Хіназолінові похідні для лікування вірусних інфекцій та подальших захворювань
Номер патенту: 114476
Опубліковано: 26.06.2017
Автори: Рабуассон П'єр Жан-Марі Бернар, Ласт Стефаан Жюльєн, Йонкерс Тім Хьюго Марія, Пітерс Серж Марія Алоїзіус, Макгован Девід, Ембрехтс Вернер

























































Формула / Реферат
1. Сполука формули (I)
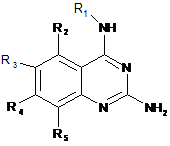 (I)
(I)
або її фармацевтично прийнятна сіль або сольват, де
R1 являє собою С4-8алкіл, заміщений гідроксилом,
R2 являє собою водень, галоген, гідроксил, амін, С1-7алкіл, С1-7алкіламіно, С1-6алкокси, (С1-4)алкоксі-(С1-4)алкіл, С3-6циклоалкіл, С4-7гетероцикл, ароматичний, біциклічний гетероцикл, арилалкіл, гетероарил, гетероарилалкіл, амід карбонової кислоти, естер карбонової кислоти, кожний з яких необов'язково заміщений одним або декількома замісниками, незалежно вибраними з галогену, гідроксилу, аміно, С1-6алкілу, ді-(С1-6)алкіламіно, С1-6алкіламіно, С1-6алкілу, С1-6алкокси, С3-6циклоалкілу, карбонової кислоти, естеру карбонової кислоти, аміду карбонової кислоти, гетероциклу, арилу, алкенілу, алкінілу, арилалкілу, гетероарилу, гетероарилалкілу або нітрилу,
R3 являє собою водень, галоген, гідроксил, амін, С1-7алкіл, С1-7алкеніл, С1-7алкініл, С1-7алкіламіно, С1-6алкокси, (С1-4)алкоксі-(С1-4)алкіл, С3-6циклоалкіл, С4-7гетероцикл, ароматичний, біциклічний гетероцикл, арилалкіл, гетероарил, гетероарилалкіл, арилокси, гетероарилокси, кетон, нітрил, кожний з яких необов'язково заміщений одним або декількома замісниками, незалежно вибраними з галогену, гідроксилу, аміно, С1-6алкілу, ді-(С1-6)алкіламіно, С1-6алкіламіно, С1-6алкілу, С1-6алкокси, С3-6циклоалкілу, карбонової кислоти, естеру карбонової кислоти, аміду карбонової кислоти, гетероциклу, арилу, алкенілу, алкінілу, арилалкілу, гетероарилу, гетероарилалкілу або нітрилу,
R4 являє собою водень, галоген, гідроксил, амін, С1-7алкіл, С1-7алкіламіно, С1-6алкокси, (С1-4)алкоксі-(С1-4)алкіл, С3-6циклоалкіл, С4-7гетероцикл, біциклічний гетероцикл, арилалкіл, гетероарилалкіл, арилокси, гетероарилокси, кожний з яких необов'язково заміщений одним або декількома замісниками, незалежно вибраними з галогену, гідроксилу, аміно, С1-6алкілу, ді-(С1-6)алкіламіно, С1-6алкіламіно, С1-6алкілу, С1-6алкокси, С3-6циклоалкілу, карбонової кислоти, естеру карбонової кислоти, аміду карбонової кислоти, гетероциклу, арилу, алкенілу, алкінілу, арилалкілу, гетероарилу, гетероарилалкілу або нітрилу, та
R5 являє собою водень, фтор, хлор або метил, за умови, що
R2, R3, R4 та R5 не можуть являти собою Н.
2. Сполука формули (І) за п. 1, де R1 являє собою одне з наступного:
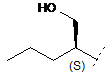 ,
,
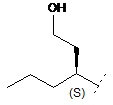 ,
,
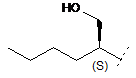 ,
,
 .
.
3. Сполука формули (І) за п. 1 або 2, де R5 являє собою водень або фтор.
4. Сполука формули (І) за п. 1-3, що має наступну структуру:
 .
.
5. Фармацевтична композиція, що містить сполуку формули (І) або її фармацевтично прийнятну сіль або сольват за будь-яким з пп. 1-4 разом із одним або декількома фармацевтично прийнятними наповнювачами, розріджувачами або носіями.
6. Сполука формули (І) або її фармацевтично прийнятна сіль або сольват за будь-яким з пп. 1-4 або фармацевтична композиція за п. 5 для застосування як лікарського препарату.
7. Сполука формули (І) або її фармацевтично прийнятна сіль або сольват за будь-яким з пп. 1-4 або фармацевтична композиція за п. 5 для застосування при лікуванні розладу, при якому передбачається модуляція TLR7 та/або TLR8.
Текст
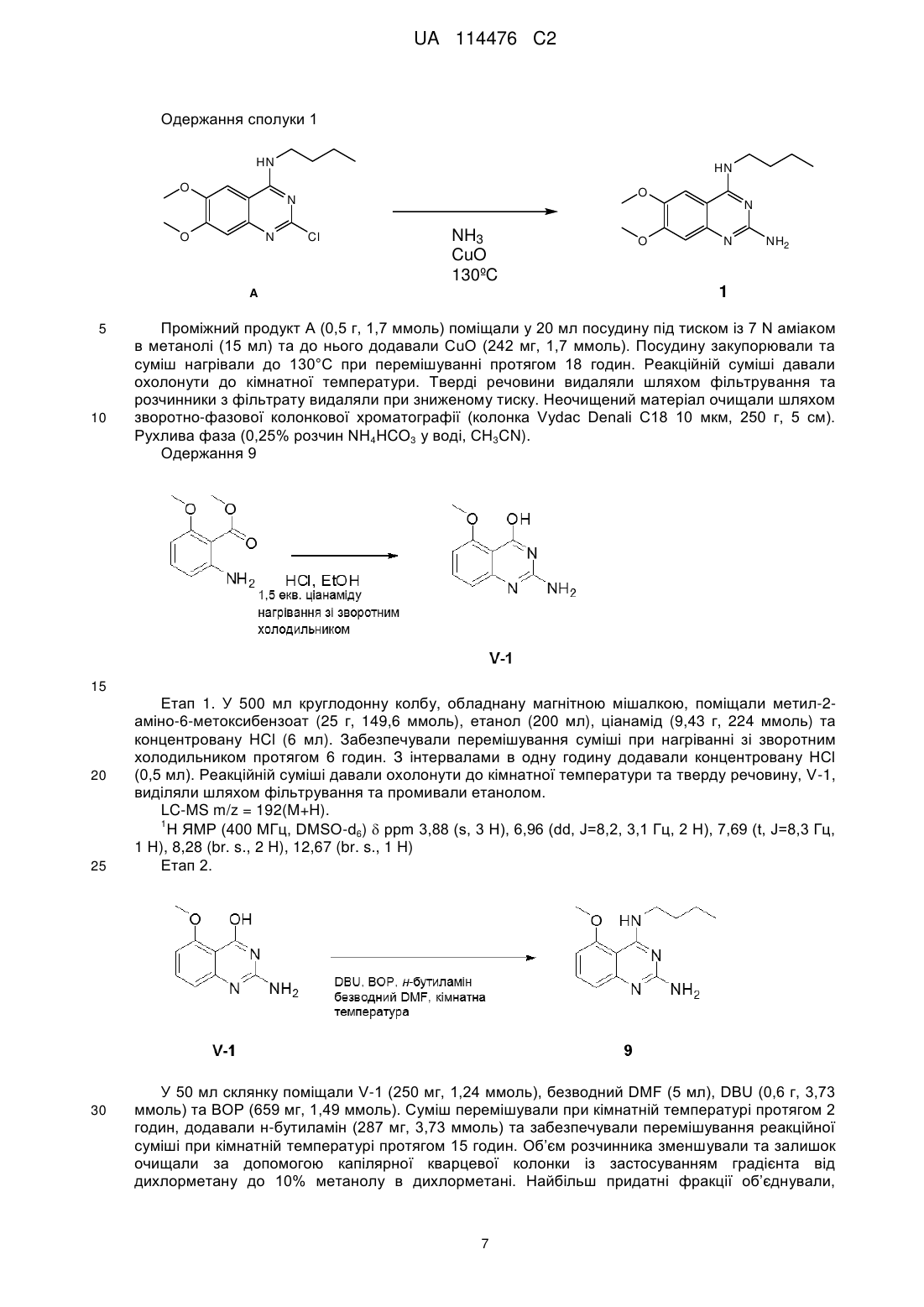
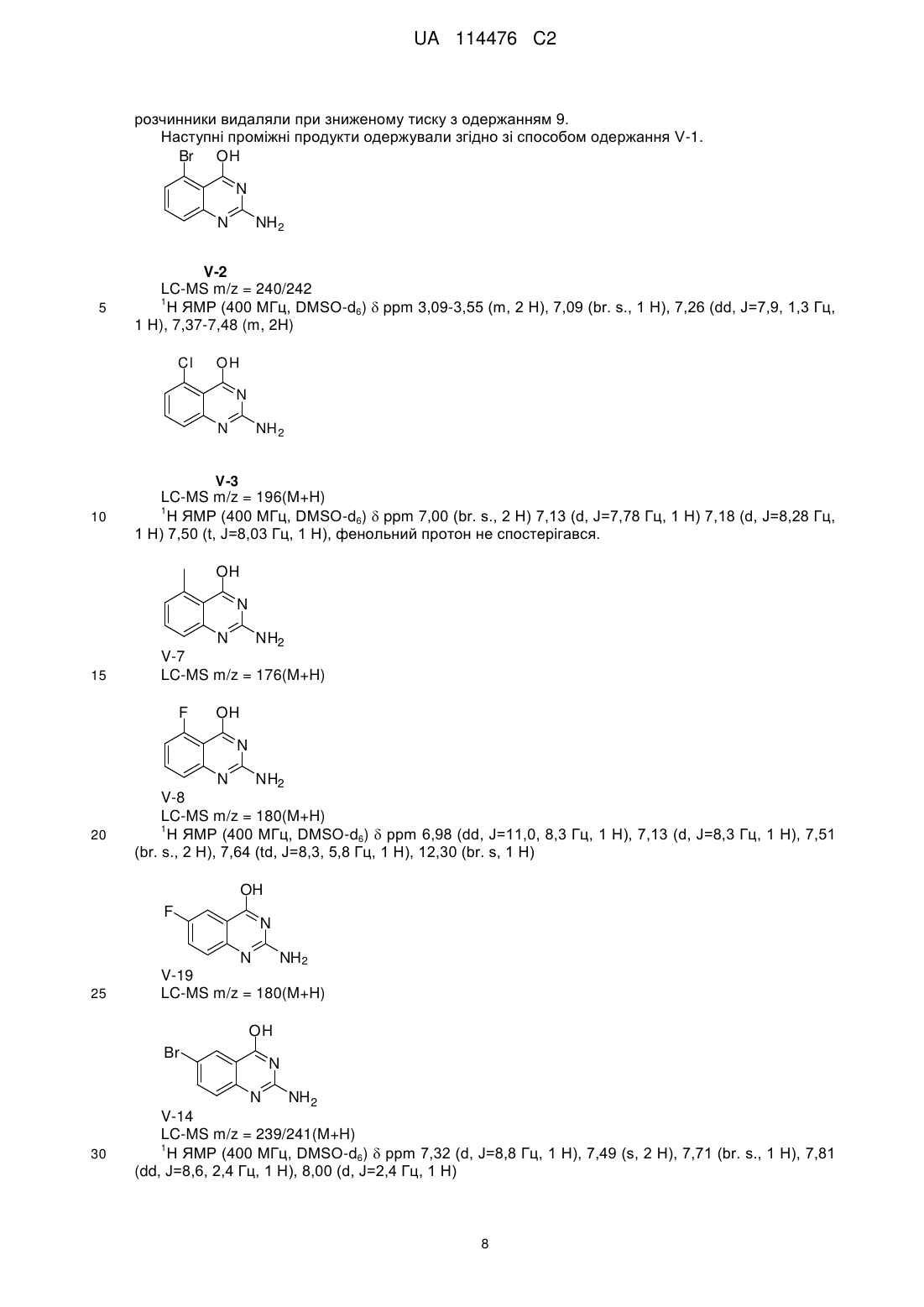
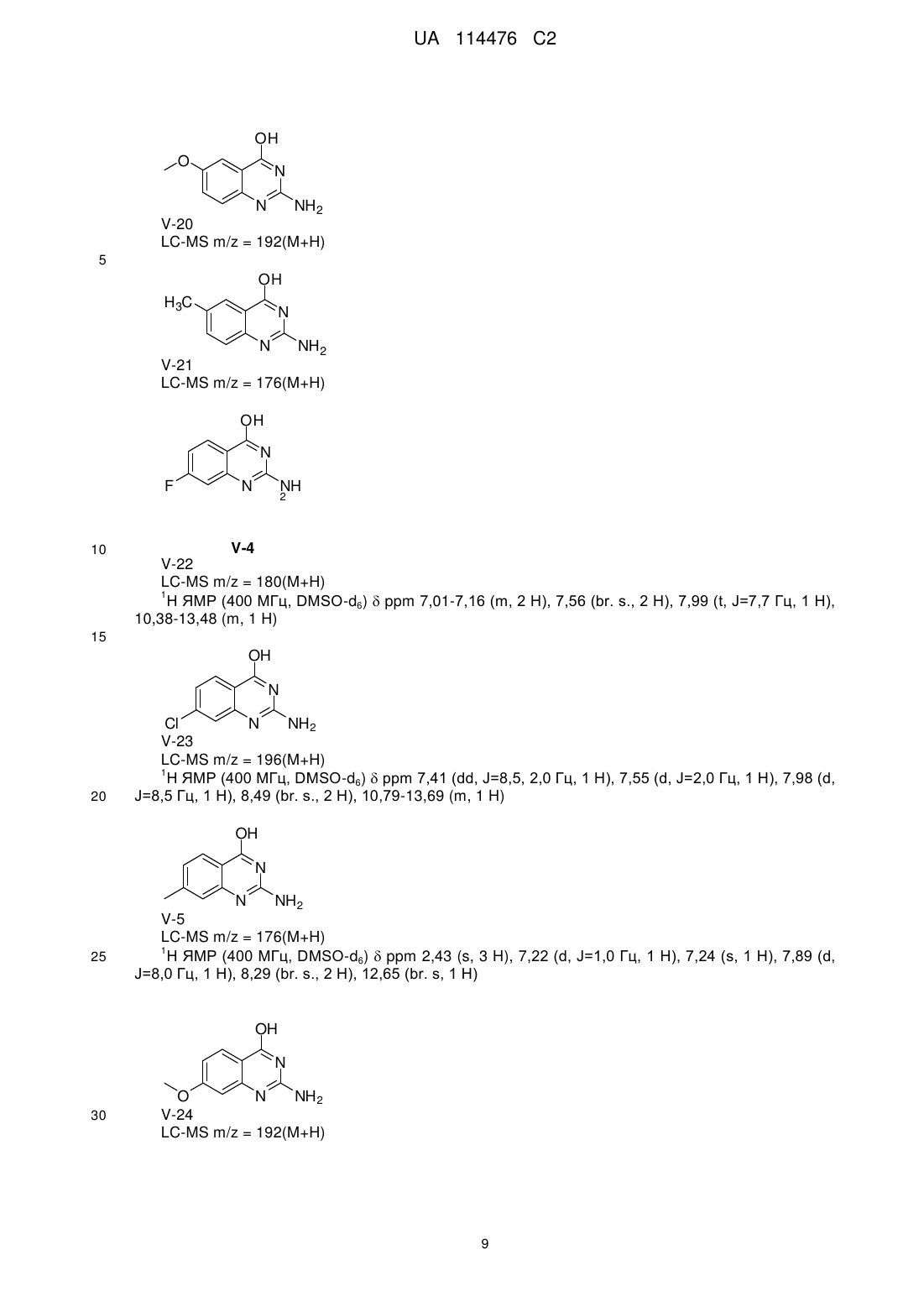
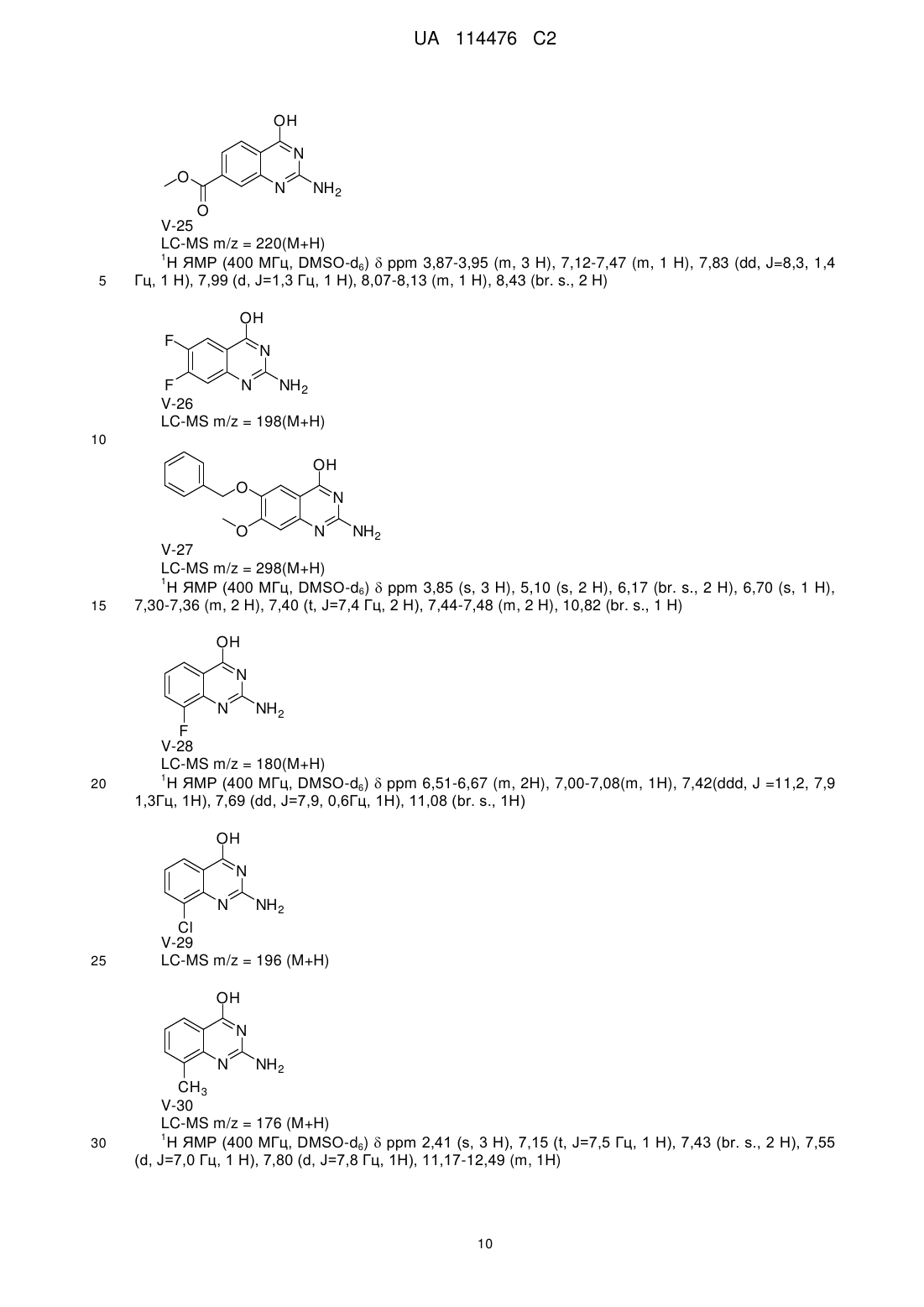
Реферат: Даний винахід стосується хіназолінових похідних, способів їх одержання, фармацевтичних композицій та їх застосування у лікуванні розладів, при яких передбачається модуляція толподібних рецепторів. R1 R2 NH R3 N R4 N R5 NH2 UA 114476 C2 (12) UA 114476 C2 UA 114476 C2 5 10 15 20 25 30 35 40 45 Даний винахід відноситься до хіназолінових похідних, способів їх одержання, фармацевтичних композицій та їх застосування при лікуванні. Дійсний винахід відноситься до застосування хіназолінових похідних при лікуванні вірусних інфекцій, імунологічних або запальних розладів, при яких передбачається модуляція або агонізм толл-подібних рецепторів (TLR). Толл-подібні рецептори являють собою основні трансмембранні білки, що характеризуються позаклітинним багатим лейцином доменом та цитоплазматичним «розширенням», яке містить консервативну ділянку. Вроджена імунна система може розпізнавати патоген-асоційовані молекулярні патерни через дані TLR, що експресуються на поверхні клітини певних типів імунних клітин. Розпізнавання чужорідних патогенів активує продукування цитокінів та апрегуляцію ко-стимулюючих молекул на фагоцитах. Це веде до модуляції поведінки T-клітин. Було встановлено, що більшість видів ссавців мають від десяти до п’ятнадцяти типів толлподібних рецепторів. Тринадцять TLR (які називаються просто TLR1-TLR13) було ідентифіковано у людей та мишей разом, та еквівалентні форми багатьох із них було виявлено в інших видів ссавців. Однак еквіваленти певних TLR, виявлених у людей, не присутні у всіх ссавців. Наприклад, ген, що кодує білок, аналогічний TLR10 у людини, присутній у мишей, але, як виявляється, він був деякою мірою пошкоджений ретровірусом в минулому. З іншого боку, у мишей експресуються TLR 11, 12 та 13, жоден з яких не представлений у людини. В інших ссавців можуть експресуватися TLR, котрі не виявлені у людини. Інші види, що не є ссавцями, можуть мати TLR, що відрізняються від таких у ссавців, як демонструється TLR14, який виявлений у риби фугу. Це може ускладнювати процес застосування експериментальних тварин у якості моделей вродженого імунітету людини. Для детального огляду толл-подібних рецепторів див. наступні журнальні статті. Hoffmann, J.A., Nature, 426, p33-38, 2003; Akira, S., Takeda, K., and Kaisho, T., Annual Rev. Immunology, 21, p335-376, 2003; Ulevitch, R. J., Nature Reviews: Immunology, 4, p512-520, 2004. Сполуки, що проявляють активність щодо толл-подібних рецепторів, було описано раніше, як наприклад, пуринові похідні в документі WO 2006 117670, аденінові похідні в документах WO 98/01448 та WO 99/28321 та піримідини в документі WO 2009/067081. Однак існує значна потреба у нових модуляторах толл-подібних рецепторів, що характеризуються переважною селективністю, більш високою активністю, більш високою метаболічною стабільністю та покращеним профілем безпеки порівняно зі сполуками з рівня техніки. При лікуванні певних вірусних інфекцій можуть призначати регулярні ін’єкції інтерферону (IFN ), як у випадку із вірусом гепатиту C (HCV), (Fried et. al. Peginterferon-alfa plus ribavirin for chronic hepatitis C virus infection, N Engl J Med 2002; 347: 975-82). Доступні для перорального застосування низькомолекулярні індуктори IFN надають можливі переваги у вигляді зниженої імуногенності та зручності введення. Отже, нові індуктори IFN являють собою потенційно ефективний новий клас лікарських засобів для лікування вірусних інфекцій. Для прикладу у літературі стосовно низькомолекулярного індуктора IFN, що має противірусний ефект, див. De Clercq, E.; Descamps, J.; De Somer, P. Science 1978, 200, 563-565. акож призначають у комбінації з іншими лікарськими засобами при лікуванні певних типів раку (для прикладів див. Eur. J. Cancer 46, 2849–57, та Cancer Res. 1992, 52, 1056). Агоністи TLR 7/8 також становлять інтерес в якості вакцинних ад’ювантів внаслідок їх здатності індукувати явно виражену Th1 реакцію. Згідно з даним винаходом забезпечується сполука формули (I), R1 R2 NH R3 N R4 N R5 50 NH2 (I), або її фармацевтично прийнятна сіль, сольват або поліморф, де R1 являє собою C3-8алкіл, C3-8алкоксі, C2-6алкеніл або C2-6алкініл, кожний з яких необов’язково заміщений одним або декількома замісниками, незалежно вибраними з галогену, гідроксилу, аміно, нітрилу, естеру, аміду, C1-3алкілу, C1-3алкоксі або C3-6циклоалкілу, R2 являє собою водень, галоген, гідроксил, амін, C1-7алкіл, C1-7алкіламіно, C1-6алкоксі, (C1 1 UA 114476 C2 4)алкоксі-(C1-4)алкіл, 5 10 15 20 25 30 C3-6циклоалкіл, C4-7гетероцикл, ароматичний, біциклічний гетероцикл, арилалкіл, гетероарил, гетероарилалкіл, амід карбонової кислоти, естер карбонової кислоти, кожний з яких необов’язково заміщений одним або декількома замісниками, незалежно вибраними з галогену, гідроксилу, аміно, C1-6алкілу, ди-(C1-6)алкіламіно, C1-6алкіламіно, C16алкілу, C1-6алкоксі, C3-6циклоалкілу, карбонової кислоти, естеру карбонової кислоти, аміду карбонової кислоти, гетероциклу, арилу, алкенілу, алкінілу, арилалкілу, гетероарилу, гетероарилалкілу або нітрилу, R3 являє собою водень, галоген, гідроксил, амін, C1-7алкіл, C1-7алкеніл, C1-7алкініл, C17алкіламіно, C1-6алкоксі, (C1-4)алкоксі-(C1-4)алкіл, C3-6циклоалкіл, C4-7гетероцикл, ароматичний, біциклічний гетероцикл, арилалкіл, гетероарил, гетероарилалкіл, арилоксі, гетероарилоксі, кетон, нітрил, кожний з яких необов’язково заміщений одним або декількома замісниками, незалежно вибраними з галогену, гідроксилу, аміно, C 1-6алкілу, ди-(C1-6)алкіламіно, C16алкіламіно, C1-6алкілу, C1-6алкоксі, C3-6циклоалкілу, карбонової кислоти, естеру карбонової кислоти, аміду карбонової кислоти, гетероциклу, арилу, алкенілу, алкінілу, арилалкілу, гетероарилу, гетероарилалкілу або нітрилу. R4 являє собою водень, галоген, гідроксил, амін, C1-7алкіл, C1-7алкіламіно, C1-6алкоксі, (C1C3-6циклоалкіл, C4-7гетероцикл, біциклічний гетероцикл, арилалкіл, 4)алкоксі-(C1-4)алкіл, гетероарилалкіл, арилоксі, гетероарилоксі, кожний з яких необов’язково заміщений одним або декількома замісниками, незалежно вибраними з галогену, гідроксилу, аміно, C 1-6алкілу, ди-(C16)алкіламіно, C1-6алкіламіно, C1-6алкілу, C1-6алкоксі, C3-6циклоалкілу, карбонової кислоти, естеру карбонової кислоти, аміду карбонової кислоти, гетероциклу, арилу, алкенілу, алкінілу, арилалкілу, гетероарилу, гетероарилалкілу або нітрилу, та R5 являє собою водень, фтор, хлор або метил за умови, що усі з R 2, R3, R4 та R5 не можуть являти собою H. У першому варіанті здійснення дійсний винахід передбачає сполуки формули (I), де R1 являє собою бутил, пентил або 2-пентил, та де R2, R3, R4 та R5 є такими, як визначено вище. У наступному варіанті здійснення дійсний винахід відноситься до сполук формули (I), де R 1 являє собою C4-8алкіл, заміщений гідроксилом, та де R2, R3, R4 та R5 є такими, як визначено вище. Інший варіант здійснення відноситься до сполук формули (I), де R 1, якщо він являє собою C4-8алкіл, заміщений гідроксилом, являє собою одне з наступного: OH HO (S) (S) OH HO (S) 35 40 45 50 (S) . В іншому варіанті здійснення дійсний винахід передбачає сполуки формули (I), де R5 являє собою переважно водень або фтор, та R1, R2, R3 та R4 є такими, як описано вище. Сполуки формули (I) та їх фармацевтично прийнятна сіль, сольват або поліморф мають активність як фармацевтичні препарати, зокрема, як модулятори активності толл-подібного рецептора (особливо TLR7 та/або TLR8). Отже, у наступному аспекті дійсний винахід передбачає фармацевтичну композицію, що містить сполуку формули (I) або її фармацевтично прийнятну сіль, сольват або поліморф разом із одним або декількома фармацевтично прийнятними наповнювачами, розріджувачами або носіями. Окрім того, сполуку формули (I) або її фармацевтично прийнятну сіль, сольват або поліморф згідно з дійсним винаходом або фармацевтичну композицію, що включає зазначену сполуку формули (I) або її фармацевтично прийнятну сіль, сольват або поліморф, можна застосовувати в якості лікарського препарату. Інший аспект дійсного винаходу полягає в тому, що сполуку формули (I) або її фармацевтично прийнятну сіль, сольват або поліморф або зазначену фармацевтичну композицію, що включає зазначену сполуку формули (I) або її фармацевтично прийнятну сіль, 2 UA 114476 C2 5 10 15 20 25 30 35 40 45 50 55 60 сольват або поліморф, можна відповідним чином застосовувати у лікуванні розладу, при якому передбачається модуляція TLR7 та/або TLR8. Термін “алкіл” відноситься до насиченого аліфатичного вуглеводню із нерозгалуженим ланцюгом або розгалуженим ланцюгом, що містить певну кількість атомів вуглецю. Термін “галоген” відноситься до фтору, хлору, брому або йоду. Термін “алкеніл” відноситься до алкілу, що визначений вище та включає щонайменше два атоми вуглецю та щонайменше один подвійний зв’язок вуглець-вуглець. Термін “алкініл” відноситься до алкілу, що визначений вище та включає щонайменше два атоми вуглецю та щонайменше один потрійний зв’язок вуглець-вуглець. Термін “циклоалкіл” відноситься до карбоциклічного кільця, що містить певну кількість атомів вуглецю. Термін “алкоксі” відноситься до алкільної групи (ланцюга з вуглецю та водню), зв’язаної одинарним зв’язком із киснем, як наприклад, метоксигрупа або етоксигрупа. Термін “арил” означає ароматичну кільцеву структуру, що необов’язково включає один або два гетероатоми, вибрані з N, O та S, зокрема, з N та O. Зазначена ароматична кільцева структура може включати 5, 6 або 7 атомів кільця. Зокрема, зазначена ароматична кільцева структура може включати 5 або 6 атомів кільця. Термін “арилоксі” відноситься до ароматичної кільцевої структури. Зазначена ароматична група зв’язана одинарним зв’язком із киснем, як наприклад, фенол. Термін “гетероарилоксі” відноситься до ароматичної кільцевої структури, що необов’язково включає один або два гетероатоми, вибрані з N, O та S. Зазначена ароматична група включає 5-7 атомів кільця, один з яких зв’язаний одинарним зв’язком із киснем, як наприклад, гідроксипіридин. Термін “біциклічний гетероцикл” означає ароматичну кільцеву структуру, що визначена для терміна “арил” та включає два конденсованих ароматичних кільця. Кожне кільце необов’язково включає гетероатоми, вибрані з N, O та S, зокрема, з N та O. Термін “арилалкіл” означає ароматичну кільцеву структуру, що визначена для терміна “арил” та необов’язково заміщена алкільною групою. Термін “гетероарилалкіл” означає ароматичну кільцеву структуру, що визначена для терміна “гетероарил” та необов’язково заміщена алкільною групою. Гетероцикл відноситься до молекул, що є насиченими або частково насиченими та включають етилоксид, тетрагідрофуран, діоксан або інші циклічні етери. Гетероцикли, що містять азот, включають, наприклад, азетидин, морфолін, піперидин, піперазин, піролідин та подібне. Інші гетероцикли включають, наприклад, тіоморфолін, діоксолініл та циклічні сульфони. Гетероарильні групи являють собою гетероциклічні групи, що за своєю природою є ароматичними. Вони є моноциклічними, біциклічними або поліциклічними та містять один або декілька гетероатомів, вибраних із N, O або S. Гетероарильні групи можуть являти собою, наприклад, імідазоліл, ізоксазоліл, фурил, оксазоліл, піроліл, піридоніл, піридил, піридазиніл, піразиніл. Фармацевтично прийнятні солі сполук формули (I) включають їх кислотно-адитивні та основні солі. Придатні кислотно-адитивні солі утворюють із кислот, що утворюють нетоксичні солі. Придатні основні солі утворюють із основ, що утворюють нетоксичні солі. Сполуки за дійсним винаходом можуть також існувати у несольватованій та сольватованій формі. Термін “сольват” використовують у даному документі для опису молекулярного комплексу, що включає сполуку за дійсним винаходом та одну або декілька молекул фармацевтично прийнятного розчинника, наприклад, етанолу. Термін “поліморф” відноситься до здатності сполуки за дійсним винаходом існувати у більш ніж одній формі або кристалічній структурі. Сполуки за дійсним винаходом можна вводити у вигляді кристалічних або аморфних продуктів. Їх можна одержати, наприклад, у вигляді твердих циліндрів, порошків або плівок за допомого способів, таких як осадження, кристалізация, ліофілізація, розпилювальне сушіння або сушіння випарюванням. Їх можна вводити окремо або в комбінації з одним або декількома іншими сполуками за дійсним винаходом або в комбінації з одним або декількома іншими лікарськими засобами. В основному, їх будуть вводити у вигляді складу у поєднанні з одним або декількома фармацевтично прийнятними наповнювачами. Термін “наповнювач” використовується у даному документі для опису будь-якого інгредієнта, що відрізняється від сполуки(сполук) за дійсним винаходом. Вибір наповнювача значною мірою залежить від факторів, таких як конкретний спосіб введення, вплив наповнювача на розчинність та стабільність та природа лікарської форми. Сполуки за дійсним винаходом або будь-яку їх підгрупу можна скласти у різноманітні 3 UA 114476 C2 5 10 15 20 25 30 35 40 45 50 55 60 фармацевтичні форми з метою введення. В якості придатних композицій можна привести усі композиції, що звичайно використовують для системного введення лікарських засобів. Для одержання фармацевтичних композицій за даним винаходом ефективну кількість конкретної сполуки, необов’язково у формі адитивної солі, в якості активного інгредієнта об’єднують в однорідній суміші з фармацевтично прийнятним носієм, причому носій може приймати різноманітні форми залежно від форми препарату, необхідної для введення. Бажано, щоб дані фармацевтичні композиції знаходились в одиничній лікарській формі, придатній, наприклад, для перорального, ректального або черезшкірного введення. Наприклад, при одержанні композицій у пероральній лікарській формі можна використовувати будь-яке із загальноприйнятих фармацевтичних середовищ, таких як, наприклад, вода, гліколі, масла, спирти та подібне у випадку пероральних рідких препаратів, таких як суспензії, сиропи, еліксири, емульсії та розчини; або твердих носіїв, таких як крохмалі, цукри, каолін, розріджувачі, змащувальні засоби, сполучні засоби, засоби для поліпшення розпадання таблеток та подібне у випадку порошків, пілюль, капсул та таблеток. Внаслідок простоти введення таблетки та капсули являють собою найбільш переважні пероральні стандартні лікарські форми, при цьому безумовно використовуються тверді фармацевтичні носії. Також включено препарати у твердій формі, які безпосередньо перед застосуванням можна перетворити у рідкі форми. У композиціях, придатних для черезшкірного введення, носій необов’язково включає засіб для посилення проникнення та/або придатний змочувальний засіб, необов’язково поєднаний із придатними добавками будь-якої природи у незначних співвідношеннях, причому добавки не завдають значного шкідливого впливу шкірі. Зазначені добавки можуть полегшувати нанесення на шкіру та/або можуть бути корисними для одержання необхідних композицій. Дані композиції можна наносити різноманітними способами, наприклад, такими як у вигляді трансдермального пластиру, у вигляді засобів для точкового нанесення, у вигляді мазі. Сполуки за дійсним винаходом можна також вводити шляхом інгаляції або інсуфляції за допомогою способів та складів, що використовуються в даній галузі для введення таким шляхом. Отже, в основному сполуки за дійсним винаходом можна вводити у легені у формі розчину, суспензії або сухого порошку. Особливо переважним є складання вищезазначених фармацевтичних композицій в одиничній лікарській формі для простоти введення та рівномірності дозування. Одинична лікарська форма, як використовується у даному документі, відноситься до фізично відокремлених одиниць, придатних у якості одиничних доз, причому кожна одиниця містить попередньо встановлену кількість активного інгредієнта, розраховану для одержання бажаного терапевтичного ефекту, у поєднанні з необхідним фармацевтичним носієм. Прикладами таких одиничних лікарських форм є таблетки (у тому числі таблетки із рискою або таблетки із покриттям), капсули, пілюлі, пакети з порошкоподібним продуктом, облатки, супозиторії, ін’єкційні розчини або суспензії та подібне та їх розділені багатократні кількості. Спеціаліст в галузі лікування інфекційних захворювань зможе визначити ефективну кількість на основі результатів тестів, представлених у даному документі далі. В основному передбачається, що ефективна добова кількість буде становити від 0,01 мг/кг до 50 мг/кг маси тіла, більш переважно від 0,1 мг/кг до 10 мг/кг маси тіла. Може бути доцільним введення необхідної дози у вигляді двох, трьох, чотирьох або більше частин дози із придатними інтервалами протягом доби. Зазначені частини дози можна скласти у вигляді одиничних лікарських форм, наприклад, таких, що містять від 1 до 1000 мг, та, зокрема, від 5 до 200 мг активного інгредієнта на одиничну лікарську форму. Точне дозування та частота введення залежать від застосовуваної конкретної сполуки формули (I), конкретного стану, що підлягає лікуванню, тяжкості стану, що підлягає лікуванню, віку, маси та загального фізичного стану конкретного пацієнта, а також іншої лікарської терапії, яку може проходити особа, як це добре відомо спеціалістам в даній галузі. Окрім того, очевидно, що ефективну кількість можна знизити або підвищити залежно від реакції суб’єкта, що проходить лікування, та/або залежно від оцінки лікаря, що призначає сполуки за дійсним винаходом. Вищезгадані діапазони ефективної кількості є лише рекомендаціями та не призначені для обмеження в тій або іншій мірі обсяг або застосування дійсного винаходу. Одержання сполук Сполуки формули (I) одержують згідно зі схемою 1. Можна забезпечити реакцію 2,4дихлорхіназолінів у декілька окремих етапів з одержанням 2,4-діамінохіназолінів із прийнятним виходом. На першому етапі 2,4-дихлорхіназолін змішують або нагрівають з аміном із каталізатором-перехідним металом або без нього з одержанням 2-хлор-4-амінохіназоліну. Після обробки неочищеного 2-хлор-4-амінохіназоліну проміжний продукт нагрівають у посудині під тиском із джерелом аміаку (наприклад, аміак в метанолі) та необов’язково з CuO. 4 UA 114476 C2 Схема 1 5 10 15 20 Сполуки формули (I) можна також одержувати згідно зі схемою 2. Заміщені естери антранілової кислоти (IV) нагрівали в кислому середовищі в присутності надлишкового ціанаміду із застосуванням спиртового розчинника (наприклад, етанолу) або дигліму згідно зі способом, описаним у літературі (O'Hara et. al. JOC (1991) 56, p776). Наступне заміщення 2аміно-4-гідроксихіназолінів (V) за аміногрупою може відбуватися декількома різними шляхами. В одному прикладі проміжні продукти V можна нагрівати в присутності оксихлориду фосфору (POCl3) із розчинником або без нього. Після видалення розчинників амін можна додавати у чистому вигляді або в присутності полярного розчинника (наприклад, ацетонітрилу) з одержанням VI при кімнатній температурі або при нагріванні. Другий спосіб полягає у забезпеченні реакції проміжних продуктів V зі сполучною речовиною, такою як BOP або PyBOP у присутності DBU та аміну. Реакція проходить у полярному розчиннику (наприклад, DMF). Третій спосіб полягає у захисті 2-аміногрупи в проміжному продукті V ацильною групою. Забезпечують реакцію проміжного продукту V з ангідридом (наприклад, оцтовим ангідридом), як правило, при нагріванні зі зворотним холодильником протягом декількох годин. Розчинники можна видаляти при зниженому тиску та неочищений продукт можна піддавати наступній реакції з POCl3, як описано вище. Поверхове видалення захисної ацильної групи здійснюють шляхом реакції в основному розчиннику (наприклад, метоксиді натрію в метанолі). 5 UA 114476 C2 Схема 2 5 10 15 Експериментальна частина. Одержання проміжного продукту А До суміші 2,4-дихлор-6,7-диметоксихіназоліну (500 мг, 1,9 ммоль), діізопропілетиламіну (0,73 мл, 4,2 ммоль) та ацетонітрилу (0,1 мл) краплинно додавали розчин н-бутиламіну (0,19 мл, 1,9 ммоль) в ацетонітрилі (5 мл) при перемішуванні. Забезпечували перемішування суміші протягом однієї доби при температурі навколишнього середовища. Додавали етилацетат, органічний шар промивали насич. водн. хлоридом амонію. Органічний шар видаляли, сушили над сульфатом магнію. Тверді речовини видаляли шляхом фільтрування з одержанням неочищеного продукту A, який без подальшого очищення використовували на наступному етапі. 6 UA 114476 C2 Одержання сполуки 1 HN HN O O N O N N Cl NH3 CuO 130ºC 10 N NH2 1 A 5 O Проміжний продукт A (0,5 г, 1,7 ммоль) поміщали у 20 мл посудину під тиском із 7 N аміаком в метанолі (15 мл) та до нього додавали CuO (242 мг, 1,7 ммоль). Посудину закупорювали та суміш нагрівали до 130°C при перемішуванні протягом 18 годин. Реакційній суміші давали охолонути до кімнатної температури. Тверді речовини видаляли шляхом фільтрування та розчинники з фільтрату видаляли при зниженому тиску. Неочищений матеріал очищали шляхом зворотно-фазової колонкової хроматографії (колонка Vydac Denali C18 10 мкм, 250 г, 5 см). Рухлива фаза (0,25% розчин NH4HCO3 у воді, CH3CN). Одержання 9 15 20 25 30 Етап 1. У 500 мл круглодонну колбу, обладнану магнітною мішалкою, поміщали метил-2аміно-6-метоксибензоат (25 г, 149,6 ммоль), етанол (200 мл), ціанамід (9,43 г, 224 ммоль) та концентровану HCl (6 мл). Забезпечували перемішування суміші при нагріванні зі зворотним холодильником протягом 6 годин. З інтервалами в одну годину додавали концентровану HCl (0,5 мл). Реакційній суміші давали охолонути до кімнатної температури та тверду речовину, V-1, виділяли шляхом фільтрування та промивали етанолом. LC-MS m/z = 192(M+H). 1 H ЯМР (400 МГц, DMSO-d6) ppm 3,88 (s, 3 H), 6,96 (dd, J=8,2, 3,1 Гц, 2 H), 7,69 (t, J=8,3 Гц, 1 H), 8,28 (br. s., 2 H), 12,67 (br. s., 1 H) Етап 2. У 50 мл склянку поміщали V-1 (250 мг, 1,24 ммоль), безводний DMF (5 мл), DBU (0,6 г, 3,73 ммоль) та BOP (659 мг, 1,49 ммоль). Суміш перемішували при кімнатній температурі протягом 2 годин, додавали н-бутиламін (287 мг, 3,73 ммоль) та забезпечували перемішування реакційної суміші при кімнатній температурі протягом 15 годин. Об’єм розчинника зменшували та залишок очищали за допомогою капілярної кварцевої колонки із застосуванням градієнта від дихлорметану до 10% метанолу в дихлорметані. Найбільш придатні фракції об’єднували, 7 UA 114476 C2 розчинники видаляли при зниженому тиску з одержанням 9. Наступні проміжні продукти одержували згідно зі способом одержання V-1. Br OH N N 5 NH 2 V-2 LC-MS m/z = 240/242 1 H ЯМР (400 МГц, DMSO-d6) ppm 3,09-3,55 (m, 2 H), 7,09 (br. s., 1 H), 7,26 (dd, J=7,9, 1,3 Гц, 1 H), 7,37-7,48 (m, 2H) Cl OH N N 10 NH 2 V-3 LC-MS m/z = 196(M+H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 7,00 (br. s., 2 H) 7,13 (d, J=7,78 Гц, 1 H) 7,18 (d, J=8,28 Гц, 1 H) 7,50 (t, J=8,03 Гц, 1 H), фенольний протон не спостерігався. OH N 15 N NH2 V-7 LC-MS m/z = 176(M+H) F OH N 20 N NH2 V-8 LC-MS m/z = 180(M+H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 6,98 (dd, J=11,0, 8,3 Гц, 1 H), 7,13 (d, J=8,3 Гц, 1 H), 7,51 (br. s., 2 H), 7,64 (td, J=8,3, 5,8 Гц, 1 H), 12,30 (br. s, 1 H) OH F 25 N N NH 2 V-19 LC-MS m/z = 180(M+H) OH Br 30 N N NH 2 V-14 LC-MS m/z = 239/241(M+H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 7,32 (d, J=8,8 Гц, 1 H), 7,49 (s, 2 H), 7,71 (br. s., 1 H), 7,81 (dd, J=8,6, 2,4 Гц, 1 H), 8,00 (d, J=2,4 Гц, 1 H) 8 UA 114476 C2 OH O N N NH 2 V-20 LC-MS m/z = 192(M+H) 5 OH H 3C N N NH 2 V-21 LC-MS m/z = 176(M+H) OH N F 10 N NH 2 V-4 V-22 LC-MS m/z = 180(M+H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 7,01-7,16 (m, 2 H), 7,56 (br. s., 2 H), 7,99 (t, J=7,7 Гц, 1 H), 10,38-13,48 (m, 1 H) 15 OH N 20 Cl N NH 2 V-23 LC-MS m/z = 196(M+H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 7,41 (dd, J=8,5, 2,0 Гц, 1 H), 7,55 (d, J=2,0 Гц, 1 H), 7,98 (d, J=8,5 Гц, 1 H), 8,49 (br. s., 2 H), 10,79-13,69 (m, 1 H) OH N N 25 NH 2 V-5 LC-MS m/z = 176(M+H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 2,43 (s, 3 H), 7,22 (d, J=1,0 Гц, 1 H), 7,24 (s, 1 H), 7,89 (d, J=8,0 Гц, 1 H), 8,29 (br. s., 2 H), 12,65 (br. s, 1 H) OH N 30 O N NH 2 V-24LC-MS m/z = 192(M+H) 9 UA 114476 C2 OH N O N NH 2 O 5 V-25 LC-MS m/z = 220(M+H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 3,87-3,95 (m, 3 H), 7,12-7,47 (m, 1 H), 7,83 (dd, J=8,3, 1,4 Гц, 1 H), 7,99 (d, J=1,3 Гц, 1 H), 8,07-8,13 (m, 1 H), 8,43 (br. s., 2 H) OH F N F N NH 2 V-26 LC-MS m/z = 198(M+H) 10 OH O 15 N O N NH 2 V-27 LC-MS m/z = 298(M+H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 3,85 (s, 3 H), 5,10 (s, 2 H), 6,17 (br. s., 2 H), 6,70 (s, 1 H), 7,30-7,36 (m, 2 H), 7,40 (t, J=7,4 Гц, 2 H), 7,44-7,48 (m, 2 H), 10,82 (br. s., 1 H) OH N 20 N NH 2 F V-28 LC-MS m/z = 180(M+H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 6,51-6,67 (m, 2H), 7,00-7,08(m, 1H), 7,42(ddd, J =11,2, 7,9 1,3Гц, 1H), 7,69 (dd, J=7,9, 0,6Гц, 1H), 11,08 (br. s., 1H) OH N N 25 NH 2 Cl V-29 LC-MS m/z = 196 (M+H) OH N N 30 NH 2 CH 3 V-30 LC-MS m/z = 176 (M+H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 2,41 (s, 3 H), 7,15 (t, J=7,5 Гц, 1 H), 7,43 (br. s., 2 H), 7,55 (d, J=7,0 Гц, 1 H), 7,80 (d, J=7,8 Гц, 1H), 11,17-12,49 (m, 1H) 10 UA 114476 C2 Одержання 10 5 10 Етап 1. Одержання V-6. У 50 мл склянку, обладнану магнітною мішалкою, поміщали V-3 (500 мг, 2,16 ммоль), фенілборонову кислоту (342 мг, 2,8 ммоль), карбонат калію (1,19 г, 8,62 ммоль), діоксан (5,5 мл), воду (1,8 мл) та тетракіс(трифенілфосфін)паладій (249 мг, 0,215 ммоль). Газоподібний азот барботували через реакційну суміш протягом 10 хвилин. Склянку герметично закривали та нагрівали до 130C. Реакційну суміш охолоджували до кімнатної температури та розчинники видаляли при зниженому тиску. Неочищений продукт очищали шляхом зворотнофазової колонкової хроматографії (RP Vydac Denali C18 - 10 мкм, 200 г, 5 см. Рухлива фаза 0,25% розчин NH4HCO3 у воді, CH3CN) з одержанням V-6. LC-MS m/z = 238 (M+H) 15 20 25 Етап 2. У 50 мл склянку, обладнану магнітною мішалкою, поміщали V-6 (148 мг, 0,624 ммоль), безводний DMF (3,5 мл), DBU (0,373 мл, 2,5 ммоль), BOP (345 мг, 0,78 ммоль), потім (S)-2-амінопентанол (322 мг, 3,12 ммоль). Перемішування реакційної суміші забезпечували при кімнатній температурі протягом 3 днів. Леткі речовини видаляли при зниженому тиску та неочищений продукт розділяли між водою та етилацетатом. Органічні шари об’єднували, сушили (сульфат магнію), тверді речовини видаляли шляхом фільтрування та розчинники з фільтрату видаляли при зниженому тиску. Неочищений продукт очищали шляхом зворотнофазової колонкової хроматографії (RP SunFire Prep C18 OBD-10 мкм, 30 x 150 мм). Рухлива фаза (0,25% розчин NH4HCO3 у воді, CH3CN) з одержанням 10. Одержання 11 11 UA 114476 C2 5 10 15 20 25 30 Етап 1. У 1 л круглодонну колбу, обладнану магнітною мішалкою, поміщали V-1 (8,8 г, 46,03 ммоль) та оцтовий ангідрид (150 мл). Колбу обладнали зворотним холодильником та суміш нагрівали зі зворотним холодильником при перемішуванні протягом 15 годин. Осад виділяли шляхом фільтрування та промивали діізопропіловим етером, потім сушили у вакуумі з одержанням білої твердої речовини, V-9. LC-MS m/z = 234 (M+H) Етап 2. У 250 мл круглодонну колбу, обладнану магнітною мішалкою, додавали V-9 (4,5 г, 19,3 ммоль) та ацетонітрил (100 мл). PoCl3 (5,56 мл, 59,8 ммоль) додавали краплинно протягом 30 хвилин, після чого додавали DIPEA (10,3 мл, 59,8 ммоль). Реакційна суміш перетворювалась на коричневий розчин, та його перемішували протягом 2 годин при кімнатній температурі. Реакційну суміш виливали у 1 М NaOH (100 мл) та екстрагували етилацетатом (2 x 100 мл). Об’єднані органічні шари сушили над MgSO 4, тверді речовини видаляли шляхом фільтрування та фільтрат без подальшого очищення використовували на наступному етапі. Етап 3. Розчину фільтрату з етапу 2 в етилацетаті обробляли DIPEA (9,2 мл, 53,6 ммоль) та н-бутиламіном (3,5 мл, 35,8 ммоль). Реакційну суміш перемішували протягом 16 годин при температурі навколишнього середовища. Розчинник видаляли при зниженому тиску та неочищений продукт повторно розчиняли в дихлорметані та промивали водою. Органічний шар сушили (MgSO4), тверді речовини видаляли шляхом фільтрування та розчинники з фільтрату випарювали до висихання з одержанням оранжевої твердої речовини, V-11. LC-MS m/z = 289 (M+H) Етап 4. У 30 мл пробірку під тиском поміщали V-11 (2,8 г, 9,71 ммоль), гідрохлорид піридину (6,73 г, 58,26 ммоль) та піридин (50 мл) та суміш нагрівали до 120°C протягом 16 годин. Піридин видаляли при зниженому тиску. Неочищений продукт розчиняли в суміші дихлорметан/метанол: 95/5 та промивали 1 N розчином HCl та водою. Органічний шар сушили (MgSO4), тверді 12 UA 114476 C2 5 10 15 речовини видаляли шляхом фільтрування та розчинники з фільтрату видаляли при зниженому тиску з одержанням VI-1. LC-MS m/z = 231 (M-H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 0,92 (t, J=7,37 Гц, 3 H) 1,33-1,43 (m, 2 H) 1,50-1,59 (m, 2 H) 3,41-3,49 (m, 2 H) 5,79-5,88 (m, 1 H) 6,02 (d, J=8,14 Гц, 1 H) 6,91 (br. s., 2 H) 6,99-7,04 (m, 1 H) 10,78 (br. s., 1 H) 13,35 (br. s., 1 H) Етап 5. У 100 мл колбу поміщали VI-1 (175 мг, 0,753 ммоль), карбонат цезію (0,74 г, 2,26 ммоль) та DMF (15 мл). Суміш перемішували при температурі навколишнього середовища протягом 30 хвилин. Додавали 2-брометил-метиловий етер (0,089 мл, 0,94 ммоль) та суміш перемішували протягом 16 годин при кімнатній температурі. Розчинник видаляли при зниженому тиску та неочищений залишок очищали за допомогою HPLC (RP Vydac Denali C18 10 мкм, 250 г, 5 см). Рухлива фаза (0,25% розчин NH 4HCO3 у воді, метанол), найбільш придатні фракції збирали та розчинники видаляли при зниженому тиску з одержанням 11 у вигляді твердої речовини. Одержання 12 20 25 30 Етап 1. V-2 розчиняли в DMF (15 мл) та продували N2 на масляній бані при 80°C протягом 10 хвилин. Потім додавали біс(трифенілфосфін)паладію(II) дихлорид (69 мг, 0,098 ммоль), трифенілфосфін (57,6 мг, 0,22 ммоль) та йодид міді (42,5 мг, 0,22 ммоль). Після 5 хвилин продувания N2 додавали діетиламін (3,15 мл, 30,31 ммоль) з наступним додаванням 2піридилетину (168 мг, 1,63 ммоль). Посудину закривали та реакційну суміш перемішували при 80°C протягом 16 годин. Реакційну суміш вливали в крижану воду та осад виділяли шляхом фільтрування, промивали водою та сушили у вакуумі. Продукт перемішували в дихлорметані протягом 30 хвилин. Осад виділяли шляхом фільтрування, промивали дихлорметаном та діізопропіловим етером та сушили у вакуумі при 50°C з одержанням V-12. LC-MS m/z = 263 (M-H) 13 UA 114476 C2 N N OH OH N N N H 2, Pd/C N NH2 V-12 5 NH2 V-13 Етап 2. У розчин V-12 (300 мг, 1,15 ммоль) в THF (50 мл) поміщали 10% Pd/C (100 мг) у атмосфері N2 (газ). Реакційну суміш перемішували протягом 16 годин при кімнатній температурі та потім фільтрували через ущільнений декаліт. Розчинник з фільтрату видаляли при зниженому тиску з одержанням неочищеного V-13, який використовували без подальшого очищення на наступному етапі. LC-MS m/z = 267 (M-H) 10 Етап 3. Сполуку-приклад 12 одержували згідно зі способом одержання 9. Одержання 14 15 Br Br Br N N HN N NH 2 HN Ac2 O N O N H N HN N O N O 13 20 VI-2 VI-3 Етап 1. Проміжні продукти VI-2 та VI-3 одержували згідно зі способом одержання VI-1. VI-3 виділяли після перемішування з діізопропіловим етером при кімнатній температурі. VI-2 : LC-MS m/z = 337 (M+H) VI-3 : LC-MS m/z = 379 (M+H) 14 UA 114476 C2 OH Br HN N HN N O N H N PdCl2(PPh 3) 2 PPh3 , HNEt2, CuI, DMF N NH2 14 VI-2 Етап 2. Сполуку 14 одержували згідно зі способом одержання проміжного продукту V-12. Одержання 15 5 10 15 Етап 1. У 500 мл круглодонну колбу, обладнану магнітною мішалкою, поміщали гідрохлорид 3-амінофталевої кислоти (25 г, 115 ммоль), етанол (250 мл), ціанамід (7,25 г, 172 ммоль) та концентровану HCl (6 мл). Колбу обладнали зворотним холодильником та забезпечували перемішування суміші при нагріванні зі зворотним холодильником протягом 6 годин. З інтервалами в одну годину скляною піпеткою додавали концентровану HCl (0,5 мл). Реакційній суміші давали охолонути до кімнатної температури, розчинники видаляли при зниженому тиску з одержанням жовтого масла. Неочищений продукт сушили над силікагелем, потім частково очищали за допомогою колонкової хроматографії на силікагелі із використанням градієнта від дихлорметану до 10% метанолу в дихлорметані. Неочищений продукт, жовту тверду речовину, V-14, використовували без подальшого очищення на наступному етапі. LC-MS m/z = 234 (M+H). 20 25 Етап 2. У 100 мл круглодонну колбу, обладнану магнітною мішалкою, поміщали V-14 (1,7 г, 7,29 ммоль), безводний DMF (25 мл), DBU (3,3 г, 21,87 ммоль) та PyBOP (4,55 г, 8,75 ммоль). Перемішування реакційної суміші забезпечували протягом 1 години при кімнатній температурі. Потім додавали н-бутиламін (2,1 г, 29,2 ммоль) та забезпечували перемішування суміші протягом 15 годин при кімнатній температурі. Розчинник видаляли при зниженому тиску та неочищений продукт фільтрували через силікагель із застосуванням 20% метанолу в 15 UA 114476 C2 дихлорметані. Розчинники з фільтрату видаляли при зниженому тиску та неочищене масло (15,4 г) очищали шляхом зворотно-фазової колонкової хроматографії (RP Vydac Denali C18 - 10 мкм, 200 г, 5 см). Рухлива фаза (0,25% розчин NH 4HCO3 у воді, CH3CN). Одержання 16 5 10 15 20 До суспензії 10% Pd/C у метанолі (25 мл) додавали сполуку 14 (111 мг, 0,39 ммоль) у атмосфері N2. Атмосферу азоту видаляли та заміняли на газоподібний водень. Перемішування суміші забезпечували при кімнатній температурі до витрати 2 еквівалентів газоподібного водню. Реакційну суміш фільтрували через ущільнений декаліт. Розчинник з фільтрату видаляли при зниженому тиску. Неочищений продукт очищали шляхом колонкової хроматографії на силікагелі із застосуванням градієнта від дихлорметану до 10% метанолу в дихлорметані з одержанням 16. Одержання 18 У безводному THF (10 мл) розчиняли 17 (625 мг, 2,28 ммоль). LAH (1 М в THF, 3,42 мл, 3,42 ммоль) додавали краплинно та реакційну суміш перемішували протягом 3 годин при кімнатній температурі. LC-MS демонструвала повне перетворення у необхідний продукт. Реакційну суміш гасили насич. водн. NH4Cl, тверді речовини видаляли шляхом фільтрування та розчинники з фільтрату видаляли при зниженому тиску. Залишок очищали шляхом препаративної HPLC з одержанням на виході продукту у вигляді білої твердої речовини. Одержання 19 25 16 UA 114476 C2 5 10 Суміш VI-2 (500 мг, 1,48 ммоль), тетракіс(трифенілфосфін)паладію (86 мг, 0,074 ммоль) та ціаніду цинку (106 мг, 0,89 ммоль) в DMF (5 мл) у 10 мл пробірці поміщали під мікрохвильове випромінювання при 160C на 10 хвилин. Суміш охолоджували до кімнатної температури та концентрували у вакуумі. Залишок розділяли між водою та дихлорметаном. Органічний шар відділяли, сушили (MgSO4), розчинники видаляли шляхом фільтрування та розчинники з фільтрату концентрували у вакуумі. Продукт розтирали в порошок в CH3CN, тверду речовину виділяли шляхом фільтрування. Зняття ацильної захисної групи забезпечували після обробки метоксидом натрію в метанолі при 60C протягом однієї години. Суміш охолоджували та продукт осаджали. Білу тверду речовину, 19, виділяли шляхом фільтрування та сушили у вакуумі. Одержання 20 15 20 25 30 35 У 50 мл склянку, обладнану магнітною мішалкою, барботували газоподібний азот та поміщали VI-3 (300 мг, 0,79 ммоль), естер боронової кислоти (198 мг, 0,95 ммоль), воду (3 мл, дегазовану) та DME (6 мл, дегазований), додавали бікарбонат натрію (199 мг, 2,37 ммоль) та PdCl2(PPh3)2 (55 мг, 0,079 ммоль) та суміш нагрівали до 90°C протягом 1 години. Суміш охолоджували та додавали етилацетат. Органічний шар відділяли, сушили (MgSO 4), тверді речовини видаляли шляхом фільтрування та розчинники з фільтрату видаляли у вакуумі. Залишок очищали шляхом колонкової хроматографії на силікагелі із застосуванням градієнта від дихлорметану до 10% метанолу в дихлорметані (міститься аміак). Фракції продукту збирали та концентрували у вакуумі. Зняття ацильної захисної групи забезпечували після обробки метоксидом натрію в метанолі при 60C протягом однієї години. Розчинники видаляли при зниженому тиску та залишок розділяли між водою та дихлорметаном. Органічний шар відділяли, сушили (MgSO4), розчинники видаляли шляхом фільтрування та розчинники з фільтрату видаляли у вакуумі. Продукт кристалізували з CH 3CN, виділяли шляхом фільтрування та сушили у вакуумі з одержанням білої твердої речовини, 20. Одержання 21 У першій склянці, обладнаній магнітною мішалкою та різьбовою кришкою із прокладкою, розчин Pd2(dba)3 (6 мг, 0,007 ммоль) та 2-ди-трет-бутилфосфіно-3,4,5,6-тетраметил-2',4',6'триізопропіл-1,1'-біфенілу (6 мг, 0,013 ммоль) в толуолі (0,5 мл) продували газоподібним N2, а 17 UA 114476 C2 5 10 потім перемішували при 120°C протягом 3 хвилин. Другу склянку, обладнану магнітною мішалкою та різьбовою кришкою із прокладкою, наповнювали 2-метилімідазолом (104 мг, 1,26 ммоль) та K3PO4 (224 мг, 1,05 ммоль), потім VI-3 (200 мг, 0,53 ммоль), а також продували N2(газ). У другу склянку за допомогою шприца додавали попередньо перемішаний розчин каталізатора з наступним додаванням безводного толуолу (0,5 мл) та трет-бутанолу (1,0 мл) (у сумі 2 мл розчину толуол:трет-BuOH 1:1). Реакційну суміш нагрівали до 120°C протягом 12 годин. Суміш охолоджували та додавали метоксид натрію (30% у метанолі). Суміш нагрівали при 60°C протягом 1 години. Суміш охолоджували до кімнатної температури та концентрували у вакуумі. Залишок розділяли між водою та дихлорметаном. Органічний шар відділяли, сушили (MgSO4), тверді речовини видаляли шляхом фільтрування та розчинники з фільтрату концентрували у вакуумі. Неочищений продукт очищали за допомогою препаративної HPLC (RP SunFire Prep C18 OBD-10 мкм, 30x150 мм). Рухлива фаза (0,25% розчин NH4HCO3 у воді, CH3CN). Фракції продукту збирали та концентрували у вакуумі з одержанням сполуки 21. Одержання 22 15 20 25 30 Суміш VI-2 (500 мг, 1,48 ммоль), трибутил(1-етоксивініл)олова (0,626 мл, 1,85 ммоль), PdCl2(PPh3)2 (220 мг, 0,31 ммоль) в DMF (10 мл) нагрівали до 80C протягом 16 годин. Реакційну суміш охолоджували та додавали HCl (1 N, 2 мл). Суміш перемішували при кімнатній температурі протягом 2 годин, потім виливали в насич. водн. NaHCO 3 (100 мл) та осад виділяли шляхом фільтрування, повторно розчиняли в дихлорметані, сушили (MgSO 4), тверді речовини видаляли шляхом фільтрування та розчинники з фільтрату концентрували у вакуумі. Продукт очищали шляхом колонкової хроматографії на силікагелі із застосуванням градієнта від дихлорметану до 5% метанолу в дихлорметані, фракції продукту збирали та концентрували у вакуумі. Продукт розтирали в порошок у DIPE, фільтрували та сушили у вакуумі до перетворення в блідо-жовту тверду речовину. До суміші додавали метанол (6 мл) та метоксид натрію (0,716 мл) та перемішували при 60°C протягом 1 години. Суміш охолоджували та концентрували у вакуумі. Залишок розділяли між водою та дихлорметаном. Органічний шар відділяли, сушили (MgSO 4), тверді речовини видаляли шляхом фільтрування та розчинники з фільтрату концентрували у вакуумі. Продукт розтирали в порошок у DIPE, виділяли шляхом фільтрування та сушили у вакуумі до перетворення в блідо-жовту тверду речовину, 22. Одержання 23 35 18 UA 114476 C2 5 Сполуку 22 (59 мг, 0,23 ммоль) суспендували в метанолі (2 мл) та додавали борогідрид натрію (9 мг, 0,23 ммоль). Суміш перемішували в N 2(газ) при кімнатній температурі протягом двох годин. Суміш розводили дихлорметаном (5 мл), потім додавали насич. водн. NH 4Cl (0,5 мл) із наступним додаванням NaHCO3. Органічний шар сушили (MgSO 4), тверді речовини видаляли шляхом фільтрування та розчинники з фільтрату концентрували у вакуумі. Продукт розтирали в порошок у DIPE, виділяли шляхом фільтрування та сушили у вакуумі до перетворення в блідо-жовту тверду речовину, 23. Одержання 24 10 15 20 Етап 1. 75 мл автоклав із нержавіючої сталі наповнювали в атмосфері азоту VI-2 (626 мг, 1,87 ммоль), Pd(OAc)2 (8 мг, 0,037 ммоль), 1,3-біс(дифенілфосфіно)пропаном (31 мг, 0,074 ммоль), ацетатом калію (364 мг, 3,71 ммоль), THF (20 мл) та метанолом (20 мл). Автоклав закривали та подавали CO(газ) під тиском 30 бар. Реакційну суміш перемішували протягом 16 годин при 120°C. Реакційній суміші давали охолонути до кімнатної температури, а потім концентрували у вакуумі. Залишок розчиняли у воді та екстрагували дихлорметаном. Органічний шар сушили (MgSO4), тверді речовини видаляли шляхом фільтрування та розчинник з фільтрату концентрували у вакуумі. Продукт очищали за допомогою капілярної кварцевої колонки із застосуванням градієнта від дихлорметану до 5% метанолу у дихлорметані. Фракції продукту збирали та концентрували у вакуумі з одержанням брудно білої твердої речовини, VI4. O OH NH O NH N N N NH 2 N LAH, THF, -75ºC VI-4 NH 2 24 25 30 35 Етап 2. У розчин VI-4 (190 мг, 0,69 ммоль) у безводному THF (20 мл) додавали LAH (1 М у THF, 1,04 мл, 1,04 ммоль) при -75°C у атмосфері азоту. Забезпечували перемішування реакційної суміші протягом двох годин при повільному нагріванні до 0°C. Потім суміш охолоджували на бані з кригою та етанолом та обережно гасили додаванням 15 мл етилацетату з наступним додаванням Na2SO4 10H2O (2 г). Суміш перемішували протягом однієї години та потім сушили над MgSO 4, тверді речовини видаляли шляхом фільтрування та розчинник з фільтрату видаляли при зниженому тиску. Залишок очищали за допомогою препаративної HPLC (RP Vydac Denali C18-10 мкм, 200 г, 5 см). Рухлива фаза (0,25% розчин NH4HCO3 у воді, CH3CN), з наступним очищенням SFC (Chiralpak Diacel AD 30 x 250 мм). Рухлива фаза (CO 2, метанол із 0,2% ізопропіламіном), необхідні фракції збирали та розчинники видаляли при зниженому тиску з одержанням 24. 19 UA 114476 C2 Одержання 25 OH Br TMS N N N NH 2 PdCl2(PPh 3) 2 PPh3, HNEt2, CuI, DMF N NH 2 V-15 V-14 5 OH TMS Етап 1. Забезпечували реакцію V-14 з триметилацетиленом згідно зі способом одержання сполуки 14 із одержанням V-15. LC-MS m/z = 258 (M+H) 10 Етап 2. VI-5 одержували згідно зі способом одержання сполуки 9. Зняття захисної TMS-групи здійснювали у суміші NaHCO3, води, метанолу. LC-MS m/z = 357(M+H) 15 Етап 3. Гідрогенізацію здійснювали згідно зі способом одержання 16. Одержання 26 20 20 UA 114476 C2 5 Етап 1. Карбонілювання 2-бром-4-ізопропіланіліну, що каталізується паладієм, здійснювали згідно з процедурою одержання VI-4, за винятком того, що реакцію проводили при 110C з одержанням 2-аміно-5-ізопропілбензойної кислоти. LC-MS m/z = 180 (M+H) 10 Етап 2. V-16 одержували згідно зі способом одержання V-1. LC-MS m/z = 204(M+H) 15 Етап 3. Сполуку-приклад 26 одержували згідно зі способом одержання 15. Одержання 27 20 Етап 1. Ціанамід розчиняли в етері та суміш перемішували у атмосфері азоту. HCl (2 М в етері) додавали краплинно в реакційну суміш при температурі навколишнього середовища та продовжували перемішування протягом 2 годин при кімнатній температурі. Осад, A-2, виділяли шляхом фільтрування та сушили у вакуумі при 50°C. O S Cl O F F Cl O O F NH2 F O F Cl O HCl H 2N N F HO N NH2 NH IV-1 V-17 25 Етап 2. SO2(CH3)2 (20,4 г, 217 ммоль) нагрівали до плавлення. Додавали A-2 (3,3 г, 29 21 UA 114476 C2 5 10 ммоль) та одержану суміш перемішували та нагрівали до 120°C для повного розчинення. Метил-5-(2-хлор-4-трифторметилфеноксі)антранілат (5 г, 14,5 ммоль) додавали однією порцією в реакційну суміш. Перемішування продовжували протягом 30 хвилин. Реакційну суміш обробляли водою (10 мл) та перемішували протягом 10 хвилин. Осад, V-17, білу тверду речовину виділяли шляхом фільтрування та сушили у вакуумній печі. LC-MS m/z = 356 (M+H) Етап 3. Сполуку 27 одержували згідно зі способом одержання 15. Одержання 28 O O O Cl O HNO 3, H2 SO4 0 ºC 25 30 O O O N+ O O O Cl O Cl O O A-4 A-3 20 N+ O O 15 O A-5 Етап 1. A-3 (101 г, 0,44 моль) розчиняли у сірчаній кислоті (850 мл). Даний розчин охолоджували до 0°C. HNO3 (18,3 мл, 0,44 моль) у сірчаній кислоті (200 мл) додавали краплинно протягом 2 годин. Реакційну суміш перемішували протягом 45 хвилин при -10°C, потім виливали у крижану воду (6 л). Розчинники зливали та залишок розчиняли у дихлорметані (1,5 л). Водний шар екстрагували дихлорметаном (1 л). Об’єднані органічні шари сушили (MgSO4), тверді речовини видаляли шляхом фільтрування та розчинник видаляли при зниженому тиску з одержанням A-4 і побічного продукту-ізомеру A-5, розділяли за допомогою колонкової хроматографії на силікагелі із застосуванням градієнта від гептану до етилацетату. Етап 2. У 500 мл колбу Ерленмеєра, обладнану магнітною мішалкою, барботували газоподібний азот та поміщали метанол (100 мл, що містить 2% тіофену), 5% Pt/C (2 г, 0,513 ммоль), після чого її розміщали у атмосфері водню. Реакційну суміш перемішували протягом 16 годин при кімнатній температурі. Каталізатор видаляли шляхом фільтрування та леткі речовини з фільтрату видаляли при зниженому тиску. Залишок очищали на діоксиді кремнію із застосуванням градієнта від дихлорметану до суміші дихлорметан:метанол 9:1 з одержанням на виході жовтого масла, IV-2. LC-MS m/z = 244 (M+H) 22 UA 114476 C2 O S O NH 2 O O O O O Cl O Cl Cl H 2N HCl NH IV-2 HO N N NH 2 V-18 Етап 3. Проміжний продукт V-18 одержували згідно зі способом одержання V-17. LC-MS m/z = 254 (M+H) 5 Етап 4. Процедуру одержання сполуки 9 використовували при синтезі 28 з V-18. Одержання сполуки 29 10 Етап 1. Сполуку-приклад 29 одержували після каталітичної гідрогенізації сполуки 27 згідно зі способом, описаним при одержанні 25. Одержання 90 15 Етап 1. Сполуку 17 (12,515 г, 45,62 ммоль) розчиняли у THF (100 мл). Додавали LiOH (3,83 г, 91,2 ммоль), розчинений у воді (20 мл), з наступним додаванням метанолу (50 мл). Реакційну 23 UA 114476 C2 5 10 15 суміш перемішували протягом ночі при кімнатній температурі. Леткі речовини видаляли при зниженому тиску, тверду речовину промивали водою та розтирали в порошок з DIPE з одержанням VI-6 у вигляді брудно білої твердої речовини. 1 H ЯМР (400 МГц, DMSO-d6) ppm 0,95 (t, J=7,4 Гц, 3 H), 1,40 (dq, J=14,9, 7,3 Гц, 2 H), 1,68 (quin, J=7,3 Гц, 2 H), 3,54-3,65 (m, 2 H), 7,89-8,05 (m, 2 H), 8,14-8,31 (m, 2 H), 9,11 (br. s., 1 H), 11,10 (br. s., 1 H), 16,37 (br. s., 1 H) Етап 2. У 50 мл склянку поміщали VI-6 (200 мг, 0,768 ммоль), DMF (10 мл), триетиламін (0,641 мл, 4,61 ммоль), 3-амінопіридин (181 мг, 1,92 ммоль) та діетил-ціанофосфонат (0,233 мл, 1,54 ммоль). Забезпечували перемішування реакційної суміші протягом 2 годин при кімнатній температурі. Розчинник видаляли при зниженому тиску та неочищений продукт очищали за допомогою зворотно-фазової колонкової хроматографії (Sunfire Prep C18, OBD 10 мкм, 30 x 150 мм). Рухлива фаза (0,25% розчин NH4HCO3 у воді, метанол) з одержанням 90. Схема синтезу для одержання AA-9 Boc HO NH2 (S) Boc 2 O, Et3N DCM AA-7 20 HO NH (S) AA-8 Синтез проміжного продукту AA-3 24 HCl/EtOAc HO NH2 HCl (S) EtOAc AA-9 UA 114476 C2 5 10 15 20 25 До розчину валеріанового альдегіду (43 г, 500 ммоль) в THF (1 л) додавали AA-2 (200 г, 532 ммоль) та реакційну суміш перемішували протягом 16 годин при кімнатній температурі. Розчинники випарювали, а залишок розводили петролейним етером та фільтрували. Розчинники з фільтрату видаляли при зниженому тиску та залишок очищали за допомогою хроматографії на діоксиді кремнію із застосуванням градієнта від петролейного етеру до 3% етилацетату в петролейному етері з одержанням AA-3 (90 г) у вигляді безбарвного масла. 1 H ЯМР (400 МГц, CDCl3): ppm 6,81-6,77 (m, 1H), 5,68-5,64 (td, J=1,2 Гц, 15,6 Гц, 1H), 2,112,09 (m, 2H), 1,406 (s, 9H), 1,38-1,26 (m, 4H), 0,85-0,81 (t, J=7,2 Гц, 3H). Синтез сполуки AA-5 Н-бутиллітій (290 мл, 725 ммоль, 1,5 екв.) додавали в перемішаний розчин AA-4 (165 г, 781 ммоль) в THF (800 мл) при -78°C. Реакційну суміш перемішували протягом 30 хвилин, потім додавали AA-3 (90 г, 488,4 ммоль) в THF (400 мл) та реакційну суміш перемішували протягом 2 годин при -78°C. Суміш гасили насич. водн. розчином NH4Cl та підігрівали до кімнатної температури. Продукт розділяли між етилацетатом та водою. Органічну фазу промивали сольовим розчином, сушили та випарювали. Залишок очищали за допомогою колонкової хроматографії при елююванні 5% етилацетатом у петролейному етері з одержанням безбарвного масла, AA-5 (132 г). 1 H ЯМР (400 МГц, CDCl3): ppm 7,36-7,16 (m, 10H), 3,75-3,70 (m, 2H), 3,43-3,39 (d, J=15,2 Гц, 1H), 3,33-3,15 (m, 1H), 1,86-1,80 (m, 2H), 1,47-1,37 (m, 2H), 1,32 (s, 9H), 1,26-1,17 (m, 7H), 0,830,79 (t, J=7,2 Гц, 3H). Синтез AA-6 O (S) O N (S) LiAlH4 (S) HO THF OºC AA-5 30 N (S) AA-6 AA-5 (130 г, 328 ммоль) розчиняли в THF (1,5 л) та додавали малими порціями LAH (20 г, 526 ммоль) при 0°C. Одержану суміш перемішували при тій же температурі протягом 2 годин, а потім дали нагрітися до кімнатної температури. Суміш гасили насич. водн. розчином NH 4Cl. Продукт розділяли між етилацетатом та водою. Органічну фазу промивали сольовим розчином, сушили та випарювали. Об’єднані органічні шари сушили над сульфатом натрію, тверді речовини видаляли шляхом фільтрування та концентрували з одержанням неочищеного AA-6 (100 г), який використовували на наступному етапі без додаткового очищення. 25 UA 114476 C2 H ЯМР (400 МГц, CDCl3): ppm 7,33-7,14 (m, 10H), 3,91-3,86 (m, 1H), 3,80-3,77 (d, J=13,6 Гц, 1H), 3,63-3,60 (d, J=13,6 Гц, 1H), 3,43-3,42 (m, 1 H), 3,15-3,10 (m, 1H), 2,70-2,63 (m, 2H), 1,65-1,28 (m, 10H), 0,89-0,81 (m, 3H). Синтез AA-9 1 5 10 15 20 25 Розчин AA-6 (38 г, 116,75 ммоль) та 10% Pd/C у метанолі (200 мл) гідрогенізували воднем при тиску 50 фунтів/кв. дюйм при 50°C протягом 24 годин. Реакційну суміш фільтрували та розчинник випарювали з одержанням неочищеного продукту AA-7 (17 г). Неочищений продукт розчиняли в дихлорметані (200 мл), триетиламін (26,17 г, 259,1 ммоль) та ди-трет-бутилдикарбонат (84,7 г, 194,4 ммоль) додавали при 0°C. Одержану суміш перемішували при кімнатній температурі протягом 16 годин. Суміш розділяли між дихлорметаном та водою. Органічну фазу промивали сольовим розчином, сушили та випарювали. Залишок очищали за допомогою хроматографії на силікагелі при елююванні 20% етилацетатом у петролейному етері з одержанням AA-8 (13 г) у вигляді безбарвного масла. 1 H ЯМР (400 МГц, CDCl3): ppm 4,08-4,03 (br, 1H), 3,68 (m, 1H), 3,58-3,55 (m, 2H), 3,20-2,90 (br, 1H), 1,80-1,73 (m, 1H), 1,42-1,17 (m, 15 H), 0,85-0,82 (t, J=6,8 Гц, 3H). AA-8 (42 г, 0,182 моль) розчиняли у діоксані (200 мл) та додавали діоксан/HCl (4 М, 200 мл) при 0°C. Одержану суміш перемішували при кімнатній температурі протягом 2 годин. Розчинник випарювали з одержанням неочищеного продукту. Суміш дихлорметан/петролейний етер (50 мл, 1:1, об./об.) додавали до неочищеного продукту та зливали супернатант. Цю процедуру повторювали два рази з одержанням масла, AA-9 (26,6 г). 1 H ЯМР (400 МГц, DMSO-d6): ppm 8,04 (s, 3H), 3,60-3,49 (m, 2H), 3,16-3,15 (m, 1H), 1,711,67 (m, 2H), 1,60-1,55 (m, 2H), 1,33-1,26 (m, 4H), 0,90-0,87 (t, J=6,8 Гц, 3H). Одержання AA-10 HO NH2 HCl (S) 30 AA-10 AA-10 одержували згідно з одержанням AA-9 із застосуванням масляного альдегіду замість валеріанового альдегіду. 1 H ЯМР (400 МГц, DMSO-d6): ppm 8,07 (s, 3H), 4,85 (br, 1H), 3,57-3,45 (m, 2H), 3,14-3,12 (m, 1H), 1,70-1,64 (m, 2H), 1,56-1,49 (m, 2H), 1,38-1,30 (m, 2H), 0,90-0,80 (t, J=6,8 Гц, 3H). 26 UA 114476 C2 Таблиця 1 Сполуки формули (I) # Структура H ЯМР Спосіб, Rt Спосіб синтезу H ЯМР (360 МГц, DMSO-d6) ppm 0,93 (t, J=7,3 Гц, 3H), 1,311,43 (m, 2H), 1,60 (t, J=7,1 Гц, 2H), 3,40-3,48 (m, 2H), 3,79 (s, A, 0,67 3H), 3,79 (s, 3H), 5,67 (s, 2H), 6,63 (s, 1H), 7,40 (s, 1H), 7,44-7,50 (m, 1H) 1 1 H ЯМР (360 МГц, DMSO-d6) ppm 0,85-0,93 (m, 3H), 1,27-1,37 (m, 4H), 1,57-1,68 (m, 2H), 3,39A, 0,86 3,49 (m, 2H), 3,78 (s, 3H), 3,79 (s, 3H), 5,67 (s, 2H), 6,63 (s, 1H), 7,40 (s, 1H), 7,47 (t, J=5,7 Гц, 1H) 1 2 H ЯМР (360 МГц, DMSO-d6) ppm 0,79-0,91 (m, 3H), 1,29 (m, J=3,3 Гц, 4H), 1,59 (m, J=6,6 Гц, 2H), 1,64-1,70 (m, 1H), 1,72-1,79 (m, 1H), 3,40-3,50 (m, 2H), 3,80 (s, 3H), 3,80 (s, 3H), 4,33-4,43 (m, 1H), 4,48 (t, J=5,1 Гц, 1H), 5,68 (s, 2H), 6,63 (s, 1H), 7,09 (d, J=8,4 Гц, 1H), 7,44 (s, 1H) 1 H ЯМР (400 МГц, хлороформ-d) ppm 0,91 (t, J=7,0 Гц, 3H), 1,281,48 (m, 5H), 1,58-1,77 (m, 2H), 3,48 (s, 1H), 3,72 (dd, J=11,0, 6,3 Гц, 1H), 3,88 (s, 3H), 3,91 (s, 3H), 4,34 (td, J=6,8, 2,8 Гц, 1H), 4,78 (br. s., 2H), 5,64 (d, J=7,0 Гц, 1H), 6,81 (s, 1H), 6,81 (s, 1H) 1 H ЯМР (360 МГц, DMSO-d6) ppm 0,88 (t, J=7,3 Гц, 3H), 1,231,42 (m, 2H), 1,48-1,81 (m, 4H), 3,39-3,48 (m, 2H), 3,79 (s, 3H), 3,80 (s, 3H), 4,38-4,46 (m, 1H), 4,49 (t, J=5,3 Гц, 1H), 5,68 (s, 2H), 6,63 (s, 1H), 7,08 (d, J=8,4 Гц, 1H), 7,44 (s, 1H) 1 H ЯМР (400 МГц, хлороформ-d) ppm 0,95 (t, J=7,3 Гц, 3H), 1,351,52 (m, 2H), 1,60-1,71 (m, 2H), 3,48 (s, 1H), 3,71 (dd, J=11,0, 6,3 Гц, 1H), 3,85 (s, 3H), 3,85-3,88 (m, 1H), 3,90 (s, 3H), 4,37 (td, J=6,7, 3,3 Гц, 1H), 4,85 (br. s., 2H), 5,82 (d, J=7,3 Гц, 1H), 6,78 (s, 1H), 6,85 (s, 1H) Такий же спосіб, як і при одержанні 1. 1 3 4 5 6 27 A, 0,74 Такий же спосіб, як і при одержанні 1. A, 0,68 Такий же спосіб, як і при одержанні 1. A, 0,69 Такий же спосіб, як і при одержанні 1. A, 0,69 Такий же спосіб, як і при одержанні 1. UA 114476 C2 Таблиця 1 Сполуки формули (I) Структура # H ЯМР 1 7 8 O HN 9 N N NH2 OH (S) HN 10 N N NH2 O 11 O HN N N 12 NH 2 Спосіб, Rt Спосіб синтезу H ЯМР (400 МГц, хлороформ-d) ppm 0,89-0,96 (m, 4H), 1,01 (d, J=1,0 Гц, 4H), 1,25 (ddd, J=13,7, Такий же спосіб, 8,5, 7,4 Гц, 1H), 1,47-1,65 (m, 1H), як і при одержанні 1,77-1,92 (m, 1H), 3,48 (s, 0H), 1. 3,81-3,84 (m, 1H), 3,87 (s, 3H), 3,87 (s, 3H), 4,21-4,31 (m, 1H), 5,15 (br. s., 2H), 6,04-6,11 (m, 1H), 6,74 (s, 1H), 6,86 (s, 1H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 0,89-0,96 (m, 3H), 1,31-1,43 (m, 2H), 1,57-1,67 (m, 2H), 3,44Такий же спосіб, 3,52 (m, 2H), 6,04 (s, 2H), 7,01 (ddd, J=8,1, 7,0, 1,0 Гц, 1H), 7,20 A, 0,64 як і при одержанні 1. (dd, J=8,4, 0,9 Гц, 1H), 7,46 (ddd, J=8,3, 6,9, 1,4 Гц, 1H), 7,75 (t, J=5,4 Гц, 1H), 7,98 (dd, J=8,2, 0,9 Гц, 1H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 0,94 (t, J=7,4 Гц, 3H), 1,291,44 (m, 2H), 1,63 (t, J=7,3 Гц, C,0,83 2H), 3,55-3,64 (m, 2H), 4,02 (s, 3H), 6,99 (dd, J=8,3, 1,8 Гц, 2H), 7,69 (t, J=8,3 Гц, 1H), 7,81-8,29 (m, 2H), 9,10 (s, 1H), 12,49 (s, 1H) 1 H ЯМР (360 МГц, DMSO-d6) ppm 0,80 (t, J=1,00 Гц, 3H) 0,830,93 (m, 1H) 0,96-1,17 (m, 2H) 1,20-1,35 (m, 1H) 3,10-3,26 (m, 2H) 3,36 (br. s., 2H) 4,12 (td, C,0,88 J=8,23, 4,39 Гц, 1H) 4,56-4,74 (m, 1H) 5,96 (d, J=8,42 Гц, 1H) 7,18 (d, J=1,00 Гц, 1H) 7,37-7,64 (m, 6H) 7,81 (t, J=1,00 Гц, 1H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 0,94 (t, J=7,28 Гц, 3H) 1,361,46 (m, 2H) 1,55-1,63 (m, 2H) Див. 3,37 (s, 3H) 3,44 (td, J=6,96, 5,14 Гц, 2H) 3,74-3,80 (m, 2H) 4,24 (dd, C,0,85 експериментальну частину J=5,27, 3,76 Гц, 2H) 6,04 (br. s, 2H) 6,57 (d, J=7,53 Гц, 1H) 6,776,81 (m, 1H) 7,34 (t, J=8,16 Гц, 1H) 7,97 (t, J=5,02 Гц, 1H) 1 H ЯМР (400 МГц, DMSO-d6) ppm 0,90 (t, J=7,37 Гц, 3H) 1,321,42 (m, 2H) 1,63-1,71 (m, 2H) Див. 3,05-3,12 (m, 2H) 3,38-3,48 (m, C,0,99 експериментальну 2H) 3,52-3,59 (m, 2H) 5,93 (s, 2H) частину 6,88 (dd, J=7,15, 1,21 Гц, 1H) 7,07 (dd, J=8,25, 1,21 Гц, 1H) 7,23-7,34 (m, 4H) 7,71-7,76 (m, 1H) 8,538,56 (m, 1H) 28
ДивитисяДодаткова інформація
Назва патенту англійськоюQuinazoline derivatives for the treatment of viral infections and further diseases
Автори англійськоюMc Gowan, David, Raboisson, Pierre Jean-Marie Bernard, Jonckers, Tim, Hugo, Maria, Last, Stefaan, Julien, Embrechts, Werner, Pieters, Serge, Maria, Aloysius
Автори російськоюМакгован Дэвид, Рабуассон Пьер Жан-Мари Бернар, Йонкерс Тим Хьюго Мария, Ласт Стефаан Жюльен, Эмбрехтс Вернер, Питерс Серж Мария Алоизиус
МПК / Мітки
МПК: A61K 31/517, C07D 239/95
Мітки: хіназолінові, вірусних, захворювань, подальших, лікування, похідні, інфекцій
Код посилання
<a href="https://ua.patents.su/61-114476-khinazolinovi-pokhidni-dlya-likuvannya-virusnikh-infekcijj-ta-podalshikh-zakhvoryuvan.html" target="_blank" rel="follow" title="База патентів України">Хіназолінові похідні для лікування вірусних інфекцій та подальших захворювань</a>
Похідні піримідину для лікування вірусних інфекцій

Номер патенту: 113956
Опубліковано: 10.04.2017
Автори: Мк Гован Давід, Ребойсон П'єр Жан-Марія Бернард, Ембрехтс Вернер, Пітерс Серж Марія Алойсіс, Ласт Стефан Джульєн, Джонкерс Тім Хьюго Марія, Влач Яромір
МПК: C07D 471/04, A61K 31/506, A61K 31/505, C07D 401/12, C07D 403/12, C07D 239/48, C07D 413/12, C07D 405/12
Мітки: піримідину, інфекцій, вірусних, похідні, лікування
Формула / Реферат:
1. Сполука формули (І)або її фармацевтично прийнятна сіль, таутомер (таутомери) або сольват, деR1 являє собою водень, С1-4алкіл, циклопропіл або С1-6алкокси, галоген, гідроксил, трифторметил,R2 являє собою С1-8алкіл, (С1-4)алкоксі-(С1-4)алкіл, С3-7циклоалкіл, С4-7гетероцикл, ароматичний, біциклічний гетероцикл, арилалкіл, гетероарил,...
Похідні піперидинопіримідину для лікування вірусних інфекцій

Номер патенту: 112668
Опубліковано: 10.10.2016
Автори: Йонкерс Тім Хьюго Марія, МакГован Девід Крейг, Рабуассон П'єр Жан-Марі Бернар, Даубі Хамлічі Мурад
МПК: A61K 31/519, C07D 471/04
Мітки: піперидинопіримідину, вірусних, інфекцій, лікування, похідні
Формула / Реферат:
1. Сполука формули (І) (I)або її фармацевтично прийнятна сіль, деА вибраний з групи, що складається з СН2 і NCOR2 в будь-якій стереохімічній конфігурації,В вибраний з групи, що складається з СН2 і NCOR4 в будь-якій стереохімічній конфігурації,за умови, що, якщо А являє собою NCOR2, тоді В не являє собою NCOR4,X вибраний з СН2 в...
Дієтична добавка – природна сила 6 – для лікування запальних захворювань лор-органів та інфекцій дихальних шляхів (ларинготрахеїти, бронхіти, пневмонії, астматичний компонент), респіраторних вірусних інфекцій

Номер патенту: 88547
Опубліковано: 25.03.2014
Автор: Матюшенко Роман Анатолійович
МПК: A61K 35/00
Мітки: природна, сила, дихальних, дієтична, запальних, інфекцій, лор-органів, бронхіті, астматичний, лікування, респіраторних, захворювань, компонент, шляхів, вірусних, пневмонії, ларинготрахеїти, добавка
Формула / Реферат:
Дієтична добавка - природна сила 6 - для лікування запальних захворювань ЛОР-органів та інфекцій дихальних шляхів (ларинготрахеїти, бронхіти, пневмонії, астматичний компонент), респіраторних вірусних інфекцій, що містить квітки бузини чорної, траву череди, яка відрізняється тим, що додатково містить листя підбілу (мати й мачуха), листя подорожника великого, листя м'яти перцевої, траву деревію, траву звіробою, квітки нагідок при наступному...
Похідні пурину або деазапурину, корисні для лікування (серед інших) вірусних інфекцій

Номер патенту: 107805
Опубліковано: 25.02.2015
Автори: Ротл Пол А., Хелкомб Рендл Л.
МПК: A61K 31/522, C07D 473/16, C07D 473/18, A61P 31/12, C07D 473/24, C07D 473/34
Мітки: інших, інфекцій, вірусних, серед, деазапурину, похідні, корисні, пурину, лікування
Формула / Реферат:
1. Сполука Формули І Iабо її фармацевтично прийнятна сіль, у якій: L1 являє собою -О-; R1 являє собою Н, С1-6алкіл, С1-6гетероалкіл або С4-20гетероциклілалкіл, де кожна гетероалкільна група включає 1 або 2 гетероатоми, вибрані з О, N або S, і де кожна гетероциклільна група включає від 1 до 6...
Похідні 5,6-дихлорбензімідазолу, способи їх одержання, фармацевтичний склад та спосіб лікування вірусних інфекцій

Номер патенту: 66744
Опубліковано: 15.06.2004
Автори: Кошалка Джордж Волтер, Чемберлен Стенлі Доз
МПК: C07D 235/04, A61K 31/7052, C07H 19/052, A61P 31/12
Мітки: одержання, лікування, вірусних, способи, похідні, спосіб, 5,6-дихлорбензімідазолу, склад, інфекцій, фармацевтичний
Формула / Реферат:
1. Соединение формулы (I):, (I)в которой R - водород или галоген, или –NR1R2, гдеR1 и R2 одинаковы или различны и независимо выбраны из группы: водород, С1-6алкил, цианоС1-6алкил, гидроксиС1-6алкил, галогеноС1-6алкил, С3-7циклоалкил, С1-6алкилС3-7циклоалкил, С2-6алкенил, С3-7циклоалкилС1-6алкил, С2-6алкинил, арил, арилС1-6алкил, гетероциклический...
Попередній патент: Муфта для з’єднання трубчастих елементів для вибійних компонувань
Наступний патент: Пристрій для інтрамедулярної фіксації переломів трубчатих кісток
Випадковий патент: Ливарний сплав на основі магнію з підвищеною жароміцністю