835 C2 5 10 15 20 25 30 35 40 45 50 маточному розчині складає приблизно 3/1, і одержують другий збір -(4) шляхом кристалізації, що має чистоту, щонайменше, порівнянну з чистотою першого збору -(4). Альтернативно, можна здійснювати одночасну кристалізацію епімеру -(4) і реакцію епімеризації -(4) в епімер -(4). В іншому варіанті здійснення даного винаходу можна почати з епімеризації -(4) в епімер -(4) і згодом кристалізувати епімер -(4). У ще одному варіанті здійснення даного винаходу можна почати з епімеризації -(4) в епімер -(4) і згодом кристалізувати епімер -(4), застосовуючи другу епімеризацію маточного розчину, що залишився, та додаткову кристалізацію, що дає другий збір епімеру -(4). По суті, в одному варіанті здійснення даного винаходу маточний розчин попередньої кристалізації сполуки формули -(4) з ізопропанолу можна епімеризувати випаровуванням ізопропанолу, перенесенням залишку в метанол і кип'ятінням зі зворотним холодильником протягом приблизно від 30 хвилин до 4 годин з MeSO 3H, краще, в кількості приблизно від 0,1 до 0,3 еквівалента. Якщо реакційну суміш згодом виливають у водний NaHCO 3, екстрагують EtOAc і органічнуфазу в розчиннику поміщають в ізопропанол, то можна одержати другу порцію чистої сполуки формули -(4) шляхом кристалізації. У кращому варіанті здійснення даного винаходу суміш епімерів -(4) і -(4) може бути трансформована в одній стадії в 100 % або майже 100 % альфа-ізомер, зі 100 % або майже 100 % виходом, тобто, без утворення побічних продуктів, шляхом прямої кристалізації епімеру -(4) та одночасної епімеризації -(4) в епімер -(4); зазначений процес відомий як індукована кристалізацією асиметрична трансформація. Індуковану кристалізацією асиметричну трансформацію можна здійснити розчиненням суміші епімерів -(4) та -(4) в метанолі у присутності приблизного 0,10 еквівалента MeSO 3H, в розрахунку на суму обох епімерів, і випаровуванням метанолу в умовах вакууму при температурі приблизно від 30 °C до 40 °C. Даний варіант здійснення даного винаходу є особливо кращим, оскільки суміш епімерів -(4) і -(4) можна трансформувати тільки в епімер -(4) в одну стадію, що знижує витрати на виробництво і дає одну партію -(4) гомогенної якості. У кращому варіанті здійснення даного винаходу нейтралізацію кислоти, такої як MeSO 3H, здійснюють до заміни розчинника з метанолу на розчинник для кристалізації, такий як ізопропанол. Зазначену нейтралізацію можна здійснювати додаванням невеликого молярного надлишку основи, в розрахунку на використану для епімеризації кислоту. Як основу можна використовувати будь-яку основу, якщо тільки сіль основи з кислотою, використаною для епімеризації, не контамінує кристали епімеру -(4). Наприклад, у разі використання MeSO3H як кислоти для епімеризації, можна використовувати третинний амін, краще, триетиламін, що дає метансульфонатну сіль триетиламонію, яка не контамінує кристали епімеру -(4) під час кристалізації з ізопропанолу. Додавання NЕt3 в невеликому надлишку порівняно з MeSO3H для нейтралізації дозволяє уникнути утворення ізопропілових ацеталів як побічних продуктів, які могли б утворитися в кислотних умовах під час подальшої заміни розчинника з метанолу на ізопропанол. Подальша заміна розчинника з метанолу на ізопропанол і кристалізації дають сполуку формули -(4) високої чистоти, без контамінації або з мінімальною контамінацією метансульфонатною сіллю триетиламонію. Сполука формули (6) Сполуку формули (6) одержують відновленням сполуки формули -(4) з подальшою реакцією циклізації. Проміжною сполукою при відновленні сполуки формули -(4) є сполука формули (5). Сполуку формули (5), краще, не ізолюють, а безпосередньо піддають циклізації, з одержанням сполуки формули (6). Стадію відновлення можна належним чином здійснити дією на проміжну сполуку формули -(4) гідридами металів, такими як борогідрид літію, борогідрид натрію, борогідрид натриюхлорид літію, у придатних безводих розчинниках. Приклади придатних безводих розчинників включають, без обмеження, дихлорметан, 17 UA 100835 C2 5 10 15 20 25 30 35 40 45 50 толуол, ксилол, бензол, пентан, гексан, гептан, петролейний ефір, 1,4-тіоксан, діетиловий ефір, діізопропіловий ефір, тетрагідрофуран, 1,4-діоксан, 1,2-диметоксіетан і загалом будь-який безводий розчинник, придатний для використання в процесі хімічного відновлення з використанням відновників, перелічених вище. Кращим розчинником є тетрагідрофуран. Згідно з кращим варіантом здійснення даного винаходу, стадію відновлення здійснюють з використанням борогідриду літію або борогідриду натрію в тетрагідрофурані. У разі використання борогідриду літію як відновника, кількість відновника знаходиться в межах приблизно від 1 до 1,5 еквівалентів в розрахунку на сполуку формули -(4), краще, приблизно від 1,1 до 1,3 еквівалента. Зазначену стадію відновлення можна здійснювати при температурі приблизно від -78 °C до 55 °C, краще, приблизно від -15 °C до 45 °C і, найкраще, приблизно від 0 °C до 40 °C. Час реакції може складати приблизно до 24 годин, і звичайно варіює приблизно між 2 і 24 годинами. Сполуку формули (5) можна конвертувати в бажану сполуку формули (6) за допомогою реакції циклізації. Реакція циклізації відбувається через інтрамолекулярну трансацеталізацію і може здійснюватися в будь-якому сумісному з кислотою органічному розчиннику або в комбінації розчинника, що змішується з водою, та води, у присутності сильної органічної або неорганічної кислоти. Зазначену реакцію відповідно здійснюють дією на сполуку формули (5) каталітичної кількості сильної кислоти. У кращому варіанті здійснення даного винаходу сильну кислоту вибирають з групи, що складається з хлористоводневої кислоти і сірчаної кислоти в тетрагідрофурані. Зазначену стадію циклізації краще здійснюють при температурі нижче приблизно 5 °C, ще краще, нижче приблизно -5°С. В особливо кращому варіанті здійснення даного винаходу на сполуку формули (5), яку після відновлення борогідридом літію або натрію в тетрагідрофурані одержують у вигляді комплексу з бором, діють концентрованою мінеральною кислотою, і декомплексацію сполуки формули (5) та циклізацію сполуки формули (5) до сполуки формули (6) здійснюють одночасно. Краще, використовують сильну мінеральну кислоту, ще краще, концентровану сірчану кислоту або концентровану хлористоводневу кислоту, найкраще, концентровану хлористоводневу кислоту. Кількість хлористоводневої кислоти може варіювати від 1,0 до 1,4 еквівалента в розрахунку на кількість використаного борогідриду літію або натрію, але краще, від 1,1 до 1,3 еквівалента. Що стосується виділення сполуки формули (6) в чистій формі, бажано видалити неорганічні солі, що утворюються з реагентів, використаних для стадій відновлення, декомплексації та циклізації. Це можна здійснити процедурою екстракції з використанням водно-органічного розчинника, але, краще, це роблять додаванням невеликого надлишку основи в порівнянні з кислотою, використаною для декомплексації сполуки формули (5) та її реакції циклізації до сполуки формули (6). Згодом розчинник заміняють на більш неполярний розчинник, що приводить до випадання в осад солей, утворюваних в результаті відновлення та декомплексації. Як основу, використовувану для обробки сполуки формули (6), можна використовувати будь-яку основу, якщо тільки розчинність її солі з мінеральною кислотою, використаною для декомплексації та реакцій циклізації сполуки формули (5) в сполуку формули (6), в кінцевому розчиннику після зміни розчинника є низькою. Наприклад, якщо борогідрид літію в тетрагідрофурані використовують для відновлення, концентровану водну HCl використовують для декомплексації/циклізації, і етилацетат є кінцевим розчинником, то третинні аміни є придатними основами для нейтралізації кислоти, особливо, триетиламін. В даному випадку солі бору і гідрохлорид триетиламіну майже повністю випадають в осад, а сполука формули (6) повністю залишається в розчині. Після відфільтровування твердих речовин залишається розчин сполуки формули (6) з високою чистотою, яка може бути перероблена в будь-яку бажану форму. Було помічено, що інший енантіомер сполуки формули (6), а саме, сполука формули (6d), (3S, 3aR, 6aS)-гексагідрофуро[2,3-b]фуран-3-ол, є також активною частиною інгібіторів ВІЛпротеїнази. 18 UA 100835 C2 5 10 15 20 25 30 По суті, способи, процедури, реагенти і умови, ідентичні описаним в даному винаході, включаючи відповідну кристалізацію і епімеризацію, можна застосовувати для одержання сполуки формули (6d), з використанням сполуки формули (1d), її попередників та інших проміжних сполук для одержання сполуки формули (6d), таких як сполука формули (4d) нижче. Сполуки формули (6) і (6d) знаходять своє особливе застосування для виготовлення лікарського засобу. Згідно з кращим варіантом здійснення даного винаходу, дані сполуки формули (6) і (6d) використовують як попередники при виготовленні противірусних лікарських засобів, зокрема, лікарських засобів проти ВІЛ, конкретніше, інгібіторів ВІЛ-протеїнази. Сполука формули (6) і всі проміжні сполук, що ведуть до утворення зазначеної стереоізомерно чистої сполуки, представляють особливий інтерес для виготовлення інгібіторів ВІЛ-протеїнази, як описано у WO 95/24385, WO 99/65870, WO 00/47551, WO 00/76961 і US 6127372, WO 01/25240, EP 0715618 і WO 99/67417, включених в цей документ як посилання, зокрема, наступних інгібіторів ВІЛ-протеїнази: (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-іловий ефір [(1S, 2R)-2-гідрокси-3-[[(4метоксифеніл)сульфоніл](2-метилпропіл)амін]-1-(фенілметил)пропіл]-карбамінової кислоти (інгібітор ВІЛ-протеїнази 1); (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-іловий ефір [(1S, 2R)-3-[[(4амінофеніл)сульфоніл](2-метилпропіл)амін]-2-гідрокси-1-(фенілметил)пропіл]-карбамінової кислоти (інгібітор ВІЛ-протеїнази 2); (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-іловий ефір [(1S, 2R)-3-[(1,3-бензодіоксол-5ілсульфоніл)(2-метилпропіл)амін]-2-гідрокси-1-(фенілметил)-пропіл]карбамінової кислоти (інгібітор ВІЛ-протеїнази 3), або будь-яка їхня фармацевтично прийнятна сіль. Таким чином, даний винахід стосується також інгібіторів ВІЛ-протеїнази 1, 2, 3 або будь-якої їхньої фармацевтично прийнятної солі або проліків, одержаних з використанням сполуки формули (6), одержаної згідно з даним винаходом, для хімічного синтезу зазначених інгібіторів ВІЛ-протеїнази. Зазначений хімічний синтез описаний в літературі, наприклад, у WO 01/25340, EP 0715618 і WO 99/67417. По суті, інгібітори протеїнази, згадані вище, можна одержати з використанням наступної загальної процедури. N-захищений аміноепоксид формули 2 35 де Р є амінозахисною групою, а R позначає радикали алкіл, арил, циклоалкіл, циклоалкілалкіл і аралкіл; зазначені радикали, необов'язково, заміщені групою, вибраною з 9 9 9 радикалів алкілу і галогену, нітро, ціано, трифторметил, -OR і –SR , де R позначає водень, алкільні і галогенові радикали; одержують з відповідного хлоркетону в присутності основи і системи розчинників. Придатні системи розчинників для одержання аміноепоксиду включають етанол, метанол, ізопропанол, тетрагідрофуран, діоксан і т.п., включаючи їхні суміші. Придатні основи для одержання епоксиду з відновленого хлоркетону включають гідроксид калію, 19 UA 100835 C2 гідроксид натрію, т-бутоксид калію, DBU і т.п. Альтернативно, захищений аміноепоксид можна одержати, починаючи з L-амінокислоти, яка взаємодіє з придатною амінозахисною групою у придатному розчиннику, з одержанням амінозахищеного ефіру L-амінокислоти формули: 5 10 15 20 де Р’’’ означає карбоксилзахисну групу, наприклад, метил, етил, бензил, третинний бутил і 2 т.п.; R визначений вище; і P" та P’’ незалежно вибрані з амінозахисних груп, включаючи, без обмеження, арилалкіл, заміщений арилалкіл, циклоалкенілалкіл і заміщений циклоалкенілалкіл, аліл, заміщений аліл, ацил, алкоксикарбоніл, аралкоксикарбоніл і силіл. Крім того, захисні групи P" та/або P’’ можуть утворювати гетероциклічне кільце з атомом азоту, до якого вони приєднані, наприклад, 1,2-біс(метилен)бензол, фталімідил, сукцинімідил, малеімідил і т.п.; причому зазначені гетероциклічні групи можуть додатково включати суміжні арильні і циклоалкільні кільця. Крім того, гетероциклічні групи можуть бути моно-, ди- або тризаміщеними, наприклад, нітрофталімідил. Амінозахищений ефір L-амінокислоти потім відновлюють до відповідного спирту. Наприклад, амінозахищений ефір L-амінокислоти можна відновити гідридом діізобутилалюмінію при -78 °C у придатному розчиннику, такому як толуол. Кращі відновники включають гідрид літію-алюмінію, борогідрид літію, борогідрид натрію, боран, гідрид літію-тритретбутоксіалюмінію, комплекс боран/ТГФ. Одержаний спирт потім конвертують, наприклад, шляхом окиснення Swern, до відповідного альдегіду формули: 2 25 30 35 40 45 50 де Р", P’’ і R визначені вище. Таким чином, розчин спирту в дихлорметані додають до охолодженого (від -75 °C до -68 °C) розчину оксалілхлориду в дихлорметані і ДМСО в дихлорметані і перемішують протягом 35 хвилин. Прийнятні окисні агенти включають, наприклад, комплекс триоксиду сірки-піридину і ДМСО, оксалілхлорид і ДМСО, ацетилхлорид або ангідрид і ДМСО, трифторацетилхлорид або ангідрид і ДМСО, метансульфонілхлорид і ДМСО або тетрагідротіафен-S-оксид, толуолсульфонілбромід і ДМСО, трифторметансульфонілангідрид (ангідрид трифторметансульфокислоти) і ДМСО, пентахлорид фосфору і ДМСО, диметилфосфорилхлорид і ДМСО та ізобутилхлорформіат і ДМСО. Альдегіди даного процесу можна також одержати шляхом відновлення захищеного фенілаланіну та аналогів фенілаланіну або їхніх амідних чи складноефірних похідних, наприклад, амальгамою натрію з HCl в етанолі або літієм чи натрієм чи калієм чи кальцієм в аміаку. Температура реакції може становити приблизно від -20 °C до 45°, краще, приблизно від 5 °C до 25°. Два додаткові способи одержання азот-захищеного альдегіду включають окиснення придатного спирту хлорним вапном в присутності каталітичної кількості вільного радикала 2,2,6,6-тетраметил-1-піридилокси. В другому способі, окиснення спирту до альдегіду здійснюють з використанням каталітичної кількості перрутенату тетрапропіламонію в присутності N-метилморфолін-N-оксиду. Альтернативно, хлорангідридне похідне захищеного фенілаланіну або похідні фенілаланіну, описані вище, можна відновлювати воднем і каталізатором, таким як Pd на карбонаті барію або сульфаті барію, з додатковим агентом, що уповільнює каталізатор, таким як сірка або тіол (відновлення Rosenmund) або без нього. Альдегід, що утворюється в результаті окиснення Swern, потім вводять в реакцію з реагентом літійгалоїдметилом, який утворюється in situ взаємодією алкіллітію або сполук 1 2 1 2 ариллітію з дигалогенметаном, представленим формулою X CH2 × , де Х і Х незалежно представляють йод, бром або хлор. Наприклад, розчин альдегіду і хлорйодметану в ТГФ охолоджують до -78 °C і додають розчин н-бутиллітію в гексані. Одержаний продукт є сумішшю діастереоізомерів відповідних амінозахищених епоксидів формул: 20 UA 100835 C2 510 15 20 25 30 35 40 45 50 55 та Діастереоізомери можна розділити, наприклад, шляхом хроматографії або, альтернативно, можна розділити діастереоізомерні продукти після того, як вони прореагували, на подальших стадіях. Для сполук, що мають (S)-стереохімію, замість L-амінокислоти можна використовувати D-амінокислоту. Додавання хлорметиллітію або бромметиллітію до хірального аміноальдегіду є високо діастереоселективним. Краще, хлорметиллітій або бромметиллітій утворюються in situ в результаті реакції дигалогенметану і н-бутиллітію. Прийнятні метиленувальні галогенметани включають хлорйодметан, бромхлорметан, дибромметан, дийодметан, бромфторметан і т.п. Сульфонатний ефір продукту приєднання, наприклад, гідроброміду до формальдегіду, також є метиленувальним агентом. Тетрагідрофуран є кращим розчинником, проте, можна використовувати альтернативні розчинники, такі як толуол, диметоксіетан, етилендихлорид, метилендихлорид, як чисті розчинники або у вигляді суміші. Біполярні апротонні розчинники, такі як ацетонітрил, ДМФ, Nметилпіролідон, є придатними як розчинники або частина суміші розчинників. Реакцію можна здійснювати в інертній атмосфері, такій як азот або аргон. Н-бутиллітій може бути заміщений іншими металорганічними реагентами, такими як метиллітій, трет-бутиллітій, втор-бутиллітій, феніллітій, фенілнатрій і т.п. Реакцію можна проводити при температурі приблизно від -80 °C до 0 °C, краще, приблизно від -80 °C до -20 °C. Конверсію альдегідів в їхні епоксидні похідні також можна здійснювати в декілька стадій. Наприклад, додавання аніону тіоанізолу, одержаного, наприклад, з реагенту бутил- або ариллітію, до захищеного аміноальдегіду, окиснення одержаного захищеного аміносульфідного спирту з використанням добре відомих окисних агентів, таких як пероксид водню, третбутилгіпохлорит, хлорне вапно або періодат натрію, дає сульфоксид. Алкілування сульфоксиду, наприклад, метилйодидом або бромідом, метилтозилатом, метилмезилатом, метилтрифлатом, етилбромідом, ізопропілбромідом, бензилхлоридом і т.п., здійснюють в присутності органічної або неорганічної основи. Альтернативно, захищений аміносульфідний спирт можна алкілувати, наприклад, алкілувальними агентами, переліченими вище, з одержанням солей сульфонію, які згодом конвертують в зазначені епоксиди третинним аміном або мінеральними основами. Бажані епоксиди утворюються при використанні найкращих умов, діастереоселективно, в кількісному співвідношенні щонайменше близько 85:15 (S:R). Продукт можна очищати хроматографією, з одержанням діастереоізомерно і енантіомерно чистого продукту, але набагато зручніше використовувати його безпосередньо, без очищення, для виготовлення інгібіторів ретровірусної протеїнази. Вищезазначений процес є застосовним до сумішей оптичних ізомерів, а також до розділених сполук. Якщо бажаним є конкретний оптичний ізомер, його можна вибрати шляхом вибору вихідного матеріалу, наприклад, L-фенілаланіну, Dфенілаланіну, L-фенілаланінолу, D-фенілаланінолу, D-гексагідрофенілаланінолу і т.п., або можна здійснювати розділення на проміжних чи кінцевих стадіях. Хіральні допоміжні речовини, такі як один або два еквіваленти камфорсульфонової кислоти, лимонної кислоти, камфорної кислоти, 2-метоксифенілоцтової кислоти і т.п., можна використовувати для утворення солей, складних ефірів або амідів сполук за даним винаходом. Зазначені сполуки або похідні можна кристалізувати або розділяти хроматографічно, з використанням хіральної або ахіральної колонок, як добре відомо фахівцям. Потім аміноепоксид взаємодіє у придатній системі розчинників з рівною кількістю або, 3 3 краще, надлишком бажаного аміну формули R NH2, де R позначає водень, алкіл, галогеналкіл, алкеніл, алкініл, гідроксіалкіл, алкоксіалкіл, циклоалкіл, циклоалкілалкіл, гетероциклоалкіл, гетероарил, гетероциклоалкілалкіл, арил, аралкіл, гетероаралкіл, аміноалкіл та моно- і дизаміщені аміноалкільні радикали, де зазначені замісники вибрані з радикалів алкілу, арилу, аралкілу, циклоалкілу, циклоалкілалкілу, гетероарилу, гетероаралкілу, гетероциклоалкілу і гетероциклоалкілалкілу, або, у разі дизаміщеного аміноалкільного радикала, зазначені замісники, разом з атомом азоту, до якого вони приєднані, утворюють гетероциклоалкільний або гетероарильний радикал. Реакцію можна проводити в широких температурних межах, наприклад, приблизно від 10 °C до 100 °C, але краще, хоч і не необхідно, проводити реакцію при температурі, при якій 21 UA 100835 C2 5 10 15 20 25 30 35 40 розчинник починає кипіти із зворотнимхолодильником. Придатні системи розчинників включають протонні, непротоннні та біполярні апротоннні органічні розчинники, такі як, наприклад, системи, в яких розчинником є спирт, такий як метанол, етанол, ізопропанол і т.п., ефіри, такі як тетрагідрофуран, діоксан і т.п., і толуол, N, Nдиметилформамід, диметилсульфоксид та їхні суміші. Кращим розчинником є ізопропанол. 3 Приклади амінів, що відповідають формулі R NH2, включають бензиламін, ізобутиламін, нбутиламін, ізопентиламін, ізоаміламін, циклогексанметиламін, нафталінметиламін і т.п. 2 3 Одержаний продукт є похідним 3-(N-захищений амін)-3-(R )-1-(NHR )пропан-2-олу, який далі в цьому документі називається аміноспиртом, і представлений формулами: 2 3 де Р, Р", P’’, R і R визначені вище. Альтернативно, замість аміноепоксиду можна використовувати галогеноспирт. Аміноспирт, визначений вище, потім взаємодіє у придатному розчиннику з 4 сульфонілхлоридом (R SO2Cl) або сульфонілангідридом в присутності акцептора кислоти. Придатні розчинники, в яких можна проводити реакцію, включають метиленхлорид, тетрагідрофуран. Придатні акцептори кислоти включають триетиламін, піридин. Кращими сульфонілхлоридами є метансульфонілхлорид і бензолсульфонілхлорид. Одержане сульфонамідне похідне може бути представлене, залежно від використаного епоксиду, формулами: 2 3 4 де Р, Р", P’’, R , R і R визначені вище. Дані проміжні сполук є придатними для виготовлення інгібіторів протеїназ, і також є активними інгібіторами ретровірусних протеїназ. 4 Сульфонілгалогеніди формули R SO2X можна одержати взаємодією придатного реагенту Ґриняра або алкіллітію із сульфурилхлоридом або діоксидом сірки, з подальшим окисненням галогеном, краще, хлором. Також можна окисляти тіоли до сульфонілхлоридів, використовуючи хлор в присутності води за ретельно контрольованих умов. Крім того, сульфонові кислоти можна конвертувати в сульфонілгалогеніди, використовуючи такі реагенти, як PCl 5, а також в ангідриди, використовуючи придатні дегідратуючі реагенти. Сульфонові кислоти, у свою чергу, можна одержати, використовуючи процедури, добре відомі фахівцям. Зазначені сульфонові кислоти також є комерційно доступними. Замість сульфонілгалогенідів можна використовувати 4 4 сульфінілгалогеніди (R SOX) або сульфенілгалогеніди (R SX) для одержання сполук, в яких частина -SO2- замінена на частину -SO- або -S-, відповідно. Після одержання сульфонамідного похідного амінозахисну групу Р або амінозахисні групи P" і P’’ видаляють за таких умов, що не впливають на частину молекули, що залишається. Зазначені способи добре відомі фахівцям і включають кислотний гідроліз, гідрогеноліз і т.п. Кращий спосіб включає видалення захисної групи, наприклад, видалення карбобензоксигрупи шляхом гідрогенолізу, з використанням паладію на вугіллі, у придатній системі розчинників, такій як спирт, оцтова кислота і т.п. або їхні суміші. Якщо захисною групою є тбутоксикарбонільна група, її можна видалити, використовуючи неорганічну або органічну кислоту, наприклад, HCl або трифтороцтову кислоту, у придатній системі розчинників, наприклад, в діоксані або метиленхлориді. Одержаний продукт є сіллю аміну формули: Зазначений амін може бути приєднаний до карбоксилату, представленого формулою: 22 UA 100835 C2 5 10 15 20 25 30 35 40 45 50 55 де R є групою (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-окси, а L є придатною відхідною групою, такою як галогенід. Розчин вільного аміну (або аміноацетатної солі) і приблизно 1,0 еквівалент карбоксилату змішують у придатній системі розчинників і, необов'язково, діють основою в кількості до п'яти еквівалентів, такою як, наприклад, N-метилморфолін, приблизно при кімнатній температурі. Придатні системи розчинників включають тетрагідрофуран, метиленхлорид або N, N-диметилформамід і т.п., включаючи їхні суміші. Альтернативно, амін може бути приєднаний до активованого сукцинімідилкарбонату (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-олу. Активацію (3R, 3aS, 6aR)-гексагідрофуро[2,3b]фуран-3-олу можна здійснити, наприклад, взаємодією з дисукцинімідилкарбонатом і триетиламіном. Приклади Наступні приклади призначені для ілюстрації даного винаходу. Зазначені приклади представлені як приклади даного винаходу і не повинні тлумачитися як такі, що обмежують обсяг винаходу. Всі реакції здійснювали в атмосфері азоту. Розчинники і реагенти використовували в тому 1 вигляді, в якому вони були придбані, без додаткового очищення. Спектри Н ЯМР записували 1 при 200 мГц в CDCl3 або ДМСО-d6 на ЯМР спектрометрі Bruker AC-200. Кількісний Н ЯМР здійснювали з хлорбензолом як внутрішнім стандартом. Всі зазначені виходи були скориговані з урахуванням домішок в продукті. Газову хроматографію (GC) і визначення S-2,3-O-ізопропіліденгліцеральдегіду з похибкою в допустимих межах в реакційних сумішах здійснювали з використанням Agilent 6890 GC (EPC) і колонки Betadex (шифр 24305, Supelco або еквівалент) 60 м та з товщиною плівки 0,25 мкм, з використанням тиску на головці колонки 26,4 кПа, об'ємній витраті в колонці 1,4 мл/хв., роздільного потоку 37,5 мл/хв. і температури інжекції 150 °C. Використовували такий температурний режим: вихідна температура 60 °C (3 хв.), швидкість 5 °C/хв., проміжна температура 130 °C (1 хв.), швидкість 25 °C/хв., кінцева температура 230 °C (8 хв.). Детектування виконували на детекторі FID при температурі 250 °C. Час утримання становив: хлорбензол (внутрішній стандарт) 13,9 хв., S-2,3-O-ізопропіліденгліцеральдегід 15,9 хв., R-2,3O-ізопропіліденгліцеральдегід 16,2 хв. Аналіз GC і визначення енантіомерного надлишку R-3-(2,2-диметил-[1,3]діоксолан-4іл)акрилової кислоти з похибкою в допустимих межах здійснювали з використанням описаного вище устаткування, але з використанням температури інжекції 250 °C. Використаний температурний режим: вихідна температура 80 °C (1 хв.), швидкість 5 °C/хв., кінцева температура 225 °C (10 хв.). Детектування здійснювали на детекторі FID при температурі 250 °C. Час утримання становив: толуол 7,3 хв., хлорбензол (внутрішній стандарт) 9,4 хв., S-2,3O-ізопропіліденгліцеральдегід 10,7 хв., R-2,3-O-ізопропіліденгліцеральдегід 10,9 хв., етиловий ефір Z-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти 20,4 хв., етиловий ефір E-R-3-(2,2диметил-[1,3]діоксолан-4-іл)акрилової кислоти 22,6 хв., етиловий ефір E-S-3-(2,2-диметил[1,3]діоксолан-4-іл)акрилової кислоти 22,9 хв., триетилфосфоноацетат (ТЕРА) 25,5 хв. Аналіз GC сполук -(4) і -(4) здійснювали з використанням Agilent 6890 GC (EPC) і колонки CP-Sil 5 CB (шифр CP7680 (Varian) або еквівалент) 25 м і з товщиною плівки 5 мкм, з використанням тиску на головці колонки 5,1 кПа, роздільного потоку 40 мл/хв. і температури інжекції 250 °C. Використаний температурний режим: вихідна температура 50 °C (5 хв.), швидкість 10 °C/хв., кінцева температура 250 °C (15 хв.). Детектування здійснювали на детекторі FID при температурі 250 °C. Час утримання становив: хлорбензол (внутрішній стандарт) 17,0 хв., -(4) 24,9 хв., -(4) 25,5 хв. Приклад 1: Одержання S-2,3-O-ізопропіліденгліцеральдегіду і конверсія в етиловий ефір R3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти До добре перемішуваної суспензії KIO4 (530 г, 2,3 моль, 2,3 экв.), KHCO3 (230 г, 2,3 моль, 2,3 экв.) у воді (1200 г) по краплях додавали розчин L-5,6-O-ізопропіліденгулоно-1,4-лактону (218,5 г, 1 моль) у воді (135 г) і тетрагідрофурані (1145 г) протягом 3 год. при 32-34 °C. Реакційну суміш перемішували протягом 4,5 год. при 32 °C. Згідно GC, окиснення було завершене, оскільки вміст S-2,3-O-ізопропіліденгліцеральдегіду складав 4,38 % мас. і більше не зростав. Реакційну суміш охолоджували до 5 °C і витримували при зазначеній температурі протягом 14 год. Тверді речовини (що складалися, головним чином, з KIO 3) видаляли фільтруванням і осад, що залишився на фільтрі, промивали тетрагідрофураном (115 мл) і ще однією порцією 23 UA 100835 C2 5 10 15 20 25 30 35 40 45 50 55 60 тетрагідрофурану (215 мл) шляхом ресуспендирования. З фільтрату (2975 г) брали зразок і 1 аналізували методом кількісного Н ЯМР (ДМСО-d6), який показав, що вміст S-2,3-Oізопропіліденгліцеральдегіду у фільтраті складав 3,69 % мас., що відповідало 109,6 г (0,843 моль) і виходу 84 %, в розрахунку на L-5,6-O-ізопропіліденгулоно-1,4-лактон. До 2953 г одержаного фільтрату (що містив 108,8 г = 0,837 моль S-2,3-Oізопропіліденгліцеральдегіду) при 13 °C по краплях додавали при перемішуванні триетилфосфоноацетат (ТЕРА, 194,7 г, 97 % чистота, 0,843 моль, 1,01 экв.) протягом 25 хв. при 13-17 °C. Потім порціями додавали K2CO3 (838 г, 6,07 моль, 7,26 экв.) протягом 30 хв. при 1725 °C. Кінцева величина рН реакційної суміші становила 11,6. Реакційну суміш перемішували протягом ще 17 год. при 20 °C. Водну і тетрагідрофуранову фази розділяли, і водну фазу екстрагували двічі 660 мл толуолу. Об'єднані тетрагідрофуранову і толуолову фази концентрували під вакуумом (260-25 мбар, температура 28-56 °C) протягом 8 год., з одержанням 175,5 г рідини світло-жовтого кольору. 1 Кількісний Н ЯМР показав вміст 78 % мас. етилового ефіру Е-R-3-(2,2-диметил[1,3]діоксолан-4-іл)акрилової кислоти, 2,5 % мас. етилового ефіру Z-3-(2,2-диметил[1,3]діоксолан-4-іл)акрилової кислоти, 4,4 % мас. ТЕРА (4,1 % мол. від вихідної кількості) і 6,8 % мас. толуолу. Це відповідає загальному виходу етилового ефіру R-3-(2,2-диметил[1,3]діоксолан-4-іл)акрилової кислоти 141,2 г (0,706 моль), що склало 71 % виходу в розрахунку на L-5,6-O-ізопропіліденгулоно-1,4-лактон і 84 % виходу в розрахунку на S-2,3-Oізопропіліденгліцеральдегіду. GC показала, що енантіомерний надлишок (е.е.) етилового ефіру Е-R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти становить >99 %. Приклад 2: Одержання суміші сполук -(4) і -(4) з етилового ефіру R-3-(2,2-диметил[1,3]діоксолан-4-іл)акрилової кислоти, з використанням різних типів і кількостей основ, без виділення сполук приєднання нітрогрупи Приклад 2А: Застосування DBU в реакції приєднання Міхаеля і NaOMe як додаткової основи в реакції Нефа До етилового ефіру R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти (21,2 г маслянистої рідини, чистота 94,5 % мас., 0,1 моль) додавали нітрометан (13,0 г 51,7 % мас. розчину в метанолі, 0,11 моль, 1,1 экв.) і розчин охолоджували до 0 °C. Потім по краплях додавали DBU (15,2 г, 0,1 моль, 1 экв.) протягом 25 хв. і воронку промивали метанолом (1 г). Реакційну суміш нагрівали до 20 °C і перемішували при вказаній температурі протягом 17 год. Одержаний розчин (50 г) розділяли на дві рівні частини; одну 25-грамову частину далі обробляли як описано в прикладі 2В. Іншу 25 г частину охолоджували до 0 °C і по краплях додавали NaOMe (10,0 г 29,6 % мас. розчину в метанолі, 0,055 моль, 1,1 экв.) протягом 10 хв. при 0 °C і воронку промивали метанолом (1,6 г). Реакційну суміш перемішували протягом 50 хв. при 0 °C, а потім гасили в розчині H2SO4 (17,9 г, 96 % мас., 0,175 моль, 3,5 экв.) в метанолі (30,4 г) при 0-5 °C шляхом додавання по краплях протягом 60 хв. при інтенсивному перемішуванні. Воронку промивали метанолом (24 г). Одержану реакційну суміш перемішували протягом 2 год. при 0 °C, а потім гасили в перемішуваній суміші насиченого водного NaHCO 3 (300 мл) і етилацетату (100 мл) при 0-5 °C шляхом додавання по краплях протягом 15 хв. Кінцева величина рН становила 6,9. Додавали ще одну порцію етилацетату (50 мл) і рН доводили до 4,2 за допомогою H2SO4 (96 % мас.). Після розділення фаз водну фазу екстрагували етилацетатом (1150 мл, 3100 мл). Об'єднані органічні фази концентрували під вакуумом при 40-50 °C, з одержанням 8,1 г твердої речовини оранжевого кольору. За даними кількісного аналізу методом 1 Н ЯМР, дана тверда речовина містила 4,2 г (0,026 моль) сполук -(4) і -(4), що відповідає загальному виходу 53 %, в розрахунку на етиловий ефір R-3-(2,2-диметил-[1,3]діоксолан-4іл)акрилової кислоти. Співвідношення -(4):-(4) склало 3,1:1. Приклад 2В: Застосування DBU в реакції приєднання Міхаеля і без додаткової основи в реакції Нефа Інші 25 г розчину, одержані в реакції приєднання Міхаеля в прикладі 2А, охолоджували до 0° і гасили в розчині H2SO4 (7,8 г, 96 % мас., 0,076 моль, 1,5 экв.) в метанолі (13,2 г) при 0 °C шляхом додавання по краплях протягом 40 хв. при інтенсивному перемішуванні. Воронку промивали метанолом (7,7 г). Одержану реакційну суміш перемішували протягом 4 год. при 0 °C, а потім обробляли, відповідно до процедури прикладу 2А, з одержанням твердої 1 речовини, яка, згідно кількісному аналізу Н ЯМР, містила 2,8 г (0,0175 моль) сполук -(4) і -(4), що відповідає виходу 35 %, в розрахунку на етиловий ефір R-3-(2,2-диметил-[1,3]діоксолан-4іл)акрилової кислоти. Приклад 2С: Застосування TMG в реакції приєднання Міхаеля і NaOMe як додаткової основи в реакції Нефа До етилового ефіру R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти (47,5 г 24 UA 100835 C2 5 10 15 20 25 30 35 40 45 50 55 маслянистої рідини, чистота 84,2 % мас., 0,2 моль) додавали нітрометан (26,0 г 51,7 % мас. розчину в метанолі, 0,22 моль, 1,1 экв.) і розчин охолоджували до 0 °C. Потім по краплях додавали TMG (23 г, 0,2 моль, 1 экв.) протягом 20 хв. і воронку промивали метанолом (2 г). Реакційну суміш нагрівали до 20 °C і перемішували при вказаній температурі протягом 22 год. Розчин охолоджували до 0 °C і по краплях додавали NaOMe (40,2 г 29,6 % мас. розчину в метанолі, 0,22 моль, 1,1 экв.) протягом 15 хв. при 0 °C і воронку промивали метанолом (6,4 г). Після перемішування протягом ще 70 хв. при 0 °C суміші гасили в розчині H2SO4 (71,6 г, 96 % мас., 0,7 моль, 3,5 экв.) в метанолі (121,6 г) при 0-5 °C шляхом додавання по краплях протягом 70 хв. при інтенсивному перемішуванні. Воронку промивали метанолом (215 г). Одержану реакційну суміш перемішували протягом 145 хв. при 0 °C, а потім гасили в перемішуваній суміші насиченого водного NaHCO3 (1200 мл) і етилацетату (400 мл) при 0 °C шляхом додавання по краплях протягом 30 хв. Кінцева величина рН становила 7,4. Після додавання ще однієї порції етилацетату (200 мл) рН доводили до 4,2 за допомогою H2SO4 (96 % мас.). Після розділення фаз водну фазу екстрагували етилацетатом (4400 мл). Об'єднані органічні фази концентрували під вакуумом при 40-50 °C, з одержанням 38,5 г твердої речовини жовто1 оранжевого кольору, яка, згідно кількісному аналізу Н ЯМР, містила -(4) (12,2 г, 0,077 моль) і -(4) (4,6 г, 0,029 моль), що відповідає загальному виходу 53 %, в розрахунку на етиловий ефір R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти, і співвідношенню -(4):-(4) 2,7:1. Приклад 2D: Застосування тільки NaOMe в реакції приєднання Міхаеля і в реакції Нефа До етилового ефіру R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти (47,5 г маслянистої рідини, чистота 84,2 % мас., 0,2 моль) в метанолі (200 г) додавали нітрометан (26,0 г 51,7 % мас. розчину в метанолі, 0,22 моль, 1,1 экв.) і розчин охолоджували до 0 °C. Додавали NaOMe (40 г 30 % мас. розчину в метанолі, 0,22 моль, 1,1 экв.) і реакційну суміш перемішували протягом 18 год. при 0 °C, а потім гасили в розчині H2SO4 (58 г, 96 % мас., 0,57 моль, 2,9 экв.) в метанолі (140 г) при -3-0 °C шляхом додавання по краплях протягом 75 хв. при інтенсивному перемішуванні. Реакційну суміш перемішували протягом 4 год. при 0 °C і згодом тримали 1 протягом 16 год. при -30 °C. Згідно з кількісним аналізом Н ЯМР, загальний вихід (у реакційній суміші) -(4) і -(4), в розрахунку на етиловий ефір R-3-(2,2-диметил-[1,3]діоксолан-4іл)акрилової кислоти, склав 45 %, а співвідношення -(4):-(4) 2,5:1. Реакційну суміш потім гасили в перемішуваному розчині NaHCO3 (80 г) у воді (1 л) при 0-5 °C шляхом додавання по краплях протягом 90 хв. До кінця гасіння додавали розчин NaHCO 3 (4 г) у воді (50 мл), щоб довести величину рН до 5-5,5. Після розділення фаз водну фазу екстрагували етилацетатом (4500 мл) і об'єднані органічні фази концентрували під вакуумом при 30-40 °C з одержанням 32 1 г маслянистої рідини червоного кольору. Згідно з кількісним аналізом Н ЯМР, дана масляниста рідина містила 13,2 г (0,084 моль) -(4) і -(4), що відповідає загальному виходу 42 %, в розрахунку на етиловий ефір R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти, і співвідношенню -(4):-(4) 3:1. Приклад 3: Одержання чистого -(4) з етилового ефіру R-3-(2,2-диметил-[1,3]діоксолан-4іл)акрилової кислоти з використанням DBU в реакції приєднання Міхаеля, NaOMe як додаткової основи в реакції Нефа і кристалізації -(4) з ізопропанолу Приклад 3А: Використання не удосконаленої процедури обробки для -(4) і -(4) До етилового ефіру R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти (42,3 г, чистота 94,5 % мас., 0,2 моль) додавали нітрометан (26,0 г 51,7 % мас. розчину в метанолі, 0,22 моль, 1,1 экв.) і розчин охолоджували до 0 °C. Потім по краплях додавали DBU (30,4 г, 0,2 моль, 1 экв.) протягом 20 хв. і воронку промивали метанолом (4 г). Реакційну суміш нагрівали до 20 °C, а потім охолоджували до 0 °C. Потім по краплях додавали NaOMe (40,4 г 29,6 % мас. розчину в метанолі, 0,22 моль, 1,1 экв.) протягом 20 хв. при 0 °C і воронку промивали метанолом (6,4 г). Реакційний розчин перемішували протягом 50 хв. при 0 °C, а потім гасили в розчині H2SO4 (71,6 г, 96 % мас., 0,7 моль, 3,5 экв.) в метанолі (121,6 г) при 0-5 °C шляхом додавання по краплях протягом 70 хв. при інтенсивному перемішуванні. Воронку промивали метанолом (216 г), а реакційну суміш перемішували протягом 2 год. при 0-2 °C, а потім гасили в перемішуваній суміші насиченого водного розчину NaHCO3 (1,2 л) та етилацетату (400 мл) при 0-9 °C шляхом додавання по краплях протягом 17 хв. Кінцева величина рН становила 7,2. Воронку промивали метанолом (40 мл) і рН доводили до 4,0 за допомогою H2SO4 (96 % мас.) при 9 °C. Після додавання етилацетату (200 мл) і розділення фаз водну фазу екстрагували етилацетатом (600 мл, 3400 мл). Об'єднані органічні фази концентрували під вакуумом при 40-50 °C, з одержанням 35,9 г напівтвердої речовини жовто-оранжевого кольору, яка, згідно з кількісним 1 аналізом Н ЯМР, містила 16,5 г (0,104 моль) -(4) і -(4), що відповідає загальному виходу 52 %, в розрахунку на етиловий ефір R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти. 25 UA 100835 C2 5 10 15 20 25 30 35 40 45 50 55 Співвідношення -(4):-(4) склало 3,0:1. Необроблений напівтвердий продукт розчиняли в ізопропанолі (69,5 г) при 80 °C. Одержаний розчин охолоджували до 60 °C, поміщали в нього затравку і охолоджували далі до 0 °C протягом 2 год., що привело до кристалізації -(4). Тверді речовини виділяли фільтруванням, промивали ізопропанолом (30 мл, 20 °C) і сушили на повітрі, з одержанням 12,0 1 г кристалічного продукту не зовсім білого кольору, який, згідно з кількісним аналізом Н ЯМР, складався з 9,8 г -(4) (вихід 31 %, в розрахунку на етиловий ефір R-3-(2,2-диметил[1,3]діоксолан-4-іл)акрилової кислоти) і 0,38 г -(4) (вихід 1,2 %, в розрахунку на етиловий ефір R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти). Це відповідає виходу кристалізації 60 % (вихід -(4)/[витрата -(4)+-(4)]) і співвідношенню -(4):-(4) 26:1. Приклад 3В: Використання удосконаленої процедури обробки для -(4) і -(4) До етилового ефіру R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти (47,5 г, чистота 84,2 % мас., 0,2 моль) додавали нітрометан (26,0 г 51,7 % мас. розчину в метанолі, 0,22 моль, 1,1 экв.) і розчин охолоджували до 0 °C. Потім по краплях додавали DBU (30,4 г, 0,2 моль, 1 экв.) протягом 30 хв. при 0-20 °C і воронку промивали метанолом (4 г). Реакційну суміш нагрівали до 20 °C, перемішували протягом ще 18 год. при вказаній температурі і охолоджували до 0°С. Потім по краплях додавали NaOMe (40 г 29,6 % мас. розчину в метанолі, 0,22 моль, 1,1 экв.) протягом 20 хв. при 0 °C і одержаний розчин перемішували протягом 1 год. при 0 °C. Потім суміш гасили в розчині H2SO4 (72 г, 96 % мас., 0,7 моль, 3,5 экв.) в метанолі (72 г) при 0-5 °C шляхом додавання по краплях протягом 3 год. при інтенсивному перемішуванні. Реакційну суміш перемішували протягом ще 2 год. при 0-5 °C, а потім гасили в перемішуваній суспензії KHCO3 (99 г) у воді (200 мл) при 0-5 °C шляхом додавання по краплях протягом 1 год. Кінцева величина рН становила 4,1. Після нагрівання до 20 °C солі видаляли фільтруванням і промивали етилацетатом (500 мл). Водний маточний розчин фільтрату (454 г) концентрували під вакуумом при 35 °C для видалення метанолу до кінцевої ваги 272 г і екстрагували етилацетатом (6500 мл; перші порції – промивною рідиною після фільтрації солей, потім – свіжим). Об'єднані органічні фази концентрували під вакуумом при 40-50 °C, з одержанням 40,4 г твердої речовини, яка, згідно GC, містила 14,5 г -(4) і 3,4 г -(4), що відповідає загальному виходу 57 %, в розрахунку на етиловий ефір R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти і співвідношенню -(4):-(4) 4,3:1. Необроблений твердий продукт розчиняли в етилацетаті (300 мл) і розчин промивали насиченим водним розчином NaCl (25 мл) і водою (10 мл). Органічний шар, який, згідно GC, містив 14,1 г -(4) і 3,4 г -(4), концентрували під вакуумом, з одержанням 42,4 г забрудненої твердої речовини. До 38 г зазначеного необробленого продукту додавали ізопропанол (62 г) і тверду речовину розчиняли шляхом нагрівання до 60 °C. Одержаний розчин охолоджували до 50 °C, поміщали в нього затравку і охолоджували далі до 0 °C протягом 2 год., що привело до кристалізації -(4). Тверді речовини виділяли фільтруванням, промивали ізопропанолом (220 мл, 0 °C) і сушили на повітрі, з одержанням 12,9 г кристалічного продукту не зовсім білого кольору, який, згідно GC, містив 12,2 г -(4). Це відповідає 39 % виходу, в розрахунку на етиловий ефір R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти) і виходу кристалізації 78 % (вихід -(4)/[витрата -(4)+-(4)]). -(4) виявити не вдалося. Приклад 4: Одержання чистого -(4) з етилового ефіру R-3-(2,2-диметил-[1,3]діоксолан-4іл)акрилової кислоти шляхом кристалізації -(4), епімеризації -(4) і другої кристалізації -(4) До етилового ефіру R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти (42,3 г, чистота 94,6 % мас., 0,2 моль) додавали нітрометан (26,0 г 51,7 % мас. розчину в метанолі, 0,22 моль, 1,1 экв.) і розчин охолоджували до 0 °C. Потім по краплях додавали DBU (30,4 г, 0,2 моль, 1 экв.) протягом 30 хв. при 0-20 °C і реакційну суміш нагрівали до 20 °C та перемішували протягом ще 18 год. при зазначеній температурі. Одержану реакційну суміш охолоджували до 0 °C і по краплях додавали NaOMe (40 г 29,6 % мас. розчину в метанолі, 0,22 моль, 1,1 экв.) при 0 °C. Одержаний розчин перемішували протягом 1 год. при 0 °C і гасили в розчині H2SO4 (72 г, 96 % мас., 0,7 моль, 3,5 экв.) в метанолі (72 г) при 0-5 °C шляхом додавання по краплях протягом 1,5 год. при інтенсивному перемішуванні. Реакційну суміш перемішували протягом 2 год. при 0-5 °C, а потім гасили в перемішуваній суспензії NaHCO 3 (100 г), води (400 мл) і етилацетату при 0-5 °C шляхом додавання по краплях протягом 1 год. По частинах додавали NaHCO3 (40 г), щоб підтримувати величину рН вище 3,5. Солі видаляли фільтруванням при 05 °C і промивали етилацетатом (300 мл). Після розділення фаз водну фазу екстрагували етилацетатом (300 мл промивної рідини після фільтрації солей, 3150 мл свіжого). Об'єднані органічні фази концентрували під вакуумом, додавали етилацетат (200 мл) і суміш концентрували під вакуумом ще раз, з одержанням 33,2 г напівтвердої речовини, яка, згідно з 26 UA 100835 C2 кількісним аналізом Н ЯМР, містила 13,5 г -(4) і 4,0 г -(4), що відповідає загальному виходу 53 % в розрахунку на етиловий ефір R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти і співвідношенню -(4):-(4) 3,5:1. Необроблений продукт розчиняли в ізопропанолі (70 г) при 60 °C. Одержаний розчин охолоджували до 50 °C, поміщали в нього затравку і охолоджували далі до 0 °C, що привело до кристалізації -(4), який виділяли фільтруванням, промивали холодним (0 °C) ізопропанолом 1 (215 мл) і сушили на повітрі. Отримували 12,3 г -(4), який, згідно з кількісним аналізом Н ЯМР, був чистим на 97,1 % мас. і не містив -(4). Це (перший збір) відповідає 38 % виходу, в розрахунку на етиловий ефір R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти) і виходу кристалізації 68 % (вихід -(4)/[витрата -(4)+-(4)]). Об'єднані маточний розчин і промивну рідину після першої кристалізації (108 г, що містять 4,0 г -(4) і 1,2 г -(4)) концентрували під вакуумом до17,9 г рідини. Згодом додавали метанол (9,05 г) та MeSO3H (0,91 г, 0,29 экв.) і суміш кип'ятили із зворотним холодильником. Після 2 год. кипіння із зворотним холодильником реакція епімеризації була завершена (співвідношення -(4):-(4) > 3. Після охолоджування до 20 °C додавали триетиламін (0,96 г, 1 экв., в розрахунку на MeSO3H) і суміш концентрували під вакуумом до 18,7 г в'язкого залишку. Залишок повторно розчиняли в ізопропанолі (13,9 г) при 50 °C. Після охолоджування до 45 °C в суміш поміщали затравку і охолоджували до 0 °C, що приводило до кристалізації -(4), який ізолювали фільтруванням, промивали холодним (0 °C) ізопропанолом (26 мл) і сушили на 1 повітрі. Це дало 2,24 г -(4), який, згідно з кількісним аналізом Н ЯМР, був чистим на 95,3 % мас. і не містив -(4). Це (другий збір) відповідає 6 % виходу, в розрахунку на етиловий ефір R3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти, і виходу кристалізації 43 % (вихід -(4)/[витрата -(4)+-(4)] після епімеризації). Таким чином, загальний вихід -(4) (перший і другий збір), в розрахунку на етиловий ефір R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти, склав 44 %. Приклад 5: Кристалізація -(4), починаючи з необроблених сумішей -(4) і -(4), з розчинників, що не є ізопропанолом Приклад 5А: З трет-бутанолу Необроблену суміш (6,5 г) -(4) і -(4), одержану в прикладі 2А (що містила 3,37 г -(4)+-(4) в співвідношенні 3,1:1) розчиняли в трет-бутанолі (16 г) при 72 °C. Охолодження до 55 °C, внесення затравки і подальше охолодження до 25 °C привело до кристалізації -(4), який ізолювали фільтруванням, промивали ізопропанолом (5 мл, 20 °C) і сушили під вакуумом. Це 1 дало сполуку -(4) (1,85 г), яка, згідно з кількісним аналізом Н ЯМР, була чистою на 82,9 % мас., що відповідає виходу кристалізації 46 % (вихід -(4)/[витрата -(4)+-(4)]) із співвідношенням -(4):-(4) 30:1. Приклад 5В: З трет-амілового спирту Необроблену суміш (7,25 г) -(4) і -(4) (яка містила 3,44 г -(4)+-(4) в співвідношенні 2,9:1) розчиняли в трет-аміловому спирті (15,7 г) при 70 °C. Охолодження до 60 °C, внесення затравки і подальше охолодження до 40 °C не привело до кристалізації. Після ще одного внесення затравки при 40 °C розчин додатково охолоджували і кристалізація -(4) почалася при 27 °C. Суміш далі охолоджували до 2 °C і кристали -(4) ізолювали фільтруванням, промивали третаміловим спиртом (7,5 мл, 20 °C) і сушили під вакуумом. Це дало 2,35 г продукту не зовсім 1 білого кольору, який згідно з кількісним аналізом Н ЯМР, складався з 1,91 г -(4) і 0,11 г -(4), що відповідає виходу кристалізації 59 % (вихід -(4)/[витрата -(4)+-(4)]) і співвідношенню -(4):-(4) 18:1. Приклад 6: Кристалізація чистого -(4) з суміші -(4) і -(4) з одночасною епімеризацією -(4) Розчин -(4) світло-коричневого кольору (5,0 г, чистота 96,6 % мас., 30,6 ммоль, не містить -(4)) і MeSO3H (0,3 г, 0,1 экв.) в метанолі (200 мл) перемішували при 20 °C протягом 92 год., що приводило до епімеризації до співвідношення -(4):-(4) 3,6:1. Реакційну суміш потім концентрували під вакуумом (20 мбар; 45 °C), з одержанням 5,2 г клейкої твердої речовини. Останню поміщали в метанол (50 мл) і ще раз концентрували під вакуумом (20 мбар; 50 °C), з одержанням 5,1 г сухої твердої речовини світло-коричневого кольору, яка, згідно з кількісним 1 аналізом Н ЯМР, містила -(4) з чистотою 90 % мас. (4,6 г, 29 ммоль). -(4) виявити не вдалося. Таким чином, майже весь (96 %) вихідний -(4) був виділений. Приклад 7: Одержання чистого -(4) зі свіжовиготовленого S-2,3-Oізопропіліденгліцеральдегіду з використанням удосконаленої процедури і кристалізації -(4), епімеризації -(4) та другої кристалізації -(4) До 175 г етилового ефіру Е-R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти, 1 5 10 15 20 25 30 35 40 45 50 55 27 UA 100835 C2 5 10 15 20 25 30 35 40 45 50 55 одержаного, як описано в прикладі 1 (чистота 78 % мас., 136,5 г, 0,68 моль) додавали нітрометан (88,6 г 51,7 % мас. розчину в метанолі, 0,75 моль, 1,1 экв.) і розчин охолоджували до 0 °C. Потім по краплях додавали DBU (103,4 г, 0,68 моль, 1 экв.) протягом 35 хв. при 10-21 °C і воронку промивали метанолом (7 г). Після перемішування протягом 18 год. при 20 °C одержаний розчин темно-червоного кольору охолоджували до 0 °C і по краплях додавали NaOMe (134,6 г 30 % мас. розчину в метанолі, 0,748 моль, 1,1 экв.) протягом 35 хв. при 0 °C і воронку промивали метанолом (10 г). Після перемішування протягом 30 хв. при 0°С реакційну суміш гасили в розчині H2SO4 (243 г, 96 % мас., 2,38 моль, 3,5 экв.) в метанолі (243 г) при 0-5 °C шляхом додавання по краплях протягом 3 год. при інтенсивному перемішуванні і воронку промивали метанолом (215 г). Після перемішування протягом 2 год. при 0-2 °C реакційну суміш гасили в перемішуваній суспензії КHCO 3 (353 г) у воді (680 мл) при 0-6 °C шляхом додавання по краплях протягом 1 год. До кінця гасіння величина рН складала 7 і її доводили до 4,1 з використанням H2SO4 (96 % мас.) при 0 °C. Після нагрівання до 20 °C солі видаляли фільтруванням і промивали етилацетатом (3375 мл). Промивну рідину пізніше використовували для екстракції. Маточну рідину фільтрату (1380 г), яка, згідно GC, містила 3,08 % мас. -(4) і 0,82 % мас. -(4) (що відповідає загальному виходу 50 % в розрахунку на етиловий ефір Е-R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти і співвідношенню -(4):-(4) 3,75:1), концентрували під вакуумом для видалення метанолу. До одержаного залишку (760 г) додавали воду (80 г) і рН доводили до 4,1 з використанням H2SO4 (96 % мас.). Одержаний водний розчин екстрагували етилацетатом (700 мл, 4500 мл). Об'єднані органічні фази концентрували під вакуумом при 35-40 °C до 181 г залишку. Леткі речовини спільно випаровували 3 з ізопропанолом (2140 г і 90 г), одержуючи залишок (146 г), що складався з необробленої суміші -(4)+-(4). Необроблену суміш (146 г) розчиняли в ізопропанолі (202 г) при 70 °C. Нерозчинний матеріал видаляли фільтруванням і промивали ізопропанолом (5 мл); вага після сушіння складала 0,33 г. Фільтрат (346 г) охолоджували до 50 °C, що привело до спонтанної кристалізації -(4). Суспензію далі охолоджували до 1 °C протягом 4 год., і кристали виділяли фільтруванням, промивали ізопропанолом (2100 мл, 0 °C) і сушили під вакуумом протягом 17 год. при 35 °C, з одержанням кристалічного продукту не зовсім білого кольору (44,2 г). Згідно з кількісним аналізом GC, він складався з 89,0 % мас. -(4) і 1,0 % мас. -(4), що відповідає загальному виходу 37 %, в розрахунку на етиловий ефір Е-R-3-(2,2-диметил-[1,3]діоксолан-4іл)акрилової кислоти), і співвідношенню -(4):-(4) 89:1. Маточний розчин і промивні рідини після першої кристалізації -(4) (всього 374 г) концентрували під вакуумом до 90,8 г, додавали метанол (120 мл) і одержану суміш концентрували до 83 г. Ще раз додавали метанол (120 мл) і суміш концентрували до 83 г. До залишку додавали метанол (45 г) і MeSO3H (2,66 г, 0,0277 моль, 0,2 экв., в розрахунку на загальну кількість -(4)+-(4), присутню в маточному розчині і промивних рідинах), і розчин кип'ятили із зворотним холодильником. Після 1 год. кипіння із зворотним холодильником (6065 °C) GC показала завершення епімеризації (співвідношення -(4):-(4) складало 3,1:1) і розчин охолоджували до 33 °C, нейтралізували триетиламіном (2,94 г, 1,05 экв., в розрахунку на MeSO3H) і концентрували під вакуумом. До одержаного залишку додавали ізопропанол (120 мл) і суміш концентрували під вакуумом, з одержанням 88 г залишку. Залишок розчиняли в ізопропанолі (37 г) при 47 °C. Одержаний розчин охолоджували до 2 °C протягом 2,5 год.; кристалізація починалася спонтанно при 30 °C. Кристалічний продукт ізолювали фільтруванням, промивали ізопропанолом (320 мл, 0 °C) і сушили під вакуумом (17 год. при 35 °C), з одержанням 10,1 г кристалічного продукту білого кольору, який, згідно GC, складався з 96,4 % мас. -(4) і 0,065 % мас. -(4), що відповідає загальному виходу 9 % в розрахунку на етиловий ефір Е-R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти і співвідношенню -(4):-(4) > 1000:1. Таким чином, загальний вихід першого і другого збору -(4), в розрахунку на етиловий ефір Е-R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти, склав 46 %. Приклад 8: Одержання чистого (3R, 3aS, 6aR)-гексагідрофуро[2,3-b]фуран-3-олу з проміжної сполуки -(4) Використовували процедуру, описану в WO 03/022853, приклад IV, остання стадія. Приклад 9: Одержання чистого -(4) з етилового ефіру R-3-(2,2-диметил-[1,3]діоксолан-4іл)акрилової кислоти шляхом прямої кристалізації -(4) з необробленої суміші -(4) і -(4) і одночасною епімеризації -(4) в -(4) До етилового ефіру R-3-(2,2-диметил-[1,3]діоксолан-4-іл)акрилової кислоти (399,5 г, чистота 75,1 % мас., 1,5 моль) додавали нітрометан (915,0 г 11 % мас. розчину в метанолі, 1,65 моль, 28
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for the synthesis of (3r,3as,6ar) hexahydro-furo [2,3-b]furan-3-ol, intermediate and process for the preparation thereof
Автори англійськоюQuaedflieg Peter Jan Leonard, Kesteleyn Bart Rudolf Romanie, Vijn Robert Jan, Liebregts Constantinus Simon Maria, Kooistra Jacob Hermanus Matheus Hero, Lommen Franciscus Alphons Mari
Назва патенту російськоюСпособ синтеза (3r,3as,6ar)-гексагидрофуро[2,3-b]фуран-3-ола, промежуточное соединение и способ его получения
Автори російськоюКваедфлиег Питер Ян Леонар, Кестелин Барт Рудольф Романи, Виджн Роберт Ян, Лиэбрегтс Константинус Симон Мария, Кооистра Якоб Херманус Матеус Херо, Ломмен Францискус Альфонс Мари
МПК / Мітки
МПК: C07D 493/04, C07D 307/20, C07H 15/04
Мітки: 3r,3as,6ar)-гексагідрофуро[2,3-b]фуран-3-олу, одержання, проміжна, синтезу, спосіб, сполука
Код посилання
<a href="https://ua.patents.su/34-100835-sposib-sintezu-3r3as6ar-geksagidrofuro23-bfuran-3-olu-promizhna-spoluka-ta-sposib-oderzhannya.html" target="_blank" rel="follow" title="База патентів України">Спосіб синтезу (3r,3as,6ar)-гексагідрофуро[2,3-b]фуран-3-олу, проміжна сполука та спосіб її одержання</a>
Спосіб одержання похідної бензімідазолу та проміжна сполука
Номер патенту: 94988
Опубліковано: 25.06.2011
Автори: Брьодер Вольфганг, Зоботта Райнер
МПК: C07D 401/12
Мітки: проміжна, спосіб, бензимідазолу, похідної, одержання, сполука
Формула / Реферат:
1. Спосіб одержання сполуки формули, 1не обов'язково у формі її адитивних солей з кислотою, який відрізняється тим, що на першій стадії діамін формули 22вводять у реакцію з карбоновою кислотою 3
Спосіб одержання похідної сполуки хіноліну та проміжна сполука

Номер патенту: 65627
Опубліковано: 15.04.2004
Автори: Янагава Йошінобу, Охара Йошіо, Такада Ясутака, Сузукі Мікіо
МПК: C07D 215/14, C07D 215/12
Мітки: проміжна, сполуки, сполука, хіноліну, одержання, похідної, спосіб
Формула / Реферат:
1. Проміжна сполука нітрилу формули (1). (1)2. Спосіб одержання похідного хіноліну (3), що проводять за допомогою сполуки нітрилу, одержуваної в результаті реакції сполуки альдегіду, представленої формулою (2), з діетилціанометилфосфонатом (2) (1) (3).
Спосіб одержання колоїдної дисперсії карбонату кальцію (варіанти), проміжна сполука, присадка до мастильних олив, мастильна олива

Номер патенту: 66931
Опубліковано: 15.06.2004
Автори: Кайбалдін Костянтин Артурович, Суховерхов Віктор Дмитрович
МПК: C07F 9/16, C10M 125/10, C10M 137/00, C10M 177/00, C07F 3/00, C10M 141/00
Мітки: одержання, олив, мастильних, спосіб, проміжна, олива, колоїдної, кальцію, мастильна, сполука, варіанти, дисперсії, присадка, карбонату
Формула / Реферат:
1) Сполука загальної формули (1) ,(1) де R1 і R2 - алкіл, що містить від 2 до 8 атомів вуглецю або алкіларил формули (2) ,(2)де R - алкіл, що містить від 9 до 12 атомів вуглецю.2. Сполука за п. 1, де R1=R2 - ізобутил.3. Сполука за п. 1, де R1 -...
Водорозчинні проліки блокованих спиртів чи фенолів, фармацевтична композиція на їх основі, спосіб їх одержання, проміжна сполука (варіанти) та спосіб її одержання (варіанти)

Номер патенту: 73479
Опубліковано: 15.08.2005
Автори: Георг Інгрід Гунда, Сафаді Мухаммед С., Зігмунт Джан Дж., Стелла Валентіно Дж.
МПК: C07F 9/655, A61K 31/437, C07C 319/00, A61K 31/661, C07D 491/052, C07F 9/6515, C07H 17/04, C07C 323/12, C07D 311/72, A61K 38/00, C07F 9/6561, A61K 31/355, C07F 9/09, A61K 31/352, C07K 7/64, A61P 25/00, A61K 31/10, A61K 31/665, A61K 31/7048, A61K 31/7042, A61K 31/675, A61P 35/00
Мітки: проміжна, спосіб, фармацевтична, одержання, сполука, блокованих, композиція, спиртів, фенолів, варіанти, проліки, основі, водорозчинні
Формула / Реферат:
1. Сполука відповідно до формули І:, IдеR-O- - залишок спиртвмісної або фенолвмісної фармацевтичної сполуки, крім таксолу і його похідних,R1 - атом водню або іон лужного металу, або протонований амін, або протонована амінокислота,R2 - атом водню або іон лужного металу, або протонований амін, або протонована амінокислота, іn являє...
Спосіб одержання 2-хлоретоксіоцтової кислоти n,n-диметиламіду, проміжна сполука та способи її одержання

Номер патенту: 98448
Опубліковано: 25.05.2012
Автори: Рейтер Йожеф, Мезей Тібор, Катона Золтан, Верецкейне Донат Дйордьі, Трінка Петер, Барта Ференц, Понгьо Ласло, Надь Кальман
МПК: C07C 231/12, C07C 235/06
Мітки: одержання, n,n-диметиламіду, проміжна, спосіб, кислоти, 2-хлоретоксіоцтової, способи, сполука
Формула / Реферат:
1. Спосіб одержання 2-хлоретоксіоцтової кислоти N,N-диметиламіду формули (І), Iпри якому піддають взаємодії розчин 2-гідроксіетоксіоцтової кислоти N,N-диметиламіду формули (II) IIу розчиннику з тіонілхлоридом при температурі між 0 і 40 °C.2. Спосіб за п. 1, який...
Попередній патент: Аксіально-поршневий насос
Наступний патент: Застосування модифікованого вірусу вісповакцини анкара (mva) для швидкої індукції імунітету проти поксвірусних або інших інфекційних агентів
Випадковий патент: Антидіабетичне білково-збивне печиво