Спосіб одержання кислотно-адитивної солі венлафаксину





Формула / Реферат
1. Спосіб одержання кислотно-адитивної солі венлафаксину, що включає
(а) перетворення попередників венлафаксину, вибраних із N,N-дидесметилвенлафаксину формули (І), його солі, спіровенлафаксину формули (II) і його солі
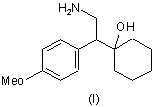 ,
,
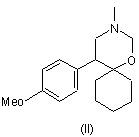 ,
,
у венлафаксин, де зазначене перетворення проводять у присутності солі мурашиної кислоти, вибраної із солі з металом або амонієвої солі мурашиної кислоти, і формальдегіду, причому молярне співвідношення солі мурашиної кислоти і попередника венлафаксину складає 0,3-10:1, і
(b) взаємодію утвореного венлафаксину з кислотою з одержанням кислотно-адитивної солі венлафаксину.
2. Спосіб за п. 1, який відрізняється тим, що молярне співвідношення солі мурашиної кислоти і попередника венлафаксину складає 0,5-3:1.
3. Спосіб за п. 1 або 2, який відрізняється тим, що сіль мурашиної кислоти з металом є сіллю мурашиної кислоти і лужного або лужноземельного металу.
4. Спосіб за п. 3, який відрізняється тим, що сіллю мурашиної кислоти і лужного металу є сіль Na, K або Li.
5. Спосіб за кожним із пп. 1-4, який відрізняється тим, що на стадії (а) N,N-дидесметилвенлафаксин (І) або його сіль перетворюють у венлафаксин у присутності формальдегіду і мурашиної кислоти, яку перетворюють у сіль.
6. Спосіб за п. 5, який відрізняється тим, що на стадії (а) N,N-дидесметилвенлафаксин (І) використовують у вигляді його адитивної солі з НСl.
7. Спосіб за п. 5 або 6, який відрізняється тим, що на стадії (а) сіль мурашиної кислоти утворюють in situ шляхом взаємодії мурашиної кислоти з гідроксидом лужного або лужноземельного металу або NH4OH у достатній для цього кількості.
8. Спосіб за п. 7, який відрізняється тим, що гідроксидом лужного металу є NaOH, що утворить in situ форміат Na.
Текст
1. Спосіб одержання кислотно-адитивної солі венлафаксину, що включає (а) перетворення попередників венлафаксину, вибраних із N,N-дидесметилвенлафаксину формули (І), його солі, спіровенлафаксину формули (II) і його солі C2 2 (19) 1 3 кількістю побічних продуктів, і до того ж для завершення реакції потрібен тривалий час. Відомі з попереднього рівня техніки способи одержання форми І венлафаксину гідрохлориду володіють, зокрема, тим недоліком, що вони не дозволяють одержувати задовільну чистоту і прийнятний вихід продукту. Крім того, у них часто використовуються розчинники, неприйнятні з екологічних розумінь, і, отже, ці способи не можна в достатньому обсязі використовувати в промислових масштабах. Нарешті, при використанні цих способів найчастіше складно контролювати реакційні умови таким чином, щоб виходила тільки необхідна поліморфна модифікація І з високим ступенем очищення. Таким чином, метою даного винаходу є розробка удосконалених способів одержання венлафаксину і форми І венлафаксину гідрохлориду, що не мають зазначених вище недоліків. Дана мета досягається за допомогою способу одержання венлафаксину за кожним із пп. 1-9 і за допомогою способу одержання форми І венлафаксину гідрохлориду за кожним із пп.10-24. Крім того, винахід включає форму І венлафаксину гідрохлориду за кожним із пп.25-26. Венлафаксин є МНН 1-(2-диметиламіно-1-(4метоксифеніл)етил)циклогексанолу. Його рацемічна сіль гідрохлорид входить до складу препарату, що надходить у продаж під торговим найменуванням Еффексор. Венлафаксин використовують у вигляді солі, оскільки це полегшує готування різних форм препаратів для перорального застосування, таких як таблетки, капсули, пастилки, порошки і т.д. Венлафаксин уперше описаний у ЕР-А-112 669. Відповідно до цього документа, венлафаксин одержують N-метилуванням попередника Ν,Νдідесметилвенлафаксину за допомогою формальдегіду і мурашиної кислоти. Венлафаксин потім відокремлюють за допомогою хроматографії, видаляючи побічний продукт спіровенлафаксин, і потім перетворюють у венлафаксину гідрохлорид, використовуючи 4Н розчин НСІ в ізопропанолі. Однак при цьому способі досягається лише дуже невеликий вихід продукту. У документі Journal of Medicinal Chemistry, 1990, vol.33, No.10 (2899-2905) розкритий синтез венлафаксину з п-метоксибензилціаніду, що включає N-метилювання за модифікованим методом Ешвейлера-Кларка за допомогою формальдегіду і мурашиної кислоти. Розчин венлафаксину в er/ілацетаті обробляють розчином НСІ у пропанолі-2, після чого отриманий венлафаксину гідрохлорид перекристалізовують із суміші метанол/етилацетат при незазначених умовах. Розрахунковий вихід технічного продукту складає 80% у перерахуванні на Ν,Ν-дідесметил венлафаксин. Про існування деяких поліморфних модифікацій венлафаксину гідрохлориду згадується в ЕР-А797 991. У цьому документі описані дві поліморфні форми, при цьому вважається, що одна з них є кінетичним продуктом процесу кристалізації. Крім того, розкривається, що при нагріванні в розчиннику кристалізації одна із поліморфних модифікацій 89037 4 трансформується в іншу поліморфну модифікацію. Однак у цьому документі не розкрито, який розчинник використовують для перекристалізації. Пізніше був опублікований цілий ряд патентних заявок, що розкривають різні поліморфні модифікації рацемічного венлафаксину гідрохлориду, наприклад, позначені як форми І, II, III і IV і А, В, С і D, а також способи їхнього одержання. У WO 02/45658 описується одержання кристалічного венлафаксину із Ν,Νдідесметилвенлафаксину гідрохлориду і спосіб одержання форм І, II, III і IV венлафаксину гідрохлориду у вигляді кристалів. Одержуваний цим способом продукт часто являє собою суміш поліморфних модифікацій, що свідчить про важливість того, при яких умовах виконується спосіб. Повідомляється, що форма І венлафаксину гідрохлориду може бути отримана шляхом реакції розчину венлафаксину в ізопропанолі і впливом на розчин газоподібним НСІ. Реакційну суміш прохолоджують, фільтрують і сушать. Вихід продукту, однак, не вказується. Альтернативний спосіб одержання форми І венлафаксину гідрохлориду включає розчинення венлафаксину гідрохлориду в метанолі при кип'ятінні зі зворотним холодильником і додавання осаджувача, обраного з етилацетата, ізопропілового ефіру або метил-трет-бутилового ефіру, і перетворення отриманої форми III у форму І шляхом сушіння при температурі близько 60°С. У WO 02/36542 розкриваються також поліморфні модифікації венлафаксину гідрохлориду, позначені як форми А, В, С і D, а також способи їхнього одержання. У WO 02/50017 розкривається N-метилування Ν,Ν-дідесметилвенлафаксину, що також може бути використаний у вигляді солі з мурашиною кислотою. У WO 03/050074 описується спосіб одержання форми І венлафаксину гідрохлориду в результаті взаємодії венлафахсину з газоподібним хлороводнем у різних розчинниках, а саме, в етилацетаті, ацетонітрилі, ацетоні і метилізобутилкетоні. Розрахунковий вихід отриманого продукту складає всього 50% у перерахуванні на вихідний Ν,Νдідесметилвенлафаксин. Крім того, показано, що тип поліморфної утворюваної модифікації та її чистота піддається впливу численних факторів. З приведених вище документів видно, що більшість відомих поліморфних модифікацій венлафаксину гідрохлориду одержують, використовуючи розчин венлафаксину гідрохлориду в різних розчинниках і при різних реакційних умовах. Очевидно, ці параметри відіграють основну роль в утворенні різних поліморфних модифікацій, що володіють різною кристалічною структурою. Серед цих параметрів може бути наявність співрозчинників, температура, при якій відбувається утворення гідрохлориду, то ж, чи піддається реакційна суміш кип'ятінню зі зворотним холодильником після утворення гідрохлориду, а також температура, при якій гідрохлорид фільтрують. Тому що венлафаксину гідрохлорид поставляють на ринок у вигляді рацемічної суміші, до поліморфізму варто відноситися дуже уважно, особливо через те, що форма, що є більш термо 5 динамічно стабільною і виявляє необхідну біодоступність, більш краща в порівнянні з іншими формами у відношенні умов збереження й терміну придатності. Термодинамічно менш стабільні форми піддаються перетворенню в більш стабільні, і тому не підходять для використання у фармацевтиці, оскільки таке перетворення буде виявлятися при збереженні матеріалу. Несподівано виявилося, що спосіб одержання форми І венлафаксину гідрохлориду відповідно до винаходу дозволяє легким і відтвореним чином одержувати даний продукт з високим виходом і, що особливо важливо, із досить високою поліморфною чистотою. Крім того, встановлено, що спосіб одержання венлафаксину відповідно до винаходу не приводить до утворення значної кількості побічних продуктів і його проведення до завершення не вимагає великих витрат часу, і, таким чином, високий вихід венлафаксину може бути досягнутий економічно вигідним чином. Відповідно до першого аспекту, винахід відноситься до способу одержання венлафаксину, що включає: (а) перетворення попередника венлафаксину, обраного з Ν,Ν-дідесметилвенлафаксину формули (І), його солі, спіровенлафаксину формули (II) і його солі Переважно використовувати сіль мурашиної кислоти в такій кількості, щоб молярне відношення солі мурашиної кислоти до попередника венлафаксину складало 0,3-10 до 1, зокрема, 0,5-3 до 1. Це приводить до особливо плавного й повного протікання реакції. Також переважно, щоб металева сіль мурашиної кислоти була сіллю мурашиної кислоти і лужного або лужноземельного металу. Прикладами найбільш кращих солей є солі Na, К або Li. Крім того, може бути використана також сіль NH4. Сіль мурашиної кислоти можуть додавати в реакційну суміш. Однак сіль мурашиної кислоти також може утворюватися in situ у реакційній суміші. При проведенні стадії (а) обраний попередник венлафаксину звичайно розчиняють у придатному розчиннику, такому як вода або інші полярні розчинники, наприклад ДМФА або ДМСО. Концентрація попередника венлафаксину в розчині переважно складає від 0,2 до 3моль/л, зокрема, від 0,5 до 1,5моль/л. Перетворення на стадії (а) переважно проводять, використовуючи Ν,Ν 89037 6 у венлафаксин, де зазначене перетворення проводять у присутності солі мурашиної кислоти, обраної із солі металу або амонієвої солі мурашиної кислоти, (b) необов'язково, взаємодія венлафаксину з кислотою з одержанням кислотно-адитивної солі венлафаксину. Несподівано було встановлено, що присутність визначеної солі мурашиної кислоти дозволяє здійснювати N-метилування попередників не тільки з високою швидкістю, але також і з високим виходом, без утворення небажаних побічних продуктів. Передбачається, що це викликано тим, що сіль мурашиної кислоти прискорює перетворення спіровенлафаксину, що є ймовірним проміжним продуктом при Ν-метилуванні Ν,Νдідесметилвенлафаксину, що дуже повільно реагує до венлафаксину. Це проілюстровано наступною реакційною схемою, що відображає кращий варіант здійснення способу. дідесметилвенлафаксин або його сіль, наприклад, адитивну сіль із соляною кислотою. Крім того, переважно проводити реакцію в присутності формальдегіду і мурашиної кислоти. Дане перетворення може бути назване N-метилуванням. Кількість мурашиної кислоти звичайно складає від 2 до 20 еквівалентів стосовно попередника венлафаксину. Кількість формальдегіду звичайно складає від 2 до 15 еквівалентів стосовно попередника венлафаксину. Також це перетворення дуже зручно проводити в присутності гідроксиду лужного або лужноземельного металу, або ΝΗ4ΟΗ, узятих у такій кількості, щоб приводити до утворення in situ солі з мурашиною кислотою. Таким чином, зазначені гідроксиди металів або NH4OH реагують із уже присутньою мурашиною кислотою з утворенням in situ відповідної солі мурашиної кислоти. При використанні кислотно-адитивної солі Ν,Νдідесметилвенлафаксину, наприклад, солі із соляною кислотою, варто враховувати, що дані гідроксиди також будуть реагувати з кислотою, що входить до складу адитивної солі. Отже, їхня кількість 7 повинна бути такою, щоб утворилася ще і необхідна кількість солі мурашиної кислоти. Переважно, щоб гідроксидом лужного металу був NaOH, що утворить in situ форміат Na. Крім того, переважно, щоб суміш попередника венлафаксину, формальдегіду і мурашиної кислоти в обраному розчиннику нагрівали зі зворотним холодильником протягом від 2 до 24 годин, переважно, протягом від 3 до 7 годин. Після завершення реакції значення рН суміші звичайно доводять приблизно до 12 за допомогою, наприклад, NaOH. Відповідно до кращого альтернативного варіанта здійснення, значення рН суміші спочатку доводять приблизно до 1 за допомогою НСІ, суміш екстрагують органічним розчинником, і потім значення її рН доводять приблизно до 12 за допомогою, наприклад, NaOH. Ця процедура дозволяє видалити домішку рожевого кольору, яка часто спостерігається. Після цього венлафаксин може бути екстрагований органічним розчинником. Кращими є такі органічні розчинники, що можуть бути використані для азеотропного відгону води і не змішуються з водою, наприклад, ізопропілацетат, пропілацетат, бутилацетат, ізобутилацетат, трет-бутилацетат і циклогексан. Після можливої додаткової обробки одержують венлафаксин, що має високу чистоту, переважно, більше 98%, обумовлену методом ВЕРХ. Отриманий у такий спосіб венлафаксин на стадії (b) можуть піддавати взаємодії з кислотою для одержання кислотно-адитивної солі венлафаксину, але це не є обов'язковим. Відповідно до другого аспекту, винахід відноситься до способу одержання форми І венлафаксину гідрохлориду, при якому форму І венлафаксину гідрохлориду кристалізують з розчину венлафаксину гідрохлориду в органічному розчиннику, при цьому розчинник містить ізопропілацетат і/або циклогексан. Несподівано було встановлено, що використання розчинника, що містить ізопропілацетат і/або циклогексан, робить можливим легке і відтворене одержання венлафаксину гідрохлориду високої поліморфної чистоти. Циклогексан дає додаткову перевагу, оскільки має високу стійкість до гідролізу. Переважно, щоб розчинник складався з ізопропілацетата і/або циклогексану. Відповідно до першого кращого варіанта здійснення даного другого аспекту винаходу, венлафаксину гідрохлорид одержують, піддаючи венлафаксин взаємодії із соляною кислотою. Кількість соляної кислоти звичайно складає від 0,85 до 1,5 еквівалентів, переважно, від 0,9 до 1,2 еквівалентів. Крім того, відповідно до даного варіанту здійснення винаходу переважно, щоб після проходження цієї реакції вміст води в розчині венлафаксину гідрохлориду складав менше 3мас.%, переважно, менше 1,5мас.%, при визначенні за методом Карла-Фішера. Це переважно досягається азеотропною перегонкою розчину. Крім того, було показано, що особливо гарні результати відповідно до даного варіанта здійс 89037 8 нення винаходу одержують при проведенні кристалізації при температурі розчину, меншої, ніж температура його кипіння, не більше ніж на ЗО °С; переважно проводити кристалізацію приблизно при температурі кипіння розчину. Крім того, відповідно до даного варіанта здійснення винаходу переважно перемішувати отриману суспензію при температурі дефлегмації, переважно протягом до трьох годин. Отриманий продукт являє собою форму І венлафаксину гідрохлориду дуже високої чистоти (більше 99,5%; ВЕРХ). Цей продукт ідентичний кристалічній формі, для якої в Acta Crystallographica (2000) С56, 1009-1010, розкриті дані рентгеноструктурного дослідження. Відповідно до другого кращого варіанта здійснення даного другого аспекту винаходу, розчин венлафаксину гідрохлориду одержують, піддаючи венлафаксин реакції з розчином НСІ у спирті. Кращими спиртами є метанол, етанол і/або ізопропанол. Після додавання розчину НСІ суміш переважно перемішують протягом до чотирьох годин. Крім того, відповідно до даного варіанта здійснення винаходу до венлафаксину переважно додають форму І венлафаксину гідрохлориду, зокрема, у кількості до 10мас.% відносно венлафаксину, переважно, до 5мас.% відносно венлафаксину. Переважно, його додають перед проведенням реакції венлафаксину з розчином НСІ у спирті. Крім того, відповідно до даного варіанта здійснення, кристалізацію переважно проводять при температурі розчину венлафаксину гідрохлориду, рівної приблизно 20°С. До того ж даний варіант здійснення способу дозволяє одержувати форму І венлафаксину гідрохлориду, що має дуже високу чистоту (більше 99,5%; ВЕРХ). Відповідно до третього аспекту, винахід відноситься до способу одержання форми І венлафаксину гідрохлориду, при якому (a) розчин венлафаксину в органічному розчиннику взаємодіє із соляною кислотою, (b) вміст води в отриманому розчині венлафаксину гідрохлориду доводять менше ніж до 3мас.%, переважно, менше ніж до 1,5мас.%, і (c) кристалізують форму І венлафаксину гідрохлориду. Вміст води на стадії (b) краще регулювати шляхом азеотропної перегонки розчину. Таким чином, даний спосіб не вимагає обов'язкового застосування саме ізопропілацетата і/або циклогексану як розчинники, на відміну від способу відповідно до другого аспекту винаходу. Кращим органічним розчинником на стадії (а) є ізопропілацетат і/або циклогексан, але ним також може бути, наприклад, пропілацетат, бутилацетат, ізобутилацетат, трет-бутилацетат. Кристалізацію на стадії (с) переважно виконують за методикою, приведеною вище для першого варіанта здійснення другого аспекту винаходу. Способи відповідно до другого і третього аспектів винаходу переважно \ здійснюють, використовуючи венлафаксин, отриманий відповідно до спо 9 89037 собу, що складає перший аспект винаходу. Це приводить в результаті до задовільного виходу форми І венлафаксину гідрохлориду, що складає більше 85% у перерахуванні на кількість використаного попередника венлафаксину. Способи відповідно до попереднього рівня техніки дозволяли одержувати в даній реакційній послідовності вихід лише близько 50%. Способи відповідно до другого і третього аспектів винаходу мають ще одну перевагу, що полягає в можливості одержання форми І венлафаксину гідрохлориду, що має середній розмір часток менше 50мкм, переважно розмір часток складає від 10 до 40мкм. Цей середній розмір часток визначають методом лазерної дифракції, наприклад, за допомогою апарата Mastersizer S фірми Malvern. Такий розмір часток є найкращим, оскільки полегшує включення форлли І венлафаксину гідрохлориду в цілий ряд різних фармацевтичних препаратів. Відповідно до четвертого аспекту, винахід також відноситься до форми І венлафаксину гідрохлориду, що може бути отримана описаними вище способами. Переважно, щоб форма І венлафаксину гідрохлориду мала чистоту більше 99,5%, обумовлену ВЕРХ. Для визначення чистоти використовували метод ВЕРХ у градієнтному режимі з наступними устаткуванням/умовами: Стовпчик: Температура стовпчика: Детекція: Prontosil 300-5-C18-aceEPS, 5мкм, 250 4,6мм 20°С УФ детектор з довжиною хвилі 227нм Потік: 1,2мл/хв. Рухлива Розчинника 0,05Μ розчин Na2PO4, фаза: pH6,5 Розчинник В ацетонітрил Далі винахід додатково проілюстрований прикладами. Приклади: Приклад 1 - Одержання венлафаксину з Ν,Νдідесметилвенлафаксину гідрохлориду 50% водяний розчин NaOH (4мл, 74ммоль) додавали при перемішуванні до розчину Ν,Ν Комп’ютерна верстка Т. Чепелева 10 дідесметилвенлафаксину гідрохлориду (5,72г, 20ммоль) у воді (16мл) при кімнатній температурі. До отриманої суміші додавали мурашину кислоту (98%, 11,5мл, 305ммоль) і 37% водяний розчин формальдегіду (8,4мл, 113ммоль). Суміш перемішували при температурі дефлегмації, перетворення завершилося через 5 годин (98,67%; ВЕРХ). Потім розчин прохолоджували до кімнатної температури й обробляли 50% водяним розчином NaOH, доводячи значення рН до 12. Суміш двічі екстрагували за допомогою 66мл ізопропілацетату. Зібрані органічні фази тричі промивали водою (66мл). Відокремлений у такий спосіб розчин основи венлафаксину мав високу чистоту (98,9%; ВЕРХ). Приклад 2 - Одержання форми І венлафаксину гідрохлориду з розчину основи венлафаксину в ізопропілацетаті До розчину основи венлафаксину в ізопропілацетаті з Приклада 1 (66мл, 10ммоль) додавали 5мл 2Μ соляної кислоти. Суміш нагрівали і видаляли воду азеотропним відгоном, використовуючи пастку Діна-Старка. Після того як воду цілком видаляли із суміші, продукт починав повільно кристалізуватися. Отриману суспензію гріли при температурі дефлегмації протягом 1,5 годин, потім прохолоджували і фільтрували. Було отримано 2,75г (88% відносно Ν,Ν-дідесметилвенлафаксину гідрохлориду) чистої форми І венлафаксину гідрохлориду (99,65%; ВЕРХ). Приклад 3 - Одержання форми І венлафаксину гідрохлориду з розчину основи венлафаксинув ізопропілацетаті Розчин венлафаксину в ізопропілацетаті з Приклада 1 (66мл, 10ммоль) концентрували до половини об'єма. Потім до розчину додавали від 10 до 50мг форми І венлафаксину гідрохлориду. Після цього протягом 30 хвилин повільно додавали 4,0мл 2,5Μ розчину НСІ в етанолі Після додавання зазначеної кількості кислоти отриману суспензію перемішували протягом ще двох годин. Потім суміш фільтрували, продукт промивали ізопропілацетатом і сушили. Одержали 2,69г (86% відносно Ν,Ν-дідесметилвенлафаксину гідрохлориду) чистої форми І венлафаксину гідрохлориду (ВЕРХ: 99,65%). Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparing venlafaxine acid addition salt
Автори англійськоюZuppancic Silvo
Назва патенту російськоюСпособ получения кислотно-аддитивной соли венлафаксина
Автори російськоюЖупанчич Силво
МПК / Мітки
МПК: C07C 217/74, C07C 213/00
Мітки: кислотно-адитивної, спосіб, солі, одержання, венлафаксину
Код посилання
<a href="https://ua.patents.su/5-89037-sposib-oderzhannya-kislotno-aditivno-soli-venlafaksinu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання кислотно-адитивної солі венлафаксину</a>
Спосіб одержання вільної основи і/або кислотно-адитивної солі рацемічного циталопрамдіолу і/або вільної основи і/або кислотно-адитивної солі r- або s-циталопрамдіолу

Номер патенту: 84859
Опубліковано: 10.12.2008
Автори: Крістіансен Брайан, Петерсен Ханс, Хумбле Рікке Ева, Дансер Роберт
МПК: C07C 253/34, C07D 307/87
Мітки: вільної, солі, одержання, кислотно-адитивної, рацемічного, циталопрамдіолу, спосіб, s-циталопрамдіолу, основі
Формула / Реферат:
1. Спосіб одержання вільної основи і/або кислотно-адитивної солі рацемічного циталопрамдіолу і/або вільної основи і/або кислотно-адитивної солі R- або S-циталопрамдіолу, який включає розділення початкової нерацемічної суміші вільної основи і/або кислотно-адитивної солі R- і S-циталопрамдіолу із вмістом одного з енантіомерів більше 50 % на фракцію, збагачену вільною основою і/або кислотно-адитивною сіллю S-циталопрамдіолу або...
Спосіб одержання заміщеного імідазолу або його нетоксичної фармацевтично прийнятної кислотно-адитивної солі

Номер патенту: 19310
Опубліковано: 25.12.1997
Автори: Маттіантеро Ляхде, Рісто Арво Сакарі Ламінтауста, Ар'ялена Кар'яланен, Ар'я Маркета Калапудас, Рейно Олаві Пєльконен, Мар'я Ліса Сєдервал, Арто Йоханес Кар'ялайнен
МПК: A61K 31/415, C07D 233/64
Мітки: кислотно-адитивної, фармацевтично, імідазолу, солі, нетоксичної, прийнятної, спосіб, одержання, заміщеного
Формула / Реферат:
Способ получения замещенного имидазола формулы:или его нетоксичной фармацевтически приемлемой кислотно-аддитивной соли, где R1, R2, R'1 и R'2, которые могут быть одинаковыми или разными, представляют Н, СН3, С2Н5, С3Н7, ОСН3, NO2, CF3, CHF2, CH2F или галоген;где R3 представляет Η, СН3 или галоген; R4 представляет Η и R5 представляет Η или ОН или R4 и R5 вместе образуют связь; X и У, которые могут...
Моногідрат гідрохлориду венлафаксину і спосіб його одержання (варіанти)

Номер патенту: 77234
Опубліковано: 15.11.2006
Автори: Лі Йонг Дзай, Хан Дзун
МПК: A61K 47/02, A61K 31/137, A61K 31/14, A61P 25/00, C07C 217/74, A61P 25/24, A61K 31/133, A61P 25/22, C07C 213/00, A61P 25/18
Мітки: спосіб, гідрохлориду, варіанти, венлафаксину, моногідрат, одержання
Формула / Реферат:
1. Моногідрат гідрохлориду венлафаксину.2. По суті чистий моногідрат гідрохлориду венлафаксину.3. Гідрохлорид венлафаксину, що має порошкову рентгенограму по суті таку ж, як показана на фіг. 1.4. Моногідрат гідрохлориду венлафаксину, що показує порошкову рентгенограму, що має характеристичні піки, виражені в градусах 2, при близько 7,45, 8,60,...
Спосіб одержання заміщеного імідазола або його нетоксичної фармацевтично прийнятної кислотно-аддитивної солі

Номер патенту: 13471
Опубліковано: 28.02.1997
Автори: Арто Йоханнес Карьялайнен, Маттіантеро Ляхде Рісто, Мар'я Лііса Седервалл, Рейно Олаві Пельконен, Ар'я Лена Кар'ялайнен, Арво Сакарі Ламміітауста, Ар'я Маркетта Калапудас
МПК: C07D 233/64, C07D 233/58, A61K 31/415, A61P 35/00, C07D 233/54, A61P 43/00, C07D 233/56
Мітки: заміщеного, солі, фармацевтично, імідазола, спосіб, нетоксичної, одержання, прийнятної, кислотно-аддітивної
Текст:
...(t, 1H), 6,7 (с, 4-(1,3-дифонилпропил)-1Н-имидазол. 1-бзнзілл-5-(1,3-ди-фенилпропил)-1Н- имиП р и м е р 7. дзгоя гидрогенизировали в смеси 2 н.рас4-(1,4-бис)4-фторфенил(0утііл)-1 Н-имитвора хлористоводородной кислоты и эта- 25 дазол. нала при температуро 60°С и использоваКонцентрированный водный раствор нии 10% Pd/C з качестве катализатора, формата аммония (0,16 г и 2 мл воды)добапПосла прекращения поглощения водолялся по каплям к кипящей...
Антрациклінові глікозиди та їх фармацевтично прийнятні кислотно-адитивні солі, спосіб їх одержання, похідні 1,6-дийод-3-оксапентану та спосіб їх одержання

Номер патенту: 27108
Опубліковано: 28.02.2000
Автори: Гранді Маріа, Суарато Антоніо, ФАІАРДІ Даніела, Барджотті Альберто
МПК: A61K 31/7034, C07H 15/252, A61K 31/70, A61K 31/704, C07C 43/313, A61K 31/7028, A61P 35/00
Мітки: 1,6-дийод-3-оксапентану, прийнятні, глікозиди, похідні, спосіб, солі, кислотно-адитивні, фармацевтично, одержання, антрациклінові
Формула / Реферат:
1. Антрациклиновые гликозиды общей формулы (А):в которой X представляет линейный или разветвленный С1-С6 алкил или бензил, который имеет (S) или (R) конфигурацию при 2"-углеродном атоме, и их фармацевтически приемлемые кислотно-аддитивные соли.2. Соединение по п.1, в котором X -метил.3. Соединение по п.1, в котором С2-С6 алкил.4. Соединение по п.1, выбранное из группы, состоящей из следующих...
Попередній патент: Спосіб нанесення накладки на край пластини
Наступний патент: Піролотриазинові сполуки як інгібітори кінази
Випадковий патент: Спосіб вдосконалення бензидинової проби при визначенні ступеня свіжості риби