Похідні амінокислот та їх кислотно-адитивні солі





Формула / Реферат
1. Производные аминокислот общей формулы
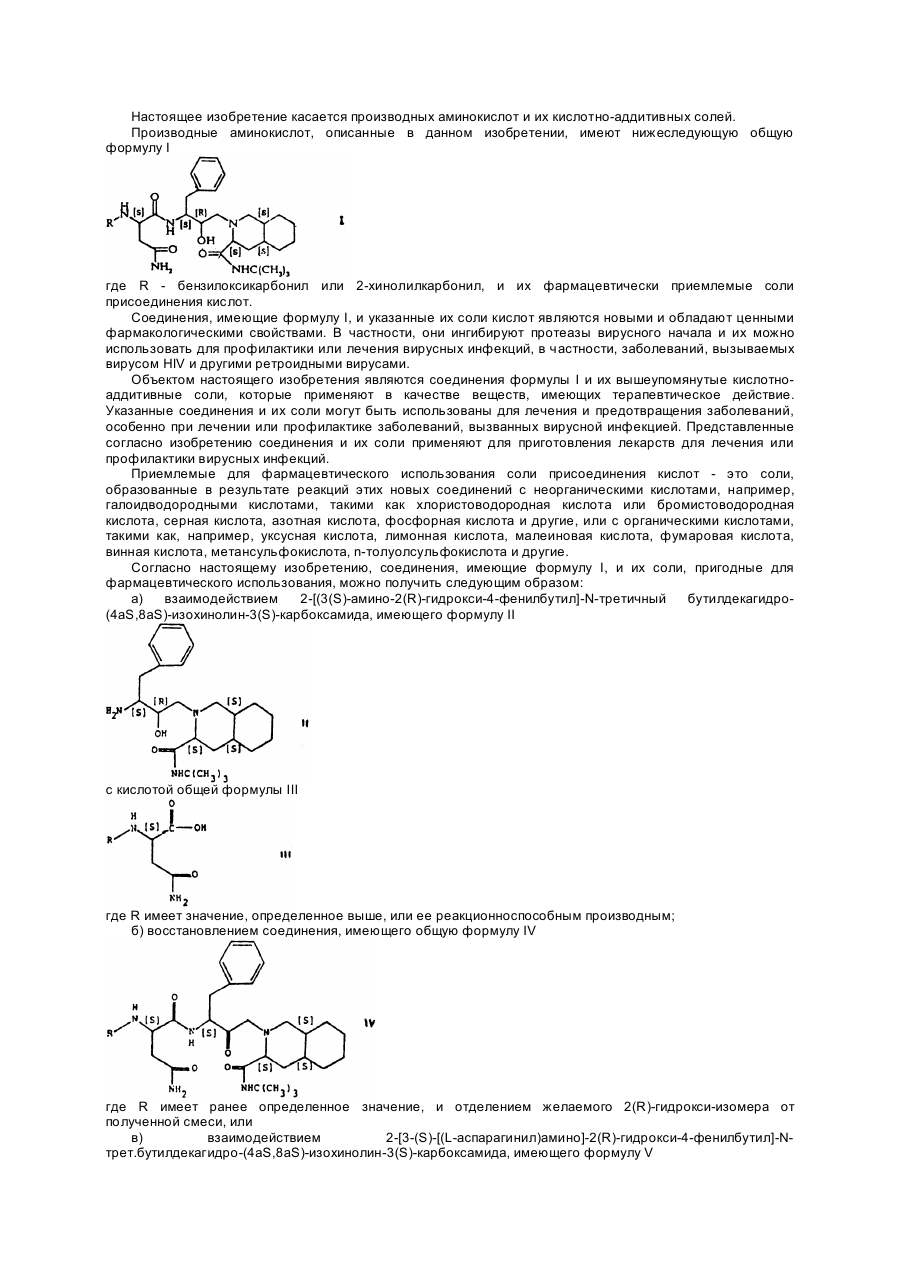
где R - бензилоксикарбонил или 2-хинолилкарбония, и их кислотно-аддитивные соли.
2. N-Трет.бутилдекагидро-2-[2(R)-гидрокси-4-фенил-3(S)-[[N-(2-хинолилкарбонил)-L-аспарагинил]амино]бутил]-(4aS,8aS)-изохинолин-3(S)-карбоксамид.
3. 2-[3(S)-Амино-2(R)-гидрокси-4-фенилбутил]-N-трет.бутилдекагидро-(4aS,8aS)-изохинолин-3(S)-карбоксамид.
4. 2-[3-(R)-[(L-Аспарагинил)амино]-2(R)-гидрокси-4-фенилбутил]-N-трет.бутилдекагидро-(4aS,8aS)-изохинолин-3(S)-карбоксамид.
Текст
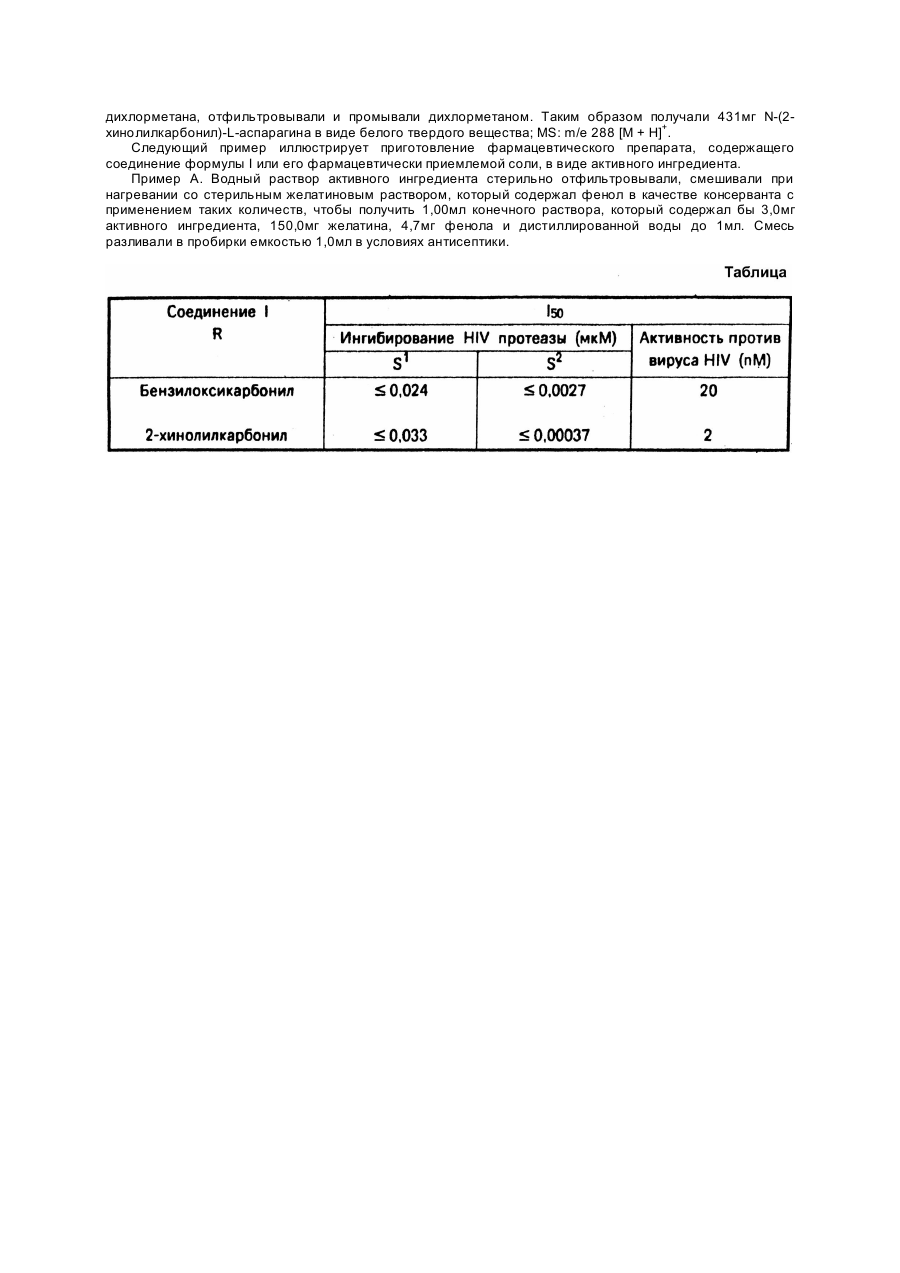
Настоящее изобретение касается производных аминокислот и их кислотно-аддитивных солей. Производные аминокислот, описанные в данном изобретении, имеют нижеследующую общую формулу I где R - бензилоксикарбонил или 2-хинолилкарбонил, и их фармацевтически приемлемые соли присоединения кислот. Соединения, имеющие формулу I, и указанные их соли кислот являются новыми и обладают ценными фармакологическими свойствами. В частности, они ингибируют протеазы вирусного начала и их можно использовать для профилактики или лечения вирусных инфекций, в частности, заболеваний, вызываемых вирусом HIV и другими ретроидными вирусами. Объектом настоящего изобретения являются соединения формулы I и их вышеупомянутые кислотноаддитивные соли, которые применяют в качестве веществ, имеющих терапевтическое действие. Указанные соединения и их соли могут быть использованы для лечения и предотвращения заболеваний, особенно при лечении или профилактике заболеваний, вызванных вирусной инфекцией. Представленные согласно изобретению соединения и их соли применяют для приготовления лекарств для лечения или профилактики вирусных инфекций. Приемлемые для фармацевтического использования соли присоединения кислот - это соли, образованные в результате реакций этих новых соединений с неорганическими кислотами, например, галоидводородными кислотами, такими как хлористоводородная кислота или бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и другие, или с органическими кислотами, такими как, например, уксусная кислота, лимонная кислота, малеиновая кислота, фумаровая кислота, винная кислота, метансульфокислота, n-толуолсульфокислота и другие. Согласно настоящему изобретению, соединения, имеющие формулу I, и их соли, пригодные для фармацевтического использования, можно получить следующим образом: а) взаимодействием 2-[(3(S)-амино-2(R)-гидрокси-4-фенилбутил]-N-третичный бутилдекагидро(4aS,8aS)-изохинолин-3(S)-карбоксамида, имеющего формулу II с кислотой общей формулы III где R имеет значение, определенное выше, или ее реакционноспособным производным; б) восстановлением соединения, имеющего общую формулу IV где R имеет ранее определенное значение, и отделением желаемого 2(R)-гидрокси-изомера от полученной смеси, или в) взаимодействием 2-[3-(S)-[(L-аспарагинил)амино]-2(R)-гидрокси-4-фенилбутил]-Nтрет.бутилдекагидро-(4aS,8aS)-изохинолин-3(S)-карбоксамида, имеющего формулу V с агентом, образующим бензилоксикарбонильную группу или 2-хинолилкарбонильную группу; г) если необходимо, осуществляют превращение полученного соединения, имеющего формулу I, в фармацевтически приемлемую соль, полученную взаимодействием с кислотой. Взаимодействие соединения, имеющего формулу II, с кислотой, формулы III, согласно способу (а), можно осуществить, применяя методы, известные из химии пептидов. Так, когда используется кислота формулы III, реакцию предпочтительно проводят в присутствии конденсирующего агента, такого как гидроксибензотриазол и дициклогексилкарбодиимид. Эту реакцию осуществляют в инертном органическом растворителе, таком как эфир (например, диэтиловый эфир; тетрагидрофуран и др.) или в диметилформамиде при низкой температуре, предпочтительно от примерно -10°C до +5°C, а особенно при температуре около 0°C. Подходящими реакционноспособными производными кислот формулы III, используемыми в данном способе, являются, например, соответствующие галоидангидриды (например, хлорангидрид), ангидриды кислот, смешанные ангидриды, активированные сложные эфиры и др. При использовании реакционноспособных производных реакция обычно проводится в инертных органических растворителях, таких как галоидзамещенные алифатические углеводороды (например, дихлорметан) или в эфире (например, диэтиловом эфире, тетрагидрофуране и др.) и, что благоприятно, в присутствии органического основания (например, N-этилморфолина, диизопропилэтиламина и др.) при низкой температуре, желательно от приблизительно -10°C до +5°C, особенно при температуре около 0°C. Реакция восстановления соединений формулы IV (согласно способу, описанному в (б), может быть осуществлена в соответствии с методами, известными для восстановления карбонильной группы до гидроксильной. Так, например, восстановление осуществляется с использованием комплексных гидридов металлов, таких как боргидрид щелочного металла, особенно боргидрид натрия, в присутствии подходящего органического растворителя, такого как алканол (например, метанол, этанол, пропанол, изопропанол и др.). Обычно реакцию восстановления проводят при комнатной температуре. Отделение желаемого 2(R)-гидроксиизомера от полученной смеси можно осуществить известными способами, например, хроматографией и т.д. Для осуществления процесса согласно (в) подходящим реагентом для получения бензилоксикарбонильной группы является бензиловый эфир хлормуравьиной кислоты. Подходящими реагентами для получения 2-хинолилкарбонильной группы являются соответствующая кислота и ее реакционноспособное производное, такое как соответствующий галоидангидрид (например, хлорангидрид кислоты), ангидрид кислоты, смешанные ангидриды, активированные сложные эфиры и т.д. Взаимодействие соединения формулы V с вышеупомянутыми реагентами осуществляется способом, описанным в стадии (а). Превращение соединений формулы I в фармацевтически приемлемые соли кислот, согласно методу (г), можно осуществить обработкой этих соединений по общепринятому методу неорганической кислотой, например, галоидводородной кислотой, такой как хлористоводородной или бромистоводородной кислотами, серной кислотой, азотной кислотой, фосфорной кислотой и др.; или органической кислотой, такой как уксусная кислота, лимонная кислота, малеиновая кислота, фумаровая кислота, винная кислота, метансульфокислота, паратолуолсульфокислота и др. Соединение формулы II, используемое в качестве исходного вещества в методе (а), является новым. Оно может быть получено, например, взаимодействием соединения, имеющего общую формулу VI, где R1 группа, защищающая аминогруппу (например, трет.бутоксикарбональная или бензилоксикарбонильная), а "X" - атом хлора или брома, с N-трет.-бутилдекагидро-(4aS,8AS)-изохинолин3(S)-карбоксамидом, имеющим формулу VII и восстановлением полученного соединения общей формулы VIII где R1 имеет значение, определенное ранее, отделением желаемого 2(R)-гидроксиизомера от полученной смеси и отщеплением группы R1 от полученного соединения, имеющего общую формулу IX в которой R1 имеет ранее определенное значение, с образованием соединения формулы II. Взаимодействие соединения формулы VI, где предпочтительно одна группа R1 означает бензилоксикарбонил, с соединением формулы VII можно осуществить известным способом, например, в инертном органическом растворителе, таком как галоидзамещенный алифатический углеводород (например, дихлорметан и т.д.) и в присутствии основания (например, триалкиламин, такой как триэтиламин и др.) обычно при комнатной температуре. Восстановление соединения формулы VIII с получением соединения формулы IX и с последующим отделением желаемого 2(R)-гидроксиизомера можно осуществить методом, описанным ранее, согласно способу (б) настоящего изобретения, то есть восстановлением соединения формулы IV и отделением 2(R)-гидроксиизомера из полученной смеси. Отщепление группы R1 от соединения формулы IX можно также осуществить известным способом, используя сильную неорганическую кислоту, такую как галоидводородная кислота или сильную органическую кислоту (например, трифторуксусная кислота и т.д.) удобнее всего при температуре от примерно 0°C до комнатной температуры. С другой стороны, аминоблокирующая группа R1, отщепляемая гидрогенолитически, может быть подвергнута отщеплению при использовании водорода в присутствии благородного металла в качестве катализатора (например, палладиевого катализатора, такого как палладий на угле) в органическом растворителе или в смеси растворителей, которые в условиях реакции инертны (например, алканол, такой как этанол, изопропанол, или сложный эфир алканкарбоновой кислоты, как, например, этилацетат и др.), обычно при комнатной температуре. Следующий метод получения соединения формулы II включает сначала взаимодействие соединения общей формулы X где R1 определен ранее, с соединением, имеющим формулу VII, описанную ранее, желательно в инертном органическом растворителе, таком как алканол (например, метанол и др.), диметилформамид или в аналогичном растворителе при повышенной температуре, лучше от приблизительно 60°C до примерно 120°C, с последующим отщеплением группы R1 в продукте реакции (соединение формулы IX), как было описано ранее. Соединения формулы IV, которые используются в качестве исходных веществ для получения по (б), можно приготовить при отщеплении аминозащитной группы R1 от соединения, имеющего формулу VIII, и взаимодействием продукта реакции с кислотой формулы III, или с ее реакционноспособным производным. Взаимодействие можно осуществлять способом, аналогичным описанному ранее в (а). Соединение, имеющее формулу V и которое используется в качестве исходного вещества в осуществлении процесса по (в), является новым и служит еще одним объектом настоящего изобретения. Соединение формулы V можно получить, например, отщеплением бензилоксикарбонильной группы R от соединения формулы I, в котором R - бензилоксикарбонильная или трет.бутоксикарбонильная группа, с образованием соединения I, но в котором R означает третичную бутоксикарбонильную группу. Это последнее соединение можно получить, например, при взаимодействии соединения формулы II с N(трет.бутоксикарбонил)-L-аспарагином, согласно методике (а). Процесс отщепления выполняется способом, аналогичным описанному ранее в связи с отщеплением группы R1 от соединений формулы VIII. Исходные вещества формулы III и их реакционноспособные производные, также как и соединения формул VI, VII и X, описанные выше, поскольку они являются новыми и не аналогичны известным соединениям, можно получить способом, аналогичным для получения известных соединений, или способом, описанным в нижеприведенных примерах или аналогичных им. Более того, реагенты, использованные в методике (в), являются большей частью известными соединениями. Как было упомянуто выше, соединения формулы I и их фармацевтически приемлемые соли ингибируют протеазы вирусного начала и поэтому пригодны для лечения и профилактики вирусных инфекций, в частности, инфекций, вызванных вирусом HIV и другими ретроидными вирусами. Ингибирование протеазы вируса HIV вне организма соединениями, которые представлены в настоящем изобретении, можно продемонстрировать посредством следующего теста: Протеаза HIV была экспрессирована в е.соlі и частично очищена от растворимых экстрактов бактерии фракционированием сульфатом аммония (0% - 30%). Активность протеазы анализировалась с применением в качестве субстрата защищенного гексапептидасукцинил-Ser-Leu-Asn-Tyr-Pro-Ile изобутиламида (S 1) или защищенного гептапептида сукцинила-Val-Ser-Gin-Asn-Phe-Pro-Ile изобутиламида (S2) в качестве субстрата. Отщепление субстрата оценивалось посредством количественного определения образовавшегося H-Pro-Ile изобутиламида при помощи спектрофото-метрического анализа Nтерминального пролина. 1,25мМ субстрата растворяли в 125мМ цитратного буфера (pH = 5,5), содержащего 0,125мг/мл Tween 20. К 80мкл вышеупомянутого буферного субстрата добавляли 10мкл раствора исследуемого соединения различных концентраций (растворенного в метаноле или в диметилсульфоксиде и разбавленного водой, содержащей 0,1% Tween 20) и 10мкл протеазы. Переваривание осуществлялось при температуре 37°C в течение установленного времени, затем процесс останавливали добавлением 1мл цветного реагента [30мкг/мл изатина и 1,5мг/мл 2-(4-хлорбензоил)бензойной кислоты в 10% ацетоне в этаноле (соотношения объем/объем)]. Раствор нагревали на водяной бане, затем пигментированный осадок подвергался вторичному растворению в 1мл 1% пирогаллола с 33% содержанием воды в ацетоне (соотношения вес/объем/объем). Оптическая плотность раствора измерялась методом спектрофотометрии при 599нм. Образование H-Pro-Ile изобутиламида в присутствии исследуемого соединения сравнивали с контрольными, концентрация исследуемого соединения, дающая 50% ингибирования (I 50), была определена графическим построением различных концентраций применяемых исследуемых соединений. Антивирусная активность соединений in vitro формулы I может быть продемонстрирована на примере анализа, описанного ниже. Активность против вируса HIV. В этой пробе были использованы HTLV-III (штамм RF), выращенные в клетках C8166 (CD4+ человеческого T-лимфобластоидного происхождения), с применением среды RPMI1640 с бикарбонатным буфером, антибиотиками и 10% сывороткой коровьего эмбриона. Суспензию клеток заражали вирусом в количестве, десятикратном TCD50, адсорбцию осуществляли при температуре 37°C в течение 90 минут. Клетки отмывались средой 3 раза. Тест выполнялся в 6мл пробирках с культурой ткани, каждая пробирка содержала 2 ´ 105 инфицированных клеток в 1,5мл среды. Анализируемые соединения растворялись в водно-эфирной середе или в диметилсульфоксиде в зависимости от растворимости, и добавлялось 15мкл раствора субстанции. Культуры инкубировали при температуре 37°C в течение 72 часов во влажной атмосфере с содержанием 5% углекислого газа. Затем культуры центрифугировали, а аликвотная проба надосадочной жидкости переводилась в растворимое состояние посредством Nonidet P40 и подвергалась действию пробы антигена, в которой была первичная антисыворотка, имеющая конкретную реактивность против белка вируса 24 и систему нахождения пероксидазы хрена обыкновенного. Окрашенное образование определялось методом спектрофотометрии и наносилось на диаграмму в зависимости от концентраций исследуемой субстанции. Концентрация, при которой наблюдалась 50% защита, определялась индексом (I 50). Проба на цитотоксичность, основанная на поглощении красителя и метаболизме или на внедрении меченого радиоактивного изотопа аминокислоты, представляет собой серию опытов, наряду с вышеуказанной пробой, для определения антивирусной селективности. Результаты, полученные при проведении вышеуказанных исследований, где используются соединения, имеющие формулу I, в качестве анализируемых соединений, объединены в следующей таблице. Соединения, описываемые формулой I, а также их фармацевтически приемлемые соли, можно использовать в качестве лекарственных препаратов. Фармацевтические препараты должны быть приготовлены для внутреннего применения, например, перорального (в форме таблеток, облаток, драже, мягких и твердых желатиновых капсул, растворов, эмульсий или суспензий), назального (в форме носовых аэрозолей) или ректального (в форме суппозиториев). Помимо этого, полученные препараты пригодны и для парентерального применения, например, внутримышечного или внутривенного (в форме растворов для инъекций). В промышленном производстве таблеток, облаток, драже и твердых желатиновых капсул соединения формулы I и их соли можно сочетать с инертными неорганическими или органическими наполнителями. В качестве таких наполнителей можно использовать лактозу, кукурузный крахмал или их производные, тальк, стеариновую кислоту и ее соли. Подходящей средой для мягких желатиновых капсул являются растительные масла, воски, жиры, полутвердые или жидкие полиолы и т.д. Подходящей средой для растворов и сиропов могут быть вода, полиолы, сахароза, инвертный сахар, глюкоза и т.д. Подходящей средой для растворов для инъекций являются вода, спирты, полиолы, глицерин, растительные масла и т.д. Подходящей средой для суппозиториев могут быть натуральные или отвержденные масла, воски, жиры, полужидкие или жидкие полиолы и т.д. Более того, в состав фармацевтических препаратов могут входить консервирующие вещества, растворители, вещества, повышающие вязкость, стабилизирующие агенты, антикоагулянты, смачивающие агенты, эмульгаторы, подслащивающие вещества, красители, ароматизирующие вещества, соли для изменения осмотического давления, буферы, покрывающие вещества (оболочки) или антиоксиданты. Кроме того, могут входить и другие вещества, имеющие терапевтическую ценность. Согласно настоящему изобретению соединения, имеющие формулу I, и их фармацевтически приемлемые соли присоединения кислот можно использовать для лечения и профилактики вирусных заболеваний, в частности, ретровирусных инфекций. Дозировка может варьироваться в широких пределах и, естественно, подбирается индивидуально в каждом конкретном случае. Обычно в случае орального применения достаточно следующей суточной дозы - примерно от 3мг до примерно 3г, предпочтительно от примерно 10мг до примерно 1г (например, приблизительно 300мг на человека), предпочтительно разделенную на прием в течение суток на 1 - 3 раза, причем каждая доза, как правило, одинаковая. Однако, следует отметить, что верхний предел может быть превышен, если это показано. Приведенные ниже примеры наглядно иллюстрируют настоящее изобретение. Пример 1. Раствор 561мг 2-[3(S)-амино-2(R)-гидрокси-4-фенилбутил]-N-трет.-бутилдекагидро(4aS,8aS)-изохинолин-3(S)-карбоксамида и 372мг N-(бензилоксикарбонил)-L-аспарагина в 20мл сухого тетрагидрофурана охлаждали в смеси льда с солью. Добавляли 189мг гидроксибензотриазола, 161мг Nэтилморфолина и 317мг дициклогексилкарбодиимида, смесь перемешивали в течение 16 часов. Затем смесь разбавляли этиловым эфиром уксусной кислоты и фильтровали. Фильтрат промывали водным раствором бикарбоната натрия и раствором хлорида натрия. Растворитель удалялся упариванием, а осадок подвергался хроматографии на силикагеле с применением смеси дихлорметана и метанола (9 : 1) для элюирования, получая 434мг 2-[3(S)-[N-(бензилоксикарбонил)-L-аспарагинил]амино]-2(R)-гидрокси-4фенилбутил-N-третичный бутилдекагидро-(4aS,8aS)-изохинолин-3(S)-карбоксамида в виде белого твердого вещества из смеси метанол / диэтиловый эфир; Масс-спектр: 650 [М + H]; ЯМР: d (d4CH3OH, 400MHz); 7,33 (5Н, m, PhCH2O), 7,25 (2Н, m); 7,18 (2Н, m); 7,09 (1Н, m), 5,05 (2Н, s, PhCH2O), 4,42 (1H, dd, Asn aJ = 7,8, 6,1), 4,22 (1H, m, -CH2CHCH(OH)-J = 10,7, около 4, около 4), 3,85 (1H, m, -CHCH(OH)CH2-J = 8,0, 6,2, около 4), 3,02 (1H, dd, PhCH(H)CHJ = -13,9, около 4), 3,02 (1H, dd, 1eqJ = -12,0, немного), 2,69 (1H, dd, PhCH(H)-J = -13,9, 10,7), 2,63 (1H, dd, -CH(OH)CH(H)N-J = -12,6, 8,0), 2,62 (1H, dd, H3axJ = около 11, немного), 2,57 (1H, dd, Asn b 1J = -15,2 6,1), 2,38 (1H, dd, Asnb 2J = -15,2, 7,8), 2,19 (1H, dd, -CH(OH)CH(H)N-J = -12,6, 6,2), 2,17 (1H, dd,1axJ = -12,0, 3,2), 2,07 (1H, m, H4axJ = -12,7, около 11, около 11,5), 1,78 (1H, m, H4aJ4a-4ax = около 11,5, J4a-4eq = немного, J4a-8a = немного), 1,63 (1H, m, H8aJ8a-1ax = 3,2, J8a-1eq = немного, J8a-4a = немного), 1,35 (1H, m, H4eqJ = -12,7, немного, немного), 1,30 (9H, s, t-бутил), 2,0 - 1,2 (8H, m). Используемый в качестве исходного материала 2-[3(S)-амино-2(R)-гидрокси-4-фенилбутил]-Nтретичный бутилдекагидро-(4aS, 8aS)-изохинолин-3(S)-карбоксамид получали следующим образом. (i) Суспензию из 12,676г (71,6 ммолей) 1,2,3,4-тетрагидро-3(S)-изохинолинкарбоновой кислоты (Chem. Pharm. Bull. 1983, 31, 312) в 200мл 90% - ной уксусной кислоты гидрогенизировали при 80°C и давлении 140атм над 5% - ным родием на угле в течение 24 часов. Смесь охлаждали до комнатной температуры, а катализатор затем отфильтровывали. Фильтрат испаряли до получения камеди, которую растворяли в 10мл этилацетата, и медленно добавляли к 100мл энергично перемешиваемого диизопропилового эфира. Получался смолистый осадок. Надосадочные жидкости удалялись декантацией, а осадок экстрагировали горячим этилацетатом. Этот горячий раствор вливали в интенсивно перемешиваемую смесь 150мл диэтилового эфира и диизопропилового эфира (1 : 1) до получения светлосерого твердого вещества, которое отфильтровывали, промывали диэтиловым эфиром и высушивали. Таким образом получали 5,209г смеси декагидроизохинолин-3(S)-карбоновых кислот, в состав которых входили в большей степени (около 65%) 4aS, 8aS изомеры вместе с 4aR, 8aR изомерами (около 25%), и около 10% трансизомеров; MS; m/e 184 [M +H]+. (ii) 9,036г (49,4 ммоля) предшествующей смеси декагидроизохинолин-3(S)-карбоновых кислот растворяли в 50мл (50 ммоль) 1М раствора гидроксида натрия; полученный раствор охлаждали до 0°C. 7,40мл (51,87 ммоля) бензилхлороформиата и 58,7мл (58,7 ммоль) 1М раствора гидроксида натрия добавляли по каплям в течение 1 часа, поддерживая температуру на уровне 0 - 5°C охлаждением. Затем смесь перемешивали еще 2 часа, за это время температура смеси доводилась до комнатной. Добавляли 100мл диэтилового эфира и смесь фильтровали, в результате чего получали нерастворимый R,R-изомер, который отделяли. Водный слой фильтрата сепарировали и доводили до pH = 1,5 - 2 добавлением концентрированной соляной кислоты, в результате чего осаждалось масло. Смесь дважды экстрагировали 100мл этил ацетата. Объединенные органические экстракты промывались водой, высушивались над безводным сульфатом натрия и упаривались до получения масла. Это масло растворяли в 35мл этилацетата с добавлением 2,85мл (25 ммолей) циклогексиламина. Белый осадок собирали фильтрованием, получая (после нескольких дробных перекристаллизаций из смеси метанол/этилацетат) 2,38г циклогексиламиновой соли 2-(бензилоксикарбонил)-декагидро-(4aS,8aS)-изохинолин-3(S)-карбоновой кислоты; MS: m/e 318 [M + H]+. (iii) 2,334г циклогексиламиновой соли 2-(бензилоксикарбонил)декагидpo-(4aS,8aS)-изохинолин-3(S)карбоновой кислоты разделили между 50мл этилацетата и 50мл 10% - ного раствора лимонной кислоты. Органическую фазу отделили, промыли водой, отфильтровали и упарили до получения 1,87г 2(бензилоксикарбонил)-декагидро-(4aS,8aS)-изохинолин-3(S)-карбоновой кислоты в виде бесцветной смолы; MS: m/e 318 [М + H]+. (iv) Раствор 0,634г (2,0 ммоля) 2-(бензилоксикарбонил)-декагидро-(4aS,8aS)изохинолин-3(S)карбоновой кислоты в 6мл диметоксиэтана обработали 0,23г (2,0 ммоля) N-гидроксисукцинимидом и 0,412г (2,0 ммоля) дициклогексилкарбодиимидом. Смесь перемешивали при комнатной температуре 18 часов. Смесь фильтровали, и фильтрат упаривали до получения 0,879г эфира N-гидроксисукцинимида в виде светло-желтого масла. Раствор 0,828г (2,0 ммоля) эфира N-гидроксисукцинимида перемешивали в 5мл дихлорэтана, охлаждали до 0°C и обрабатывали 0,219г (3,0 ммоля) третичного бутиламина. Смесь перемешивали при 0°C в течение 2 часов, затем в течение 4,5 часов при комнатной температуре. Затем смесь промывали 2М соляной кислотой, раствором карбоната натрия и раствором хлорида натрия, высушивали безводным сульфатом магния и упаривали. Осадок растворяли в 20мл диэтилового эфира и отфильтровывали. Фильтрат упаривали до получения 0,712г 2-(бензилоксикарбонил)-N-третичный бутилдекагидро-(4aS,8aS)-изохинолин-3(S)-карбоксамида в виде белого твердого вещества; MS: m/e 373 [M + H]+. (v) Растврр 0,689г (1,85 ммоля) 2-(бензилоксикарбонил)-N-третичный бутилдекагидро-(4aS,8aS)изохинолин-3(S)-карбоксамида в 20мл этанола гидрогенизировали в присутствии 0,01г 10% - ного палладия на угле при комнатной температуре и при атмосферном давлении в течение 18 часов. Катализатор удаляли фильтрацией, а растворитель удаляли упариванием до получения количественного выхода N-третичного бутилдекагидро-(4aS,8aS)-изохинолин-3(S)-карбоксамидав виде чистого масла; MS: m/e 239 [M + H]+, которое использовалось на следующей стадии без дальнейшей очистки. (vi) Раствор 440мг N-третичного бутилдекагидро-(4aS,8aS)-изохинолин-3(S)-карбоксамида и 549мг 3(S)-бензилоксиформамидо)-1,2(S)-эпокси-4-фенилбутана в 6мл этанола перемешивали при 60°C в течение 7 часов. Затем добавляли дополнительные 54мг 3(S)-(бензилоксиформамидо)-1,2(S)-эпокси-4фенилбутана, раствор перемешивали при 20°C в течение 16 часов. Растворитель удаляли упариванием, а осадок подвергали хроматографии на силикагеле с использованием смеси диэтилового эфира : н-гексана : метанола (47,5 : 47,5 : 5) для элюирования с получением 771мг 2-[3(S)-(бензилоксиформамидо)-2(R)гидрокси-4-фенилбутил]-N-третичного бутилдекагидpo-(4aS,8aS)-изохинолин-3(S)-карбоксамида в виде белого твердого осадка; MS: m/e 536 [M + H]+. (vii) Раствор 747мг 2-[3(S)-(бензилоксиформамидо)-2(R)-гидрокси-4-фенилбутил)-N-трет.бутилдекагидро-(4aS,8aS)-изохинолин-3(S)-карбоксамида в 40мл этанола гидрогенизировали на 10% палладия на угле при 20°C, атмосферном давлении в течение 5 часов. Катализатор удаляли фильтрацией, а фильтрат упаривали до получения 561мг 2-[3-(S)-амино-2(R)-гидрокси-4-фенилбутил]-N-трет.бутилдекагидро-(4aS,8aS)-изохинолин-3(S)-карбоксамида в виде светло-желтого твердого вещества, которое использовалось на следующих стадиях без дальнейшей очистки. Пример 2. Раствор 154мг 2-[3(S)-[(l-аспарагинил)-амино]-2(R)-гидрокси-4-фенилбутил]-N-третичного бутилдекагидро-(4aS,8aS)-изохинолин-3(S)-карбоксамида и 52мг хинальдиновой кислоты в 6мл сухого тетрагидрофурана охлаждали в смеси льда и соли. Добавляли 41мг гидроксибензотриазола, 35мг Nэтилморфолина и 68мг дициклогексилкарбодиимида, смесь перемешивали в течение 64 часов. Смесь разбавляли этилацетатом и отфильтровывали. Фильтрат промывали водным раствором бикарбоната натрия и раствором хлорида натрия, затем испаряли. Осадок хроматографировался на силикагеле с применением дихлорметана и метанола (9 : 1) для элюирования с целью получения 50мг N-третичного бутилдекагидро-2-[2(R)-гидрокси-4-фенил-3-(S)-[{N-(2-хинолилкарбонил)-L-аспарагинил]-амино]-бутил](4aS,8aS)-изохинолин-3(S)-карбоксамида в виде белого осадка; MS: m/e 671 [M + H]+; ЯМР: d (d4 CH3OH, 400VHz): 8,52 (1Н, m), 8,18 (1H, m), 8,14 (1Н, m), 8,02 (1H, m), 7,84 (1H, m), 7,69 (1H, m), 7,18 (2H, m); 6,90 (2H, m), 6,72 (1Н; m), 4,93 (1Н, dd, Asna CHJ = 6,6, 6,8), 4,27 (1H, m, -CH2CHCH(OH)-J = 3,8, 3,8, 11,0), 3,89 (1H, m, CHCH(OH)CH2-J = 7,2, 6,4, 3,8), 3,06 (1H, dd, H1 eqJ = -12,0, 3,0), 3,02 (1H, dd, PhCH(H)CH-J = -14,0, 3,8), 2,77 (1H, dd, Ash b 1J = -15,6, 6,6), 2,68 (1H, dd, Asnb 2J = -15,6, 6,8), 2,68 (1H- dd- PhCH(H)CH-J = -14,0, 11,0), около 2,68 (1H, dd, -CH(OH)CHN-J = 12,0, 7,2), 2,63 (1H, dd, H3axJ = 11,0, 2,2), 2,22 (1H, dd, -CH(OH)CH(H)NJ = -12,0, 6,4), 2,18 (1H, dd, H1axJ = -12,0, 2,2), 2,06 (1H, m, H4axJ = -11,0, 11,0, 11,0), 1,78 (1H, m, 4 aJ4a-4ax = 11,0, J4a-4eq = около 4, J4a-8a = около 4), 1,65 (1H, m, 8aJ8a-1ax = 2,2, J8a-1eq = 3,0, J8a-4a = около 4), 1,37 (1H, m, H4eqJ = -11,0, 2,2, около 4), 1,30 (9H, s, t-бутил), 2,0 - 1,2 (8H, m). Используемый в качестве исходного 2-[3(S)-[(L-аспарагинил)-амино]-2(R)-гидрокси-4-фенилбутил]-Nтретичный бутилдекагидро-(4aS,8aS)-изохинолин-3(S)-карбоксамид получали следующим методом. Раствор 195мг 2-[3(S)-[[N-(бензилоксикарбонил)-L-аспарагинил]-амино]-2(R)-гидрокси-4-фенилбутил]N-третичного бутилдекагидро-(4aS,8aS)-изохинолин-3(S)-карбоксамида в 20мл этанола гидрировали при комнатной температуре и атмосферном давлении в течение 18 часов над 10% - ным палладием на угле. Катализатор отфильтровывали, а фильтрат упаривали при пониженном давлении до получения 154мг 2[3(S)-[(L-аспарагинил)амино)-2(R)-гидрокси-4-фенилбутил)-N-третичного бутилдекагидро-(4aS,8aS)изохинолин-3(S)-карбоксамида, который использовали на следующей стадии без дальнейшей очистки. Пример 3. Раствор 287мг N-(2-хинолилкарбонил)-L-аспарагина и 401мг 2-(3(S)-амино-2(R)-гидрокси-4фенилбутил]-N-третичного бутилдекагидро-(4aS,8aS)-изохинолин-3(S)-карбоксамида [полученного способом, описанным в примере 1 (i) - (vii)] в 3мл тетрагидрофурана охлаждали до -10°C и добавляли 163мг 3-гидрокси-1,2,3-бензотриазин-4(3H)-она и 220мг дициклогексилкарбодиимида. Смесь перемешивали при -10°C в течение 2 часов и при температуре 20°C в течение 16 часов, затем разбавляли этилацетатом и отфильтровывали. Фильтрат промывали насыщенным раствором бикарбоната натрия, насыщенным раствором хлорида натрия, а затем упаривали. Осадок подвергали хроматографии на силикагеле с использованием 4% (по объему) метанола в дихлорметане для элюции с получением 537мг N-третичного бутилдекагидро-2-[2(R)-гидрокси-4-фенил-3(S)-[[N-(2-хинолилкарбонил)-Lаспарагинил]амино]бутил]-(4aS,8aS)-изохинолин-3(S)-карбоксамида, который был идентичен продукту, полученному в первом абзаце примера 2. Вследствие реакции вышеуказанного свободного основания с паратолуолсульфоноксикислотой и кристаллизации из метанол / этилацетата получали паратолуолсульфонат N-трет.-бутилдекагидро-2-[2(R)гидрокси-4-фeнил-3(S)-[[N-(2-xинoлилкapбoнил)-L-acпapaгинил]амино]бутил]-(4aS,8aS)-изохинолин3(S)карбоксамида в виде белого твердого вещества с точкой плавления 246 - 248°C (разложение). N-(2-хинолилкарбонил)-L-аспарагин, используемый в качестве исходного материала, получали следующим образом. Смесь 540мг сложного эфира амида янтарной кислоты и хинальдиновой кислоты и 300мг L-аспарагин моногидрата в 2мл диметилформамида перемешивали при 20°C в течение 96 часов. Растворитель удаляли испарением до получения белого твердого осадка, который интенсивно перемешивали в 10мл дихлорметана, отфильтровывали и промывали дихлорметаном. Таким образом получали 431мг N-(2хинолилкарбонил)-L-аспарагина в виде белого твердого вещества; MS: m/e 288 [M + H]+. Следующий пример иллюстрирует приготовление фармацевтического препарата, содержащего соединение формулы I или его фармацевтически приемлемой соли, в виде активного ингредиента. Пример А. Водный раствор активного ингредиента стерильно отфильтровывали, смешивали при нагревании со стерильным желатиновым раствором, который содержал фенол в качестве консерванта с применением таких количеств, чтобы получить 1,00мл конечного раствора, который содержал бы 3,0мг активного ингредиента, 150,0мг желатина, 4,7мг фенола и дистиллированной воды до 1мл. Смесь разливали в пробирки емкостью 1,0мл в условиях антисептики.
ДивитисяДодаткова інформація
МПК / Мітки
МПК: A61K 31/47, C07D 401/12, A61P 31/12, C07D 217/26
Мітки: солі, похідні, кислотно-адитивні, амінокислот
Код посилання
<a href="https://ua.patents.su/7-26029-pokhidni-aminokislot-ta-kh-kislotno-aditivni-soli.html" target="_blank" rel="follow" title="База патентів України">Похідні амінокислот та їх кислотно-адитивні солі</a>
Спосіб одержання 2-(4-метоксіфенілпропіоніл)аміно-6-н-пропіламіно-4,5,6,7-тетрагідробензотіазола у вигляді рацемата або (-)-енантіомера або його кислотно-аддітивної солі

Номер патенту: 8032
Опубліковано: 26.12.1995
Автори: Герберт Мерц, Рудольф Бауер, Іоахім Мірау, Клаус Шнейдер, Гюнтер Шингнітц, Райнер Соботта
МПК: C07D 277/82, A61K 31/425, A61K 31/428, A61P 25/00, A61P 5/00, A61P 25/18
Мітки: кислотно-аддітивної, спосіб, 2-(4-метоксіфенілпропіоніл)аміно-6-н-пропіламіно-4,5,6,7-тетрагідробензотіазола, енантіомера, вигляді, одержання, солі, рацемата
Формула / Реферат:
Способ получения 2-(4-метоксифенилпропионил)амино-6-н-пропиламино-4,5,6,7-тетра-гидробензотиазола формулыв виде рацемата или (-)-энантиомера или его кислотно-аддитивной соли, отличающийся тем, что 2-(4-метоксифенилпропионил)амино-6-оксо-те-трагидробензотиазол формулыподвергают взаимодействию с н-пропиламином в присутствии восстановителя с последующим выделением целевого продукта в виде рацемата или...
Спосіб одержання заміщеного імідазолу або його нетоксичної фармацевтично прийнятної кислотно-адитивної солі

Номер патенту: 19310
Опубліковано: 25.12.1997
Автори: Ар'ялена Кар'яланен, Арто Йоханес Кар'ялайнен, Рейно Олаві Пєльконен, Маттіантеро Ляхде, Ар'я Маркета Калапудас, Мар'я Ліса Сєдервал, Рісто Арво Сакарі Ламінтауста
МПК: A61K 31/415, C07D 233/64
Мітки: одержання, спосіб, імідазолу, заміщеного, фармацевтично, кислотно-адитивної, прийнятної, нетоксичної, солі
Формула / Реферат:
Способ получения замещенного имидазола формулы:или его нетоксичной фармацевтически приемлемой кислотно-аддитивной соли, где R1, R2, R'1 и R'2, которые могут быть одинаковыми или разными, представляют Н, СН3, С2Н5, С3Н7, ОСН3, NO2, CF3, CHF2, CH2F или галоген;где R3 представляет Η, СН3 или галоген; R4 представляет Η и R5 представляет Η или ОН или R4 и R5 вместе образуют связь; X и У, которые могут...
Спосіб одержання заміщеного імідазола або його нетоксичної фармацевтично прийнятної кислотно-аддитивної солі

Номер патенту: 13471
Опубліковано: 28.02.1997
Автори: Арво Сакарі Ламміітауста, Арто Йоханнес Карьялайнен, Маттіантеро Ляхде Рісто, Мар'я Лііса Седервалл, Ар'я Маркетта Калапудас, Ар'я Лена Кар'ялайнен, Рейно Олаві Пельконен
МПК: C07D 233/54, A61P 35/00, A61P 43/00, C07D 233/64, C07D 233/58, C07D 233/56, A61K 31/415
Мітки: прийнятної, заміщеного, нетоксичної, імідазола, фармацевтично, кислотно-аддітивної, солі, спосіб, одержання
Текст:
...(t, 1H), 6,7 (с, 4-(1,3-дифонилпропил)-1Н-имидазол. 1-бзнзілл-5-(1,3-ди-фенилпропил)-1Н- имиП р и м е р 7. дзгоя гидрогенизировали в смеси 2 н.рас4-(1,4-бис)4-фторфенил(0утііл)-1 Н-имитвора хлористоводородной кислоты и эта- 25 дазол. нала при температуро 60°С и использоваКонцентрированный водный раствор нии 10% Pd/C з качестве катализатора, формата аммония (0,16 г и 2 мл воды)добапПосла прекращения поглощения водолялся по каплям к кипящей...
Похідні аміноалкілбензолу оба їх фармацевтично прийнятні солі з кислотами

Номер патенту: 18867
Опубліковано: 25.12.1997
Автори: Філіп Гері, Сінез Жолідон, Рене Цурфлю
МПК: A61K 9/00, A61P 31/10, C07C 211/28, C07C 225/00
Мітки: прийнятні, фармацевтично, аміноалкілбензолу, похідні, оба, солі, кислотами
Формула / Реферат:
Производные аминоалкилбензола общей формулыR1где R1 и R2 каждый обозначают водород, низший алкил или вместе образует неразветвленный алкилен С2-С4; Q-С4-С11- алкилен, минимум с двумя атомами углерода между двумя свободными валентностями; Υ - простая связь, R - обозначает, что кольцо незамещено или замещено галогеном, трифторметилом, нитрогруппой или низшим алкилом, или их фармацевтически приемлемые соли с...
Похідні глутатіону або їх фармацевтично прийнятні солі, які проявляють протизапальну, протиалергійну та гепатотропну активність, і спосіб їх одержання

Номер патенту: 21900
Опубліковано: 30.04.1998
Автори: Казумі Огата, Такахіро Сакау, Сіндзі Охморі
МПК: A61K 38/00, C07K 5/02
Мітки: протиалергійну, прийнятні, солі, одержання, протизапальну, проявляють, фармацевтично, гепатотропну, активність, похідні, глутатіону, спосіб
Формула / Реферат:
1. Производные глутатиона общей формулыгде n = 0 или 1;r1 - водород или низший алкил;R2 и R3, одинаковые или различные, - гидроксил, низший алкоксил, амино- или иминогруппа, при условии, что r1 - алкил, когда n = 0, и R2 и R3, одинаковые или различные, гидроксил или низший алкоксил, или их фармацевтически приемлемые соли, обладающие противовоспалительной, противоаллергической и гепатотропной активностью.2....
Попередній патент: Спосіб одержання похідних 2,5-дихлорфенолу
Наступний патент: Консоль склоочисника для автомобілів
Випадковий патент: Спосіб боротьби або попередження пошкодження рослини, спосіб захисту матеріалу для розмноження рослин та композиція, що містить абамектин і цифлуметофен