Спосіб одержання форми і венлафаксину гідрохлориду




Формула / Реферат
1. Спосіб одержання форми І венлафаксину гідрохлориду, який відрізняється тим, що
(a) розчин венлафаксину в органічному розчиннику, що є ізопропілацетатом і/або циклогексаном або пропілацетатом, або бутилацетатом, або ізобутилацетатом, або трет-бутилацетатом, піддають взаємодії з водним розчином НСl, і
(b) вміст води в одержаному розчині венлафаксину гідрохлориду доводять менш ніж до 3 мас. %, і
(c) кристалізують форму І венлафаксину гідрохлориду.
2. Спосіб за п. 1, який відрізняється тим, що вміст води в одержаному розчині венлафаксину гідрохлориду на стадії (b) доводять менш ніж до 1,5 мас. %.
3. Спосіб за п. 1, який відрізняється тим, що вміст води на стадії (b) регулюють шляхом азеотропної перегонки розчину.
4. Спосіб за будь-яким з пп. 1-2, який відрізняється тим, що одержана форма І венлафаксину гідрохлориду має середній розмір частинок менше 50 мкм, переважний середній розмір частинок в інтервалі від 10 до 40 мкм.
Текст
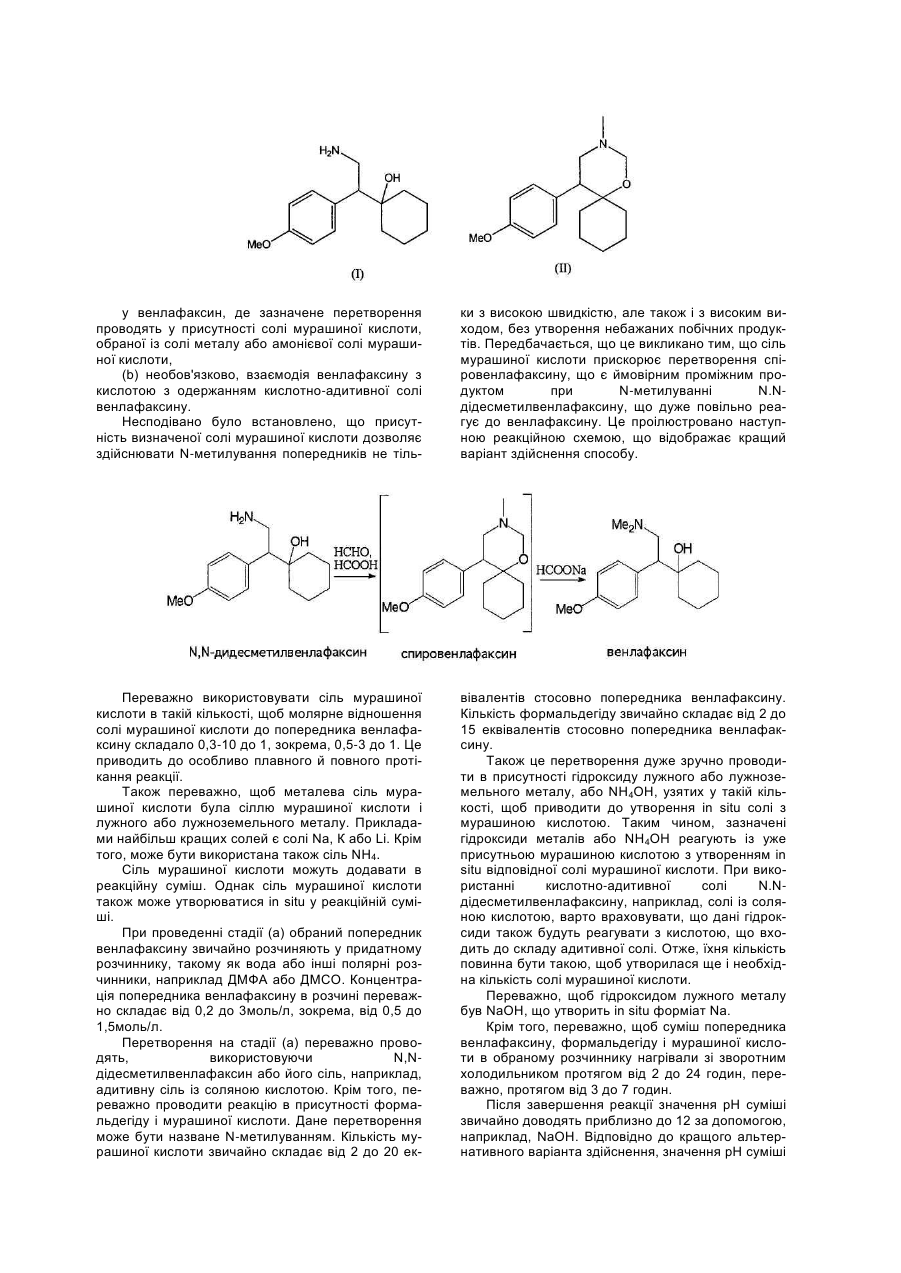
1. Спосіб одержання форми І венлафаксину гідрохлориду, який відрізняється тим, що (a) розчин венлафаксину в органічному розчиннику, що є ізопропілацетатом і/або циклогексаном C2 1 3 90630 4 Венлафаксин є МНН 1-(2-диметиламіно-1-(4ру, і перетворення отриманої форми III у форму І метоксифеніл)етил)циклогексанолу. Його рацемічшляхом сушіння при температурі близько 60°С. на сіль гідрохлорид входить до складу препарату, У WO 02/36542 розкриваються також поліщо надходить у продаж під торговим найменуванморфні модифікації венлафаксину гідрохлориду, ням Еффексор. Венлафаксин використовують у позначені як форми А, В, С і D, а також способи вигляді солі, оскільки це полегшує готування різїхнього одержання. них форм препаратів для перорального застосуУ WO 02/50017 розкривається N-метилування вання, таких як таблетки, капсули, пастилки, поN,N-дідесметилвенлафаксину, що також може рошки і т.д. бути використаний у вигляді солі з мурашиною Венлафаксин уперше описаний у ЕР-А-112 кислотою. 669. Відповідно до цього документа, венлафаксин У WO 03/050074 описується спосіб одержання одержують N-метилуванням попередника N,Nформи І венлафаксину гідрохлориду в результаті дідесметилвенлафаксину за допомогою формальвзаємодії венлафаксину з газоподібним хлороводдегіду і мурашиної кислоти. Венлафаксин потім нем у різних розчинниках, а саме, в етилацетаті, відокремлюють за допомогою хроматографії, виацетонітрилі, ацетоні і метилізобутилкетоні. Роздаляючи побічний продукт спіровенлафаксин, і рахунковий вихід отриманого продукту складає потім перетворюють у венлафаксину гідрохлорид, всього 50% у перерахуванні на вихідний N.Nвикористовуючи 4Н розчин НСІ в ізопропанолі. дідесметилвенлафаксин. Крім того, показано, що Однак при цьому способі досягається лише дуже тип поліморфної утворюваної модифікації та її невеликий вихід продукту. чистота піддається впливу численних факторів. У документі Journal of Medicinal Chemistry, З приведених вище документів видно, що бі1990, vol.33, No.10 (2899-2905) розкритий синтез льшість відомих поліморфних модифікацій венлавенлафаксину з п-метоксибензилціаніду, що вклюфаксину гідрохлориду одержують, використовуючи чає N-метилювання за модифікованим методом розчин венлафаксину гідрохлориду в різних розЕшвейлера-Кларка за допомогою формальдегіду і чинниках і при різних реакційних умовах. Очевидмурашиної кислоти. Розчин венлафаксину в етино, ці параметри відіграють основну роль в утволацетаті обробляють розчином НСІ у пропанолі-2, ренні різних поліморфних модифікацій, що після чого отриманий венлафаксину гідрохлорид володіють різною кристалічною структурою. Серед перекристалізовують із суміші метанол/етилацетат цих параметрів може бути наявність співрозчиннипри незазначених умовах. Розрахунковий вихід ків, температура, при якій відбувається утворення технічного продукту складає 80% у перерахуванні гідрохлориду, то ж, чи піддається реакційна суміш на N.N-дідесметил венлафаксин. кип'ятінню зі зворотним холодильником після Про існування деяких поліморфних модифікаутворення гідрохлориду, а також температура, при цій венлафаксину гідрохлориду згадується в ЕР-Аякій гідрохлорид фільтрують. 797 991. У цьому документі описані дві поліморфні Тому що венлафаксину гідрохлорид поставформи, при цьому вважається, що одна з них є ляють на ринок у вигляді рацемічної суміші, до кінетичним продуктом процесу кристалізації. Крім поліморфізму варто відноситися дуже уважно, того, розкривається, що при нагріванні в розчинниособливо через те, що форма, що є більш термоку кристалізації одна із поліморфних модифікацій динамічно стабільною і виявляє необхідну біодострансформується в іншу поліморфну модифікацію. тупність, більш краща в порівнянні з іншими форОднак у цьому документі не розкрито, який розмами у відношенні умов збереження й терміну чинник використовують для перекристалізації. придатності. Термодинамічно менш стабільні фоПізніше був опублікований цілий ряд патентрми піддаються перетворенню в більш стабільні, і них заявок, що розкривають різні поліморфні мотому не підходять для використання у фармацевдифікації рацемічного венлафаксину гідрохлориду, тиці, оскільки таке перетворення буде виявлятися наприклад, позначені як форми І, II, III і IV і А, В, С при збереженні матеріалу. і D, а також способи їхнього одержання. Несподівано виявилося, що спосіб одержання У WO 02/45658 описується одержання кристаформи І венлафаксину гідрохлориду відповідно до лічного венлафаксину із N,Nвинаходу дозволяє легким і відтвореним чином дідесметилвенлафаксину гідрохлориду і спосіб одержувати даний продукт з високим виходом і, одержання форм І, II, III і IV венлафаксину гідрохщо особливо важливо, із досить високою полілориду у вигляді кристалів. Одержуваний цим споморфною чистотою. собом продукт часто являє собою суміш поліморКрім того, встановлено, що спосіб одержання фних модифікацій, що свідчить про важливість венлафаксину відповідно до винаходу не привотого, при яких умовах виконується спосіб. Повідодить до утворення значної кількості побічних промляється, що форма І венлафаксину гідрохлориду дуктів і його проведення до завершення не вимаможе бути отримана шляхом реакції розчину венгає великих витрат часу, і, таким чином, високий лафаксину в ізопропанолі і впливом на розчин вихід венлафаксину може бути досягнутий еконогазоподібним НСІ. Реакційну суміш прохолоджумічно вигідним чином. ють, фільтрують і сушать. Вихід продукту, однак, Відповідно до першого аспекту, винахід відноне вказується. Альтернативний спосіб одержання ситься до способу одержання венлафаксину, що форми І венлафаксину гідрохлориду включає розвключає: чинення венлафаксину гідрохлориду в метанолі (а) перетворення попередника венлафаксину, при кип'ятінні зі зворотним холодильником і додаобраного з N,N-дідесметилвенлафаксину формули вання осаджувана, обраного з етилацетата, ізоп(І), його солі, спіровенлафаксину формули (II) і ропілового ефіру або метил-трет-бутилового ефійого солі у венлафаксин, де зазначене перетворення проводять у присутності солі мурашиної кислоти, обраної із солі металу або амонієвої солі мурашиної кислоти, (b) необов'язково, взаємодія венлафаксину з кислотою з одержанням кислотно-адитивної солі венлафаксину. Несподівано було встановлено, що присутність визначеної солі мурашиної кислоти дозволяє здійснювати N-метилування попередників не тіль ки з високою швидкістю, але також і з високим виходом, без утворення небажаних побічних продуктів. Передбачається, що це викликано тим, що сіль мурашиної кислоти прискорює перетворення спіровенлафаксину, що є ймовірним проміжним продуктом при N-метилуванні N.Nдідесметилвенлафаксину, що дуже повільно реагує до венлафаксину. Це проілюстровано наступною реакційною схемою, що відображає кращий варіант здійснення способу. Переважно використовувати сіль мурашиної кислоти в такій кількості, щоб молярне відношення солі мурашиної кислоти до попередника венлафаксину складало 0,3-10 до 1, зокрема, 0,5-3 до 1. Це приводить до особливо плавного й повного протікання реакції. Також переважно, щоб металева сіль мурашиної кислоти була сіллю мурашиної кислоти і лужного або лужноземельного металу. Прикладами найбільш кращих солей є солі Na, К або Li. Крім того, може бути використана також сіль NH4. Сіль мурашиної кислоти можуть додавати в реакційну суміш. Однак сіль мурашиної кислоти також може утворюватися in situ у реакційній суміші. При проведенні стадії (а) обраний попередник венлафаксину звичайно розчиняють у придатному розчиннику, такому як вода або інші полярні розчинники, наприклад ДМФА або ДМСО. Концентрація попередника венлафаксину в розчині переважно складає від 0,2 до 3моль/л, зокрема, від 0,5 до 1,5моль/л. Перетворення на стадії (а) переважно проводять, використовуючи N,Nдідесметилвенлафаксин або його сіль, наприклад, адитивну сіль із соляною кислотою. Крім того, переважно проводити реакцію в присутності формальдегіду і мурашиної кислоти. Дане перетворення може бути назване N-метилуванням. Кількість мурашиної кислоти звичайно складає від 2 до 20 ек вівалентів стосовно попередника венлафаксину. Кількість формальдегіду звичайно складає від 2 до 15 еквівалентів стосовно попередника венлафаксину. Також це перетворення дуже зручно проводити в присутності гідроксиду лужного або лужноземельного металу, або NH4OH, узятих у такій кількості, щоб приводити до утворення in situ солі з мурашиною кислотою. Таким чином, зазначені гідроксиди металів або NH4OH реагують із уже присутньою мурашиною кислотою з утворенням in situ відповідної солі мурашиної кислоти. При використанні кислотно-адитивної солі N.Nдідесметилвенлафаксину, наприклад, солі із соляною кислотою, варто враховувати, що дані гідроксиди також будуть реагувати з кислотою, що входить до складу адитивної солі. Отже, їхня кількість повинна бути такою, щоб утворилася ще і необхідна кількість солі мурашиної кислоти. Переважно, щоб гідроксидом лужного металу був NaOH, що утворить in situ форміат Na. Крім того, переважно, щоб суміш попередника венлафаксину, формальдегіду і мурашиної кислоти в обраномурозчиннику нагрівали зі зворотним холодильником протягом від 2 до 24 годин, переважно, протягом від 3 до 7 годин. Після завершення реакції значення рН суміші звичайно доводять приблизно до 12 за допомогою, наприклад, NaOH. Відповідно до кращого альтернативного варіанта здійснення, значення рН суміші 7 90630 8 спочатку доводять приблизно до 1 за допомогою (2000) С56, 1009-1010, розкриті дані рентгеноструНСІ, суміш екстрагують органічним розчинником, і ктурного дослідження. потім значення її рН доводять приблизно до 12 за Відповідно до другого кращого варіанта здійсдопомогою, наприклад, NaOH. Ця процедура донення даного другого аспекту винаходу, розчин зволяє видалити домішку рожевого кольору, яка венлафаксину гідрохлориду одержують, піддаючи часто спостерігається. венлафаксин реакції з розчином НСІ у спирті. Після цього венлафаксин може бути екстрагоКращими спиртами є метанол, етанол і/або ізопваний органічним розчинником. Кращими є такі ропанол. Після додавання розчину НСІ суміш пеорганічні розчинники, що можуть бути використані реважно перемішують протягом до чотирьох годля азеотропного відгону води і не змішуються з дин. водою, наприклад, ізопропілацетат, пропілацетат, Крім того, відповідно до даного варіанта здійсбутилацетат, ізобутилацетат, mpem-бутилацетат і нення винаходу до венлафаксину переважно доциклогексан. дають форму І венлафаксину гідрохлориду, зокПісля можливої додаткової обробки одержурема, у кількості до 10мас.% відносно ють венлафаксин, що має високу чистоту, перевавенлафаксину, переважно, до 5мас.% відносно жно, більше 98%, обумовлену методом ВЕРХ. венлафаксину. Переважно, його додають перед Отриманий у такий спосіб венлафаксин на проведенням реакції венлафаксину з розчином стадії (b) можуть піддавати взаємодії з кислотою НСІ у спирті. для одержання кислотно-адитивної солі венлафаКрім того, відповідно до даного варіанта здійсксину, але це не є обов'язковим. нення, кристалізацію переважно проводять при Відповідно до другого аспекту, винахід віднотемпературі розчину венлафаксину гідрохлориду, ситься до способу одержання форми І венлафакрівної приблизно 20°С. сину гідрохлориду, при якому форму І венлафакДо того ж даний варіант здійснення способу сину гідрохлориду кристалізують з розчину дозволяє одержувати форму І венлафаксину гідвенлафаксину гідрохлориду в органічному розчинрохлориду, що має дуже високу чистоту (більше нику, при цьому розчинник містить ізопропілацетат 99,5%; ВЕРХ). і/або циклогексан. Відповідно до третього аспекту, винахід відноНесподівано було встановлено, що викорисситься до способу одержання форми І венлафактання розчинника, що містить ізопропілацетат сину гідрохлориду, при якому і/або циклогексан, робить можливим легке і відт(a) розчин венлафаксину в органічному розворене одержання венлафаксину гідрохлориду чиннику взаємодіє із соляною кислотою, високої поліморфної чистоти. Циклогексан дає (b) вміст води в отриманому розчині венлафадодаткову перевагу, оскільки має високу стійкість ксину гідрохлориду доводять менше ніж до до гідролізу. 3мас.%, переважно, менше ніж до 1,5мас.%, і Переважно, щоб розчинник складався з ізоп(c) кристалізують форму І венлафаксину гідроропілацетата і/або циклогексану. хлориду. Відповідно до першого кращого варіанта здійВміст води на стадії (b) краще регулювати снення даного другого аспекту винаходу, венлашляхом азеотропної перегонки розчину. факсину гідрохлорид одержують, піддаючи венТаким чином, даний спосіб не вимагає обов'ялафаксин взаємодії із соляною кислотою. Кількість зкового застосування саме ізопропілацетата і/або соляної кислоти звичайно складає від 0,85 до 1,5 циклогексану як розчинники, на відміну від способу еквівалентів, переважно, від 0,9 до 1,2 еквіваленвідповідно до другого аспекту винаходу. Кращим тів. органічним розчинником на стадії (а) є ізопропілаКрім того, відповідно до даного варіанту здійсцетат і/або циклогексан, але ним також може бути, нення винаходу переважно, щоб після проходженнаприклад, пропілацетат, бутилацетат, ізобутиланя цієї реакції вміст води в розчині венлафаксину цетат, mpem-бутилацетат. гідрохлориду складав менше 3мас.%, переважно, Кристалізацію на стадії (с) переважно виконуменше 1,5мас.%, при визначенні за методом Карють за методикою, приведеною вище для першого ла-Фішера. Це переважно досягається азеотропваріанта здійснення другого аспекту винаходу. ною перегонкою розчину. Способи відповідно до другого і третього аспеКрім того, було показано, що особливо гарні ктів винаходу переважно здійснюють, використорезультати відповідно до даного варіанта здійсвуючи венлафаксин, отриманий відповідно до спонення винаходу одержують при проведенні криссобу, що складає перший аспект винаходу. Це талізації при температурі розчину, меншої, ніж приводить в результаті до задовільного виходу температура його кипіння, не більше ніж на 30°С; форми І венлафаксину гідрохлориду, що складає переважно проводити кристалізацію приблизно більше 85% у перерахуванні на кількість викориспри температурі кипіння розчину. таного попередника венлафаксину. Способи відКрім того, відповідно до даного варіанта здійсповідно до попереднього рівня техніки дозволяли нення винаходу переважно перемішувати отримаодержувати в даній реакційній послідовності вихід ну суспензію при температурі дефлегмації, перелише близько 50%. важно протягом до трьох годин. Способи відповідно до другого і третього аспеОтриманий продукт являє собою форму І венктів винаходу мають ще одну перевагу, що полялафаксину гідрохлориду дуже високої чистоти (бігає в можливості одержання форми І венлафаксильше 99,5%; ВЕРХ). Цей продукт ідентичний крисну гідрохлориду, що має середній розмір часток талічній формі, для якої в Acta Crystallographica менше 50мкм, переважно розмір часток складає від 10 до 40мкм. Цей середній розмір часток ви 9 90630 10 значають методом лазерної дифракції, наприклад, лориду, що може бути отримана описаними вище за допомогою апарата Mastersizer S фірми способами. Переважно, щоб форма І венлафаксиMalvern. Такий розмір часток є найкращим, оскільну гідрохлориду мала чистоту більше 99,5%, обуки полегшує включення форми І венлафаксину мовлену ВЕРХ. Для визначення чистоти викорисгідрохлориду в цілий ряд різних фармацевтичних товували метод ВЕРХ у градієнтному режимі з препаратів. наступними устаткуванням/умовами: Відповідно до четвертого аспекту, винахід також відноситься до форми І венлафаксину гідрохСтовпчик: Температура стовпчика: Детекція: Потік: Розчинника Рухлива фаза: Розчинник В Prontosil 300-5-C18-ace-EPS, 5мкм, 250x4,6мм 20°С УФ детектор з довжиною хвилі 227нм 1,2мл/хв 0,05 М розчин Na2PO4, pH 6,5 ацетонітрил Далі винахід додатково проілюстрований прикладами. Приклади: Приклад 1 - Одержання венлафаксину з N.Nдідесметилвенлафаксину гідрохлориду 50% водяний розчин NaOH (4мл, 74 ммоль) додавали при перемішуванні до розчину N,Nдідесметилвенлафаксину гідрохлориду (5,72г, 20 ммоль) у воді (16мл) при кімнатній температурі. До отриманої суміші додавали мурашину кислоту (98%, 11,5мл, 305 ммоль) і 37% водяний розчин формальдегіду (8,4мл, 113 ммоль). Суміш перемішували при температурі дефлегмації, перетворення завершилося через 5 годин (98,67%; ВЕРХ). Потім розчин прохолоджували до кімнатної температури й обробляли 50% водяним розчином NaOH, доводячи значення рН до 12. Суміш двічі екстрагували за допомогою 66мл ізопропілацетату. Зібрані органічні фази тричі промивали водою (66мл). Відокремлений у такий спосіб розчин основи венлафаксину мав високу чистоту (98,9%; ВЕРХ). Приклад 2 - Одержання форми І венлафаксину гідрохлориду з розчину основи венлафаксину в ізопропілацетаті До розчину основи венлафаксину в ізопропілацетаті з Приклада 1 (66мл, 10ммоль) додавали Комп’ютерна верстка В. Мацело 5мл 2М соляної кислоти. Суміш нагрівали і видаляли воду азеотропним відгоном, використовуючи пастку Діна-Старка. Після того як воду цілком видаляли із суміші, продукт починав повільно кристалізуватися. Отриману суспензію гріли при температурі дефлегмації протягом 1,5 годин, потім прохолоджували і фільтрували. Було отримано 2,75г (88% відносно N,N-дідесметилвенлафаксину гідрохлориду) чистої форми І венлафаксину гідрохлориду (99,65%; ВЕРХ). Приклад 3 - Одержання форми І венлафаксину гідрохлориду з розчину основи венлафаксинув ізопропілацетаті Розчин венлафаксину в ізопропілацетаті з Приклада 1 (66мл, 10 ммоль) концентрували до половини об'єма. Потім до розчину додавали від 10 до 50мг форми І венлафаксину гідрохлориду. Після цього протягом 30 хвилин повільно додавали 4,0мл 2,5М розчину НСІ в етанолі Після додавання зазначеної кількості кислоти отриману суспензію перемішували протягом ще двох годин. Потім суміш фільтрували, продукт промивали ізопропілацетатом і сушили. Одержали 2,69г (86% відносно N.N-дідесметилвенлафаксину гідрохлориду) чистої форми І венлафаксину гідрохлориду (ВЕРХ: 99,65%). Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparing venlafaxine hydrochloride of form i
Автори англійськоюSilvo Zupancic
Назва патенту російськоюСпособ получения формы и венлафаксина гидрохлорида
Автори російськоюСилво Жупанчич
МПК / Мітки
МПК: C07C 217/00, C07C 213/00
Мітки: спосіб, форми, гідрохлориду, венлафаксину, одержання
Код посилання
<a href="https://ua.patents.su/5-90630-sposib-oderzhannya-formi-i-venlafaksinu-gidrokhloridu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання форми і венлафаксину гідрохлориду</a>
Моногідрат гідрохлориду венлафаксину і спосіб його одержання (варіанти)

Номер патенту: 77234
Опубліковано: 15.11.2006
Автори: Хан Дзун, Лі Йонг Дзай
МПК: A61P 25/24, A61K 31/14, C07C 213/00, A61P 25/18, A61K 31/133, C07C 217/74, A61P 25/00, A61K 31/137, A61P 25/22, A61K 47/02
Мітки: венлафаксину, спосіб, моногідрат, варіанти, гідрохлориду, одержання
Формула / Реферат:
1. Моногідрат гідрохлориду венлафаксину.2. По суті чистий моногідрат гідрохлориду венлафаксину.3. Гідрохлорид венлафаксину, що має порошкову рентгенограму по суті таку ж, як показана на фіг. 1.4. Моногідрат гідрохлориду венлафаксину, що показує порошкову рентгенограму, що має характеристичні піки, виражені в градусах 2, при близько 7,45, 8,60,...
Спосіб одержання кислотно-адитивної солі венлафаксину

Номер патенту: 89037
Опубліковано: 25.12.2009
Автор: Жупанчіч Сілво
МПК: C07C 217/74, C07C 213/00
Мітки: спосіб, венлафаксину, кислотно-адитивної, солі, одержання
Формула / Реферат:
1. Спосіб одержання кислотно-адитивної солі венлафаксину, що включає(а) перетворення попередників венлафаксину, вибраних із N,N-дидесметилвенлафаксину формули (І), його солі, спіровенлафаксину формули (II) і його солі , , у венлафаксин, де зазначене...
Форма пролонгованого вивільнення гідрохлориду венлафаксину

Номер патенту: 82597
Опубліковано: 25.04.2008
Автори: Бхаттачар'я Сампад, Джоші Маянк, Пандіта Сандіп, Кширсагар Раджеш
МПК: A61K 31/137, A61K 9/36, A61P 25/18, A61K 9/48, A61K 9/32
Мітки: венлафаксину, пролонгованого, форма, вивільнення, гідрохлориду
Формула / Реферат:
1. Форма пролонгованого вивільнення гідрохлориду венлафаксину у формі міні-таблеток, якими заповнена тверда желатинова капсула; причому зазначені міні-таблетки мають ядро і зовнішню оболонку, що складає 2-15 % загальної ваги міні-таблеток, ядро зазначених міні-таблеток включає гідрохлорид венлафаксину, мікрокристалічну целюлозу і полівінілпіролідон, а зазначена оболонка включає полімер, нерозчинний у воді, і полімер, розчинний у...
Поліморфні кристалічні форми гідрохлориду лерканідипіну та спосіб їх одержання

Номер патенту: 82988
Опубліковано: 10.06.2008
Автори: Кампана Франческо, Де Іасі Джанлука, Леонарді Амедео, Боніфачо Фаусто
МПК: B01D 9/02, C07D 211/90, A61K 31/4422, A61P 9/12
Мітки: кристалічні, одержання, гідрохлориду, поліморфні, лерканідипіну, спосіб, форми
Формула / Реферат:
1. Неочищена тверда Форма (А) гідрохлориду лерканідипіну, яка має температуру плавлення приблизно 150-152 °С (пік ДСК) та містить приблизно 3-4 % (мас.) етилацетату.2. Неочищена тверда Форма (В) гідрохлориду лерканідипіну, яка має температуру плавлення приблизно 131-135 °С (пік ДСК) та містить приблизно 0,3-0,7 % (мас.) етилацетату.3. Спосіб одержання неочищеної Форми гідрохлориду лерканідипіну за п. 1, який включає такі...
Спосіб одержання гідрохлориду 2-(1′ – аміноетил) біцикло [2.2.1] гептану
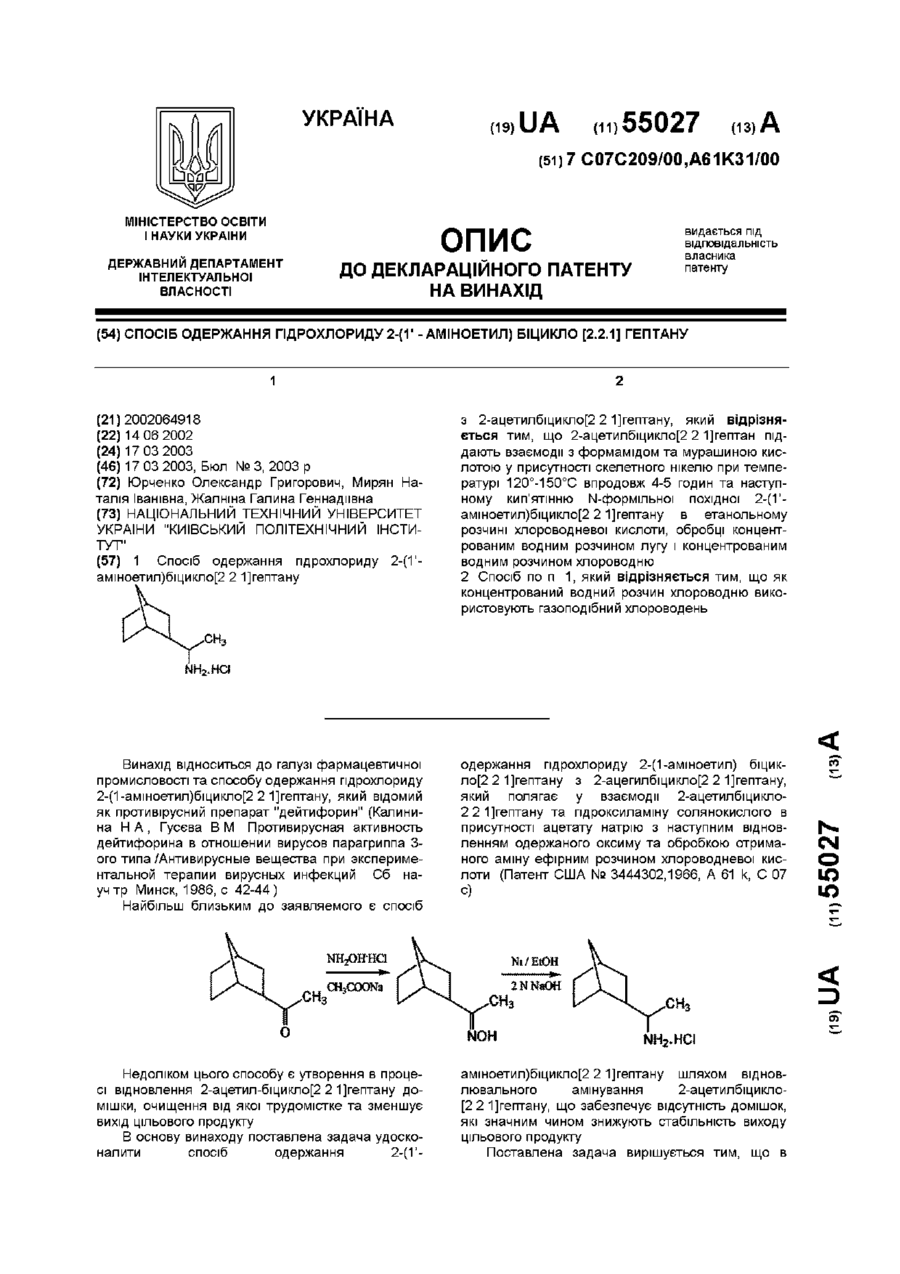
Номер патенту: 55027
Опубліковано: 17.03.2003
Автори: Жалніна Галина Геннадіївна, Мирян Наталія Іванівна, Юрченко Олександр Григорович
МПК: A61K 31/00, C07C 209/00, B01J 23/755, C07C 13/00
Мітки: біцикло, спосіб, 2-(1, аміноетил, 2,2,1, гептану, одержання, гідрохлориду
Формула / Реферат:
1. Спосіб одержання гідрохлориду 2-(1’-аміноетил)біцикло[2.2.1]гептануз 2-ацетилбіцикло[2.2.1]гептану, який відрізняється тим, що 2-ацетилбіцикло[2.2.1]гептан піддають взаємодії з формамідом та мурашиною кислотою у присутності скелетного нікелю при температурі 120°-150°С впродовж 4-5 годин та наступному кип’ятінню N-формільної похідної 2-(1’-аміноетил)біцикло[2.2.1]гептану в етанольному розчині хлороводневої кислоти, обробці...
Попередній патент: Стенд для оцінки функцій штучних стоп протезів
Наступний патент: Відцентровий млин ударної дії
Випадковий патент: Приймально-передавальний пристрій абонентської мережі