Сульфоксимінзаміщені анілінопіримідинові похідні як cdk-інгібітори, їх одержання й застосування як лікарських засобів
Номер патенту: 103500
Опубліковано: 25.10.2013
Автори: Люккінг Ульріх, Шульце Юліа, Лінау Філіп, Яутелат Рольф, Зімайстер Герхард





Формула / Реферат
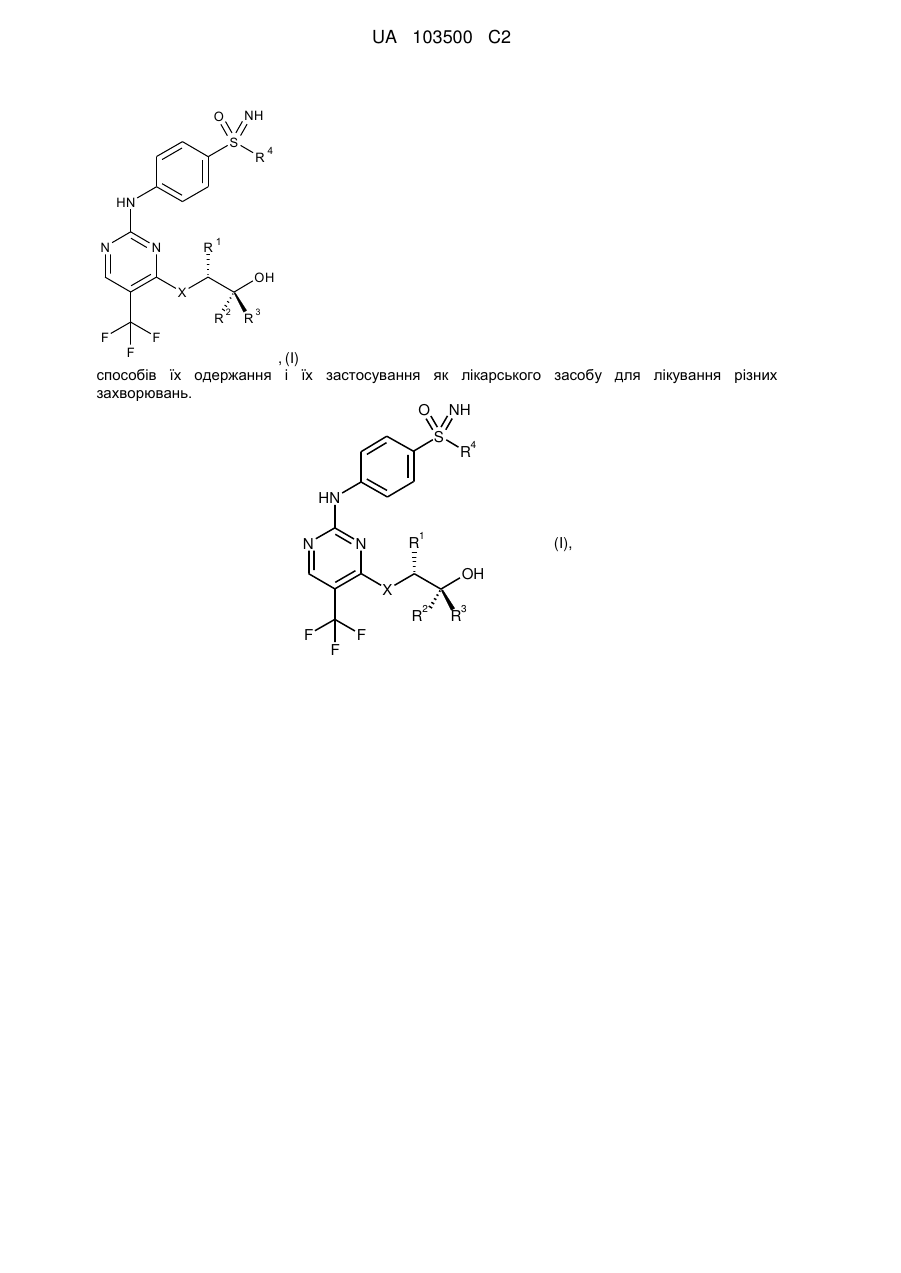
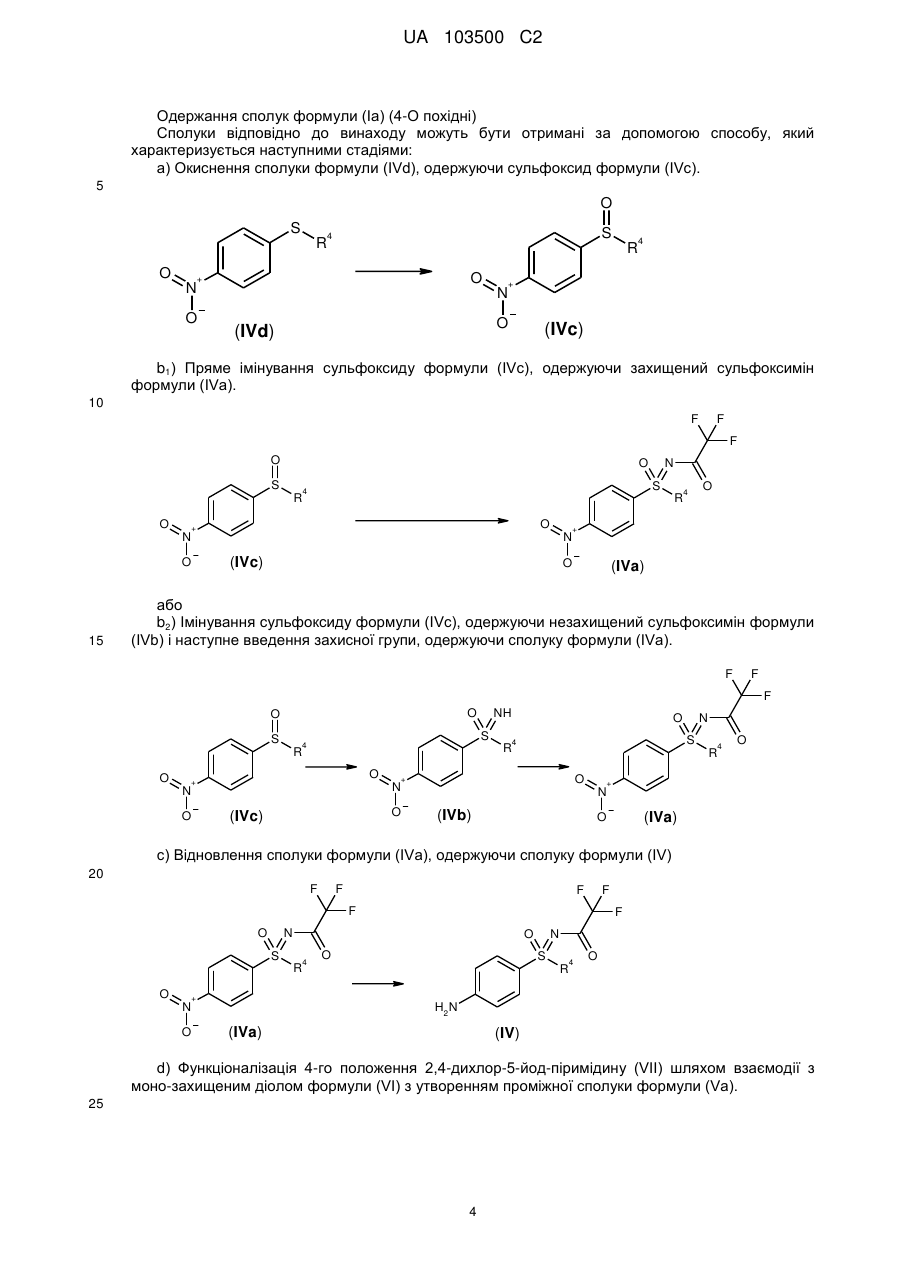
1. Сполука загальної формули (І)
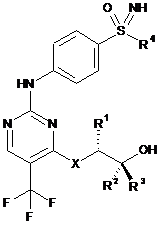 , (І)
, (І)
у якій
X являє собою -О- або -NH-, і
R1 являє собою метильну, етильну, пропільну або ізопропільну групу, і
R2 і R3, незалежно один від одного, являють собою водень, метильну або етильну групу, і
R4 являє собою С1-С6-алкільну групу або С3-С7-циклоалкільне кільце, або її солі, діастереомери й енантіомери.
2. Сполука, як заявлено в пункті 1, де X являє собою -О-, або її солі, діастереомери й енантіомери.
3. Сполука, як заявлено в будь-якому з пунктів 1 або 2, де R1 являє собою метильну групу, або її солі, діастереомери й енантіомери.
4. Сполука, як заявлено в будь-якому з пунктів 1-3, де R2 являє собою метильну групу, або її солі, діастереомери й енантіомери.
5. Сполука,як заявлено в будь-якому з пунктів 1-4, де R3 являє собою водень або метильну групу, або її солі, діастереомери й енантіомери.
6. Сполука, як заявлено в будь-якому з пунктів 1-5, де R4 являє собою метильну або етильну групу або являє собою циклопропільне кільце, або її солі, діастереомери й енантіомери.
7. Сполука загальної формули (І), як заявлено в пункті 1, де
X являє собою -О- або -NH-, і
R1 являє собою метильну групу, і
R2 являє собою метильну групу, і
R3 являє собою водень або метильну групу, і
R4 являє собою метильну або етильну групу або являє собою циклопропільне кільце,
або її солі, діастереомери й енантіомери.
8. Спосіб одержання сполук загальної формули (Іа), в якому здійснюють принаймні одну зі стадій a)-h):
а) окиснення сполуки формули (IVd), з одержанням сульфоксиду формули (IVc):
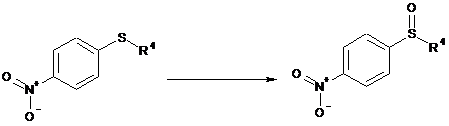 ,
,
(IVd)
(IVc)
b1) пряме імінування сульфоксиду формули (IVc), з одержанням захищеного сульфоксиміну формули (IVa):
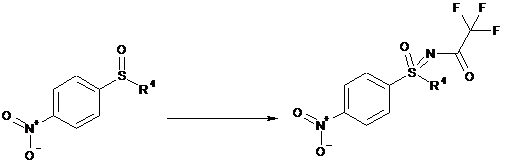 ,
,
(IVc)
(IVa)
або
b2) імінування сульфоксиду формули (IVc), з одержанням незахищеного сульфоксиміну формули (IVb), і наступне введення захисної групи, з одержанням сполуки формули (IVa):
 ,
,
(IVc)
(IVb)
(IVa)
c) відновлення сполуки формули (IVa), з одержанням сполуки формули (IV):
 ,
,
(IVa)
(IV)
d) функціоналізація 4-го положення 2,4-дихлор-5-йодпіримідину (VII) шляхом взаємодії з монозахищеним діолом формули (VI), з утворенням проміжної сполуки формули (Va):
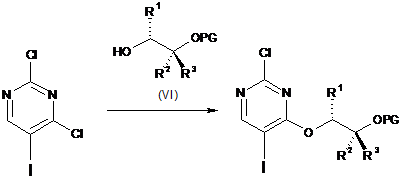 ,
,
(VII)
(Va)
e) одержання 5-CF3 проміжної сполуки (V):
 ,
,
(Va)
(V)
f) сполучення сполук формули (IV) і (V), з одержанням проміжної сполуки формули (III):
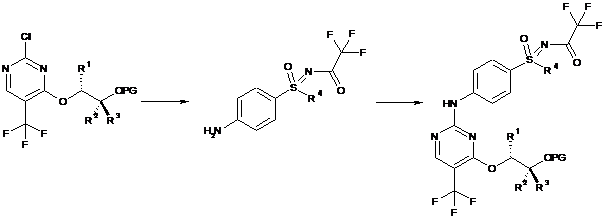 ,
,
(V)
(IV)
(III)
g) відщеплення захисної групи PG з утворенням (II):
 ,
,
(III)
(II)
h) відщеплення захисної групи на сульфоксиміні з утворенням (Іа):
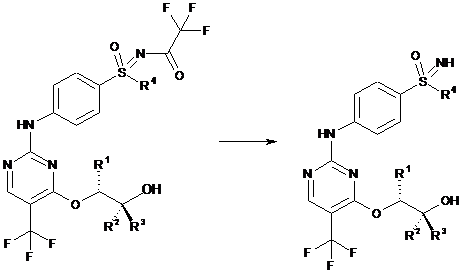 ,
,
(II)
(Ia)
де замісники R1, R2, R3 i R4 мають значення, наведені в загальній формулі (І) пунктів 1-7.
9. Спосіб одержання сполук загальної формули (Іb), в якому здійснюють принаймні одну зі стадій a)-f):
а) окиснення сполуки формули (IVd), з одержанням сульфоксиду формули (IVc):
,
(IVd)
(IVc)
b1) пряме імінування сульфоксиду формули (IVc), з одержанням захищеного сульфоксиміну формули (IVa):
,
(IVc)
(IVa)
або
b2) імінування сульфоксиду формули (IVc), з одержанням незахищеного сульфоксиміну формули (IVb), і наступне введення захисної групи, з одержанням сполуки формули (IVa):
,
(IVc)
(IVb)
(IVa)
c) відновлення сполуки формули (IVa), з одержанням сполуки формули (IV):
,
(IVa)
(IV)
d) функціоналізація 4-го положення 2,4-дихлор-5-трифторметилпіримідину (VІІb) шляхом взаємодії з аміном формули (VІa), з утворенням проміжної сполуки формули (Vb):
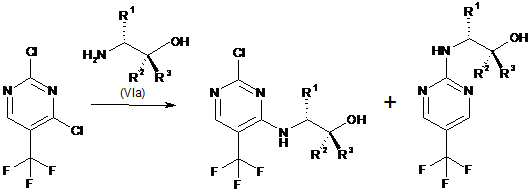 ,
,
(VIIb)
(Vb)
(Vc)
e) сполучення сполук формули (Vb) і (IV) з одержанням проміжної сполуки формули (ІІb):
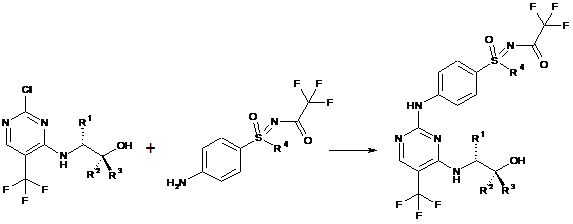 ,
,
(Vb)
(IV)
(IIb)
f) відщеплення захисної групи на сульфоксиміні з утворенням (Іb):
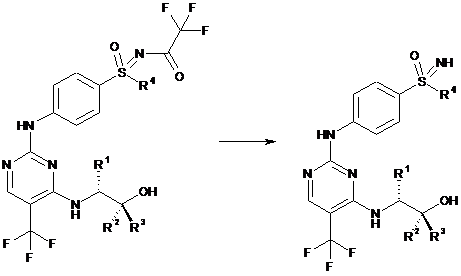 ,
,
(IIb)
(Ib)
де замісники R1, R2, R3 і R4 мають значення, наведені в загальній формулі (І) пунктів 1-7.
10. Сполуки, як заявлено в будь-якому з пунктів 1-7, для застосування як лікарських засобів.
11. Застосування сполуки, як заявлено в будь-якому з пунктів 1-7, для одержання лікарського засобу для лікування злоякісного новоутворення.
12. Сполуки, як заявлено в будь-якому з пунктів 1-7, для застосування як лікарських засобів проти злоякісного новоутворення.
13. Фармацевтичний склад, що містить сполуку, як заявлено в будь-якому з пунктів 1-7.
Текст
Реферат: Винахід стосується сульфоксимінзаміщених анілінопіримідинових похідних формули (І) ЇХ UA 103500 C2 (12) UA 103500 C2 NH O S R 4 HN N N R 1 OH X R F F 2 R 3 F , (І) способів їх одержання і їх застосування як лікарського засобу для лікування різних захворювань. O NH S 4 R HN N 1 (I), R N OH X 2 R F F F 3 R UA 103500 C2 5 10 15 20 25 30 35 Даний винахід стосується вибраних сульфоксимін-заміщених аніліно-піримідинових похідних, способів їх одержання і їх застосування як лікарського засобу для лікування різних захворювань. Циклін-залежні кінази (CDK) представляють собою сімейство ферментів, які відіграють важливу роль у регуляції клітинного циклу й, таким чином, представляють собою надзвичайно цікаву мішень для розвитку низькомолекулярних інгібіторних молекул. Селективні інгібітори CDK можуть використовуватися для лікування злоякісного новоутворення або інших захворювань, які викликані розладами клітинної проліферації. Піримідини й аналоги вже були описані як активні речовини, наприклад, 2-аніліно-піримідини як фунгіциди (DE 4029650) або заміщені піримідинові похідні для лікування неврологічних або нейродегенеративних захворювань (WO 99/19305). Дуже різні піримідинові похідні описані як CDK інгібітори, наприклад, 2-аміно-4-заміщені піримідини (WO 01/14375), пурини (WO 99/02162), 5-ціано-піримідини (WO 02/04429), анілінопіримідини (WO 00/12486) і 2-гідрокси-3-N,Nдиметиламінопропокси-піримідини (WO 00/39101). Зокрема, похідні піримідину описані в WO 02/096888 і WO 03/076437, які мають інгібуючу дію по відношенню до CDK. Сполуки, які містять фенілсульфонамідну групу, є відомими інгібіторами карбоангідрази людини (особливо, карбоангідрази-2) і застосовуються як діуретики, зокрема, для лікування глаукоми. Атом азоту й атоми кисню сульфонаміду зв'язуються за допомогою водневих містків з 2+ іоном цинку і амінокислотою Thr 199 в активному центрі карбоангідрази-2 і в такий спосіб блокують його ферментативну функцію (A. Casini, F. Abbate, A. Scozzafava, C.T. Supuran, Bioorganic. Med. Chem. Lett. 2003, 1, 2759). Клінічне застосування CDK інгібіторів, що містять фенілсульфонамідну групу, може бути обмежене внаслідок можливості інгібування карбоангідраз і отриманого у результаті цього спектра побічних дій. Прикладами сульфоксимінових активних речовин є сульфонимідоїл-модифіковані триазоли як фунгіциди (H. Kawanishi, H. Morimoto, T. Nakano, T. Watanabe, K. Oda, K. Tsujihara, Heterocycles 1998, 49, 181) або аралкілсульфоксиміни як гербіциди і пестициди (Shell International Research, Ger. P. 2 129 678). В WO 2005/037800 описані незамкнуті сульфоксимін-заміщені аніліно-піримідинові похідні як інгібітори циклін-залежних кіназ. Представлені приклади представляють собою структури, які або незаміщені, або заміщені галогеном, зокрема, бромом в 5-ому положенні піримідину. Ніякі з конкретно описаних структур не мали 5-трифторметильного замісника. З урахуванням розкритого відомого рівня техніки, задачею, що лежить в основі даного винаходу, є забезпечення сполук, які є не лише ефективними CDK інгібіторами, але також можуть ефективно інгібувати ріст пухлини. У дійсності, ефективне CDK інгібування є необхідною, але не достатньою вимогою для ефективного інгібування пухлини. Для структур також необхідні інші властивості, наприклад, властивості проникнення в пухлинну клітину. Зараз було виявлено, що сполуки загальної формули (I) O NH S R 4 HN N R N 1 (I), OH X R F 40 45 2 R 3 F F у яких X являє собою –O– або –NH–, і 1 R являє собою метильну, етильну, пропільну або ізопропільну групу, і 2 3 R і R являють собою, незалежно один від одного водень, метильну або етильну групу, і 4 R являє собою C1-C6-алкільну групу або C3-C7-циклоалкільне кільце, і їх солі, діастереомери й енантіомери, не тільки ефективно інгібують CDK, але також надзвичайно ефективно інгібують ріст 1 UA 103500 C2 пухлини. Сполуки, у яких X представляє собою –O-, згруповані разом з формулою (Ia). O NH S R 4 HN N R N 1 (Ia), OH O R F 2 R 3 F F 5 Сполуки, у яких X представляє собою –NH-, згруповані разом з формулою (Ib). O NH S R 4 HN N R N 1 (Ib), OH N H F R 2 R 3 F F 10 15 20 25 30 35 Заявка ґрунтується на наступних визначеннях: C1-C6-алкіл C1-C6-алкільна група розуміється в кожному випадку як лінійний або розгалужений алкільний залишок, наприклад, метильний, етильний, пропільний, ізопропільний, бутильний, ізобутильний, втор-бутильний, трет-бутильний, пентильний, ізопентильний або гексильний залишок. C3-C7-циклоалкіл C3-C7-циклоалкільне кільце розуміється як циклопропільне, циклобутильне, циклопентильне, циклогексильне або циклогептильне кільце. У загальній формулі (I) X може представляти собою –O- або –NH-. Переважно X представляє собою –O-. 1 У загальній формулі (I) R може представляти собою метильну, етильну, пропільну або ізопропільну групу. 1 Переважно R представляє собою метильну групу. 2 3 У загальній формулі (I), R і R , незалежно один від одного, можуть представляти собою водень, метильну або етильну групу. 2 3 Переважно R і R представляють собою, незалежно один від одного водень або метильну групу. 2 3 Особливо переважно R представляє собою метильну групу й R представляє собою водень або метильну групу. 4 У загальній формулі (I) R може представляти собою C1-C6-алкільний залишок або C3-C7циклоалкільне кільце. 4 Переважно R представляє собою метильну або етильну групу або представляє собою циклопропільне кільце. Краща підгрупа сполук відповідно до загальної формули (I) включає сполуки відповідно до загальної формули (I), у яких 2 UA 103500 C2 5 10 15 20 25 30 35 40 45 50 55 60 X являє собою –O– або –NH–, і 1 R являє собою метильну групу, і 2 R являє собою метильну групу, і 3 R являє собою водень або метильну групу, і 4 R являє собою метильну або етильну групу або представляє собою циклопропільне кільце, і їх солі, діастереомери й енантіомери. Сполуки відповідно до винаходу придатні для лікування - злоякісного новоутворення, такого як сóлідні пухлини, пухлинні метастази, і гематологічні пухлини, зокрема: пухлини голови й шиї; легеневі й бронхіальні пухлини; пухлини шлунково-кишкового тракту, наприклад, рак шлунка, колоректальна карцинома, карцинома підшлункової залози, печінковоклітинний рак; ендокін-активні пухлини; рак молочної залози й гінекологічні пухлини; пухлини сечостатевої системи, наприклад, карцинома нирки, карцинома сечового міхура, рак передміхурової залози; пухлини шкіри; саркоми; лейкози й лімфоми. - вірусних захворювань, і - серцево-судинних захворювань, таких як стенози, артеріосклерози й рестенози, рестенози, індуковані стентом. Приготування лікарських форм сполук відповідно до винаходу у вигляді фармацевтичних препаратів здійснюють за допомогою способу, відомого per se, шляхом перетворення активної речовини або речовин зі звичайними наповнювачами, використовуваними для галенових препаратів з одержанням бажаної дозованої форми. Як наповнювачі можна використовувати, наприклад, носії, заповнювачі, агенти, що викликають дезінтеграцію, сполучні, гігроскопічні речовини, речовини, що сприяють ковзанню, абсорбенти й адсорбенти, розріджувачі, розчинники, співрозчинники, емульсифікатори, солюбілізатори, коригенти, барвники, консерванти, стабілізатори, змочувальні речовини, солі для модифікації осмотичного тиску або буфери. Як посилання можна навести Remington's Pharmaceutical Science, 15-е вид. Mack Publishing Company, East Pennsylvania (1980). Лікарські засоби можуть бути представлені у твердій формі, наприклад, у вигляді таблеток, таблеток із цукровим покриттям, пігулок, супозиторіїв, капсул, трансдермальних систем або в напівтвердій формі, наприклад, у вигляді мазей, кремів, гелів, супозиторіїв, емульсій або в рідкій формі, наприклад, у вигляді розчинів, настойок, суспензій або емульсій. Наповнювачі в контексті винаходу можуть представляти собою, наприклад, солі, сахариди (моно-, ди-, три-, оліго-, та/або полісахариди), білки, амінокислоти, пептиди, жири, воски, масла, вуглеводні і їх похідні, де наповнювачі можуть бути природного походження або можуть бути отримані синтетично або частково синтетично. Таблетки, таблетки із цукровим покриттям, капсули, пігулки, порошки, гранули, пастилки, суспензії, емульсії або розчини можуть, зокрема, розглядатися для орального або перорального застосування. Суспензії, емульсії й, головним чином, розчини, можуть, зокрема, розглядатися для парентерального застосування. Одержання сполук відповідно до винаходу Сульфоксиміни, як правило, мають високу стабільність відносно структури й конфігурації (C. Bolm, J.P. Hildebrand, J. Org. Chem. 2000, 65, 169). Ці властивості функціональної групи часто представляють можливість навіть сильнодіючих умов реакції й дозволяють здійснити просту дериватизацію сульфоксимінів на іміному азоті й αвуглеці. Енатіомерно чисті сульфоксиміни також використовуються як допоміжні речовини при діастереоселективному синтезі ((a) S.G. Pyne, Sulfur Reports 1992, 12, 57; (b) C.R. Johnson, Aldrichimica Acta 1985, 18, 3). Описане одержання енантіомерно чистих сульфоксимінів, наприклад, шляхом розділення рацемату з енантіомерно чистою камфор-10-сульфоновою кислотою ((a) C.R. Johnson, C.W. Schroeck, J. Am. Chem. Soc. 1973, 95, 7418; (b) C.S. Shiner, A.H. Berks, J. Org. Chem. 1988, 53, 5542). Інший спосіб одержання оптично активних сульфоксимінів представляє собою стереоселективне імінування оптично активних сульфоксидів ((a) C. Bolm, P. Müller, K. Harms, Actchem. Scand. 1996, 50, 305; (b) Y. Tamura, J. Minamikawa, K. Sumoto, S. Fujii, M. Ikeda, J. Org. Chem. 1973, 38, 1239; (c) H. Okamura, C. Bolm, Org. Lett. 2004, 6, 1305). Для огляду статей відносно сульфоксимінів, див., наприклад: a) M. Regglin, C. Zur, Synthesis 2000, 1; (b) C.R. Johnson, Aldrichimica Acta 1985, 18, 3). Наступні приклади пояснюють приготування сполук відповідно до винаходу, не обмежуючи обсяг заявлених сполук цими прикладами. 3 UA 103500 C2 Одержання сполук формули (Ia) (4-O похідні) Сполуки відповідно до винаходу можуть бути отримані за допомогою способу, який характеризується наступними стадіями: a) Окиснення сполуки формули (IVd), одержуючи сульфоксид формули (IVc). 5 O S R O N S 4 R O + O N + O (IVd) 4 (IVc) b1) Пряме імінування сульфоксиду формули (IVc), одержуючи захищений сульфоксимін формули (IVa). 10 F F F O O S N S 4 R O O + N O 15 4 O R + N (IVc) O (IVa) або b2) Імінування сульфоксиду формули (IVc), одержуючи незахищений сульфоксимін формули (IVb) і наступне введення захисної групи, одержуючи сполуку формули (IVa). F F F O O S S 4 O + 4 O R O + O (IVc) N S 4 N N O O R R O NH + N (IVb) (IVa) O c) Відновлення сполуки формули (IVa), одержуючи сполуку формули (IV) 20 F F F F F O F N S O 4 O R O + N O N S 4 O R H2 N (IVa) (IV) d) Функціоналізація 4-го положення 2,4-дихлор-5-йод-піримідину (VII) шляхом взаємодії з моно-захищеним діолом формули (VI) з утворенням проміжної сполуки формули (Va). 25 4 UA 103500 C2 1 R OPG HO Cl 2 R 3 Cl R (VI) N N N N 1 R Cl OPG O 2 3 R I I (VII) R (Va) e) Одержання 5-CF3 проміжної сполуки (V). Cl Cl N N N 1 N 1 R R OPG O 2 R OPG O 2 R 3 R I F 3 R F F (Va) (V) 5 f) Сполучення сполук формул (IV) і (V), одержуючи проміжну сполуку формули (III). F F N O N O 2 R 3 R + 4 O HN H2 N N N F OPG 2 R (IV) F F F 10 1 R O F (V) 4 R R OPG N S N S 1 R O F F F F Cl g) Відщеплення захисної групи (PG) з утворенням (II). 5 F 3 R (III) O UA 103500 C2 F F F F F O F N O S O 4 N S 4 R HN N HN N N 1 R O N OPG 2 R F O R 1 R O 3 R OH 2 3 R F F F R F F (III) (II) h) Відщеплення захисної групи на сульфоксиміні з утворенням (Ia). F F F O N O S O 4 NH S R HN N HN N N 1 R O R 15 20 25 30 OH 2 R F F 3 R F F (II) (Ia) 1 10 1 R O 3 F 5 N OPG 2 R F 4 R 2 3 4 де замісники R , R , R і R мають значення, вказані в загальній формулі (I). Стадія a) Сполуку формули (IVd) окиснюють, одержуючи сульфоксид формули (IVc). Різні методи, включаючи стереоселективні методи, доступні для перетворення простого тіоефіру, одержуючи сульфоксид (див., наприклад: (a) M.H. Ali і ін., Synthesis 1997, 764; b) M.C. Carreno, Chem. Rev. 1995, 95, 1717; c) I. Patel і ін., Org. Proc. Res. Dev. 2002, 6, 225; c) N. Khiar і ін., Chem. Rev. 2003, 103, 3651). Описане застосування перйодної кислоти / хлориду заліза (III) надзвичайно придатне для одержання сполук формули (IVc). Стадія b1) Взаємодія сульфоксиду формули (IVc) із трифторацетамідом у комбінації з діацетатом йодбензолу, оксидом магнію й каталітичними кількостями димеру ацетату родію (II) надає можливість одержання захищеного сульфоксиміну формули (IVa). Ця реакція є стереоспецифічною й здійснюється зі збереженням конфігурації в стереоцентрі (див.: (a) H. Okamura, C. Bolm, Org. Lett. 2004, 6, 1305). Стадія b2) Спочатку здійснюють реакцію сульфоксиду формули (IVc), одержуючи незахищений сульфоксимін формули (IVb). Підходящими методами є, наприклад: a) Взаємодія сульфоксиду з азидом натрію / конц. сірчаною кислотою (див., наприклад: (a) C.R. Johnson, P.E. Rogers, J. Org. Chem. 1973, 38, 1793). b) Взаємодія сульфоксиду з азидом натрію / димлячою сірчаною кислотою (див., наприклад: (a) N.V. Kondratenko, O.A. Radchenko, L.M. Yagupol'ski, J. Org. Chem. USSR 1984, 20, 2051). c) Взаємодія сульфоксиду з o-мезитилен сульфонілгідроксиламіном (MSH) (див., наприклад: (a) C.R. Johnson, R.A. Kirchhoff, H.G. Corkins, J. Org. Chem. 1974, 39, 2458). Наступне введення захисної групи з утворенням сполук формули (IVa) можна здійснювати, 6 UA 103500 C2 5 10 15 20 25 30 35 40 наприклад, як описано, шляхом взаємодії із трифтороцтовим ангідридом у лужних умовах. Стадія c) Для наступного відновлення ароматичної нітро групи з одержанням сполуки формули (IV), доступні, у принципі, різні умови реакцій (див., наприклад: R.C. Larock, Comprehensive Organic Transformations, VCH, New York, 1989, 411). Надзвичайно придатним є описане застосування хлориду титану (III) або гідрування, використовуючи паладій на вугіллі. Стадія d) Взаємодія 2,4-дихлор-5-йод-піримідину (VII) зі спиртом формули (VI) надає можливість одержання проміжної сполуки формули (Va) (див., наприклад: U. Lücking і ін., WO2007/071455). Стадія e) По суті, доступні різні методи для заміни галогену на трифторметильну групу в азот-вмісній гетероароматичній сполуці (див., наприклад: a) G.E. Carr, R.D. Chambers, T.F. Holmes, J. Chem. Soc. Perkin Trans. 1, 1988, 921; b) F. Cottet, M. Schlosser, Eur. J. Org. Chem. 2002, 327; c) F.G. Njoroge і ін., J. Med. Chem 1997, 40, 4290). Зокрема, описане застосування йодиду міді (I), фториду калію й (трифторметил)триметилсилану в N-метил-2-піролідиноні й ТГФ придатне для введення піримідину (Va) в 5-е положення з утворенням проміжної сполуки (V). Стадія f) 2-хлор-піримідин формули (V) можна піддавати реакції з аніліном формули (IV), одержуючи проміжну сполуку формули (III) (див., наприклад: (a) J. Bryant і ін., WO 2004/048343). Стадія g) Відщеплення захисної групи (PG) від проміжної сполуки (III) приводить до одержання проміжної сполуки (II) (див., наприклад: P.J. Kocienski, Protecting Groups, Georg Thieme Verlag Stuttgart, New York, 1994). Описане гідрування надзвичайне придатне для описаного відщеплення бензильної групи. Відщеплення ТГП групи, можна, при необхідності, уже здійснювати в умовах стадії f). Стадія h) Відщеплення трифторацето групи на сульфоксиміні (II) приводить до одержання сполуки формули (Ia). Описана техніка, з використанням карбонату калію в метанолі при кімнатній температурі, надзвичайно придатна для цього (див., наприклад: (a) H. Okamura, C. Bolm, Org. Lett. 2004, 6, 1305). Загальна інформація Усі реакції зі сполуками, чутливими до окиснення або чутливими до гідролізу, здійснювали в атмосфері аргону, з безводними розчинниками. За винятком сульфоксимінових похідних, речовини називали за допомогою програми Autonom 2000 Name, яка реалізована в MDL ISIS Draw. Autonom 2000 Name не приймає будьяких сульфоксимінів, отже, сульфоксиміни називали згідно із правилами ІЮПАК (IUPAC, Nomenclature of Organic Chemistry, 1979 видання, C-6.3. Sulfoxides and Sulfones, Rule C-633, 633.1 Sulfimide and Sulfoximide). Скорочення Скорочення Ac Aloc Boc BOM br CI d dd ДХМ ДМФА ДМСО ESI ВЕРХ m MEM MOM МС MTM Значення Ацетил Алілоксикарбоніл трет-Бутилоксикарбоніл Бензилоксиметил Широкий Хімічна іонізація Дублет Дублет дублетів Дихлорметан N,N-Диметилформамід Диметилсульфоксид Іонізація методом електророзпилення Високоефективна рідинна хроматографія Мультиплет (2-Метоксіетокси)метил Метоксиметил Мас-спектрометрія Метилтіометил 7 UA 103500 C2 NMP N-Метил-2-піролідинон Ядерна магнітно-резонансна спектроскопія: хімічний зсув (δ) представлений у част. на млн. Захисна група, що включає такі групи, як TMS, TES, TBDMS, TBDPS, TIPS, Бензил, PMB, Тритил, Аліл, Aloc, MOM, MTM, MEM, BOM, SEM, ТГП п-Метоксибензил квартет Синглет β-(Триметилсиліл)етоксиметил трет.-Бутилсилілдиметил трет.-Бутилсилілдифеніл Триетиламін Триетилсиліл Тетрагідрофуран Тетрагідропіраніл Триізопропіл Триметилсиліл Триплет ЯМР Pg PMB q s SEM TBDMS TBDPS TEA TES ТГФ ТГП TIPS TMS tr Приклад 1 (RS)-S-циклопропіл-S-(4-{[4-{[(1R,2R)-2-гідрокси-1-метилпропіл]окси}-5(трифторметил)піримідин-2-іл]аміно}феніл)сульфоксимід 5 O NH S HN N N OH O F F F 10 1a) Одержання проміжних сполук Сполука 1.1 1-циклопропілсульфаніл-4-нітробензол S O + N O 15 20 25 1,78 г (44,6 ммоль) гідриду натрію (60 %) порціями додавали до розчину 3,00 г (40,5 ммоль) циклопропан тіолу (одержання відповідно до: E. Block і ін., J. Am. Chem. Soc. 1992, 114, 3492) в 100 мл ТГФ / 100 мл простого діетилового ефіру й перемішували протягом 30 хвилин при кімнатній температурі. Потім порціями додавали 6,00 г (38,7 ммоль) 1-фтор-4-нітробензолу. Суміш перемішували протягом 2 годин при 40 °C. Після її охолодження, суміш вводили у воду й екстрагували бензолом (3x). Об'єднані органічні фази концентрували шляхом упарювання й залишок очищали хроматографічно (гексан / етилацетат 95:5). Одержували 4,6 г (23,6 ммоль; вихід: 61 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,12 (m, 2H), 7,54 (m, 2H), 2,35 (m, 1H), 1,16 (m, 2H), 0,61 (m, 2H). Сполука 1.2 (RS)-1-циклопропан сульфініл-4-нітробензол 8 UA 103500 C2 O S O + N O 5 10 179 мг (1,11 ммоль) хлориду заліза (III) додавали до суміші 7,2 г (36,88 ммоль) 1циклопропілсульфаніл-4-нітробензолу в 140 мл ацетонітрилу і її перемішували протягом 15 хвилин при кімнатній температурі. Потім порціями додавали 9,25 г (40,57 ммоль) перйодної кислоти при 25 °C. Суміш перемішували протягом 30 хвилин і після цього додавали, при перемішуванні, до охолодженого насиченого розчину тіосульфату натрію. Після цього її екстрагували етилацетатом (2x). Об'єднані органічні фази висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Отриманий залишок очищали хроматографічно (гексан / етилацетат 1:1). Одержували 5,93 г (28,07 ммоль; вихід: 76 %) продукту. 1 H-ЯМР (400 МГц, ДМСО):δ = 8,41 (m, 2H), 7,98 (m, 2H), 2,60 (m, 1H), 1,01 (m, 3H), 0,86 (m, 1H). Сполука 1.3 (RS)-S-циклопропіл-S-(4-нітрофеніл)-N-(трифторацетил)сульфоксимід 15 F F F O N S O O + N O 20 25 1,58 г (3,58 ммоль) димеру ацетату родію (II) додавали, в атмосфері аргону, до суспензії 15,1 г (71,53 ммоль) (RS)-1-циклопропан сульфініл-4-нітробензолу, 17,8 г (157,37 ммоль) трифторацетаміду, 38,0 г (118,02 ммоль) діацетату йодбензолу й 12,7 г (314,73 ммоль) оксиду магнію в 801 мл ДХМ і перемішували протягом ночі при кімнатній температурі. Суміш фільтрували через целіт із відсмоктуванням і концентрували шляхом упарювання. Залишок, який залишився, очищали хроматографічно (гексан / етилацетат 2:1). Одержували 18,0 г (55,97 ммоль; вихід: 78 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,49 (m, 2H), 8,25 (m, 2H), 3,56 (m, 1H), 1,51 (m, 1H), 1,41 (m, 1H), 1,18 (m, 2H). Сполука 1.4 (RS)-S-(4-амінофеніл)-S-циклопропіл-N-(трифторацетил)сульфоксимід F F F O N S 30 35 O H2N 1,4 г паладію на вугіллі (10 % / 50 % вологість) додавали до розчину 6,9 г (21,44 ммоль) (RS)-S-циклопропіл-S-(4-нітрофеніл)-N-(трифторацетил)сульфоксиміду в 214 мл етанолу й 39 мл ТГФ і гідрували протягом 1 години при атмосферному тиску при 25 °C. Додатково додавали 1,4 г паладію на вугіллі, і його гідрували додатково протягом 4,5 годин при атмосферному тиску. Суміш фільтрували, до фільтрату знову додавали 1,4 г паладію на вугіллі й на завершення гідрували протягом 45 хвилин. Суміш фільтрували й концентрували шляхом упарювання. Одержували 5,8 г (19,91 ммоль; вихід: 93 %) продукту. 9 UA 103500 C2 1 H-ЯМР (400 МГц, ДМСО): δ = 7,53 (m, 2H), 6,71 (m, 2H), 6,40 (br, 2H), 3,21 (m, 1H), 1,28 (m, 2H), 1,08 (m, 2H). Сполука 1.5 (2R,3R)-3-бензилокси-бутан-2-ол 5 O HO 10 15 20 5,00 г (44,6 ммоль) трет-бутилату калію додавали до розчину 4,00 г (44,4 ммоль) (2R,3R)бутан-2,3-діолу в 300 мл ТГФ при кімнатній температурі й суміш нагрівали в колбі зі зворотним холодильником протягом 15 хвилин. Суміш охолоджували приблизно до 50 °C і додавали 5,3 мл (44,6 ммоль) бензил броміду. Її нагрівали в колбі зі зворотним холодильником протягом 3 годин, і після цього її перемішували протягом ночі при кімнатній температурі. Суміш розводили етилацетатом і розчином хлориду натрію й після цього промивали за допомогою 1 н. розчину соляної кислоти (1x) і розчину хлориду натрію (2x). Органічну фазу висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Отриманий залишок очищали хроматографічно (гексан / етилацетат 1:1). Одержували 3,41 г (18,9 ммоль; вихід: 43 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 7,35 (m, 4H), 7,28 (m, 1H), 4,52 (m, 3H), 3,67 (m, 1H), 3,37 (m, 1H), 1,05 (d, 3H), 1,01 (d, 3H). Сполука 1.6 4-((1R,2R)-2-бензилокси-1-метил-пропокси)-2-хлор-5-йод-піримідин Cl N N O O I 25 30 35 2,07 г гідриду натрію (55 %) порціями додавали до 8,55 г (47,4 ммоль) (2R,3R)-3-бензилоксибутан-2-олу в 56 мл простого діетилового ефіру при 0 °C при перемішуванні. Через 10 хвилин льодяну баню видаляли й суміш перемішували додатково протягом 3 хвилин при кімнатній температурі. Утворену суспензію додавали при 0 °C до розчину 6,52 г (23,7 ммоль) 2,4-дихлор5-йод-піримідину. Суміш перемішували протягом 4 годин при 40 °C і після цього додавали розведений розчин хлориду натрію. Після цього її екстрагували етилацетатом (2x). Об'єднані органічні фази висушували (Na2SO4), фільтрували й концентрували шляхом упарювання. Отриманий залишок піддавали хроматографії (гексан / етилацетат 4:1). Одержували 4,12 г (9,8 ммоль; вихід: 41 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,78 (s, 1H), 7,29 (m, 5H), 5,27 (m, 1H), 4,64 (d, 1H), 4,53 (d, 1H), 3,73 (m, 1H), 1,30 (d, 3H), 1,19 (d, 3H). Сполука 1.7 4-((1R,2R)-2-бензилокси-1-метил-пропокси)-2-хлор-5-трифторметил-піримідин Cl N N O O F F F 40 3,82 г (20,0 ммоль) йодиду міді (I), 0,97 г (16,7 ммоль) фториду калію й 2,47 мл (16,7 ммоль) (трифторметил)-триметилсилану додавали, при перемішуванні, до розчину 4,66 г (11,1 ммоль) 10 UA 103500 C2 5 10 4-((1R,2R)-2-бензилокси-1-метил-пропокси)-2-хлор-5-йод-піримідину в 15,8 мл NMP і 15,8 мл ТГФ при кімнатній температурі. Суміш перемішували протягом 5,5 годин при 80 °C. Після охолодження, суміш додавали до розведеного розчину хлориду натрію й екстрагували етилацетатом (2x). Об'єднані органічні фази висушували (Na2SO4), фільтрували й концентрували шляхом упарювання. Отриманий залишок піддавали хроматографії (гексан / етилацетат 4:1). Одержували 2,17 г (6,0 ммоль; вихід: 54 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,81 (s, 1H), 7,21 (m, 5H), 5,40 (m, 1H), 4,57 (d, 1H), 4,42 (d, 1H), 3,70 (m, 1H), 1,28 (d, 3H), 1,13 (d, 3H). Сполука 1.8 (RS)-S-(4-{[4-{[(1R,2R)-2-(Бензилокси)-1-метилпропіл]окси}-5-(трифторметил)піримідин-2іл]аміно}феніл)-S-циклопропіл-N-(трифторацетил)сульфоксимід F F F O N O S HN N N O O F F F 15 20 25 0,96 мл 4 н. розчину хлористого водню в діоксані додавали до 1,39 г (3,85 ммоль) 4-((1R,2R)2-бензилокси-1-метил-пропокси)-2-хлор-5-трифторметил-піримідину й 1,35 г (4,62 ммоль) (RS)S-(4-амінофеніл)-S-циклопропіл-N-(трифторацетил)сульфоксиміду в 18,8 мл ацетонітрилу й перемішували протягом 5 годин при 80 °C. Після охолодження, суміш розводили етилацетатом і промивали насиченим розчином гідрокарбонату натрію й насиченим розчином хлориду натрію, висушували (Na2SO4), фільтрували й концентрували шляхом упарювання. Отриманий залишок очищали хроматографічно (гексан / етилацетат 4:1). Одержували 1,32 г (2,14 ммоль, вихід: 56 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 10,71 (s, 1H), 8,84 (s, 1H), 8,08 (m, 2H), 7,93 (m, 2H), 7,26 (m, 5H), 5,52 (m, 1H), 4,62 (d, 1H), 4,51 (d, 1H), 3,78 (m, 1H), 3,35 (m, 1H), 1,37 (m, 5H), 1,16 (m, 5H). Сполука 1.9 (RS)-S-циклопропіл-S-(4-{[4-{[(1R,2R)-2-гідрокси-1-метилпропіл]окси}-5(трифторметил)піримідин-2-іл]аміно}феніл)-N-(трифторацетил)сульфоксимід F F F O N O S HN N N OH O F F F 30 1,64 г паладію на вугіллі (10 %) додавали до розчину 1,31 г (2,12 ммоль) (RS)-S-(4-{[4{[(1R,2R)-2-(бензилокси)-1-метилпропіл]окси}-5-(трифторметил)піримідин-2-іл]аміно}феніл)-S 11 UA 103500 C2 5 10 15 циклопропіл-N-(трифторацетил)сульфоксиміду в 66 мл етанолу й гідрували при атмосферному тиску при кімнатній температурі. Суміш фільтрували й концентрували шляхом упарювання. Одержували 0,88 г (1,67 ммоль; вихід: 79 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 10,65 (s, 1H), 8,58 (s, 1H), 8,04 (m, 2H), 7,89 (m, 2H), 5,28 (m, 1H), 4,86 (d, 1H), 3,82(m, 1H), 3,35 (m, 1H), 1,45 (m, 5H), 1,15 (m, 5H). 1b) Одержання кінцевого продукту 1130 мг (8,20 ммоль) карбонату калію додавали до 863 мг (1,64 ммоль) (RS)-S-циклопропілS-(4-{[4-{[(1R,2R)-2-гідрокси-1-метилпропіл]окси}-5-(трифторметил)піримідин-2-іл]аміно}феніл)N-(трифторацетил)сульфоксиміду в 35 мл метанолу й перемішували протягом 1,5 години при кімнатній температурі. Суміш розводили за допомогою насиченого розчину хлориду натрію й екстрагували етилацетатом (3x). Об'єднані органічні фази висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Одержували 709 мг (1,64 ммоль) неочищеного продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 10,50 (s, 1H), 8,59 (s, 1H), 7,94 (m, 2H), 7,84 (m, 2H), 5,32 (m, 1H), 4,91 (d, 1H), 4,07 (s, 1H), 3,86 (m, 1H), 2,63 (m, 1H), 1,30 (d, 3H), 1,11 (m, 4H), 0,91 (m, 3H). МС: 431 (ES+). Суміш діастереомерів розділяли на чисті стереоізомери шляхом препаративної ВЕРХ: Колонка: Chiralpak IA 5µ 250 × 30 мм Елюенти: Гексан / етанол 8:2 Потік: 40,0 мл/хв. Детектор: УФ 254 нм Температура: Кімнатна температура Час утримання: 10,8-13,4 хв.; стереоізомер 1 (= приклад 1-SI-1) 13,6-18,5 хв.; стереоізомер 2 (= приклад 1-SI-2) Приклад 2 (RS)-S-(4-{[4-{[(1R,2R)-2-гідрокси-1-метилпропіл]окси}-5-(трифторметил)піримідин-2іл]аміно}феніл)-S-метилсульфоксимід 20 O NH S HN N N OH O F F F 25 2a) Одержання проміжних сполук Сполука 2.1 (RS)-S-(4-нітрофеніл)-S-метилсульфоксимід O NH S O + N O 30 35 0,70 г (10,76 ммоль) азиду натрію додавали до 1,56 г (8,42 ммоль) 1-(метилсульфініл)-4нітробензолу в 20 мл ДХМ. 2,3 мл концентрованої сірчаної кислоти повільно додавали до суміші при 0 °C і потім її нагрівали до 45 °C. Через 16 годин суміш охолоджували до кімнатної температури, і після додавання води її екстрагували за допомогою ДХМ. Водну фазу доводили до pН 11 за допомогою 15 % розчину гідроксиду натрію й екстрагували за допомогою ДХМ (2x). Об'єднані органічні фази висушували (Na2SO4), фільтрували й концентрували шляхом упарювання. Одержували 1,08 г (5,39 ммоль; вихід: 63 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,43 (m, 2H), 8,17 (m, 2H), 4,62 (s, 1H), 3,18 (s, 3H). Сполука 2.2 12 UA 103500 C2 (RS)-S-метил-S-(4-нітрофеніл)-N-(трифторацетил)сульфоксимід F F F O N S O O + N O 5 10 15 1,00 мл (7,08 ммоль) трифтороцтового ангідриду додавали по краплях, при охолодженні за допомогою льоду, до розчину 1000 мг (4,99 ммоль) (RS)-S-(4-нітрофеніл)-Sметилсульфоксиміду, 55 мг (0,45 ммоль) DMAP і 0,76 мл (5,49 ммоль) триетиламіну в 32 мл ДХМ. Суміш перемішували додатково протягом 2 годин на льодяній бані. Її розводили за допомогою ДХМ і промивали за допомогою напівконцентрованого розчину хлориду натрію. Органічну фазу висушували (Na2SO4), фільтрували й концентрували шляхом упарювання. Отриманий залишок очищали хроматографічно (гексан / етилацетат 60:40). Отриманий продукт на завершення перемішували із простим діізопропіловим ефіром. Тверду речовину відфільтровували з відсмоктуванням і висушували. Одержували 1444 мг (4,87 ммоль; вихід: 98 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,50 (m, 2H), 8,24 (m, 2H), 3,87 (s, 3H). Сполука 2.3 (RS)-S-(4-амінофеніл)-S-метил-N-(трифторацетил)сульфоксимід F F F O N S O H2N 20 25 30 292 мг паладію на вугіллі (10 % / 50 % вологість) додавали до розчину 1,34 г (4,52 ммоль) (RS)-S-метил-S-(4-нітрофеніл)-N-(трифторацетил)сульфоксиміду в 45 мл етанолу й 8 мл ТГФ і гідрували протягом 45 хвилин при атмосферному тиску при 24 °C. Суміш фільтрували й концентрували шляхом упарювання. Отриманий залишок перемішували із простим діізопропіловим ефіром. Тверду речовину відфільтровували з відсмоктуванням і висушували. Одержували 1,07 г (4,03 ммоль; вихід: 89 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 7,54 (m, 2H), 6,67 (m, 2H), 6,35 (s, 2H), 3,55 (s, 3H). Сполука 2.4 (RS)-S-(4-{[4-{[(1R,2R)-2-(Бензилокси)-1-метилпропіл]окси}-5-(трифторметил)піримідин-2іл]аміно}феніл)-S-метил-N-(трифторацетил)сульфоксимід 13 UA 103500 C2 F F F O N O S HN N N O O F F F 5 10 15 0,97 мл 4 н. розчину хлористого водню в діоксані додавали до 1,40 г (3,88 ммоль) 4-((1R,2R)2-бензилокси-1-метил-пропокси)-2-хлор-5-трифторметил-піримідину й 1,20 г (4,51 ммоль) (RS)S-(4-амінофеніл)-S-метил-N-(трифторацетил)сульфоксиміду в 19,0 мл ацетонітрилу й після цього суміш перемішували протягом 6 годин при 80 °C. Після охолодження, суміш розводили етилацетатом і промивали насиченим розчином гідрокарбонату натрію й насиченим розчином хлориду натрію, висушували (Na2SO4), фільтрували й концентрували шляхом упарювання. Отриманий залишок очищали хроматографічно (гексан / етилацетат 1:1). Одержували 1,76 г (2,98 ммоль, вихід: 77 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 10,66 (s, 1H), 8,60 (s, 1H), 8,02 (m, 2H), 7,93 (m, 2H), 7,21 (m, 5H), 5,46 (m, 1H), 4,57 (d, 1H), 4,46 (d, 1H), 3,72 (m, 1H), 3,68 (s, 3H), 1,31 (d, 3H), 1,16 (d, 3H). Сполука 2.5 (RS)-S-(4-{[4-{[(1R,2R)-2-гідрокси-1-метилпропіл]окси}-5-(трифторметил)піримідин-2іл]аміно}феніл)-S-метил-N-(трифторацетил)сульфоксимід F F F O N O S HN N N OH O F F F 20 25 0,18 г паладію на вугіллі (10 %) додавали до розчину 1,75 г (2,96 ммоль) (RS)-S-(4-{[4{[(1R,2R)-2-(бензилокси)-1-метилпропіл]окси}-5-(трифторметил)піримідин-2-іл]аміно}феніл)-Sметил-N-(трифторацетил)сульфоксиміду в 35 мл етанолу й його гідрували при атмосферному тиску при кімнатній температурі. Суміш фільтрували й концентрували шляхом упарювання. Одержували 1,40 г неочищеного продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 10,65 (s, 1H), 8,58 (s, 1H), 8,04 (m, 2H), 7,93 (m, 2H), 5,28 (m, 1H), 4,86 (d, 1H), 3,83 (m, 1H), 3,70 (s, 3H), 1,26 (d, 3H), 1,07 (d, 3H). 2b) Одержання кінцевого продукту 1,92 г (13,89 ммоль) карбонату калію додавали до 1,39 г (2,78 ммоль) (RS)-S-(4-{[4-{[(1R,2R)2-гідрокси-1-метилпропіл]окси}-5-(трифторметил)піримідин-2-іл]аміно}феніл)-S-метил-N(трифторацетил)сульфоксиміду в 60 мл метанолу й перемішували протягом 1,5 години при 14 UA 103500 C2 5 10 кімнатній температурі. Суміш розводили за допомогою насиченого розчину хлориду натрію й екстрагували за допомогою етилацетату (2x). Об'єднані органічні фази висушували (Na2SO4), фільтрували й концентрували шляхом упарювання. Залишок очищали хроматографічно (ДХМ / EtОН 9:1). Одержували 862 мг (2,13 ммоль; вихід: 77 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 10,47 (s, 1H), 8,55 (s, 1H), 7,90 (m, 2H), 7,83 (m, 2H), 5,27 (m, 1H), 4,86 (d, 1H), 4,04 (s, 1H), 3,82 (m, 1H), 3,00 (s, 3H), 1,26 (d, 3H), 1,07 (d, 3H). Суміш діастереомерів розділяли на чисті стереоізомери шляхом препаративної ВЕРХ: Колонка: Chiralpak IC 5µ 250 × 20 мм Елюенти: Гексан / етанол 8:2 Буфер: Гексан/0,1 % DEA Потік: 25,0 мл/хв. Детектор: УФ 280 нм Т емпература: Кімнатна температура Час утримання: 9,5-12,1 хв.; стереоізомер 1 (= приклад 2-SI-1) 13,1-16,0 хв.; стереоізомер 2 (= приклад 2-SI-2) Приклад 3 (RS)-S-(4-{[4-{[(R)-2-гідрокси-1,2-диметилпропіл]окси}-5-(трифторметил)піримідин-2іл]аміно}феніл)-S-метилсульфоксимід O NH S HN N N OH O F F F 15 3a) Одержання проміжних сполук Сполука 3.1. (R)-2-метил-бутан-2,3-діол OH HO 20 25 Розчин 10,0 г (96,1 ммоль) (R)-(+)-метил лактату в 20 мл ТГФ повільно додавали по краплях до 160 мл (480,0 ммоль) охолодженому на льоді 3 н. розчину хлориду метилмагнію в ТГФ. Суміш спочатку повільно нагрівали до кімнатної температури й після цього нагрівали зі зворотним холодильником протягом 30 хвилин. Після охолодження, суміш додавали до насиченого розчину хлориду амонію й екстрагували етилацетатом (3x). Об'єднані органічні фази фільтрували через фільтр Ватману й концентрували шляхом упарювання. Одержували 4,5 г (43,1 ммоль) неочищеного продукту, і використовували без додаткового очищення. 1 H-ЯМР (400 МГц, ДМСО): δ = 4,21 (d, 1H), 3,93 (s, 1H), 3,29 (m, 1H), 0,97 (m, 9H). Сполука 3.2. (R)-3-(2-хлор-5-йод-піримідин-4-ілокси)-2-метил-бутан-2-ол 30 Cl N N O OH I 15 UA 103500 C2 5 10 1,84 г (42,3 ммоль) гідриду натрію (55 %) порціями додавали, при перемішуванні при 0 °C, до розчину 4,40 г (42,3 ммоль) (R)-2-метил-бутан-2,3-діолу в 83 мл простого діетилового ефіру й перемішували протягом 10 хвилин. Її перемішували додатково протягом 3 хвилин при кімнатній температурі й після цього суміш додавали до охолодженого на льоді розчину 9,68 г (35,2 ммоль) 2,4-дихлор-5-йод-піримідину в 97 мл ацетонітрилу. Суміш перемішували протягом 4 годин при 40 °C і, після охолодження, додавали лід і насичений розчин NaСl. Після цього її екстрагували етилацетатом (3x). Об'єднані органічні фази висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Отриманий залишок очищали хроматографічно (гексан / етилацетат 4:1). Одержували 4,96 г (14,5 ммоль; вихід: 41 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,73 (s, 1H), 4,96 (q, 1H), 4,62 (s, 1H), 1,21 (d, 3H), 1,13 (s, 6H). ES: 343 (CI+). Сполука 3.3. 2-хлор-4-[(R)-1,2-диметил-2-(тетрагідро-піран-2-ілокси)-пропокси]-5-йод-піримідин Cl N N O O O I 15 20 25 2,64 мл (29,0 ммоль) дигідропірану й 0,36 г (1,5 ммоль) тозилату піридинію додавали до розчину 4,96 г (14,5 ммоль) (R)-3-(2-хлор-5-йод-піримідин-4-ілокси)-2-метил-бутан-2-олу в 30 мл ДХМ і перемішували протягом 22 годин при кімнатній температурі. Суміш розводили за допомогою ДХМ і промивали насиченим розчином гідрокарбонату натрію. Органічну фазу висушували (Na2SO4), фільтрували й концентрували шляхом упарювання. Отриманий залишок очищали хроматографічно (гексан / етилацетат 4:1). Одержували 5,50 г (12,9 ммоль; вихід: 89 %) суміші діастереомерів. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,75 (s, 1H), 8,74 (s, 1H), 5,15 (m, 2H), 4,91 (m, 2H), 3,70 (m, 2H), 3,30 (m, 2H), 1,31 (m, 30H). Сполука 3.4. 2-хлор-4-[(R)-1,2-диметил-2-(тетрагідро-піран-2-ілокси)-пропокси]-5-трифторметил-піримідин Cl N N O F F O O F 30 35 40 45 1,61 г (8,44 ммоль) йодиду міді (I), 0,41 г (7,03 ммоль) фториду калію й 1,04 мл (7,03 ммоль) (трифторметил)-триметилсилану додавали при кімнатній температурі до розчину 1,00 г (2,34 ммоль) 2-хлор-4-[(R)-1,2-диметил-2-(тетрагідро-піран-2-ілокси)-пропокси]-5-йод-піримідину в 3,3 мл NMP і 3,3 мл ТГФ. Суміш перемішували протягом 2 годин при 90 °C. Після охолодження, суміш додавали до розведеного розчину хлориду натрію й екстрагували етилацетатом (3x). Об'єднані органічні фази висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Отриманий залишок очищали хроматографічно (гексан / етилацетат 4:1). Одержували 0,53 г (1,43 ммоль; вихід: 61 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,84 (s, 1H), 5,32 (m, 1H), 4,85 (m, 1H), 3,68 (m, 1H), 3,30 (m, 1H), 1,31 (m, 15H). 3b) Одержання кінцевого продукту 200 мг (0,54 ммоль) 2-хлор-4-[(R)-1,2-диметил-2-(тетрагідро-піран-2-ілокси)-пропокси]-5трифторметил-піримідину й 87 мг (0,33 ммоль) (RS)-S-(4-амінофеніл)-S-метил-N(трифторацетил)сульфоксиміду в 5 мл етанолу перемішували протягом 6 годин при 70 °C. Суміш упарювали насухо на роторному випарнику й залишок ресуспендували в 11,6 мл. 373 мг 16 UA 103500 C2 5 10 (2,70 ммоль) карбонату калію додавали до розчину і його перемішували протягом 1,5 години при кімнатній температурі. Суміш розводили за допомогою насиченого розчину хлориду натрію й екстрагували етилацетатом (2x). Об'єднані органічні фази висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Залишок очищали шляхом ВЕРХ. Одержували 31 мг (0,07 ммоль; вихід: 14 %) продукту. Колонка: XBridge C18 5µ 100 × 30 мм Елюент A: H2O / 0,1 % HCOOH Елюент B: Ацетонітрил Градієнт: 0 хв. 70 %A 30 %B 1,00 хв. 70 %A 30 %B 7,50 хв. 40 %A 60 %B 7,52 хв. 1 %A 99 %B 10,00 хв. 1 %A 99 %B Потік: 50,0 мл/хв. Виявлення: DAD діапазон сканування 210-400 нм; МС ESI+, ESI-, діапазон сканування 160-1000 m/z Температура: КT 1 H-ЯМР (400 МГц, ДМСО): δ = 10,48 (s, 1H), 8,56 (s, 1H), 7,90 (m, 2H), 7,83 (m, 2H), 5,13 (q, 1H), 4,67 (s, 1H), 4,06 (s, 1H), 3,01 (s, 3H), 1,28 (d, 3H), 1,12 (m, 6H). Одержання сполук загальної формули (Ib) (4-N похідні) Сполуки відповідно до винаходу можуть бути отримані за допомогою способу, який характеризується наступними стадіями: a) Окиснення сполуки формули (IVd), одержуючи сульфоксид формули (IVc). O S R O N R O + N O 15 S 4 + O (IVd) 4 (IVc) b1) Пряме імінування сульфоксиду формули (IVc), одержуючи захищений сульфоксимін формули (IVa). F F F O S O 4 R O N S O 4 R O + N + N (IVc) O O (IVa) або 20 b2) Імінування сульфоксиду формули (IVc), одержуючи незахищений сульфоксимін формули (IVb) і наступне введення захисної групи, одержуючи сполуку формули (IVa). F F F O O S S 4 O + O (IVc) O 17 4 R + N (IVb) N S O + N N O 4 R R O NH O (IVa) O UA 103500 C2 c) Відновлення сполуки формули (IVa), одержуючи сполуку формули (IV) F F F F F O F N O S O 4 S R O N O 4 R + N H2 N (IVa) O (IV) 5 d) Функціоналізація 4-го положення 2,4-дихлор-5-трифторметил-піримідину (VIIb) шляхом взаємодії з аміном формули (VIa) з утворенням проміжної сполуки формули (Vb). 1 R H2 N Cl 1 R OH 2 R 3 R Cl N N N + 1 R N F F F 3 R N OH N H R2 R3 Cl F 2 R (VIa) N OH HN Cl F F F F F (VIIb) (Vc) (Vb) 10 e) Сполучення сполук формули (Vb) і (IV), одержуючи проміжну сполуку формули (IIb). F F F F O N N H R2 R3 4 O NH N H2 N N 1 R OH N H R2 R3 F F F F F (Vb) 15 4 R R + N S N S 1 R OH F O F Cl N F (IV) (IIb) f) Відщеплення захисної групи на сульфоксиміні з утворенням (Ib). 18 O UA 103500 C2 F F F O N O S 4 O NH S R NH N NH N N 1 R N OH N H R2 R3 F F F (Ib) 1 15 20 F F (IIb) 10 1 R OH N H R2 R3 F 5 4 R 2 3 4 де замісники R , R , R і R мають значення, наведені в загальній формулі (I). Стадії a)-c) Ці стадії ідентичні до стадій a)-d) для одержання сполук відповідно до загальної формули (Ia). Стадія d) Взаємодія 2,4-дихлор-5-трифторметил-піримідину (VIIb) з аміном формули (VIa) забезпечує суміш продуктів (Vb) і (Vc). Бажаний продукт (Vb) може бути виділений, наприклад, хроматографічно (див., наприклад: (a) J. Bryant і ін., WO 2004/048343). Стадія e) 2-хлор-піримідин формули (Vb) можна піддавати реакції з аніліном формули (IV), одержуючи проміжну сполуку формули (IIb) (див., наприклад: (a) J. Bryant і ін., WO 2004/048343). Стадія f) Відщеплення трифторацето групи на сульфоксиміні (IIb) забезпечує одержання сполуки формули (Ib). Описана методика з використанням карбонату калію в метанолі при кімнатній температурі надзвичайно придатна для цього. Приклад 4 (RS)-S-циклопропіл-S-(4-{[4-{[(1R,2R)-2-гідрокси-1-метилпропіл]аміно}-5(трифторметил)піримідин-2-іл]аміно}феніл)сульфоксимід O NH S HN N N OH N H F F F 25 4a) Одержання проміжних сполук Сполука 4.1 (2R,3R)-3-(2-хлор-5-трифторметил-піримідин-4-іламіно)-бутан-2-ол Cl N N OH N H F F F 19 UA 103500 C2 5 10 15 20 25 30 72,2 мл (520,71 ммоль) триетиламіну додавали по краплях при 0 °C до 56,5 г (260,35 ммоль) 2,4-дихлор-5-трифторметил-піримідину й 32,7 г (260,35 ммоль) гідрохлориду (2R,3R)-3-амінобутан-2-олу в 1035 мл ацетонітрилі. Суміш повільно нагрівали протягом ночі до кімнатної температури. Суміш додавали до напівконцентрованого розчину хлориду натрію й екстрагували етилацетатом (2x). Об'єднані органічні фази висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Залишок, який залишився, очищали хроматографічно (гексан / етилацетат 0-100 %). Одержували 18,6 г (68,97 ммоль; вихід: 27 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,38 (s, 1H), 6,71 (d, 1H), 5,00 (d, 1H), 4,08 (m, 1H), 3,71 (m, 1H), 1,12 (d, 3H), 1,01 (d, 3H). Одержання (RS)-S-(4-амінофеніл)-S-циклопропіл-N-(трифторацетил)сульфоксиміду здійснювали, як описано для Сполуки 1.4. 4b) Одержання кінцевого продукту 0,21 мл 4 н. розчину хлористого водню в діоксані додавали до 250 мг (0,86 ммоль) (RS)-S-(4амінофеніл)-S-циклопропіл-N-(трифторацетил)сульфоксиміду й 231 мг (0,86 ммоль) (2R, 3R)-3(2-хлор-5-трифторметил-піримідин-4-іламіно)-бутан-2-олу в 3,75 мл ацетонітрилу й потім перемішували протягом 3,5 годин при 60 °C. Суміш упарювали насухо. 18,4 мл метанолу й 590 мг (4,28 ммоль) карбонату калію додавали і її перемішували протягом однієї години при кімнатній температурі. Суміш розводили за допомогою насиченого розчину хлориду натрію й екстрагували етилацетатом (2x). Об'єднані органічні фази висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Залишок, який залишився, очищали хроматографічно (ДХМ / MeОН 4:1). Одержували 242 мг (0,56 ммоль; вихід: 65 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 10,04 (s, 1H), 8,25 (s, 1H), 7,91 (m, 2H), 7,74 (m, 2H), 6,05 (d, 1H), 5,07 (d, 1H), 4,14 (m, 1H), 3,97 (s, 1H), 3,77 (m, 1H), 2,56 (m, 1H), 1,19 (d, 3H), 1,05 (m, 4H), 0,86 (m, 3H). МС: 430 (ESI+). Суміш діастереомерів розділяли на чисті стереоізомери шляхом препаративної ВЕРХ: Колонка: Chiralpak IA 5µ 250 × 20 мм Елюенти: Гексан / 2-пропанол 50:50 Буфер: Гексан/ 0,1 % DEA Потік: 15,0 мл/хв. Детектор: УФ 254 нм Температура: Кімнатна температура Час утримання: 5,9-6,6 хв.; стереоізомер 1 (= приклад 4-SI-1) 7,1-8,8 хв.; стереоізомер 2 (= приклад 4-SI-2) Приклад 5 (RS)-S-циклопропіл-S-(4-{[4-{[(R)-2-гідрокси-1,2-диметилпропіл]аміно}-5(трифторметил)піримідин-2-іл]аміно}феніл)сульфоксимід O NH S HN N N OH N H F F F 35 5a) Одержання проміжних сполук Сполука 5.1 (R)-3-(2-хлор-5-трифторметил-піримідин-4-іламіно)-2-метил-бутан-2-ол 20 UA 103500 C2 Cl N N OH N H F F F 5 10 15 20 25 30 3,6 г (35,03 ммоль) (R)-3-аміно-2-метил-бутан-2-олу додавали по краплях до розчину 7,6 г (35,03 ммоль) 2,4-дихлор-5-трифторметил-піримідину в 139 мл ацетонітрилу. Зараз додавали 9,7 мл (70,1 ммоль) триетиламіну по краплях при 0 °C і суміш повільно нагрівали протягом ночі до кімнатної температури. Її перемішували додатково протягом 2 днів при кімнатній температурі. Суміш додавали до напівконцентрованого розчину хлориду натрію й екстрагували етилацетатом (2x). Об'єднані органічні фази висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Залишок, який залишився, очищали шляхом препаративної ВЕРХ. Одержували 3,0 г (10,65 ммоль; вихід: 30 %) продукту. Колонка: XBridge C18 5µ 150 × 20 мм Елюент A: H2O / 0,2 % NH3 Елюент B: Ацетонітрил Градієнт: 70 %A+30 %B(2') 30→60 %B(10') 60→99 %B(0,1') Потік: 50,0 мл/хв. Детектор: DAD (200-400 нм) TAC; МС-ESI+ (160-1000 m/z) TIC Температура: Кімнатна температура Час утримання: 5,6-6,4 хв. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,42 (s, 1H), 6,52 (d, 1H), 5,01 (s, 1H), 4,10 (m, 1H), 1,11 (m, 9H). Одержання (RS)-S-(4-амінофеніл)-S-циклопропіл-N-(трифторацетил)сульфоксиміду здійснювали, як описано для Сполуки 1.4. 5b) Одержання кінцевого продукту 0,34 мл 4 н. розчину хлористого водню в діоксані додавали до 400 мг (1,37 ммоль) (RS)-S-(4амінофеніл)-S-циклопропіл-N-(трифторацетил)сульфоксиміду й 388 мг (1,37 ммоль) (R)-3-(2хлор-5-трифторметил-піримідин-4-іламіно)-2-метил-бутан-2-олу в 6,0 мл ацетонітрилу й перемішували протягом 3,5 годин при 60 °C. Суміш упарювали насухо. 29,4 мл метанолу й 950 мг (6,84 ммоль) карбонату калію додавали і її перемішували протягом однієї години при кімнатній температурі. Суміш розводили за допомогою насиченого розчину хлориду натрію й екстрагували етилацетатом (2x). Об'єднані органічні фази висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Одержували 600 мг (1,35 ммоль) неочищеного продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 10,08 (s, 1H), 8,30 (s, 1H), 7,94 (m, 2H), 7,80 (m, 2H), 6,07 (d, 1H), 4,95 (s, 1H), 4,16 (m, 1H), 4,02 (s, 1H), 2,62 (m, 1H), 1,20 (m, 6H), 1,10 (m, 4H), 0,89 (m, 3H). МС: 444 (ESI+). Суміш діастереомерів розділяли на чисті стереоізомери шляхом препаративної ВЕРХ: Колонка: Chiralpak AD-H 5µ 250 × 20 мм Елюенти: Гексан / 2-пропанол 60:40 Буфер: Гексан/ 0,1 % DEA Потік: 20,0 мл/хв. Детектор: УФ 280 нм Температура: Кімнатна температура Час утримання: 5,1-6,3 хв.; стереоізомер 1 (= приклад 5-SI-1) 8,0-10,8 хв.; стереоізомер 2 (= приклад 5-SI-2) Приклад 6 (RS)-S-етил-S-(4-{[4-{[(1R,2R)-2-гідрокси-1-метилпропіл]аміно}-5-(трифторметил)піримідин-2іл]аміно}феніл)сульфоксимід 21 UA 103500 C2 O NH S HN N N OH N H F F F 5 6a) Одержання проміжних сполук Сполука 6.1 1-етилсульфаніл-4-нітробензол S O + N O 10 15 16,56 г (106,72 ммоль) 4-нітротіофенолу додавали, при охолодженні за допомогою води, до розчину 4,27 г (106,76 ммоль) гідроксиду натрію в 320 мл етанолу і його перемішували протягом 15 хвилин при кімнатній температурі. Потім, при охолодженні за допомогою води, додавали 8,63 мл (106,79 ммоль) етил йодиду й суміш перемішували протягом ночі при кімнатній температурі. Суміш додавали до насиченого розчину хлориду натрію й екстрагували етилацетатом (2x). Об'єднані органічні фази висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Потім їх розчиняли в ДХМ і знову фільтрували й упарювали насухо. Одержували 16,86 г (92,02 ммоль) неочищеного продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,14 (m, 2H), 7,49 (m, 2H), 3,14 (q, 2H), 1,31 (tr, 3H). Сполука 6.2 (RS)-1-етилсульфініл-4-нітробензол 20 O S O + N O 25 30 35 428 мг (2,64 ммоль) хлориду заліза (III) додавали до суміші 16,86 г (92,02 ммоль) 1етилсульфаніл-4-нітробензолу в 75 мл ацетонітрилу і її перемішували протягом 10 хвилин при кімнатній температурі. Потім порціями додавали 22,44 г (98,44 ммоль) перйодної кислоти, таким чином, що температура не перевищувала 30 °C. Суміш перемішували протягом 50 хвилин і після цього додавали, при перемішуванні, до суміші 170 мл ДХМ, 500 мл суміші води з льодом і 100 г пентагідрату тіосульфату натрію. Її екстрагували за допомогою ДХМ (2x). Об'єднані органічні фази висушували (Na2SO4), фільтрували й концентрували шляхом упарювання. Отриманий залишок перекристалізовували з етилацетату / гексану. Одержували 12,49 г (62,69 ммоль; вихід: 68 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,35 (m, 2H), 7,88 (m, 2H), 3,12 (m, 1H), 2,84 (m, 1H), 0,99 (tr, 3H). Сполука 6.3 (RS)-S-етил-S-(4-нітрофеніл)сульфоксимід 22 UA 103500 C2 O NH S O + N O 5 10 30,5 мл димлячої сірчаної кислоти (20 % SO3) додавали обережно, на льодяній бані, до 6,00 г (30,12 ммоль) (RS)-1-етилсульфініл-4-нітробензолу. Потім, в атмосфері аргону, обережно додавали 2,35 г (36,14 ммоль) азиду натрію, порціями й при перемішуванні, і суміш потім нагрівали до 45 °C. Через 6 годин суміш охолоджували до кімнатної температури й обережно вливали в лід. Суміш підлуговували за допомогою гідрокарбонату натрію й екстрагували етилацетатом (2x). Об'єднані органічні фази висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Одержували 5,74 г (26,79 ммоль; вихід: 89 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,37 (m, 2H), 8,09 (m, 2H), 4,56 (s, 1H), 3,18 (q, 2H), 1,04 (tr, 3H). Сполука 6.4 (RS)-S-етил-S-(4-нітрофеніл)-N-(трифторацетил)сульфоксимід F F F O N S O 15 20 O + N O 4,53 мл (32,04 ммоль) трифтороцтового ангідриду додавали по краплях, при охолодженні на льоді, до розчину 5,72 г (26,70 ммоль) (RS)-S-етил-S-(4-нітрофеніл)сульфоксиміду й 4,07 мл (29,37 ммоль) триетиламіну в 175 мл ДХМ. Суміш перемішували додатково протягом 3 годин на льодяній бані, де температура підвищувалася прибл. до 10 °C. Її розводили за допомогою ДХМ і промивали за допомогою напівконцентрованого розчину хлориду натрію. Органічну фазу висушували (Na2SO4), фільтрували й концентрували шляхом упарювання. Одержували 8,17 г (26,33 ммоль) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 8,52 (m, 2H), 8,22 (m, 2H), 3,99 (m, 2H), 1,16 (tr, 3H). Сполука 6.5 (RS)-S-(4-амінофеніл)-S-етил-N-(трифторацетил)сульфоксимід 25 F F F O N S O H2N 30 35 40 88,5 мл 15 % розчину хлориду титану (III) у прибл. 10 % соляній кислоті повільно додавали, при охолодженні на льоді, до розчину 4,05 г (13,05 ммоль) (RS)-S-етил-S-(4-нітрофеніл)-N(трифторацетил)сульфоксиміду в 191 мл ТГФ. Суміш перемішували протягом 3,5 годин при кімнатній температурі, розводили за допомогою етилацетату й після цього промивали напівконцентрованим розчином хлориду натрію (3x). Органічну фазу висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Одержували 3,17 г (11,31 ммоль) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 7,48 (m, 2H), 6,68 (m, 2H), 3,64 (m, 2H), 1,06 (tr, 3H). Одержання (2R,3R)-3-(2-хлор-5-трифторметил-піримідин-4-іламіно)-бутан-2-олу здійснювали, як описано для Сполуки 4.1. 6b) Одержання кінцевого продукту 0,36 мл 4 н. розчину хлористого водню в діоксані додавали до 400 мг (1,43 ммоль) (RS)-S-(4амінофеніл)-S-етил-N-(трифторацетил)сульфоксиміду й 385 мг (1,43 ммоль) (2R, 3R)-3-(2-хлор5-трифторметил-піримідин-4-іламіно)-бутан-2-олу в 7,0 мл ацетонітрилу й перемішували 23 UA 103500 C2 5 10 протягом 4,5 годин при 60 °C. Суміш упарювали насухо. Додавали 19,0 мл метанолу й 608 мг (4,40 ммоль) карбонату калію, і її перемішували протягом 1 години при кімнатній температурі. Суміш розводили за допомогою насиченого розчину хлориду натрію й екстрагували етилацетатом (2x). Об'єднані органічні фази висушували (Na2SO4), фільтрували й концентрували шляхом упарювання. Одержували 590 мг (1,41 ммоль) неочищеного продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 10,10 (s, 1H), 8,30 (s, 1H), 7,96 (m, 2H), 7,78 (m, 2H), 6,10 (d, 1H), 5,11 (d, 1H), 4,19 (m, 1H), 4,00 (s, 1H), 3,82 (m, 1H), 3,07 (q, 2H), 1,24 (d, 3H), 1,06 (m, 6H). МС: 418 (ESI+). Суміш діастереомерів розділяли на чисті стереоізомери шляхом препаративної ВЕРХ: Колонка: Chiralpak AD-H 5µ 250 × 20 мм Елюенти: Гексан / 2-пропанол 60:40 Буфер: Гексан/ 0,1 % DEA Потік: 20,0 мл/хв. Детектор: УФ 280 нм Температура: Кімнатна температура Час утримання: 6,2-6,8 хв.; стереоізомер 1 (= приклад 6-SI-1) 7,2-8,9 хв.; стереоізомер 2 (= приклад 6-SI-2) Приклад 7 (RS)-S-етил-S-(4-{[4-{[(R)-2-гідрокси-1,2-диметилпропіл]аміно}-5-(трифторметил)піримідин-2іл]аміно}феніл)сульфоксимід O NH S HN N N OH N H F F F 15 20 25 30 7a) Одержання проміжних сполук Одержання (RS)-S-(4-амінофеніл)-S-етил-N-(трифторацетил)сульфоксиміду здійснювали, як описано для Сполуки 6.5. Одержання (R)-3-(2-хлор-5-трифторметил-піримідин-4-іламіно)-2-метил-бутан-2-олу здійснювали, як описано для Сполуки 5.1. 7b) Одержання кінцевого продукту 0,36 мл 4 н. розчину хлористого водню в діоксані додавали до 400 мг (1,43 ммоль) (RS)-S-(4амінофеніл)-S-етил-N-(трифторацетил)сульфоксиміду й 405 мг (1,43 ммоль) (R)-3-(2-хлор-5трифторметил-піримідин-4-іламіно)-2-метил-бутан-2-олу в 7,0 мл ацетонітрилу й перемішували протягом 4,5 годин при 60 °C. Суміш упарювали насухо. Додавали 25,0 мл метанолу й 788 мг (5,70 ммоль) карбонату калію і її перемішували протягом однієї години при кімнатній температурі. Суміш розводили за допомогою насиченого розчину хлориду натрію й екстрагували етилацетатом (2x). Об'єднані органічні фази висушували (Na2SO4), фільтрували й концентрували шляхом упарювання. Одержували 620 мг (1,43 ммоль) неочищеного продукту. 1 H-ЯМР (400 МГц, ДМСО): δ =10,06 (s, 1H), 8,28 (s, 1H), 7,92 (m, 2H), 7,74 (m, 2H), 6,03 (d, 1H), 4,90 (s, 1H), 4,12 (m, 1H), 3,96 (s, 1H), 3,03 (q, 2H), 1,16 (m, 6H), 1,08 (m, 3H), 1,02 (tr, 3H). МС: 432 (ESI+). Суміш діастереомерів розділяли на чисті стереоізомери шляхом препаративної ВЕРХ: Колонка: Chiralpak AD-H 5µ 250 × 20 мм Елюенти: A:Гексан B:2-пропанол Буфер: Гексан/ 0,1 % DEA Градієнт: 20→40 %B(20')+40 %B(5') Потік: 10,0 мл/хв. Детектор: УФ 280 нм Температура: Кімнатна температура Час утримання: 17,5-19,8 хв.; стереоізомер 1 (= приклад 7-SI-1) 20,1-22,0 хв.; стереоізомер 2 (= приклад 7-SI-2) 24 UA 103500 C2 Приклад 8 (RS)-S-(4-{[4-{[(1R,2R)-2-гідрокси-1-метилпропіл]аміно}-5-(трифторметил)піримідин-2іл]аміно}феніл)-S-метилсульфоксимід O NH S HN N N OH N H F 10 15 20 F F 5 8a) Одержання проміжних сполук Одержання (RS)-S-(4-амінофеніл)-S-метил-N-(трифторацетил)сульфоксиміду здійснювали, як описано для Сполуки 2.3. Одержання (2R,3R)-3-(2-хлор-5-трифторметил-піримідин-4-іламіно)-бутан-2-олу здійснювали, як описано для Сполуки 4.1. 8b) Одержання кінцевого продукту 0,38 мл 4 н. розчину хлористого водню в діоксані додавали до 399 мг (1,50 ммоль) (RS)-S-(4амінофеніл)-S-метил-N-(трифторацетил)сульфоксиміду й 404 мг (1,50 ммоль) (2R,3R)-3-(2-хлор5-трифторметил-піримідин-4-іламіно)-бутан-2-олу в 7,3 мл ацетонітрилу й перемішували протягом 9 годин при 60 °C. Суміш упарювали насухо. 32,2 мл метанолу й 1040 мг (7,50 ммоль) карбонату калію додавали і її перемішували протягом 1,5 години при кімнатній температурі. Суміш розводили за допомогою насиченого розчину хлориду натрію й екстрагували етилацетатом (3x). Об'єднані органічні фази висушували (Na2SO4), фільтрували й концентрували шляхом упарювання. Одержували 565 мг (1,40 ммоль) неочищеного продукту. 1 H-ЯМР (400 МГц, ДМСО): 10,09 (s, 1H), 8,30 (s, 1H), 7,96 (m, 2H), 7,83 (m, 2H), 6,10 (d, 1H), 5,11 (d, 1H), 4,18 (m, 1H), 4,03 (s, 1H), 3,82 (m, 1H), 3,03 (s, 3H), 1,25 (d, 3H), 1,10 (d, 3H). Суміш діастереомерів розділяли на чисті стереоізомери шляхом препаративної ВЕРХ: Колонка: Chiralpak IC 5µ 250 × 20 мм Елюенти: Гексан / етанол 50:50 Буфер: Гексан/ 0,1 % DEA Потік: 20,0 мл/хв. Детектор: УФ 254 нм Температура: Кімнатна температура Час утримання: 5,1-5,8 хв.; стереоізомер 1 (= приклад 8-SI-1) 6,1-6,7 хв.; стереоізомер 2 (= приклад 8-SI-2) Приклад 9 25 O NH S HN N N OH N H F F F 30 9a) Одержання проміжних сполук Одержання (RS)-S-(4-амінофеніл)-S-метил-N-(трифторацетил)сульфоксиміду здійснювали, як описано для Сполуки 2.3. 25 UA 103500 C2 5 10 15 20 Одержання (R)-3-(2-хлор-5-трифторметил-піримідин-4-іламіно)-2-метил-бутан-2-олу здійснювали, як описано для Сполуки 5.1. 9b) Одержання кінцевого продукту 0,38 мл 4 н. розчину хлористого водню в діоксані додавали до 399 мг (1,50 ммоль) (RS)-S-(4амінофеніл)-S-метил-N-(трифторацетил)сульфоксиміду й 425 мг (1,50 ммоль) (R)-3-(2-хлор-5трифторметил-піримідин-4-іламіно)-2-метил-бутан-2-олу в 7,3 мл ацетонітрилу і її перемішували протягом 4 годин при 60 °C. Суміш упарювали насухо. Додавали 32,2 мл метанолу й 1040 мг (7,50 ммоль) карбонату калію і її перемішували протягом 1,5 години при кімнатній температурі. Суміш розводили за допомогою насиченого розчину хлориду натрію й екстрагували етилацетатом (2x). Об'єднані органічні фази висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Одержували 600 мг (1,44 ммоль) неочищеного продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 10,05 (s, 1H), 8,26 (s, 1H), 7,91 (m, 2H), 7,79 (m, 2H), 6,03 (d, 1H), 4,91 (s, 1H), 4,11 (m, 1H), 3,99 (s, 1H), 2,99 (s, 3H), 1,16 (m, 6H), 1,10 (m, 3H). МС: 418 (ESI+). Суміш діастереомерів розділяли на чисті стереоізомери шляхом препаративної ВЕРХ: Колонка: Chiralpak IC 5µ 250 × 20 мм Елюенти: Гексан / етанол 80:20 Потік: 30,0 мл/хв. Детектор: УФ 254 нм Температура: Кімнатна температура Час утримання: 6,0-6,7 хв.; стереоізомер 1 (= приклад 9-SI-1) 7,1-8,9 хв.; стереоізомер 2 (= приклад 9-SI-2) Одержання порівняльних речовин Сполуки відповідно до винаходу, які характеризуються, зокрема, 5-CF3 замісником в 5-ому положенні піримідину, порівнювали, відносно їх ефективності в умовах in-vitro і in-vivo, з них 5-Br аналогами, які або розкриті докладно в WO 2005/037800 або альтернативно охоплюються її загальним розкриттям. V11=5-Br порівняльна речовина в прикладі 11 O NH S HN N N OH N H Br 25 30 35 Порівняльна речовина в прикладі 11 представляє собою більш активний стереоізомер суміші діастереомерів (RS)-S-[4-({5-бром-4-[(R)-(2-гідрокси-1,2-диметилпропіл)аміно]піримідин2-іл}аміно)феніл]-S-циклопропіл-сульфоксимід, який описаний у вигляді прикладу 1.6 у заявці WO 2005/037800 (с. 35). Для порівняння в умовах in-vivo, діастереомер (R)-S-[4-({5-бром-4-[(R)-(2-гідрокси-1,2диметилпропіл)аміно]піримідин-2-іл}аміно)феніл]-S-циклопропіл-сульфоксимід, який має більш високу ефективність в умовах in-vitro у порівнянні з (S)-S-[4-({5-бром-4-[(R)-(2-гідрокси-1,2диметилпропіл)аміно]піримідин-2-іл}аміно)феніл]-S-циклопропіл-сульфоксимідом, використовували в прикладі 11. Для цієї мети, V11 одержували за допомогою наступного способу: V11a) Одержання проміжної сполуки 26 UA 103500 C2 O O N S O HN N N OH N H Br 5 10 15 20 0,07 мл 4 н. розчину хлористого водню в діоксані додавали до 988 мг (3,35 ммоль) (R)-3-(5бром-2-хлор-піримідин-4-іламіно)-2-метил-бутан-2-олу й 750 мг (2,79 ммоль) (R)-S-(4амінофеніл)-N-(етоксикарбоніл)-S-циклопропілсульфоксиміду (одержання відповідно до: U. Lücking і ін., WO 2007 / 071455, с. 112, Приклад 4) в 16,50 мл бутанолу й 1,65 мл метанолу й перемішували протягом 3 днів при 60 °C. Після охолодження, суміш упарювали насухо й очищали хроматографічно (ДХМ / EtОН 9:1). Одержували 319 мг (0,61 ммоль, вихід: 22 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 9,96 (s, 1H), 8,13 (s, 1H), 7,92 (m, 2H), 7,73 (m, 2H), 6,28 (d, 1H), 4,05 (m, 1H), 3,84 (m, 2H), 2,96 (m, 1H), 1,05 (m, 16H). V11b) Одержання кінцевого продукту 2,0 мл свіжоприготовленого 1,5M розчину етаноляту натрію додавали до 319 мг (0,61 ммоль) проміжної сполуки в 4,3 мл етанолу й перемішували протягом 18 годин при 60 °C. Після охолодження, суміш додавали до насиченого розчину хлориду натрію й екстрагували етилацетатом (3x). Об'єднані органічні фази висушували (Na 2SO4), фільтрували й концентрували шляхом упарювання. Після кінцевої перекристалізації (ДХМ / етилацетат), одержували 215 мг (0,47 ммоль; вихід: 78 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 9,68 (s, 1H), 8,08 (s, 1H), 7,86 (m, 2H), 7,70 (m, 2H), 6,07 (d, 1H), 4,82 (s, 1H), 4,05 (m, 1H), 3,93 (s, 1H), 2,55 (m, 1H), 1,15 (m, 6H), 1,10 (m, 3H), 1,03 (m, 1H), 0,83 (m, 3H). Колонка: Chiralpak AD-H 5µ 150 × 4,6 мм Елюенти: Гексан / етанол 80,20 Потік: 1,0 мл/хв. Виявлення: PDA 280 нм Температура: 25 °C Час утримання: 11,64 хв. V12=5-Br порівняльна речовина в прикладі 12 O NH S HN N N OH N H Br 25 Порівняльну речовину в прикладі 12 одержували відповідно до прикладу 1.52 у заявці WO 2005/037800. Приклад 1.52 має більш високу ефективність в умовах in-vitro у порівнянні із прикладом 1.53. V13=5-Br порівняльна речовина в прикладі 13 30 27 UA 103500 C2 O NH S HN N N O OH Br 5 Порівняльну речовину в прикладі 13 одержували відповідно до прикладу 3.13 у заявці WO 2005/037800. Приклад 3.13 має більш високу ефективність в умовах in-vitro у порівнянні із прикладом приклад 3.12. V14=5-Br порівняльна речовина в прикладі 14 O NH S HN N N OH O Br 10 V14a) Одержання проміжної сполуки F F F O N O S HN N N OH O Br 15 20 25 30 1,5 мл 4 н. розчину хлористого водню в діоксані додавали до 845 мг (3,00 ммоль) (2R,3R)-3(5-бром-2-хлор-піримідин-4-ілокси)-бутан-2-олу (одержання відповідно до: WO 2005 / 037800, с. 93) і 877 мг (3,00 ммоль) (RS)-S-(4-амінофеніл)-S-циклопропіл-N(трифторацетил)сульфоксиміду в 13,1 мл ацетонітрилу й перемішували протягом 5 годин при 80 °C. До суміші додатково додавали 422 мг (1,50 ммоль) (2R,3R)-3-(5-бром-2-хлор-піримідин-4ілокси)-бутан-2-олу і її перемішували додатково при 80 °C. Через 3 години, знову додавали 0,75 мл 4 н. розчину хлористого водню в діоксані й перемішували додатково при 80 °C. Через 33 годин, знову додавали 175 мг (0,60 ммоль) (RS)-S-(4-амінофеніл)-S-циклопропіл-N(трифторацетил)сульфоксиміду й суміш перемішували на закінчення протягом 18 годин при 80 °C. Після охолодження, суміш концентрували шляхом упарювання й залишок, який залишився, очищали хроматографічно (ДХМ / EtОН 9:1). Одержували 740 мг (1,38 ммоль, вихід: 46 %) продукту. 1 H-ЯМР (400 МГц, ДМСО): δ = 10,31 (s, 1H), 8,44 (s, 1H), 8,01 (m, 2H), 7,84 (m, 2H), 5,19 (m, 1H), 4,88 (d, 1H), 3,81 (m, 1H), 3,31 (m, 1H), 1,40 (m, 1H), 1,29 (m, 4H), 1,08 (m, 5H). V14b) Одержання кінцевого продукту 950 мг (6,84 ммоль) карбонату калію додавали до 735 мг (1,37 ммоль) проміжної сполуки в 29 мл метанолу й перемішували протягом 1,5 години при кімнатній температурі. Суміш розводили за допомогою насиченого розчину хлориду натрію й екстрагували етилацетатом (3x). 28
ДивитисяДодаткова інформація
Назва патенту англійськоюSulfoxivine-substituted anilinopyrimidine derivatives as cdk inhibitors, the production thereof, and use as medicine
Автори англійськоюLucking, Ulrich, Jautelat, Rolf, Siemeister, Gerhard, Schulze, Julia, Lienau, Philip
Автори російськоюЛюккинг Ульрих, Яутелат Рольф, Зимайстер Герхард, Шульце Юлиа, Линау Филип
МПК / Мітки
МПК: C07D 239/47, A61K 31/505, A61P 35/00
Мітки: анілінопіримідинові, похідні, одержання, лікарських, сульфоксімінзаміщені, застосування, засобів, cdk-інгібітори
Код посилання
<a href="https://ua.patents.su/49-103500-sulfoksiminzamishheni-anilinopirimidinovi-pokhidni-yak-cdk-ingibitori-kh-oderzhannya-jj-zastosuvannya-yak-likarskikh-zasobiv.html" target="_blank" rel="follow" title="База патентів України">Сульфоксимінзаміщені анілінопіримідинові похідні як cdk-інгібітори, їх одержання й застосування як лікарських засобів</a>
Сульфоксімінзаміщені піримідини як інгібітори cdk та/або vegf, їх одержання та застосування як лікарських засобів

Номер патенту: 86041
Опубліковано: 25.03.2009
Автори: Зімайстер Герхард, Люкінг Ульріх, Крюгер Мартін, Яутелат Рольф
МПК: C07D 239/48, C07D 239/47
Мітки: застосування, піримідини, лікарських, vegf, інгібітори, засобів, сульфоксімінзаміщені, одержання
Формула / Реферат:
1. Сполуки загальної формули (І) (І),в якій Q означає групу або ,D, Е, G, L, М та Т, у кожному випадку незалежно один від одного, означаютьвуглець, кисень, азот або сірку,R1 означає водень,...
Похідні сульфонаміду, їх одержання та застосування як лікарських засобів

Номер патенту: 78252
Опубліковано: 15.03.2007
Автори: АНДАЛУЗ-МАТАРО Блас, ФРІГОЛА КОНСТАНСА Хорді, МЕРСЕ-ВІДАЛЬ Рамон
МПК: A61P 25/18, C07D 401/14, A61K 31/437, A61K 31/4439, C07D 401/12, A61K 31/454, A61K 31/4709, A61K 31/496, C07D 403/06, A61P 25/24, A61K 31/433, C07D 513/04, C07D 471/04, C07D 401/04, A61K 31/4045, A61K 31/404, C07D 409/14, C07D 417/12, A61K 31/429, A61P 25/22, A61K 31/5377, C07D 209/14, A61P 43/00, A61P 25/28, C07D 409/12
Мітки: застосування, засобів, похідні, одержання, сульфонаміду, лікарських
Формула / Реферат:
1. Похідна сульфонаміду загальної формули (I), (I)деA є замісник, вибраний з групи, яку складають:- гетероароматичний 5- або 6-членний цикл, який містить 1 гетероатом або 2 гетероатоми, вибрані з кисню, азоту та сірки, факультативно заміщений одним або двома атомами галогену, C1-C4-алкілом або фенілом чи 5- або 6-членним гетероарилом, який...
1-феніл-2-гетероарилзаміщені похідні бензимідазолу, їх застосування для одержання лікарських засобів, фармацевтичний засіб, який містить ці похідні

Номер патенту: 81243
Опубліковано: 25.12.2007
Автори: ХАЛЬФБРОДТ Вольфганг, Блюме Торстен, Шнайдер Херберт, Мьоннінг Урсула, Кунке Йоахім, Ельгер Бернд
МПК: A61P 37/02, A61P 29/00, A61K 31/5377, C07D 401/04, C07D 417/14, A61K 31/4439, A61P 43/00, C07D 409/04, C07D 413/14, A61K 31/4184, A61P 37/08
Мітки: засіб, містить, фармацевтичний, похідні, одержання, 1-феніл-2-гетероарилзаміщені, застосування, бензимідазолу, лікарських, засобів
Формула / Реферат:
1. Похідні бензимідазолу загальної формули І, (I)R1 означає фенільну групу, яка необов'язково містить до трьох замісників, незалежно один від одного вибраних із групи, яка включає F, Сl, Br, J, ОН, OR4, OCOR4, SR4, SOR4, SO2R4, R4, NH2, NHR4 і NR4R4', або два суміжні замісники при R1 спільно утворюють групу -О-СН2-О-, -О-СН2-СН2-О- або...
Похідні 6-триазолпіридинсульфанілбензотіазолу і -бензімідазолу, спосіб їх одержання (варіанти), застосування їх як лікарських засобів, фармацевтична композиціїя та застосування як інгібіторів met

Номер патенту: 101328
Опубліковано: 25.03.2013
Автори: Немесек Консепсьон, Бак Ерік, Юголіні Антоніо, Альбер Ева, Венслер Сільві
МПК: A61K 31/5025, A61P 35/00, C07D 487/04
Мітки: лікарських, спосіб, 6-триазолпіридинсульфанілбензотіазолу, фармацевтична, засобів, одержання, композиція, варіанти, застосування, бензимідазолу, інгібіторів, похідні
Формула / Реферат:
1. Продукт формули (I):, (I)в якій:означає простий або подвійний зв'язок,Ra означає атом водню; атом галогену; радикал алкокси, необов'язково заміщений атомом хлору, гідроксильним радикалом або гетероциклоалкільним радикалом, який сам необов'язково заміщений; О-циклоалкільний радикал; гетероарильний радикал, необов'язково заміщений; фенільний радикал, необов'язково заміщений; радикал NHCOалк або NHCOциклоалк або...
Похідні бензоксазинону, спосіб їх одержання, проміжна сполука, лікарський засіб на їх основі та їх застосування як лікарських засобів

Номер патенту: 79460
Опубліковано: 25.06.2007
Автори: Мас Пріо Хосеп, Дордал Зуерас Альберто, Ангелес Фісас Ескасані Маріа, Фрігола Констанца Хорді, Ауреліо Кастрілло Перез Хосе, Торренс Ховер Антоні
МПК: A61P 9/10, A61P 25/00, A61P 29/00, A61P 25/08, A61P 37/00, A61K 31/536, A61P 25/24, A61P 25/22, C07D 413/04, A61P 3/10, A61P 9/12, A61P 1/14, C07D 413/14, A61P 3/04, C07D 417/14, A61P 25/04, A61P 25/02, A61P 3/00, A61P 43/00, A61P 25/28, A61P 19/02
Мітки: засобів, сполука, застосування, спосіб, лікарський, проміжна, засіб, бензоксазинону, одержання, основі, похідні, лікарських
Формула / Реферат:
1. Похідне бензоксазинону загальної формули (І), (I)деR1, R2, R3, R4 кожен незалежно вибраний із групи, яка складається з водню, галогену, нерозгалуженого або розгалуженого, насиченого або ненасиченого, необов’язково принаймні монозаміщеного аліфатичного радикала, насиченого або ненасиченого, необов’язково принаймні монозаміщеного, який необов’язково...
Попередній патент: Антитіло проти інтерлейкіну-17 (іл-17) людини та його застосування
Наступний патент: Вірус бичачої вірусної діареї з модифікованим erns білком
Випадковий патент: Спосіб вимірювання величини подвійного променезаломлення