Похідне 10,11,12,13-тетрагідродесмікозину, проміжні сполуки та спосіб одержання похідного 10,11,12,13-тетрагідродесмікозину




Текст
Изобретение относится к производным тилозина, а именно: к новым соединениям из ряда макролидных антибиотиков, обладающих антибактериальной активностью. В частности, изобретение относится к производным 10,11,12,13-тетрагидродесмикозина формулы (I): (I) где R1 означает Н; R2 означает Н; и его фармацевтически приемлемым солям с органическими или неорганическими кислотами, обладающими антибактериальной активностью и способу их получения. Изобретение также относится к производным 10,11,12,13-тетрагидродесмикозина формулы (III): R означает О или (ОС2Н5)2; R1 означает Si(СНз)з или Н; R2 означает Н, ОН, I или ОSО2 СНз; являющихся промежуточными соединениями для получения производных 10,11,12,13-тетрагидродесмикозина формулы (I). Десмикозин представляет собой 16-членный макролидный антибиотик получаемый посредством гидролиза микарозы в положении С-4' исходного антибиотика тилозина (R.L. Hamill, Antibiotics and Chemotherapy, 11, 328, 1961). 10,11,12,13-тетрагидродесмикозин получали посредством каталитического гидрирования десмикозина (Narandja et al., DD 272304 A5, 1989). Известно, что 4-дезоксидесмикозин получают в последовательности реакций, которая отличается тем, что реакционные группы, не требующиеся для той или иной реакции, предварительно защищают избирательными блокаторами, снимая защиту при проведении желаемой реакции. Так, известно, получение 4-дезокcидесмикозина, путем следующей последовательности реакций: ацелирование десмикозина в положениях С-2' и С-4', тетрагидрофуранизация в положениях С-3 и 4", метанолиз ацетила в положениях С-2' и С-4', избирательное сульфонирование в положении С-4', замещение йодом, дойодирование посредством обработки трибутилгидридом олова и гидролиз тетрагидрофуранильной и ацетильной защитных группировок (J. Antibiot, 34, 1381, 1981). Известен также способ получения 4-дезоксидесмикозина с помощью последовательности реакций, включающей следующие процедуры: избирательное ацелирование тилозина в положении С-2', гидролиз микарозы, ацелирование полученного десмикозина в положении С-4", сульфонирование в положении С-4', замещение сульфонильной группировки йодом, дезацелирование в положении С-4"; дейодирование посредством обработки трибутилгидридом олова и гидролиз ацетильной группы в положении С-2' (патент США № 4421911 (1983). Известен также способ получения 19-деформил-4-десмикозина, путем деформилирования тилозина катализатором Уилкинсона, силилирование гидроксильных групп в положениях С-3, 2' и 4", сульфонирование в положении С-4', замещение сульфонила йодом, восстановление йода трибутилгидридом олова и гидролиз силила (J. Antibiot, 42, 903, 1989). Анализ имеющихся материалов показывает, что 4'-дезокcи-10,11,12,13-тетрагидродeсмикозин (I), а также продукты присоединения в форме солей с органическими или неорганическими кислотами и промежуточные продукты (1а-1е) реакции его получения, представляют собой новые, ранее не описанные в литературе соединения . Первым предметом настоящего изобретения являются новые производные 10,11,12,13- тeтрагидродесмикозина формулы (I): и их пригодные для фармацевтических целей соли с органическими и неорганическими кислотами. Другим предметом изобретения является способ получения производных 10,11,12,13-тетрагидродесмикозина посредством ацилирования в этаноле 10,11,12,13-тетрагидродесмикозина формулы (II): в присутствии эквимолярного количества п-толуолсульфоновой кислоты. Окончание реакции регистрируют посредством хроматографирования в системе В. После частичной нейтрализации триэтиламином, растворитель упаривают до четверти его первоначального объема, добавляют насыщенный раствор гидрокарбоната натрия и экстрагируют хлороформом, с последующим выделением образующегося 10,11,12,13-тетрагидродесмикозин диэтила1 2 цеталя (la, R - (OC2H5)2, R - Н, R - ОН). Соединение la избирательно силилируют в положениях С-3, 2' и 4" 6-8 эквивалентами триметилхлорсилана в присутствии трет.-органического амина, такого как пиридин, триэтиламин или диметиланилин, в таком органическом растворителе как дихлорметан, дихлорэтан или хлороформ, при температуре 0-5°С. Окончание реакции контролируют хроматографическим методом (система А), добавляют смесь воды со льдом и после экстракции хлороформом при рН 8-9 выделяют 3,2',4"-три-О-триметилсилил-10,11,12,13-тетрагидродесмикозин диэтилацеталь (Ib, R - (ОС2Н5)2, R1 - Si(СНз)з, R2 ОН). Неочищенное соединение разводят в пиридине и подвергают сульфонированию в положении С-4' 5-7 эквивалентами соответствующего сульфохлорида, например, метан сульфонилхлорида или бензилсульфонилхлорида, при температуре от - 5 до 15°С. Окончание реакции контролируют хроматографическим методом (система А). После добавления смеси воды со льдом и доведения рН до 9-9,5, в осадок выпадает 4'метансульфонил-3,2',4"-три-О-триметилсилил-10,11,12,13-тетрагидродесмикозин диэтилацеталь (Ic, R (ОС2Н5)2, R1 - Si(СНз)з, а R2 - OSO2СНз). Влажный осадок разводят в хлороформе и обрабатывают насыщенным раствором хлористого натрия. После высушивания экстракта и упаривания растворителя при пониженном давлении соединение Ic быстро переводят в 4'-йодпроизводное, разводя его в сухом инертном растворителе и добавляя 4-6 эквивалентов йодида щелочного металла, с непрерывным перемешиванием при повышенной температуре (максимум - при температуре кипения) вплоть до полного исчезновения исходного соединения (система С). В качестве инертного растворителя можно использовать диметоксиэтан или метилэтилкетон, тогда как в качестве йодида щелочного металла применяют Kl, Nal или Lil. Концентрируют реакционную смесь и после добавления в нее воды со льдом экстрагируют хлороформом рН 9-9,5. Экстракты обрабатывают водным раствором тиосульфата натрия (10%), и после упаривают раствор при пониженном давлении, получая 4'-деокси-4'-йод-3,2',4"-три-О-триметилсилил-10,11,12,13-тетрагидродесмикозин диэтилацеталь (Id, R - (ОС2Н5)2, R1 - Si(СНз)з, R2 - йод). В 50 %-нoм растворе ацетонитрила и 0,2 N HCl проводят гидролиз защитной группы в 4'-йодпроизводном (Id). Гидролиз проводят при комнатной температуре а после его окончания (система А, система В) и доведения рН до 9-9,5, посредством экстракции выделяют 4'-деокси-4'-йод-10,11,12,13-тетрагидродесмикозин (Ie, R - О, R1 - Н, а R2 - йод). Неочищенный продукт очищают на силикагеле в системе А и С. Фракции с величиной Rf 0,85 (система А) упаривают при пониженном давлении. Образующийся при этом аморфный продукт желтого цвета разводят в сухом этаноле и подвергают дейодированию в присутствии каталитических количеств 5-10%-ного палладия на угле (01-0,2 % по весу) при давлении водорода 0,1-0,2 МПа и комнатной температуре в течение 2-8 часов. Продолжительность восстановления оценивают хроматографически (система А и С). После отделения катализатора фильтрованием, к продукту добавляют два объема воды, экстрагируют смесь до рН 8-9. Экстракт промывают насыщенным раствором хлористого натрия. После упаривания при пониженном давлении получают белое кристаллическое вещество 4'-деокси-10,11,12,13-тетрагидродесмикозин (I, R - О, R1=R2=H). Другой предмет настоящего изобретения - получаемые присоединением кислые соли 4'-дезокси10,11,12,13-тетрагидродесмикозина, кото-рые получают путем взаимодействия 4'-дезокси-10,11,12,13-тетрадосмикозина в реакционно-инертном растворителе, по крайней мере, с эквимолярным количеством органической или неорганической кислоты, такой как соляная, серная, фосфорная, уксусная, пропионовая, лимонная или винная кислота. Продукт реакции выделяют путем осаждения нерастворимого реагента или, чаще путем лиофилизации. 4'-дезокси-10,11,12,13-тетрагидродесмико-зин и его соли обладают высокой антибактериальной активностью. Результаты ее оценки ин витро представлены в таблицах 1 и 2, в сопоставлении с активностью исходного антибиотика тилозина и его 10,11,12,13- тетрагидропроизводных. Полученные результаты показывают, что 4'-дезокси-10,11,12,13-тетрагидродесмикозин можно применять в качестве антибиотика для лечения многих инфекционных заболеваний в составе обычных лекарственных форм. Изобретение иллюстрируется следующими не ограничивающими примерами. Пример 1. 10,11,12,13-тетрагидродесмикозин-диэтилацеталь (la). 10,11,12,13-тетрагидродесмикозин (50г, 64,4 ммоль) разводили в 500 мл сухого этанола и в полученную смесь добавляли п-толуолсульфокислоту (12,5 г, 65 ммоль). После перемешивания в течение 2 часов при комнатной температуре добавляли 6 мл триэтиламина, этанол упаривали при пониженном давлении до 1/4 первоначального объема, а затем добавляли 700 мл насыщенного раствора гидрокарбоната натрия и экстрагировали хлороформом (дважды по100 мл). Полученные экстракты подсушивали над углекислым калием и упаривали при пониженном давлении до сухого остатка. Получали 50,9 г конечного продукта с выходом 93 %, при Rf (A) 0,30, Rf (В) 0,50. ИК (КВr) см-1 3480, 2970, 1720, 1460, 1380, 1265, 1170, 1085, 1060, 1010, 960. 1 H-ЯМР (CDCI3) мч: 3,61 (ЗН, 3"ОСНз), 3,56 (2Н, 20-OCH2-), 3,50 (ЗН, 2"ОСНз), 3,45 (2Н, 20-ОСН2-), 2,49 (6H, N(CH3)2). 13 С-ЯМР (CDCl3) мч: 215,07 (С-9), 172,52 (С-1), 105,31 (С-1'), 102,31 (С-20), 100,62 (С-1"), 61,80 (20ОСН2-), 61,63 (3"-ОСНз), 60,62 (20-ОСН2-), 59,31 (2"-ОСНз). Пример 2. 3,2,4"-три-O-триметилсилил-10,11,12,13-тетрагидродесмикозин-диэтилацеталь (1b). 10,11,12,13-тетрагидродесмикозин-даэтилацеталь (la) (10 г, 11,7 ммоль) разводили в 200 мл сухого метиленхлорида и 7,8 мл (96,6 ммоль) сухого пиридина. Смесь охлаждали до 0°С, после чего добавляли в нее по каплям 11 мл триметилхлорсилана (87 ммоль). После перемешивания на протяжении 2 часов при 5°С смесь переливали в сосуд, содержавший 400 мл воды со льдом, доводили ее рН до 9 добавлением концентрированного раствора HCl и экстрагировали хлороформом (дважды по 100 мл). Экстракты промывали насыщенным раствором хлористого натрия, подсушивали над углекислым калием и упаривали при пониженном давлении. В результате получали 12,0 г искомого продукта с выходом 96 %. При Rf (А) 0,75. ИК (КВr) см-1 2970, 1730, 1460, 1380, 1265, 1255, 1170, 1100, 1085, 1060, 1010, 970, 885, 842, 775. 1 Н-ЯМР (СDСl3) мч: 3,59 (5Н, 3"ОСНз, 20-ОСН2), 3,51 (5Н, 2"ОСНз, 20-ОСН2), 2,52 (6H, N(СНз)2), 0,17 (27Н, ЗхSi(СНз)з). Пример 3. 4'-метансульфонил-3,2-4"-три-O-триметилсилил-10,11,12,13-тетрагидродесмикозин-диэтилацеталь (Ic). Продукт 1b (12 г, 11,25 ммоль) разбавляли в 100 мл пиридина. Затем в раствор, охлажденный до 10°С, добавляли 5,2 мл (67 ммоль) метансульфохлорида и оставляли при помешивании в условиях охлаждения на 4 часа. Затем реакционную смесь выливали в 1500 мл воды со льдом и добавлением концентрированного раствора NaOH доводили рН до 9. Спустя 30 мин осадок отделяли фильтрованием и быстро разбавляли 100 мл хлороформа. Раствор тщательно отмывали насыщенным раствором хлористого натрия, подсушивали над MgS04 и упаривали до сухого остатка. Получали 12,1 г искомого продукта с выходом 94%, при величине Rf (A) 0,90. ИК (КВr) см-1 2970, 1730, 1460, 1380, 1265, 1255, 1170, 1100, 1085, 1060, 965, 885, 842,755. 1 Н-ЯМР (CDCl3) мч: 3,59 (5Н, 3"ОСНз, 20-ОСН2), 3,51 (5Н, 2"ОСНз, 20-OCH2), 3,15 (ЗН, -SO2--СНз), 2,54 и 2,49 (6Н, N(CH3)2), 0,16 (27Н, ЗхSi(СНз)з). Пример 4. 4'-дезокси-4'-иод-3,2',4"-три-O-триметилсилил-10,11,12,13-тетрагидродесмикозин-диэтилацеталь (1d). Соединение 1с (12 г, 10,5 ммоль) разводили в 120 мл метилэтилкетона, в полученную смесь добавляли 7,8 г йодистого натрия и нагревали в условиях очень мягкой возгонки в течение 2 часов. Растворитель упаривали при пониженном давлении до 1/10 первоначального объема после чего в оставшуюся жидкость добавляли 100 мл хлороформа и 200 мл воды, доводили рН до 9 добавлением щелочи и разделяли образовавшиеся слои. Органический материал отмывали 10 %-ным раствором тиосульфата натрия (дважды по 100 мл). Упариванием при пониженном давлении получали 11,22 г искомого продукта с выходом 90,9 % в виде желтого вещества с Rf (А) 0,95, Rf (C) 0,85. ИК (КВr) см-1 2970, 1725, 1460, 1380, 1265, 1255, 1170, 1100, 1085, 1060, 965, 885, 842, 755. 1 Н-ЯМР (СDСlз) мч: 3,59 (5Н, 3"ОСНз, 20-ОСНз), 3,50 (5Н, 2"ОСНз, 20-ОСН2), 2,54 и 2,49 (6Н, N(СНз)2), 0,16 (27Н, ЗхSi(СНз)з). Пример 5. 4'-дезокси-4'-иод-10,11,12,13-тетрагидроде-смикозин (1е). Соединение 1d (11 г, 9,3 ммоль) разводили в 110 мл смеси ацетонитрила со 110 мл 0,2 N HCI, перемешивая в течение 2 часов при комнатной температуре. После добавления твердого гидрокарбоната натрия для доведения рН до 9 смесь дважды экстрагировали хлороформом по 60 мл. Экстракты промыва ли насыщенным раствором гидрокарбоната натрия и упаривали при пониженном давлении. В результате получали 7,3 г сырого продукта (выход 88,6 %), который разбавляли небольшим количеством метиленхлорида и подвергали очистке путем хроматографирования на колонке с силикагелем 60 фирмы Мерк (70-230 меш) в системе растворителей А. Упариванием фракций с Rf (А) 0,86 получали 3,44 г хроматографически чистого продукта с Rf (С) 0,30 с выходом 55%. ИК (КВr) см-1 3480, 2970, 1720, 1460, 1380, 1260, 1170, 1085, 1060, 960. Н-ЯМР (СDClз) мч: 9,67 (1Н, 20-СНО), 3,61 (ЗН, 3"ОСНз), 2,49 (ЗН, 2"ОСНз), 2,58 и 2,56 (6Н, N(СНз)2). 13 С-ЯМР (СDСlз) мч: 214,68 (С-9), 201,60 (С-20), 172,35 (С-1), 61,63 (3"-ОСНз), 59,43 (2"-ОСНз), 30,70 1 (С-4'). Пример 6. 4'-дезокси-10,11,12,13-тетрагидродесмико-зин (1f). Процедура А. Соединение 1е (1 г, 1,1 ммоль) разводили в 50 мл сухого этанола. После добавления 0,2 г 10 %-ного палладия на угле смесь гидрировали в течение 2 часов при комнатной температуре и давлении водорода 0,2 МПа. По окончании реакции катализатор удаляли фильтрованием, этанол упаривали при пониженном давлении до образования маслянистого продукта, добавляли 100 мл воды и, наконец, экстрагировали хлороформом при рН 8,5. Экстракты подсушивали над углекислым калием и упаривали при пониженном давлении до образования кристаллического продукта белого цвета. Получали 0,73 г искомого продукта с Rf (А) 0,35 и Rf 759 при выходе 85,9 %. ИК (КВr) см-1 3480, 2970, 1720, 1460, 1380, 1260, 1170, 1085, 1060, 960. 1 Н-ЯМР (CDCl3) мч: 9,67 (1Н, 20-СНО), 3,62 (ЗН, 3"ОСНз), 3,50 (ЗН, 2"ОСНз), 2,26 (6Н, N(СН3)2), 1,25 (1Н, 4'), 1,18 (1Н, 4'). 13 С-ЯМР (CDCl3) мч: 214,79 (С-9), 202,99 (С-20), 172,37 (С-1), 61,74 (3"-ОСНз), 59,43 (2"-ОСНз), 28,27 (С-4'). Процедура В. Соединение 1е (1 г, 1,1 ммоль) разбавляли в 50 мл сухого этанола, добавляли в полученный раствор 0,1 г 5 %-ного палладия на угле и подвергали гидрированию в течение 8 часов при комнатной температуре и давлении водорода 0,1 МПа. После полного гидрирования катализатор удаляли фильтрованием, после чего выделение проводили, как описано в разделе Процедура А. Получали продукт с характеристиками, указанными в том же разделе. Таблица 1 Антибактериальная активность ин витро а) Соединение ед/мг Тилозин 983 10,11,12,13-тетрагидротилозин 1138 10,11,12,13-тетрагидродесмикозин 1118 4'-дезокси-10,11,12,13-тетрагидродесмикозин 1358 а) тестирование проводили на Sarcina lutea ATCC 9341. После синтеза новых производных 10,11,12,13-тетрагидродесмикозина проводили хроматографирование в тонком слое силикагеля в следующих системах растворителей: система А: метиленхлорид-метанол-концентрированная NH40H (90:9:1,5), система В: этилацетат-метанол-концентрированная NH40H (85:10:5), система С: бензол-ацетон (4:1). Таблица 2 Минимальные ингибирующие концентрации (МИК, мгг/ мл) Микроорганизмы I II Sarcina lutea ATCC 9341 0,39 0,39 Mycroc. pyog. vr. ST. Aureus ATCC 6538 0,78 0,09 Strep. b. hem. A 0,05 0,05 Strep. b. hem. В 3,12 3,12 Strep. pyog. animales 0,19 0,05 Pasteurella bem. L-314 12,50 6,25 Pasteurella multocida L-315 12,50 3,12 Microc. flavus ATCC 10240 0,78 0,05 I = 10,11,12,13-тетрагидродесмикозин. II = 4'-дезокси-10,11,12,13-тетрагидродесмикозин. Тираж 50 екз. Відкрите акціонерне товариство «Патент» Україна, 88000, м. Ужгород, вул. Гагаріна, 101 (03122) 3 – 72 – 89 (03122) 2 – 57 – 03
ДивитисяДодаткова інформація
Назва патенту англійською10,11,12,13-tetrahydrodesmycosin derivative, intermediates and process for the preparation of 10,11,12,13-tetrahydrodesmycosin derivative
Автори англійськоюNarandja Amalija, Djokic Slobodan
Назва патенту російськоюПроизводные 10,11,12,13-тетрагидродесмикозина, промежуточные соединения и способ получения производного 10,11,12,13-тетрагидродесмикозина
Автори російськоюНаранджя Амалия, Джекич Слободан
МПК / Мітки
МПК: C07F 7/18, A61K 31/7042, C07H 17/08, A61P 31/04, A61K 31/70, A61K 31/7048
Мітки: одержання, сполуки, спосіб, похідного, проміжні, 10,11,12,13-тетрагідродесмікозину, похідне
Код посилання
<a href="https://ua.patents.su/6-34418-pokhidne-10111213-tetragidrodesmikozinu-promizhni-spoluki-ta-sposib-oderzhannya-pokhidnogo-10111213-tetragidrodesmikozinu.html" target="_blank" rel="follow" title="База патентів України">Похідне 10,11,12,13-тетрагідродесмікозину, проміжні сполуки та спосіб одержання похідного 10,11,12,13-тетрагідродесмікозину</a>
Похідне прегна-4,9(11),17(20)-трієн-3-она, спосіб його одержання, спосіб одержання 21-ацилоксипрегна-4, 9(11),16-трієн-3,20-діону та проміжні сполуки

Номер патенту: 29395
Опубліковано: 15.11.2000
Автори: Бріон Франсі, Бюендіа Жан, Віва Мішель, Діолез Крістіан
МПК: C07J 7/00, C07J 33/00, C07J 75/00, C07J 21/00, C07J 13/00
Мітки: похідне, сполуки, прегна-4,9(11),17(20)-трієн-3-она, 21-ацилоксипрегна-4, 9(11),16-трієн-3,20-діону, спосіб, проміжні, одержання
Текст:
...2 или 3, его индивидуальных изомеров или их смесей, отли чающийся тем, что соединение общей формулы (II) 01) обрабатывают уксусным ангидридом в присутствии сильной кислоты, а затем подвергают кислотному гидролизу, с целью получить соединение общей формулы (III): m О где Hal - а том хлора или брома, R - (Сі-Сє)алкил, (С7-Сі5)аралкил, силил, К - гр уппировка, защищающая 3-оксогр уппу, фор м улы -°>сн2)п, -°>снл. rScHA. где п...
Спосіб одержання похідних хінолінкарбонової кислоти або її фармацевтично придатних солей та проміжні сполуки для їх одержання
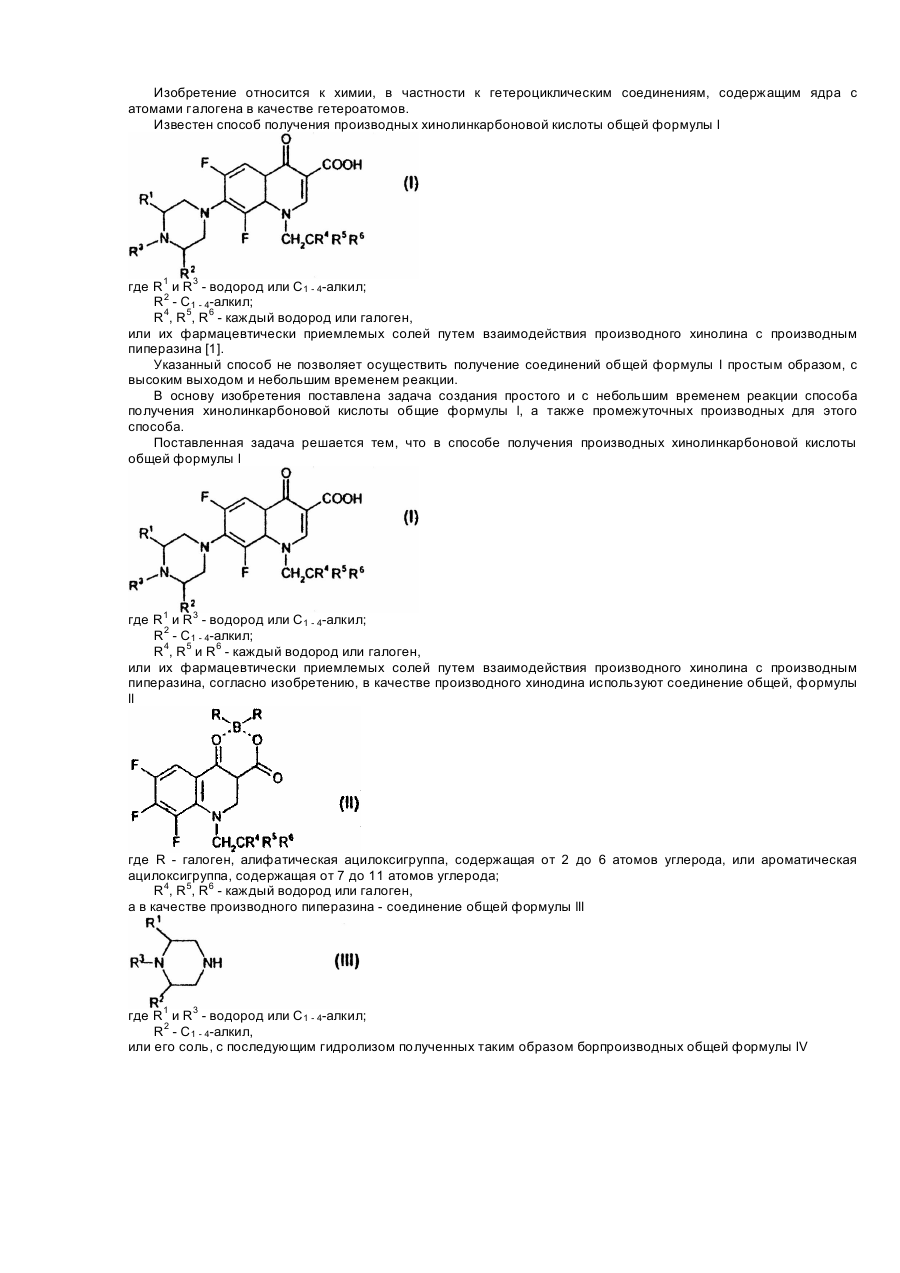
Номер патенту: 26568
Опубліковано: 11.10.1999
Автори: Вашварі Лелле, Керестурі Геза, Шіпош Юдіт, Хорват Агнеш, Пайор Аніко, Рітлі Петер, Хермец Іштван, Балог Марія
МПК: C07D 401/04, C07D 215/56, A61K 31/495, C07F 5/00, A61P 31/04
Мітки: сполуки, похідних, спосіб, солей, придатних, проміжні, кислоти, хінолінкарбонової, фармацевтично, одержання
Формула / Реферат:
1. Способ получения производных хинолинкарбоновой кислоты общей формулы lгде R1 и R3 - водород или C1 - 4-алкил;R2 - C1 - 4-алкил;R4, R5 и R6 - каждый - водород или галоген,или ее фармацевтически приемлемых солей, взаимодействием производного хинолина с производным пиперазина, отличающийся тем, что в качестве производного хинолина используют соединение общей формулы llгде R - галоген,...
Спосіб одержання гетероциклічних сполук та проміжні сполуки для їх одержання

Номер патенту: 26678
Опубліковано: 12.11.1999
Автори: ЛЕННБЕРГ Карі Калєві, Пюстюнен Ярмо Йохан, Норе Пентті Тапіо, Луіро Анне Марія, Хонканен Ерккі Юхані, Хайкала Хеймо Олаві, ПІППУРІ Айно Кюллікі
МПК: A61K 31/54, C07D 237/32, C07D 401/12, C07D 253/00, A61K 31/53, C07D 403/12, A61K 31/50, C07D 237/14, C07D 285/16, A61P 9/04, A61P 9/12, A61P 9/08, C07D 273/00, A61K 31/502, C07D 413/12, C07D 237/04, C07D 417/12, A61K 31/535
Мітки: одержання, сполук, проміжні, спосіб, сполуки, гетероциклічних
Формула / Реферат:
1. Способ получения гетероциклических соединений формулы (I)в которой Het представляет одну из следующих групп:где R11, R13 и R14 представляют независимо водород или низшую алкильную группу;Z представляет собой S, О или NH;A представляет валентную связь, -CH-CH- или CH2-CH2-группу;R1 и R2 независимо представляют нитро, циано, галогеновую, амино, карбоксамидо, фенильную, бензоильную,...
Заміщені фенілімідазолідини, спосіб їх одержання, фармацевтична композиція, проміжні сполуки

Номер патенту: 34428
Опубліковано: 15.03.2001
Автори: ТЕТШ Жан-Жорж, Гейар-Келлі Мартін, ФІЛІБЕР Даніель, ГУБЕ Франсуа
МПК: A61P 15/00, A61P 43/00, A61P 13/02, C07D 233/84, A61K 31/4166, C07D 233/86, A61P 35/00, C07D 233/80, A61K 31/415, C07D 233/88, C07D 233/74, C07D 233/76
Мітки: одержання, сполуки, заміщені, проміжні, фенілімідазолідини, фармацевтична, спосіб, композиція
Текст:
...(Г) в которой R"i, R^, - А"—В"— имеют значения, указанные выше для Ri, R2 и —А—В—, с учетом того, что когда —А"—В"— представляет собой группу —СО—N(RMt3)—. в которой R"'3 представляет собой атом водорода или линейный или разветвленный алкильнмй радикал, включающий не более 7 атомов yf лерода, Y является атомом кислорода, R"i представляет собой цианильный радикал, причем данный метод отличается тем, что продукт формулы (V). Hal 34428 в...
Похідні хроману, активні стосовно 5-нт рецептора, спосіб їх одержання та проміжні сполуки

Номер патенту: 34416
Опубліковано: 15.03.2001
Автори: Росс Сванте Бертіл, Рені Люсі Анна, Норен Рольф, Сонн Даніель Дунган, Свенссон Бьорн Ерік, Ларссон Ларс-Гюннар, ТОРБЕРГ Сет-Олов
МПК: C07D 409/12, A61K 31/38, A61K 31/382, C07D 407/04, A61K 31/352, A61P 25/20, A61K 31/40, C07D 409/04, C07D 417/12, C07D 413/06, C07D 311/58, A61K 31/4025, C07D 335/00, C07D 405/12, A61P 25/24, A61P 25/00, A61K 31/35, C07D 409/06, C07D 413/04, C07D 405/04, A61P 25/26, A61P 25/28
Мітки: рецептора, спосіб, проміжні, 5-нт, похідні, активні, одержання, стосовно, хроману, сполуки
Текст:
...(29 мг). Получающаяся смесь снова подвергалась действию атмосферы СО и нагревалась до 80°С при перемешивании в течение 4 часов. Раствор охлаждался, выпаривался в вакууме (вакуумный насос), затем разбавлялся эфиром. Смесь промывалась 2 М раствором аммиака (х2), обрабатывалась насыщенным солевым раствором, сушилась (сульфатом натрия) и выпаривалась в вакууме, давая сырое вещество. Хроматография на двуокиси кремния (элюент: 30%...
Попередній патент: Спосіб водопідготовки
Наступний патент: Спосіб визначення лігніну у розчинах хімічної переробки целюлозовмісних матеріалів
Випадковий патент: Заміщені піперидини, які підвищують активність p53, і їх застосування