Спосіб отримання азинілхлорангідридних сполук



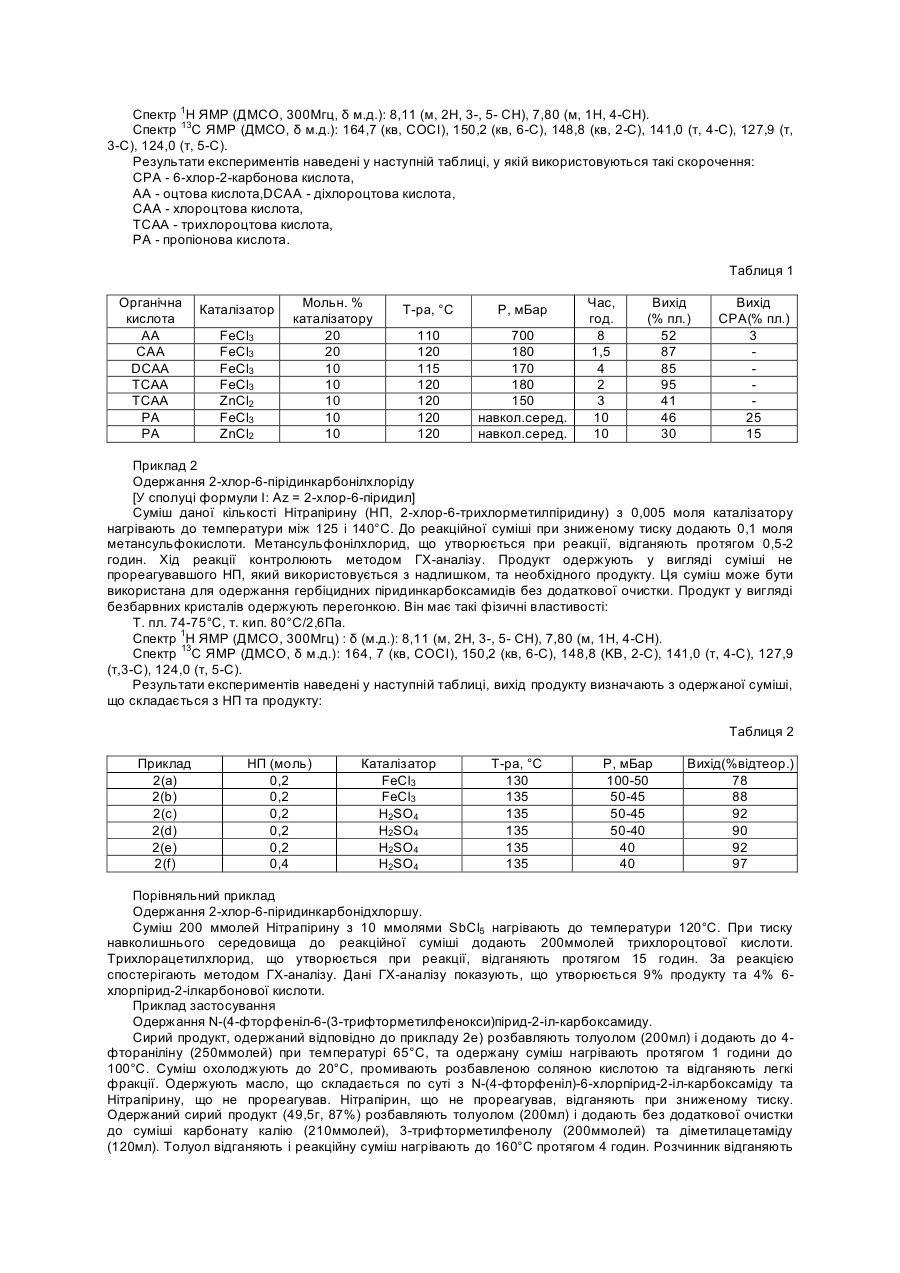

Формула / Реферат
1.Спосіб отримання азинілхлорангідридних сполук формули І,
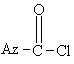 , (І)
, (І)
де замісник Az представляє необов'язково заміщену азинільну групу, який включає нагрівання трихлорметилазину формули ІІ
Az - CCl3, (II)
де замісник Az приймає значення, що визначені вище, з кислотою, що утворює хлорангідрид, який можна відганяти під час реакції при зниженому тиску, у присутності кислого каталізатора.
2. Спосіб згідно з п. 1, у якому вказана кислота являє собою сполуку формули III
R1 – X – OH, (III)
де замісник R1 являє собою С1-6-алкільну або С1-6-галогеналкільну групу і Х являє собою СO або SO2.
3. Спосіб згідно з п. 1, де замісник Аz являє собою азинільну групу, заміщену одним атомом галогену і необов'язково заміщену одною алкільною або галогеналкільною групою.
4. Спосіб згідно з п. 1, у якому кислий каталізатор вибирають з сірчаної кислоти, FeCl3 та ZnCl2.
5. Спосіб згідно з п.1, де замісник Az являє собою заміщену піридильну групу формули V
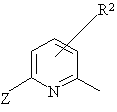 , (V)
, (V)
де замісник R2 являє собою атом водню або алкільну групу, або галогеналкільну групу, та замісник Z являє собою атом галогену.
6. Спосіб згідно з п. 5, де Az являє собою 6-галогенпірид-2-ільну групу.
7. Спосіб згідно з п. 5, у якому 1 моль трихлорметилазину формули II оброблюють 0,4-1,2 моля кислоти формули III.
8. Спосіб згідно з п.5, у якому 1 моль трихлорметиларену формули II оброблюють кислотою формули III у присутності 0,01-0,10 моля кислого каталізатора.
Текст
Даний винахід стосується способу одержання азинілхлорангідридних сполук з трихлорметилазину, в якому трихлорметилазин обробляють кислотою, здатною утворювати хлорангідрид, який по ходу реакції можна відганяти при зниженому тиску, у присутності кислого каталізатору. Азинілхлорангідридні сполуки є придатними проміжними сполуками для одержання широкого ряду сполук, які можуть бути використані як агрохімікати, лікарські препарати або рідкі кристали. Зокрема, вони є ключовими проміжними сполуками при одержанні гербіцидних шридинкарбоксамідів, які описані, наприклад, в ЕР 0 447 004 А. У патенті США № 3875226 описано спосіб, в якому трихлорметильні сполуки обробляють діоксидом сірки у присутності кислоти Льюіса з одержанням хлорангідридних сполук та тіонілхлориду. Однак, цей спосіб важко здійснити у промисловому масштабі, оскільки діоксид сірки є газоподібним при нормальних умовах і з ним необхідно працювати при охолодженні та/або під тиском, а ці умови непридатні у широкомасштабному виробництві. У заявці ЕР 0 646 666 А пропонується гідролізувати трихлорметилазини водою у присутності хлорованих вуглеводнів та кислоти Льюісу. Однак, при такому способі виникають проблеми, пов'язані зі швидкістю дозування і точного еквімолярного дозування води. Будь-який надлишок води викликає гідроліз цільової хлорангідридної сполуки, зменшуючи таким чином вихід. Крім того, у даний час використання хлорованих вуглеводнів небажано з точки зору захисту навколишнього середовища, а кількість розчинника, яка використовується за методикою попереднього рівня, висока. Більше того, час реакції, необхідний при використанні системи вода/1,2-діхлоретан3 дуже значний (24 години). В ЕР 0 091 022 описується одержання ізоксазол-5-карбонової кислоти з 5-трихлорметилізоксазолу з використанням трихлороцтової кислоти та пентахлориду сурми або хлориду залізу (3) у якості кислоти Льюіса. Однак, відсутні припущення про можливість використання цього способу для інших трихлорметилазинів та іншої карбонової кислоти. У той час, як реакція з використанням пентахлориду сурми закінчується за 2 години, при використанні хлориду залізу (3) необхідно 8 годин. Одним з недоліків цього способу є те, що дорогий пентахлорид сурми отруйний і, отже, цей спосіб не може бути використаний у промисловому масштабі. Крім того, при використанні пентахлориду сурми для виготовлення азиноїлхлоридів досягаються тільки низькі виходи. Німецька патентна заявка DE 30 04 693 описує спосіб одночасного одержання ароматичних сульфонілгалогенидів і бензоїлгалогенидів реакцією ароматичних сульфонових кислот з трихлорметиларенами. Однак, розділення цих продуктів вимагає дуже важких технологій перегонки. Завдання, яке необхідно вирішити, полягає у тому, щоб забезпечити спосіб одержання азинілхлорангідридів з високими виходами, в якому не використовуються розчинники, які викликають проблеми з навколишнім середовищем, та який не потребує тривалого часу на проведення реакції. Суть винаходу Несподівано було виявлено, що азинілхлорангідридьгі сполуки формули І (I) де замісник Az являє собою необов'язково заміщену азинільну групу, можуть бути легко одержані з високими виходами способом, який включає нагрівання трихлорметилазину формули II (II) де замісник Az має значення, визначені вище, з кислотою, яка створює хлорангідрид, який можна відганяти по ходу реакції при зниженому тиску, в присутності кислого каталізатора, переважно нагріванням названого трихлорметилазину формули II з кислотою формули ІІІ (IІІ) 1 де замісник R являє собою С1-6-алкільну або С1-6-галогеналкільну групу, а X являє собою CO або SO2, в присутності кислого каталізатора при пониженому тиску. Таким чином, мета даного винаходу полягає у створенні нового ефективного способу одержання азиноїлхлоридних сполук. Ще одна мета даного винаходу полягає у використанні азиноїлхлоридних сполук, одержаних згідно зі способом даного винаходу, для одержання (гетеро)-арилоксиазинілкарбоксамідів. Інші об'єкти та переваги даного винаходу будуть очевидні для спеціаліста в даній галузі з наступного опису та прикладеної формули винаходу. Докладний опис винаходу. Якщо не оговорено спеціально, то у загальному випадку термін "необов'язково заміщена азинільна група", що застосовується при описі щодо замісника Az, стосується 6-членної гетероциклічної групи з, принаймні, одним атомом азоту, зокрема, піридинової або піримідинової групи, необов'язково заміщеної одним або більше атомами галогену, нітро-, ціано-групами, алкілом, переважно С1-6-алкілом, алкоксигрупою, переважно С1-6-алкокси, 4-алкілциклогексилом, переважно 4-С1-6-алкілциклогексилом, або галогеналкілом, переважно С1-4-галогеналкільними групами. Як правило, переважними є ті гетероароматичні групи, які заміщені принаймні одною електронакцепторною групою, зокрема, одним або більше атомами галогену, нітро-, піано-, або галогеналкільними групами. В особливо переважному варіанті здійснення винаходу замісник Az являє собою необов'язково заміщену піридильну групу формули V, (V) в якій замісник R2 являє собою атом водню або галогену або алкільну або галогеналкільну групу, та замісник Ζ являє собою атом галогену. Якщо не оговорено спеціально, то у загальному випадку термін "алкільна або галогеналкільна група", що застосовується в описі щодо радикалу або залишку, стосується прямого або розгалуженого радикала або залишка. Як правило, такі радикали мають до 10, зокрема до 6, атомів вуглецю. Відповідно, алкільний або галогеналкільний залишок містить від 1 до 6 атомів вуглецю, переважно від 1 до 3 атомів вуглецю. Переважним алкільним залишком є етильна або особливо метальна група. Переважними галогеналкільними групами є полі- або пергалогеновані алкільні групи формули -(CX2)n-Y де n являє собою ціле число від 1 до 10, переважно від 1 до 6, зокрема від 1 до 3, замісник X являє собою атом фтору або хлору та замісник Υ являє собою водень або X. Преважна полігалогенована алкільна група являє собою пентафторетил, пентахлоретил або особливо діфтор- або трифторметильну групу або діхлорабо трихлорметильну групу. Необов'язково заміщені групи можуть бути незаміщеними або містити від одного до максимально можливого числа замісників. Як правило, присутні від 0 до 2 замісників.Додаткові переважні варіанти здійснення способу відповідно до даного винаходу представляють собою спосіб, у якому (а) Спосіб, де замісник Az являє собою азинільну групу, яка заміщена одним атомом галогену та необов'язково заміщена одною алкільною або галогеналкільною групою, переважно заміщену піридильну групу формули V, (V) в якій замісник R2 являє собою атом водню або алкільну або галогеналкільну групу, та замісник Ζ являє собою атом галогену, зокрема 6-галогенпірид-2-ільну групу. (b) Спосіб, де замісник R1 являє собою метильну групу, необов'язково заміщену одним або більше атомами хлору. (c) Спосіб, де кислий каталізатор вибирають з сірчаної кислоти, FeCl3 і ZnCl3. (d) Спосіб, де нагрівають реакційну суміш, яка складається по суті з трихлорметилазину формули II, кислоти формули IIІ та кислого каталізатору, та хлорангідрид формули IV, (IV) де замісник R1 і X визначені вище, який утворюється у ході реакції, відганяють при зниженому тиску. (e) Спосіб, у якому 1 моль трихлорметилазину формули ІІ обробляють від 0,4 до 1,2 моля кислоти формули ІІІ. (f) Спосіб, у якому X являє собою S02 і кислий каталізатор являє собою сірчану кислоту з вмістом води не менше 5% вагових. (g) Спосіб, у якому 1 моль трихлорметиларену формули II обробляють кислотою формули ІІІ у присутності від 0,01 до 0,10 моля кислого каталізатору. Реакцію проводять при температурі між температурою навколишнього середовища та температурою перегонки реакційної суміші, переважно при підвищеній температурі, особливо при температурі перегонки, переважно між 75 та 160°С, зокрема при температурі між 85 та 130°С. Інший об'єкт даного винаходу являє собою застосування сполуки формули І для одержання (гетеро)арилоксигетероарилкарбоксамидів формули VI (VI) Де Az являє собою азинільну групу, необов'язково заміщену одною алкільною або галогеналкільною групою, переважно необов'язково заміщену піридильну групу; Аг являє собою необов'язково заміщену арильну або гетероарильну групу, переважно фенильну групу, заміщену, принаймні, одним атомом галогену або галогеналкільною або галогеналкоксигрупою, зокрема, піридин-2,6-діїльну групу, зокрема 3-трифторметилфенильну групу; R3 являє собою атом водню або алкільну групу, переважно атом водню; і R4 являє собою необов'язково заміщену алкільну, арильну, гетероарильну або циклоалкільну групу, переважно фенильну групу, заміщену, принаймні, одним атомом галогену або галогеналкільною або галогеналкоксигрупою, зокрема 4-фторфенильну групу, де (а) моногалогенований азиноїлхлорид формули І, що одержується з моногалогенованого азинілтрихлорметану формули II відповідно до будь-якого з пунктів 1-10, (b) реагує з аміном формули VII (VII) 3 4 де замісники R та R приймають значення, визначені вище, необов'язково у присутності інертного розчинника та/або основи, та(с) одержуваний моногалогенований азинілкарбоксамид реагує з ароматичною або гетероароматичною гідроксил-сполукою формули VIII, (VIII) в якій замісник Аг приймає визначені вище значення,у присутності основи, зокрема де галогенований азинілхлорангідрид формули І, одержаний відповідно до будь-якого з пунктів 1-10, реагує з аміном формули VII без додаткової очистки. Як правило, реакцію проводять при зниженому тиску з метою полегшення відгонки сполуки формули IV, що створюється по ходу реакції. Найбільш переважним є проведення реакції при тисках між 20 та 400мбар (2 і 40кПа), зокрема між 25 та 250мбар (2,5 та 25кПа). В особливо переважному варіанту здійснення способу у відповідності з даним винаходом 1 еквівалент трихлорметилазину формули ІІ, переважно в якому замісник Az являє собою заміщену піридинову групу, зокрема Нітрапірин, змішують з 0,05-0,15 еквіваленту каталізатору, зокрема FeCl3, і нагрівають до температури 80-150°С, зокрема 110-130°С. Потім до цієї реакційної суміші, яку тримають при зниженому тиску, додають 0,8-1,2 еквіваленту кислоти формули III, де X являє собою CO, зокрема хлороцтову кислоту, діхлороцтову кислоту або трихлороцтову кислоту. Відповідний ацилхлорид формули IV відганяють до тих пір, поки реакція не закінчиться. За цих переважних умов реакція, як правило, закінчується за час від 1 до 5 годин, зокрема від 1,5 до 4 годин. В іншому особливо переважному варіанті здійснення способу у відповідності з даним винаходом 1 еквівалент трихлорметилазину формули ІІ, переважно в якому замісник Az являє собою заміщену піридинову групу, зокрема Нітрапірин, змішують з 0,01-0,05 еквіваленту каталізатора, зокрема H2SO4, і нагрівають до температури 80-150°С, зокрема 110-130°С. Потім до цієї реакційної суміші, яку підтримують при пониженому тиску, додають 0,2-0,8 еквіваленту кислоти формули III, де X являє собою SO2, зокрема метансульфокислоту. Відповідний алкансульфонілхлорид відганяють до тих пір, поки реакція не закінчиться. Коли реакцію проводять з еквімолярними кількостями гетероциклічного трихлорметилазину формули ІІ і алкансульфокислоти формули ІІІ, можуть бути одержані смолоподібні побічні продукти, які зменшують виходи, що досягаються, та викликають труднощі під час процедури очистки. Таким чином, використання надлишку трихлорметилазину може бути вигідним, оскільки при цьому виключається утворення цих побічних продуктів. В особливо переважному варіанті здійснення даного винаходу надлишок 1-4 еквівалентів Нітрапірину реагує з 1 молем метансульфокислоти у присутності 0,01-0,05 моля H2SO4 (96-99% вагових). При переважних умовах реакція, як правило, закінчується за час 0,25-5 годин, зокрема 0,3-3 години. Азиноїлхлорид, що залишається, може бути використаний як проміжна сполука для одержання необхідних кінцевих продуктів без подальшої очистки, їх можна також очистити з використанням звичайних методик; таких як, наприклад, кристалізація або перегонка при зниженому тиску, зокрема, при тисках між 1 і 100мбар (0,1 і 10кПа). Новий спосіб дозволяє здійснення одержання азинілхлорангідридів у промисловому масштабі та з високими виходами, з використанням дешевих, легкодоступних виділених речовин. Більше того, кислоти формули ІІІ, які використовують як реагенти у новій методиці, можуть бути рециркульовані шляхом додавання води до відповідного хлорангідрида, що утворюється в ході реакції. Таким чином, по новій методиці необхідна лише невелика кількість реагенту. Для полегшення розуміння винаходу нижче приведені наступні ілюстративні приклади. Винахід не обмежується конкретними описаними або наведеними варіантами, а охоплює весь об'єм робіт представленої формули винаходу. Приклад 1 Одержання 2-хлор-6-пірідинкарбонілхдориду [У сполуці формули І: Az = 2-хлор-6-піридил] Суміш 200ммолей Нітрапірину (2-хлор-6-трихлорметилпіридину) з даною кількістю каталізатора нагрівають до температури між 90 і 130°С. До реакційної суміші при зниженому тиску додають 200ммолей органічної кислоти. При проведенні реакції хлорангідрид органічної кислоти, що утворюється в ході реакції, відганяють. Хід реакції контролюють за допомогою газової хроматографії (ГХ). Продукт у вигляді безбарвних кристалів одержують за допомогою звичайної переробки та перегонки при зниженому тиску. Він має наступні фізичні властивості: Т. пл. 74-75°С, т. кип. 80°С/2,6Па. Спектр 1Н ЯМР (ДМСО, 300Мгц, δ м.д.): 8,11 (м, 2Н, 3-, 5- СН), 7,80 (м, 1Н, 4-СН). Спектр 13С ЯМР (ДМСО, δ м.д.): 164,7 (кв, СОСІ), 150,2 (кв, 6-С), 148,8 (кв, 2-C), 141,0 (т, 4-С), 127,9 (т, 3-С), 124,0 (т, 5-С). Результати експериментів наведені у наступній таблиці, у якій використовуються такі скорочення: СРА - 6-хлор-2-карбонова кислота, АА - оцтова кислота,DCAA - діхлороцтова кислота, САА - хлороцтова кислота, ТСАА - трихлороцтова кислота, РА - пропіонова кислота. Таблиця 1 Органічна кислота АА САА DCAA ТСАА ТСАА РА РА Каталізатор FeCl3 FeCl3 FeCl3 FeCl3 ZnCl2 FeCl3 ZnCl2 Мольн. % каталізатору 20 20 10 10 10 10 10 Т-ра, °С Р, мБар 110 120 115 120 120 120 120 700 180 170 180 150 навкол.серед. навкол.серед. Час, год. 8 1,5 4 2 3 10 10 Вихід (% пл.) 52 87 85 95 41 46 30 Вихід СРА(% пл.) 3 25 15 Приклад 2 Одержання 2-хлор-6-пірідинкарбонілхлоріду [У сполуці формули І: Az = 2-хлор-6-піридил] Суміш даної кількості Нітрапірину (НП, 2-хлор-6-трихлорметилпіридину) з 0,005 моля каталізатору нагрівають до температури між 125 і 140°С. До реакційної суміші при зниженому тиску додають 0,1 моля метансульфокислоти. Метансульфонілхлорид, що утворюється при реакції, відганяють протягом 0,5-2 годин. Хід реакції контролюють методом ГХ-аналізу. Продукт одержують у вигляді суміші не прореагувавшого НП, який використовується з надлишком, та необхідного продукту. Ця суміш може бути використана для одержання гербіцидних піридинкарбоксамидів без додаткової очистки. Продукт у вигляді безбарвних кристалів одержують перегонкою. Він має такі фізичні властивості: Т. пл. 74-75°С, т. кип. 80°С/2,6Па. Спектр 1Н ЯМР (ДМСО, 300Мгц) : δ (м.д.): 8,11 (м, 2Н, 3-, 5- СН), 7,80 (м, 1Н, 4-СН). Спектр 13С ЯМР (ДМСО, δ м.д.): 164, 7 (кв, СОСІ), 150,2 (кв, 6-С), 148,8 (KB, 2-C), 141,0 (т, 4-С), 127,9 (т,3-С), 124,0 (т, 5-С). Результати експериментів наведені у наступній таблиці, вихід продукту визначають з одержаної суміші, що складається з НП та продукту: Таблиця 2 Приклад 2(а) 2(b) 2(с) 2(d) 2(е) 2(f) НП (моль) 0,2 0,2 0,2 0,2 0,2 0,4 Каталізатор FeCl3 FeCl3 H2SO4 H2SO4 H2SO4 H2SO4 T-pa, °С 130 135 135 135 135 135 Ρ, мБар 100-50 50-45 50-45 50-40 40 40 Вихід(%відтеор.) 78 88 92 90 92 97 Порівняльний приклад Одержання 2-хлор-6-піридинкарбонідхлоршу. Суміш 200 ммолей Нітрапірину з 10 ммолями SbCl5 нагрівають до температури 120°С. При тиску навколишнього середовища до реакційної суміші додають 200ммолей трихлороцтової кислоти. Трихлорацетилхлорид, що утворюється при реакції, відганяють протягом 15 годин. За реакцією спостерігають методом ГХ-аналізу. Дані ГХ-аналізу показують, що утворюється 9% продукту та 4% 6хлорпірид-2-ілкарбонової кислоти. Приклад застосування Одержання N-(4-фторфеніл-6-(3-трифторметилфенокси)пірид-2-іл-карбоксамиду. Сирий продукт, одержаний відповідно до прикладу 2е) розбавляють толуолом (200мл) і додають до 4фтораніліну (250ммолей) при температурі 65°С, та одержану суміш нагрівають протягом 1 години до 100°С. Суміш охолоджують до 20°С, промивають розбавленою соляною кислотою та відганяють легкі фракції. Одержують масло, що складається по суті з N-(4-фторфеніл)-6-хлорпірид-2-іл-карбоксаміду та Нітрапірину, що не прореагував. Нітрапірин, що не прореагував, відганяють при зниженому тиску. Одержаний сирий продукт (49,5г, 87%) розбавляють толуолом (200мл) і додають без додаткової очистки до суміші карбонату калію (210ммолей), 3-трифторметилфенолу (200ммолей) та діметилацетаміду (120мл). Толуол відганяють і реакційну суміш нагрівають до 160°С протягом 4 годин. Розчинник відганяють і залишок розбавляють толуолом. Суміш промивають NaHCO3, сушать і концентрують. Залишок перекристалізовують з метанолу, одержують названу сполуку (58,2г, 85%), т.пл. 105-107°С.
ДивитисяДодаткова інформація
Назва патенту англійськоюA method to obtain azinyl acid chloride combinations
Автори англійськоюKnell Marcus, Brink Monika
Назва патенту російськоюСпособ получения азинилхлорангидридных соединений
Автори російськоюКнелль Маркус, Бринк Моника
МПК / Мітки
МПК: C07D 213/78, C07D 213/46, C07C 51/58
Мітки: сполук, спосіб, азинілхлорангідридних, отримання
Код посилання
<a href="https://ua.patents.su/5-51641-sposib-otrimannya-azinilkhlorangidridnikh-spoluk.html" target="_blank" rel="follow" title="База патентів України">Спосіб отримання азинілхлорангідридних сполук</a>
Спосіб одержання 1-(тіометил)циклопропаноцтової кислоти та проміжних сполук для її одержання

Номер патенту: 50730
Опубліковано: 15.11.2002
Автори: Кінг Стівен, Конлон Девід Е., Піпік Бренда
МПК: C07D 327/00, C07C 253/16, C07C 319/00, C07C 327/00
Мітки: 1-(тіометил)циклопропаноцтової, спосіб, одержання, сполук, проміжних, кислоти
Формула / Реферат:
1. Спосіб одержання циклічного сульфіту 1,1-циклопропандиметанолу формули,який передбачає:а) взаємодію 1,1-циклопропандиметанолу з діалкілсульфітом в присутності кислоти або основи таb) вилучення з реакційної суміші спиртового побічного продукту реакції.2. Спосіб за п. 1, за яким реакцію проводять в присутності основи.3. Спосіб за...
Спосіб отримання чотирьохкомпонентного твердого розчину на основі сполук а-4, в-6.

Номер патенту: 31810
Опубліковано: 15.12.2000
Автори: Фреїк Дмитро Михайлович, Запухляк Руслан Ігорович, Варшава Славомир Степанович
МПК: C30B 11/02
Мітки: спосіб, сполук, розчину, в-6, а-4, отримання, твердого, основі, чотирьохкомпонентного
Текст:
...-768 с). Однак, ці способи їх отримання складні, дорогі, не дозволяють плавно керувати електричними і термоелектричними параметрами. Найбільш близькими до запропонованого винаходу є спосіб отримання чотирьохкомпонентного твердого розчину на основі сполук А^В^ , який полягає в тому, що вихідну речовину, розташовану в кварцевій вакуумованій ампулі, поміщають у двохзонну піч, температура першої зони якої є вищою від температури плавлення...
Спосіб отримання ароматичних сполук

Номер патенту: 35553
Опубліковано: 16.04.2001
Автори: Ревелант Деніс, Вакус Паскаль, Джулак Фаузі
МПК: C07C 265/00, C07C 263/00
Мітки: отримання, спосіб, сполук, ароматичних
Формула / Реферат:
1. Способ получения ароматических соединений, замещенных, по меньшей мере, двумя группами изоцианатов, путем контакта соответствующего ароматического амина с фосгеном в газовой фазе, отличающийся тем, что используют, по меньшей мере, одно соединение (А), относящееся к диамину, соответствующее формуле (1):H2N-R-NH2 , (1)в которой R является ароматическим звеном с С6-С14, предпочтительно с С6-С10,...
Спосіб отримання арилпіперазиніл-гетероциклічних сполук

Номер патенту: 40005
Опубліковано: 16.07.2001
Автори: БУШ ФРЕНК Р., ГОДЕК Денніс М., БОУЛС Поль, АЛЛЕН Дуглас Дж. М., ДІРОМА Сабето А.
МПК: C08F 236/00, C08C 19/00, C08F 8/04
Мітки: отримання, спосіб, арилпіперазиніл-гетероциклічних, сполук
Формула / Реферат:
1. Способ получения арилпиперазинил-гетероциклических соединений общей формулы (I) или их фармацевтически приемлемых кислотно-аддитивных солей, гдеАг представляет нафтил, необязательно замещенный от одного до четырех заместителями независимо выбранными из фтора, хлора, трифторметила, метокси, циано и нитро, хинолил, 6-гидрокси-8-хинолил, изохинолил, хиназолил, бензоизотиазолил и его оксиды или диоксиды,...
Спосіб отримання термоелектричних сплавів на основі сполук аiv bvi

Номер патенту: 46281
Опубліковано: 15.05.2002
Автори: Никируй Любомир Іванович, Довгий Олег Ярославович, Фреїк Дмитро Михайлович, Межиловська Любов Йосипівна, Іванишин Ірина Мирославівна
МПК: C30B 1/00
Мітки: термоелектричних, сполук, сплавів, основі, спосіб, отримання
Формула / Реферат:
Спосіб отримання термоелектричних сплавів на основі сполук АIVВIV, який полягає в тому, що вихідну речовину розташовують в кварцовій вакуумованій ампулі, поміщають у піч, температура якої є вищою від температури плавлення вихідних елементів, ампулу з вихідними елементами витримують при цій температурі, після чого охолоджують до кімнатної температури, який відрізняється тим, що як вихідну речовину використовують окремі елементи, співвідношення...
Попередній патент: Спосіб утруднення встановлення або реактивації латентної інфекції, яка викликається вірусами герпесу
Наступний патент: Дезоксирибонуклеїнова кислота, яка кодує білок глутатіон-s-трансферазу, білок
Випадковий патент: Розділювана галенова форма, яка робить можливим модифіковане вивільнення активного інгредієнта