Кристалічна форма 1-хлор-4-(b-d-глюкопіраноз-1-ил)-2-[4-((s)-тетрагідрофуран-3-ілокси)-бензил]-бензолу, спосіб її одержання та її застосування при приготуванні лікарських засобів
Номер патенту: 91546
Опубліковано: 10.08.2010
Автори: Зік Сандра, Шюле Мартін, Хіммельсбах Франк, Мартін Ханс-Юрген, Екхардт Маттіас







Формула / Реферат
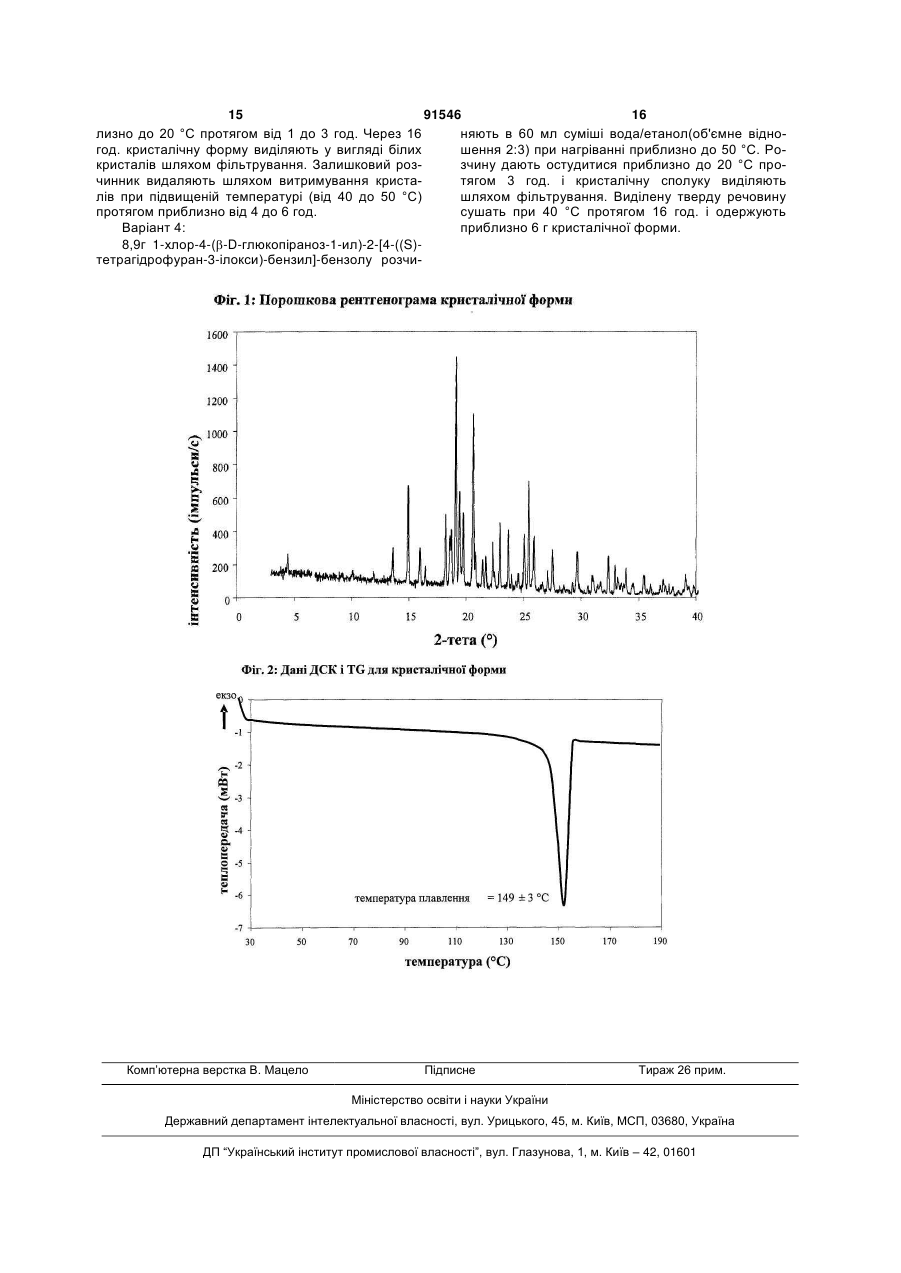
1. Кристалічна форма 1-хлор-4-(b-D-глюкопіраноз-1-ил)-2-[4-((S)-тетрагідрофуран-3-ілокси)-бензил]-бензолу, що має порошкову рентгенограму, що включає піки при 18,84, 20,36 і 25,21° 2θ (±0,05° 2θ), і зазначена порошкова рентгенограма отримана з використанням випромінювання СuКa1.
2. Кристалічна форма за п. 1, порошкова рентгенограма якої додатково включає піки при 14,69, 19,16 і 19,50° 2θ (±0,05° 2θ), і зазначена порошкова рентгенограма отримана з використанням випромінювання СuКa1.
3. Фармацевтична композиція, що включає кристалічну форму за п. 1 або 2.
4. Застосування кристалічної форми за п. 1 або 2 для одержання фармацевтичної композиції, що придатна для лікування або попередження метаболічних порушень, переважно - метаболічного порушення, вибраного з групи, що включає цукровий діабет типу 1 і 2, ускладнення при діабеті, метаболічний ацидоз або кетоз, реактивну гіпоглікемію, гіперінсулінемію, порушення метаболізму глюкози, резистентність до інсуліну, метаболічний синдром, дисліпідемії різної етіології, атеросклероз і родинні захворювання, ожиріння, високий артеріальний тиск, хронічну серцеву недостатність, набряк і гіперурікемію.
5. Спосіб одержання кристалічної форми за п. 1 або 2, де зазначений спосіб включає наступні стадії:
(a) розчинення 1-хлор-4-(b-D-глюкопіраноз-1-ил)-2-[4-((S)-тетрагідрофуран-3-ілокси)-бензил]-бензолу в розчиннику або суміші розчинників з одержанням насиченого або майже насиченого розчину;
(b) витримування розчину для осадження кристалічної форми за пп. 1, 2 або 3 з розчину й внаслідок цього утворення суспензії;
(c) виділення осаду з суспензії; і
(d) сушіння осаду до видалення надлишку зазначеного розчинника або суміші розчинників.
Текст
1. Кристалічна форма 1-хлор-4-(-Dглюкопіраноз-1-ил)-2-[4-((S)-тетрагідрофуран-3ілокси)-бензил]-бензолу, що має порошкову рентгенограму, що включає піки при 18,84, 20,36 і 25,21° 2θ (±0,05° 2θ), і зазначена порошкова рентгенограма отримана з використанням випромінювання СuК1. 2. Кристалічна форма за п. 1, порошкова рентгенограма якої додатково включає піки при 14,69, 19,16 і 19,50° 2θ (±0,05° 2θ), і зазначена порошкова рентгенограма отримана з використанням випромінювання СuК1. C2 2 (11) 1 3 91546 4 стабільності й характеристик кристалічної активної речовини пред'являються строгі вимоги. Стабільність фармацевтично активної речовини у фармацевтичних композиціях також важлива для встановлення строку придатності конкретного лікарського препарату; строк придатності є проміжком часу, протягом якого лікарський препарат можна вводити без якого-небудь ризику. Тому висока стабільність лікарської речовини в зазначеСполуки, описані в даному винаході, мають них вище фармацевтичних композиціях при різних корисний інгібувальний вплив на натрійзалежний умовах зберігання є додатковою перевагою, як співпереносник глюкози SGLT, особливо SGLT2. для пацієнта, так і для виготовлювача. Спосіб одержання сполуки А, описаний у даному Поглинання вологи зменшує вміст фармацеввинаході, не приводить до кристалічної форми. тично активної речовини внаслідок збільшення Зрозуміло, що наявність певної фармацевтичмаси, обумовленої надходженням води. Фармаценої активності є головною попередньою умовою втичні композиції, схильні до поглинання вологи, для того, щоб фармацевтично активний засіб був при зберіганні необхідно захищати від впливу возатверджений для продажу як лікарський препалоги, наприклад, шляхом додавання придатних рат. Однак існує безліч додаткових вимог, яким засобів, що осушують, або шляхом зберігання ліповинний відповідати фармацевтично активний карського засобу в середовищі, що захищене від засіб. Ці вимоги відносяться до різних параметрів, надходження вологи. Тому переважно, щоб фарпов'язаних з природою самого активного засобу. мацевтично активна речовина була лише незначПрикладами цих параметрів є, але не обмежуютьно гігроскопічною. ся тільки ними, стабільність активного засобу при Крім того, наявність строго певної кристалічної різних умовах навколишнього середовища, його форми дозволяє виконати очищення лікарської стабільність під час виготовлення фармацевтичної речовини шляхом перекристалізації. композиції й стабільність активного засобу в готоПоряд з зазначеними вище вимогами в загавих лікарських композиціях. Фармацевтично актильному випадку також варто враховувати, що вна речовина, що застосовується для одержання будь-яка зміна фізичного стану фармацевтичної фармацевтичних композицій, повинна бути як мокомпозиції, що може підвищити її фізичну й хімічну жна більше чистою і повинна бути гарантованою стабільність, забезпечує значну перевагу в порівйого стабільність при тривалому зберіганні при нянні з менш стабільними формами того ж самого різних умовах навколишнього середовища. Це лікарського засобу. необхідно для того, щоб виключити використання В основу даного винаходу покладене завдання фармацевтичних композицій, які на додаток до одержання стабільної кристалічної форми сполуки конкретної активної речовини містять, наприклад, А, що відповідає зазначеним вище вимогам, які продукти її розкладання. У таких випадках вміст пред'являються до фармацевтично активних реактивного засобу в лікарському засобі може бути човин. менше заданого. Об'єкти винаходу Рівномірний розподіл лікарського засобу в Першим об'єктом даного винаходу є кристалікомпозиції є критично важливим фактором, особчна форма сполуки А. ливо коли необхідно вводити невеликі дози лікарДругим об'єктом даного винаходу є кристалічського засобу. Для забезпечення рівномірного рона форма сполуки А, що має порошкову рентгенозподілення необхідно зменшити розмір часток граму, що включає піки при 18,84,20,36 і 25,21° 2θ активної речовини до придатних значень, напри(±0,05° 2θ), і зазначена порошкова рентгенограма клад, шляхом розмелу. Оскільки необхідно по моотримана з використанням випромінювання СuК1. жливості зменшити розкладання фармацевтично Третім об'єктом даного винаходу є сполука А, активної речовини, що є побічним ефектом розмеу якій не менш 50% зазначеної сполуки міститься у лу (або мікронізації), незважаючи на жорсткі умовигляді кристалічної форми, визначеної вище й ви, необхідні для його проведення, важливо, щоб нижче в даному винаході. активна речовина під час розмелу мала високу Внаслідок фармацевтичної ефективності спостабільність. Тільки якщо активна речовина є долуки А четвертим об'єктом даного винаходу є фасить стабільною під час розмелу, можна відтворермацевтична композиція або лікарський засіб, що ним образом одержувати однорідні до фармацеввключає кристалічну форму, визначену вище й тичної композиції, які завжди містять задану нижче в даному винаході. кількість активної речовини. П'ятим об'єктом даного винаходу є застосуІншим утрудненням, що може виникнути під вання кристалічної форми, визначеної вище або час розмелу при одержанні фармацевтичної комнижче в даному винаході, для одержання фармапозиції, є підведення енергії, обумовлене розмецевтичної композиції, що придатна для лікування лом, і тиск на поверхні кристалів. За певних умов або попередження захворювань або патологічних це може привести до поліморфних перетворень, станів, на які можна вплинути шляхом інгібування утворення аморфної речовини або до змін кристанатрійзалежного співпереносника глюкози SGLT, лічних ґраток. Оскільки для забезпечення фармапереважно - SGLT2. цевтичної якості фармацевтичної композиції необШостим об'єктом даного винаходу є застосухідно, щоб кристали активної речовини завжди вання кристалічної форми, визначеної вище або мали однакову морфологію, з цього погляду до нижче в даному винаході, для одержання фарма 5 91546 6 цевтичної композиції, що придатна для лікування Інші об'єкти даного винаходу стануть зрозуміабо попередження метаболічних порушень. лими для фахівця в даній галузі техніки з наступСьомим об'єктом даного винаходу є застосуного докладного опису даного винаходу й приклавання кристалічної форми, визначеної вище або дів. нижче в даному винаході, для приготування фарКороткий опис креслень мацевтичної композиції, Призначеної для інгібуНа фіг. 1 наведена порошкова рентгенограма вання натрійзалежного співпереносника глюкози кристалічної форми. SGLT2. На фіг. 2 наведені результати термічного анаВосьмим об'єктом даного винаходу є застосулізу й визначення температури плавлення криставання кристалічної форми, визначеної вище або лічної форми за допомогою ДСК. нижче в даному винаході, для приготування фарДокладний опис винаходу мацевтичної композиції для попередження дегеВідповідно до винаходу несподівано було нерації бета-клітин панкреатичних острівців і/або встановлено, що існує кристалічна форма сполуки для поліпшення й/або відновлення функціональА, що відповідає важливим вимогам, зазначеним ності бета-клітин панкреатичних острівців. вище в даному винаході. Відповідно до цього даДев'ятим об'єктом даного винаходу є застосуний винахід відноситься до кристалічної форми вання кристалічної форми, визначеної вище або сполуки А. нижче в даному винаході, для приготування фарЦю кристалічну форму можна ідентифікувати мацевтичної композиції для попередження, уповіза її характеристичною порошковою рентгеногральнення, затримування або лікування захворюмою (ПРРГ). вань або патологічних станів, обумовлених Кристалічна форма характеризується порошаномальним накопиченням жиру в печінці, у нужковою рентгенограмою, що включає піки при 18,84, денні в ньому пацієнта. 20,36 і 25,21° 2θ (±0,05° 2θ), і зазначена порошкоДесятим об'єктом даного винаходу є спосіб ва рентгенограма отримана з використанням виодержання кристалічної форми, визначеної вище промінювання СuК1. або нижче в даному винаході, зазначений спосіб Переважно, якщо зазначена порошкова рентвключає наступні стадії: генограма включає піки при 14,69, 18,84, 19,16, (a) розчинення сполуки А у розчиннику або су19,50, 20,36 і 25,21° 2θ (±0,05° 2θ), і зазначена міші розчинників з одержанням насиченого або порошкова рентгенограма отримана з використанмайже насиченого розчину; ням випромінювання СuК1. (b) витримування розчину, переважно - з охоБільш переважно, якщо кристалічна форма лодженням, для осадження кристалічної форми й характеризується порошковою рентгенограмою, внаслідок цього утворення суспензії; отриманою з використанням випромінювання (c) виділення осаду з суспензії; СuК1, що включає піки при 2θ (±0,05° 2θ), наве(d) сушіння осаду до видалення надлишку задені в таблиці 1. значеного розчинника або суміші розчинників. Таблиця 1: Порошкова рентгенограма кристалічної форми (наведені тільки піки аж до 30° 2θ): 2θ [°] 4,46 9,83 11,68 13,35 14,69 15,73 16,20 17,95 18,31 18,43 18,84 19,16 19,50 20,36 20,55 21,18 21,46 22,09 22,22 22,71 23,44 23,72 24,09 24,33 Міжплощинна відстань [Å] 19,80 8,99 7,57 6,63 6,03 5,63 5,47 4,94 4,84 4,81 4,71 4,63 4,55 4,36 4,32 4,19 4,14 4,02 4,00 3,91 3,79 3,75 3,69 3,66 Інтенсивність І/І0 [%] 8 4 4 14 42 16 8 30 22 23 100 42 31 74 13 11 13 19 4 28 27 3 3 7 7 24,81 25,21 25,65 26,40 26,85 27,26 27,89 28,24 29,01 29,41 91546 3,59 3,53 3,47 3,37 3,32 3,27 3,20 3,16 3,08 3,03 Ще більш переважно, якщо кристалічна форма характеризується порошковою рентгенограмою, отриманою з використанням випромінювання СuК1, що включає піки при 2θ (±0,05° 2θ), наведені на фіг. 1. Крім того, кристалічна форма сполуки А характеризується температурою плавлення, рівною приблизно 149°С ± 3°С (визначена за допомогою ДСК; за неї прийнята температура, що відповідає настанню переходу; швидкість нагрівання 10 К/хв.). Отримана за допомогою ДСК залежність наведена на фіг. 2. Порошкові рентгенограми в даному винаході знімали за допомогою дифрактометра STOE STADIР у режимі пропущення, постаченому детектором положення (OED) і анодом з Сu як джерело рентгенівського випромінювання (випромінювання СuК1, = 1,54056 Å, 40 кВ, 40 мА). У наведеній вище таблиці значення "2θ [°]" означають кут дифракції в градусах і значення "d [Å]" означають зазначені міжплощинні відстані в Å. Інтенсивності, зазначені на фіг. 1, наведені в імпульсах у секунду. Для обліку експериментальної погрішності, описані вище значення 2θ варто вважати точними з відхиленнями ± 0,05° 2θ. Це означає, що при рішенні питання про те, чи є даний зразок кристалів сполуки А кристалічною формою пропонованою в даному винаході, значення 2θ, отримане експериментально для зразка, варто вважати співпадаючим з характеристичним значенням, зазначеним вище, якщо воно відрізняється від характеристичного значення не більше ніж на ± 0,05° 2θ. Температуру плавлення визначають за допомогою ДСК (диференціальна сканувальна калориметрія) з використанням приладу ДСК 821 (Mettler Toledo). Іншим об'єктом даного винаходу є спосіб одержання кристалічної форми сполуки А, визначеної вище й нижче в даному винаході, зазначений спосіб включає наступні стадії: (a) розчинення сполуки А у розчиннику або суміші розчинників з одержанням насиченого або майже насиченого розчину; (b) витримування розчину для осадження кристалічної форми з розчину; (c) витяг осаду з розчину; і (d) сушіння осаду до видалення надлишку зазначеного розчинника або суміші розчинників. Терміни "насичений" або "майже насичений" означають вихідну сполуку А, що використається на стадії (а). Наприклад, розчин, що насичений 8 24 46 23 2 8 17 2 3 4 18 вихідною сполукою А, може бути перенасиченим його кристалічною формою. Придатні розчинники переважно вибрані з групи, що включає С1-С4-алканоли, воду, етилацетат, ацетонітрил, ацетон, діетиловий ефір і суміш одного або більшої кількості цих розчинників. Більш переважні розчинники вибрані з групи, що включає метанол, етанол, ізопропанол, етилацетат, діетиловий ефір, ацетон, воду й суміш одного або більшої кількості цих розчинників, переважно - суміші одного або більшої кількості зазначених органічних розчинників з водою. Особливо кращі розчинники вибрані з групи, що включає етилацетат, етанол, ізопропанол і суміші етанолу й/або ізопропанолу з водою. У випадку використання суміші води й одного або більшої кількості С1-С4-алканолів, переважно метанолу, етанолу й/або ізопропанолу, найбільше переважно етанолу краще об'ємне відношення вода: алканол знаходиться в діапазоні приблизно від 1:4 до 4:1; більш переважно - приблизно від 1:2 до 2:1; ще більш переважно - приблизно від 2:3 до 3:2. Стадію (а) переважно проводити приблизно при температурі (приблизно 20°С) або при підвищеній температурі, приблизно до температури кипіння розчинника або суміші розчинників. Для зниження розчинності сполуки А у розчиннику, на стадії (а) і/або на стадії (b) можна додати один або більшу кількість антирозчинників або нерозчинників, переважно під час стадії (а) або на початку стадії (b).Прикладом придатного антирозчинника або нерозчинника є вода. Кількість антирозчинника або нерозчинника або їх суміші переважно вибирати так, щоб одержати перенасичений або майже перенасичений розчин. На стадії (b) розчин витримують протягом періоду часу, достатнього для одержання осаду. Температура розчину на стадії (b) є приблизно такою ж або нижче, ніж на стадії (а). Під час витримування температуру розчину, що містить сполуки А, переважно понизити, переважно - до температури в діапазоні від 20 до 0°С або навіть більше низкою. Стадію (b) можна проводити з перемішуванням або без нього. Як відомо фахівцеві в даній галузі техніки, шляхом зміни періоду часу й різниці температур на стадії (b) можна міняти розміри, форму і якість одержуваних кристалів. Крім того, кристалізацію можна ініціювати за методиками, відомими у даній галузі техніки, наприклад, шляхом дряпання або розтирання. В (майже) насичений розчин необов'язково можна внести кристали затравки. На стадії (с) розчинник (розчинники) можна видалити з осаду за відомими методиками, такими 9 91546 10 як, наприклад, фільтрування, фільтрування з відсШляхом введення кристалічної форми, пропомоктуванням, декантація або центрифугування. нованої в даному винаході, можна зменшити або На стадії (d) надлишок розчинника (розчиннипригнічити аномальне накопичення жиру в печінці. ків) видаляють з осаду за методиками, відомими Тому іншим об'єктом даного винаходу є спосіб фахівцеві в даній галузі техніки, такими як, наприпопередження, уповільнення, затримування або клад, зниження парціального тиску розчинника лікування захворювань або патологічних станів, (розчинників), переважно - у вакуумі, і/або шляхом обумовлених аномальним накопиченням жиру в нагрівання до температури вище приблизно 20°С, печінці, у пацієнта, що потребує цього, що харакпереважно - у температурному діапазоні нижче теризується тим, що вводять фармацевтичну ком80°С, ще більш переважно - нижче 50°С. позицію, пропоновану в даному винаході. ЗахвоСполуки А можна синтезувати за методиками, рювання або патологічні стани, які обумовлені зокрема й/або в загальному вигляді описаними аномальним накопиченням жиру в печінці, переабо цитованими у заявці WO 2005/092877. Крім важно вибрані з групи, що включає генералізовану того, біологічні характеристики сполуки А можна жирову інфільтрацію печінки, неалкогольну жирову досліджувати так, як це описано в заявці WO інфільтрацію печінки (НАІП), неалкогольний стеа2005/092877, що у всій своїй повноті включена в тогепатит (НАСГ), викликану переїданням жирову даний винахід як посилання. інфільтрацію печінки, діабетичну жирову інфільтКристалічну форму, пропоновану в даному вирацію печінки, алкогольну жирову інфільтрацію наході, переважно використати, як лікарську актипечінки й токсичну жирову інфільтрацію печінки. вну речовину в основному в чистій формі, тобто Зокрема, кристалічна форма, пропонована в яка в основному не містить інших кристалічних даному винаході, є застосовною для приготування форм сполуки А. Проте, даний винахід також фармацевтичних композицій, призначених для включає кристалічну форму, визначену в даному попередження або лікування діабету, переважно винаході, у суміші з іншою кристалічною формою цукрового діабету типу 1 і 2 і/або ускладнень при або формами. Якщо активна лікарська речовина діабеті. повинна бути сумішшю кристалічних форм, то пеКрім того, кристалічна форма, пропонована в реважно, якщо ця речовина включає не менш 50% даному винаході, є особливо придатною для покристалічної форми, описаної в даному винаході. передження або лікування надлишкової маси, Внаслідок своєї здатності інгібувати активність ожиріння (включаючи ожиріння класу І, класу II SGLT, кристалічна форма, пропонована в даному і/або класу III), вісцерального ожиріння й/або абвинаході, придатна для приготування фармацевдомінального ожиріння. тичних композицій, призначених для лікування Дози, необхідні для забезпечення відповідної й/або попереджувального лікування всіх тих патоактивності з метою лікування або попередження, логічних станів або захворювань, на які можна звичайно залежать від пацієнта, характеру й ваги вплинути шляхом інгібування активності SGLT, захворювання або патологічного стану й методики переважно - активності SGLT-2. Тому кристалічна й частоти введення й визначаються лікарем. Доціформа є особливо придатною для приготування льна доза може становити від 1 до 100 мг, перефармацевтичних композицій, призначених для важно - від 1 до 30 мг при внутрішньовенному попередження або лікування захворювань, перевведенні й від 1 до 1000 мг, переважно - від 1 до важно - метаболічних порушень або патологічних 100 мг при пероральному введенні й у кожному станів, таких як цукровий діабет типу 1 і 2, ускладвипадку засіб уводять від 1 до 4 разів на добу. Для нення при діабеті (таких як, ретинопатія, нефтопацього фармацевтичні композиції, пропоновані в тія або невропатії, діабетична стопа, виразки, макданому винаході, переважно включають кристаліроангіопатії), метаболічний ацидоз або кетоз, чну форму разом з одним або більшою кількістю реактивна гіпоглікемія, гіперінсулінемія, порушензвичайних інертних наповнювачів і/або розріджуня метаболізму глюкози, резистентність до інсулівачів. Такі фармацевтичні композиції можна пригону, метаболічний синдром, дисліпідемії різної етіотувати у вигляді звичайних галенових препаратів, логії, атеросклероз і родинні захворювань, таких як таблетки без покриття або з покриттям, ожиріння, високий артеріальний тиск, хронічна капсули, порошки, суспензії або супозиторії. серцева недостатність, набряк і гіперурикемія. Наведені нижче приклади синтезу призначені Кристалічна форма також є придатною для пригодля ілюстрації методики одержання сполуки А і тування фармацевтичних композицій, призначених його кристалічної форми. їх варто розуміти тільки, для попередження дегенерації бета-клітин, такої як можливі методики, наведені як приклад, а не як наприклад, апоптоз або некроз бета-клітин пандля обмеження даного винаходу їх описом. креатичних острівців. Кристалічна форма також є Одержання вихідних сполук: придатною для приготування фармацевтичних Приклад І композицій, призначених для поліпшення або відновлення функціональності клітин підшлункової залози, а також для збільшення кількості й розміру бета-клітин панкреатичних острівців. Кристалічну форму, пропоновану в даному винаході, також можна використати для приготування фармацевтичних композицій, застосовних у якості діуретиків або гіпотензивних засобів і придатних для попередження й лікування гострої серцевої недостатнос(5-бром-2-хлорфеніл)-(4-метоксифеніл)ті. метанон 11 91546 12 38,3 мл оксалілхлориду й 0,8 мл диметилформаміду додають до суміші 100 г 5-бром-24-(5-бром-2-хлорбензил)-фенол хлорбензойної кислоти з 500 мл дихлорметану. Розчин 14,8 г 4-бром-1-хлор-2-(4Реакційну суміш перемішують протягом 14 год., метоксибензил)-бензолу в 150 мл дихлорметану потім фільтрують і за допомогою роторного випарохолоджують у бані з льодом. Потім додають 50 ника відокремлюють від всіх летких компонентів. мл 1 М розчину триброміду бору в дихлорметані, і Залишок розчиняють в 150 мл дихлорметану, розрозчин перемішують протягом 2 год. при темперачин охолоджують до -5 °С і додають 46,5 г анізолу. турі навколишнього середовища. Потім розчин Потім порціями додають 51,5 г трихлориду алюміповторно охолоджують у бані з льодом і по крапнію, так щоб температура не перевищувала 5 °С. лях додають насичений водяний розчин карбонату Розчин перемішують протягом ще 1 год. при темкалію. При температурі навколишнього середовипературі від 1 до 5 °С і потім виливають на здрібща значення рН суміші доводять до 1 за допомонений лід. гою 1 М водяного розчину хлористоводневої кисОрганічну фазу відокремлюють і водну фазу лоти, органічну фазу відокремлюють і водну фазу ще тричі екстрагують дихлорметаном. ще тричі екстрагують етилацетатом. Об'єднані органічні фази промивають 1 М воОб'єднані органічні фази сушать над сульфадяним розчином хлористоводневої кислоти, двічі 1 том натрію й розчинник повністю видаляють. М водяним розчином гідроксиду натрію й розсоВихід: 13,9 г (98% від теоретичного) лом. Потім органічну фазу сушать, розчинник виМас-спектр (ESI-): m/z = 295/297/299 (Br+Cl) даляють і залишок перекристалізують з етанолу. [M-H]Вихід: 86,3 г (64% від теоретичного) Мас-спектр (ESI+): m/z - 325/327/329 (Br+Cl) [M+H]+ 4-бром-1 -хлор-2-(4-метоксибензил)-бензол Розчин 86,2 г (5-бром-2-хлорфеніл)-(4метоксифеніл)-метанону й 101,5 мл триетилсилану в 75 мл дихлорметану й 150 мл ацетонітрилу охолоджують до 10°С. Потім при перемішуванні додають 50,8 мл ефірату трифториду бору, так щоб температура не перевищувала 20°С. Розчин перемішують протягом 14 год. при температурі навколишнього середовища, а потім додають ще 9 мл триетилсилану й 4,4 мл ефірату трифториду бору. Розчин перемішують протягом ще 3 год. при температурі від 45 до 50°С і потім охолоджують до температури навколишнього середовища. Додають розчин 28 г гідроксиду калію в 70 мл води й отриману суміш перемішують протягом 2 год. Потім органічну фазу відокремлюють і водну фазу ще тричі екстрагують діізопропіловим ефіром. Об'єднані органічні фази двічі промивають 2 М водяним розчином гідроксиду калію й один раз розсолом і потім сушать над сульфатом натрію. Після видалення розчинника залишок промивають етанолом, повторно відокремлюють і сушать при 60°С. Вихід: 50,0 г (61% від теоретичного) Мас-спектр (ESI+): m/z = 310/312/314 (Br+Cl) [M+H]+ [4-(5-бром-2-хлорбензил)-фенокси]-третбутилдиметилсилан Розчин 13,9 г 4-(5-бром-2-хлорбензил)-фенолу в 140 мл дихлорметану охолоджують у бані з льодом. Потім додають 7,54 г третбутилдиметилсилілхлориду в 20 мл дихлорметану, а після цього 9,8 мл триетиламіну й 0,5 г 4диметиламінопіридину. Розчин перемішують протягом 16 год. при температурі навколишнього середовища й потім розбавляють за допомогою 100 мл дихлорметану. Органічну фазу двічі промивають 1 М водяним розчином хлористоводневої кислоти й один раз водяним розчином гідрокарбонату натрію й потім сушать над сульфатом натрію. Після видалення розчинника залишок фільтрують через силікагель (циклогексан/етилацетат 100:1). Вихід: 16,8 г (87% від теоретичного) Мас-спектр (El): m/z = 410/412/414 (Br+Cl) [M]+ 2, 3, 4, 6-тетракіс-O-(триметилсіліл)-Dглюкопіранон Розчин 20 г D-глюконо-1,5-лактону й 98,5 мл N-метилморфоліну в 200 мл тетрагідрофурану охолоджують до -5 °С. Потім по краплях додають 85 мл триметилсілілхлориду, так щоб температура 13 91546 14 не перевищувала 5 °С. Потім розчин перемішують тані й перекристалізації продукту з етанолу. Отрипротягом 1 год. при температурі навколишнього маний у такий спосіб продукт перетворюють у висередовища, протягом 5 год. при 35 °С і повторно хідну сполуку шляхом дезацетилування в метанолі протягом 14 год. при температурі навколишнього 4 М водяним розчином гідроксиду калію. середовища. Після додавання 300 мл толуолу Вихід: 1,6 г (46% від теоретичного) + + розчин охолоджують у бані з льодом і додають 500 Мас-спектр (ESI ): m/z = 398/400 (СІ) [М+Н] мл води, так щоб температура не перевищувала 10°С. Потім органічну фазу відокремлюють і по одному разу промивають водяним розчином дигідрофосфату натрію, водою й розсолом. Розчинник видаляють, залишок розчиняють в 250 мл толуолу й розчинник повторно повністю видаляють. Вихід: 52,5 г (чистота приблизно 90%) Мас-спектр (ESI+): m/z = 467 [М+Н]+ 1-хлор-4-(-D-глюкопіраноз-1-ил)-2-(4гідроксибензил)-бензол Розчин 4,0 г [4-(5-бром-2-хлорбензил)фенокси]-трет-бутилдиметилсилану в 42 мл сухого діетилового ефіру охолоджують до -80 °С в атмосфері аргону. До охолодженого розчину по краплях повільно додають 11,6 мл 1,7 М розчину третбутиллітію в пентані й потім перемішують протягом 30 хв. при -80 °С. Потім цей розчин за допомогою шприца, який охолоджують твердим діоксидом вуглецю, по краплях додають до розчину 4,78 г 2, 3, 4, 6-тетракіс-О-(триметилсіліл)-D-глюкопіранону в 38 мл діетилового ефіру, охолодженого до -80 °С. Отриманий розчин перемішують протягом 3 год. при -78 °С. Потім додають розчин 1,1 мл метансульфонової кислоти в 35 мл метанолу й розчин перемішують протягом 16 год. при температурі навколишнього середовища. Потім розчин нейтралізують гідрокарбонатом натрію, додають етилацетат і метанол видаляють разом з ефіром. До розчину, що залишився, додають водяний розчин гідрокарбонату натрію й отриману суміш чотири рази екстрагують етилацетатом. Органічні фази сушать над сульфатом натрію й випарюють. Залишок розчиняють в 30 мл ацетонітрилу й 30 мл дихлорметану й розчин охолоджують до -10 °С. Після додавання 4,4 мл триетилсилану по краплях додають 2,6 мл ефірату трифториду бору, так щоб температура не перевищувала -5 °С. Після завершення додавання розчин перемішують протягом ще 5 год. при температурі від -5 до -10 °С і потім реакцію зупиняють шляхом додавання водяного розчину гідрокарбонату натрію. Органічну фазу відокремлюють і водну фазу чотири рази екстрагують етилацетатом. Об'єднані органічні фази сушать над сульфатом натрію, розчинник видаляють і залишок очищають за допомогою хроматографії на силікагелі (дихлорметан/метанол 1:03:1). Одержують продукт у вигляді суміші складу приблизно 6:1 /, яку можна перетворити в чистий Раномер шляхом повного ацетилування гідроксигруп оцтовим ангідридом і піридином у дихлорме 1-хлор-4-(-D-глюкопіраноз-1-ил)-2-[4-((S)тетрагідрофуран-3-ілокси)-бензил]-бензол 0,19 г (R)-3-(4-метилфенілсульфонілокси)тетрагідрофуран додають до суміші 0,20 г 1-хлор4-(-D-глюкопіраноз-1-ил)-2-(4-гідроксибензил)бензолу й 0,29 г карбонату цезію в 2,5 мл диметилформаміду. Суміш перемішують при 75 °С протягом 4 год., а потім додають ще 0,29 г карбонату цезію й 0,19 г (R)-3-(4-метилфенілсульфонилокси)тетрагідрофурану. Після ще 14 год. перемішування при 75 °С суміш охолоджують до температури навколишнього середовища й додають розсіл. Отриману суміш екстрагують етилацетатом, об'єднані органічні екстракти сушать над сульфатом натрію й розчинник видаляють. Залишок очищають за допомогою хроматографії на силікагелі (дихлорметан/метанол 1:0 5:1). Вихід: 0,12 г (49% від теоретичного) Масспектр (ESI+): m/z = 451/453 (СІ) [М+Н]+ Одержання кристалічної форми: Варіант 1: 30мг 1-хлор-4-(-D-глюкопіраноз-1-ил)-2-[4((S)-тетрагідрофуран-3-ілокси)-бензил]-бензолу (отриманого, як описано вище) розчиняють в 0,8 мл етилацетату (що містить 0,5-3% води) при нагріванні приблизно до 50 °С. Розчину дають повільно остудитися (приблизно від 1 до 3 год.) приблизно до 20 °С. Через 48 год. кристалічну форму виділяють у вигляді білих кристалів шляхом фільтрування. Надлишок розчинника видаляють шляхом витримування кристалів при підвищеній температурі (від 40 до 50 °С) протягом приблизно від 3 до 4 год. при зниженому тиску. Варіант 2: 1г 1-хлор-4-(-D-глюкопіраноз-1-ил)-2-[4-((S)тетрагідрофуран-3-ілокси)-бензил]-бензолу розчиняють в 5 мл суміші вода/етанол (об'ємне відношення 2:3) при нагріванні приблизно до 50 °С. Додають 8 мл води й розчину дають остудитися приблизно до 20 °С протягом від 1 до 3 год. Через 16 год. кристалічну форму виділяють у вигляді білих кристалів шляхом фільтрування. Надлишок розчинника видаляють шляхом витримування кристалів при підвищеній температурі (від 40 до 50 °С) протягом приблизно від 4 до 6 год.. Варіант 3: 1г 1-хлор-4-(-D-глюкопіраноз-1-ил)-2-[4-((S)тетрагідрофуран-3-ілокси)-бензил]-бензолу розчиняють в 11 мл ізопропанолу при нагріванні приблизно до 50 °С. Розчину дають остудитися приб 15 91546 16 лизно до 20 °С протягом від 1 до 3 год. Через 16 няють в 60 мл суміші вода/етанол(об'ємне відногод. кристалічну форму виділяють у вигляді білих шення 2:3) при нагріванні приблизно до 50 °С. Рокристалів шляхом фільтрування. Залишковий роззчину дають остудитися приблизно до 20 °С прочинник видаляють шляхом витримування кристатягом 3 год. і кристалічну сполуку виділяють лів при підвищеній температурі (від 40 до 50 °С) шляхом фільтрування. Виділену тверду речовину протягом приблизно від 4 до 6 год. сушать при 40 °С протягом 16 год. і одержують Варіант 4: приблизно 6 г кристалічної форми. 8,9г 1-хлор-4-(-D-глюкопіраноз-1-ил)-2-[4-((S)тетрагідрофуран-3-ілокси)-бензил]-бензолу розчи Комп’ютерна верстка В. Мацело Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюCrystalline form of 1-chloro-4-(я-d-glucopyranos-1-yl)-2-[4-((s)-tetrahydrofuran-3-yloxy)-benzyl]-benzene, a method for its preparation and the use thereof for preparing medicaments
Автори англійськоюEckhardt Matthias, Himmelsbach Frank, Sick Sandra, Schuehle Martin, Martin Hans-Juergen
Назва патенту російськоюКристаллическая форма 1-хлор-4-(b-d-глюкопираноз-1- ил)-2-[4-((s)- тетрагидрофуран-3-илокси)-бензил]-бензола, способ ее получения и ее применение при приготавлении лекарственных средств
Автори російськоюЭкхардт Маттиас, Химмельсбах Франк, Зик Сандра, Шюле Мартин, Мартин Ханс-Юрген
МПК / Мітки
МПК: A61P 3/00, A61K 31/351, A61P 9/00, C07D 309/10
Мітки: форма, спосіб, лікарських, кристалічна, застосування, одержання, засобів, 1-хлор-4-(b-d-глюкопіраноз-1-ил)-2-[4-((s)-тетрагідрофуран-3-ілокси)-бензил]-бензолу, приготуванні
Код посилання
<a href="https://ua.patents.su/8-91546-kristalichna-forma-1-khlor-4-b-d-glyukopiranoz-1-il-2-4-s-tetragidrofuran-3-iloksi-benzil-benzolu-sposib-oderzhannya-ta-zastosuvannya-pri-prigotuvanni-likarskikh-zasobiv.html" target="_blank" rel="follow" title="База патентів України">Кристалічна форма 1-хлор-4-(b-d-глюкопіраноз-1-ил)-2-[4-((s)-тетрагідрофуран-3-ілокси)-бензил]-бензолу, спосіб її одержання та її застосування при приготуванні лікарських засобів</a>
Дималеат 4-[(3-хлор-4-фторфеніл)аміно]-6-{[4-(n,n-диметиламіно)-1-оксо-2-бутен-1-іл]аміно}-7-((s)-тетрагідрофуран-3-ілокси)хіназоліну, спосіб його одержання та застосування
Номер патенту: 91401
Опубліковано: 26.07.2010
Автори: Ралль Вернер, Кулінна Крістіан, Зойка Райнер, Зігер Петер, Шнаубельт Юрген
МПК: A61K 31/505, A61P 35/00, C07D 405/12
Мітки: дималеат, застосування, 4-[(3-хлор-4-фторфеніл)аміно]-6-{[4-(n,n-диметиламіно)-1-оксо-2-бутен-1-іл]аміно}-7-((s)-тетрагідрофуран-3-ілокси)хіназоліну, спосіб, одержання
Формула / Реферат:
1. Спосіб одержання дималеату 4-[(3-хлор-4-фторфеніл)аміно]-6-{[4-(N,N-диметиламіно)-1-оксо-2-бутен-1-іл]аміно}-7-((S)-тетрагідрофуран-3-ілокси)хіназоліну, який включає наступні стадії:а) сполуку загальної формули (V) (V)піддають у відповідних розчинниках після відповідної активації взаємодії з ді-(С1-С4алкіл)фосфонооцтовою кислотою таб)...
Кристалічна форма осанетанту (варіанти), спосіб її одержання (варіанти) та фармацевтична композиція

Номер патенту: 72890
Опубліковано: 16.05.2005
Автори: Гросклод Патрік, Алькад Ален, Мон'є Олів'є, Рош Жером, Анн-Аршар Жіль
МПК: A61K 31/4468, A61K 31/451, C07D 211/58, A61P 43/00
Мітки: кристалічна, варіанти, фармацевтична, одержання, форма, спосіб, осанетанту, композиція
Формула / Реферат:
1. Спосіб кристалізації осанетанту, який відрізняється тим, що:і) осанетант, який містить менше 20% забруднень, кристалізують із суміші етанол/вода або з ізопропанолу, отримуючи кристалічну форму І, абоіі) осанетант, який містить менше 20% забруднень, кристалізують із суміші етанол/ізопропіловий етер/вода, отримуючи кристалічну форму II.2. Спосіб за п. 1, який відрізняється тим, що осанетант до кристалізації містить...
Азабіциклічні сполуки, спосіб їх одержання і їх застосування як лікарських засобів, зокрема, як антибактеріальних засобів

Номер патенту: 73791
Опубліковано: 15.09.2005
Автори: Лампіла Максім, Асзоді Жозеф, Роулендс Девід Ален, Фроментен Клод
МПК: A61P 11/00, C07B 61/00, A61P 31/04, C07D 487/08, C07D 498/08, A61K 31/439, A61P 31/00, C07D 471/08, A61K 31/5386, A61K 31/529, A61P 17/00, A61P 9/10, A61K 31/4188
Мітки: спосіб, лікарських, азабіциклічні, антибактеріальних, сполуки, одержання, застосування, зокрема, засобів
Формула / Реферат:
1. Сполука загальної формули (І) або одна з її солей з основою або з кислотою:в якій:R1 означає атом водню, радикал СООН, CN, COOR, CONR6R7, (СН2)n'R5 або радикал ,де- R вибирають з групи, що складається з алкілу з 1-6 атомами вуглецю, можливо заміщеного...
Стабільна поліморфна модифікація флібансерину, спосіб її промислового одержання та її застосування для одержання лікарських засобів

Номер патенту: 76767
Опубліковано: 15.09.2006
Автори: Бомбарда Карло, Ежая Антоіне, Дубіні Енріка, ШНАЙДЕР Хайнріх
МПК: A61P 25/18, A61K 31/496, A61P 25/20, A61P 25/24, A61P 25/22, A61P 15/00, A61P 25/00, C07D 235/26, A61P 25/28, C07D 233/26, A61P 25/16
Мітки: лікарських, засобів, поліморфна, одержання, стабільна, застосування, модифікація, флібансерину, спосіб, промислового
Формула / Реферат:
1. Кристалічна поліморфна модифікація А (форма А) флібансерину формули 1 1,у якої ендотермічний максимум при її термічному аналізі диференціальною сканувальною калориметрією припадає на температуру 161°С.2. Флібансерин формули 1 у формі А за п. 1.3. Спосіб промислового одержання флібансерину формули 1 за п. 1 або п. 2
Альфа-кристалічна форма ранелату стронцію, спосіб її одержання і фармацевтична композиція, яка її містить, і її застосування у виробництві ліків

Номер патенту: 80008
Опубліковано: 10.08.2007
Автори: Дам'єн Жерар, Демюінк Ізабель, Орват Стефан
МПК: A61P 19/02, A61K 31/38, A61K 33/24, A61F 3/00, C07D 333/38, A61K 31/381
Мітки: містить, ранелату, виробництві, альфа-кристалічна, форма, композиція, яка, одержання, ліків, фармацевтична, спосіб, стронцію, застосування
Формула / Реферат:
1. Альфа-кристалічна форма ранелату стронцію формули (І): , (I)яка відрізняється тим, що вміст води складає від 22 до 24%, і вона має наступну порошкову рентгеноструктурну дифракційну діаграму, виміряну з використанням дифрактометра PANalytical X'Pert Pro разом з детектором X'Celerator і виражену в одиницях положення променя (брегівський кут 2 тета, який...
Попередній патент: Пестицидна комбінація, спосіб боротьби з ураженням патогенами або шкідниками та спосіб захисту матеріалу для розмноження рослин
Наступний патент: Спосіб одержання cu/zn/al-каталізатора, каталізатор та його застосування
Випадковий патент: Свердловинний асиметричний кумулятивний заряд