Спосіб отримання похідних цефалоспорину чи їх легко гідролізуємих скадних ефірів або їх солей з лужними металами







Формула / Реферат
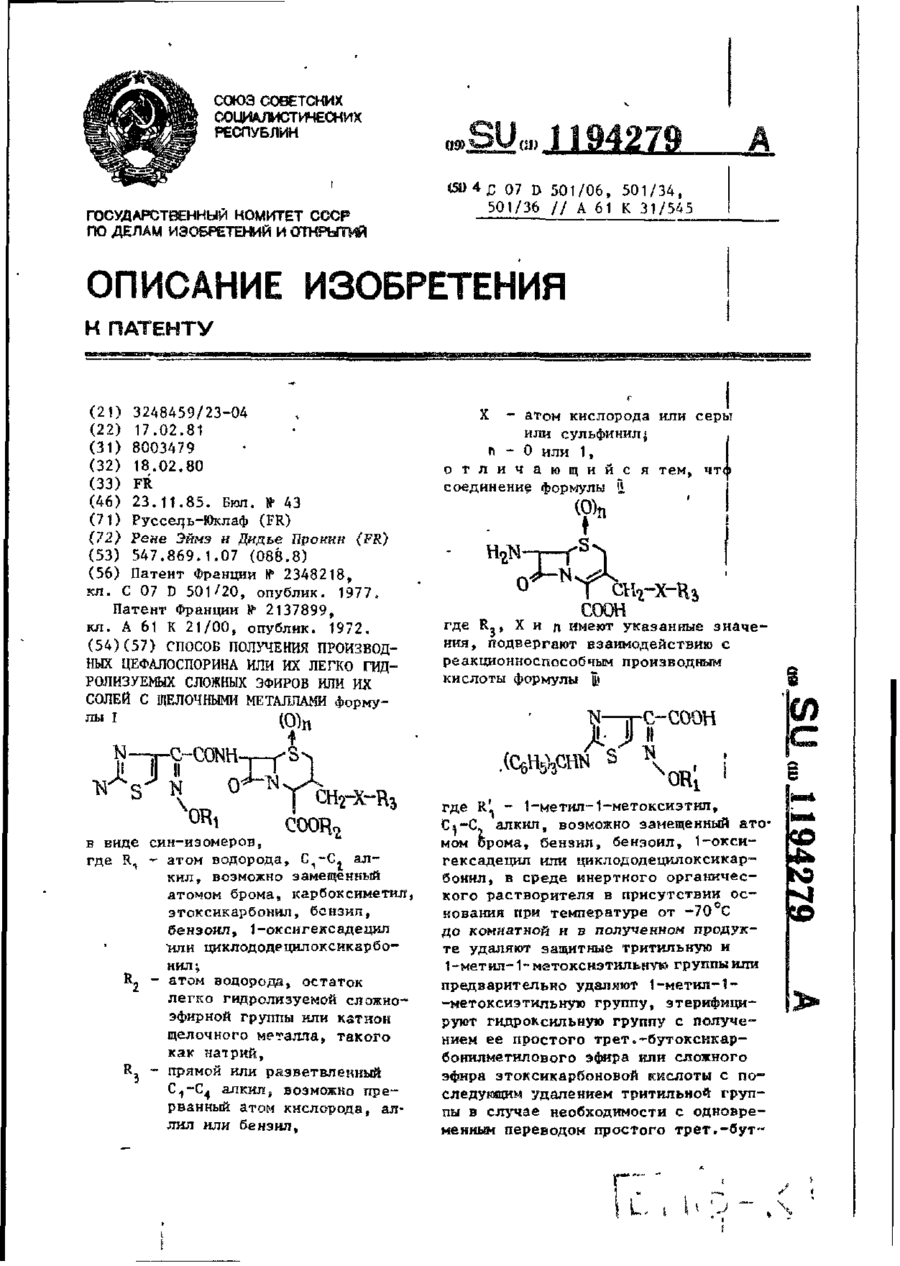
Способ получения производных цефалоспорина или их легко гидролизусмых сложных эфиров, или их солей с щелочными металлами
в виде син-изомеров,
где R1- атом водорода, С1-С2 алкил, возможно замещенный атомом брома, карбоксиметил, этоксикарбонил, бензил, бензоил, 1-оксигексадецил или циклододецилокси карбонил,
R2 - атом водорода, остаток легко гидролизируемой сложноэфирной группы или катион щелочного металла, такого как натрий,
R3 - прямой или разветвленный С1-С4 алкил, возможно прерванный атом кислорода, аллил или бензил;
Х- атом кислорода или серы или сульфинил;
n - 0 или 1, отличающийся тем, что соединение формулы II
где R3, Х и n имеют указанные значения, подвергают взаимодействию с реакционноспособным производным кислоты формулы III
где R'1 - 1-метил-1-метоксизтил, С1-С2 алкил, возможно замещенный атомомброма, бензил, бензоил, 1-оксигексадецил или циклододецилоксикарбонил, в среде инертного органического растворителя в присутствии основания при температуре от -70°С до комнатной и в полученном продукте удаляют защитные тритильную и 1-метил-1-метоксиэтильную группы, или предварительно удаляют 1-метил-1-метоксиэтильную группу, этерифицируют гидроксильную группу с получением ее простого трет.-бутоксикарбонилметилового эфира или сложного эфира этоксикарбоновой кислоты с последующим удалением тритильной группы в случае необходимости с одновременным переводом простого трет.-бутоксикарбонилметилового эфира в простой карбоксиметиловый эфир и выделяют целевой продукт в виде свободной кислоты или ее сложного легко гидролизуемого эфира, или ее соли с щелочным металлом, таким как натрий.
Текст
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЦЕФАЛОСПОРИНА ИЛИ ИХ ЛЕГКО ГИДРОЛИЗУЕКЫХ СЛОЖНЫХ ЭФИРОВ ИЯИ ИХ СОЛЕЙ С ЩЕЛОЧНЫМИ МЕТАЛЛАМИ формулы I (0)П і X - атом кислорода или серы или сульфиншц п - 0 или 1, о т л и ч а ю щ и й с я тем, что соединение формулы ІІ CHr-X-Rj Й-і где R_, X и п имеют указанные значения, подвергают взаимодействию с реакционноспособчым производным кислоты формулы jt [ N—n-Cс-соон «ami \ в виде син-изомеров, где R , - атом водорода, С^-С. ал. кил, возможно замещенный атомом брома, карбоксиметил, зтоксикарбонил, бензил, бензоил, 1-окснгексадецкл "или циклододецилоксикарбонил^ R^ - атом водородаs остаток легко гидролизуемой сложноэфирной группы кли катион щелочного металла, такого как натрий, Rj - прямой или разветвленный С ? -С 4 алкил, возможно прерванный атом кислорода, аллил или бенэнл. где Ц - 1-метил-1-метоксиэтил, Cs-C алкил, возможно замещенный атомом >рома, бензил, бензоил, 1-оксигексадецил или циклододецилоксикар бонил, в среде инертного органичес кого растворителя в присутствии основания при температуре от -70 С до комнатной и в полученном продукте удаляют защитные тритильную и 1-метил-1-метоксиэтильну» группы или предварительно удаляют 1-метил-1-метоксиэтильную группу, этерифициї>уют гидроксильную группу с получением ее простого трет.-бутоксикарбонилметилового эфира кли сложного эфира этоксикарбоновой кислоты с последующим удалением тритильной группы в случае необходимости с одновременным переводом простого трет.-бут I— (О 1194279 оксикарбонилметилового эфира в проскислоты или ее сложного легко гидротой карбоксиметиловый эфир и выделялизуемого эфира или ее соли с щелочют целевой продукт в виде свободной ным металлом, таким как натрий. 1 водой, сушат, концентрируют досуха, Изобретение относится к способу растирают с этиловым эфиромз отсасыполучения новых антибиотиков цефаловают и получают 3,07 г сырого проспоринового ряда, а именно новых продукта, который перекристаллизуют в изводных цефалоспорина или их легко 5 метаноле для получения 1,21 г целагидролизуемых сложных эфиров или их вого продукта. солей с щелочными металлами,» которые обладают, антимикробной активностью ЯМР (CDC! 3 ) S ч . / м л н , : 1,52 (двойи. могут найти применение в мепицине. ные СН 3 ); 3,22-3,25 (-0-СН 5 У, 3*45 1 (-CH 2 S); 4,25 (-СН2ОМе), 4,99 (d , Цель изобретения - получение ноW j = 5; Н6), 5,73 (dd, J = 5, J = 8*f вых антибиотиков цефалоспоринового H7); 6,70 (Hj тиазола, с и н ) ; 7 S 2B ряда, обладающих повышенной активностью как против грамположительных, так и против грамотрицательных бакСтадия В: 3-метоксиметил-7-[2~(2терий, в том числе устойчивых по от-аминотиазол-4-ил)-2-гидроксииминоношению к известным антибиотикам. 1S ацетамидо ]-цеф-3-ем~4-карбоновая кисл о т а , син-иэомер, П р и м е р 1; З-МетоксиметилПри 45-50°С перемешивают в т е ч е -7-[2-(2-аминотиазол-4-ил)-2-(гидрокние 10 мин 1,1 г полученного продуксиимино)-ацетамидо 3~цеФ~3-ем-4-карбота с 5 см 3 водкой муравьиной кислоновая кислота, син-изомер. Стадия А: п-толуолсульфоновый 226 ты, прибавляют 2 см 3 воды, отсасыва- ( 2~тритиламинотиаэол-4--ил)-2~(1ют, концентрируют досуха фильтрат -метил-1-метоксиэтоксиимино) уксуспод уменьшенным давлением при 30°С, ный ангидрид, син-изомер. отгоняя воду этанолом. Остаток кристаллизуют в воде. Отсасывают и Суспендируют 3,01 г соли триэтиламина 2-(2-тритиламинотиазол-4-ил)25 получают 0 9 54 г сырого продукта, к о -2-(1~метил-1-метоксиэтоксиимино) торый растворяют при помощи триэтилуксусной кислоты, син-изомера, в амика в 5 см3 50%-кого водного э т а 3 15 см ацетона. Прибавляют 1,05 г нола. Подьисляют до рН 2-3 прибавлетозилхлорида и перемешивают в т е ч е нием муравьиной кислоты, отсасывают ние 1 9 5 ч . Вводят 20 ем3 этилового 30 кристаллизованный продукт^ промывают эфира в смесь, охлаждают до -10°С, этанолом, а затем эфиром и получают отсасывают, промывают эфиром и прлу0 F 44 г сольвата. чагот 2,90 г продукта, состоящего из Вычислено,%: С 39,9; И 3,82; целевого ангидрида и хлоргвдрата N 16,58; S 15,18. триэтиламина. ,. С 1 4 Н ifO 4 N 5 S z - 1/2 Н г 0 (мол.м. Стадия Б: метоксиметил-7-[2-(2422,43). -тритиламинотиазол-4-ил)-2-(1-метилНайдено,%: С 40,0^ Н 4 , 0 ; -1-метокси)-этоксннмнно/-апетамидо1 N 16,1» S 14,8. -цеф-З-ем-4-карбоновая кислота, синИК-спектр (вазелиновое масло), изомер. см"1: 1757 (С=0 /*-л£ктам,) 1637-1638 т Растворяют 0,732 г 7-амино~3-мет(ОС„ С*Ю; 1605-1572-1488 (ароматиоксиметилцеф-3-ем-4~карбоновой кисло ческое соединение)о ты в 10 см 3 хлористого метилена и УФ-спектр (ЕтОН, НСЇ 0,1 н . ) . 0,84 см 3 триэтиламина, охлаждают до Перегиб 220 MMKS Е^ - 288; ^ м а к с -20°С и прибавляют 2„4 г полученной 262 ммк, EJ = 421, t= 17400. . 4й в стадии А смеси, дают температуре Спектр ЯМР (DMSO), ч./млн; 3f20 подняться до комнатной, прибавляют ( О С Н І ) , 3,50 (S-C42); 4,17 (СН 2 О)^ 0,5 см 3 уксусной кислоты,' промывают 5,14 (d! J* 5, Н6); 5,77 (dd; J = 5, \ З 1 194279 , 4 раствора муравьиной кислоты. Р а з б а в ляют с 90 см 3 воды в горячем с о с т о я нии, отсасывают и отгоняют фильтрат под уменьшенным давлением при темпе5 ратуре ниже 30°С. Остаток забирают в 100 см 3 хлористого метилена, промывают разбавленным до 1/10 насыщенным раствором хлористого натрия и 7 см3 нормального раствора бикарбо10 ната натрия, а затем разбавленным до 1/10 насыщенным раствором хлористого натрия. Органический слой сушат, п е регоняют досуха под уменьшенным д а в лением и остаток забирают в 100 см 3 15 этилового эфира, отсасывают и получают 2,10 г сырого продукта. Забирают его в 15 см 3 этилового эфира уксусной кислоты, перемешивают 30 мин, отсасывают, прополаскивают 20 этиловым эфиром уксусной кислоты, а затем этиловым эфиром и получают 1,69 г продукта. Растворяют 1,57 г этого продукта в 15 см 3 хлористого метилена, отфильтровывают, перегоня25 ют под уменьшенным давлением,остаток , забирают в 10 см 3 этилового эфира уксусной кислоты, перемешивают 30 мин, отсасывают, прополаскивают этиловым эфиром уксусной кислоты, Зо а затем эфиром и получают 1,29 г целевого продукта (оС)з - + 5 2 в ± 1 ; С = 1,5% DMSO. / = 8; Н7); 6,65 (Н 5 т и а з о л а ) , 7,1 (NH 2 )i 9,43 (d: J= 8, NHCO) t П р и м е р 2. 1-Оксопропоксиметиловый эфир 3-метоксиметил-7-[2-(2-аминотиазол-4-ил)-2-гидроксииминоацетамидо]-цеф-3-ем-4-карбоновой кислоты, син-изомер. Стадия А: 1-оксопропоксиметиловый эфир 3-метоксиметил-7-[2-(2-тритиламинотиаэол-4-ил)-2-/(1-метил- 1-метокси)-этоксиимино/-ацетамидо}-цеф-3-ем-4-карбоновой кислоты, син-изомер. При комнатной температуре р а с т воряют 4,15 г 3-метоксиметил-7-[2-(2-тритиламинотиазол-4-ил)-2-(/1-метоксиметил/-этоксиимино)-ацетамидо]-цеф-3-ем-4-карбоновой кислоты, син-изомер, и 0,456 г сухого карбоната калия в 14 см 3 безводного диметилформамида. Охлаждают до 0°С, вводят в течение 10 мин приготовленную, как это указано ниже, суспензию иодметилового эфира пропионовой кислоты и перемешивают 30 мин при 0 в С, а затем 30 мин при 20 е С. Р е а к ционную среду выливают в смесь, с о стоящую из 340 см 5 воды, 17 см3 нормального водного раствора бикарбоната натрия и 50 см 3 этилового эфира уксусной кислоты. Перемешивают, д е кантируют, экстрагируют этиловым эфиром уксусной кислоты, промывают водой, сушат, концентрируют досуха под уменьшенным давлением при температуре ниже 35 С. Остаток забирают в 25 см 3 иэопропилового эфира и отсасывают 4,42 г целевого продукта. ЯМР (CDCI 3 ), ч./млн: 1,15 ( t , J - 7 ) ; 2,40 (q, J= 7, С2Н 5)-, 3,34 ( О С Н ^ ) І 3,55 (5 SCH 2 ), 4,33 (CH.OCHj) j 5,05 (d, J = 5, H6); 6,71 (Ну син- т и а з о л а ) ; 7,33 (тритил). Получение иодметилового эфира пропионовой кислоты. Нагревают с обратным холодильником в течение 10 мин 1,4 г хлорметилового эфира пропионовой кислоты, 1,71 г йодида натрия и 23 см 3 б е з водного ацетона. Получают суспензию, которую употребляют сразу. Стадия Б: 1-оксопропоксиметиловый эфир 3-метоксиметил-7[2~(2-аминотиазол-4~ил)~2-гидроксииминоацетамидо]- цеф-З-ем-4-карбоновоЙ кислоты, син-изомер. В течение 15 мин при 45-50 С перемешивают 4,37 г полученного про3 дукта з 22 см 65%-ного водного ЯМР (CDC1,), ч./млн: 1,13 ( Т І J - 7 ) І 2,41 ( q i J= 7; C ? H f f )i 3,30 (OCH-)-, 3,53 (SCH 2 5i 4,3 (CH2-OCH3)1, 5,02 (d; J - 5; H6); 6,92 (Ну т и а з о ла, син). П р и м е р 3. 3-Meтоксйметил-7-[2-(2~аминотиазол-4-ил)-2-меток._ сииминоацетамидо]-цеф-3-ем-4-карбоновая кислота, син-изомер. Стадия А: h-толуолсульфоновый 2-(2-тритиламинотиазол-4-ил)-2-метоксииминоуксусный ангидрид, синизомер. [ 45 Перемешивают 1,80 г триэтиламиновой соли 2-(2-тритиламинотиаэол— -4-ил)-2-метоксниминоуксусной к и с лоты, син-изомер, и 0,63 г хлорида тоэила в 20 см 3 безводного ацетона. Перемешивают в течение 1 ч при 20 С и получают суспензию, которая сразу употребляется на следующей стадии. Сталин Б: 3-метоксиметил-7-[2-(2~тритиламинотиазол-4-ил)-2-метэксииминоацетамидо ]-цеф-3-ем-4-карДоновая кислота, син-изомер. Приготавливают перед употреблением следующий раствор при 20 С при $ 1194279 перемешивании в инертной атмосфере: C 1 ? H 1 1 O 4 N 5 S 2 (мол.м. 427,459). 0,732 г З-метоксиметил-7-аминоНайдено,%: С 42$ Н 3,9, N. 15,8І -цеф-З-ем-4-карбоновоЙ кислоты, S 15,2. 6,6 см3 молярного раствора бикарбоИК-спектр (вазелиновое масло), ната натрия и 3,4 см3 воды. Охлажсм*1: 1756 ( 0 0 /j-лактам) і 1660 (амид) 5 дают до 5°С и эа 5 мин вводят полуС=С 1637 ченную в стадии А суспензию сметанСопряж» ного ангидрида. Перемешивают C=N 1623 1 ч при 0-5 й С а затем 1 ч при 20°С. + амид 11 1562 Отфильтровывают нерастворимое веще10 ство и перегоняют ацетон под уменьшенным давлением при максимум 30 е С. ONOR 1031 Подкисляют прибавкой 0,7 см3 муравьиной кислоты, экстрагируют хлорисУФ-спектр [ЕтОН - НСІ/10] Перетым метиленом, промывают водой, суt5 гиб 244 ммк, Е\ = 355;, ммк шат и концентрируют досуха под Е; = 437, £= 18700. уменьшенным давлением. ОстатокзабиСпектр Я Р (DMSO)S ч./млиї 3 S 22 М рают в 10 см3 этилового эфира, отса(OCH3)i 3,85 (N-O-CHj); 3,53 (CH 7 S); сывают и получают 1,75 г продукта, 4,18 (СН20СНэ),- 5,t4 (d; J= 5; H6), кбторый употребляют в данном виде -на 20 5,76 (d,d; J = 5, 3 - 8; H7); 6,76 следующей стадии. (Hj тиазола, син), 9,60 (d; J = 8J Стадия В: 3-метоксиметил-7-[2NHCO). -(2-аминотиаэол-4~ил)-2-метоксиимиП р и м е р 4. З-Метилтиометил-ноацетамидо}~цеф-3-ем-4-карбоновая -7~[2-(2-аминотиазол-4-ил)-2-гидроккислота, син-нзомер. 25 сииминоацетамидо ]~цеф-3-ем-4-карбо~ При 45-50 С в инертной атмосфере новая кислота, син-изомер. перемешивают в течение 12 мин 1,07 г Стадия А: 3-метилтиометил-7-[2полученного на предыдущей стадии -(2-тритштаминотиазол-4-ил)-2-(/1продукта и 8 Я 4 см3 66%-ной водной -метил-1-метокси/-этоксиимино)-ацетмуравьиной кислоты, при 45-50°С амидо]-цеф-1~3-ем-4-карбоновая кисприбавляют 3,4 см3 воды и сразу лота, син-изомер» отсасывают. Фильтрат перегоняют под Перемешивают 2,60 г 3-метилтиоуменьшенным давлением, сушат переметил-7-аминоцеф-3-ем-4-карбоновой киспотьц 37,5 см3 хлористого метилена гонкой с этанолом, растирают сухой 3 и 2,8 см3 триэтиламина, охлаждают до экстракт с 10 см воды. Отсасывают, 35 -20°С и вводят 9,5 г паратолуолсульпрополаскивают водой, а затем этилофонового 2-(2-тритиламинотиазол-4вым эфиром и получают 0,436 г целе-ил)-2-(1-метил-1-метоксиэтоксиимивого продукта. Из маточных растворов но) уксусного ангидрида, полученнорекуперируют после очистки- 0,111 г го как это указано в стадии А приидентичного продукта. Забирают мера 1. Перемешивают при 0 С в тече0 s 542 г этого продукта в 5,5 см3 воние 2 ч, подкисляют прибавкой ды в течение 1 ч . Отсасываютs пропо1,75 см3 уксусной кислоты, промываласкивают водой, а затем эфиром и получают 0 Р 453 г продукта, который ют водой органический слой, сушат сгущают в ацетоне. Отсасываютя сушат и концентрируют досуха под уменьшен" и получают 0,379 г продукта. Этот иым давлением. Остаток забирают в продукт забирается в 10 см3 воды. 50 см3 этилового эфира, отсасывают, Медленно прибавляют 0,83 см3 молярнопрополаскивают эфиром и получают го раствора бикарбоната нгтрия, р а з 8,45 г сырого продукта. Забирают бавляют 1 см3 2 М раствора хлористоего в 45 см3 метанола и перемешивают го натрия, отсасывают, прополаскива- 50 в течение 30 мин. Затравляют крисют водой. Фильтрат подкисляют таллизацию, отсасывают, прополаски0,5 см3 2 н. раствора соляной кисловают метанолом, а затем этиловым ты до рН - 3, отсасывают, прополасэфиром и получают 5,2 г целевого кивают водой, эфиром, сгущают ацепродукта» тоном и получают 0,227 г очищенного 55 Я Р (CDCI,), ч./млн: 1S85 (CH^S); М продукта. 5,05 (d; J=J5-> H6); 5,70 (d,d, J = Вычислено,%: С 42,15; Н 4 , 0 1 ; = 5, 3 = 8 , H7V, 6,71 (H 5 тиазола, N 16,38; S 15,00. син), 7,28-тритил. т 8 7 11 94279 приготовленной перед употреблением, Стадия Б: 3-метилтиометил-7-[2исходя из 1,7 15 г хлорметилового -эфи-(2-аминотиазол-4-ил)-2-(гидроксира пропионовой кислоты. Перемешивают имино) ацетамидо]-цеф-3-ем--4-карбо30 мин при 5°С, а затем 30 мин при новая кислота, син-иэомер. 5 При 50 С и в течение 10 мин пере- 5 20 С. Реакционную смесь выпивают в раствор, образованный 260 см 3 воды мешивают 3,72 г полученного выше про э при 10-15°С, 20 см3 н. водного дукта и 18,6 см 66%-ной водной муэ раствора бикарбоната натрия и 50 см 3 равьиной кислоты. Прибавляют 7,4 см этилового эфира уксусной кислоты, певоды и сразу отсасывают. Фильтрат перегоняют под уменьшенным давлени- 10 ремешивают, декантируют, экстрагируют ем при максимум 30°С, перегоняют этиловым эфиром уксусной кислоты, 2 раза со смесью этанол-вода ( 2 - 1 ) , промывают водой, сушат и перегоняют забирают остаток в 10 см3 воды, отдосуха под уменьшенным давлением. сасывают, прополаскивают водой, а Остаток забирают в 50 см 3 нзопропилозатем этиловым эфиром и получают (5 вого эфира, отсасывают и получают 1,945 г сырого продукта. Забирают 5,53 г целевого продукта. его в 136 см 3 50%-ного водного э т а Стадия Б: 1-оксопропоксиметиловый нола и медленно прибавляют 0,63 смэ эфир З-метилтнометил-7-[2-(2-аминотриэтиламина. Отсасывают нерастворитиаэол-4-ил)-2-гкдроксинминоацетамимое вещество, подкисляют фильтрат 20 до]~цеф-3-ем-4-карбоновой кислоты, до рН х 3-4 прибавкой 0,45 см 5 син-иэомер. 50%-ной водной муравьиной кислоты. і При 50°С в течение 10 мин перемеОтсасывают, прополаскивают 50%-ным шивают 3,75 г полученного выше проводным этанолом, безводным этанолом дукта в 18да7 см3 66Х-ной водной муа затем этиловым эфиром и получают 25 равьиной кислоты. Прибавляют 7,5 см3 1,392 г очищенного продукта. воды, отсасывают, перегоняют фильтИК-спектр (вазелиновое масло), рат под уменьшенным давлением при см' 1 : 1772 (С=0/5-лактам), 1693 амид; максимум 30°С и производят две пере1619 (NH^, образование). гонки со смесью этанол-вода ( 2 - 1 ) . Ароматическое Г1595 30 Остаток забирают в 10 см3 воды, отсоединение і 1535 сасывают, прополаскивают водой, а УФ-спектр [ЕтОН - 1/10 н.НСІ]. затем этиповым эфиром и получают Перегиб 220 ммк, Е^ = 320; Д 2,17 г сырого продукта. Этот последС 262 ммк, Е!, = 444, £ = 19100. ний хроматографируют на двуокиси Спектр ЯМР (DMSO), ч./млн: 1,98~' 3 5 кремния, элюнруют смесью этиловый (CH3S)-, 3,58 (CH 2 S), 5,17 (d,- J = 5, эфир уксусной кислоты - ацетон ( 3 - 1 ) , H6)-, 5,72 ( d , d , J = 5, J = 8; H7); забирают сухой остаток в 10 см 3 6,66 (H 5 тиазола, с и н ) ; 9,43 (d, изопропнлового эфира, отсасывают J= 8, NHCO). 1,55 г продукта, который забирают в П р и м е р 5. 1-0ксопропокси4Q 4,5 см 3 этилового эфира уксусной метиловый эфир З-метилтнометил-7кислоты, прибавляют 7,5 см3 этилово-[2-(2-аминотиазол-4-ил)-2-гидроксиго эфира уксусной кислоты, отсасываиминоацетамидо]-цеф-3-ем-4-карбоноют, прополаскивают этиловым эфиром вой кислоты, снн-изомер. уксусной кислоты, а затем нэопропиСтадия А: 1-оксопропоксиметиловый повым эфиром к получают 0,536 г це45 эфир 3-метиотиометил-7-С2-(2-тритиллевого продукта. аминотиаэол-4-ил)-2-(1-метил-1-метУФ-спектр (этанол) . Д ^ к с 222 ммк, оксиэтоксиимино)-ацетамидоЗ-цеф-3ЕІ = 384; 1= 19,800; Л ^ я к с 262 ммк, -ем-4-карбоновой кислоты, син-изомер Е^ = 271, £ - 14,000, (этанол, ' При 20^0 перемешивают 5,2 г 30,1 н.НСІ). Перегиб 218ММК, Е{ ~ 50 -метилтиометил-7-Г2-(2~тритилакиноти - 293, Д МЧКС 263 ммк, ЕІ = 382, азол-4-ил)-2-(1-метил-1-метоксиЄ- 19,700. -карбоновой кислоты, снн-изомер, и 0,56 г карбоната калия в 20 см3 55 безводного диметилформамида, охлаждают до 5°С и прибавляют при 5-10°С 28 см3 ацетоновой суспензии йодометилового эфира пропионовой кислоты, Спектр ЯМР 1,17 ( т ; J = 7) 2,42 ( * І J - 7) 2,1 , 3,6~ 3,8 (-Р (ОСН^ ) . П р и м е р 89. 2 - 0 , 3 - б и с ) -Ацетилоксипропнловый эфир 3-метоксиметил-7-[2-(2-аминотиазол-4-ил)-2~оксиимииоадетамидоЗ~цеф-3-ем~4-карбоновэй кислоты, син-изомер. Стадия А: 2~{\ Э 3-бис)-ацетилоксипропиловнй эфир З-метоксиметіш-Т-СЗ-(2~трктилаі>оінотиазап-4-ил)-2-(1-метокси-1-метилэтокси)-иминоацет- • амидоЗ'~цеф-'3-ем-4-карбоновой кислоты, син-изомер, • 3,63 г 3-метоксиметил-7-[2-(2окси-1-метмлзгокси~и№їноацета*£идо]-цеф-З-ем-4-карбоновой кислоты, 1,, 55 г глицерин-1 ,,3-ци ацетата,, 30 кг 4-диметилаі'*мнопмридигіа раство3 ряют в 36 ск метиленхлор-ида. Охлаждают до 5-Ю °С и добавляют 1,2 дкітклогексилкарбодиимида о Доводят до комнатной температуры и выдержи
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparation of cephalosporin derivatives or their easily hydrolyzable esters, or their salts with the alkali metals
Автори англійськоюEjme Rene, Pronin Dide
Назва патенту російськоюСпособ получения производных цефалоспорина или их легко гидролизуемых сложных эфиров, или их солей со щелочными металлами
Автори російськоюРене Эйме, Дидье Пронин
МПК / Мітки
МПК: C07D 501/00, C07D 277/20, A61K 31/546, A61P 31/04, A61K 31/545
Мітки: легко, отримання, металами, солей, лужними, похідних, цефалоспорину, ефірів, скадних, гідролізуємих, спосіб
Код посилання
<a href="https://ua.patents.su/52-5579-sposib-otrimannya-pokhidnikh-cefalosporinu-chi-kh-legko-gidrolizuehmikh-skadnikh-efiriv-abo-kh-solejj-z-luzhnimi-metalami.html" target="_blank" rel="follow" title="База патентів України">Спосіб отримання похідних цефалоспорину чи їх легко гідролізуємих скадних ефірів або їх солей з лужними металами</a>
Спосіб одержання похідних 7-[2-/2-амінотіазоліл/2-оксиіміноацетамідо]-3-цефєм-4-карбонових кислот або їх складних ефірів, або їх солей з лужними металами

Номер патенту: 1749
Опубліковано: 25.10.1994
Автори: Кіесі Цудзі, Тосіюкі Тіба, Такао Такая, Хісасі Такасугі
Мітки: кислот, металами, складних, одержання, лужними, спосіб, солей, похідних, ефірів
Формула / Реферат:
Формула изобретенияСпособ получения производных 7-[2-(2-аминотиазолил)-2-оксиимино- ацетамидо ]-3-цефем-4-карбоновых кислот формулыв виде син-изомеров, где R1 - свободная аминогруппа или защищенная формильной или трифторацетильной группой аминогруппа;R2 - водород, C7-C8 -алкил, аллил, пропаргил, С5-С6-циклоалкил, карбоксиметил, этоксикарбонилметил или моно- или тризамещенный хлор или фтор этил;R3 -...
Спосіб одержання похідних цефалоспорину або їх складних ефірів, простих ефірів або солей, або їх гідратів, або гідратів їх складних ефірів, простих ефірів або солей

Номер патенту: 4199
Опубліковано: 27.12.1994
Автори: Марк Монтафон, Роланд Рейнер
МПК: C07D 501/00, A61K 31/545
Мітки: простих, складних, ефірів, спосіб, гідратів, солей, одержання, цефалоспорину, похідних
Формула / Реферат:
1. Способ получения производных цефалоспорина общей формулы І в которой X - 2,5-дигидро-6-оксо-2-метил-5-оксо-астриазин-З-илгруппа, которая находится в таутомерном равновесии с 1,2,5,6-тетрагидро-2-мстил-5,6-диоксо-астриазин-3-илгруппой, или 1,4,5,6-тетрагидро-4-метил-5,6-диоксо-астриазин-З-илгруппа, или их сложных эфиров, простых эфиров или солей, или их гидратов или гидратов их сложных эфиров,...
Спосіб отримання похідних 7-[2-(2-амітіазоліл-4)2-оксііміноацетамідо]-3-ацетоксіметил-3-цефем-4карбонової кислоти чи її солей з лужними металами

Номер патенту: 4796
Опубліковано: 28.12.1994
Автори: Андре Лютц, Рене Ейме
МПК: A61K 31/545, C07D 501/00
Мітки: металами, солей, отримання, кислоти, спосіб, 7-[2-(2-амітіазоліл-4)2-оксііміноацетамідо]-3-ацетоксіметил-3-цефем-4карбонової, похідних, лужними
Спосіб отримання похідних бензаміду, або їх солей, або рацемічних сумішей, або стерєоізомерів

Номер патенту: 4753
Опубліковано: 28.12.1994
Автори: Свен Ове Егрен, Геста Леннарт Фрорвалл
МПК: C07C 51/347, C07C 51/00, A61P 25/18, C07C 65/00, A61K 9/20, C07D 207/09, A61K 31/40, A61K 9/48, C07C 67/00
Мітки: спосіб, сумішей, солей, похідних, бензаміду, стерєоізомерів, отримання, рацемічних
Формула / Реферат:
Способ получения производных бензамида общей формулыгде R1 - алкил с 1-3 С;R2 и R3 - одинаковые или различные и означают водород, хлор или бром,или их солей, или рацемических смесей, или сте-реоизмеров, отличающийся тем, что соединение общей формулыгде R1, R2 и R3 имеют вышеуказанные значения, подвергают взаимодействию с амином общей формулыв среде органического растворителя и целевой...
Спосіб отримання похідних імідазола або його фармацевтично прийнятих солей

Номер патенту: 2697
Опубліковано: 26.12.1994
Автори: Дейвід Джон Керіні, Джон Джонес Вітотес Данкіе
Мітки: спосіб, прийнятих, похідних, фармацевтично, імідазола, отримання, солей
Текст:
...5 Гц). пластинках для ТСХ. Масс-спектр 325. ЯМР (200 мГц, Е. 1~(4~Аминобензил)~5-гидроксиме3 э 8,00-6,80 (м, 8Н), тил-2-(2*-метоксиэтил~-4-«зшоримидазол. 5,15 ( с , 2Н>, 4,45 ( с , 2Н), 3,60 ( т , Соединение СИНТеЗИРУЮТ П Примеру О 2Н, 5 Г ц ) , 3,15 ( с , ЗН), 2,75 54, Е из 5-тидроксимет'ил—2~(2-меток~ ( т , 2Н, 5 Г ц ) 0 П р и м е р ы сиэгил)— 1 — (4—нитроб ензил) — 4—хлорими— 57—71„ Соединения, синтезированные по дазола ( 2 , 2 г, 6,75...