Спосіб одержання заміщеного імідазола або його нетоксичної фармацевтично прийнятної кислотно-аддитивної солі
Номер патенту: 13471
Опубліковано: 28.02.1997
Автори: Мар'я Лііса Седервалл, Арто Йоханнес Карьялайнен, Ар'я Лена Кар'ялайнен, Арво Сакарі Ламміітауста, Ар'я Маркетта Калапудас, Рейно Олаві Пельконен, Маттіантеро Ляхде Рісто







Текст
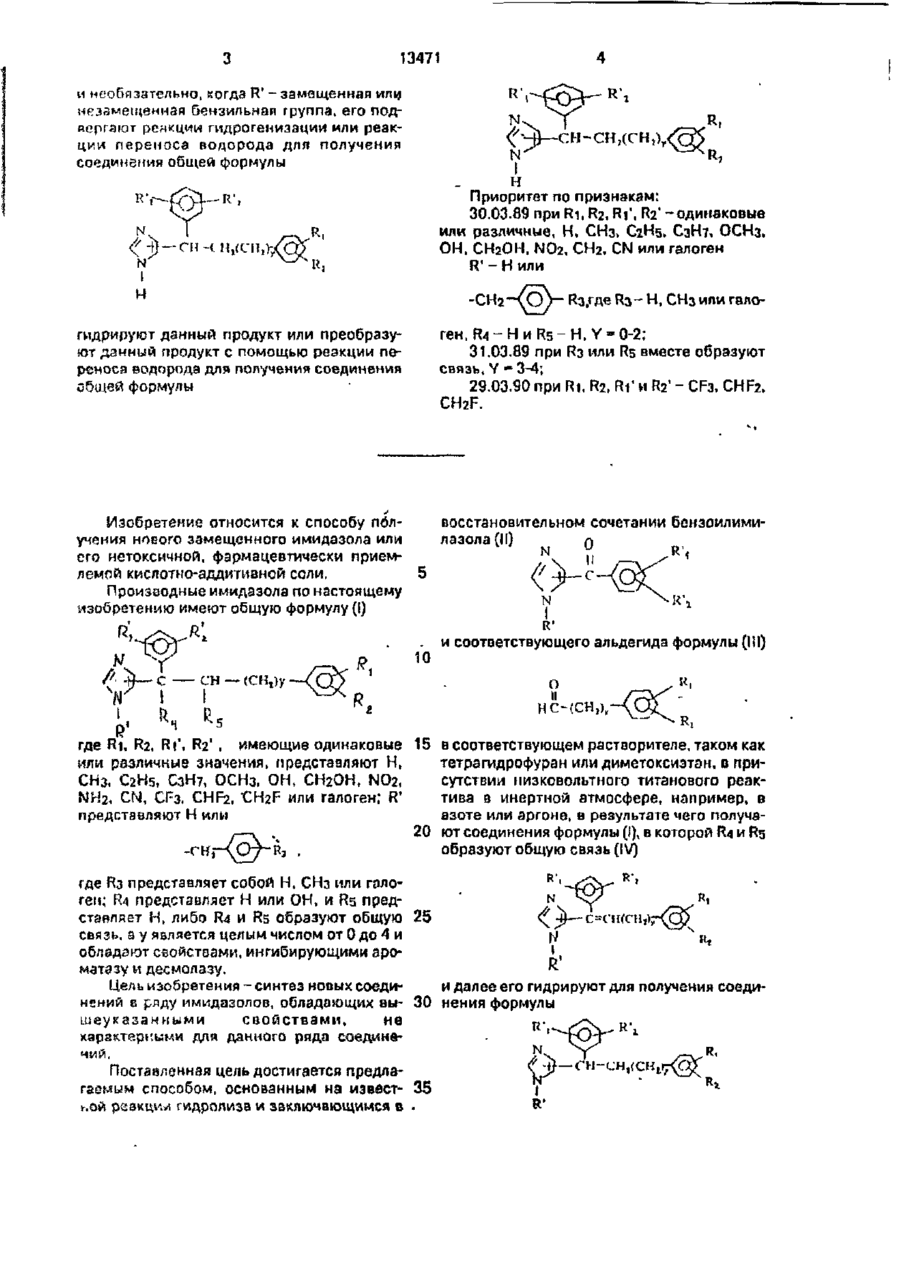
УКРАЇНА (19) (5і)5 C07D233/5S-.A61 К 31/415 ОПИС ДО ПАТЕНТУ ДЕРЖАВНЕ ПАТЕНТНЕ ВІДОМСТВО НА ВИНАХІД (54) СПОСІБ ОДЕРЖАННЯ ЗАМІЩЕНОГО ІМІДАЗОЛА АБО ЙОГО НЕТОКСИЧНО! ФАРМАЦЕВТИЧНО ПРИЙНЯТНОЇ КИСЛОТНО-АДИТИВНОТ СОЛІ 1 (20)95320801.20.09.93 (21)4895673/SU (22)25.06.91 (24)28.02.97 (62)4743449/04.29.03.90 (31)8907218.8907309 (32)30.03.89,31.03.89 (33) G В (46)28.02.97. Бюл. Г* 1 (56) Бюлер К., Пирсок Д. Органические синтезы, ч. 1.М.: Мир, 1973, с 496. (72) Арто Йохаинес Кар'ялайнен (F1), Рейно ОлаоІ Пельконен (FI), Мар'я ЛПса Седервалл (FI), Маттіантеро Ляхде РІсто (FI). Ароо Сзкарі ЛзммНтаустя (F/), Ар'я Лена Кар"ялайнен (FI). Ар'я Маркетта Калапудас^І) (73)Оріоіі-ІхтюмяОй (FI) (57) Способ получения замещенного имидазопэ общей формулы CH — (СН,)у о т л и ч а ю щ и й с я тем, что беиэоилимидазол общей формулы О N R К'. 1 с где R' имеет указанное значение, R j ' и Иг' одинаковые или разные, Н, СНз. С2Н5. СзН?. ОСНз, N02. СРз, CHF2, CH 2 F или галогон, подвергают взаимодействию с альдегидом "~ общей формулы В і и Вг-одиивкооые или разные, Н, СНз, С2Н5, СгН7. ОСНз. ОН, СН2ОН, NO2, NH2, CN, СГз. CHFa, CH2F или гаьоген; Y имеет указанное значение, в присутствии титанового реагента с низкой валентностью для получений соединений общей формули или его нетоксичной фармацевтически приемлемой кислотно-аддитионой соли, где Ri. R2, RJ' и R2' - одинаковые или разные, Н. СНз. С2Н5. СзН7. СОНз. ОН, СН2ОР, NOz. NH2. CN, CF3. CHF2. CH2F или галоген; R'-H или •енг где R3 - Н, СНз или галоген; Гїз - Н; Rs ~ Н, или Rfl и Rs вместе образуют связь; У = 0-4, и необязательно гидрируют данный продукт для получении соединения общей формулы N t R' 13471 и необязательно, когда R' - замещенная или незамещенная бензильнзп группа, его подкоргагот реакции гидрогенизации или реакции переноса водорода для получения соединения общей формулы СН-СН,(СН,)ГН Приоритет по признакам: 30.03.89 при Ri, R2, Ri', R2' -одинаковые или различные, Н, СНз, С2Н5, C3H7, ОСНз, ОН. СНгОН, N02, СНг. CN или галоген 1 R - Н или Кз.где Ra- H, СНз или гало гидрируют данный продукт или преобразуют данный продукт с помощью реакции переноса водорода для получения соединения общей формулы - Н и R5 - Н, Y - 0-2; 31.03.89 при R3 или Rs вместе образуют связь, Y -3-4; 29.03.90 при Ri, R2, R11 и R2' - CF3, CHF2, CH2F. Изобретение относится к способу получения ноеого замещенного имидазола или его нетоксичной, фармацевтически приемлемой кислотно-аддитивной соли, Производные имидазола по настоящему изобретению имеют общую формулу (I) восстановительном сочетании бензоилимилазола (II) л ген, НА N и . R', R' 10 сн — (cnt)y и соответствующего альдегида формулы (III) о и НС-(СН,), R где Ri, R2, Rt\ R2' , имеющие одинаковые или различные значения, представляют Н, СНз, С2Н5, СзН7, ОСНз. ОН, СНгОН, N02, NH 2 , CN, СРз. CHF2, -CH2F или галоген; R' представляют Н или -С К," 15 в соответствующем растворителе, таком как тетрагидрофуран или диметоксиэтэн, в присутствии низковольтного титанового реактива в инертной атмосфере, например, в азоте или аргоне, в результате чего получа20 ют соединения формулы (І), в которой R4 и Rs образуют общую связь (IV) где Из представляет собой Н, СНз или галоген; R4 представляет Н или ОН, и Rs представляет Н, либо R4 и Rs образуют общую 25 связь, а у является целым числом от 0 до 4 и обладают свойствами, ингибирующими аромата зу и десмолазу. Цель изобретения-синтез новых соедии далее его гидрируют для получения соединений в ряду имидазолов, обладающих вы- 30 нения формулы шеуказанными свойствами, не RY характерными для данного ряда соединений, Поставленная цель достигается предлагаемым способом, основанным на извест- 35 ной реакции гидролиза и заключающимся в 13471 D которых R\ Ri, R2. R11, R2' и у имеют указанные выше значения. Fc/ш Н' представляет замещенный или незамещенный бензил, то эту группу также можно удалить путем гидрогенизации. В этом случае гидрогенизацию осуществляют • кислой среде, такой как смесь хлористоводородной кислоты и этанола, при повышенной температуре. Другим способом удаления бенэильной R' группы является реакция переноса подорода, при осуществлении которой исходное соединение {Vlil) нагревают с обратным холодильником вместе с формиатом аммония и 10% Pcf/C в соответствующем спирте, таком как метанол или этанол, либо о его водном растворе. Соединения (ЇХ)такжо можно получить непосредственно из соединений (IV) D результате осуществления реакции переноса водорода при использоознии формиэта аммония или путем одновременной гидрогенизации двойной связи и защитной бензильиой группы, для получения соединений формулы: 1ST Rr. w П р и м е р 1. 1-Бензил-5(1-}4-фторфенил)-3-фснил-1пропенил)-1 Н-имидазол. Четыреххлорисшй титан (0,03 ммоля) по каплям добавляли к перемешанной суспензии порошкообразного цинка (0,06 ммолл) в тетрагидрофуране (30 мл) при температуре -10°С и атмосфере сухого азота. Эту смесь нагревали до кипения с обратным холодильником и продолжали нагревать с обратмцм холодильником D течение 1 часа. Этот раствор охлаждали до 0°С, после чего в эту смесь доба оля л и 1-6ензил-5(1-)4-фторфонил}-1-оксометил)-1Н-имидазол (0,005 моля) в тетрагидрофуране (10 мл) и фемилацетальдегид (0,006 моля) в тетрагидрофуране. Эту смесь нагревали с обратным холодильником при перемешивании в течение 1 часа. Темную смесь выливали в воду (60 мл), тетрагидрофурэи выпзрипали, а смесь экстрагировали метиленхлоридом (2xtOO мл), раствор метиленхлорида промыоали 1 н.раствором гидроокиси натрия и водой, высушивали с помощью сульфата натрия и выпаривали до сухого состояния. Остаток очищали при помощи испарительной хроматографии. Спектр Н ЯМР (в виде основания, CDCb): 3,35 и 3,45 (2cl, 28), 4,62 и 4,65 (2S, 2Н), 6,03 и 6,20 (2t. 11 і), 6,8-7,3 (m, 16Н). 7,50 и 7,64 (2S. Ж). П р и м е р 2. 4-{1,3 6ис(4-нитрофе5 нил)пропил}-1 Н-имидазол. 1,8 г (14,8 ммоля) нитрата мочевины добавляли небольшими порциями к с*лес\л 2,7 г (7,3 ноля) 4-(1,3-дифенилпропил)-1Н-имидазола в 6,4 мл концентрированной серной 10 кислоты при температуре 10°С. Реакционную смесь перемешивали е течение 2 часов при комнатной температуре. Эту смесь подщелачивали 2М раствором гидроокиси натрия и целевой продукт очищали при 15 помощи испарительной хроматографии, используя смесь метиленхлорида и метанола (95:5) о качестве зпюентз. П р и п е р 3. 4-{1-(4-фіорфенил)-5-фснилпснтил)-1Н20 имидазол. Хлоргидрат 4 (1^-фтсрфенил)-5-фенил1-пентенил)-1Н-имидазола (2,0 г) растворяли а этаноле и добавляли кртзлптическоа количество 10 Pcl/C. Реакционную смесь 25 интенсивно перемешивали при комнатной температуре Б атмосфере водорода до тех пор, пока не прекращалось поглощение водорода. Эту смесь фильтровали, в филырзт выпаривали до сухого состояния. Остаток, 30 представляющий деловой продукт, очищали при помощи испаритепьиой хроматографии, производят элюирование смесью метиленхлорида и метанола. Выход составил 82%. Споктр И ЯМР (в виде основания, 35 CDCb): 1,1-2,7 ( т . ОН). 3,84 (t, 1H), 0,7? (широкий S. 1Н), 7,70-7,38 ( т , 9Н), 7.47 (широкий S. 1 И), 0,22 (широкий S, 111). При использовании аналогичного мсто40 да были получены следующие соединения, входящие в оОъем настоящего изобретения: 4-[И^-фторфенил)-5-(3-метлфепил}пе11тил}-1 Н-имидазол, Масс-спектр: 322 (20. М\ 189 (28), 176 45 (38), 175(72), И9(їОО), 125(20), 121 (И), 109 (42), 105(16) 97(21). Спектр Н ЯМР (в виде хлористозодородноп сопи, МеОН-сі4): 1,t-2J (m, BH), 2,27 (S, ЗН), 4.06 (t, 1Н), 6,7-7,5 ( т . Ш І), 7,37 (d, 1Н). 50 8.77 (d, 1H). 4-{1-{4-фторфеііил)-5-{4-метилфенил /пеншл]-1 Н-имидазол. Спектр И ЯМР (и виде хлористоводородной соли, МеОН-сМ): 55 1,1-2,7 (гп, 8Н). 2,26 (S, ЗН), 4,05 (t, IH), 6,8-7,6 (m. 9H), 3,70 (d, 1H). 4-(1-(4-фторфсііил)-5-{2-метилфанил(пйнтил)-1 Н-имидазол. 13471 8 Масс-спектр: 322 (53. М4), 189 (30), 176 ную смесь перемешивали при комнатной (55). 175(100). 118 (IB). 121(12), 105(42), 101 температуре в течение 3 часов. Добавляли (KJ. 79 (15), 77(13). воду и отделяли слой толуола. Фазу толуола промывали водой и выпариаали до сухого Сгижір 'H ЯМР (я виде хлоригловодо* родной соли, МеОН,сі*): 1,1-2,7 (m. 8H), 2,24 5 состояния. Остаток содержал изомеры 1бєнзил^і.З-дифенилпропил^іН-имидазо (S, ЗН), 4.11 (t. 1Н), 6,9-7.5(т, 9Н), B.79(d, 1H). ла и 1-бензил-5-(1,5-дифенилпропил)-1НА~{їі (3.5-диметилфепил)}-{4-фторфенил) имидэзола. Первый изомер отделяли и очи-пентил)-1 Н-имидазол. щали при помощи испарительной Масс-спектр: 336 (47, М"1). 189 (90), 176 (07), 175(100), 166(13), 148 (1G), 121(12), 119 10 хроматографии (смесь мотиленхлорида и метанола, 9,5:0,5)06). 91(14). Спектр И ЯМР (в виде хлористоводоП р и м е р 6. родной соли, MeOH-cU): 4-{1,4-Сн1с(4-фторфенил)6утил)}-1 Н-имидазол. 1,1-2,6 (m, BH), 2,22 (S, 6H), 4,09 (t, 1H). 6,6-7,4 (m, 7H). 7,40 (широкий S, 1H), 8,80 15 1-бензил-5-(1,4-бис(4-фторфенил}-1-бу(широкий S, Ж). тенил)-1 Н-имидазол (0,2 г) п виде гидрохлоридной соли гидрировался, как описано в ен-тил}-1Н-имиддзол. примере 2. 4-{5-(3,5-диметокг.ифенип)-1-(1-фторфе *Н ЯМР (в виде основании. CDCb): нип)пентип]-Ш-имидазол. 20 1,4-1.88 (m,2H), 1,8-1,95 (МИН), 2,05-2,2 П р и м е р 4. (m, 1H), 2,5-2,65 (m, 2M). 3,87 (t, 1H), 6,7 (с, 4-(1,3-дифонилпропил)-1Н-имидазол. 1-бзнзілл-5-(1,3-ди-фенилпропил)-1Н- имиП р и м е р 7. дзгоя гидрогенизировали в смеси 2 н.рас4-(1,4-бис)4-фторфенил(0утііл)-1 Н-имитвора хлористоводородной кислоты и эта- 25 дазол. нала при температуро 60°С и использоваКонцентрированный водный раствор нии 10% Pd/C з качестве катализатора, формата аммония (0,16 г и 2 мл воды)добапПосла прекращения поглощения водолялся по каплям к кипящей смеси 1-Бензилродз реакционную смесь охлаждали, фильтровали и выпаривали до сухого состояния. 30 дазела (0,21 г) и 10% палладия на угле (0,02 г) в 20 мл этанола и поды (3:1). Смесь нагреДобавляли поду и смесь подщелачивали гидвалась с обратным холодильником в течение роокисью натрии. Полученный продукт экс2 часов. Катализатор отфильтровывался и трагировали о метиленхлориде, промывали растворитель выпаривался. Добаолялся 2М водой, высушивали с помощью сульфата натрия и выпаривали до сухого состояния. Ос- 35 NaOH и продукт экстрагировался в метиленхлорид. Метиленхлормдная фаза сушилась и таток очиіцзли при помощи испарительной выпаривалась досуха, даоая продукт, хроматографии, используя смесь метииенхСоединения по настоящему изобретеяорида и метанола (9,5:0,5) в качестве элюнию являются особенно полезным D качестонта. Выход составил 73%, Спектр 'Н fiMP (в виде основания, 40 ве ингибиторов ароматазы и поэтому успешно применяются при лечении болезCDCb): ней вызываемых экстрогенами, например, 2,1-2,7(m, 4 Н), 3,88 (t, 1H), 6,71 (широкий рака молочной железы. S, 1Н), 7,01-7,26 (m, ЮН), 7,30 (d, 1H), 10,5 (широкий 5, 1H). Экстрогены являются важными стероиПри использовании аналогичного мето 45 дами в физиологии и нормальном развитии да было получено следующее соединение; молочной железы и половых органов у женщин. С другой стороны, экстрогены стимули4-(1 ДгдмфеиилпентилН Н-иммдазол. руют рост злокачестпенных опухолей, Спектр *Н ЯМР (в виде хлористоводородной соли, MeOH-cU): вызываемых эксгрогемами, о частности зло1,2-2,3 (пі, ЄН), 2,57 (искаженный t. 2H), БО качественных опухолей молочной железы и 4,05 (t. 1H). 7,05-7.40 (m, iH).0,73(d, 1H). матки, причем они могут увеличивать опасП р и м е р 5. ность развития злокачественной опухоли 1-Бенэил-4-(1,3-дифенилпропил)-1 Н-имимолочной железы при введении в виде фардазол. мацевтических доз а течение длительного 2,0 г Єемзнлбромида в 5 мл толуола по 55 периода времени. Избыточное образование каплям добавляли к смеси 4-(1,3-дифенилпэкстрадиола также может вызывать другие ропил)-1Р-имидазола (2,6 г), 48% (аОН (10 доброкачественные нарушения в гормомомл), толуола (20 мл) и бромида тетрабутиламзависимых органах. Важное значение эксмопия (0,2 г) при комнатной температуре. трогеноа а качестве стимуляторов и/или После окончания добавления эту реакцион- регуляторов роста злокачестпенных опухо 13471 лей подчеркивается тем фактом, что энтизкстрогены занимают центральное место при лечении злокачественных опухолей молочной железы, имеющей многочисленные рецепторы экстрогена. Действие антиэкстро- 5 генов выражается в связывании рецепторов экстрогана, п результате чего ингибируется биологическое воздействие зкстрогеноа. Другим подходом, направленным на блокирование действия экстрогенов, является ин- 10 гибирование синтеза экстрогенов. Это было достигнуто в клинических условиях с помощью аминоглгатемида, ингибирующего синтез вредных стероидов. Синтез экстрогенов можно блокировать путем ингибирова- 15 ния зроматазы фермента, которая является ключевым ферментом в биохимическом синтезе зкстрогенов. Ингибировзние ароматазы имеет важное значение потому, что некоторые опухоли молочной железы синте- 20 зируют зкетродиол и экстрен в этом месте, в результате чего стимулируется постоянный рост опухоли (Алан Липтон и др. Са се" 69:779-782, 1987 г.). Способность соединений по этому изо- 25 бретению ингибировать йроматазу фермента испытывалась в лабораторных условиях по методу М.Пасанена ("Biological Research In Pregnancy", т. 6, № 2, 1985 г., стр. 94-99). В ходе исследования использовали фермент 30 с ароматазой человека. Этот фермент готовили из плаценты человека, которая отличается большим содержанием этого фермента. Микросомную фракцию (100000 х г осадка) готовили центрифугированием, Фермент- 35 иый препарат использовали без дальнейшей очистки. Испытуемые соединения добавляли вместе с 1,2/3Н/-андростен-3,17дионом, скорость распада которого составляла 100000 распадов в минуту, и системой 40 генерации А РН. Концентрации испытуемых соединений равнялись 0,001;0,01; 0,1 и 1,0 ммоль, Инкубацию производили при темперагуре 37°С в течение 40 минут. Ароматизация 1,2/3Н/-адростен-3,17-диана 45 привела к образованию НгО. Насыщенную тритием зоду и насыщенный тритием субстрат легко разделяли при помощи миниколонны "Sep-Pak", в которой происходило поглощение стероида и элюированив сво- 50 бодной воды. Счет рздиоактипности вели при помощи жидкостного сцинтилляционного счетчика. Степень ингибирооания аромзтазы определяли при сравнении радиоактивности НгО у образцов, обрабо- 55 тайных ингибитором, и у контрольных образцов, но содержащих ингибиторов. Значения средней концентрации ингибироеания (1С-50) высчитывали о виде концентраций, ингибирующих активность фермента Ю на 50%. Эти концентрации предегаолрлы н таблице 2. Актионость о отношении р.ісщоплвния боковой цепи холестерина (CSCC) десмолаза) измеряли по методу Пазанена и Пелкомена ("Стероиды") 43:517-527, 1984). Инкубацию производили я !,5 м/т пластиковых пробирках Эппондорфа с использованием в качество единого устройства вибратора Эппандорфа, центрифуги и термостата. В 300 мл инкубированном объеме готовили субстрат [5 ммолей) по методу Хакукоглу и Джефкоута (l.Chromatogr, 190:256262, 1980), в который затем добавляли радиоактивный 3Н-4-холестерин, скорость распада которого составляла 100000 распадов а минуту (чистоту этого соединения проверял и с помощью тонкослойной хроматографии)в0,5% "Тонне20", Юммол МоСіг. 5 мкмолей цизмокетона и 2 ссоля Л РН. Контрольные образцы содержали все вышеуказанные вещества, но ферментный препарат инактивирова/ш да инкубации посредством добавления 900 мхл метанола. В качестве источника фермента использовали митохондриальную фракцию (1 кг белка) из плаценты человека или надпочечников коров. После инкубации в течение 30 минут при температуро 37°С реакцию завершали путем добавлення 900 мкл метанола; в каждый термостат добавляли маркер, представляющий 14С-4-прегиенолон со скоростью распада 1500 распадов в минуту, после чего пробирки интенсивно встряхивали. После достижения равновесного СОСТОЯНИЙ а течение 10 минут осажденные метанолом белки отделяли путем центрифугирования (8000 х г о течение 2 мин), а всплывающий слой засасывали в 1 мл пластиковый шприц и переносили в предварительно уравновешенную (75% метанол) миниколонну. Эту колонну промывали одним мл 75% метанола, а затем 3 мл В0% этанола. Элюат 80% метанола помещали в счетную пробирку и добавляли 10 мл сцинтилляционной жидкости. Счет радиоактивности вели с использованием программы с двумя метками в жидкостном сцинтилляционпом счетчике (LKB RackBeta). Типичные значения активности для ферментного препарата плаценты человека и надпочечников коровы соответственно предстазляли 0,5-3 и 50-100 кмолей прегнеколона, образованного на мг белка в минуту. В эксперимеитбх по ингибировямию полученное вещество (в интервале концентраций от 1 до 100 мкмолей) добавляли к инкубационной смеси о объеме 110-20 мкл обычно в виде раствора в метаноле или зтаноле. Такой же обьом растворенного веще 12 1347 ной дозы испытуемых соединений формулы {1) равнялись - ^00 мг/кг иаи больше. Суточная доза для больного изменяется от примерно 20 до 200 мг при введении перорзльиым способом. Это изобретение иллюстрируется следующими примерами. Спектры 1 Н ЯМР определяли при помощи аппарата Брукера VVP 80 (80 мгц). Вещество, выбранное для сравОструю токсичность, среднюю легальную доз/, определяли с использованием мо- 10 нения, представляло тетраметнлеилан. лодых взрослых самок мышей вида МР1. Масс-спектры опродоляли при помощи апВведение испытуемых соединений произвопарата Кратоса М 80РГ с автоматическим дили переральмо. Значения средней летальпультом. стеа добавляли в контрольную инкубационную пробирку, Значенич сродней концентрации иншбировэния (концентрация, вызывающая 50% ингийироаание) опредеяч/іи графически, и получонныа результаты представлены в таблице 2. В таблице 1 указаны соединения, которые были испытаны. Таблица 1 15 Испытание соединений Название 6 4-(1,Ь-дифенилпентил)-1Н-имилазол 4-{1,3-дифснигіпропмл)-1Н-имидазал 1-бє.ізпл В-(1,3-дифенилпропііл}-1Н-ґмидазол 4-МЧ4-фторфснил)-3-фе(иіяпропил)-1Н-имидазол ^-(і-Н-фгорфенилН'феиилбутил^іН-имидазол 4-(Ь)4-фторфені1л)-5-)3-нетоксифенил)пентил)-1Н-имидазол 4-(1-)4-фторфенил}5-}4-мстоксифенил)пеитиі)-1Н-имчдазол 4-{1-}4-фторфенил)-5-)2,6-лиметилфенил)пентил)-1Н-ипидззол 4-{1,3-бис)4-фторфенил)пропил)-1Н-имидазал 4-{1.4-бис)4-фгорфснил)бутил)-1Н-имидазол 9 10 11 12 13 14 15 Таблица 2 рромзгазы и диомолззы о ирганизме человека (ССС) под действием испытуемых соединений. Средняя концентрация иигибировапия (1С-50) предсіавляет концентрацию, которая ингибирует 50 % фермента № соединения 2 3 4 5 6 7 8 9 К) И 12 13 14 1С-50 мкмоль/л 1C-50cSOC 2,8 3,5 3,8 19 16 57 170 80 8,5 7,5 25 4,5 * 7,5 30 0,72 0,70 2,6 3 7J 2,2 0.G3 22 31 12 22,5 31 22 29 13471 Упорядник Замовлення Техред М.Моргентал 4117 Коректор Л.Лукач Тираж Підписне Державне патентне відомство України, 254655, ГСП, КиГв-53, Львівське пл., 8 Відкрите акціонерне товариство "Патент", м. Ужгород, оул.ГагарІна, 101
ДивитисяДодаткова інформація
МПК / Мітки
МПК: A61P 43/00, C07D 233/58, C07D 233/64, C07D 233/54, A61P 35/00, A61K 31/415, C07D 233/56
Мітки: фармацевтично, нетоксичної, заміщеного, спосіб, імідазола, прийнятної, кислотно-аддітивної, одержання, солі
Код посилання
<a href="https://ua.patents.su/8-13471-sposib-oderzhannya-zamishhenogo-imidazola-abo-jjogo-netoksichno-farmacevtichno-prijjnyatno-kislotno-additivno-soli.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання заміщеного імідазола або його нетоксичної фармацевтично прийнятної кислотно-аддитивної солі</a>
Спосіб отримання поліциклічного ефірного антибіотика або його фармацевтично прийнятної катіонної солі

Номер патенту: 8042
Опубліковано: 26.12.1995
Автори: Алєксандр Кроссан Гоуді, Найджел Дерек Артур Волш
МПК: C12P 19/60, A61P 31/04, C12P 19/00, C07D 493/10, A61K 31/335, A61P 33/02, C12P 19/02, C07H 19/01
Мітки: ефірного, антибіотика, фармацевтично, отримання, прийнятної, поліциклічного, спосіб, солі, катіонної
Формула / Реферат:
Способ получения полициклического эфирного антибиотика формулы I:или его фармацевтически приемлемой катионной соли, отличающийся тем, что соединение формулы IIподвергают контролируемому кислотному гидролизу действием 1.1 мол. эквивалента п-толуол-сульфоновой кислоты на 1 мол. эквивалент соединения формулы II в среде растворяющей смеси, содержащей 95 мас. % ацетонитрила и 5 мас. % воды, и, если необходимо,...
Спосіб отримання циклоалкомедів (8-beta)-1алкіл-6-(заміщеного) ерголіну або їх фармацевтично придатних кислотно-адитивних солей

Номер патенту: 6022
Опубліковано: 29.12.1994
Автори: Віллям Лі Гарбрехт, Марк Мортенсен Форман, Джіффорд Пурнелл Марзоні, Кетлін Роуз Віттен
МПК: C07D 457/00
Мітки: отримання, спосіб, солей, фармацевтично, ерголіну, 8-beta)-1алкіл-6-(заміщеного, кислотно-адітивних, придатних, циклоалкомедів
Формула / Реферат:
Способ получения циклоалкиламидов (8-b)-І-алкил-6-(замещенного) эрголина общей формулыгде R1 - С1-С4-алкил; R2 - С1-С4-алкил с прямой цепью; R3 - водород или С1-С4-алкил с прямой цепью; R4 -водород, С1-С4-алкил, оксигруппа или С1-С4-алкоксигруппа; m=0,1 или 2,при условии, что когда R1 и R2каждый оначает метил, а R3 и R4каждый оначает водород, m не равно 0; или их фармацевтически...
Спосіб одержання 2-(2,4-діфторфеніл)-1,3-біс-(1н-1,2,4-тріазол-1-іл)-пропан-2-ола або його фармацевтично допустимій солі

Номер патенту: 8019
Опубліковано: 26.12.1995
Автор: Кеннет Річардсон
МПК: A61K 31/41, C07D 249/08
Мітки: солі, спосіб, одержання, допустимій, 2-(2,4-діфторфеніл)-1,3-біс-(1н-1,2,4-тріазол-1-іл)-пропан-2-ола, фармацевтично
Формула / Реферат:
Способ получения 2- (2,4-дифторфенил)-1,3-бис-(1Н-1,2,4-триазол-1-ил)-пропан-2-ола формулыили его фармацевтически допустимой соли, отличающийся тем, что 1,2,4-триазол при 50-120°С вводят в присутствии основания, такого как карбонат калия, во взаимодействие с соединением общей формулыгде R-ОН;Х и Y каждый - галоид, или R и Х - вместе кислород, а Y - радикал формулыи в случае, когда Х и Y -...
Спосіб отримання похідних імідазола або його фармацевтично прийнятих солей

Номер патенту: 2697
Опубліковано: 26.12.1994
Автори: Джон Джонес Вітотес Данкіе, Дейвід Джон Керіні
Мітки: фармацевтично, солей, спосіб, імідазола, прийнятих, отримання, похідних
Текст:
...5 Гц). пластинках для ТСХ. Масс-спектр 325. ЯМР (200 мГц, Е. 1~(4~Аминобензил)~5-гидроксиме3 э 8,00-6,80 (м, 8Н), тил-2-(2*-метоксиэтил~-4-«зшоримидазол. 5,15 ( с , 2Н>, 4,45 ( с , 2Н), 3,60 ( т , Соединение СИНТеЗИРУЮТ П Примеру О 2Н, 5 Г ц ) , 3,15 ( с , ЗН), 2,75 54, Е из 5-тидроксимет'ил—2~(2-меток~ ( т , 2Н, 5 Г ц ) 0 П р и м е р ы сиэгил)— 1 — (4—нитроб ензил) — 4—хлорими— 57—71„ Соединения, синтезированные по дазола ( 2 , 2 г, 6,75...
Спосіб одержання 2-(4-метоксіфенілпропіоніл)аміно-6-н-пропіламіно-4,5,6,7-тетрагідробензотіазола у вигляді рацемата або (-)-енантіомера або його кислотно-аддітивної солі

Номер патенту: 8032
Опубліковано: 26.12.1995
Автори: Райнер Соботта, Рудольф Бауер, Гюнтер Шингнітц, Клаус Шнейдер, Іоахім Мірау, Герберт Мерц
МПК: A61P 25/18, A61P 5/00, A61K 31/428, A61P 25/00, C07D 277/82, A61K 31/425
Мітки: спосіб, вигляді, 2-(4-метоксіфенілпропіоніл)аміно-6-н-пропіламіно-4,5,6,7-тетрагідробензотіазола, рацемата, одержання, кислотно-аддітивної, солі, енантіомера
Формула / Реферат:
Способ получения 2-(4-метоксифенилпропионил)амино-6-н-пропиламино-4,5,6,7-тетра-гидробензотиазола формулыв виде рацемата или (-)-энантиомера или его кислотно-аддитивной соли, отличающийся тем, что 2-(4-метоксифенилпропионил)амино-6-оксо-те-трагидробензотиазол формулыподвергают взаимодействию с н-пропиламином в присутствии восстановителя с последующим выделением целевого продукта в виде рацемата или...
Попередній патент: Спосіб ремонту ушкоджень емалевого покриття
Наступний патент: Спосіб складання електричної машини
Випадковий патент: Спосіб очищення тритієвої води