Спосіб одержання (-)-(s)-3-[1-(диметиламіно)етил]феніл-n-етил-n-метилкарбамату та його гідротартрату
Номер патенту: 79972
Опубліковано: 10.08.2007
Автори: Гайічек Йосеф, Сімек Станіслав, Степанкова Гана







Формула / Реферат
1. Спосіб одержання (-)-(S)-3-[1-(диметиламіно)етил]феніл-N-етил-N-метилкарбамату, тобто ривастигміну, формули II
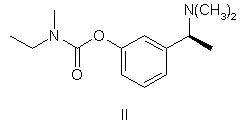 ,
,
який відрізняється тим, що метоксіацетофенон формули VI
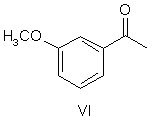
піддають відновному амінуванню з отриманням сполуки формули V
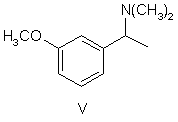 ,
,
яку потім О-дезалкілують з отриманням рацемічного аміну формули IV
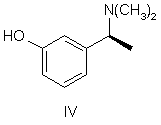 ,
,
який далі розділяють шляхом взаємодії з оптично активною кислотою, після чого бажаний відповідний діастереоізомер кристалізують і, нарешті, перетворюють в сполуку формули III
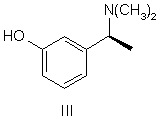 ,
,
яку, в свою чергу, піддають взаємодії, необов'язково в формі її лужної солі, із сполукою формули VII
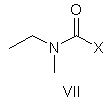 ,
,
де X являє собою групу, що видаляється.
2. Спосіб одержання (-)-(S)-3-[1-(диметиламіно)етил]феніл-N-етил-N-метилкарбамату гідротартрату формули І
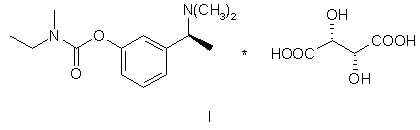 ,
,
який відрізняється тим, що метоксіацетофенон формули VI
піддають відновному амінуванню з отриманням сполуки формули V
,
яку потім О-дезалкілують з отриманням рацемічного аміну формули IV
,
який далі розділяють шляхом взаємодії з оптично активною кислотою, після чого бажаний відповідний діастереоізомер кристалізують і, нарешті, перетворюють в сполуку формули III
,
яку, в свою чергу, піддають взаємодії, необов'язково в формі її лужної солі, із сполукою формули VII
,
де X являє собою групу, що видаляється, з одержанням сполуки формули (ІІ)
,
яку далі перетворюють у відповідну сіль формули (І)
.
3. Спосіб за п. 1, який відрізняється тим, що рацемічний амін формули IV розділяють шляхом взаємодії з (S)-(+)-камфор-10-сульфокислотою.
4. Спосіб за п. 3, який відрізняється тим, що рацемічний амін формули IV розділяють шляхом взаємодії з 1 еквівалентом (S)-(+)-камфор-10-сульфокислоти.
5. Спосіб за п. 3, який відрізняється тим, що рацемічний амін формули IV розділяють шляхом взаємодії з 0,6 еквівалента (S)-(+)-камфор-10-сульфокислоти.
6. Спосіб за будь-яким з попередніх пунктів, який відрізняється тим, що отриманий бажаний відповідний діастереоізомер додатково перекристалізовують.
7. Спосіб за п. 6, який відрізняється тим, що отриманий бажаний відповідний діастереоізомер додатково перекристалізовують з етилацетату, необов'язково в суміші з етанолом.
Текст
УКРАЇНА (19) UA (11) 79972 (13) C2 (51) МПК (2006) C07C 215/00 C07C 271/44 (2007.01) МІНІСТЕРСТВО ОСВІТИ І НАУКИ УКРАЇНИ ДЕРЖАВНИЙ Д ЕПАРТАМЕНТ ІНТЕЛ ЕКТУАЛЬНОЇ ВЛАСНОСТІ ОПИС ДО ПАТЕНТУ НА ВИНАХІД (54) СПОСІБ ОДЕРЖАННЯ (-)-(S)-3-[1-(ДИМЕТИЛАМІНО)ЕТИЛ]ФЕНІЛ-N-ЕТИЛ-N-МЕТИЛКАРБАМАТУ ТА ЙОГО ГІДРОТАРТРАТУ 1 N(CH 3 )2 HO IV , який далі розділяють шляхом взаємодії з оптично активною кислотою, після чого бажаний відповідний діастереоізомер кристалізують і, нарешті, перетворюють в сполуку формули III N(CH 3 )2 HO O N X VII , де X являє собою гр упу, що видаляється. O II , який відрізняється тим, що метоксіацетофенон формули VI O 2. Спосіб одержання (-)-(S)-3-[1(диметиламіно)етил]феніл-N-етил-Nметилкарбамату гідротартрату формули І N(CH3) 2 H 3CO N OH O * O VI піддають відновному амінуванню з отриманням сполуки формули V N(CH3 )2 COOH HOOC OH I , який відрізняється тим, що метоксіацетофенон формули VI O H3CO V , яку потім О-дезалкілують з отриманням рацемічного аміну формули IV піддають відновному амінуванню з отриманням сполуки формули V (19) H 3CO (13) N(CH 3)2 N 79972 O (11) , яку, в свою чергу, піддають взаємодії, необов'язково в формі її лужної солі, із сполукою формули VII C2 III UA (21) a200500538 (22) 21.10.2003 (24) 10.08.2007 (86) PCT/CZ03/00058, 21.10.2003 (31) PV-2002-3555 (32) 24.10.2002 (33) CZ (46) 10.08.2007, Бюл. № 12, 2007 р. (72) Степанкова Гана , CZ, Гайічек Йосеф , CZ, Сімек Станіслав , CZ (73) ЗЕНТІВА, А.С., CZ (56) Chen Chung-Pin: tetrahedron letters, vol.32, no.49, 1991, pages 7175-7178 US, 5602176, A, 11.02.1997 EP, 0193926, A, 10.09.1986 Ciszewska Grazyna: J.Labelled compd. radiopharm., vol.39, no.8, 1997, pages 651-668 (57) 1. Спосіб одержання (-)-(S)-3-[1(диметиламіно)етил]феніл-N-етил-Nметилкарбамату, тобто ривастигміну, формули II 2 VI 3 79972 4 N( CH3)2 N(CH3 )2 N H 3CO O O V , яку потім О-дезалкілують з отриманням рацемічного аміну формули IV N(CH 3)2 HO II , яку далі перетворюють у відповідну сіль формули (І) N(CH3) 2 N OH O * O IV , який далі розділяють шляхом взаємодії з оптично активною кислотою, після чого бажаний відповідний діастереоізомер кристалізують і, нарешті, перетворюють в сполуку формули III N(CH 3)2 HO III , яку, в свою чергу, піддають взаємодії, необов'язково в формі її лужної солі, із сполукою формули VII O N X VII , де X являє собою групу, що видаляється, з одержанням сполуки формули (ІІ) Винахід відноситься до способу отримання (-)(S)-3-[1-(диметиламіно)етил]феніл-N-етил-Nметилкарбамату і його гідротартрату. Тартрат (-)-(S)-3-[1-(диметиламіно)етил]фенілN-етил-N-метилкарбамату формули І COOH HOOC OH I . 3. Спосіб за п.1, який відрізняється тим, що рацемічний амін формули IV розділяють шляхом взаємодії з (S)-(+)-камфор-10-сульфокислотою. 4. Спосіб за п. 3, який відрізняється тим, що рацемічний амін формули IV розділяють шляхом взаємодії з 1 еквівалентом (S)-(+)-камфор-10сульфокислоти. 5. Спосіб за п.3, який відрізняється тим, що рацемічний амін формули IV розділяють шляхом взаємодії з 0,6 еквівалента (S)-(+)-камфор-10сульфокислоти. 6. Спосіб за будь-яким з попередніх пунктів, який відрізняє ться тим, що о триманий бажаний відповідний діастереоізомер додатково перекристалізовують. 7. Спосіб за п.6, який відрізняється тим, що отриманий бажаний відповідний діастереоізомер додатково перекристалізовують з етилацетату, необов'язково в суміші з етанолом. тенті ЕР 193926]. Спосіб його отримання був оснований на взаємодії Nгідроксифенілетилдиметиламіну формули IV з карбамоїлгалогенідом формули VII відомий під назвою INN ривастигмін, [описаний в патентній заявці CS No. PV 1991-4110] як речовина, яка індукує селективне інгібування активності ацетилхолінестерази в головному мозку. Вказана якість нарівні з хорошою переносимістю організмом людини, можливістю вибору доставки в формі таблеток (пероральна ефективність) і тривалою дією зумовлюють можливість використання ривастигміну для лікування захворювань, пов'язаних з порушенням холінергічної системи, зокрема, хвороби Альцгеймера. Рацемічний 3-[1-(диметиламіно)етил]феніл-Nетил-N-метилкарбамат (далі рацемічний ривастигмін), як сполука, що володіє можливою активністю проти хвороби Альцгеймера, був [описаний в па де X являє собою гр упу, що видаляється. Способи отримання цих рацемічних проміжних продуктів були описані в більш ранній літературі, але ніде не вказувалася можливість їх перетворення в оптично активні енантіомери. [У процитованій заявці CS PV 1991-4110] було продемонстровано в експериментах як "in vitro", так і "in vivo", що оптично активний (S)-iзомep є набагато більше ефективним і селективним інгібітором ацетилхолінестерази, ніж рацемічна суміш двох ізомерів. 5 79972 У процитованій заявці описаний спосіб отримання ривастигміну з рацемічної суміші, що включає отримання діастереоізомерних солей з (+)О,О-ди(паратолуїл)-D-винною кислотою і їх розділення кристалізацією. (S)-енантіомер ривастигміну був вивільнений з отриманої солі за допомогою розчину гідроксиду натрію. Основним технологічним недоліком цього способу є те, що оптичне розділення здійснюють тільки на кінцевій стадії синтезу. Це означає, що щонайменше 50% отриманого рацемічного ривастигміну (тобто, (R)-енантіомеру) являє собою некорисний відхід; насправді ж, вказані відходи набагато більші, оскільки при оптичному розділенні енантіомери ніколи не розділяються кількісно. Це робить загальний вихід синтезу низьким, а весь спосіб економічно невигідним. Інший недолік полягає в наявності втрат на окремих стадіях. Звичайно втрати проміжного продукту на більш пізніх стадіях є такими, що більш дорого коштують, ніж на початкових стадіях. Повертаючись до розгляду вказаного способу, потрібно зазначити, що розщеплення не приводить до досягнення задовільної оптичної чистоти, і речовину потрібно додатково перекристалізовувати (порів. посилальний приклад 1). Іншим недоліком є необхідність використання дорогої і канцерогенної сполуки формули VII (найбільш конкретно, N-етил-N-метилкарбамоїлхлориду) приблизно в 300%-ному надлишку. Розділення на більш ранній стадії синтезу здається на перший погляд бажаним, але далеким від здійсненності. Залишається питання, чи можна отримувати енантіомерно чисті проміжні продукти і, зокрема, чи можуть вказані продукти бути використані в подальшому синтезі без того, що вони будуть зазнавати рацемізації. Необхідність перекристалізації ставить під сумнів переваги вказаного способу. У цей час повертаються до того, що оптичне розділення проміжних продуктів (тобто, проведення операції на більш ранній стадії отримання) і проведення кінцевої стадії з оптично чистою речовиною дозволяє отримувати дуже хороший вихід (5)-ривастигміну із збереженням високої аналітичної чистоти. Даний винахід відноситься до способу отримання (-)-(S)-3-[1-(диметиламіно)етил]феніл-Nетил-N-метилкарбамату (ривастигміну) формули II або його гідротартрату формули І згідно з яким метоксіацетофенон формули VI 6 піддають відновному амінуванню з отриманням сполуки формули V яку потім О-дезалкілують з отриманням рацемічного аміну формули IV який далі розділяють шляхом взаємодії з оптично активною кислотою, після чого бажаний відповідний діастереоізомер кристалізують і, нарешті, перетворюють в сполуку формули III яку, в свою чергу, піддають взаємодії, необов'язково у вигляді її лужної солі, із сполукою формули VII де X являє собою гр упу, що видаляється. Отриману сполуку формули II перетворюють шляхом взаємодії з винною кислотою у відповідну сіль формули І. Переважно, фенол формули III перетворюють за допомогою сильної основи в інертному розчиннику в фенолят, і його піддають взаємодії з карбамоїлгалогенідом формули VII. Як сильна основа можуть бути використані гідриди лужних металів, такі як гідрид натрію, або сполуки алкіллітію, такі як бутиллітій. Інертний розчинник, переважно, вибраний з групи простих діалкілових ефірів, таких як тетрагідрофуран або 1,2-диметоксіетан. Відновне амінування проводять за допомогою диметиламіну або його гідрохлориду і відновника, звичайно це гідрид, такий як боргідрид натрію. Агенти О-дезалкілування можуть бути вибрані з сильних кислот, таких як, наприклад, бромисто 7 79972 воднева кислота, або з галогенідів бору, таких як бромід бору. Рацемічний амін формули IV, переважно, розділяють шляхом взаємодії з (S)-(+)-камфор-10сульфоновою кислотою. Отриманий бажаний відповідний діастереоізомер може бути потім перекристалізований, переважно, з етилацетату, необов'язково, в суміші з етанолом. Як показано в прикладах особливо переважних варіантів виконання, даний спосіб робить можливим отримання продукту формули І особливо високої оптичної чистоти. Відтворення раніше відомого способу, навіть з перекристалізацією, не приводить до досягнення високої оптичної чистоти. Додатковим результатом порівняння є те, що використання оптично активної сполуки формули III приводить до зменшення на 2/3 споживання і канцерогенного N-етил-N-метилкарбамоїлхлориду, що дорого коштує (відповідного загальній формулі VII, де Х=С1). Об'єкт винаходу більш детально продемонстрований в наступних прикладах. Приклад 1 Отримання гідротартрату (S)-(-)-ривастигміну (І) (схема 1) Розділення рацемічної сполуки (IX) за допомогою (+)-О,О'-дитолуїлвинної кислоти 49,6г рацемічної сполуки (IX) розчиняють в 100мл метанолу і до розчину додають розчин 75,8 г (+)-О,О'-дитолуїлвинної кислоти в 200мл метанолу. До прозорого розчину при перемішуванні поступово додають 150мл води. Розчин стає каламутним і починає кристалізуватися на холоді. Отримують 81,3г бажаної солі у ви гляді білих кристалів з т.пл.=145-147°С. Вказану кількість першої фракції кристалів перекристалізовують з суміші 200мл метанолу і 100мл води. Отримують другу фракцію кристалів (49,6г, т.пл.=155-156°С), яку знов перекристалізовують з суміші (метанол:вода (2:1), 200мл). Отримують третю фракцію з т.пл.=159-162°С. Її перекристалізовують з 150мл вищезгаданої суміші розчинників і виділяють четверту кінцеву фракцію кристалів (33,0г, т.пл.=1567°С), що відповідає виходу 26,9% від теоретичного, [a]D=+78,5° (с=1, етанол). Вивільнення основи (II) Сіль S-(-)-ривастигміну (II) з (+)-О,О'дитолуїлвинною кислотою (33,0г) повільно додають до суміші 150мл дихлорметану і 150мл 1н розчину NaOH, що добре перемішується. Після 8 розчинення усієї твердої речовини шари розділяють; дихлорметановий шар двічі екстрагують 100мл води і сушать сульфатом магнію. Розчинник упарюють у вакуумі і залишок від упарювання переганяють у вакуумі (т.кип.=135-8°С, 40Па). Отримують 10,72г безбарвного масла, що відповідає 85%-ному виходу від теоретичного ([a]D=-24,5°C; c=3,5, метанол). Отримання гідротартрату (S)-(+)ривастигміну (І) 8,86г основи (II) і 5,31г L-(+)-винної кислоти розчиняють при нагріванні і перемішуванні в 20мл етанолу, і до прозорого розчину для осадження додають етилацетат (250мл). Отриману суміш залишають охолоджуватися до +5°С. Кристали, що випали, відсмоктують і промивають етилацетатом. Отримують 9,7 г бажаного гідротартрату ривастигміну (І) (тобто ви хід 68,4% від теоретичного) з т.пл.=124-126°С. Вміст небажаного (ІІ)енантіомеру у вказаній фракції становив 1% (за даними капілярного електрофорезу). 0,3г кислого тартрату перекристалізовують з суміші етанол/етилацетат вищезгаданим чином, і отримують 0,24г перекристалізованої фракції (І) (80%). Вміст небажаного (R)-енантіомеру знизився до 0,69% (капілярний електрофорез). Загальний вихід, з розрахунку на вихідн у речовину (IV), становив 12,5%. Приклад 2 Отримання гідротартрату (S)-(+)ривастигміну (схема 2) Отримання [1-(3-метоксифеніл)етил] диметиламіну (V) згідно з літературним посиланням 2 0,5л ізопропоксиду титану повільно (5 хвилин) додають в інертній атмосфері (N2, аргон) до розчину 75,5г диметиламіну в 1,5л етанолу, охоло 9 79972 дженого до 10°С на водяній бані з льодом і вміщеного в 6-літрову тригорлу колбу, забезпечену мішалкою KPG, впуском і випуском для інертного (газу) і термометром, і потім додають 148,4г 3метоксіацетофенону (VI) (5 хвилин). Додавання ізопропоксиду слабо екзотермічно. Результуюча температура реакційної суміші після додавання досягає 35°С. Реакційну суміш потім перемішують при кімнатній температурі від 9 до 10 годин. Під час протікання реакції суміш стає злегка каламутною. Після вказаного періоду до реакційної суміші повільно і обережно додають 56,6г боргідриду натрію. Тривалість вказаного додавання становить приблизно 2 години. Реакційна суміш гусне в суспензію і піниться, і її необхідно вельми інтенсивно перемішува ти. Температуру підтримують в інтервалі від 25°С до 30°С слабким охолоджуванням льодом. Якщо реакційну суміш переохолоджують нижче 20°С після додавання боргідриду, утворюється щільна піна, що важко перемішується. Після додавання борогідриду отриману білу суспензію перемішують від 10 до 12 годин при кімнатній температурі. Потім подачу інертного (газу) припиняють, і до реакційної суміші повільно (більше 10 хвилин) підливають 800мл водного розчину гідроксиду амонію (2:1). Отриману суміш перемішують 20 хвилин. Тонкі білі кристали неорганічної речовини відсмоктують і ретельно промивають метанолом (близько 1 л). Всю спиртову фракцію упарюють від фільтрату на роторному вакуумному випарнику. Залишок від упарювання розбавляють 1000мл води і екстрагують етилацетатом (3×300мл). Об'єднаний етилацетатний екстракт промивають один раз 100 мл води і екстрагують соляною кислотою (3×200мл) (5:2). Кислі водні екстракти об'єднують, підлуговують 20%-ним NaOH (близько 1л) до рН від 12 до 14 і екстрагують етилацетатом (3×300мл). Органічну фракцію промивають 100мл води і 150мл насиченого сольового розчину. Сушать безводним сульфатом натрію. Осушувач відфільтровують і фільтрат упарюють досуха на роторному вакуумному випарнику. Сирий продукт переганяють і отримують близько 60% бажаного продукту у вигляді безбарвного масла. Т.кип.=68°С при 400Па, 108°С при 800Па. Отримання 3-(1-диметиламіноетил)фенолу (IV) згідно з літературним посиланням 3. 94г [1-(3-метоксифеніл)етил]диметиламіну (V) розчиняють в 285мл азеотропної бромистоводневої кислоти, і отриманий розчин кип'ятять при перемішуванні із зворотним холодильником 12 годин (температура бані 145-150°С). У процесі кип'ятіння реакційна суміш темніє. Розчин потім залишають остигати до кімнатної температури. Надлишок бромистоводневої кислоти упарюють з використанням роторного вакуумного випарника, і залишок від упарювання розчиняють в 200мл води. Розчин екстрагують етилацетатом (3×100мл). Водну фракцію потім поступово підлуговують насиченим розчином карбонату натрію при постійному перемішуванні (пінення). Розчин стає молочно каламутним, і його екстрагують етилацетатом (3×200мл). Етилацетатну фракцію струшують 1 раз із водою, 1 раз із насиченим розчином солі і 10 сушать над безводним сульфатом магнію. Перед фільтрацією осушувача додають активоване вугілля і осушувач відфільтровують разом з вугіллям. Етилацетатний розчин сполуки (IV) використовують на наступній стадії. Отримання S-(-)-3-(1-диметиламіно)фенолу (III) Розділення сполуки (IV) 1 еквівалентною кількістю S-(+)-камфор-10-сульфокислоти Розчин сполуки (IV) в етилацетаті (0,505моль) (вміст сполуки (IV) визначали титруванням) в 500 мл етилацетату вміщують в 1-літрову круглодонну колбу з магнітною мішалкою і додають розчин S(+)-камфор-10-сульфокислоти (117,4г (0,505моль) в 250мл безводного етанолу, отриманий при нагріванні. В розчин вносять затравку і залишають стояти в холодильнику (+5°С) протягом ночі. Кристали, що випали, відсмоктують через пористий скляний фільтр і залишають сушитися на повітрі протягом ночі. 1) Отримують 82,2г білих кристалів з т.пл.=165-171°С, які розчиняють в 190мл абсолютного етанолу при кип'ятінні із зворотним холодильником. Додають 380мл етилацетату і здійснюють кристалізацію при нагріванні згідно з вищезгаданою методикою. 2) Отримують 64,1г білих кристалів з т.пл.=174-176°С, які розчиняють в 150мл етанолу (абс.) при кип'я тінні із зворотним холодильником, і при нагріванні додають 300мл етилацетату. 3) Отримують 56,5г білих кристалів з т.пл.=177-179°С, які розчиняють в 130мл етанолу (абс.) при кип'я тінні із зворотним холодильником, і при нагріванні додають 260 мл етилацетату. 4) Отримують 51,6г білих кристалів з т.пл. 179181°С, тобто 25,7% від теоретичної кількості. Розділення сполуки (IV) 0,6 еквівалентною кількістю S-(+)-камфор-10-сульфокислоти 100г (0,605моль) сполуки (IV) розчиняють в 600мл етилацетату при перемішуванні і кип'ятінні із зворотним холодильником. Додають при перемішуванні при 70°С розчин S-(+)-камфор-10сульфокислоти (84,3г (0,363моль) в 125мл безводного етанолу. В розчин вносять затравку, залишають остигати до кімнатної температури при перемішуванні, охолоджують насиченим сольовим розчином до температури від -10 до -15°С і залишають кристалізуватися щонайменше на 12 годин без доступу вологи повітря. Першу фракцію кристалів, що випала, відсмоктують і сушать на повітрі. 1) Отримують 95,0г білих кристалів з т.пл.=173-175°С, які розчиняють в 175мл етанолу при перемішуванні і кип'ятінні із зворотним холодильником і додають 350мл етилацетату при температурі розчину від 60 до 70°С. Камфорсульфонат починає кристалізуватися, і його залишають кристалізуватися при температурі від -5 до -10°С щонайменше на 12 годин. Фракцію, що випала, відсмоктують, промивають етилацетатом (2×50мл) і сушать на повітрі. 2) Отримують 79,5г другої фракції з т.пл.=176178°С, яку знов перекристалізовують з суміші етанол:етилацетат (150мл:300мл) згідно з описаним вище способом. Після промивання етилацетатом (2×50мл) продукт сушать на повітрі. 11 79972 3) Отримують 74,6г третьої фракції з т.пл.=177-179°С, тобто вихід 31,0% від теоретичного. Вивільнення S-(-)-3-(1-диметиламіно)фенолу (III) 4л води вміщують в 10-літрову тонкостінну хімічну склянку з KPG-мішалкою. Додають і розчиняють при перемішуванні 250 г карбонату натрію. Частинами при перемішуванні додають кристали камфорсульфонату (517,5г). Коли додана приблизно половина всієї кількості, додають 2 літри дихлорметану. Інший камфорсульфонат додають при постійному перемішуванні. Період додавання складав близько 0,5 години. Отриману суміш перемішують ще 0,5 години. Потім шари розділяють в 10-літровій ділильній лійці. Водну фракцію екстрагують ди хлорметаном (2×1,5 літри). Об'єднані органічні фракції екстрагують 1,5 літрами води і сушать над 600г безводного сульфату натрію. Осушувач відфільтровують, і фільтрат упарюють досуху. Отриманий від упарювання залишок потім сушать на роторному вакуумному випарнику до постійної маси при 50 °С і 2,7кПа. Утворюється біла кристалічна речовина (187,0г; 87%), яку використовують на наступній стадії без очищення, [a]D=-55,7; с=1,55, метанол. Вміст небажаного (ІІ)-енантіомеру
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod of production of (-)-(s)-3-[1-(dimethylamino)ethyl]phenyl-n-ethyl-n-methylcarbamate and hydrogen-tartrate thereof
Назва патенту російськоюСпособ получения (-)-(s)-3-[1-(диметиламино)этил] фенил-n-этил-n-метилкарбамида и его гидротартрата
МПК / Мітки
МПК: C07C 271/44, C07C 215/00
Мітки: одержання, s)-3-[1-(диметиламіно)етил]феніл-n-етил-n-метилкарбамату, гідротартрату, спосіб
Код посилання
<a href="https://ua.patents.su/7-79972-sposib-oderzhannya-s-3-1-dimetilaminoetilfenil-n-etil-n-metilkarbamatu-ta-jjogo-gidrotartratu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання (-)-(s)-3-[1-(диметиламіно)етил]феніл-n-етил-n-метилкарбамату та його гідротартрату</a>
Спосіб одержання n-метил-n-[(1s)-1-феніл-2-((3s)-3-гідроксипіролідин-1-іл)етил]-2,2-дифенілацетаміду або n-метил-n-[(1r)-1-феніл-2-((3r)-3-гідроксипіролідин-1-іл)етил]-2,2-дифенілацетаміду

Номер патенту: 73472
Опубліковано: 15.08.2005
Автори: Акерманн Карл-Оугест, Готшліх Рудольф, Будак Дженс, Хелферт Берн, Бате Андрес, Стейн Інгеборг
МПК: A61P 29/00, C07D 207/12, A61K 31/40, A61P 7/10
Мітки: n-метил-n-[(1s)-1-феніл-2-((3s)-3-гідроксипіролідин-1-іл)етил]-2,2-дифенілацетаміду, спосіб, n-метил-n-[(1r)-1-феніл-2-((3r)-3-гідроксипіролідин-1-іл)етил]-2,2-дифенілацетаміду, одержання
Формула / Реферат:
1. Спосіб одержання N-метил-N-[(1S)-1-феніл-2-((3S)-3-гідроксипіролідин-1-іл)етил]-2,2-дифенілацетаміду або N-метил-N-[(1R)-1-феніл-2-((3R)-3-гідроксипіролідин-1-іл)етил]-2,2-дифенілацетаміду, який відрізняється тим, щоа) N-заміщену похідну фенілгліцину формули І, (І)в якійR означає OR1 або SR1,R1 означає А, арил, гетероарил, Si(R3)3,...
Спосіб одержання (1r, 2s, 4r)-(-)-2-[(2′-{n,n-диметиламіно}-етокси)]-2-[феніл]-1, 7, 7-три-[метил]-біцикло[2.2.1]гептану і його фармацевтично прийнятних кислотних адитивних солей
Номер патенту: 49974
Опубліковано: 15.10.2002
Автори: ДОНАТ ВЕРЕЦКЕІ Дьйордьі, Краснаі Дьйордь, НЕМЕТ Норберт, Мезеі Тібор, Лукач Дьюла, Порч-Маккаі Марта, САБО Тібор, ШІМІГ Дьюла, Будаі Золтан, Суладьі Янош, Надь Кальман
МПК: A61K 31/135, A61P 25/22, C07C 217/02, C07B 53/00, C07C 217/12, C07C 213/00
Мітки: прийнятних, кислотних, солей, 7-три-[метил]-біцикло[2.2.1]гептану, спосіб, одержання, адитивних, фармацевтично, 4r)-(-)-2-[(2'-{n,n-диметиламіно}-етокси)]-2-[феніл]-1
Формула / Реферат:
1. Спосіб одержання (1R, 2S, 4R)-(-)-2-[(2'-{N,N-диметиламіно}-етокси)]-2-[феніл]-1,7,7-три-[метил]-біцикло[2.2.1]гептану формулиі його фармацевтично прийнятних кислотних адитивних солей, що включає перетворення (+)-1,7,7-три-[метил]-біцикло[2.2.1]гептан-2-ону {(+)-камфори} формули
(1r,2s,4r)-(-)-2-[(2′-{n,n-диметиламіно}-етокси)]-2-[феніл]-1,7,7-три-[метил]-біцикло[2,2,1]гептан високого ступеня чистоти, його фармацевтично прийнятні кислотні адитивні солі, спосіб одержання цих сполук та лікарські засоби

Номер патенту: 61122
Опубліковано: 17.11.2003
Автори: Порч-Маккаі Марта, Лукач Дьюла, Будаі Золтан, Мезеі Тібор, ДОНАТ ВЕРЕЦКЕІ Дьйордьі, САБО Тібор, Краснаі Дьйордь, НЕМЕТ Норберт, Суладьі Янош, Надь Кальман, ШІМІГ Дьюла
МПК: C07C 217/12
Мітки: лікарські, спосіб, високого, засоби, фармацевтично, прийнятні, одержання, солі, ступеня, кислотні, 1r,2s,4r)-(-)-2-[(2'-{n,n-диметиламіно}-етокси)]-2-[феніл]-1,7,7-три-[метил]-біцикло[2,2,1]гептан, чистоти, адитивні, сполук, цих
Формула / Реферат:
1. (1R,2S,4R)-(-)-2-[(2'-{N,N-диметиламіно}-етокси)]-2-[феніл]-1,7,7-три-[метил]-біцикло[2,2,1]гептан формули (І)і його фармацевтично прийнятні кислотно-адитивні солі, який відрізняється тим, що він містить не більше 0,2% (1R,3S,4R)-3-[(2'-{N,N-диметиламіно}-етил)]-1,7,7-три-[метил]-біцикло[2,2,1]гептан-2-ону формули V
Спосіб одержання [(1s)-[1a,2b,3b,4a(s)]]-4-[7-[[1-(3-хлор-2-тієніл)метил] пропіл]аміно]-3н-імідазо[4,5-b]піридин-3-іл]-n-етил-2,3-дигідроксициклопентанкарбоксаміду, способи одержання проміжних сполук для його одержання та проміжні сполуки

Номер патенту: 54479
Опубліковано: 17.03.2003
Автори: Паунер Торі Х., Ванасс Бенуа Дж., Томпсон Майкл Д., Рейллі Лоренс, Цуей Чінг Т., Леон Патрік, Шах Харшавадан К., О'Брайєн Майкл К., Гарсіа Ерве, Вальтер Френсіс Л.
МПК: C07D 213/69, A61P 3/06, A61P 9/12, C07D 471/04, C07D 409/12, A61P 9/00, C07D 409/14
Мітки: спосіб, пропіл]аміно]-3н-імідазо[4,5-b]піридин-3-іл]-n-етил-2,3-дигідроксициклопентанкарбоксаміду, способи, сполук, одержання, проміжних, 1s)-[1a,2b,3b,4a(s)]]-4-[7-[[1-(3-хлор-2-тієніл)метил, проміжні, сполуки
Формула / Реферат:
1. Спосіб одержання [(1S)-[1a,2b,3b,4a(S*)]]-4-[7-[[1-(3-хлор-2-тієніл)метил]пропіл]аміно]-3H-імідазо[4,5-b]піридин-3-іл]-N-етил-2,3-дигідроксициклопентанкарбоксаміду, який включає взаємодію [(1S)-[1a,2b,3b,4a(S*)]]-4-[[3-аміно-4-[[1-(3-хлор-2-тієніл)метил]пропіл]аміно]-2-піридиніл]аміно]-N-етил-2,3-дигідроксициклопентанкарбоксаміду зі складним ефіром ортоформіату, ацетатом формамідину або диметилацеталем диметилформаміду.2. Спосіб за...
(-)цис-6(s)-феніл-5(r)[4-(2-піролідин-1-ілетокси)феніл]-5,6,7,8-тетрагідронафталін-2-ол d-тартрат, спосіб його одержання, спосіб лікування захворювань, що піддаються лікуванню агоністами естрогену, та фармацевтична композиція

Номер патенту: 51676
Опубліковано: 16.12.2002
Автори: Чіу Чарльз К., Мельтц Морган
МПК: A61K 31/40, C07D 295/092, C07D 295/08, C07D 319/00
Мітки: лікування, композиція, одержання, цис-6(s)-феніл-5(r)[4-(2-піролідин-1-ілетокси)феніл]-5,6,7,8-тетрагідронафталін-2-ол, лікуванню, фармацевтична, d-тартрат, естрогену, спосіб, піддаються, захворювань, агоністами
Формула / Реферат:
1. (-)Цис-6(S)-феніл-5(R)-[4-(2-піролідин-1-ілетокси)феніл]-5,6,7,8-тетрагідронафталін-2-ол D-тартрат.2. Спосіб одержання сполуки (-)цис-6(S)-феніл-5(R)-[4-(2-піролідин-1-ілетокси)феніл]-5,6,7,8-тетрагідронафталін-2-ол D-тартрату, який відрізняється тим, що він включає:а) розчинення рацемічного або частково оптично збагаченого цис-6-феніл-5-[4-(2-піролідин-1-ілетокси)феніл]-5,6,7,8-тетрагідронафталін-2-олу в киплячому водному...
Попередній патент: Стійкий до повзучості магнієвий сплав
Наступний патент: Пристрій індуктивного нагріву деталей складної форми
Випадковий патент: Спосіб лікування перелому нижньої щелепи