Спосіб одержання нуклеозидів
Номер патенту: 63956
Опубліковано: 16.02.2004
Автори: Бандару Раджанікант, Еверетт Деврон, Рамасамі Кандасамі





Формула / Реферат
1. Спосіб одержання нуклеозиду, який відповідає структурі А, за яким
,
забезпечують сполукою, яка відповідає структурі Б
,
L структури Б піддають реакції з утворенням структури Г, що має гетероциклічне кільце, в одну стадію та
ароматизують це гетероциклічне кільце в одну стадію, де А являє собою О, де L являє собою -CN; Χ являє собою S; R1, R2 і R3 являють собою Н; R4 являє собою Η або нижчий алкіл; В1, B2 і В3 незалежно являють собою блокувальні групи або нижчий алкіл та Z1, Z2 і Z3 являють собою Н та де стадія реакції L структури Б з утворенням структури Γ містить в собі взаємодію структури Б зі структурою В, де XY являє собою SH і R4 являє собою нижчий алкіл
.
2. Спосіб за п. 1, який відрізняється тим, що сполука, яка відповідає структурі Б, являє собою структуру Д
3. Спосіб за п. 2, який відрізняється тим, що стадія реакції L з утворенням структури Г містить в собі взаємодію структури Д зі структурою В
,
де Y являє собою H або нижчий алкіл.
4. Спосіб за п. 1, який відрізняється тим, що далі сполуку структури Е
ароматизують.
5. Спосіб за п. 4, який відрізняється тим, що стадія ароматизування містить в собі обробку сполуки структури Е
активованим діоксидом марганцю.
6. Спосіб за п. 5, який відрізняється тим, що зазначена сполука містить ізопропіліденову групу і цю ізопропіліденову групу видаляють шляхом обробки реагентом, вибраним із групи, що складається з трифторооцтової кислоти, мурашиної кислоти, оцтової кислоти, Н+-смоли в органічному розчиннику і йоду в метанолі.
7. Спосіб за будь-яким з пп. 1-6, який відрізняється тим, що зазначений нуклеозид далі піддають реакції з утворенням тіазофурину.
8. Спосіб за будь-яким з пп. 1-6, який відрізняється тим, що сполука структури Б є бета-ізомером.
9. Спосіб одержання нуклеозиду, за яким цукор структури Ж піддають взаємодії з етилбромпіруватом згідно з реакцією Б
.
10. Спосіб за п. 9, який відрізняється тим, що зазначений нуклеозид далі піддають реакції з утворенням тіазофурину.
11. Спосіб за п. 9, який відрізняється тим, що сполука структури Ж є бета-ізомером.
Текст
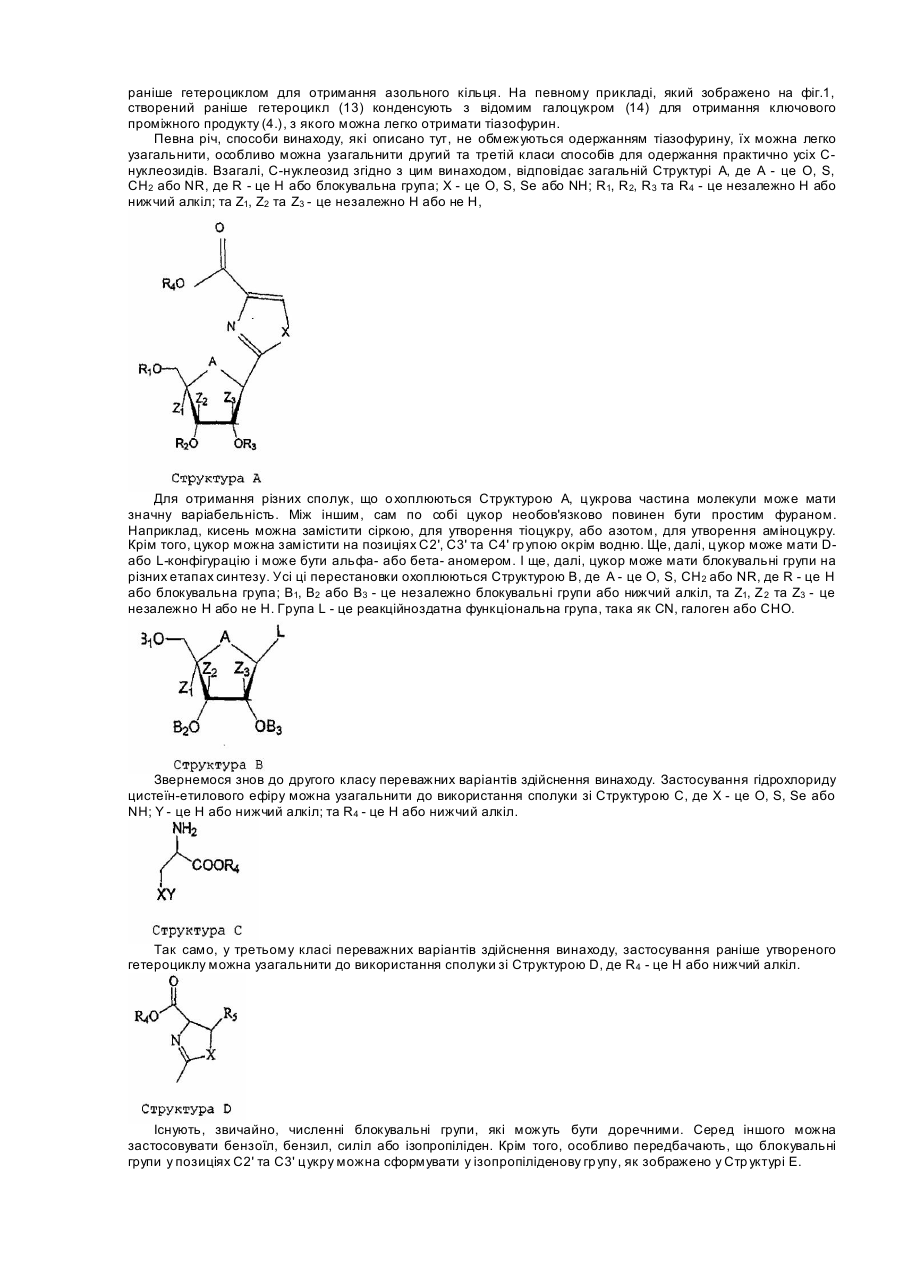
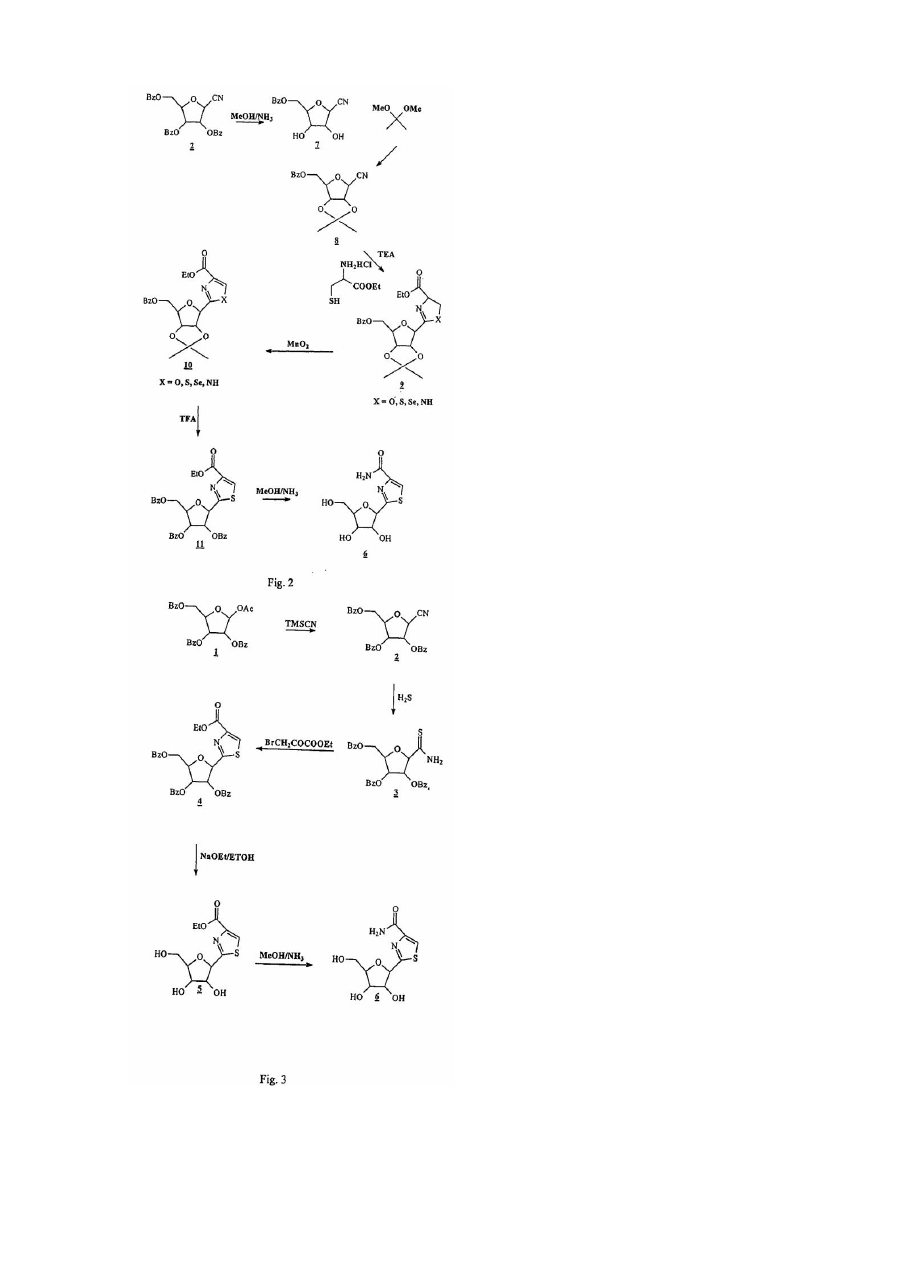
С-нуклеозиди - це цікаві сполуки, що мають потенційну активність як фармацевтичні агенти. Одна з цих сполук, тіазофурин, [6,2-(β-D-рибофуранозил)тіазол-4-карбоксамід)], має значну активність як проти лімфоїду (F.Earle and R.I.Glazer, Cancer Res., 1983, 43 133), клітинних ліній пухлини легенів (D.N.Carnex, G.S.Abluwalia, Η.Ν.Jayaram, D.A.Cooney and D.G.Johns, J.Clin. Invest., 1985, 75 175) людини, так і проти раку яєчників людини, імплантованого щурам (J.P.Micha, P.R.Kucera, С.N.Preve, Μ.Α.Rettenmaier, J.A.Stratton, P.J.DiSaia, Gynecol. Oncol. 1985, 21, 351). Тіазофурин також був ефективним під час лікування гострого лейкозу кісткового мозку (G.Т.Tricot, Η.N.Jasyaram, С.R.Nichols, К.Pennington, Ε.Lapis, G.Weber and R.Hoffman, Cancer Res. 1997, 47 4988). Крім того, за останній час дослідження викликали інтерес до тіазофурину, як можливого лікувального засобу для пацієнтів з хронічним лейкозом кісткового мозку (CML) у бластному кризі (G.Weber, патент США №5,405,837; 1995). У клітинах тіазофурин перетворюється у свій активний метаболіт, тіазол-4-карбоксамід аденін динуклеотид (TAD), який інгібує IMP дегідрогеназу, внаслідок чого виснажує нуклеотидні пули гуанозину (Е.Olah, Y.Natusmeda, Т.Ikegami, Z.Kote, Μ.Horanyi, I.Szelenye, Ε.Paulik, Τ.Kremmer, S.R.Hollan, J.Sugar and G.Weber, Proc. Natl Acad. Sci. USA, 1988, 85, 6533). Незважаючи на те, що тіазофурин був відомим протягом більш ніж 15 років, і у наш час знаходиться на ІІ/ІІІ фазі випробувань на людині, досі не існує придатного способу синтезу для широкомасштабного виробництва. Спочатку тіазофурин синтезували переважно за М.Fuertes та інші (J.Org. Chem., 1976, 41, 4076) та за Srivastava та інші (J.Med. Chem., 197'7, 20, 256) у невеликій кількості. З використанням обох способів автори отримували побічні продукти (а саме сполуку 12) і для очищення цих продуктів застосовували хроматографію на колонках на кожному етапі. Принциповим недоліком цих методів є утворення похідної фурану, а також застосування високотоксичного сірководневого газу. W.J.Harmon та інші (J.Org. Chem., 1985, 50, 1741) розробили декілька інший спосіб, під час якого вихід тіазофурину становив 19%. Внаслідок застосування способу Hannon вихід тіазофурину також є низьким, використовують H2S і очищення шляхом хроматографії. Пізніше P.Vogel та інші (Helv. Chem. Acta., 1989, 72, 1825) синтезували тіазофурин способом, що складався з дев'яти етапів, з виходом 25%. Ще пізніше D.С.Humber та інші (J.Chem. Soc. Perkin Trans. 1, 1990, 283) розробили синтез для Тізофурину, почавши з бензил(2,3,5-три-О-бензиоїл-b-D-рибофуранозил)пеніцилінату. Єдиний відомий спосіб, що є практично придатним для крупномасштабного виробництва, розроблено Parsons та іншими (US 4,451,684). Нажаль, спосіб Parsons застосовує як ціанід ртуті, так і сірководень, з якими пов'язані проблеми безпечності та навколишнього середовища. Спосіб за Parsons також дає суміш продуктів. Проблеми, що обговорюються вище стосовно великомасштабного виробництва тіазофурину, є актуальними і для великомасштабного виробництва інших С-нуклеозидів. Під час виробництва тіокарбоксамідів, наприклад, у більшості відомих способів застосовується газоподібний сірководень як реагент для перетворення ціаногрупи у відповідну тіокарбоксамідну групу. Цим способам притаманні екологічні проблеми. У виробництві С-нуклеозидів практично більшість або усі відомі способи синтезу дають суміш продуктів під час етапу замикання циклу. Отже, досі існує потреба у новому способі для великомасштабного виробництва тіазофурину та інши х С-нуклеозидів. Суть винаходу Цим винаходом пропонується новий спосіб синтезування С-нуклеозидів, у якому позицію С 1 цукру перетворюють у похідне у єдиному етапі з отриманням гетероциклу, а потім гетероцикл ароматизують у іншому єдиному етапі. Згідно з одним класом переважних варіантів здійснення винаходу ціаноцукор перетворюють у тіокарбоксамід, а потім конденсують для утворення азольного кільця. Згідно з другим класом переважних варіантів здійснення винаходу ціаноцукор конденсують амінокислотою для створення азольного кільця. Згідно з третім класом переважних варіантів здійснення винаходу галоцукор конденсують з попередньо утвореним гетероциклом для отримання азольного кільця. Цей винахід має багато переваг. Однією з переваг є те, що цей спосіб не потребує застосування газоподібного сірководню, що є небезпечним для навколишнього середовища. Інша перевага полягає у тому, що ви хід від такого способу є значно кращим порівняно з попередніми способами. Третя перевага полягає у тому, що цей спосіб виключає необхідність у процедурі хроматографічного очищення, що знижує вартість виробництва. Ці та різні інші цілі, ознаки, аспекти та переваги цього винаходу стануть більш зрозумілими завдяки наступному докладному опису переважних варіантів здійснення винаходу разом із супровідним ілюстративним матеріалом, у якому однакові номери відповідають однаковим компонентам. Стислий опис ілюстративного матеріалу Фіг.1 - це ряд охем реакції, що демонструє різні варіанти здійснення цього винаходу. Фіг.2 - це інший ряд схем реакції, що демонструє різні варіанти здійснення цього винаходу. Фіг.3 - це інший ряд схем реакції, що демонструє різні варіанти здійснення цього винаходу. Докладний опис Існує три переважних класи способів для здійснення цього винаходу; приклади кожного з них щодо виробництва тіазофурину наведено у фіг.1, 2 та 3. Згідно з першим переважним класом варіантів здійснення винаходу ціаноцукор перетворюють у тіокарбоксамід, а потім конденсують для утворення азольного кільця. На певному прикладі, який зображено на фіг.1, блокований ціаноцукор (2) перетворюють у тіокарбоксамід (3), а потім конденсують з етилбромпіруватом, внаслідок чого отримують проміжний продукт (4). Спосіб, який зображено, дає тіазофурин у кількісному виході без будь-яких побічних продуктів (12 або a-аномеру 4). Згідно з другим переважним класом варіантів здійснення винаходу ціаноцукор конденсують з амінокислотою для отримання азольного кільця. На певному прикладі, який зображено на фіг. 2, відомий ціано (8) конденсують з гідрохлоридом цистеїн-етилового ефіру, внаслідок чого отримують продукт (9) з замкнутим циклом, який потім ароматизують активованим діоксидом марганцю для отримання проміжного продукту (10). Цей ключовий проміжний продукт (10) звичайним чином перетворюють у тіазофурин з гарним виходом. Згідно з третім переважним класом варіантів здійснення винаходу галоцукор конденсують з утвореним раніше гетероциклом для отримання азольного кільця. На певному прикладі, який зображено на фіг.1, створений раніше гетероцикл (13) конденсують з відомим галоцукром (14) для отримання ключового проміжного продукту (4.), з якого можна легко отримати тіазофурин. Певна річ, способи винаходу, які описано тут, не обмежуються одержанням тіазофурину, їх можна легко узагальнити, особливо можна узагальнити другий та третій класи способів для одержання практично усіх Снуклеозидів. Взагалі, С-нуклеозид згідно з цим винаходом, відповідає загальній Структурі А, де А - це O, S, СН2 або NR, де R - це Η або блокувальна група; X - це O, S, Se або NH; R1, R2, R3 та R4 - це незалежно Η або нижчий алкіл; та Z1, Z2 та Z3 - це незалежно Η або не Н, Для отримання різних сполук, що о хоплюються Структурою А, цукрова частина молекули може мати значну варіабельність. Між іншим, сам по собі цукор необов'язково повинен бути простим фураном. Наприклад, кисень можна замістити сіркою, для утворення тіоцукру, або азотом, для утворення аміноцукру. Крім того, цукор можна замістити на позиціях С2', С3' та С4' гр упою окрім водню. Ще, далі, цукор може мати Dабо L-конфігурацію і може бути альфа- або бета- аномером. І ще, далі, цукор може мати блокувальні групи на різних етапах синтезу. Усі ці перестановки охоплюються Структурою В, де А - це O, S, СН2 або NR, де R - це Η або блокувальна група; Β1, Β2 або В3 - це незалежно блокувальні групи або нижчий алкіл, та Ζ1, Ζ 2 та Ζ3 - це незалежно Η або не Н. Група L - це реакційноздатна функціональна група, така як CN, галоген або СНО. Звернемося знов до другого класу переважних варіантів здійснення винаходу. Застосування гідрохлориду цистеїн-етилового ефіру можна узагальнити до використання сполуки зі Структурою С, де X - це O, S, Se або ΝΗ; Υ - це Η або нижчий алкіл; та R4 - це Η або нижчий алкіл. Так само, у третьому класі переважних варіантів здійснення винаходу, застосування раніше утвореного гетероциклу можна узагальнити до використання сполуки зі Структурою D, де R 4 - це Η або нижчий алкіл. Існують, звичайно, численні блокувальні групи, які можуть бути доречними. Серед іншого можна застосовувати бензоїл, бензил, силіл або ізопропіліден. Крім того, особливо передбачають, що блокувальні групи у позиціях С2' та С3' цукру можна сформувати у ізопропіліденову гр упу, як зображено у Стр уктурі Е. Цю ізопропіліденову гр упу можна видалити багатьма способами, до яких належить обробка реагентом, що обирають з групи, що складається з трифторооцтової кислоти, мурашиної кислоти, оцтової кислоти, полімеру Н+ в органічному розчиннику або йоду у метанолі. Внаслідок застосування цього способу до Структури Ε можна отримати сполуку зі Структурою F, де R5 - це Н, нижчий алкіл, амін або арил. Варіант здійснення винаходу також включає ароматизацію Структури F активованим діоксидом марганцю або іншими реагентами з наступним деблокуванням захисних гр уп для отримання тіазофурину або споріднених С-нуклеозидів. Особливо переважні варіанти здійснення згідно зі змістом цього винаходу включають Реакцію А або Реакцію В, які зображено нижче. Ці та інші ознаки можна зрозуміти за допомогою наступних робочих прикладів, які пояснюють різні аспекти суті винаходу, проте вони не обмежують об'єм формули винаходу. Експериментальна частина Приклад №1 2,3,5-Три-O-бензоїл-b-D-рибофуранозил-1-карбонітрил (2): Суміш 1-O-ацетил-2,3,5-три-O-бензоїл-b-D-рибофуранози (висушеної при 60°С, 1мм, 12 годин) (630г, 1,249 моль), триметилсилілціаніду (висушеного над молекулярним ситом, 24 години) (250мл, 1,875моль) та дихлорометану (висушеного над сульфатом магнію і витриманого над молекулярним ситом) (1,25л) перемішали та охолодили до 0-2°С. Хлорид олова (50мл, 0,425моль) повільно додавали (1,5 годину), підтримуючи реакційну температуру 0-2°С, отриману суміш перемішували і залишили ще на 1,5 години при -50°С. Реакційну суміш повільно додавали (30 хвилин), сильно перемішуючи, у холодний (5°С) 10% розчин гідроксиду натрію (1,5л), температура якого під час додавання залишилася у межах 5-8°С. Шари відокремили і органічний шар промили водою (3x500мл), доки він не-став нейтральним, а потім висушили над безводним сульфатом магнію (приблизно 150г). Суміш профільтрували і сушильний агент промили дихлорометаном (3x500мл). Фільтрат і змиви об'єднали і розчин концентрували (
ДивитисяДодаткова інформація
Назва патенту англійськоюA method for preparing thiazofurine and other c-nucleosides
Назва патенту російськоюСпособ получения тиазофурина и других с-нуклеозидов
МПК / Мітки
МПК: A61P 35/02, C07H 19/04, C07H 9/00, A61K 31/427, C07D 417/04
Мітки: спосіб, одержання, нуклеозидів
Код посилання
<a href="https://ua.patents.su/7-63956-sposib-oderzhannya-nukleozidiv.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання нуклеозидів</a>
Спосіб одержання збагачених бета-аномером нуклеозидів

Номер патенту: 41261
Опубліковано: 17.09.2001
Автори: Ларрі Вейн Хертел, Кора Сю Гроссман, Річард Елмер Холмс, Лаурі Мішель Потіт, Томас Едвард Мебрі, Дуглас Петтон К'єлл, Чарлз Девід Джоунс, Та-Сен Чоу
МПК: C07H 19/06, C07H 19/056, C07H 19/14, C07H 19/04, C07H 1/00, C07H 19/16
Мітки: одержання, бета-аномером, нуклеозидів, збагачених, спосіб
Формула / Реферат:
1. Способ получения обогащенных бета-аномером нуклеозидов общей формулы (I):где Т- фтор,R представляет нуклеозид, выбранный из группы, состоящей из радикаловгде R1 выбран из группы, состоящей из водорода, алкила, замещенного алкила и галогена,R2 выбран из группы, состоящей из гидрокси, галогена, первичного амина или вторичного амина,R4 , R5 и R6 независимо выбраны из группы, состоящей из...
Блок-сополімер, що містить полісилоксан та спосіб його одержання

Номер патенту: 62981
Опубліковано: 15.01.2004
Автори: Руохонен Яркко, Ала-Сорварі Юха, Юкарайнен Харрі, Лехтінен Матті, Сеппяля Юкка
МПК: C08G 77/00, C08G 81/00, C08F 290/00
Мітки: спосіб, одержання, полісилоксан, містить, блок-сополімер
Формула / Реферат:
1. Блок-сополімер, що містить полісилоксан та характеризується формулоюТ(АВ)х(АТ) (І),в якійА = -(SiR'R"O)nSiR'R"-, де R' та R" однакові або різні і являють собою нижчу алкільну або фенільну групу, де зазначена алкільна або фенільна група може бути заміщеною або незаміщеною;В - поліалкіленоксид загальної формули
Трициклічні бензазепіни як судинозвужувальні антагоністи, спосіб їх одержання, фармацевтична композиція та спосіб лікування захворювань

Номер патенту: 54383
Опубліковано: 17.03.2003
Автори: Сум Фук-Вах, Венкатесан Аранапакам Мудумбай, Дусза Джон Пол, Олбрайт Джей Дональд
МПК: A61K 31/495, C07D 487/04, C07D 487/14, A61P 9/12, C07D 471/04, C07D 471/14, A61P 1/16, A61P 7/08, A61K 31/55, A61P 9/00
Мітки: антагоністи, бензазепіни, фармацевтична, захворювань, одержання, лікування, судинозвужувальні, композиція, трициклічні, спосіб
Формула / Реферат:
1. Трициклічні бензазепіни загальної формули (І): (I),де Y означає фрагмент, вибраний з: -(СН2)n-, де n означає ціле число від 0 до 2,>СН-нижчий алкіл (C1-С3) і -С(O)-;А-В означає фрагмент, вибраний з -(CH2)m-NR3- і -NR3-(CH2)m-,де m означає ціле число від 1 до 2 за умови, що, коли Y означає -(СН2)n-, і n означає 2, m може бути 0, і коли n означає 0, то m може також означати 3, за умови, що, коли Y...
Тієнілазолілалкоксіетанаміни, спосіб їх одержання, проміжні продукти для їх одержання та фармацевтична композиція

Номер патенту: 58589
Опубліковано: 15.08.2003
Автори: ФРІГОЛА КОНСТАНСА Хорді, МЕРСЕ-ВІДАЛЬ Рамон, АНДАЛУЗ-МАТАРО Блас
МПК: A61K 31/381, A61K 31/4155
Мітки: проміжні, тієнілазолілалкоксіетанаміни, продукти, фармацевтична, спосіб, одержання, композиція
Формула / Реферат:
1. Похідна тієнілазолілалкоксіетанаміну загальної формули (І):, (І)деR1 - атом водню або атом галогену, або алкільний радикал з 1-4 атомами вуглецю;R2, R3 та R4, незалежно, - атом водню або алкільний радикал з 1-4 атомами вуглецю; таAz являє собою азотвмісне гетероциклічне ароматичне п'ятичленне N-метил-заміщене кільце, до складу якого входять від одного до трьох атомів азоту, загальної формули...
Похідні циклопептидів, спосіб їх одержання, фармацевтична композиція, спосіб її одержання

Номер патенту: 55439
Опубліковано: 15.04.2003
Автори: ФІТТШЕН Клаус, ГОДМАН Сімон, ХЬОЛЬЦЕМАНН Гюнтер
МПК: C07K 7/56
Мітки: фармацевтична, композиція, одержання, циклопептидів, спосіб, похідні
Формула / Реферат:
1. Сполуки формули Іцикло(Arg-X-Asp-R1), (I)в якійХ являє собою Gly, Ala або NH-NH-CO,причому можуть братися до уваги також похідні вказаних амінокислот, і залишки амінокислот з'єднані один з одним через -аміно- і -карбоксигрупи по типу пептидного зв'язку,R1 являє собою залишок формули II(II),R2, R3, R4 кожний незалежно один від одного являє собою Н, А, Аr, R5-Ar, Het абоR5-Het,А...
Попередній патент: Спосіб приготування будівельної суміші на основі гіпсу-напівгідрату
Наступний патент: Роликовий вузол ввідної роликової коробки
Випадковий патент: Спосіб діагностики розвитку атеросклерозу