Спосіб одержання заміщених 2-(a,b,w-аміноалкіл)імідазолів
Номер патенту: 106083
Опубліковано: 25.07.2014
Автори: Завада Оксана Олександрівна, Борисов Олександр Володимирович, Коваленко Сергій Миколайович, Журавель Ірина Олександрівна






Формула / Реферат
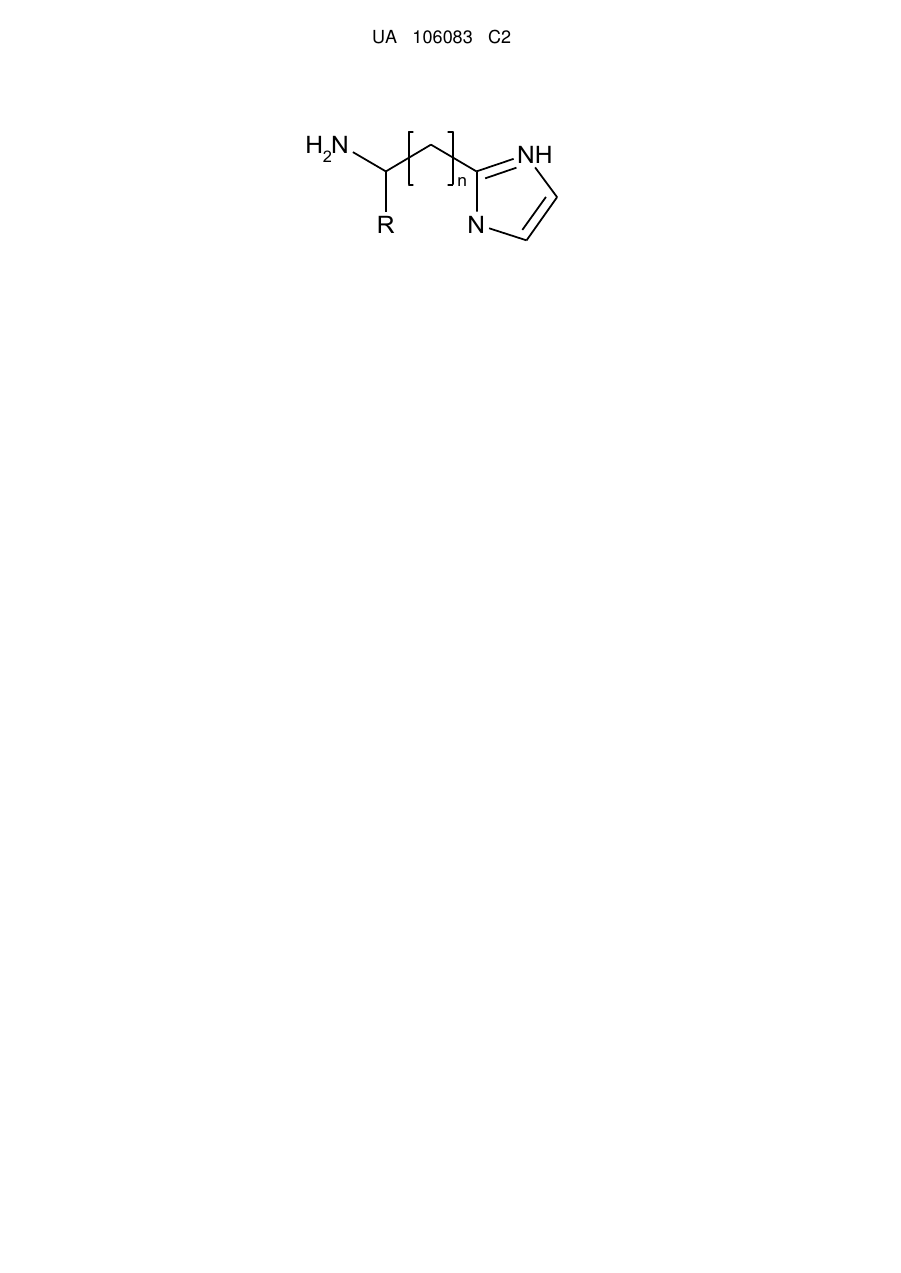
1. Спосіб одержання заміщених 2-(α,β,ω-аміноалкіл)імідазолів загальної формули 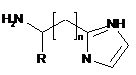 ,
,
де R - Н або Alk, або Ph, або Аr та n = 0…5,
шляхом взаємодії амінокислоти з захищеною аміногрупою з ацеталем амінооцтового альдегіду, реакції утвореної сполуки з ацетатом амонію з подальшим відновленням захищеної аміногрупи, який відрізняється тим, що реакції піддають α- або β-, або ω-амінокислоту, захист реакційно здатної аміногрупи здійснюють додаванням фталевого ангідриду у крижаній оцтовій кислоті при кип'ятінні протягом 2 годин, до утвореної сполуки додають 10 % надлишок 1,1-карбодіімідазолу в апротонному розчиннику при нагріванні реакційної суміші до 60-80 °C з наступним проведенням реакції з диметокси- або діетоксіацеталем амінооцтового альдегіду, одержану сполуку сплавляють з ацетатом амонію протягом 4 годин при 120 °C з подальшим відновленням захищеної аміногрупи з використанням гідразину гідрату.
2. Спосіб за п. 1, який відрізняється тим, що як апротонний розчинник використовують діоксан або хлороформ, або дихлорметан, або толуол, або диметилформамід.
3. Спосіб за п. 1, який відрізняється тим, що α- або β-, або ω-амінокислоту з захищеною аміногрупою піддають взаємодії з диметокси- або діетоксіацеталем амінооцтового альдегіду у еквімолярному співвідношенні.
Текст
Реферат: Винахід належить до органічної хімії, а саме до способів синтезу органічних сполук, зокрема заміщених 2-(α,β,ω-аміноалкіл)імідазолів. Згідно з заявленим способом реакції піддають α- або β-, або ω-амінокислоту, захист реакційно здатної аміногрупи здійснюють додаванням фталевого ангідриду у крижаній оцтовій кислоті при кип'ятінні протягом 2 годин, до утвореної сполуки додають 10% надлишок 1,1-карбодіімідазолу в апротонному розчиннику при нагріванні реакційної суміші до 60-80 °C з наступним проведенням реакції з диметокси- або діетоксіацеталем амінооцтового альдегіду, одержану сполуку сплавляють з ацетатом амонію протягом 4 годин при 120 °C з подальшим відновленням захищеної аміногрупи з використанням гідразину гідрату. UA 106083 C2 (12) UA 106083 C2 H2N NH n R N UA 106083 C2 5 10 Винахід належить до органічної хімії, а саме до способів синтезу органічних сполук, зокрема заміщених 2-(α,β,ω-аміноалкіл)імідазолів. Заміщені 2-(α,β,ω-аміноалкіл)імідазолів за будовою є структурними аналогами відомого біогенного аміну гістаміну (4(5)-(β-аміноетил)імідазолу) та синтетичного аміну ізогістаміну (2-(βаміноетил)імідазолу) і тому є перспективними для скринінгу на біологічну активність. Відомий на сьогодні спосіб одержання 2-(α-аміноалкіл)імідазолів [1], а саме 2-(1-аміно-3метилбутил)імідазолу та 2-(1-аміно-2-(індоліл-3)етил)імідазолу, полягає у використанні вихідних α-амінокислот з захищеною аміногрупою (з використанням карбоксибензилхлориду) для проведення реакції гетероциклізації під дією диметилацеталю амінооцтового альдегіду. Отримані продукти реакції піддають подальшій взаємодії з 25 % надлишком розчину ацетату амонію у середовищі оцтової кислоти, реакційну масу кип'ятять упродовж 5-8 годин та отримують цільові продукти з виходом 20-30 %. Для одержання відповідного 2-αаміноалкілімідазолу захисну групу видаляють відновленням на каталізаторі (Pd(C)). Спосіб здійснюють за наступною схемою: CBZ NH CBZ NH O R O NH2CH2CH(OCH3)2 EDCI/HOBt/NMM/NH CH 2CH(OCH 3)2 2 R OH NH N H2N N R CBZ N H R N H NH 4Ac HOAc R=CH 2CHMe2, 3-indollymethil 15 CBZ=carboxybenzyl . Недоліками наведеного способу можна вважати обмеженість набору кінцевих сполук, що обумовлено використанням вихідних лише α-амінокислот, та низькі виходи продуктів реакції. Задачею винаходу є розробка способу одержання 2-(α,β,ω-аміноалкіл)імідазолів загальної формули: H2N NH n N R 20 25 , де R - H або Alk, або Ph, або Аr та n = 0…5, який шляхом взаємодії α-амінокислот, βамінокислот, ω-амінокислот із фталемідною захисною групою з 1,1-карбодіімідазолом, диметокси- або діетоксіацеталем амінооцтового альдегіду та ацетатом амонію з подальшою обробкою гідразингідратом забезпечує одержання відповідного 2-(α,β,ω-аміноалкіл)імідазолу з будь-яким вуглецевим ланцюгом, достатнього ступеня чистоти. Поставлена задача вирішується таким чином, що у способі одержання заміщених 2-(α,β,ωаміноалкіл)імідазолів загальної формули H2N NH n N R 30 35 , де R - Н або Alk, або Ph, або Аr та n = 0…5, шляхом взаємодії амінокислоти з захищеною аміногрупою з ацеталем амінооцтового альдегіду, реакції утвореної сполуки з ацетатом амонію з подальшим відновленням захищеної аміногрупи, на відміну від прототипу згідно з винаходом реакції піддають α- або β-, або ω-амінокислоту, захист реакційно здатної аміногрупи здійснюють додаванням фталевого ангідриду у крижаній оцтовій кислоті при кип'ятінні протягом 2 годин, до утвореної сполуки додають 10 % надлишок 1,1-карбодіімідазолу в апротонному розчиннику при нагріванні реакційної суміші до 60-80 °C з наступним проведенням реакції з диметокси- або діетоксіацеталем амінооцтового альдегіду, одержану сполуку сплавляють з ацетатом амонію 1 UA 106083 C2 5 протягом 4 годин при 120 °C з подальшим відновленням захищеної аміногрупи з використанням гідразину гідрату. Винаходом передбачено, що як апротонний розчинник використовують діоксан або хлороформ, або дихлорметан, або толуол, або диметилформамід. У відповідності з винаходом α- або β-, або ω-амінокислоту з захищеною аміногрупою піддають взаємодії з диметокси- або діетоксіацеталем амінооцтового альдегіду у еквімолярному співвідношенні. Заявлений спосіб здійснюють за наступною схемою: CH3 H3C H2N O n OH R N N n R 10 15 20 25 30 35 40 45 H2C N O n CDI R OH NH 2NH 2 O N H H3C PG NH 4Ac PG N n R O H PG O O CH3 O N H2N n N H R N H , де R - H або Alk, або Ph, або Аr та n = 0…5, PG - фталемідна захисна група. На першій стадії синтезу проводять захист реакційноздатної аміногрупи амінокислоти з використанням фталевого ангідриду. Далі для активації карбоксильної групи використовують 10 % надлишок 1,1-карбодіімідазолу в апротонному розчиннику (хлороформ, діоксан, дихлоретан, толуол, диметилформамід) при 60-80 °C. При додаванні диметокси- або діетоксиацеталю амінооцтового альдегіду в еквімолярних співвідношеннях і подальшому сплавленні з ацетатом амонію відбувається формування імідазольного циклу. Умови проведення реакції на етапі сплавлення (4 години 120 °C) визначені експериментальним шляхом і є оптимальними для даного винаходу. Для одержання відповідного 2-(α,β,ω-аміноалкіл)імідазолу захисну групу видаляють за стандартними процедурами з використанням гідразину гідрату. Заявлений спосіб здійснюють наступним чином. Вихідні α- або β-, або ω-амінокислоти піддають взаємодії з фталевим ангідридом, захищені амінокислоти виділяють згідно описаних [2] процедур, висушують у вакуумному ексикаторі над Р2О5. Одержані амінокислоти із захисною групою розчиняють у діоксані або хлороформі, або дихлоретані, або толуолі, або диметилформаміді, до розчину додають 10 % надлишок 1,1карбодіімідазолу, реакційну суміш нагрівають протягом 1 години при 60-80 °C, при інтенсивному перемішуванні (перебіг реакції контролюють за допомогою методу ТШХ (елюент-етилацетат). Далі до реакційного середовища додають надлишок диметокси- або діетоксіацеталю амінооцтового альдегіду, нагрівають протягом 3-4 годин при перемішуванні (перебіг реакції контролюють ТШХ (елюент-етилацетат). До реакційної суміші додають воду та екстрагують хлороформом (тричі по 100 мл), хлороформний шар промивають розчином карбонату калію та водою. Органічний шар висушують поташем, розчинник видаляють у вакуумі. Для подальшої циклізації виділений амід сплавлюють з ацетатом амонію протягом 4 годин при 120 °C і постійному перемішуванні (перебіг реакції контролюють ТШХ). До реакційної суміш додають п'ятикратний надлишок води та підкисляють розчином хлористоводневої кислоти (до рН~2). Розчин екстрагують етилацетатом, водний шар відокремлюють та нейтралізують розчином карбонату калію (30 %). Цільовий продукт екстрагують хлороформом, який відділяють та упарюють. Видаляють захисні групи за стандартними процедурами [2]. Вихід готового продукту становить 25-45 %. Всі параметри заявленого способу визначені дослідним шляхом і є необхідними та достатніми для одержання цільових сполук. Винахід ілюструється прикладами. Приклад 1. Одержання 2-β-аміноетил-1H-імідазолу R = H, n = 1 наступної формули 2 UA 106083 C2 H N H2N N 5 10 15 20 25 . До 0,125 моль (18,5 г) фталевого ангідриду у 100 мл крижаної оцтової кислоти додають 0,125 моль (11,13 г) β-аланіну. Реакційну суміш кип'ятять протягом 2 годин при інтенсивному перемішуванні. Перебіг реакції контролюють ТШХ (елюент-етилацетат). Реакційну масу охолоджують, розбавляють водою (1:1). Осад, що утворився, відфільтровують, промивають на фільтрі водою (двічі по 30 мл) і висушують в ексикаторі над Р2О5. Одержану речовину 3-(1,3-діоксо-1,3-дигідро-2H-ізоіндол-2-іл)пропанову кислоту у кількості 0,1 моль (21,919 г) розчиняють у 250 мл діоксану, додають 10 % надлишок 1,1-карбодіімідозолу 0,11 моль (18,05 г) і нагрівають протягом 1 години при 80 °C при інтенсивному перемішуванні. Перебіг реакції контролюють ТШХ (елюент-етилацетат). Після завершення реакції до реакційної суміші додають 15 % надлишок (2,2-диметоксіетил)аміну 0,15 моль (15,7 г) та нагрівають 3-4 години при перемішуванні. До реакційної суміші додають воду та екстрагують хлороформом (тричі про 100 мл), хлороформний шар промивають розчином карбонату калію та водою. Органічний шар висушують поташем, розчинник видаляють у вакуумі. Суміш одержаної речовини (N-(2,2-диметоксіетил)-3-(1,3-діоксо-1,3-дигідро-2H-ізоіндол-2-іл)пропанаміду) 0,1 моль (30,61 г) та 0,3 моль (19,2 г) ацетату амонію нагрівають протягом 4 години при 120 °C і інтенсивному перемішуванні (перебіг реакції контролюють методом ТШХ). Після закінчення реакції, до реакційної суміші додають п'ятикратний надлишок води та 100 мл хлороформу, підкисляють до рН~2. Водний шар відокремлюють та нейтралізують розчином карбонату калію. Осад цільової сполуки відфільтровують, промивають водою та висушують. Суміш 0,05 моль (13,46 г) 2-[3-(2H-імідазол-2-іл)-3-оксопропіл]-1H-ізоіндол-1,3(2H)-діону та 0,055 моль (2,75 г) гідразингідрату, нагрівають при кип'ятінні в пропанолі-2 протягом 15 хвилин. Після закінчення реакції до реакційної суміші додають 100 мл води та 2,5 екв. хлористоводневої кислоти до утворення осаду (гідразид фталевої кислоти), який відфільтровують. Фільтрат упарюють під вакуумом, заливають 40 % розчином лугу, та екстрагують етером, який потім упарюють у вакуумі, отримують 2-(β-аміноетил)-1Н-імідазол. 1 Вихід 31 %. Тпл. - 110-114 Н-ЯМР, δ, м. ч.: 3,35 (с, 4Н, 2СН2), 7,58 (с, 2Н), 8,53 (уш.с, 3Н, + NH3 ), 14,81 (с, 2Н, NH2+). Анал. для C5H9N3, 2HCl: розр. N - 22,83 %; експ.: N - 22,86 %. Приклад 2. Одержання 2-(γ-амінопропіл)-1H-імідазолу R = H, n = 2 наступної формули H N H2N 30 35 40 45 50 N . До 0,125 моль (18,5 г) фталевого ангідриду у 100 мл крижаної оцтової кислоти додають 0,125 моль (12,87 г) 4-амінобутанової кислоти. Реакційну суміш кип'ятять протягом 2 годин при інтенсивному перемішуванні. Перебіг реакції контролюють ТШХ (елюент-етилацетат). Реакційну масу охолоджують, розбавляють водою (1:1). Осад, що утворився, відфільтровують, промивають на фільтрі водою (двічі по 30 мл) і висушують в ексикаторі над Р 2О5. Одержану речовину (4-(1,3-діоксо-1,3-дигідро-2H-ізоіндол-2-іл)бутанову кислоту) у кількості 0,1 моль (23,32 г) розчиняють у 250 мл діоксану 10 % надлишок 1,1-карбодіімідозолу 0,11 моль (18,05 г). Реакційну суміш нагрівають протягом 1 години при 80 °C при інтенсивному перемішуванні. Перебіг реакції контролюють за допомогою методу ТШХ (елюент-етилацетат). Після завершення реакції до реакційної суміші додають 15 % надлишок (2,2диметоксіетил)аміну 0,15 моль (15,7 г) та нагрівають 3-4 години при перемішуванні. До реакційної суміші додають воду та екстрагують хлороформом (тричі по 100 мл), хлороформний шар промивають розчином карбонату калію та водою. Органічний шар висушують поташем, розчинник видаляють у вакуумі. Суміш одержаної речовини 0,1 моль (32,03 г) (N-(2,2-диметоксіетил)-4-(1,3-діоксо-1,3дигідро-2H-ізоіндол-2-іл)бутанамід) та 0,3 моль (19,2 г) ацетату амонію нагрівають протягом 4 години при 120 °C, при інтенсивному перемішуванні (перебіг реакції контролюють за допомогою методу ТШХ). Після закінчення реакції, до реакційної суміші додають п'ятикратний надлишок води та 100 мл хлороформу, підкисляють до рН~2. Водний шар відокремлюють та нейтралізують розчином карбонату калію. Осад цільової сполуки відфільтровують, промивають водою та висушують. Суміш 0,05 моль (12,75 г) 2-[3-(2Н-імідазол-2-іл)-3-пропіл]-1Н-ізоіндол-1,3(2Н)-діон та 0,055 моль (2,75 г) гідразингідрату, нагрівають при кип'ятінні в пропанолі-2 протягом 15 хвилин. Після 3 UA 106083 C2 5 закінчення реакції до реакційної суміші додають 100 мл води та 2,5 екв. хлористоводневої кислоти до утворення осаду (гідразид фталевої кислоти), який відфільтровують. Фільтрат упарюють під вакуумом, заливають 40 % розчином лугу, та екстрагують етером, який потім упарюють у вакуумі отримують 2-(γ-амінопропіл)-1Н-імідазол. Вихід 45 %. 1 Тпл. 95-106 Н-ЯМР, δ, м. ч.: 2,05 (т, 2Н, СН2), 2,85 (м, 2Н, СН2), 2,95 (т, 2Н, СН2), 7,58 (с, + + 2Н), 8,44 (уш.с, 3Н, NH3 ), 13,81 (уш.с, 2Н, NH2 ). Анал. для C6H11N35, 2HCl: розр. N - 21,21 %; експ.: N - 21.22 % Приклад 3. Одержання 2-(ω-амінобутил)-1H-імідазолу R = H, n = 3 наступної формули H N H2N 10 15 20 25 30 35 40 N . До 0,125 моль (18,5 г) фталевого ангідриду у 100 мл крижаної оцтової кислоти додають 0,125 моль (14,65 г) 5-амінопентанової кислоти (117,15). Реакційну суміш кип'ятять протягом 1 години при інтенсивному перемішуванні. Перебіг реакції контролюють ТШХ (елюентетилацетат). Реакційну масу охолоджують, розбавляють водою. Осад, що утворився, відфільтровують, промивають на фільтрі водою (двічі по 30 мл) і висушують в ексикаторі над Р2О5. Одержану речовину (5-(1,3-діоксо-1,3-дигідро-2H-ізоіндол-2-іл)пентанову кислоту) у кількості 0,1 моль (23,32 г) розчиняють у 250 мл діоксану додають 10 % надлишок 1,1-карбодіімідозолу 0,11 моль (18,05 г). Реакційну суміш нагрівають протягом 1 години при 80 °C при інтенсивному перемішуванні. Перебіг реакції контролюють за допомогою методу ТШХ (елюент-етилацетат). Після завершення реакції до реакційної суміші додають 15 % надлишок (2,2диметоксіетил)аміну 0,15 моль (15,7 г) та нагрівають 3-4 години при перемішуванні. До реакційної суміші додають воду та екстрагують хлороформом (тричі по 100 мл), хлороформний шар промивають розчином карбонату калію та водою. Органічний шар висушують поташем, розчинник видаляють у вакуумі. Суміш одержаної речовини 0,1 моль (33,44 г) (N-(2,2-диметоксіетил)-5-(1,3-діоксо-1,3дигідро-2H-ізоіндол-2-іл)пентанамід) та 0,3 моль (19,2 г) ацетату амонію нагрівають протягом 4 години при 120 °C, при інтенсивному перемішуванні (перебіг реакції контролюють за допомогою методу ТШХ). Після закінчення реакції, до реакційної суміші додають п'ятикратний надлишок води та 100 мл хлороформу, підкисляють до рН~2. Водний шар відокремлюють та нейтралізують розчином карбонату калію. Осад цільової сполуки відфільтровують, промивають водою та висушують. Суміш 0,05 моль (13,45 г) 2-[4-(1H-імідазол-2-іл)бутил]-1H-ізоіндол-1,3(2H)-діон та 0,055 моль (2,75 г) гідразингідрату, нагрівають при кип'ятінні в пропанолі-2 протягом 15 хвилин. Після закінчення реакції до реакційної суміші додають 100 мл води та 2,5 екв. хлористоводневої кислоти до утворення осаду (гідразид фталевої кислоти), який відфільтровують. Фільтрат упарюють під вакуумом, заливають 40 % розчином лугу, та екстрагують етером, який потім упарюють у вакуумі, отримують 2-(ω-амінобутил)-1Н-імідазол. 1 Вихід 25 %. Блідо-жовте масло H-ЯМR ДМСО-∂6, δ, м. ч.: 1,62 (т, 2Н, СН2), 1,72 (м, 2Н, + + СН2), 2,75 (м, 2Н, СН2) 3,00 (т, 2Н, СН2), 7,68 (с, 2Н), 8,35 (уш.с, 3Н, NH3 ), 13,56 (уш.с, 2Н, NH2 ). Анал. для C7H13N3*2HCl: розр. N - 19,81 %; експ.: N - 19,92 %. Приклад 4. Одержання 2-(1-аміно-2-метилпропіл-1)-1H-імідазолу R = CH(CH3)2, n = 0 наступної формули HN N H2N CH3 H3C 45 . До 0,125 моль (18,5 г) фталевого ангідриду у 100 мл крижаної оцтової кислоти додають 0,125 моль (18,13 г) 2-(амінометил)-4-метилпентанової кислоти (145,20). Реакційну суміш кип'ятять протягом 2 годин при інтенсивному перемішуванні. Перебіг реакції контролюють ТШХ (елюент-етилацетат). Реакційну масу охолоджують, розбавляють водою (1:1). Осад, що 4 UA 106083 C2 5 10 15 20 25 30 35 утворився, відфільтровують, промивають на фільтрі водою (двічі по 30 мл) і висушують в ексикаторі над Р2О5. Одержану речовину (2-(1,3-діоксо-1,3-дигідро-2H-ізоіндол-2-іл)метилбутанову кислоту) у кількості 0,1 моль (23,32 г) розчиняють у 250 мл діоксану додають 10 % надлишок 1,1карбодіімідозолу 0,11 моль (18,05 г). Реакційну суміш нагрівають протягом 1 години при 80 °C при інтенсивному перемішуванні. Перебіг реакції контролюють за допомогою методу ТШХ (елюент-етилацетат). Після завершення реакції до реакційної суміші додають 15 % надлишок (2,2-диметоксіетил)аміну 0,15 моль (15,7 г) та нагрівають 3-4 години при перемішуванні. До реакційної суміші додають воду та екстрагують хлороформом (тричі по 100 мл), хлороформний шар промивають розчином карбонату калію та водою. Органічний шар висушують поташем, розчинник видаляють у вакуумі. Суміш одержаної речовини 0,1 моль (33,44 г) (N-(2,2-диметоксіетил)-5-(1,3-діоксо-1,3дигідро-2H-ізоіндол2-іл)метилбутанамід) та 0,3 моль (19,2 г) ацетату амонію нагрівають протягом 4 години при 120 °C, при інтенсивному перемішуванні (перебіг реакції контролюють за допомогою методу ТШХ). Після закінчення реакції, до реакційної суміші додають п'ятикратний надлишок води та 100 мл хлороформу, підкисляють до рН~2. Водний шар відокремлюють та нейтралізують розчином карбонату калію. Осад цільової сполуки відфільтровують, промивають водою та висушують. Суміш 0,05 моль (13,45 г) 2-[1-(1H-імідазол-2-іл)метилпропіл]-1H-ізоіндол-1,3(2H)-діону та 0,055 моль (2,75 г) гідразингідрату нагрівають при кип'ятінні в пропанолі-2 протягом 15 хвилин. Після закінчення реакції до реакційної суміші додають 100 мл води та 2,5 екв. хлористоводневої кислоти до утворення осаду (гідразид фталевої кислоти), який відфільтровують. Фільтрат упарюють під вакуумом, заливають 40 % розчином лугу та екстрагують етером, який потім упарюють у вакуумі, отримують [2-(2-(1-аміно-2-метилпроіл-1)-1Н-імідазол. 1 Вихід 45 %. Блідо-жовте масло. Н-ЯМР, (CDCl3) δ, м.ч.: 0.92 (д, 6Н, 2СН3), 1.90 (уш.с, 2Н, NH2), 2.15-2.25 (м, 1H, СН), 4.10 (д, 1Н, СН), 7.22 (с, 1H, СН), 7.70 (с, 1Н, СН), Анал. для C 7H13N3: розр. N, 30.19; експ.: N, 30.17. Таким чином, заявлено новий спосіб одержання заміщених 2-(α,β,ω-аміноалкіл)імідазолів. Даний спосіб відзначається рядом переваг: - універсальність методу одержання 2-(α,β,ω-аміноалкіл)імідазоліл; - проста технологія синтезу та високі виходи кінцевих продуктів; - низька собівартість та доступність вихідних реагентів. Джерела інформації: 1. J.J. Chen, Y. Zhang, S. Hammond, N. Dewdney, T. Ho, X. Lin, M.F. Browner, and A.L. Castelhano // Bioorganic & Medicinal Chemistry Letters – 1996. - Vol. 6, No. 13, 1601-1606. 2. Комарова Б.М. // Защитная группа в органической химии: Пер. с англ. - М.: Мир, 1976 - С. 60-62. ФОРМУЛА ВИНАХОДУ 40 1. Спосіб одержання H2N заміщених 2-(α,β,ω-аміноалкіл)імідазолів загальної формули NH n R 45 50 N , де R - Н або Alk, або Ph, або Аr та n = 0…5, шляхом взаємодії амінокислоти з захищеною аміногрупою з ацеталем амінооцтового альдегіду, реакції утвореної сполуки з ацетатом амонію з подальшим відновленням захищеної аміногрупи, який відрізняється тим, що реакції піддають α- або β-, або ω-амінокислоту, захист реакційно здатної аміногрупи здійснюють додаванням фталевого ангідриду у крижаній оцтовій кислоті при кип'ятінні протягом 2 годин, до утвореної сполуки додають 10 % надлишок 1,1-карбодіімідазолу в апротонному розчиннику при нагріванні реакційної суміші до 60-80 °C з наступним проведенням реакції з диметокси- або діетоксіацеталем амінооцтового альдегіду, одержану сполуку сплавляють з ацетатом амонію протягом 4 годин при 120 °C з подальшим відновленням захищеної аміногрупи з використанням гідразину гідрату. 2. Спосіб за п. 1, який відрізняється тим, що як апротонний розчинник використовують діоксан або хлороформ, або дихлорметан, або толуол, або диметилформамід. 5 UA 106083 C2 3. Спосіб за п. 1, який відрізняється тим, що α- або β-, або ω-амінокислоту з захищеною аміногрупою піддають взаємодії з диметокси- або діетоксіацеталем амінооцтовогоальдегіду у еквімолярному співвідношенні. Комп’ютерна верстка С. Чулій Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 6
ДивитисяДодаткова інформація
Автори англійськоюKovalenko Serhii Mykolaiovych, Zhuravel` Iryna Oleksandrivna, Borysov Oleksandr Volodymyrovych
Автори російськоюКоваленко Сергей Николаевич, Журавель Ирина Александровна, Борисов Александр Владимирович
МПК / Мітки
МПК: C07D 233/64, C07D 233/54
Мітки: заміщених, 2-(a,b,w-аміноалкіл)імідазолів, одержання, спосіб
Код посилання
<a href="https://ua.patents.su/8-106083-sposib-oderzhannya-zamishhenikh-2-abw-aminoalkilimidazoliv.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання заміщених 2-(a,b,w-аміноалкіл)імідазолів</a>
Спосіб одержання заміщених 2-(a,b,w-аміноалкіл)імідазолів

Номер патенту: 75486
Опубліковано: 10.12.2012
Автори: Журавель Ірина Олександрівна, Борисов Олександр Володимирович, Завада Оксана Олександрівна, Коваленко Сергій Миколайович
МПК: C07D 233/54, C07D 231/10, C07D 209/00, C07D 233/64, C07D 233/00
Мітки: спосіб, одержання, 2-(a,b,w-аміноалкіл)імідазолів, заміщених
Формула / Реферат:
1. Спосіб одержання заміщених 2-(a,b,w-аміноалкіл)імідазолів загальної формули,де R - Η або Alk, або Ph, або Аr та n=0…5, шляхом взаємодії амінокислоти з захищеною аміногрупою з ацеталем амінооцтового альдегіду, реакції утвореної сполуки з ацетатом амонію з подальшим відновленням захищеної аміногрупи, який відрізняється тим, що реакції піддають a-, або...
Спосіб одержання заміщених імідазолів або їх нетоксичних фармацевтично прийнятних адитивних солей

Номер патенту: 13218
Опубліковано: 28.02.1997
Автори: Арто Йоханнес Карьялайнен, Кауко Ойва Антеро Куркела, Лаурі Вейро Матті Кангас
МПК: C07D 233/64
Мітки: спосіб, одержання, імідазолів, заміщених, адитивних, нетоксичних, фармацевтично, солей, прийнятних
Формула / Реферат:
(57) Способ получения замещенных имидазолов общей формулыгде R1 R2 R1' и R2' которые могут быть одинаковыми или различными -Н СН3, С2Н5 ОСН3 или галогеномR1-H илигде R3-H СН3 или галогеномR4-HR5-H или ОН или R4 и R5 взятые вместе образуют связь и X и Y которые могут быть одинаковыми или различными представляют собой связь или прямой алкил С1-2 или их нетоксичных фармацевтически приемлемых...
Спосіб одержання заміщених 3-алкіл/арилсульфоніл та 3-сульфонамідо-хінолін-4-онів

Номер патенту: 65628
Опубліковано: 12.12.2011
Автори: Гудіна Вікторія Юріївна, Сілін Олексій Віталійович, Коваленко Сергій Миколайович
МПК: C07C 317/02, C07C 311/10, C07D 215/00
Мітки: 3-сульфонамідо-хінолін-4-онів, спосіб, заміщених, одержання
Формула / Реферат:
1. Спосіб одержання заміщених 3-алкіл/арилсульфоніл та 3-сульфоніламідо-хінолін-4-онів шляхом проведення реакції у середовищі киплячого дифенілового етеру, який відрізняється тим, що реакції піддають у еквімолярній кількості ортоетер, відповідні сульфонілацетат та ариламін при кип'ятінні протягом 2 годин, до реакційної суміші додають дифеніловий етер з подальшим кип'ятінням протягом 1 години при температурі 255 °С, охолодженням, фільтрацією...
Спосіб одержання заміщених 3-, 4-, 5-, 6-нітро-2-n-фенілантранілових кислот

Номер патенту: 33114
Опубліковано: 15.02.2001
Автори: Яременко Віталій Дмитрович, Ісаєв Сергій Григорович, Минько Людмила Миколаївна, Павлій Олександр Іванович, Зупанець Ігор Альбертович, Ткач Андрій Олександрович, Павлій Олег Олександрович
МПК: A61K 31/196, C07C 205/00, C07C 229/58
Мітки: спосіб, кислот, заміщених, 6-нітро-2-n-фенілантранілових, одержання
Текст:
...(0,01 моль), 0,040 г (0,0005 моль) міді окису кип'ятять 2 години при температурі 180-220°С. Після охолодження додаюіь 10 ші40-50% водневого етанолу або суміші діоксан - вода(1 : 1,5) або диметилформамід - вода (1 : 1,5) із активованим вугіллям. Після чого реакційну суміш кип'ятять 10-15 хвилин, відфільтровують та підкисляють НСІ до рН=3. Осад знову відфільтровують, сушать. Вихід цільового продукту 2,71 г (90%). Одержана...
Спосіб одержання заміщених 2-ароїл-3-аміно-5-ариламінотіофенів

Номер патенту: 91223
Опубліковано: 12.07.2010
Автори: Коваленко Сергій Миколайович, Пархоменко Олексій Олександрович, Власенко Юрій Дмитрович, Черних Валентин Петрович
МПК: C07D 333/36
Мітки: 2-ароїл-3-аміно-5-ариламінотіофенів, заміщених, спосіб, одержання
Формула / Реферат:
1. Спосіб одержання заміщених 2-ароїл-3-аміно-5-ариламінотіофенів, шляхом проведення реакції похідних ацетонітрилу з фенілізотіоціанатом у рідкому середовищі з наступним додаванням a-галогенпохідних, фільтрацією кінцевого продукту з очищенням перекристалізацією, який відрізняється тим, що реакції піддають заміщені фенілсульфонілацетонітрил або алкілсульфонілацетонітрил, або малонодинітрил, або ціанацетамід з заміщеним фенілізотіоціанатом у...
Попередній патент: Амінопіразол триазолотіадіазольні інгібітори протеїнкінази c-мет
Наступний патент: Спосіб розламування підземних формацій
Випадковий патент: Спосіб прижиттєвого вивчення поверхневої структури твердих тканин зубів за допомогою знятих з них реплік