Спосіб синтезу енантіомерно чистого і хімічно чистого 3,4-метанопроліну
Номер патенту: 33424
Опубліковано: 25.06.2008
Автори: Комаров Ігор Володимирович, Гвоздовська Наталія Петрівна






Формула / Реферат
1. Спосіб синтезу оптично чистого 3,4-метанопроліну, який включає наступні стадії:
a) захист карбоксильної групи 4-гідрокси-(2S)-карбоксипіролідину
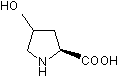 (I)
(I)
придатним захисним агентом з одержанням відповідної карбоксизахищеної похідної формули (II):
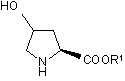 (II),
(II),
де R1 являє собою придатну захисну групу, що використовується в синтезі пептидів;
b) захист атому азоту похідної формули (II) придатним захисним агентом з одержанням відповідної азотозахищеної похідної формули (III):
НО
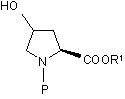 (III),
(III),
де Р являє собою придатну захисну групу, що використовується в синтезі пептидів;
c) реакцію гідроксипохідної формули (III) з придатним сульфонілюючим агентом з одержанням відповідної 4-сульфонілоксипохідної формули (IV):
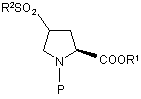 (IV),
(IV),
де R2 являє собою С1-С6-алкіл, С1-С6-галоалкіл або арил;
d) реакцію 4-сульфонілоксипохідної формули (IV) з йодид-іонами в диметилформаміді з утворенням захищеного дегідропіролідину формули:
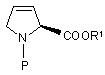 (V);
(V);
e) реакцію захищеного дегідропроліну формули (V) з циклопропанілюючим агентом з утворенням суміші захищених 3,4-метанопролінів формул (VIa) та (VIb):
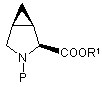 (VIa) та
(VIa) та 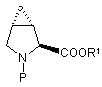 (VIb),
(VIb),
де R1 та Р є такими як визначено вище,
які промивають окислюючим агентом та розділяють суміш 3,4-метанопролінів формул (VIa) та (VIb) на індивідуальні ізомери та f) зняття захисту з карбоксильної групи захищених 3,4-метанопролінів формул (VIa) та (VIb) з одержанням відповідних N-захищених 3,4-метанопролінів формул (VIIa) та (VIIb):
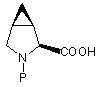 (VIIa) та
(VIIa) та 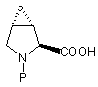 (VIIb);
(VIIb);
g) можливу обробку N-захищених 3,4-метанопролінів формул (VIIa) та (VIIb) кислотою з одержанням відповідних 3,4-метанопролінів формул (VIIIa) та (VIIIb) у формі кислотно адитивних солей:
 (VIIIa) та
(VIIIa) та 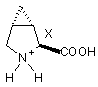 (VIIIb).
(VIIIb).
2. Спосіб згідно з п.1, де захисною групою R1 є С1-С6алкіл.
3. Спосіб згідно з п. 2, де R1 є метил.
4. Спосіб згідно з п. 1, де захисною групою Р є Вос.
5. Спосіб згідно з п. 1, де на стадії с) як сульфонілюючий агент використовують мезилхлорид.
6. Спосіб згідно з п. 1, де реакцію стадії d) проводять в присутності КІ.
7. Спосіб згідно з п. 1, в якому як окислюючий агент на стадії e) використовують перманганат калію.
8. Спосіб згідно з п. 7, в якому як окислюючий агент використовують 2-5% розчин перманганату калію у воді.
9. Спосіб згідно з п. 8, в якому реакційний розчин промивають водним розчином перманганату калію.
10. Спосіб згідно з п. 9, де суміш захищених 3,4-метанопролінів формул (VIa) та (VIb) після промивання окислюючим агентом розділяють хроматографічно.
11. Спосіб згідно з п. 1, де зняття захисту з карбоксильної групи на стадії f) проводять шляхом обробки основою.
Текст
1. Спосіб синтезу оптично чистого 3,4метанопроліну, який включає наступні стадії: a) захист карбоксильної групи 4-гідрокси-(2S)карбоксипіролідину HO 2 3 33424 мул (VIIIa) та (VIIIb) у формі кислотно адитивних солей: X N+ H X COOH H N+ (VIIIa) та H COOH H (VIIIb). 2. Спосіб згідно з п.1, де захисною групою R1 є С1С6алкіл. 3. Спосіб згідно з п. 2, де R1 є метил. 4. Спосіб згідно з п. 1, де захисною групою Р є Вос. 5. Спосіб згідно з п. 1, де на стадії с) як сульфонілюючий агент використовують мезилхлорид. 6. Спосіб згідно з п. 1, де реакцію стадії d) проводять в присутності КІ. Корисна модель стосується способу промислового синтезу енантіомерно чистого і хімічно чистого 3,4-метанопроліну. Виділення 3,4-метанопроліну (1) з Aesculus parviflora було описано Fowden, L; Smith, A.; Millington, D.S.; Sheppard, R.C. Phytochemistry, 1969- 8, 437. COOH N H (1) Було показано, що 3,4-метанопролін є сильним регулятором росту рослин SU965337 та інгібітором метаболізму проліну Machackova, I.; Zmrhal, Z. Biol. Plant., 1983, 25 (5), 394. Крім того, амідні похідні метанопроліну є інгібіторами ангіотензинперетворюючого фермерменту Rowland, I.; Tritham, H. J.Bact., 1975, 123, 871. Також, 3,4метанопролін знаходить широке застосування при синтезі різноманітних модифікованих пептидів та біологічно активних речовин. Однак, на сьогоднішній день існує досить обмежений перелік методик, придатних для одержання енантіомерно чистого і хімічно чистого 3,4метанопроліну. Так, в Sagnard, I.; Sasaki, A.; Chiaroni, A.; Riche, С; Potier, P. Tetrahedron Lett., 1995, 36, 3149 описується спосіб одержання 3,4-метанопроліну шляхом конденсування двох хіральних вихідних матеріалів: (2) SO2Ph PTHP BocHN H (2) та трифлату (2R)-гліцидилу з наступною циклізацією і взаємним перетворенням функціональної групи з одержанням N-Boc-захищеного 3,4метанопроліну формули (1) після семи стадій із загальним виходом менше, ніж 25%. 4 7. Спосіб згідно з п. 1, в якому як окислюючий агент на стадії e) використовують перманганат калію. 8. Спосіб згідно з п. 7, в якому як окислюючий агент використовують 2-5% розчин перманганату калію у воді. 9. Спосіб згідно з п. 8, в якому реакційний розчин промивають водним розчином перманганату калію. 10. Спосіб згідно з п. 9, де суміш захищених 3,4метанопролінів формул (VIa) та (VIb) після промивання окислюючим агентом розділяють хроматографічно. 11. Спосіб згідно з п. 1, де зняття захисту з карбоксильної групи на стадії f) проводять шляхом обробки основою. Недоліком запропонованого способу є досить малий загальний вихід та складність методики, що перешкоджає її промисловому застосуванню. В Fujimoto, Y.; Irreverre, F.; Karie, J.M.; Karle, I.L.; Witkop, B. J.Amer.Chem.Soc, 1971, 93, 3471 описується спосіб одержання 3,4-метанопроліну шляхом циклопропанування дегідропроліну в реакції, яка не є діастереоселективною і в якій основним продуктом є транс-ізомер 3,4-метанопроліну формули (3): COOH N H (3) Однією з перешкод можливості використання цієї методики, окрім розділення циста трансізомерів, є присутність в продукті реакції дегідропроліну, що не прореагував, який досить складно відокремити від одержаних метанопролінів із застосування простих методик (наприклад, кристалізація, колонкова хроматографія), що потребує застосування спеціальних методів розділення, яке при промисловому (масштабному) виробництві значно збільшує трудовитрати та вартість одержання метанопроліну. Тому, все ще існує потреба в промисловому способі одержання хімічно чистого та енантіомерно чистого 3,4-метанопроліну, виходячи з дешевих, комерційно доступних реагентів. Задачею винаходу є промисловий спосіб одержання 3,4-метанопроліну. Поставлена задача була вирішена за рахунок удосконалення відомих методик шляхом їх оптимізації і пристосуванням до потреб даного синтезу. Так, одним з об’єктів винаходу є спосіб синтезу оптично чистого 3,4-метанопроліну, який включає наступні стадії: а) захист карбоксильної групи 4-гідрокси-(2S)карбоксипіролідину (І): 5 33424 HO COOH N H (I) придатним захисним агентом з одержанням відповідної карбоксизахищеної похідної формули (II): HO 6 які промивають окислюючим агентом та розділяють суміш 3,4-метанопролінів формул (VIa) та (VIb) на індивідуальні ізомери; та, f) зняття захисту з карбоксильної групи захищених 3,4-метанопролінів формул (VIa) та (VIb) з одержанням відповідних N-захищених 3,4метанопролінів формул (VIIa) та (VIIb): COOH N COOR1 N H (II) де R1 являє собою придатну захисну групу, що використовується в синтезі пептидів; b) захист атому азоту похідного формули (II) придатним захисним агентом з одержанням відповідної азотзахищеної похідної формули (III): HO P COOH N P (VIIa) та (VIIb) g) необов’язково, обробку N-захищених 3,4метанопролінів формул (VIIa) та (VIIb) кислотою з одержанням відповідних 3,4-метанопролінів формул (VIIIa) та (VIIIb) у формі кислотно-адитивних солей: X N+ COOR1 N H P (III) де Р являє собою придатну захисну групу, що використовується в синтезі пептидів; c) реакцію гідроксипохідної формули (III) з придатним сульфонілюючим агентом з одержанням відповідної 4-сульфонілоксипохідної формули (IV): R2SO2 N COOR1 P (IV) де R2 являє собою С1-С6-алкіл, С1-С6галоалкіл або арил; d) реакцію 4-сульфонілоксипохідної формули (IV) з йодид-іонами в диметилформаміді з утворенням захищеного дегідропіролідину формули: N COOR1 X COOH H COOH N+ (VIIIa) та H H (VIIIb) Прикладами придатної захисної групи R1, що використовуються для захисту карбоксильної групи є, наприклад, С1-С6 алкільна група, С3-С8 циклоалкільна група і С7-С14 аралкільна, аліл, 2адамантил, 4-нітробензил, 4-метоксибензил, 4хлорбензил, фенацильна група, бензилоксикарбонілгідразид, трет-бутоксикарбонілгідразид, тритилгідразид і т.і. В переважному втіленні винаходу, R1 є С1-С6 алкільною групою. В найбільш переважному втіленні винаходу, R1 є метилом. Прикладами придатної захисної групи Р, що використовується для захисту атому азоту піролідину, є Z, Вос, трет-пентилоксикарбоніл, ізоборнілоксикарбоніл, 4-метоксибензилоксикарбоніл, ClZ, Br-Z, адамантилоксикарбоніл, трифторацетил, фталоїл, форміл, 2-нітрофенілсульфеніл, дифенілфосфінотіоїл, Fmoc, тритил і т.і. В переважному втіленні винаходу, Р є Вос. 3,4-Метанопроліни формул (VIIIa) та (VIIIb) можуть бути одержані у вигляді відповідних кислотно-адитивних солей формул P (V) e) реакцію захищеного дегідропроліну формули (V) з циклопропанілюючим агентом з утворенням суміші захищених 3,4-метанопролінів формул (VIa) та (VIb): N P COOR1 N (VIa) та COOR1 P (VIb) де R1 та Р є такими, як визначено вище, X N+ H H X COOH N+ (VIIIa) та H COOH H (VIIIb) де X означає придатний аніон відповідної кислоти, що використовується для видалення захисної групи Р та одержання відповідної кислотноадитивної солі. Таким придатним аніоном може бути залишок такої кислоти, як фторид водню, хлороводнева кислота, метансульфонова кислота, трифторметансульфонова кислота, трифтороцтова кислота, триметилсиланбромід (TMSBr), три 7 33424 фторметансульфонат триметилсилілу, тетрафторборна кислота, трис(трифтор)бор, трибромід бору і т.і.. В переважному втіленні винаходу X може бути іоном хлору і, відповідно, видалення захисної групи Р в 3,4-метанопролінах формул (VIIIa) та (VIIIb) проводять шляхом обробки розчину N-захищених 3,4-метанопролінів розчином хлорводневої кислоти. Слід зазначити, що стадія д) може не проводитись, якщо необхідним є одержання Nзахищених 3,4-метанопролінів. Іншим об’єктом винаходу є спосіб одержання захищених 3,4-метанопролінів формул (VIa) та (VIb): 8 дної речовини повного хроматографічного розділення компонентів реакційної суміші здійснити до цього не вдавалось, як і не вдавалось одержати ізомери, не забруднені похідним дегідропроліну. Далі приводиться загальна схема здійснення способу одержання оптично чистих 3,4метанопролінів та її детальний опис. Схема 1 HO N H (I) COOH MeOH SOCl2 HO MeOH SOCl2 COOMe N H (II) a) N+ P N (VIa) та COOR1 P N COOR1 P (V) з циклопропанілюючим агентом, наступним промиванням реакційної суміші окислюючим агентом з одержанням суміші 3,4-метанопролінів формул (VIa) та (VIb), які розділяють на індивідуальні ізомери. Реакцію циклопропанілювання дегідропроліну формули (V) можна проводити з використанням різноманітних циклопропанілюючих агентів, наприклад, діазометан в присутності хлориду міді, реагент Сімонса - Сміта (CH2I 2/Et2Zn) і т.і. Після завершення реакції циклопропанілювання одержаний реакційний розчин фільтрують і обробляють розчином окислюючого агенту, такого як 2-5% розчин перманганату калію у воді. Обробка одержаної суміші перманганатом калію дозволяє окислити дегідропролін, що залишився в реакційній суміші, у відповідний дигідроксипролін, який легко відокремлюється від 3,4метанопролінів завдяки добрій розчинності у воді. Його видалення в значній мірі спрощує розділення одержаних захищених 3,4-метанопролінів формул (VIa) та (VIb): N COOR1 N COOR1 P P (VIa) та (VIb) на окремі діастереомери, і дозволяє успішно розділити всі ізомери за допомогою кристалізації або колонкової хроматографії. В присутності вихі COOH g) c) N N H (VIIIb) f) g) N COOMe Boc (VIa) (IV) N COOH Boc (VIIb) f) e) 1) CH2 2) KMnO 4 хроматографічне розділення 1. LiOH 2. H+ HCl COOH COOMe Boc KI ДМФА 1. LiOH 2. H + COOH Boc (VIIa) H (VIIIa) N+ H (VIb) де R1 та Р є придатними захисними групами, що використовуються в хімії пептидів, який полягає у взаємодії захищеного дегідропроліну формули (V): COOMe N MsO d) H COOR1 b) MsCl Net3 Boc (III) HCl N HO N N COOMe Boc (V) COOMe Boc (VIb) Синтез 3,4-метанопролінів (VIIIa) та (VIIIb) був здійснений з використанням відомих реакцій та через речовини, що були описані в літературі. Однак послідовність реакцій, що зображена на Схемі 1, була використана для синтезу вперше. На стадії а) сполуку формули (І) обробляють сульфонілхлоридом в присутності метанолу при температурі від -10 до 0°С, витримують при кімнатній температурі декілька годин і потім нагрівають із зворотнім холодильником до 10 годин, одержуючи сполуку формули (II). Замість сульфонілхлориду може бути використаний будь-який придатний активуючий агент, що використовується в хімії пептидів. Замість метанолу може бути використаний будь-який інший спирт, що дозволяє ввести будь-яку з груп, згаданих вище як R1. На стадії b) сполуку формули (II) обробляють Вос2О в присутності основи та розчинника, одержуючи сполуку формули (III). Реакцію проводять при температурі від приблизно 0 до приблизно температури кипіння розчинника. Час реакції становить від 30 хвилин до 10 годин. Замість Вос20 може бути використаний будь-який Інший реагент, що дозволяє ввести будь-яку групу згадану вище як Р. Як розчинник може бути використаний будьякий придатний розчинник, такий як метиленхлорид, хлороформ, діоксан та ін. Як основа може бути використаний триетиламін, триізопропіламін, піридин, N-метилморфолін і т.і. На стадії с) сполуку формули (III) обробляють сульфонілхлоридом в присутності основи, одержуючи сполуку формули (IV). Як основа може бути використана будь-яка придатна органічна основа, така як триетиламін, триізопропіламін, піридин, Nметилморфолін та ін. В переважному втіленні винаходу використовують триетиламін. Реакцію проводять при температурі від приблизно -30 до приблизно 50°С. Час реакції становить від 30 хвилин до 12 годин. На стадії d) сполуку формули (IV) розчиняють в диметилформаміді та обробляють йодидом калію при нагріванні реакційної суміші з одержанням сполуки формули (V). Реакцію проводять при тем 9 пературі 100-180°С протягом часу від приблизно 16 годин до приблизно 2 діб. На стадії e) сполуку формули (V) в придатному розчиннику обробляють пропанілюючим агентом і після завершення реакції реаційну суміш промивають 2-5% водним розчином перманганату калію і водою, а виділену цільову речовину піддають хроматографічному розділенню на силікагелі, використовуючи як елюент суміш етилацетату з гексаном, одержуючи сполуку формули (VIa) та сполуку формули (VIb). На стадії f) сполуки формул (VIa) та (VIb) обробляють розведеним лугом в придатному розчиннику з одержанням відповідних N-захищених 3,4-метанопролінів (VIIa) та (VIIb), відповідно. В якості лугу може бути використаний 20% водний розчин гідроксиду літію, а як розчинник може бути використаний будь-який придатний розчинник або їх суміш, наприклад, метанол, тетрагідрофуран, діоксан, метанол-тетрагідрофуран і т.і. На стадії д) N-захищені 3,4-метанопроліни формул (VIIa) та (VIІb) обробляють в розчиннику придатною кислотою з одержанням кислотноадитивних солей 3,4-метанопролінів формул (VIIIa) та (VIIIb). Оброблення проводять при кімнатній температурі в придатному розчиннику, такому як діоксан, тетргідрофуран або їх суміш. Як кислоту використовують розведений водний розчин хлорводневої кислоти. Вихід даної реакції є кількісним. При бажанні одержані солі можуть бути виділені у формі вільних основ після обробки відповідних солей основою в придатному розчиннику. Приведений вище спосіб синтезу 3,4метанопролінів формул (VIIIa) та (VIIIb) був здійснений з використанням відомих реакцій та виходячи з речовин, що були описані в літературі. Однак послідовність реакцій, що зображенні на Схемі 1, була використана для синтезу вперше. Новизна полягає в застосуванні реакції дегідросульфонілювання сполуки формули (IV), що дозволило значно масштабувати та спростити синтез, не використовуючи дорогих реагентів. Однак ще одним суттєвим поліпшенням методики є використання водного розчину перманганату калію для видалення дегідропроліну, що не прореагував, з реакційної суміші на стадії циклопропанілювання. Це дозволило успішно розділити ізомери формул (VIa) та (VIb) за допомогою колонкової хроматографії або кристалізації, в присутності вихідних речовин повного розділення компонентів реакційної суміші здійснити не вдавалось. Далі приводяться приклади одержання цільових 3,4-метанопролінів формул (VIIIa) та (VIIIb). Приклад 1 Одержання 1-трет-бутил-2-метил-4гідроксипіролідін-1,2-дикарбоксилату (III) Свіжеперегнаний тіонілхлорид (73,25мл, 1 моль) по краплям додавали до розчину 4-гідроксиL-проліну (131,62г, 1 моль) і безводного метанолу (1л) при 0°С. Розчин перемішували при кімнатній температурі протягом 2 годин і потім кип’ятили із зворотнім холодильником протягом 8 годин. Розчинник видаляли при пониженому тиску. Неочищене масло розчиняли в метанолі, після чого ви 33424 10 даляли розчинник при пониженому тиску. Цю процедуру повторювали три рази, одержуючи гідрохлоридну сіль метилового естеру 4гідроксипроліну (173г, 95%). Неочищений продукт (173г, 0,95 моль) розчиняли в метиленхлориді (5л). До цього розчину додавали триетиламін (383,5мл, 2,85 моль) і ди-трет-бутилдикарбонат (239,2г, 1,1 моль). Реакційну суміш перемішували при кімнатній температурі протягом 10 годин. Розчин екстрагували 1N KHSO4 (1л´3), насиченим NaHCO3 (1л), 10% розчином лимонної кислоти (1л) і насиченим хлоридом натрію (1л). Органічну частину сушили над сульфатом магнію і розчинник видаляли у вакуумі. Очищали колонковою хроматографією одержуючи вказану в заголовку сполуку як білу речовину (209,5г, 90%). Синтез 1-третбутил-2-метил-4-гідроксипіролідін-1,2дикарбоксилату (III) проводили, використовуючи методику, описану Schumacher K.K. at el. Tetrahedron: Asymmetry 9 (1998) 47-53. Фізикохімічні дані одержаної сполуки повністю збігалися з опублікованими в літературі. Приклад 2 Одержання 1-трет-бутил-2-метил-4[(метилсульфоніл)окси]піролідін-1,2дикарбоксилату (IV). Сполуку формули (III) (245г, 1 моль), одержану в попередньому прикладі, розчиняли в метиленхлориді (3л), розчин охолоджували до -30°С і при сильному перемішуванні додавали триетиламін (202мл, 1,5 моль) та по краплях мезилхлорид (137,5г, 1,2 моль). Під час додавання мезилхлориду температуру підтримували близько -30°С і після завершення додавання реакційній суміші давали нагрітися до кімнатної температури при постійному перемішуванні. Через 8 годин, реакційну суміш промивали водою, 10% розчином лимонної кислоти, знову водою, сушили над сульфатом магнію. Розчинник видаляли на роторному випаровувачі, залишок кристалізували з тетрахлоретану одержуючи 1-трет-бутил-2-метил-4[(метилсульфоніл)окси]піролідін-1,2дикарбоксилат (IV). Вихід - 98%. Спектральні дані отриманого продукту повністю збігалися з опублікованими в літературі. Приклад 3 Одержання 1-трет-бутил-2-метил-2,5-дигідро1Н-піррол-1,2-дикарбоксилату (V) До розчину сполуки формули (IV) (10 ммоль) в N,N-диметилформаміді (80мл) в атмосфері аргону додавали КІ (50 ммоль). Суміш перемішували в атмосфері аргону 24 години при 110°С, диметилформамід упарювали у вакуумі, залишок розчиняли в діетиловому етері (200мл). Розчин промивали водою (3´50мл), сушили Na2SO4, упарювали. Залишок 1-трет-бутил-2-метил-2,5-дигідро-1Нпіррол-1,2-дикарбоксилат (V) - був достатньо чистим для наступної стадії. Його спектральні характеристики повністю співпадають з літературними даними. Приклад 4 Одержання (1R,2S,5S)-3-трет-бутил-2-метил3-азабіцикло[3.1.0]гексан-2,3-дикарбоксилату (VIa) та (1S,2S,5R)-3-трет-бутил-2-метил-3азабіцикло[3.1.0]гексан-2,3-дикарбоксилату (VIb). 11 33424 У трьохгорлу круглодонну колбу на 500мл, оснащену магнітною мішалкою, шнековою лійкою, трубкою для пропускання аргону та газовідвідною трубкою, поміщали 200мл 50%-ного водного розчину КОН і 80мл додекану. Газовідвідну трубку через камеру з осушувачем (КОН) приєднували до реактора, оснащеного магнітною мішалкою, в яку поміщали похідну 3,4-дегідропроліну (V). В реактор додавали 1г CuCl. До суміші розчину КОН і додекану при перемішуванні магнітною мішалкою маленькими порціями зі шнекової лійки додавали N-нітрозометилсечовину. При цьому через суміш пропускали струмінь аргону. Діазометан, що утворювався, барботували при перемішуванні через суспензію дегідропроліну і СuСl, причому спочатку він проходив через трубку з осушувачем, а потім через реактор із сполукою (V). Пропускання діазометану здійснювали не менше 100 годин, доки конверсія в реакторі не досягне 60-70% (ЯМРконтроль). Після цього реакційну суміш розбавляли діетиловим етером, фільтрували, промивали 5% розчином перманганату калію (обережно - розігрівання), а потім водою. Сушили над безводним сульфатом натрію, упарювали, а залишок хроматографували, використовуючи як нерухому фазу силікагель, а як елюент суміш етилацетату з гексаном. Першим з колонки виходить сполука (VIb) транс-ізомер (Rf ~ 0,3), наступною (VIa) цис-ізомер (Rf ~ 0,27). Вихід: (VIb) ~ 50%; (VIa) ~ 15%. Сполука (VIb) (2 ротамери, безбарвна в’язка рідина): 1Н-ЯМР (CDCl3): d 0,33 (м, 1Н), 0,75 (м, 1Н), 1,39 (с, 5Н), 1,44 (с, 4Н), 1,52 (м, 1Н), 1,60 (м, 1Н), 3,53 (м, 1Н), 3,59 (м, 1Н), 3.74 (с, 3Н), 4,72 (с, 0,53Н), 4,40 (с, 0,38Н). Сполука (VIa) (2 ротамери, безбарвна в’язка рідина): 1Н-ЯМР (CDCl3): d 0,65 (м, 1Н), 0,75 (м, 1Н), 1,42 (с, 5Н), 1,48 (с, 4Н), 1,61 (м, 1Н), 1,83 (м, 1Н), 3,50-3,65 (м, 2Н), 3,74 та 3.75 (2с, 3Н), 4,33 (д, J = 3Гц, 0,50Н), 4,38 (д, J = 3Гц, 0,38Н). Приклад 5 Комп’ютерна верстка Л. Купенко 12 Одержання (1R,2S,5S)-3-(третбутоксикарбоніл)-3-азабіцикло[3.1.0]гексан-2карбонової кислоти (VIIa) та (1S,2S,5R)-3-(тpeтбyтоксикарбоніл)-3-азабіцикло[3.1.0]гексан-2карбонової кислоти (VIIb). Сполуку (VIa) або (VIb) (10 ммоль) розчиняли в суміші метанол-тетрагідрофуран (2:3, 50мл) і при перемішуванні додавали 20% водний розчин LiOH (40 ммоль). Суміш перемішували 8 годин при кімнатній температурі, випарювали в роторному випаровувачі на дві третини, розбавляли водою (50мл) і отриманий розчин промивали діетиловим етером (2´50мл). Водну фазу підкислювали лимонною кислотою, екстрагували продукт етилацетатом (3´50мл), органічну фазу промивали водою (50мл), сушили (Na2SO4), упарювали. Залишок розтирали з холодним етером, фільтрували. Вихід 80%. Сполука (VIIb) (2 ротамери, безбарвні кристали, Т. топл. 145°С): 1Н-ЯМР (CDCl3): d 0,31-0,36 (м, 1Н), 0,78 (м, 1Н), 1,41 і 1,46 (2с, 9Н), 1,53 (м, 1Н), 1,69-1,78 (м, 1Н), 3,55-3,61 (м, 2Н), 4,30 і 4,41(2с, 1Н), 9,68 (шир.с, 1Н). Сполука (VIIa) (2 ротамери, безбарвні кристали, Т. топл. 101°С): 1Н-ЯМР (CD3OD): d 0,57 (шир. с, 1Н), 0,72 (шир. с, 1Н), 1,38 (с., 9Н), 1,47 (шир. с, 1Н), 1,91 (шир. с, 1Н), 3,45-3,51 (шир. м., 2Н), 4,25 (шир. с, 1Н). Приклад 6 Одержання (1R,2S,5S)-3азабіцикло[3.1.0]гексан-2-карбонової кислоти (VIIa) та (1S,2S,5R)-3-азабіцикло[3.1.0]гексан-2карбонової кислоти (VIIIb). Сполуку (VIIa) або (VIIb) (10 ммоль) розчиняли в 3,5-4М розчині НСl в діоксані (50мл) і цей розчин витримували при кімнатній температурі 0,5-1 годину (контроль ТШХ). Розчин випарювали в вакуумі. Вихід кількісний. Спектральні дані отриманих гідрохлоридів кислот повністю співпадають з опублікованими в літературі. Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod ofsynthesis of enantiopure and optically pure 3,4-methanoproline
Автори англійськоюKomarov Ihor Volodymyrovych, Hvozdovska Natalia Petrivna
Назва патенту російськоюСпособ синтеза энантиомерно чистого и химически чистого 3,4-метанопролина
Автори російськоюКомаров Игорь Владимирович, Гвоздовская Наталия Петровна
МПК / Мітки
МПК: C07B 57/00, C07D 209/00, C07B 53/00
Мітки: синтезу, хімічної, спосіб, чистого, 3,4-метанопроліну, енантіомерно
Код посилання
<a href="https://ua.patents.su/6-33424-sposib-sintezu-enantiomerno-chistogo-i-khimichno-chistogo-34-metanoprolinu.html" target="_blank" rel="follow" title="База патентів України">Спосіб синтезу енантіомерно чистого і хімічно чистого 3,4-метанопроліну</a>
Застосування енантіомерно чистого есциталопраму для лікування помірної або сильної депресії

Номер патенту: 82828
Опубліковано: 26.05.2008
Автори: Люнґ Йенсен Йеспер, Мьорк Арне, Санчес Конні
МПК: A61K 31/343, A61P 25/24
Мітки: есциталопраму, помірної, чистого, застосування, депресії, лікування, сильної, енантіомерно
Формула / Реферат:
1. Застосування есциталопраму, що містить менше ніж 3 мас. % R-циталопраму, для приготування фармацевтичної композиції, призначеної для лікування помірної або сильної депресії.2. Застосування за п. 1, яке відрізняється тим, що есциталопрам використовується для приготування фармацевтичної композиції для лікування сильної депресії.3. Застосування за п. 1 або 2, яке відрізняється тим, що есциталопрам використовується у вигляді...
Спосіб промислового синтезу периндоприлу і його фармацевтично прийнятних солей

Номер патенту: 75070
Опубліковано: 15.03.2006
Автори: Ланглуа Паскаль, Турбе Юге
МПК: C07D 209/42, C07K 5/06
Мітки: синтезу, прийнятних, периндоприлу, промислового, солей, фармацевтично, спосіб
Формула / Реферат:
1. Спосіб промислового синтезу периндоприлу формули (I) (І)і його фармацевтично прийнятних солей, який відрізняється тим, що бензиловий ефір формули (IV): ,(IV)де Вn представляє бензильну групу,реагує зі сполукою формули (V):
Спосіб модернізації in situ реактора гетерогенного екзотермічного синтезу, реактор гетерогенного екзотермічного синтезу та спосіб здійснення гетерогенних екзотермічних реакцій синтезу з високою продуктивністю

Номер патенту: 73466
Опубліковано: 15.08.2005
Автори: ПАГАНІ Джорджіо, Філліппі Ерманно
МПК: C01C 1/04, B01J 8/00, B01J 35/00, B01J 8/04
Мітки: реакцій, модернізації, гетерогенних, реактора, здійснення, екзотермічного, екзотермічних, високою, реактор, продуктивністю, синтезу, спосіб, гетерогенного
Формула / Реферат:
1. Спосіб модернізації in situ реактора гетерогенного екзотермічного синтезу, що включає зовнішній кожух, в якому розміщені один на одному в просторовому взаємозв'язку каталітичні шари, за яким попередньо встановлюють принаймні перший каталітичний шар у верхній частині згаданого кожуха та принаймні другий шар каталізатора в нижній частині цього кожуха, потім перший та другий шари завантажують першим каталізатором із завчасно...
Спосіб синтезу тропенолу для його одержання в промисловому масштабі

Номер патенту: 76761
Опубліковано: 15.09.2006
Автори: Зоботта Райнер, Рапп Армін Вальтер
МПК: C07B 61/00, C07D 451/10, C07D 451/06
Мітки: масштабі, синтезу, спосіб, промисловому, одержання, тропенолу
Формула / Реферат:
1. Спосіб одержання тропенолу формули (I) (I)необов’язково у вигляді його кислотно-адитивних солей, який відрізняється тим, що скопіновий ефір формули (II) , (II)у якій R являє собою залишок, вибраний із групи, яка включає С1-С4алкіл і С1-С4алкіленфеніл, кожний з яких може...
Спосіб одержання чистого циталопраму

Номер патенту: 71634
Опубліковано: 15.12.2004
Автори: Вілла Марко, Дансер Роберт, Сброджіо Федеріко
МПК: A61P 25/24, C07B 61/00, C07D 307/87
Мітки: спосіб, одержання, циталопраму, чистого
Формула / Реферат:
1. Спосіб одержання циталопраму формули І, (I)за яким сполуку формули II, (II)в якій Z означає йод, бром, хлор або CF3-(CF2)n-SO2-O-, n дорівнює 0, 1, 2, 3, 4, 5, 6, 7 або 8, піддають реакції ціанідного обміну з ціанідним джерелом;одержаний сирий продукт - циталопрам,...
Попередній патент: Спосіб осадження зливка увігнутими плитами з отворами
Наступний патент: Спосіб переробки анодних шламів
Випадковий патент: Конвеєр гвинтовий горизонтальний