Азабіциклічні сполуки, спосіб їх одержання і їх застосування як лікарських засобів, зокрема, як антибактеріальних засобів
Номер патенту: 73791
Опубліковано: 15.09.2005
Автори: Роулендс Девід Ален, Фроментен Клод, Асзоді Жозеф, Лампіла Максім








Формула / Реферат
1. Сполука загальної формули (І) або одна з її солей з основою або з кислотою:
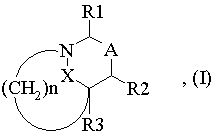
в якій:
R1 означає атом водню, радикал СООН, CN, COOR, CONR6R7, (СН2)n'R5 або радикал
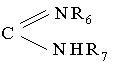 ,
,
де
- R вибирають з групи, що складається з алкілу з 1-6 атомами вуглецю, можливо заміщеного піридилом або карбамоїлом; -СН2-алкенілу, що містить в цілому 3-9 атомів вуглецю; арилу з 6-10 атомами вуглецю або аралкілу з 7-11 атомами вуглецю, причому арильне або аралкільне ядро може бути заміщене радикалом ОН, NH2, NO2, алкілом з 1-6 атомами вуглецю, алкоксилом з 1-6 атомами вуглецю або одним або декількома атомами галогену;
- R6 i R7, однакові або різні, вибирають з групи, що складається з атома водню, алкілу з 1-6 атомами вуглецю, арилу з 6-10 атомами вуглецю і аралкілу з 7-11 атомами вуглецю, можливо заміщених карбамоїлом, уреїдогрупою або диметиламіногрупою, і заміщеного піридилом алкілу з 1-6 атомами вуглецю;
- n' дорівнює 1 або 2;
- R5 вибирають з групи, що складається з радикала СООН, CN, ОН, NH2, СО-NR6R7, COOR, OR, OCOH, OCOR, OCOOR, OCONHR, OCONH2, NHR, NHCOH, NHCOR, NHSO2R, NH-COOR, NH-CO-NHR або NHCONH2, причому R, R6 і R7 мають вищезгадані значення;
- R2 означає атом водню або групу (СН2)n'1R5, де n'1 дорівнює 0, 1 або 2, і R5 має вищезгадане значення;
- R3 означає атом водню або алкіл з 1-6 атомами вуглецю;
- А означає зв'язок між двома атомами вуглецю, що несуть радикали R1 i R2,
або групу
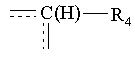 ,
,
де R4 означає атом водню або групу (СН2)n'1R5, причому n'1 і R5 мають вищезгадані значення, пунктирна лінія означає можливий зв'язок з одним або іншим з атомів вуглецю, що несуть замісники R1 і R2;
- n дорівнює 1 або 2;
- X означає двовалентну групу -С(O)-В-, пов'язану з атомом азоту за допомогою атома вуглецю, де
- В означає двовалентну групу -O-(СН2)n''-, пов'язану з карбонілом за допомогою атома кисню; групу -NR8-(CH2)n''- або -NR8-O-, пов'язану з карбонілом за допомогою атома азоту; n'' дорівнює 0 або 1; і R8 вибирають з групи, що складається з атома водню, радикала ОН, R, OR, Y, OY, Y1, OY1, Y2, OY2, Y3, OCH2CH2SOmR, OSiRaRbRc і SiRaRbRc, причому Ra, Rb і Rc означають, індивідуально, лінійний або розгалужений алкіл з 1-6 атомами вуглецю або арил з 6-10 атомами вуглецю, R має вищезгадане значення і m дорівнює 0, 1 або 2;
- Y вибирають з групи, що складається з радикалів СОН, COR, COOR, CONH2, CONHR, CONHOH, CONHSO2R, CH2СООН, CH2СООR, СН2СОNНОН, CH2CONHCN, СН2-тетразоліл, захищений СН2-тетразоліл, СН2SO3Н, CH2SO2R, CH2PO(OR)2, CH2PO(OR)(OH), CH2PO(R)(OH) i CH2PO(OH)2;
- Y1 вибирають з групи, що складається з радикалів SO2R, SO2NHCOH, SO2NHCOR, SO2NHCOOR, SO2NHCONHR, SO2NHCONH2 і SO3Н;
- Y2 вибирають з групи, що складається з радикалів РО(ОН)2, PO(OR)2, PO(OH)(OR) i PO(OH)(R);
- Y3 вибирають з групи, що складається з тетразолілу, заміщеного радикалом R тетразолілу, скварат-радикала, NH- або NR-тетразолілу, заміщеного радикалом R NH- або NR-тетразолілу, NHSO2R і NRSO2R, причому R має вищезгадане значення; маючи на увазі, що, коли n дорівнює 1 і А означає групу
,
в якій R4 означає атом водню, і або Х означає групу -С(O)-O-(СН2)n'', в якій n'' дорівнює 0 або 1, або Х означає групу -CO-NR8-(CH2)n'', в якій n'' дорівнює 1 і R8 означає ізопропіл, або Х означає групу -CO-NR8-(CH2)n'', в якій n'' дорівнює 0 і R8 означає атом водню або феніл, тоді R1, R2 і R3 всі три одночасно не можуть означати атом водню.
2. Сполука за п. 1, яка відрізняється тим, що n дорівнює 1.
3. Сполука за пп. 1 або 2, яка відрізняється тим, що А означає групу
,
визначену в п. 1.
4. Сполука за п. 3, яка відрізняється тим, що R4 означає атом водню.
5. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що Х означає двовалентну групу -СО-В-, в якій В означає групу –NR8-(CH2)n''-, визначену в п. 1.
6. Сполука за будь-яким з пп. 1-5, яка відрізняється тим, що група R8 означає групу Y1 або OY1, в якій Y1 вибирають з груп SO2R, SO2NHCOR, SO2NHCOOR, SO2NHCONHR і SO3Н, і R є таким, як визначено в п. 1.
7. Сполуки формули (І) за п. 1, вибрані з групи, що містить:
транс-7-оксо-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема натрієва сіль; транс-7-оксо-N-(фенілметил)-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема натрієва сіль; транс-7-оксо-N-(4-піридинілметил)-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема натрієва сіль; транс-7-оксо-N-(3-піридинілметил)-6-(сульфокси)-1,6-діазабіцикло-[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема натрієва сіль; транс-7-оксо-N-(2-аміно-2-оксоетил)-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема натрієва сіль; фенілметил-транс-7-оксо-6-(сульфокси)-1,6-діазабіцикло-[3,2,1]октан-2-карбоксилат.
8. Спосіб одержання сполуки за будь-яким з пп. 1-7, який відрізняється тим, що проводять:
(а) стадію, протягом якої сполуку формули (II):
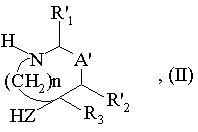
де
- R'1 означає атом водню або радикала CN, захищений СООН, COOR', (CH2)n'R'5, CONR6R7 або захищений радикал 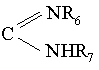 ,
,
де
- n', R6 і R7 мають значення за п. 1 і
- R' і R'5 мають, відповідно, значення, вказані для R і R5 в п. 1, в яких можливо наявні реакційноздатні захищені групи ;
- R'2 означає атом водню або групу (CH2)n'R'5, де n'1 має значення, вказане в п. 1, і R'5 має вищезгадане значення;
- R3 має вказане в п. 1 значення;
- А' означає зв'язок між двома атомами вуглецю, що несуть радикали R'1 i R'2, або групу
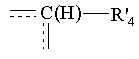 ,
,
де R'4 означає атом водню або групу (CH2)n'1R'5, причому n'1 і R'5 мають вищезгадані значення, пунктирна лінія означає можливий зв'язок з одним або іншим з атомів вуглецю, що несуть замісники R'1 і R'2;
- n має вказане в п. 1 значення;
- HZ означає групу НО-(СH2)n''-, HNR'8-(CH2)n''- або HNR'8-O-, причому n'' має вищезгадане значення і R'8 означає атом водню, захищений гідроксил R', OR', радикал Y1 або OY', причому Y' вибирають з груп: СОН, COR', COOR', CONH2, CONHR', захищена CONHOH, CONHSO2R', захищена CH2COOH, CH2COOR', захищена CH2CONHOH, CH2CONHCN, заміщений радикалом R' СН2-тетразоліл, CH2SO2R', CH2PO(OR')2, захищена СН2SО3, захищена CH2PO(OR')OH, захищена CH2PO(R')OH, захищена СН2РО(ОН)2; радикал Y'1 або OY'1, причому Y'1 вибирають з груп: SO2R', SO2NHCOH, SO2NHCOR', SO2NHCOOR', SO2NHCONH2, SO2NHCONHR' і захищена група SO2Н; радикал Y'2 або OY'2, причому Y'2 означає захищену групу РО(ОН)2, захищену групу PO(OH)(OR'), захищену групу PO(OH)R' або групу PO(OR')2; або радикал Y'3, причому Y'3 вибирають із захищеного тетразолілу, заміщеного радикалом R' тетразолілу, захищеного NH- або NR'-тетразолілу, заміщеного радикалом R' NH- або NR'-тетразолілу, NHSO2R' і NR'SO2R', де R' має вищезгадане значення;
вводять у взаємодію з карбонілуючим агентом, у разі необхідності, в присутності основи, з одержанням проміжної сполуки формули (III):
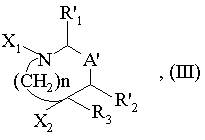
в якій R'1, R'2, R3, А' і n мають вищезгадані значення і або Х1 означає атом водню і Х2 означає групу -Z-CO-Х3, де Х3 означає залишок карбонілуючого агента, або Х2 означає групу -ZH і Х1 означає групу Х3, де Х3 має вищезгадане значення;
(b) стадію, протягом якої одержану вище проміжну сполуку циклізують в присутності основи; і
(c) у разі необхідності, перед стадією (а) і/або після стадії (b) здійснюють одну або декілька наступних реакцій у відповідному порядку: захист реакційноздатних функціональних груп; видалення захисних груп від реакційноздатних функціональних груп; етерифікація; омилення; сульфатація; фосфатація; амідування; ацилювання; сульфонілування; алкілування; введення подвійного зв'язку; утворення карбамідної групи; введення тетразолілу; відновлення карбонових кислот; дегідратація аміду до нітрилу; солеутворення; іонообмін; розщеплення або розділення діастереоізомерів; окислення сірки до сульфоксиду і/або сульфону.
9. Спосіб за п. 8, який відрізняється тим, що карбонілуючий агент вибирають з групи, що складається з фосгену, дифосгену, трифосгену, арил-, аралкіл-, алкіл- і -алкенілхлорформіатів, алкілдикарбонатів, карбонілдіімідазолу і їх сумішей.
10. Спосіб за п. 8 або 9, який відрізняється тим, що реакцію карбонілування проводять в присутності основи.
11. Спосіб за п. 10, який відрізняється тим, що основою є амін.
12. Спосіб за п. 8, який відрізняється тим, що в стадії (b) основу вибирають з групи, що складається з амінів, гідридів, алкоголятів, амідів або карбонатів лужних або лужноземельних металів.
13. Лікарський засіб, що являє собою продукт формули (І), вказаний за п. 1, а також його солі з фармацевтичнo прийнятними кислотами і основами.
14. Лікарський засіб, що являє собою продукт, визначений за будь-яким з пп. 2-7, а також його солі з фармацевтичнo прийнятними кислотами і основами.
15. Фармацевтична композиція, що містить як діюче начало щонайменше один лікарський засіб за будь-яким з пп. 13 або 14.
16. Сполука загальної формули (III) або одна з її солей з кислотою, зокрема її гідрохлорид:
в якій R'1, R'2, R3, A', n, X1 і Х3 мають такі ж значення, як вказані в п. 8.
Текст
Винахід відноситься до нових гетероциклічних сполук, до способу їх одержання і їх застосування як лікарських засобів, зокрема, як антибактеріальних засобів. У журналі J. Org. Chem., 37, № 5,697-699 (1972) описане, зокрема, одержання біциклічного похідного брутто-формули C10H18N20 . У журналі J. Org. Chem., 45, № 26, 5325-5326 (1980) описане, зокрема, одержання біциклічних похідних брутто-формул C6H9NO 2 і С7Н11NO2 . У огляді, опублікованому в Chemical Reviews, 83, № 5, 549-555 (1983), описане, зокрема, одержання біциклічних похідних брутто-формул C 10H18N2O і C 7H12N2 O. У журналі Angew. Chem. Int. Ed., 39, № 3, 625-628 (2000) описане, зокрема, одержання сполуки бруттоформули C12H12N2O. У цих документах немає ніякої інформації про конкретне використання цих сполук в області терапії. Об'єктом винаходу є сполуки, що відповідають наступній формулі (І): в якій: - R1 означає атом водню, радикал СООН, CN, COOR, CONR 6R7, (CH2)nR5 або радикал дє - R вибирають з групи, що складається з алкілу з 1-6 атомами вуглецю, можливо заміщеного піридилом або карбамоїлом; -СН2-алкенілу загалом з 3-9 атомами вуглецю; арилу з 6-10 атомами вуглецю або аралкілу з 7-11 атомами вуглецю, причому арильне або аралкільне ядро може бути заміщене радикалом OH, NH2, NO2, алкілом з 1-6 атомами вуглецю, алкоксилом з 1-6 атомами вуглецю або одним або декількома атомами галогену; - R6 і R7, однакові або різні, вибирають з групи, що складається з атома водню, алкілу з 1-6 атомами вуглецю, арилу з 6-10 атомами вуглецю і аралкілу з 7-11 атомами вуглецю, можливо заміщених карбамоїлом, уреїдогрупою або диметиламіногрупою, і заміщеного піридилом алкілу з 1-6 атомами вуглецю; - n дорівнює 1 або 2; і - R5 вибирають з групи, що складається з радикала СООН, CN, OH, NH2, СО-NReRy, COOR, OR, ОСОН, OCOR, OCOOR, OCONHR, OCONH 2, NHR, NHCOH, NHCOR, NHSO2R, NH-COOR, NH-CO-NHR або NHCONH2, причому R, Rg і R7 мають вищезгадані значення; - R2 означає атом водню або групу (CH2)n'iR5, де ηΊ дорівнює 0, 1 або 2 і R5 має вищезгадане значення; - R3 означає атом водню або алкіл з 1-6 атомами вуглецю; - А означає зв'язок між двома атомами вуглецю, що несуть радикали R1 і R2, або групу де R4 означає атом водню або групу (CH2)n'1R5, причому n’1 і R5 мають вищезгадані значення, пунктирна лінія означає можливий зв'язок з одним або іншим з атомів вуглецю, що несуть замісники R1 і R2; - n дорівнює 1 або 2; - X означає двовалентну гр упу -С(О)-В-, пов'язану з атомом азоту за допомогою атома вуглецю; де - В означає двовалентну груп у -О-(СН2)n '-, пов'язану з карбонілом за допомогою атома кисню; групу -NR8(CH2)n·- або -NR8-O-, пов'язану з карбонілом за допомогою атома азоту; n" дорівнює 0 або 1; і R 8 вибирають з групи, що складається з атома водню, радикала ОН, R, OR, Υ, ΟΥ, Yb OYb Y2, OY2 , Y3, OCH2CH2SOmR, OSiRaRbRc і SiRaRbRc, причому Ra, Rb і Re означають, індивідуально, лінійний або розгалужений алкіл з 1-6 атомами вуглецю або арил з 6-10 атомами вуглецю, R має вищезгадане значення і m дорівнює 0, 1 або 2; - Υ вибирають з групи, що складається з радикалів СОН, COR, COOR, CONH 2; CONHR, CONHOH, CONHSO2R, CH2COOH, CH2COOR, CH2CONHOH, CH2CONHCN, СН2-тетразоліл, захищений СН2-тетразоліл, CH2SO3H, CH2SO 2R, CH2PO(OR)2 , CH2PO(OR)(OH), CH 2PO(R)(OH) і СН2РО(ОН)2; - Y1 вибирають з групи, що складається з радикалів SO2R, SO 2NHCOH, SO2NHCOR, SO 2NHCOOR, SO2NHCONHR, SO2NHCONH2 і SO3H; - Y2 вибирають з групи, що складається з радикалів РО(ОН)2, PO(OR)2, PO(OH)(OR) і PO(OH)(R); - Y3 вибирають з групи, що складається з тетразолілу, заміщеного радикалом R тетразолілу, скваратрадикала, ΝΗ- або NR-тетразолілу, заміщеного радикалом R ΝΗ- або NR-тетразолілу, NHSO2R і NRSO2R, причому R має вищезгадане значення; маючи на увазі, що, коли η дорівнює 1 і А означає групу в якій R4 означає атом водню, і або X означає групу -С(О)-О-(СН2)n .., в якій n" дорівнює 0 або 1, або X означає групу -CO-NR8-(CH)2n’’ в якій n" дорівнює 1 і R8 означає ізопропіл, або X означає групу -CO-NR8-(CH2)n', в якій n" дорівнює 0 і R8 означає атом водню або феніл, тоді R1 R2 і R3 всі три одночасно не можуть означати атом водню. Об'єктом винаходу є також солі цих сполук, які можуть бути одержані з основами або неорганічними або органічними кислотами, а також внутрішні солі, у формі яких, у разі необхідності, можуть знаходитися деякі сполуки. Асиметричні атоми вуглецю, що містяться в сполуках формули (І), незалежно один від одного, можуть знаходитися в конфігурації R, S або RS, і, отже, об'єктом винаходу є також сполуки формули (І), що знаходяться у формі чистих енантіомерів або чистих діастереоізомерів або у формі суміші енантіомерів, зокрема, у вигляді рацематів, або сумішей діастереоізомерів. З вищезгаданого слідує, що замісники R1, R2, R4, кожний окремо, з одного боку, і X, з іншого боку, можуть знаходитися в цис- і/або трансі-положенні по відношенню до циклу, з яким вони пов'язані, і, отже, об'єктом винаходу є сполуки формули (І), що знаходяться у формі цис- або транс-ізомерів або сумішей. Під алкілом з 1 -6 атомами вуглецю розуміють метил, етил, пропіл, ізопропіл, а також лінійний або розгалужений бутил, пентил або гексил. Під -СН2-алкенілом з 3-9 атомами вуглецю розуміють, наприклад, аліл або бутеніл, пентеніл або гексеніл. Під арилом з 6-10 атомами вуглецю розуміють феніл або нафтил. Під аралкілом з 7-11 атомами вуглецю розуміють бензил, фенетил або мети л нафтил. Під алкіл оксигрупою з 1-6 атомами вуглецю розуміють, зокрема, метоксигрупу, етоксигрупу, пропоксигрупу, ізопропоксигрупу, а також бутоксигрупу, ізобутоксигрупу, втор-бутоксигрупу або трет-бутоксигрупу. Під атомом галогену розуміють атом фтору, хлору, брому або йоду. Під скварат-радикалом розуміють радикал формули: З солей з кислотами продуктів формули (І) можна назвати, нарівні з іншими солі, утворені з неорганічними кислотами, такими, як соляна кислота, бромоводнева кислота, йодоводнева кислота, сірчана кислота або фосфорна кислота, або з органічними кислотами, такими, як мурашина кислота, оцтова кислота, трифтороцтова кислота, пропіонова кислота, бензойна кислота, малеїнова кислота, фумарова кислота, янтарна кислота, винна кислота, лимонна кислота, щавлева кислота, гліоксилова кислота, аспарагінова кислота, алкансульфокислоти, такі, як метансульфокислота і етансульфокислота, арилсульфокислоти, такі, як бензолсульфокислота і п-толуолсуль фокислота. З солей з основами продуктів формули (І) можна назвати, нарівні з іншими, солі, утворені з неорганічними основами, такими, як, наприклад, гідроксид натрію, гідроксид калію, гідроксид літію, гідроксид кальцію, гідроксид магнію або гідроксид амоній, або з органічними основами, такими, як, наприклад, метиламін, пропіламін, триметиламін, діетиламін, триетиламін, Ν,Ν-диметилетаноламін, трис(гідроксиметил)амінометан, етаноламін, піридин, піколін, дициклогексиламін, морфолін, бензиламін, прокаїн, лізин, аргінін, гістидин, N-метилглюкамін, або ж фосфонієві солі, такі, як алкілфосфонієві, арилфосфонієві, алкіларилфосфонієві, алкеніларилфосфонієві солі, або четвертинні амонієві солі, такі, як тетра-н-бутиламонієва сіль. Зі сполук формули (І) об'єктом винаходу є особливо сполуки, в яких η дорівнює 1, а також сполуки, де А означає групу описану вище , і, зокрема, серед них такі, в яких R4 означає атом водню. Зі сполук формули (І) об'єктом винаходу є, зокрема, сполуки, в яких X означає двовалентну гр упу -СО-В-, в якій В означає групу -NR8-(CH2)n' -, описану вище, і з них, більш конкретно, сполуки, в яких R8 означає групу Υ1 або OY1 в якій Y1 має вищезгадане значення. Зі сполук формули (І) винахід відноситься більш конкретно до наступних сполук: транс-7-оксо-6(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема, натрієва сіль; транс-7оксо-М-(фенілметил)-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема, натрієва сіль; транс-7-оксо-К-(4-піридинілметил)-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема, натрієва сіль; транс-7-оксо-N-(3-піридинілметил)-6-(сульфокси)-1,6діазабіцикло[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема, натрієва сіль; транс-7-оксо-1Ч-(2аміно-2-оксоетил)-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема, натрієва сіль; фенілметил-транс-7-оксо-6-(сульфокси)-1,6-діазабіцикло-[3,2,1 ]октан-2-карбоксилат. Іншим об'єктом винаходу є спосіб одержання сполук формули (І). Цей спосіб полягає в тому, що здійснюють: (а) стадію, на якій сполуку формули (II): в якій: - R’1 означає атом водню або радикал CN, захищений СООН, COOR', (CH2)n'R'5, CONR6R7 або захищений радикал дє - n', R’ 5 і R7 мають вищезгадані значення і - R' і R'5 мають, відповідно, значення, вказані вище для R і R5, в яких реакційноздатні групи, що можливо є захищені; - R'2 означає атом водню або групу (СН2)n' 1R5, де n'1 і R' 5 мають вищезгадані значення; - R3 має вищезгадані значення; - А1 означає зв'язок між двома атомами вуглецю, що несуть радикали R’1 і R'2, або груп у де R'4 означає атом водню або групу (CH2)n'1R' 5, причому n’1 і R'5 мають вищезгадані значення, пунктирна лінія означає можливий зв'язок з одним або іншим з атомів вуглецю, що несуть замісники R'1 і R'2; - n має вищезгадане значення; - ΗΖ означає групу HO-(CH2)n'-, HNR'8-(CH)n"- або HNR'8-O-, причому n" має вищезгадане значення і R'8 означає атом водню, захищений гідроксил, R', OR1, радикал Y або ОY, причому У вибирають з груп: СОН, COR1, COOR', CONH 2, CONHR', захищена CONHOH, CONHSO2R', захищена СН 2СООН, CH2COOR1, захищена CH2CONHOH, CH2CONHCN, заміщений радикалом R' СН2-тетразоліл, CH2SO2R', CH2PO(OR') 2, за хищена CH2SO3, захищена CH2PO(OR')OH, захищена CH2PO(R')OH, захищена СН2РО(ОН)2 ; радикал У, або ОУ Ь причому ΥΊ вибирають з груп: SO2R’, SO2NHCOH,SO2NHCOR', SO2NHCOOR', SO2NHCONH2, SO2NHCONHR' і захищена група SO3H; радикал Y'2 або ΟΥ' 2, причому Υ'2 означає захищену груп у РО(ОН)2, за хищену гр упу PO(OH)(OR'), захи щену гр упу PO(OH)R' або групу PO(OR')2 ; або радикал У 3, причому У 3 вибирають із захищеного тетразолілу, заміщеного радикалом R' тетразолілу, захищеного NH- або NR1-тетразолілу, заміщеного радикалом R' NH- або NR'-тетразолілу, NHSO2R' і NR'SO 2R, де R' має вищезгадане значення; вводять у взаємодію з карбонілуючим агентом, у разі необхідності, в присутності основи, для одержання проміжної сполуки формули (III): в якій R'1, R’2, R’3, А' і n мають ви щезгадані значення і або Х1 означає атом водню і Х2 означає груп у -Z-COХ3 , де Х3 означає залишок карбонілуючого агента, або Х2 означає групу -ΖΗ і X! означає групу СО-Х3 , де Х3 має вищезгадане значення; (b) стадію, на якій одержану проміжну сполуку циклізують в присутності основи; і при цьому (c) при бажанні, стадії (а) передує і/або за стадією (b) слідує одна або декілька наступних реакцій у відповідному порядку: захист реакційноздатних функціональних груп; видалення захисних груп від реакційноздатних функціональних груп; етерифікація; омилення; сульфатація; фосфатація; амідування; ацилювання; сульфонілування; алкілування; введення подвійного зв'язку; утворення карбамідної групи; введення тетразолільної групи; відновлення карбонових кислот; дегідратація аміду до нітрилу; солеутворення; іонообмін; розщеплення або розділення діастереоізомерів; окислення сірки до сульфоксиду і/або суль фону. Як карбонілуючий агент можна використати реагент, такий, як фосген, дифосген, трифосген, арилхлорформіат, такий як фенілхлорформіат або п-нітрофенілхлорформіат, аралкілхлорформіат, такий, як бензилхлорформіат, алкіл-або алкенілхлорформіат, такий, як метил- або алілхлорформіат, алкілдикарбонат, такий, як трет-бутилдикарбонат, карбонілдіімідазол і їх суміші. Реакцію проводять переважно в присутності основи або суміші основ, яка нейтралізує кислоту, що утворюється. Основою, зокрема, може бути амін, такий, як триетиламін, діізопропілетиламін, піридин, диметиламінопіридин. Однак, можна також працювати при використанні початкового продукту формули (II) як основи. Цей продукт тоді використовують в надлишку. Цей варіант наводиться в експериментальній частині. У разі необхідності, продукт формули (II) використовують у формі солі з кислотою, як, наприклад, гідрохлорид або трифторацетат. Як основу в стадії (Ь) можна також використати аміни або ж гідриди, алкоголяти, аміди або карбонати лужних або лужноземельних металів. Аміни можуть бути вибрані, наприклад, з вищенаведеного переліку. Як гідрид можна використати, зокрема, гідрид натрію або калію. Як алкоголят лужного металу переважно використовують трет-бутилат калію. Як амід лужного металу можна використати, зокрема, біс(триметилсиліл)амід літію. Як карбонат можна використати, зокрема, карбонат або гідрокарбонат натрію або калію. У разі необхідності, проміжна сполука формули (III) може бути одержана у формі солі з кислотою, що утворюється під час реакції карбонілування, і, зокрема, у вигляді гідрохлориду. Його потім використовують в реакції циклізації в цій формі. У разі необхідності, реакцію циклізації можна здійснювати без виділення проміжної сполуки формули (III). Вказані в стадії (с) реакції являють собою, як правило, добре відомі фахівцеві класичні реакції. Реакційноздатними функціональними групами, які, у разі необхідності, треба захищати, є карбоксильні групи, аміногрупи, амідні групи, гідроксильні групи або гідроксиламіногрупи. Захист кислотної функціональної групи здійснюють, зокрема, у формі складних алкілових, алілових, бензилових, бензгідрильних або п-нітробензилових ефірів. Видалення захисних груп здійснюють шляхом омилення, кислотного гідролізу, гідрогенолізу або ж розщеплення за допомогою розчинних комплексів паладію-(О). Приклади цих захистів і видалень захисних груп приводяться нижче в експериментальній частині. Захист амінів і амідів здійснюють, зокрема, у формі бензильованих похідних, у формі карбаматів, зокрема, аліл-, бензил-, феніл- або трет-бутилкарбамату, або ж у формі силілованих похідних, таких, як третбутилдиметилсилільні, триметилсилільні, трифенілсилільні або ж дифеніл-трет-бутилсилільні похідні. Видалення захисних груп, в залежності від природи захисної групи, здійснюють за допомогою натрію або літію в рідкому аміаку, шляхом гідрогенолізу або за допомогою розчинних комплексів паладію-(О), шляхом впливу кислоти або шляхом впливу тетрабутиламонійфториду. Приклади приводяться нижче в експериментальній частині. Захист гідроксиламінів здійснюють, зокрема, у формі простих бензилових або алілових ефірів. Розщеплення простих ефірів здійснюють шляхом гідрогенолізу або за допомогою розчинних комплексів паладію-(О). Цей метод ілюструється в експериментальній частині. Захист спиртів здійснюють класичним чином у формі простих ефірів, складних ефірів або карбонатів. Про стими ефірами можуть бути прості алкілові або алкоксіалкілові ефіри, переважно прості метилові або метоксіетоксиметилові ефіри, прості арильні або переважно аралкілові, наприклад, бензилові, ефіри або прості силіловані ефіри, наприклад, вищезгадані силіловані похідні. Складними ефірами можуть бути будь-які, відомі фа хівцеві, складні ефіри, що розщеплюються, і переважно ацетат, пропіонат або бензоат або п-нітробензоат. Карбонати можуть являти собою, наприклад, метил-, трет-бутил, аліл-, бензил- або п-нітробензилкарбонат. Видалення захисних груп здійснюють відомими фахівцеві способами, як, зокрема, омилення, гідрогеноліз, розщеплення за допомогою розчинних комплексів паладію-(О), гідроліз в кислому середовищі або ж, у разі силілованих похідних, обробка тетрабутиламонійфторидом. Приклади приводяться в експериментальній частині. Реакцію сульфатації здійснюють шляхом впливу комплексів 8О3-аміни, таких, як 8О3-піридин або БОздиметилформамід, працюючи в піридині, причому сіль, що утворилася, наприклад, сіль піридину, потім може бути обмінена на сіль іншого аміну, четвертинну амонієву сіль або сіль лужного металу. Приклади приводяться в експериментальній частині. Реакцію фосфатації здійснюють, наприклад, шляхом впливу хлорфосфату, такого, як диметил-, дибензилабо дифенілхлорфосфат. Реакцію амідування здійснюють, виходячи з карбонової кислоти, за допомогою активатора, такого, як алкілхлорформіат або EDCI, шляхом впливу гідроксиду амонію або відповідного аміну або їх солей з кислотами. Приклади приводяться нижче в експериментальній частині. Реакції ацилювання і сульфонілування здійснюють при використанні гідроксисечовин шляхом впливу, відповідно, галоїдангідриду або ангідриду відповідної карбонової кислоти або галоїдангідриду відповідної сульфокислоти. Декілька прикладів приводиться нижче в експериментальній частині. Реакцію алкілування здійснюють шляхом впливу на гідроксильовані похідні алкілгалогеніду або заміщеного алкілгалогеніду, зокрема, за допомогою вільного або етерифікованого карбоксильного радикала. Пояснення приводяться нижче в експериментальній частині. Можливе кінцеве введення подвійного зв'язку, який тоді переважно знаходиться між атомами вуглецю, що несуть радикали R4 і R1, здійснюють шляхом впливу галогенованого похідного селену, потім окислення, згідно з відомими фахівцеві способам. Приклад приводиться нижче в експериментальній частині. Утворення карбамідної групи, яка відноситься до замісника R8, досягають переважно шляхом впливу відповідного ізоціанату на вільну NH-груп у. Приклад приводиться нижче в експериментальній частині. Введення тетразолільної групи здійснюють шляхом впливу захи щеного або заміщеного галогенованого, переважно фторованого, похідного тетразолу. Видалення захисних груп може бути здійснене шляхом гідрогенолізу. Відновлення кислот в спирти може бути здійснене шляхом впливу борану або через проміжний змішаний ангідрид шляхом впливу боргідриду лужного металу. Змішаний ангідрид одержують, наприклад, за допомогою алкілхлорформіату. Ілюстрація цієї реакції дана в експериментальній частині. Дегідратацію аміду до утворення нітрилу можна здійснювати в умовах реакцій карбонілування і циклізації. Окислення сульфідів з утворенням сульфоксиду і/або сульфону можна здійснювати шляхом впливу надкислоти, такої, як м-хлорнадбензойна кислота або надфталева кислота, або будь-якого іншого реагенту, відомого фахівцеві. Солеутворення за допомогою кислот, у разі необхідності, реалізовують шля хом додавання кислоти в розчинній формі до сполуки. Солеутворення за допомогою основ може стосуватися або сполук з кислотною групою, зокрема, з карбоксильною групою, або кислот з сульфоксигрупою або групою, що походить від фосфорної кислоти, або кислот, що включає гетероцикл з кислотним характером. У першому випадку діють шляхом додавання відповідної основи, такого, як вищеописані основи. У другому випадку піридинієву сіль одержують безпосередньо під час впливу комплексу SО3-піридин і інші солі одержують з цієї піридинієвої солі. У тому або іншому випадку можна діяти шляхом іонообміну при використанні смоли. Приклади солеутворень представлені нижче в експериментальній частині. Розділення енантіомерів і діастереоізомерів може бути здійснене відомими фахівцеві методами, зокрема, шляхом хроматографії. Крім синтезу вищеописаними способами, сполуки формули (І), зрозуміло, можуть бути одержані способами, в яких використовують як вихідні сполуки сполуку формули (II), в якій R'1, A’, R'2, R3 і HZ мають значення, які приводять прямо (без перетворення) до таких значень сполук, які бажають одержати. У разі необхідності, ті значення, які включають реакційноздатні функціональні групи, вказані вище, захищають, а видалення захисних гр уп здійснюють в кінці стадії циклізації (b) або в будь-який інший відповідний для цього момент синтезу. Введення і видалення захисних груп тоді здійснюють, як описано вище. Такі способи представлені нижче в експериментальній частині. Продукти загальної формули (І) володіють дуже хорошою антибіотичною активністю відносно грампозитивних бактерій, таких, як стафілококи. Особливо значною є їх ефективність відносно грамнегативних бактерій, зокрема, відносно колі-подібних бактерій. Ці властивості дозволяють використати вищезгадані продукти, а також їх фармацевтично прийнятні солі з кислотами і основами, як лікарські засоби при лікуванні захворювань, викликаних чутливими до медикаментів мікроорганізмами, і, зокрема, при лікуванні стафілококових інфекцій, таких, як стафілококові сепсиси, злоякісні стафілококові інфекції особи або шкіри, піодерміти, септичні або рани, що гнояться, карбункули, флегмони, бешихове запалення, гострі первинні або постгрипозні стафілококові інфекції, бронхопневмонії, легеневі нагноєння. Ці продукти також можуть бути використані як лікарські засоби у разі лікування колі-бактеріозів і суп утніх інфекцій, інфекції, що викликаються Proteus, клебсіелою і сальмонелою і при лікуванні інших захворювань, викликаних грамнегативними бактеріями. Отже, об'єктом даного винаходу є також застосування, як лікарських засобів, і, зокрема, як антибіотичних лікарських засобів, продуктів формули (І), вказаної вище, а також їх фармацевтично прийнятних солей з кислотами і основами. Більш переважно, об'єктом винаходу є застосування, як лікарських засобів, продуктів формули (І), вказаної вище, в якій η дорівнює 1, а також продуктів, в яких А означає групу вказану вище, і, зокрема, сполук, в яких R4 означає атом водню. Зокрема, об'єктом винаходу є застосування, як лікарських засобів, сполуки формули (І), в якій X означає двовалентну груп у -СО-В, в якій В означає групу -NR8-(CH2)n'-, вказану вище, і більш конкретно, сполук, в яких R8 означає групу Y1 або OY1 , в якій Υ1 має вищезгадане значення. Зі сполук формули (І) ще більш конкретно можна назвати як лікарські засоби, наступні сполуки: транс-7оксо-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема, натрієва сіль; транс-7-оксо-N-(фенілметил)-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема, натрієва сіль; транс-7-оксо-К-(4-піридинілметил)-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2карбоксамід і його солі з основами, зокрема, натрієва сіль; транс-7-оксо-М-(3-піридинілметил)-6-(сульфокси)1,6-діазабіцикло[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема, натрієва сіль; транс-7-оксо-К-(2аміно-2-оксоетил)-6-(сульфокси)-1,6-діазабіцикло-[3,2,1]октан-2-карбоксамід і його солі з основами, зокрема, натрієва сіль; фенілметил-транс-7-оксо-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксилат. Об'єктом винаходу є також фармацевтичні композиції, що включають як діючий початок щонайменше одну зі сполук згідно з винаходом, визначених вище. Ці композиції можуть бути введені перорально, ректально, парентерально, зокрема, внутрішньом'язово, або локально шляхом місцевого нанесення на шкіру і слизові оболонки. Композиції згідно з винаходом можуть бути твердими або рідкими і можуть знаходитись у фармацевтичних формах, що звичайно використовуються в медицині людини, як, наприклад, прості або дражовані таблетки, желатинові капсули, гранули, супозиторії, препарати для ін'єкцій, мазі, креми, гелі; їх можна одержувати звичайними способами. Діючий початок або діючі початки можуть бути включені в них з ексципієнтами, що звичайно використовуються у цих фармацевтичних композиціях, такими, як тальк, гуміарабік, лактоза, крохмаль, стеарат магнію, масло какао, водні або неводні наповнювачі, жирові речовини тварини або рослинного походження, парафінові похідні, гліколі, різні змочувачі, диспергатори або емульгатори, консерванти. Ці композиції можуть знаходитись, зокрема, у формі порошку, призначеного для розчинення в момент вживання у відповідному розчиннику, наприклад, в стерильній апірогенній воді. Доза, що вводиться, є змінною в залежності від захворювання, що виліковується, суб'єкта, який піддається лікуванню , шля ху введення і продукту, що розглядається. Вона може становити, наприклад, 0,250-10г на добу при пероральному введенні людині з використанням продукту, описаного в прикладі 1, або 0,25-10г на добу при внутрішньом'язовому або внутрішньовенному введенні. Продукти формули (І) також можуть бути використані як дезинфікуючі засоби для хірургічних інстр ументів. Нарешті, об'єктом винаходу є нові промислові продукти формули (III) і, зокрема, проміжні продукти, необхідні для одержання продуктів формули (І), а також їх солі з кислотами і, зокрема, їх гідрохлориди. Продукти формули (II) відомі або їх одержують відомими фахівцеві способами. Літературні посилання, а також способи одержання приведені нижче в експериментальній частині. Наступні приклади пояснюють винахід, однак не обмежують його об'єму охорони. ПРИКЛАДИ У нижченаведених прикладах використані наступні скорочення: DEAD: діетилазодикарбоксилат TEA: триетиламін DMAP: 4-диметиламінопіридин EDCI: 1-(3-диметиламінопропіл)-3-етилкарбодіімідгідрохлорид ТГФ: тетрагідрофуран АІБН: 2,2'-азобісизобутиронітрил Μ: молекулярна молярна маса МС: мас-спектрометрія ЕУ: електронний удар SIMS: мас-спектрометрія вторинних іонів FAB: бомбардування швидкими атомами ПРИКЛАД 1 Дифенілметил-цис-7-оксо-6-окса-1-азабіцикло[3,2,1]октан-4-пропаноат Змішують 3,16г (10,6ммоль) гідрохлориду 3-оксо-1-(фенілметил)-4-піперидинпропанової кислоти (М=297,7 г) (описується в заявці на патент Японії J54098-772) зі 100 мл етанолу і охолоджують до температури 10°С. У струмі азоту протягом 15 хвилин додають 1,84 г NaBH4, підтримуючи температуру при 8-13°С. Залишають температуру підвищуватися аж до кімнатної температури і залишають контактувати протягом 1 години ЗО хвилин. Додають ще 380 мг NaBtLt і залишають реакцію на всю ночі при кімнатній температурі. Розчинник випаровують при зниженому тиску, обробляють за допомогою 50 мл води і значення рН доводять з 10 до 2 за допомогою концентрованої соляної кислоти. Знов випаровують при зниженому тиску. Твердий залишок (приблизно 10,8 г) промивають два рази за допомогою 100 мл етанолу, потім розчинник випаровують при зниженому тиску. Таким чином одержують 3,10 г гідрохлориду 3-гідрокси- 1-(фенілметил)-4-піперидинпропанової кислоти (М= 299,7г), що відповідає виходу 97%. 3,10 г (10,3 ммоль) одержаної вище сполуки розбавляють в 100 мл етанолу, потім додають до нього 900 мг заздалегідь гідрогенізованого 10мас.%-ного паладію-на-вугіллі і 30мл етанолу. Всю суміш витримують протягом ночі в атмосфері водню при нормальному тиску, потім каталізатор видаляють шляхом відфільтровуван ня і етанол видаляють шляхом випаровування при зниженому тиску. Одержують 1,90 г гідрохлориду транс-Згідрокси-4-піперидинпропанової кислоти (М=209,6 г), або вихід становить 88%. Змішують 1,79г (8,54ммоль) одержаної вище сполуки з 20мл етанолу і 20мл води. Потім додають концентрований розчин гідроксиду натрію аж до досягнення значення рН приблизно 8,5. Після цього додають 1 мл алілхлор форміату і концентрований розчин гідроксиду натрію для підтримання значення рН від 8 до 9. Реакційну суміш екстрагують етилацетатом, потім водну фазу підкисляють до значення рН=2 шляхом додавання концентрованої соляної кислоти і повторно екстрагують етилацетатом. Після висушування і випаровування розчинника при зниженому тиску одержують 1,69г сирого продукту, який обробляють сумішшю дихлорметану і етанолу, потім відфільтровують і розчинник знов випаровують при зниженому тиску. Таким чином одержують 1,40 г транс-3-гідрокси-1-[(2-пропенілокси)карбоніл]-4-піперидинпропанової кислоти (М=257 г), або вихід становить 60%. При температурі 0°С і в атмосфері азоту, 3,24г (12,6ммоль) вищеодержаної гідроксикислоти і 6,4г трифенілфосфіну розчиняють в 60мл тетрагідрофурану. Потім додають 2,5 мл DEAD і через 15 хвилин реакційну суміш випаровують при зниженому тиску, одержуючи 12 г сирого продукту. Очищають шля хом хроматографії на діоксиді кремнію, поступово елююючи сумішшю дихлорметану і етилацетату у співвідношенні 9:1, 8:2, 7:3 для розділення цис- і трансу-лактонів. Таким чином одержують 2,72 г цис-лактону в суміші з відновленим DEAD і фосфіноксидом. Цей продукт знов розчиняють в 10мл диметоксіетану і додають 8 мл 1н розчину гідроксиду натрію. Після витримування в контакті протягом 1 години реакційну суміш екстрагують два рази етилацетатом, потім підкисляють аж до значення рН=2 за допомогою 2н соляної кислоти і повторно екстрагують етилацетатом. Після висушування і випаровування розчинника при зниженому тиску одержують 1,07 г гідроксикислоти. 1,0 г сирий гідроксикислоти розчиняють в суміші з 5 мл дихлорметану і 2 мл метанолу, потім обробляють надлишком дифенілдіазометану в дихлорметані аж до зникнення вихідного продукту. Розчинник випаровують при зниженому тиску і продукт очищають шляхом хроматографії, одержуючи 1,39 г дифенілметил-цис-3гідрокси-1-[(2-пропенілокси)карбоніл]-4-піперидинпропаноату (М=423 г), або загальний вихід становить 26%. Потім, в атмосфері азоту, 1,2 г (2,83 ммоль) одержаного вище продукту розчиняють в 23 мл дихлорметану. Після цього додають 390 мкл оцтової кислоти, потім 860мкл (С4Н9)3SnН і 70мг Рd[Р(С6Н5)3]4-· Розчинник випаровують при зниженому тиску, одержуючи 3,82 г сирого продукту, який промивають петролейним ефіром. Одержують 1,27 г продукту, який хроматографують на діоксиді кремнію, елююючи дихлорметаном, потім сумішшю дихлорметану і метанолу у співвідношенні 95:5, потім 90:10. Таким чином одержують 0,87 г дифенілметид-цис-3-гідрокси-4-піперидинпропаноату (М=339 г), або вихід становить 77%. 400мг (1,00ммоль) одержаної вище сполуки розчиняють в 25 мл дихлорметану, додають 80 мкл дифосгену (Сl3СОСОСl), 336мкл триетиламіну, 144мг DMAP. Залишають реакцію при кімнатній температурі протягом 5 годин 30 хвилин, потім розбавляють за допомогою дихлорметану. Промивають 10%-ним водним розчином винної кислоти, потім буферним розчином фосфату натрію з рН=7, органічну фазу суша ть над сульфатом натрію, після чого розчинник випаровують при зниженому тиску. Таким чином одержують 380 мг сирого продукту. Очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю ди хлорметану і етилацетату у співвідношенні 95:5 з 0,1% води. Одержують 184мг вказаної цільової сполуки (М=365,43г), або вихід становить 50%. 1 H-ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультиплетність): 1,60-1,88 (м): NCH2-CH2-CH; 2,48 (м): СН2-СН2-СО; 2,78 (д)-2,90 (м)-3,33-3,47 (м): CH2-N-CH2; 4,50 (д): СНО-СН2; 6,89 (с): СО2СН(С6Н5)2; 7,33 (м): (С6Н5)2 . ІЧ-спектр (СНСІ3): 1784, 1734, 1600, 1585, 1496 см-1. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [М]+=365. ПРИКЛАД 1bis цис-7-Оксо-6-окса-1 -азабіцикло[3,2,1 ]октан-4-пропанова кислота 176мг (0,482ммоль) одержаного вище продукту розчиняють в 10мл ацетону. Додають 90мг 10 мас.%-ного паладію-на-вугіллі. Залишають реакцію в атмосфері водню при нормальному тиску протягом 3 годин. Додають ще 25мг каталізатора і реакцію продовжують протягом 1 години 15 хвилин. Каталізатор відфільтровують, потім розчинник випаровують при зниженому тиску, одержуючи 146мг продукту. Знов проводять реакцію в 10 мл ацетону при використанні 35мг 10мас.%-ного паладію-на-вугіллі в атмосфері водню і витримують додатково протягом 1 години для повноти протікання реакції. Каталізатор потім відфільтровують і фільтрат випаровують при зниженому тиску. Одержують 137мг сирого продукту, який кристалізують з суміші діетилового ефіру і петролейного ефіру. Таким чином одержують 75мг цільового продукту (М=199 г), або ви хід становить 78%. 1 Н-ЯМР-спектр (250 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультипл етність): 1,30-1,63 (м) і 1,88 (м): NCHa-Qb-CH; 2,25 (τ): СН2-СН2-СО; 3,06 (м) і 3,38 (м): CH 2-N-CH2; 4,65 (д): С-СНОСН2; 12,08 (с): Η рухомий. ІЧ-спектр (вазелінове масло): 1785, 1717 см -1. МС (FAB) m/z:[M+H]+=200; 159. ПРИКЛАД 2 Дифенілметил-транс-7-оксо-6-окса-1 -азабіцикло[3,2,1]октан-4-ацетат В інертній атмосфері 94 мг (0,259ммоль) гідрохлориду дифенілметил-транс-З-гідрокси-4-піперидинацетату (М=361,87г) (описується в Eur. J. Med. Chem.-Chim. Ther., 17 (6), 531-535 (1982)) змішують з 7мл дихлорметану. Охолоджують за допомогою бані з льодом і інжектують 19 мкл дифосгену. Перемішують протягом 25 хвилин, потім інжектують 72мкл триетиламіну. Перемішують при кімнатній температурі протягом 30 хвилин і розчинник випаровують при зниженому тиску. Після цього обробляють за допомогою 7 мл толуолу. Додають 36мкл триетиламіну, потім 31 мг DMAP. Нагрівають протягом 15 хвилин при температурі 100°С, потім залишають повертатися до кімнатної температури. Після цього промивають 2 рази за допомогою 4 мл 10%-ного водного розчину винної кислоти, потім за допомогою 4 мл водного насиченого розчину хлориду натрію. Сушать над сульфатом магнію, фільтрують і розчинник випаровують при зниженому тиску. Одержують 78 мг ма сла, якого хроматографують на діоксиді кремнію при використанні як елююючого засобу суміші дихлорметану і етилацетату у співвідношенні 95:5. Таким чином одержують 35,7мг цільової сполуки (М=351,405 г) у вигляді кристалів білого кольору, або ви хід становить 39%. ПРИКЛАД 2bis транс-7-Оксо-6-окса-1-азабіцикло[3,2,1 ]октан-4-оцтова кислота В інертній атмосфері 38,7 мг (0,110 ммоль) одержаного в прикладі 2 продукту змішують з 2 мл ацетону і 38 мг каталізатора 10 мас.%-ного паладію-на-вугіллі. Вміщують в атмосферу водню при нормальному тиску. Залишають реакцію протягом 45 хвилин, після чого каталізатор видаляють шляхом відфільтровують і розчинник випаровують при зниженому тиску. Таким чином одержують 32,6 мг сирого продукту. Перекристалізовують з діетилового ефіру, одержуючи 14,2 мг кристалів білого кольору цільової сполуки (C8H10NO4 M=185,181 г), або вихід становить 69%. ПРИКЛАД 3 Дифенілметил-цис-7-оксо-6-окса-1-азабіцикло[3,2,1]октан-4-ацетат Змішують 1,5г (5,78ммоль) транс-1-[(1,1-диметилетокси)карбоніл]-3-гідрокси-4-піперидиноцтової кислоти (описується в Eur. J. Med. Chem. - Chim. Ther., 17 (6), 531-535 (1982)), 7мл дихлорметану, 3,03г трифенілфосфіну і 22 мл тетрагідрофурану. Додають розчин 0,91мл DEAD в 2,5мл тетрагідрофурану. Залишають реакцію протягом 3 годин 20 хвилин, потім додають 8,7мл 1 н розчину гідроксиду натрію і перемішують протягом 1 години 15 хвилин. Реакційну суміш екстрагують два рази етилацетатом, потім доводять до значення рН=2 за допомогою 2н соляної кислоти. Після цього екстрагують три рази етилацетатом. Органічні фази об'єднують і промивають водним насиченим розчином хлориду натрію, потім сушать над суль фатом магнію, фільтрують і розчинник випаровують при зниженому тиску. Таким чином одержують 1,37 г кристалів білого кольору 1,1 диметилетил-(Заа,7аа)-гексагідро-2-оксофуро[2,3-с]піридин-6(2Н)-карбоксилату (C12H21NO 5-M=259,304 г), або вихід становить 91%. В інертній атмосфері 1,37 г (5,28 ммоль) одержаної вище сполуки змішують з 32 мл ди хлорметану. Вводять надлишок розчину дифенілдіазометану в дихлорметані аж до зникнення вихідного продукту. Потім розчинник випаровують при зниженому тиску і таким чином одержують 2,81 г сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, використовуючи як елююючий засіб дихлорметан, потім суміш дихлорметану і етилацетату у співвідношенні 95:5. Одержують 2,00 г дифенілметил-цис-1-[(1,1-диметилетокси)карбоніл]-3-гідрокси-4-піперидинацетату у ви гляді кристалів білого кольору (М=425,528 г), або вихід становить 89%. Вводять 0,6 г (1,41 ммоль) одержаної вище сполуки і 1,93мл розчину хлороводню в метанолі з концентрацією 7,3 моль/л. Перемішують при кімнатній температурі і через 15 хвилин додають 1 мл дихлорметану. Через ще 15 хвилин реакційне середовище випаровують при зниженому тиску. Додають ще дихлорметан, після чого знов випаровують. Цю операцію повторюють декілька разів. Потім продукт кристалізують з діетилового ефіру. Таким чином одержують 0,44 г гідрохлориду ди фенілметил-цис-З-гідрокси-4-піперидинацетату бруттоформули C20H23NO3.HCI (М=361,871 г), або вихід становить 86%. Ця реакція приводить також до утворення змінних кількостей гідрохлориду лактону -(Заа,7аа)гексагідрофуро[2,3-с] піридин-2(ЗН)-ону (М=177,6 г). В інертній атмосфері 0,28 г (0,77 ммоль) одержаної вище сполуки C20H23NO3 .HCI змішують з 19 мл дихлорметану. При температурі 0°С додають 60 мкл дифосгену і перемішують. Через 25 хвилин вводять 0,32 мл триетиламіну. Потім додають 94 мг DMAP і залишають до повернення до кімнатної температури. Перемішують протягом 4 годин 15 хвилин, після чого промивають послідовно 10%-ним водним розчином винної кислоти, потім водним насиченим розчином хлориду натрію. Після цього сушать над сульфатом магнію, фільтрують і розчинник випаровують при зниженому тиску. Таким чином одержують 0,265 г цільової сполуки бруттоформули C21H21NO4 (М=351,405 г), або вихід становить 98%. 1 H-ЯМР-спектр (250 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультиплетність): 1,82 (м): NCH 2-CH2; 2,30-2,70 (м): СО-СН 2-СН; 2,93 (д)-2,99 (д) і 3,45 (м): CH 2-N-CH2; 4,60 (д): СН-СНО-СН 2; 6,87 (с): СО2СН(С6Н5)2; 7,10-7,35 (м): (С6Н5)2. ІЧ-спектр (СНСІ3): 1786, 1734, 1600, 1587, 1496 см-1. МС (SIMS) m/z: [M+Na]+=374+. ПРИКЛАД 3bis цис-7-Оксо-6-окса-1-азабіцикло[3,2,1]октан-4-оцтова кислота Змішують 55 мг (0,156 ммоль) одержаного в прикладі 3 продукту, 3 мл етилацетату і 55 мг каталізатора 10 мас.%-ного паладію-на-вугіллі. Вміщують в атмосферу водню при нормальному тиску. Залишають реакцію протягом 1 години 30 хвилин, потім каталізатор відфільтровують і розчинник випаровують при зниженому тиску. Таким чином одержують 38 мг сирого продукту, який кристалізують з суміші пентану і діетилового ефіру. Таким чином одержують 16 мг цільової сполуки у вигляді кристалів білого кольору (М=185,181 г), або вихід становить 55%. 'Н-ЯМР-спектр (250 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ул ьтиплетність): 1,63-1,86 (м) і 1,91 (м): NCH2-CH2; 2,27-2,49 (м) і 2,54 (дд): СО-СН2-СН; 2,98 (д) і 3,54 (д): CH2-N-CH2-CH2; 3,04 (д) і 3,41 (дд): CH 2-N-CH2-CH2; 4,71 (д): СН-СНО-СН 2. ІЧ-спектр (вазелінове масло): 1784, 1734, 1686 см -1. МС (SIMS) m/z: [M+H]+ = 186+, 167+. ПРИКЛАД 3ter Метил-цис-7-оксо-6-окса-1 -азабіцикло[3,2,1 ]октан-4-ацетат 78мг (0,421 ммоль) одержаної в прикладі 3bis сполуки розчиняють в 1 мл дихлорметану. Додають краплями надлишок діазометану доти, поки залишається жовте фарбування, потім розчинник випаровують при зниженому тиску. Таким чином одержують 80мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 95:5. Таким чином одержують 8,2 г цільової сполуки (М=199,208 г), або вихід становить 10%. ПРИКЛАД 4 цис-7-Оксо-б-окса-1 -азабіцикло[3,2,1 ]октан-4-ацетонітрил 67мг (0,38ммоль) гідрохлориду (3аа,7аа)-гексагідрофуро[2,3-е]піридин-2(3Н)-ону (М=177,6г), одержаного в прикладі 3, розчиняють в 1 мл розчину аміаку в метанолі з концентрацією 4,17 моль/л. Перемішують протягом 5 годин, розчинник випаровують при зниженому тиску, потім знов додають 1 мл розчину аміаку в метанолі і залишають реакцію продовжуватися протягом 18 годин. Розчинник випаровують при зниженому тиску і таким чином одержують 79 мг цис-3-гідрокси-4-піперидинацетаміду брутто-формули C7H14 O2N2 (M=158 г). В інертній атмосфері 75 мг одержаної вище сполуки розчиняють в 9 мл дихлорметану. Охолоджують на бані з льодом і вводять 30 мкл дифосгену. Витримують при температурі 0-5°С протягом 40 хвилин, після чого вводять 0,16 мл триетиламіну і, через 5 хвилин, 46 мг DMAP. Перемішують протягом 4 годин при кімнатній температурі. Промивають два рази за допомогою 2 мл 10%-ного водного розчину винної кислоти, потім за допомогою 2 мл водного насиченого розчину хлориду натрію. Сушать над сульфатом магнію, фільтрують, розчинник випаровують при зниженому тиску. Таким чином одержують 35 мг сирого продукту, який обробляють сумішшю етилацетату і ди хлорметану у співвідношенні 30:70. Домішки відфільтровують і фільтрат випаровують при зниженому тиску. Таким чином одержують 23 мг цільової сполуки (М=166,18 г) у вигляді масла, або вихід складає близько 26%. ІЧ-спектр (вазелінове масло): 2241, 1777 см -1. МС (ЕУ) m/z: [M]+=166, 137, 82, 55, 42. ПРИКЛАД 5 3-Бензоїл-1,3-діазабіцикло[2,2,1]гептан-2-он В інертній атмосфері 1,01 г (5,43 ммоль) 1,1-диметилетил-3-аміно-1-піролідинкарбоксилату (М= 186,25 г) (описується в міжнародній заявці на патент WO-9801426) змішують з 10 мл дихлорметану, розчин охолоджують до температури 0°С, потім додають краплями 0,76 мл триетиламіну. Перемішують протягом 15 хвилин, підтримуючи температуру 0°С, потім додають 0,63 мл бензоїлхлориду. Залишають до повернення до кімнатної температури, потім розбавляють шляхом додавання 10 мл дихлорметану. Після цього промивають за допомогою 10%-ного водного розчину винної кислоти, потім за допомогою 10 мл води. Сушать над сульфатом магнію, фільтрують і ди хлорметан видаляють шляхом випаровування при зниженому тиску. Таким чином одержують 1,30 г 1,1-диметилетил-3-(бензоїламіно)-1 -піролідинкарбоксилату (М=292,36 г) у вигляді масла жовтого кольору. Відповідний вихід становить 82%. 1,30 г (4,46 ммоль) цієї сполуки змішують з 10 мл метанолу. Розчин охолоджують до температури 0°С, потім поступово вводять 6,12 мл розчину хлороводню в метанолі з концентрацією 7,3 моль/л. Після цього розчинник випаровують при зниженому тиску. Таким чином одержують 1,01 г гідрохлориду N-(3піролідиніл)бензаміду (М=226,707 г), що знаходиться у вигляді масла каштанового кольору, або вихід складає близько 100%. В інертній атмосфері 1,01 г (4,46 ммоль) одержаної вище сполуки змішують з 10 мл дихлорметану. Охолоджують до температури 0°С, потім додають краплями 1,36 мл триетиламіну. Перемішують протягом 15 хвилин, потім додають краплями 1,44мл дифосгену. Витримують при температурі 0°С протягом 30 хвилин, після чого залишають до повернення до кімнатної температури. Після цього розбавляють за допомогою дихлорметану, промивають 10%-ним водним розчином винної кислоти, потім водою. Сушать над сульфатом магнію, фільтрують і концентрують шляхом випаровування розчинника при зниженому тиску, одержуючи 0,615 г сирого продукту. Очи щають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і ацетону у співвідношенні 90:10. Таким чином виділяють 0,320 г хлорангідриду 3-(бензоїламіно)-1піролідинкарбонової кислоти, який кристалізується. Відповідний вихід становить 28%. Потім, в інертній атмосфері 0,585 г (2,31 ммоль) одержаної сполуки розчиняють в 18 мл тетрагідрофурану. Розчин охолоджують до температури -78°С, потім додають краплями 2,55 мл 1 Μ розчину біс(триметилсиліл)аміду літію в тетрагідрофурані. Одержують розчин жовтого кольору, який витримують при температурі -78°С протягом 20 хвилин, потім продовжують перемішувати протягом 1 години при підвищенні температури. При температурі 0°С додають 350 мкл оцтової кислоти, потім 5 мл 10%-ного водного розчину винної кислоти. Розбавляють за допомогою етилацетату, після чого промивають 10%-ним розчином винної кислоти, потім розчином фосфатного буфера з рН=7, потім водою. Органічну фазу суша ть над сульфатом магнію, фільтрують і концентрують шляхом випаровування розчинника при зниженому тиску. Таким чином одержують 0,315 г сирого продукту у вигляді твердої речовини жовтого кольору. Цей сирий продукт очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю ди хлорметану і етилацетату у співвідношенні 90:10. Таким чином одержують 0,140 г цільової сполуки C12H12N2O2 (М=216,24 г) у вигляді твердої речовини білого кольору, або вихід становить 28%. ІЧ-спектр (СНСl3): 1801, 1775, 1675; 1620, 1603, 1582 см'1. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [М]+=216, 105,77. ПРИКЛАД 6 транс-6- [(Фенілметокси)карбоніл]-2-оксо-1,3 -діазабіцикло[2,2,1 ]гептан-3 -ацетат калію Змішують 1 г (3,12 ммоль-М=186,25 г) 1-(1,1-диметилетил)- і 2-(фенілметил)-транс-4-аміно-1,2піролідиндикарбоксилату (описується в J. Org. Chem., 56, 3009-3016 (1991)), 10 мл тетрагідрофурану, 560 мкл алілбромацетату і 660 мкл триетиламіну. Залишають реакцію при перемішуванні і при кімнатній температурі протягом 14 годин, потім протягом 3 годин при температурі 50°С. Після цього розбавляють етилацетатом і промивають 10%-ним водним розчином винної кислоти, потім водним насиченим розчином хлориду натрію. Органічну фазу сушать над сульфатом магнію, фільтрують, після чого розчинник випаровують при зниженому тиску. Таким чином одержують 1,21 г сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 80:20. Таким чином одержують 0,99 мг 1-(1,1-диметилетил)і 2-(фенілметил)-транс-4-[(2-пропенілокси)карбоніл]метил]аміно]-1,2піролідиндикарбоксилату брутто-формули С 12Н30О 6 (М=418 г). До 0,99 г (2,36 ммоль) одержаної вище сполуки додають, в атмосфері азоту і при температурі 0°С, 6 мл 4 Μ розчину хлороводню в етилацетаті. Потім залишають реакцію при кімнатній температурі протягом 15 хвилин. Розчинник випаровують при зниженому тиску. Одержують сирий продукт, який кристалізують з діетилового ефіру, одержуючи 0,95 г дигідрохлориду фенілметил-транс-4-[(2-пропенілокси)карбоніл]метил]аміно]-2піролідинкарбоксилату бр утто-формули C17H23N2 O4.2HC1 (M=394 г). 0,5 г цього продукту розчиняють в 20 мл дихлорметану і додають 1,3 мл 2н розчину гідроксиду натрію і 3 мл води. Декантують, екстрагують дихлорметаном, сушать над сульфатом магнію, потім фільтрують і розчинник випаровують при зниженому тиску. Таким чином одержують 339 мг вільного діаміну. Відповідний вихід становить 83%. 100 мг (0,314 ммоль) одержаного вище діаміну розчиняють, при температурі 0°С і в атмосфері азоту, в 5 мл ацетонітрилу. Додають 21 мкл дифосгену. Після контактування протягом 15 хвилин, цей розчин, в атмосфері азоту і протягом 4 годин, додають до нагрітої до температури 70°С суміші, що містить 38 мг DMAP, 88 мкл триетиламіну в 10 мл ацетонітрилу. По закінченні додавання, реакційну суміш нагрівають ще протягом однієї години, потім охолоджують, розбавляють етилацетатом і промивають послідовно 10%-ним водним розчином винної кислоти, потім водним насиченим розчином хлориду натрію. Після висушування над сульфатом натрію, фільтрації і випаровування розчинників при зниженому тиску одержують 58 мг сирого продукту. Цей продукт очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю ди хлорметану і етилацетату у співвідношенні 8:2, одержуючи 19 мг 2-пропеніл-транс-6-[(фенілметокси) карбоніл]-2-оксо-1,3-діазабіцикло[2,2,1]гептан-3ацетату бр утто-формули C18H20N2 O5 (M=344,57 г), або вихід становить 17%. Потім 24 мг (0,069 ммоль) одержаної сполуки розчиняють в 250 мкл дихлорметану. У атмосфері азоту вводять 3 мг Pd[P(C6H5)3]4 5 потім додають 150 мкл 0,5 Μ розчину 2-етилгексаноату калію в е тилацетаті. Через декілька хвилин випадає осад, який відділяють шляхом центрифугування і промивають два рази за допомогою 500 мкл етилацетату. Одержують 24 мг цільової сполуки Q5H15KN2O5 (М=342 г), або ви хід є кількісним. 1 Н-ЯМР-спектр (300 МГц; диметилсульфоксид; хімічні зсуви піків в м.ч. і мультиплетність): 1,83 (ддд) і 2,56: N-CH2-CHN-CH2; 2,50 і 2,79 (д): N-CH2-CHN-CH2; 3,23 (д) і 3,41 (д): =C-N-CH 2-C=O; 3,62 (ддд): O=C-CHN-CH 2; 4,13 (с): N-CH 2-CHN-CH2; 5,16 (с): =С-О-СН 2-С6Н5; 7,38 (м): С6Н5-СН2. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [2МК+Н]+=723; [2MK+Na]+=707; [MK+K]+=381; [MK+Na]+=365; [MK+H]+=343. ПРИКЛАД 7 Метил-транс-3-бензоїл-2-оксо-1,3-діазабіцикло[2,2,1]гептан-6-карбоксилат В атмосфері азоту, 0,471г (1,93ммоль) 1-(1,1-диметилетил)і 2-метил-транс-4-аміно-1,2піролідиндикарбоксилату (описується в J. Org. Chem., 56, 3009-3016 (1991)) змішують з 3,5 мл безводного дихлорметану для його розчинення. Розчин охолоджують до температури 0°С, потім додають краплями 269мкл триетиламіну. Перемішують протягом 15 хвилин при підтриманні температури 0°С, потім додають краплями 224мкл бензоїлхлориду. Після цього залишають для підвищення до 20°С протягом однієї години. Розбавляють за допомогою 30мл дихлорметану, після чого промивають 10%-ним водним розчином винної кислоти, потім насиченим розчином гідрокарбонату натрію, потім водою. Сушать над сульфатом магнію, фільтрують, концентрують шляхом випаровування дихлорметану при зниженому тиску. Таким чином одержують 0,6г масла жовтого кольору, які очищають шляхом хроматографії на діоксиді кремнію, використовуючи як елююючий засіб суміш дихлорметану і метанолу у співвідношенні 99:1. Таким чином виділяють 0,499г 1-(1,1-диметилетил)- і 2метил-транс-4-(бензоїламіно)-1,2-піролідиндикарбоксилату брутто-формули C18H24N2O 5 (М=348 г), або вихід становить 74%. Змішують, в атмосфері азоту, 0,400г (1,15ммоль) одержаної вище сполуки з З мл етилацетату для розчинення сполуки, потім розчин охолоджують до температури 0°С, додають 2,89 мл розчину хлороводню в етилацетаті з концентрацією 4моль/л. По закінченні 15 хвилин продовжують перемішувати при кімнатній температурі протягом 1 години. Після цього розчинник видаляють шляхом випаровування при зниженому тиску. Таким чином одержують 0,350г гідрохлориду метил-транс-4-(бензоїламіно)-2-піролідинкарбоксилату брутто-формули C13H15N2 O3.HCI (М=284,744 г) у вигляді твердої речовини бежевого кольору. В атмосфері азоту, 0,327 г (1,15 ммоль) одержаної вище сполуки змішують з 4 мл дихлорметану. Після цього суспензію охолоджують до температури 0°С, потім додають 352 мкл триетиламіну. Перемішують протягом 15 хвилин при температурі 0°С, потім додають 138 мкл дифосгену. Продовжують перемішування протягом 5 хвилин при температурі 0°С, потім реакційну суміш залишають до повернення до кімнатної температури. Залишають реакцію протягом 30 хвилин. Після цього розбавляють за допомогою дихлорметану і промивають 10%-ним водним розчином винної кислоти, потім водою і сушать над сульфатом магнію. Фільтрують і видаляють розчинник шляхом випаровування при зниженому тиску. Таким чином одержують 0,360 г сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і ацетону у співвідношенні 95:5. Таким чином одержують 93,7 мг гідрохлориду метил-транс-4-(бензоїламіно)-1-(хлоркарбоніл)2-піролідинкарбоксилату (С14Н 14N2O 4-НСІ-М=310,74 г), або вихід становить 26%. В атмосфері азоту, 93,7 мг (0,301 ммоль) одержаної вище сполуки змішують з 3мл тетрагідрофурану. Знижують температуру розчину до -78°С, потім додають краплями 332мкл 1 Μ розчину біс(триметилсиліл)аміду літію в тетрагідрофурані і реакційне середовище витримують при температурі -78 °С ще протягом 5 хвилин. Протягом 30 хвилин перемішують при кімнатній температурі. Потім розчин охолоджують до температури 0°С і додають 55 мкл оцтової кислоти. Додають 20 мл етилацетату і 3 мл фосфатного буфера з рН=7,0. Декантують, промивають водою, сушать над сульфа том магнію, фільтрують, концентрують шляхом випаровування. Таким чином одержують 76 мг піни, яку очищають шля хом хроматографії на діоксиді кремнію, елююючи сумішшю ди хлорметану і ацетону у співвідношенні 97:3. Виділяють 5 мг чистої цільової сполуки брутто-формули C14H14N 2O4-HCl (M=274,279 г), або вихід становить 6%. ІЧ-спектр (СНСІ3): 1805, 1779, 1743, 1669, 1603, 1589, 1486 см 4. МС (ЕУ) m/z: [M]+=274, 215, 169, 105, 77. ПРИКЛАД 7bis Фенілметил-транс-3-бензоїл-2-оксо-1,3-діазабіцикло[2,2,1]гептан-6-карбоксилат Слідують методиці, подібній такій, вказаній в прикладі 7, виходячи з 0,92 г 1-(1,1 -диметилетил)- і 2фенілметил-транс-4-аміно-1,2-піролідиндикарбоксилату (описується в J. Org. Chem., 56, 3009-3016 (1991)) і одержуючи цільову сполуку із загальним виходом, в розрахунку на 4 стадії, 5,4%. ПРИКЛАД 8 Фенілметил-транс-2-оксо-3-(фенілсульфоніл)-1,3-діазабіцикло[2,2,1]гептан-6-карбоксилат В атмосфері азоту, змішують 2,97 г (9,26 ммоль) 1-( 1,1-диметилетил)- і 2-(фенілметил)-транс-4-аміно-1,2піролідиндикарбоксилату (описується в J. Org. Chem., 56, 3009-3016 (1991)) брутто-формули С 17Н24^О4 (М=320,392 г) з 25 мл дихлорметану. Охолоджують до температури 5°С і додають 1,3 мл триетиламіну. Перемішують протягом 10 хвилин, потім додають 1,63 г бензолсульфонілхлориду. Перемішують при температурі 5 °С протягом 15 хвилин, потім температуру реакційної суміші доводять до 20°С протягом 45 хвилин. Розбавляють за допомогою дихлорметану, промивають 10%-ним водним розчином винної кислоти, потім фосфатним буфером з рН=7,0, після цього водним насиченим розчином хлориду натрію. Суша ть над сульфатом магнію і розчинник випаровують при зниженому тиску. Таким чином одержують 4,5 г сирого продукту, який хроматографують на діоксиді кремнію, елююючи сумішшю ди хлорметану і етилацетату у співвідношенні 90:10. Таким чином одержують 4,06 г 1-(1,1-диметилетил)- і 2-(фенілметил)-транс-4-[(фенілсульфоніл)аміно]-1,2піролідиндикарбоксилату брутто-формули C23H28N2O 6S (M=460,552 г), що відповідає виходу 95%. 3,83 г (8,31 ммоль) одержаного вище сульфонаміду змішують з 10мл безводного метанолу. Розчин охолоджують до температури 0°С і при цій температурі додають 8,2мл розчину хлороводню в метанолі з концентрацією 10 моль/л. Перемішують при температурі 0°С протягом 5 хвилин, потім залишають до підвищення температури до кімнатної температури. Через 30 хвилин метанол випаровують при зниженому тиску, залишок обробляють декілька разів метанолом, потім дихлорметаном. Після цього гідрохлорид кристалізують з діетилового ефіру. Таким чином одержують 3,2 г гідрохлориду фенілметил-транс-4-[(фенілсульфоніл)аміно]-2піролідинкарбоксилату бр утто-формули C18H20N2 O4S.HCl (M=396,896 г), що відповідає виходу 96%. В інертній атмосфері 2,78 г (7 ммоль) одержаного вище гідрохлориду змішують з 28 мл дихлорметану. Потім охолоджують до температури близько 0-5°С, після чого додають 2,15 мл триетиламіну. Продовжують перемішування протягом 15 хвилин при температурі 0-5°С, потім додають 0,46 мл дифосгену. Витримують при цій температурі протягом 4 хвилин, потім додають 10%-ний водний розчин винної кислоти, розбавляють дихлорметаном, декантують, промивають водним насиченим розчином хлориду натрію, сушать над сульфатом магнію і концентрують при зниженому тиску. Таким чином одержують 3,1г масла жовтого кольору, які очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 9:1. Одержують 1,82 г фенілметил-транс-1-(хлоркарбоніл)-4-[(фенілсульфоніл)аміно]-2піролідинкарбоксилату бр утто-формули C19H19CIN2 O5S (М=422,89 г), що відповідає виходу 61%. В інертній атмосфері 1,81 г (4,28 ммоль) одержаного вище карбамоїлхлориду змішують з 31 мл тетрагідрофурану. Одержаний розчин охолоджують до температури -70°С, потім при цій температурі протягом 10 хвилин додають 4,7 мл 1 Μ розчину біс(триметилсиліл)аміду літію в тетрагідрофурані. Перемішують протягом 45 хвилин при температурі -70°С, потім температуру підвищують до приблизно 0°С. Реакційне середовище витримують при цій температурі протягом 2 годин ЗО хвилин. Після цього додають 295 мкл оцтової кислоти. Розбавляють дихлорметаном, потім промивають 10%-ним водним розчином винної кислоти, розчином фосфатного буфера з рН=7 і водним насиченим розчином хлориду натрію. Сушать над сульфатом магнію і концентрують досуха при зниженому тиску. Сирий продукт очищають шляхом хроматографії на діоксиді кремнію, використовуючи як елююючий засіб суміш дихлорметану і етилацетату у співвідношенні 95:5. Таким чином одержують 244 мг цільової сполуки брутто-формули Ci9H18ClN2O5S (M=386,429 г), що відповідає виходу 14%. 1 H-ЯМР-спектр (400 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ул ьтиплетність): 2,15 (м): ОС-СН-СH2,85 (д) і 3,08 (д): O=C-N-CH2; 3,62 (м): O=C-CH-N-СН2; 4,94 (с): O2S-N-CH-CH 2; 5,16: СО2СН2С6Н5; 7,34 (м): С6Н5; 7,57 (м) - 7,68 (м) і 8,03 (м): SO2C6H5. ІЧ-спектр (СНСІ3): 1780, 1743; 1586, 1499 см -1. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [2 M+Na]+=795; [M+Na+CH3CN]+=450; [M+Na]+=409; [M+H]+=387. ПРИКЛАД 9 Фенілметил-транс-3-бензоїл-4-метил-2-оксо-1,3-діазабіцикло[2,2,1]гептан-6-карбоксилат В інертній атмосфері 18,69 г (58,52 ммоль) 1-(1.1-диметилетил)- і 2-(фенілметил)-4-оксо-1,2піролідиндикарбоксилату (описується в Chem. Pharm. Bull., 43 (8), 1302-1306 (1995)) брутто-формули C17H21NO 5 (M=319,361 г) змішують з 500 мл безводного діетилового ефіру. До одержаного розчину додають суспензію 10 г СеСl3 в 50 мл безводного діетилового ефіру. С успензію перемішують протягом 30 хвилин при температурі 20°С, потім охолоджують до температури -60°С. Після цього додають 3Μ розчин метилмагнійброміду в діетиловому ефірі. Залишають реакцію протягом 1 години при температурі -60°С, потім температуру підвищують до 0°С протягом 30 хвилин. Нейтралізують за допомогою 10%-ного водного розчину хлориду амонію. Екстрагують дихлорметаном, фільтрують, органічну фазу промивають водою, сушать над сульфатом магнію і концентрують досуха при зниженому тиску. Таким чином одержують 19,33 г масла, яке очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і трет-бутилметилового ефіру у співвідношенні 90:10. Одержують 7,21 г 1-(1,1-диметилетил)- і 2-(фенілметил)- цис-4-гідрокси-4-метил-1,2піролідиндикарбоксилату брутто-формули C18H25NO5 (M=335,404 г), або вихід становить 36%, а також 2,5 г епімерного спирту. В інертній атмосфері 3,17 г (9,45 ммоль) одержаної вище сполуки змішують з 70 мл дихлорметану. Охолоджують до температури 5°С і додають краплями 2,3 мл триетиламіну, потім 1,28 мл метансульфонілхлориду. Перемішують протягом 45 хвилин при температурі 5°С. Промивають 10%-ним водним розчином винної кислоти, потім розчином фосфатного буфера з рН=7, потім водою. Органічну фазу, сушать над сульфатом магнію і концентрують досуха при зниженому тиску. Таким чином одержують 3,9 г масла, яке очищають шля хом хроматографії на діоксиді кремнію, елююючи сумішшю ди хлорметану і етилацетату у співвідношенні 90:10. Одержують 2,75 г 1-(1,1-диметилетил)- і 2-(фенілметил)-цис-4-метил-4-[(метилсульфоніл)окси]-1,2піролідиндикарбоксилату брутто-формули C19H27NO7S (М=413,494 г), що відповідає виходу 70%. Готують розчин 2,54 г (6,14 ммоль) одержаного вище мезилату в 40 мл диметилформаміду. Потім, при температурі 20°С, додають 519 мг (7,98 ммоль) NaN3, нагрівають при температурі 50°С протягом 2 годин. Після охолоджування виливають в 250 мл води і екстрагують за допомогою 250 мл дихлорметану. Органічну фазу промивають водою, сушать над сульфатом магнію і випаровують досуха при зниженому тиску. Одержують 2,4 г сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію при використанні як елююючого засобу суміші дихлорметану і етилацетату у співвідношенні 95:5. Таким чином одержують 1,66 г 1-(1,1диметилетил)- і 2-(фенілметил)- транс-4-азидо-4-метил-1,2-піролідиндикарбоксилату брутто-формули C18H24N4 O4 (M=360,42 г) (зміст близько 30 мас.%), що відповідає виходу приблизно 25%. 1,85 г (або близько 1,7 ммоль) одержаного вище азиду розчиняють в 18 мл толуолу. Потім, при температурі 20°С, додають 1,38 мл (С4Н9)з8пН і 84 мг АІБН. Доводять до температури 75 °С і витримують при цій температурі протягом 2 годин. Толуол випаровують і залишок знов розчиняють в етилацетаті. Додають водний насичений розчин фториду калію і перемішують протягом ЗО хвилин при кімнатній температурі. Фільтрують на clarcel, декантують і органічну фазу сушать над сульфатом магнію. Після випаровування розчинника при зниженому тиску одержують 3 г масла, якого хроматографують на діоксиді кремнію, елююючи сумішшю ди хлорметану і метанолу у співвідношенні 9:1. Одержують 560 мг 1-(1,1-диметилетил)- і 2-(фенілметил)- транс-4аміно-4-метил-1,2-піролідиндикарбоксилату бр утто-формули C18H26N2O 4 (M=334,419 г). Отже, вихід є кількісним. В інертній атмосфері 578 мг (1,72 ммоль) одержаного вище аміну змішують з 30 мл дихлорметану. Охолоджують до температури 5°С і додають краплями 290 мкл триетиламіну, потім 240 мкл бензоїлхлориду. Продовжують перемішування при температурі 5°С протягом 30 хвилин. Розбавляють дихлорметаном, промивають 10%-ним водним розчином винної кислоти, водним насиченим розчином карбонату натрію, потім водою, органічну фаз у сушать над сульфа том магнію і розчинник випаровують при зниженому тиску. Таким чином одержують 950 мг масла, яке очищають шляхом хроматографії, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 90:10. Таким чином одержують 732 мг 1-(1,1-диметилетил)- і 2-(фенілметил)- транс-4(бензоїламіно)-4-метил-1,2-піролідиндикарбоксилату бр утто-формули C25H30N2 O5 (М=43 8,528 г), що відповідає виходу 97%. 636 мг (1,45 ммоль) одержаного вище аміду розчиняють в 1.9 мл етилацетату, охолоджують до температури близько 0-5°С на бані з льодом, потім додають 3,2 мл розчину хлороводню в етилацетаті з концентрацією 4,6 моль/л. Температуру підви щують до 20 °С, потім, через 1 годину, розчинник випаровують при зниженому тиску. Після цього гідрохлорид кристалізують з діетилового ефіру. Таким чином виділяють 570 мг гідрохлориду фенілметил-транс-4-(бензоїламіно)-4-метил-2-піролідинкарбоксилату брутто-формули C20H22N2 O3. HCl (М=374,87 г) у ви гляді порошку білого кольору. Отже, вихід є кількісним. В інертній атмосфері 100 мг (0,267 ммоль) одержаного вище гідрохлориду розчиняють в 1,5 мл дихлорметану. Охолоджують до температури близько 0-5°С, потім додають 90 мкл триетиламіну. Перемішують протягом 15 хвилин при температурі 5°С, потім додають 20 мкл дифосгену. Продовжують перемішування протягом 30 хвилин при температурі 5°С. Після цього обробляють за допомогою 10%-ного водного розчину винної кислоти, екстрагують ди хлорметаном, органічну фазу промивають водним насиченим розчином хлориду натрію, сушать над сульфатом магнію і розчинник випаровують при зниженому тиску. Таким чином одержують 130 мг масла, яке очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 9:1. Тоді одержують 72 мг фенілметил-транс-4-(бензоїламіно)-1-(хлоркарбоніл)-4-метил2-піролідинкарбоксилату брутто-формули C21H21N2O 4CI (М=400,865 г), що відповідає виходу 67%. 373 мг (0,930 ммоль) одержаної вище сполуки розчиняють в 9мл тетрагідрофурану. Потім розчин охолоджують до температури -70°С і протягом 5 хвилин додають 1 мл 1 Μ розчину біс(триметилсиліл)аміду літію в тетрагідрофурані. Реакційне середовище залишають нагріватися до температури 0°С протягом 45 хвилин, потім додають 69 мкл оцтової кислоти. Після цього розбавляють дихлорметаном, промивають 10%-ним водним розчином винної кислоти, потім розчином фосфатного буфера з рН=7,0 і водним насиченим розчином хлориду натрію. Органічну фазу сушать над сульфатом магнію, концентрують досуха при зниженому тиску, одержуючи 330 мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 98:2, що містить 0,1 об'ємн.% триетиламіну. Таким чином одержують 123 мг цільової сполуки брутто-формули C2iH2oN204(M=364,404 г), що відповідає виходу 36%. 1 H-ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультиплетність): 1,76 (с): СН3; 2,11 (дд) і 2,73 (ддд): N-CH-CH 2; 2,93 (д) і 3,00 (д): N-CH2; 3,96 (ддд): N-CH-CH2; 5,21: СО2СН2С6Н5; 7,36 (м): СН2С6Н5; 7 ,43 (т) і 7,57 (т) і 7,72 (д): СОС6Н5 . ІЧ-спектр (СНСІ3): 1776, 1745, 1682; 1601, 1580, 1498 см'1. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [2 M+Na]+=751; [2М+Н]+=729; [M+Na]+=387; [M+H]+=365. ПРИКЛАД 10 1 -Пропенілтрифенілфосфонієва сіль фенілметил-транс-2-оксо-3-(сульфокси)-1,3діазабіцикло[2,2,1]гептан-6-карбоксилату В інертній атмосфері 15 г (46,71 ммоль) 1-(1,1-диметилетил)- і 2-(фенілметил)-цис~4-гідрокси-1,2піролідиндикарбоксилату (торговий продукт) брутто-формули C17H23NO5 (M=321,377 г) розчиняють в 225 мл безводного дихлорметану. До розчину додають 5,42 мл 2,6-лутидину. Охолоджують до температури -70°С, потім протягом 5 хвилин вводять 8,25 мл ангідриду трифторметансульфокислоти. Перемішують протягом 10 хвилин при температурі -70°С, потім при температурі -70°С вводять 4,43 г О-алілгідроксиламіну. Після цього реакційну суміш, доводять до кімнатної температури протягом 27 годин. Розбавляють дихлорметаном, потім промивають 10%-ним водним розчином винної кислоти, водним насиченим розчином гідрокарбонату натрію і водою. Органічну фазу сушать над сульфатом натрію і розчинник випаровують при зниженому тиску. Таким чином одержують 23 г сирого масла, яке очищають шляхом хрома тографії на діоксиді кремнію, причому елюювання здійснюють послідовно за допомогою суміші дихлорметану і етилацетату у співвідношенні 95:5, 90:10, потім 80:20. Виділяють 7,18 г 1-(1,1-диметилетил)- і 2-(фенілметил)транс-4-[(2-пропенілокси)аміно]-1,2-піролідиндикарбоксилату брутто-формули C20H28N2O5 (М=376,456 г), що відповідає виходу 40%. 3,25 г (8,63 ммоль) одержаної вище сполуки розчиняють в 3,5 мл етилацетату. Охолоджують до температури близько 0-5°С, потім додають 19 мл розчину хлороводню в етилацетаті з концентрацією 4,6 моль/л. Реакцію проводять при температурі близько 0-5°С протягом 40 хвилин при постійному перемішуванні. Розчинник випаровують при зниженому тиску, потім обробляють декілька разів діетиловим ефіром, відбираючи рідину над осадом. Таким чином одержують 2,54 г гідрохлориду у вигляді осаду білого кольору, який розчиняють при перемішуванні в 55 мл дихлорметану. Додають 7,3 мл 2н розчину гідроксиду натрію. Після декантації органічну фазу суша ть над сульфатом натрію. Дихлорметан випаровують при зниженому тиску. Таким чином одержують 2,12 г фенілметил-транс-4-[(2-пропенілокси)аміно]-2-піролідинкарбоксилату бр утто-формули C15H20N2 O3 (М=276,337 г) у вигляді масла, або вихід становить 89%. В інертній атмосфері 4,14 г (15 ммоль) одержаної вище сполуки розчиняють в 1,5 л ацетонітрилу. Охолоджують до температури близько 0-5°С і додають 1,14 мл дифосгену. Перемішують протягом 15 хвилин при підтриманні температури 0-5°С, потім додають послідовно 4,6 мл триетиламіну і 1,83 г DMAP в 80 мл ацетонітрилу. Температур у підвищують до кімнатної температури і реакцію проводять протягом 26 годин, потім випаровують половину розчинника при зниженому тиску. Після цього обробляють 10%-ним водним розчином винної кислоти, потім екстрагують дихлорметаном. Органічну фаз у промивають за допомогою водного насиченого розчину хлориду натрію, сушать її над сульфатом магнію і розчинник випаровують при зниженому тиску. Таким чином одержують 43 г сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 90:10, що містить 0,1% триетиламіну. Одержують 312 мг фенілметил-транс-2-оксо-3-(2пропенілокси)-1,3-діазабіцикло[2,2,1]гептан-6-карбоксилату бр утто-формули C16Η18Ν2Ο 4(Μ=302,33 г), що відповідає виходу 7%. В інертній атмосфері 70,2 мг (0,232 ммоль) одержаної вище сполуки розчиняють в 2,3 мл дихлорметану. Потім вводять 26,5 мкл оцтової кислоти і 134 мг Ра[Р(С 6Н5)з]4· Реакцію проводять протягом 40 хвилин при кімнатній температурі, потім температур у знижують до-20°С і додають 2,96 мл розчину комплексу SО3-піридин з концентрацією 0,314 моль/л. Реакцію проводять протягом 2,5 годин, потім додають дихлорметан і реакційне середовище випаровують при зниженому тиску. Після цього обробляють за допомогою 40 мл дихлорметану і промивають всі за допомогою 5 мл води. Органічну фазу відділяють і сушать над сульфатом натрію, потім розчинник випаровують при зниженому тиску. Таким чином одержують 280 мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи послідовно сумішшю дихлорметану і ацетону у співвідношенні 80:20, що містить 0,1% триетиламіну, потім сумішшю дихлорметану і ацетону у співвідношенні 50:50, що містить 0,1% триетиламіну. Виділяють 34,0 мг цільової сполуки брутто-формули C34H33N2O7SP (М=644,689) у ви гляді масла жовтого кольору, або ви хід становить 23%. 1 Н-ЯМР-спектр (400 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультипл етність): 2,00 (м) і 2,48(м): CHj-CH-CO; 2,72(д) і 3,12(с): CH-CH2-N; 3,75(м): СН2-СН-О02; 4,71(с) СН-СН 2-N; 5,18 [АВ] CH.2-C6H5; 7,35(м): СН2-С6Н5 і 2,29(м): СН3-CH=CH; 6,62 і 7,21 СН3-СН=СН; 7,60-7,85 Р(С6Н5)3. МС (іонізація електронним розпиленням з утворенням позитивних і негативних іонів) m/z: [Μ аніон]"=341; [Μ катіон]+=303. ПРИКЛАД 11 1-Пропенілтрифенілфосфонієва сіль метил-транс-2-оксо-3-(сульфокси)-1,3-діазабіцикло[2,2,1]гептан-6карбоксилату Слідують методиці прикладу 10, але виходячи з 207 мг 1-(1,1-диметилетил)- і 2-метил-цис-4-гідрокси-1,2піролідиндикарбоксилату. Таким чином одержують 12 мг цільового продукту формули C7H10N2O 7S (М=266,231 г). МС (іонізація електронним розпиленням з утворенням позитивних і негативних іонів) m/z: [Μ аніон]~=265; [Μ катіон]+=303. ПРИКЛАД 12а Дифенілметил-транс-7-оксо-6-окса-1-азабіцикло[3,2,1]октан-3-карбоксилат В інертній атмосфері змішують 8 мл дихлорметану і 347 мг (1 ммоль) гідрохлориду дифенілметил-цис-5гідрокси-З-піперидинкарбоксилату (описується в Acta Chem. Scand., серія В, 35 (4), 289-294). Охолоджують до температури 0°С, потім додають 346 мкл триетиламіну і 72 мкл дифосгену. Реакцію проводять протягом 15 хвилин при підтриманні температури 0°С, потім розчинник випаровують при зниженому тиску. Обробляють за допомогою 25 мл безводного толуолу. Фільтрують для видалення триетиламінгідрохлориду. До фільтрату додають 553 мкл триетиламіну і протягом 4 годин кип'ятять із зворотним холодильником. Потім розбавляють за допомогою етилацетату і промивають 10%-ним водним розчином винної кислоти, потім водним насиченим розчином хлориду натрію і органічну фазу сушать над сульфа том магнію. Потім випаровують при зниженому тиску і виділяють 339 мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю толуолу і етилацетату у співвідношенні 70:30. Таким чином одержують 146 мг цільової сполуки (М=337,378 г), що відповідає виходу 43%. 1 Н-ЯМР-спектр (250 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультипл етність): 2,15 (ддд) і 2,73 (д квадруплет): N-CH2CHO-CH2; 2,92 (τ): О2С-СН-; 3,00 (д) і 3,45 (д): N-CH 2-CHO; 3,48 (дд) і 4,07 (дд): N-CH2-CH-C02; 4,79 (д): N-CH2CHO; 6,90 (с): СО2-СН-(С6Н5)2; 7,33 (м): (С6Н5)2. ІЧ-спектр (СНСІ3): 1792, 1734; 1600, 1585, 1497 см-1. МС (ЕУ) m/z: [М]+=337, 292, 183, 167. ПРИКЛАД 12b транс-7-Оксо-6-окса-1-азабіцикло[3,2,1]октан-3-карбонова кислота Змішують 320 мг одержаної в прикладі 12а сполуки, 17 мл ацетону і 70 мг каталізатора 20%-ного паладію на-вугіллі в масі. Перемішують в атмосфері водню при нормальному тиску. Через 2 години ЗО хвилин додають 70 мг каталізатора і проводять реакцію протягом ще 1 години 30 хвилин, потім реакційне середовище фільтрують. Розчинник випаровують при зниженому тиску і таким чином одержують 350 мг сирого продукту, який кристалізують з пентану. Відфільтровують і таким чином виділяють 158 мг цільового продукту брутто-формули C7H9NO4 (М= 171,154 г) у вигляді твердої речовини сірого кольору. Відповідний вихід становить 89%. 1 H-ЯМР-спектр (300 МГц; диметилсульфоксид; хімічні зсуви піків в м.ч. і мул ьтиплетність): 2,10 (ддд) і 2,43 (дм): N-CH2-CHO-CH2; 2,83 (τ): О2С-СН-; 3,13 (д) і 3,27 (дм): N-CH2-CHO; 3,40 (дд) і 3,72 (д): N-CH2-CH-CO2H; 4,81 (м): N-CH 2-CHO; 12,54 (с шир.): СО2Н. ІЧ-спектр (вазелінове масло): 1782, 1692 см"1. МС (ЕУ) m/z: [М]+=177, 155, 127, 82, 70. ПРИКЛАД 12с (4-Нітрофеніл)метил-транс-7-оксо-6-окса-1 -азабіцикло[3,2,1]октан-3-карбоксилат В інертній атмосфері змішують 30 мг (0,175 ммоль) одержаної в прикладі 12Ь кислоти і 0,5 мл дихлорметану. Після цього додають 26,8 мг 4-нітробензилового спирту, 2,2 мг DMAP і 37 мг EDCI. Реакцію проводять при перемішуванні протягом 2 годин при кімнатній температурі. Органічну фазу потім розбавляють дихлорметаном, промивають 10%-ним водним розчином винної кислоти і розчином фосфатного буфера з рН=7. Після висушування органічної фази над сульфатом натрію і випаровування розчинника призниженому тиску одержують 57 мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю толуолу і етилацетату у співвідношенні 85:15. Продукт потім кристалізують з суміші діетилового ефіру і пентану, одержуючи 34 мг цільової сполуки у вигляді кристалів білого кольору (М=306,277 г). Відповідний вихід становить 63,5%. 1 H-ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультиплетність): 2,14 (ддд) і 2,84 (дм): N-CH2-CHO-CH2; 2,90 (τ): О2С-СН-; 3,10 і 3,49 (дм): Ν-СН2-СНО; 3,43 (дд) і 4,14 (уш.д): N-CH 2-CH-CO2; 5,27 [АВ]: СО2-СН2-С6Н5; 7,56 і 8,24 [АА'ВВ1 ]: C-C 6H5-NO 2. ІЧ-спектр (СНСl3): 1799, 1789, 1741; 1609, 1526, 1495 см 4. МС (ЕУ) m/z: [M]+ = 306, 170, 136, 126, 106, 82. ПРИКЛАД 13 6-(Фенілметил)- 1,6-діазабіцикло[3,2,1]октан-7-он Стадія А: До розчину 20,71 г 3-амінопіридину в 200 мл дихлорметану при температурі близько 0-5°С додають 30,7 мл триетиламіну. Потім додають краплями протягом 15 хвилин 25,5 мл бензоїлхлориду і залишають для підвищення до кімнатної температури. Після перемішування протягом 1 години промивають водою, потім насиченим розчином гідрокарбонату натрію, після чого органічну фазу сушать над сульфатом натрію і розчинник випаровують при зниженому тиску. Одержують 42,29 г кристалічного цільового продукту (М=198,226г). Стадія В: До розчину 10г одержаного на стадії А продукту в 200 мл метанолу додають 4,3 мл концентрованої соляної кислоти і 500мг 5 мас.%-ного родію-на-оксиді алюмінію. Витримують в атмосфері водню при тиску 60-110 бар протягом 15 годин. Реакційну суміш фільтр ують, промивають метанолом, потім фільтрат концентрують при зниженому тиску. Гідрохлорид цільового продукту одержують в суміші з 10% гідрохлориду вихідного продукту. Продукт обробляють за допомогою 250 мл дихлорметану і додають 1,1 еквіваленти 1н розчину гідроксиду натрію. Після перемішування протягом 15 хвилин дихлорметан декантують, органічну фазу промивають водою, сушать і випаровують при зниженому тиску. Залишок хроматографують на діоксиді кремнію, елююючи сумішшю ди хлорметану, метанолу і триетиламіну у співвідношенні 92:8:3. Одержують 7,4 г кристалічного цільового продукту, або вихід становить 72%. Стадія С: IЧ-(фенілметил)-3-пшеридинамін 20 г одержаного на стадії В продукту розчиняють в 600 мл 1,2-диметоксіетану. До розчину протягом 30 хвилин додають 14,86 г літійалюмінійгідриду. Нагрівають, при перемішуванні і в атмосфері інертного газу, при температурі 75-80°С протягом 16 годин, потім охолоджують до температури 0°С і протягом 45 хвилин, не перевищуючи температуру 12°С, додають 11 мл води. Перемішують протягом 10 хвилин, фільтрують і осад промивають дихлорметаном. Фільтрат концентрують при зниженому тиску. Одержують 17,8 г цільового продукту у вигляді масла, яке переганяють при зниженому тиску (температура кипіння 114-121°С при 0,8 мбар). Одержують 16 г цільового продукту, або ви хід становить 86%. Стадія D: 6-(фенілметил)-1,6-діазабіцикло[3,2,1]октан-7-он 1,06 г одержаного на стадії 3 продукту розчиняють в 28 см 3 толуолу, потім охолоджують до температури 0°С і в атмосфері інертного газу додають 337мкл дифосгену. Залишають температуру підвищуватися і розчин витримують протягом 2 годин при температурі 20°С. Концентрують при зниженому тиску, потім залишок хроматографують на діоксиді кремнію, елююючи послідовно за допомогою суміші дихлорметану і ацетону у співвідношенні 95:5, потім 80:20, і, нарешті, за допомогою суміші дихлорметану, метанолу і триетиламіну у співвідношенні 92:8:3, і одержують 362 мг цільового продукту C13H16N2O (M=216,85 г), або ви хід становить 30%. CPV/MC (ЕУ) m/z: [M]+=216, 125, 91. ІЧ-спектр (СНС13): 1718; 1498 см-1. ПРИКЛАД 14 6-Бензоїл-1,6-діазабіцикло[3,2,1]октан-7-он Стадія А: 3-(Бензіламіно)-1 -піперидинкарбонова кислота В атмосфері азоту, 5 г продукту, одержаного на стадії В прикладу 13, розчиняють в 1,25 л безводного толуолу, потім додають 3,4 мл триетиламіну і при температурі 0-5°С протягом 3 хвилин вводять 1,47 мл дифос гену. Після витримування протягом 20 хвилин при температурі 0-5°С суміш залишають для підвищення температури до 20°С, перемішують протягом 75 хвилин, потім розчинник випаровують при зниженому тиску. Залишок хроматографують на діоксиді кремнію, елююючи сумішшю дихлорметану і ацетону у співвідношенні 8:2. Одержують 3,44 г цільового продукту (вихід становить 52,6%). Стадія В: 6-Бензоїл-1,6-діазабіцикло[3,2Д]октан-7-он В атмосфері азоту, вводять 48 мг гідриду натрію у вигляді 50%-ний дисперсії в маслі і 20 мл тетрагідрофурану. Охолоджують до температури близько 0-5°С, потім за один раз додають 266 мг одержаного на стадії А продукту. Суміш залишають для підвищення температури до кімнатної, потім додають 60 мкл оцтової кислоти і 10 мл фосфатного буфера з рН=7. Після цього додають трохи етилацетату, потім декантують і повторно екстрагують етилацетатом. Органічну фаз у суша ть над сульфатом магнію, після чого розчинники випаровують при зниженому тиску. Сирий продукт хроматографують на діоксиді кремнію, елююючи дихлорметаном, що містить 2% ацетону. Таким чином одержують 143 мг цільового продукту C13H12N2O 2 (М=228,25 г). Відповідний вихід становить 62%. 1 Н-ЯМР-спектр (250 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ул ьтиплетність): 1,20 - 2,15 (м) і 2,42 (м): NCH-CH2-CH2S 2,80 (д) - 2,93 (д); 3,11 (м); 3,28-3,58 (м): CH2-N; 4,54 (м): CH-N; 7,43 (м); 7,55 (м); 7,69 (м.): С6Н5. ІЧ-спектр (СНСl3): 1758, 1672; 1605, 1586, 1492 cм -1. МС (ЕУ) m/z: [M]+=230, 125, 105, 77. ПРИКЛАД 15 7-Оксо-1,6-діазабіцикло[3,2,1]октан-6-оцтова кислота Стадія А: 5-[(1,1-Диметилетил)диметилсиліл]-1,6-діазабіцикло[3,2,1]октан-7-он Вміщують в атмосферу азоту 843 мг літію і конденсують при температурі -70°С 320 мл аміаку. При температурі -70°С протягом 10 хвилин додають 7,56 г (34,8 ммоль) одержаного в прикладі 13 продукту в 160 мл тетрагідрофурану. Перемішують протягом 5 хвилин, потім аміак відганяють в струмі азоту при повільному нагріванні до температури 20°С. При температурі 20°С до одержаної суспензії повільно додають 7,9 г (ІД-диметилетил)диметилсилілхлориду в 10 см 3 тетрагідрофурану, потім перемішують протягом 10 хвилин. Після цього додають 160 см етилацетату, потім 60 см 10%-ного водного розчину винної кислоти. Декантують, повторно екстрагують етилацетатом, органічну фазу промивають водою, сушать її над сульфатом натрію і розчинник випаровують при зниженому тиску. Одержане масло хроматографують на діоксиді кремнію з 10% води, елююючи дихлорметаном, потім сумішшю дихлорметану і ацетону у співвідношенні 8:2 і одержують 3,04 г цільового продукту (ви хід становить 36,2%). 1 H-ЯМР-спектр (250 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультиплетність) : 0,21 (с) і 0,40 (с): SiCH 3; 0,97 (с): SitBu; 1,5-1,8 (м) і 2,07(м): N-CH-CH2-CH2; 2,85 (д) і 3,32 (м); -CH-CH.2-N: 2,93 (д) і 3,32 (м): -CH 2-CH2-N; 3,65 (м): CH-N. ІЧ-спектр (СНСl3): 1710, 842 см -1. МС (ЕУ) m/z: [M]+-240, 225, 183, 100, 83, 57. Стадія В: Фенілметил-7-оксо-1,6-діазабіцикло[3,2,1]октан-6-ацетат В атмосфері азоту, 1,44 г (5,99 ммоль) одержаного на стадії А продукту розчиняють в 14,4 мл тетрагідрофурану, потім додають 941 мкл фенілметилбромацетату і потім, краплями, 6 мл 1 Μ розчину тетра-н-бутиламонійфториду в тетрагідрофурані. Перемішують протягом 10 хвилин при температурі 20°С, потім розбавляють за допомогою 15 мл етилацетату і додають 5 мл водного розчину фосфа тного буфера з рН=7. Декантують, повторно екстрагують етилацетатом, органічну фазу промивають водою, сушать її над сульфатом натрію і розчинник випаровують при зниженому тиску. Маслянистий залишок хроматографують на діоксиді кремнію з 10% води, елююючи сумішшю дихлорметану і ацетону у співвідношенні 8:2. Одержують 140 мг цільового продукту. Відповідний вихід становить 9%. ІЧ-спектр (СНСl3): 1746, 1720 см-1. МС (ЕУ) m/z: [M]+=274, 183, 155, 139, 91, 83. Стадія С: 7-оксо-1,6-діазабіцикло[3,2,1]октан-6-оцтова кислота 137 мг одержаного на стадії В продукту розчиняють в 1,5 мл етилацетату, потім до розчину додають 14 мг 10%-ного паладію-на-вугіллі і вміщують в атмосферу водню. Через 15 хвилин знов додають 15 мг паладію-навугіллі і витримують при перемішуванні протягом 15 хвилин. Каталізатор відфільтровують, промивають етилацетатом, потім ацетоном і метанолом і розчинник випаровують при зниженому тиску. Одержують в цілому 68 мг сирого продукту, який кристалізують з простого ефіру. Одержують 58 мг цільового продукту бруттоформули C15H18N2O 3 (M=274,321 г). Відповідний вихід становить 63%. 1 H-ЯМР-спектр (400 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультиплетність): 1,48 (м), 1,63 (м), 1,73 (м) і 1,86 (м): N-CH-CH2-CH2 2,85-3,00 (м), 3,14 (дм) і 3,64 (м): CH2-N-CH2 і CH-N; 3,78 і 4,14 [АВ]: CON-СН2-СО. МС (ЕУ) m/z: [M]+=184, 139, 125, 111, 97, 83. ПРИКЛАД 16 7-Оксо-М-феніл-1,6-діазабіцикло[3,2,1 ]октан-6-карбоксамід В атмосфері інертного газу змішують 1 мл тетрагідрофурану і 99 мг (0,41 ммоль) одержаної на стадії А прикладу 15 сполуки. Послідовно додають 50 мкл фенілизоціанату, потім 450 мкл 1 Μ розчину тетрабутиламонійфториду в тетрагідрофурані. Реакцію проводять протягом 10 хвилин, потім розбавляють етилацетатом, промивають водою. Органічну фазу декантують і сушать над сульфатом магнію. Розчинник випаровують при зниженому тиску. Таким чином одержують 140 мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, використовуючи як елююючий засіб суміш дихлорметану і етилацетату у співвідношенні 90:10. Одержують 21 мг цільової сполуки брутто-формули C13H15N3O (M=245,283г), що відповідає виходу 20%. 1 H-ЯМР-спектр (250 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультиплетність): 1,78 (м), 2,02 (м) і 2,17 (м): N-CH-CH2CH2; 2,88 (д), 3,13 (д) і 3,42 (м): CH.2-N-СН3; 4,49 (м): CH-N; 7,11 (т), 7,34 (т) і 7,54 (д): С6Н5; 10,05: NH. ІЧ-спектр (СНСІ3): 3302, 3266; 1734; 1700; 1602, 1553, 1501 cm 4. МС (ЕУ) m/z: [М]+=245, 153, 126, 119, 98, 92. ПРИКЛАД 17а 6-[1-(Фенілметил)-1 Н-тетразол-5-іл]-1,6-діазабіцикло [3,2,1 ]октан-7-он Вміщують в атмосферу інертного газу 480 мг (2 ммоль) сполуки, одержаної на стадії А прикладу 15. Потім додають розчин 712 мг 5-фтор-1-(фенілметил)-1Н-тетразолу в 1,5 мл тетрагідрофурану, потім 2 мл 1 Μ розчину тетрабутиламонійфториду в тетрагідрофурані. Реакцію проводять протягом 1 хвилини. Потім розбавляють етилацетатом, промивають водою, декантують, органічну фаз у суша ть над сульфатом магнію і розчинник випаровують при зниженому тиску. Одержують 1,06 г маслянистого продукту, який хроматографують на діоксиді кремнію при використанні суміші дихлорметану і етилацетату у співвідношенні 90:10. Таким чином одержують 143 мг цільової сполуки брутто-формули C14H16N 6O (М=284,324 г) у вигляді аморфного продукту білого кольору. Відповідний вихід становить 25%. 1 Н-ЯМР-спектр (250 МГц; CDCд3; хімічні зсуви піків в м.ч. і мультиплетність): 1,80 (м), 2,04 (м) і 2,67 (м): N-CH-CH2-CH2; 2,83 (д), 2,85 (дм), 3,10 (дд) і 3,44 (дд): CH2-N-CH2; 3,99 (м): CHN; 5,63 і 5,88 [АВ]: С6Н5-СН2; 7,18 (м) і 7,32 (м): С6Н5. ПРИКЛАД 17b 6-(1Н-Тетразол-5-іл)-1,6-діазабіцикло [3,2,1 ]октан-7-он Змішують 120 мг продукту, одержаного в прикладі 17а, і 2,4 мл суміші метанолу і етилацетату у співвідношенні 90:10, потім додають 2,4 мл тетрагідрофурану аж до досягнення повного розчинення. Після цього додають 24 мг каталізатора 10%-ного паладію-на-вугіллі, потім перемішують в атмосфері водню. Після протікання реакції протягом 3 годин каталізатор відфільтровують, промивають сумішшю тетрагідрофурану і метанолу, потім розчинник випаровують при зниженому тиску. Після цього продукт кристалізують з діетилового ефіру. Таким чином одержують 72 мг цільової сполуки брутто-формули C7Hi0N6O (M= 194,198 г) у вигляді кристалічного продукту білого кольору. Відповідний вихід становить 88%. 1 H-ЯМР-спектр (300 МГц; диметилсульфоксид; хімічні зсуви піків в м.ч. і мультиплетність): 1,63 (м), 1,89 (м) і 2,07 (м): N-CH-CH 2-CH2; 3,14-3,20 (м) і 3,43 (м): CH 2-N-CH2;4,51(m)CH-N. ІЧ-спектр (вазелінове масло): 1744; 1594 см -1. МС (ЕУ) m/z: [M]+=194, 165, 124, 111, 98, 83,68, 56, 41. ПРИКЛАД 18 6-Ацетил-1,6-діазабіцикло[3,2,1]октан-7-он У 1,4 мл тетрагідрофурану розчиняють 140 мг (0,582 ммоль) сполуки, одержаної на стадії А прикладу 15. До одержаного розчину послідовно додають 55 мкл оцтового ангідриду, потім 0,58 мл 1 Μ розчину тетрабутиламонійфториду в тетрагідрофурані. Після цього розбавляють етилацетатом, промивають водою, декантують, органічну фазу сушать над сульфатом магнію, потім розчинник випаровують при зниженому тиску. Таким чином одержують 116 мг сирого масла, яке хроматографують. на діоксиді кремнію при використанні суміші дихлорметану і ацетону у співвідношенні 80:20. Таким чином одержують 18 мг цільової сполуки брутто-формули C8H12N2O 2 (M= 168,196 г), що відповідає виходу 18%. 1 Н-ЯМР-спектр (300 МГц; диметилсульфоксид; хімічні зсуви піків в м.ч. і мультиплетність): 1,65-2,20 (м): N-CH-CH2-CH2; 2,54 (с): CH 3CO-N; 2,83 (д), 3,33 (дм), 3,10 (м) і 3,45 (дд) CH2-N-CH2; 4,55 (м): OC-N-CH. ІЧ-спектр (СНС13): 1758, 1696 см-1. МС (ЕУ) m/z: [M]+=168, 140, 126, 98, 43. ПРИКЛАД 19а 6-(Фенілметокси)-1,6-діазабіцикло[3,2,1]октан-7-он Розчиняють 44,02 г (0,22 моль) 1Д-диметилетил-3-оксо-1-піперидинкарбоксилату (C10H17NO3; М=199,251 г) (описується в J. Med. Chem., 29, 224-229 (1986)) в 440 мл етанолу. Потім додають 38,79 г 0бензилгідроксиламінгідрохлориду. Після цього в суспензію краплями вводять 54 мл піридину. Реакцію проводять при перемішуванні протягом 4 годин при температурі близько 25°С, потім розчинник випаровують при зниженому тиску. Обробляють сумішшю дихлорметану і етилацетату, потім фільтрують і промивають дихлорметаном, потім - сумішшю ди хлорметану і етилацетату. Фільтрат після цього концентрують досуха при зниженому тиску. Таким чином одержують 69,8 г масла світло-жовтого кольору, які очищають шляхом хроматографії на діоксиді кремнію. Елююючим засобом, що використовується, є суміш циклогексану і етилацетату у співвідношенні 80:20. Одержують 57,21 г 1,1-диметилетил3-[(фенілметокси)іміно]-1 -піперидинкарбоксилату брутто-формули C17H24N3 O3 (М=304,39 г) у вигляді масла дуже блідо-жовтого кольору. Відповідний вихід становить 85%. В атмосфері азоту 24,82 г (0,0815 ммоль) одержаного вище оксиму розчиняють в 163 мл охолодженого до температури -10°С етанолу. Після цього додають 25 мл комплексу боран-піридин, потім, краплями, протягом 1 години 15 хвилин, 204 мл 2н соляної кислоти. Розчин перемішують протягом 1 години 15 хвилин при температурі -5°С, потім обробляють за допомогою 100 мл насиченого розчину гідрокарбонату натрію, потім за допомогою 35г карбонату натрію, які додають маленькими порціями. Значення рН тоді становить 7-8. Реакційне середовище екстрагують етилацетатом. Органічні фази об'єднують, сушать над сульфатом натрію, розчинник випаровують при зниженому тиску. Таким чином одержують 39,0 г безбарвної маслянистої рідини, яку обробляють за допомогою 400 мл етилацетату. Розчин промивають за допомогою 0,05 н соляної кислоти, потім органічні фази об'єднують і розчинник випаровують при зниженому тиску. Виділяють 35,5 г маслянистої безбарвної рідини, яку очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 95:5, потім сумішшю ди хлорметану і етилацетату у співвідношенні 80:20. Таким чином одержують 17,89 г 1,1-диметилетил-З-[(фенілметокси)аміно]-1 -піперидинкарбоксилату брутто-формули С17Н26О 3 (М=306,41 г) у вигляді безбарвного масла. Відповідний вихід становить 72%. 6,72 г (21,9 ммоль) одержаного вище піперидину розчиняють в 22 мл етилацетату, о холодженого до температури -10°С. Протягом 30 хвилин додають краплями 28 мл розчину безводного хлороводню в етилацетаті з концентрацією 4,0 моль/л. Після витримування протягом 1 години при температурі 0°С додають 40 мл діетилового ефіру, осад дигідрохлориду відфільтровують і промивають його діетиловим ефіром. Таким чином одержують 3,87 г твердої речовини білого кольору. Шляхом кристалізації фільтрату одержують ще 1,80 г цільового продукту. Одержаний продукт обробляють за допомогою 60 мл 1н розчину гідроксиду натрію і 120 мл етилацетату. Після декантації водну фазу насичують хлоридом натрію, потім екстрагують два рази етилацетатом. Органічні фази об'єднують і суша ть над сульфатом магнію, потім концентрують досуха при зниженому тиску. Таким чином одержують 3,67 г М-(фенілметокси)-3-піперидинаміну брутто-формули Ci2Hi8N2O (M=206,29 г), що відповідає виходу 81%. 518 мг (2,5 ммоль) одержаної вище сполуки розчиняють в 5 мл безводного дихлорметану, потім додають 0,5 мл триетиламіну. Одержану суспензію білуватого кольору охолоджують до температури -65 °С, потім протягом 15 хвилин додають 12,5 мл розчину дифосгену в дихлорметані з концентрацією 0,10 моль/л. Після протікання реакції протягом 45 хвилин безбарвний розчин розбавляють за допомогою 15 мл дихлорметану і обробляють за допомогою 15 мл води. Середовище декантують, потім водну фазу екстрагують за допомогою 20 мл дихлорметану. Об'єднані органічні фази сушать над сульфатом магнію, потім концентрують досуха при зниженому тиску. Таким чином одержують масло блідо-жовтого кольору, яке очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 90:10, потім сумішшю дихлорметану і етилацетату у співвідношенні 80:20. Таким чином одержують 196 мг цільової сполуки бруттоформули C13H16N2O 2 (М=232,28 г) у ви гляді безбарвного масла. Відповідний вихід становить 34%. 1 Н-ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультипл етність): 1,59 (м) і 1,93-2,18 (м): N-CH-CH2-CH2; 2,73 (д), 2,94 (д), 3,17 (д) і 3,40 (дд): CH2-N-CH2; 3,29 (τ): N-CH; 4,89 (д): N-O-CH2-(C6H5); 7,38: С 6Н5. ІЧ-спектр (СНСІ3): 1747; 1498 см -1. МС (ЕУ) m/z: [M]+=232,91. ПРИКЛАД 19b 6-(Ацетилокси)-1,6-діазабіцикло[3,2,1]октан-7-он Розчиняють 95 мг (0,41 ммоль) сполуки, одержаної в прикладі 19а, в 5 мл метанолу, додають 8 мг 10 мас.%-ного паладію-на-вугіллі, потім суспензію витримують в атмосфері водню при нормальному тиску протягом 1 години при температурі 25°С, після чого каталізатор відфільтровують. Після випаровування розчинника при зниженому тиску одержують 70 мг кристалів білого кольору. Кристали обробляють за допомогою 2 мл безводного дихлорметану. Розчин охолоджують до температури -10°С в атмосфері азоту. Потім додають 70 мкл піридину, потім 40 мкл оцтового ангідриду і перемішують протягом 20 хвилин. Концентрують при зниженому тиску і одержують 75 мг кристалів білого кольору, які очищають на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 80:20. Одержують 49 мг цільової сполуки (М= 184,20 г) у вигляді твердої речовини білого кольору. Відповідний вихід становить 65%. 1 H-ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультиплетність): 1,60-2,2: N-CH-CH2-CH2; 2,24 (с): СН3; 2,95 (д) і 3,54 (дм): N-CH.2-CH; 3,07 (д) і 3,54 (уш.дд): N-CH 2-CH2; 3,94 (уш.т): OC-N-CH. ІЧ-спектр (СНСІ3): 1798; 1764 см -1. МС (ЕУ) m/z: [M]+=184, 142, 125, 43. ПРИКЛАД 19с 6-(Бензоїлокси)-1,6-діазабіцикло[3,2,1]октан-7-он Слідують методиці, подібній такій, описаній в прикладі 19Ь, виходячи з 205 мг сполуки, одержаної в прикладі 19а, і 200 мг бензойного ангідриду. Таким чином одержують 64 мг цільової сполуки брутто-формули C13H14N2 O3 (М=246,27 г), або вихід становить 30%. 1 H-ЯМР-спектр (250 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультиплетність): 1,64-1,95 (м) і 2,10-2,35 (м): СН-СН2-СH 3,02 (д) і 3,65 (дм): N-CH2-CH; 3,13 (д) і 3,55 (уш.дд): N-CH 2-CH2; 4,09 (уш.т): OC-N-CH; 7,49 (м): 7,65 (т); 8,12 (м): С6Н5. ІЧ-спектр (СНСІ3): 1774, 1756; 1602, 1585, 1495 см-1. МС (ЕУ) m/z: [M]+=246, 105, 77. ПРИКЛАД 19d 6-(1-Оксопропокси)-1,6-діазабіцикло[3,2,1]октан-7-он Слідують методиці, подібній такій, описаній в прикладі 19с, виходячи з 163 мг сполуки, одержаної в прикладі 19а, і 70 мкл пропіонілхлориду. Таким чином одержують 17 мг цільової сполуки брутто-формули C9H14N2O3 (M=l98,23 г), або вихід становить 12%. 1 Н-ЯМР-спектр (300МГц; CDCl3; хімічні зсуви піків в м.ч. і мул ьтипл етність): 1,25 (т): О=С-СН 2-СН3; 1,65 (м), 1,78 (м) і 2,10 (м): N-CH-CH2-CH2; 2,52 (м) О=С-СН 2-СН3; 2,94 (д) і 3,55 (уш.д): N-CH 2-CH; 3,07 (д) і 3,48 (дд): N-CH 2-CH2; 3,93 (м): N-CH 2-CH. ІЧ-спектр (СНСІ3): 1792; 1763 см"1. МС (ЕУ) m/z: [М]+=198, 170, 142, 125, 97, 57. ПРИКЛАД 19е 6-[[(4-Метилфеніл)сульфоніл]окси]-1,6-діазабіцикло[3,2,1]октан-7-он Слідують методиці, подібній такій, описаній в прикладі 19d, виходячи з 139 мг сполуки, одержаної в прикладі 19а, і 126 мг тозилхлориду. Таким чином одержують 77 мг цільової сполуки брутто-формули C13H16N2 O4S (M=296,35 г), або ви хід становить 44%. 1 H-ЯМР-спектр (250 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультиплетність): 1.55 і 2,99 (м): N-CH-CH 2-CH2; 2,45 (с): СН3; 2,89 (д), 3,00 (д), 3,29 (д) і 3,39 (дд): CH 2-N-CH2; 4,04 (м): N-CH; 7,35 і 7,91 [АА'ВВ 1] CH3-C6H4-SO2. ІЧ-спектр (СНСІ3): 1775; 1599, 1495, 1383; 1193, 1180 см"1. МС (ЕУ) m/z: [M]+=296, 155, 141, 125, 91. ПРИКЛАД 19f 6-[(Метилсульфоніл)окси]-1,6-діазабіцикло[3,2,1]октан-7-он Слідують методиці, подібній такій, описаній в прикладі 19е, виходячи з 211 мг сполуки, одержаної в прикладі 19а, і 80 мкл мезилхлориду. Таким чином одержують 50 мг цільової сполуки брутто-формули C17H12N2 O4S (M=220,25 г), або вихід становить 25%. 1 H -ЯМР-спектр (250 МГц; CDCl3; хімічні зсуви піків в м.ч. і мультипл етність): 1.56 і 2,38 (м): N-CH-CH 2-CH; 3,00 (д), 3,12 (д) і 3,49 (м): N-CCH^J 3,26 (с): СН3; 4,12 (м): N-CHІЧ-спектр (СНСІ3): 1775; 1381, 1187 см-1. МС (ЕУ) m/z: [M]+=220, 141, 125, 97, 79. ПРИКЛАД 19g 6-[[(4-Нітрофеніл)сульфоніл]окси]-1,6-діазабіцикло[3,2,1]октан-7-он Слідують методиці, подібній такій, описаній в прикладі 19f, виходячи з 270 мг сполуки, одержаної в прикладі 19а, і 283 мг 4-нітробензолсульфонілхлориду. Таким чином одержують 205,5 мг цільової сполуки бруттоформули C12H13N3O 6S (М=327,32 г), або ви хід становить 54%. 1 H -ЯМР-спектр (250 МГц; CDCl3; хімічні зсуви піків в м.ч. і мультиплетність): 1,64 (д), 1,84 (м), 1,99 (м), 2,31 (дм): NCH-CH2-CH2; 2,94 (д), 3,30 (д), 3,04 (д), 3,40 (уш.дд): N(CH2)2; 4,14: O=C-N-CH; 8,25 і 8,41 [АА'ВВ1 ]: NO 2-C6H4SO 2. ІЧ-спектр (СНСl3): 1776; 1610, 1590, 1538; 1393, 1191 см"1. МС (EY)m/z: [M]+=327, 186, 141, 125, 111. ПРИКЛАД 20 6-[(4-Метилфеніл)суль фоніл]аміно]-1,6-діазабіцикло[3,2,1]октан-7-он 5 г (25,1 ммоль) 1,1-диметилетил-3-оксо-1-піперидинкарбоксилату (описується в J. Med. Chem., 29, 224229 (1986)) (C10H17NO3; М=199,251 г) розчиняють в 50 мл дихлорметану. Потім до розчину додають 4,67 г тозилгідразину і залишають реагувати протягом 2 годин при перемішуванні, потім розчинник випаровують при зниженому тиску. Таким чином одержують, з кількісним виходом, 9,56 г 1,1-диметилетил-3-[2-[(4метилфеніл)сульфоніл]гідразоно]-1 -піперидинкарбоксилату брутто-формули C17H25N3 O4S (M=367,47 г). В атмосфері інертного газу змішують 4,5 г (12,2 ммоль) одержаної вище сполуки, 90 мл суміші метанолу і тетрагідрофурану у співвідношенні 50:50 і декілька крупинок бромкрезолового зеленого. Потім додають 1,62 г NaBH3CN, потім охолоджують до температури близько 0-5°С і вводять, з метою збереження значення рН середовища в межах від 3,8 до 5,4, розчин газоподібного хлороводню в метанолі з концентрацією 0,7 моль/л. Реакцію проводять при перемішуванні протягом 2,5 годин. Випаровують при зниженому тиску 2/3 розчинників, потім додають 200 мл дихлорметану і промивають насиченим водним розчином гідрокарбонату натрію. Органічну фаз у сушать над суль фатом натрію і розчинник випаровують при зниженому тиску. Таким чином одержують 4,48 г 1,1-диметилетил-3-[2-[(4-метилфеніл)сульфоніл]гідразино]-1-піперидинкарбоксилату бруттоформули C17H27N3O 4S (М=369,486 г). Відповідний вихід становить 99%. В атмосфері інертного газу і при температурі 0°С змішують 4,48 г одержаної вище сполуки і 9 мл етилацетату. Додають ЗО мл розчину газоподібного хлороводню в етилацетаті з концентрацією 4 моль/л, перемішують протягом 15 хвилин, потім фільтрують і гідрохлорид промивають етилацетатом. Висушують при зниженому тиску і одержують 3,48 г дигідрохлориду 2-(3-піперидинил)гідразиду 4-метилбензолсульфо-кислоти бруттоформули C12H19N3O 2S.2HCl (М=342,289 г). Відповідний вихід становить 84%. Потім 3,48 г одержаної вище сполуки розчиняють в 5 мл демінералізованій води. При інтенсивному перемішуванні додають 10,2 мл 2н водного розчину гідроксиду натрію. Після контактування протягом 1-2 хвилин випадає осад. Потім перемішують протягом 10 хвилин, після чого осад відфільтровують і промивають його водою, потім етилацетатом. Одержану тверду речовину висушують при зниженому тиску. Таким чином одержують 2,21 г 2-(3-піперидиніл)гідразиду 4-метилбензолсульфокислоти брутто-формули C12H19N3O2S (М=269,328 г). Відповідний вихід становить 81%. В атмосфері інертного газу змішують 500 мг (1,85 ммоль) одержаного вище аміну і 20 мл тетрагідрофурану. До одержаної суспензії, при температурі від 0°С до 5°С, додають 112 мкл дифосгену, потім 517 мкл триетиламіну і 23 мг DMAP. Реакцію проводять при перемішуванні і підвищенні температури до 20°С. Потім розбавляють етилацетатом, після чого промивають 10%-ним водним розчином винної кислоти, потім демінералізованою водою. Органічну фазу сушать над сульфатом магнію, потім розчинник випаровують при зниженому тиску. Одержують 769 мг сирого продукту, який розчиняють в 7 мл дихлорметану і 517 мкл триетиламіну. Реакцію проводять протягом ночі при перемішуванні. Розбавляють дихлорметаном, промивають водою, сушать над сульфатом натрію і розчинник випаровують при зниженому тиску. Одержану піну (395 мг) очищають шляхом хроматографії на діоксиді кремнію за допомогою суміші дихлорметану і етилацетату у співвідношенні 80:20. Одержують 44 мг цільової сполуки брутто-формули C13H17N 3O2S (M=295,362 г). Відповідний вихід становить 8%. 1 H-ЯМР-спектр (250 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультиплетність): 1,55-1,80 (м) і 2,18 (м): N-CH-CH2-CH2; 2,42 (с): СН3; 2,88 (д) і 2,93 (м); N-СН2-СН; 3,18-3,32 (м): N-CH2-CH2; 4,08 (м): N-CH-CH2; 6,98 (уш.с): NH. ІЧ-спектр (СНСl3): 3264, 1737, 1599, 1490 см-1. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [M+Na]+=318; [М+Н]+=296. ПРИКЛАД 21 6-[(4-Метилфеніл)суль фоніл]-1,6-діазабіцикло[3,2Д]октан-7-он У 3 мл безводного дихлорметану розчиняють 305 мг (1,52 ммоль) 1,1-диметилетил-3-аміно-1 піперидинкарбоксилату (описується в J. Med. Chem., 35, 4334-4343 (1992)) брутто-формули C10H20N2 O2 (М=200,282 г). Потім додають 212мкл триетиламіну, після чого охолоджують до температури 5°С і додають 278 мг тозилхлориду. Перемішують до досягнення температури 20°С і залишають реакцію протягом 2 годин. Потім розбавляють дихлорметаном і промивають спочатку 10%-ним водним розчином винної кислоти, потім розчином фосфатного буфера з рН=7. Органічну фазу відділяють і сушать над сульфатом магнію, потім розчинник випаровують при зниженому тиску. Таким чином одержують масло, яке очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 9:1. Одержують 440 мг 1,1-диметилетил-3-[(4-метилфеніл)сульфоніл]аміно]-1-піперидинкарбоксилату (описується в J. Med. Chem., 35, 4334-4343 (1992)) брутто-формули C17H26N2O 4S (M=354,472 г). Відповідний вихід становить 82%. Суміш з 425 мг одержаної вище сполуки і 2,1 мл суміші трифтороцтової кислоти і дихлорметану у співвідношенні 50:50 охолоджують до температури 0-5°С. Перемішують протягом 30 хвилин при температурі 5°С. Потім розчинник випаровують при зниженому тиску, одержуючи 403 мг трифторацетату 4-метил-(3піперидиніл)бензолсульфонаміду брутто-формули C14H19 F3N2 O4S (M=368,377 г). 228 мг одержаної вище сполуки суспендують в 2 мл метанолу. Обробляють за допомогою надлишку активованої гідроксидом натрію смоли DOWEX 21 К, 20-50 меш. Відфільтровують, промивають смолу метанолом, потім фільтрат випаровують при зниженому тиску. Таким чином одержують 123 мг 4-метил-М-(3піперидиніл)бензолсульфонаміду брутто-формули C12H18N 2O2S (M=254,353 г). В атмосфері інертного газу 118 мг одержаного вище аміну розчиняють в 1,2 мл дихлорметану. Після цього послідовно вводять 98 мкл триетиламіну, потім 28 мкл дифосгену. Залишають реакцію при перемішуванні протягом 30 хвилин при температурі 0-5°С. Розбавляють дихлорметаном, органічну фазу промивають 10%ним водним розчином винної кислоти, потім водою. Після висушування над сульфатом натрію, фільтрації і випаровування розчинника при зниженому тиску сирий продукт очищають шляхом хроматографії на діоксиді кремнію, використовуючи як елююючий засіб суміш дихлорметану і ацетону у співвідношенні 95:5. Таким чином одержують 112 мг хлорангідриду 3-[[{4-метилфеніл)сульфоніл]аміно]-1-піперидинкарбонової кислоти брутто-формули C13H17CIN2O 3N (М=316,308 г). Відповідний вихід становить 76%. В інертній атмосфері 10 мг гідриду натрію (у вигляді 55-65%-ної суспензії в маслі) змішують з 2 мл безводного тетрагідрофурану. Потім додають 71 мг одержаних вище продукти. Перемішують при кімнатній температурі протягом 15 хвилин, потім додають 12 мкл оцтової кислоти і 2 мл розчину фосфатного буфера з рН=7. Перемішують ще протягом 5 хвилин, потім додають 5 мл етилацетату, декантують, потім повторно екстрагують етилацетатом. Після цього органічну фазу відділяють і сушать над сульфатом магнію, фільтрують і розчинник випаровують при зниженому тиску. Таким чином одержують 65 мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і ацетону у співвідношенні 95:5. Таким чином одержують 40 мг цільової сполуки брутто-формули C 13H16N2O 3S (М=280,348 г). Відповідний вихід становить 64%. 1 H -ЯМР-спектр (250 МГц; CDCI3; хімічні зсуви піків в м.ч. і мультиплетність) (в присутності двох ізомерів у співвідношенні 90/10): 1,46 (м), 1,76 (м) і 2,08 (дм): NCH-CH2-CH2; 2,44 (с) і 2,45 (с): СН3; 2,82 (д) і 2,98 (м) і 3,28-3,50 (м): -N(CH2)2; 4,55 (м) і 4,65(м): CO-N-CH; 7,33 і 7,78, 7,35 і 8,02 [АА'ВВ'1 CH 2G;H4-SCl2. ІЧ-спектр (СНСl3): 1758, 1598, 1995, 1367, 1169 см -1. МС (ЕУ) m/z: [M]+=280, 216, 155, 125, 97, 91. ПРИКЛАД 22 6-Окса-1 -азабіцикло [3,2,1 ]окт-3-ен-7-он В атмосфері інертного газу змішують 5 мл дихлорметану і 68 мг гідрохлориду 1, 2, 3,6-тетрагідропіридинЗ-олу (М=135,5 г) (описується в Chem. Pharm. Bull., 30 (10), 3617-3623 (1982)). Додають 33 мкл дифосгену і перемішують протягом 5 хвилин при температурі 0°С. Потім додають 140 мкл триетиламіну і 61 мг DMAP. Залишають реакцію при кімнатній температурі протягом 2 годин, потім розбавляють дихлорметаном і промивають 10%-ним водним розчином винної кислоти, потім водою. Органічну фазу декантують і сушать над сульфатом магнію. Розчинник випаровують при зниженому тиску. Таким чином одержують 5 мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи дихлорметаном, потім сумішшю дихлорметану і етилацетату у співвідношенні 95:5. Таким чином одержують 3 мг цільової сполуки брутто-формули C6H7NO2(M=125 г). Відповідний вихід становить 5%. ПРИКЛАД 23 Фенілметил-транс-3-бензоїл-2-оксо-4-окса-1,3-діазабіцикло[3,2,1]октан-7-карбоксилат В атмосфері інертного газу 5,50 г (13,7 ммоль) 1-( 1,1-дим етил етил)- і 2-(фенілметил)-цис-4[(метилсульфоніл)окси]-1,2-піролідиндикарбоксилату (описується в J. Org. Chem., 56, 3009-3016 (1991)) брутто-формули C18H25NO7S (М=399,466 г) змішують зі ПО мл диметилформаміду, потім додають 2,58 г Nгідроксифталиміду, потім 1,52 г гідрокарбонату калію. Нагрівають при перемішуванні до температури 100°С і реакційну суміш витримують при цій температурі протягом 4 годин. Охолоджують до температури 20°С, додають 220 мл води і льоду, потім екстрагують діізопропіловим ефіром. Сушать над сульфатом магнію, потім випаровують досуха при зниженому тиску. Залишок хроматографують на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 90:10. Таким чином одержують 3,06 г 1-(1,1-диметилетил)- і 2(фенілметил)-транс-4-[(1,3-дигідро-1,3-диоксо-2Н-ізоіндол-2-іл)окси]-1,2-піролідиндикарбоксилату бруттоформули C25H26N2O 7 (М=466,494 г). Відповідний вихід становить 47%. 3,24 г (6,94 ммоль) одержаного вище фталіміду розчиняють в 33 мл дихлорметану. Додають 372 мкл гідразингідрату. Перемішують ще протягом 2 годин ЗО хвилин при температурі 20°С. Осад, що випав, відфільтровують, промивають дихлорметаном, потім розчинник випаровують при зниженому тиску. Одержують 2,91 г сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю ди хлорметану і етилацетату у співвідношенні 90:10, потім 80:20 і 50:50. Таким чином виділяють в цілому 942 мг 1 -(1,1 диметилетил)і 2-(фенілметил)-транс-4-(аміноокси)-1,2-піролідин-дикарбоксилату брутто-формули C17H24N2O5 (М=336,39 г). Відповідний вихід становить 40%. В атмосфері інертного газу 853 мг (2,53 ммоль) одержаної вище сполуки змішують з 8,5 мл безводного дихлорметану. Охолоджують до температури близько 0-5°С, потім додають 706 мкл триетиламіну і 588 мкл бензоїлхлориду. Перемішують протягом 10 хвилин при температурі 0-5°С, потім залишають знов нагріватися до температури 20°С і залишають реакцію протягом 30 хвилин. Органічну фазу промивають 10%-ним водним розчином винної кислоти, потім водою. Органічну фазу декантують і сушать над сульфатом натрію, розчинник випаровують при зниженому тиску. Таким чином одержують 1,38 г продукти, які змішують з 25 мл дихлорметану. Охолоджують до температури 10-15°С і додають 123 мкл гідразингідрату. Реакцію проводять при перемішуванні і при температурі 20 °С протягом двох з половиною годин. Розчинник випаровують при зниженому тиску. Таким чином одержують 1,13 г сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 80:20. Одержують 948 мг 1-( 1,1 диметилетил)- і 2-(фенілметил)-транс-4-[(бензоїламіно)окси]-1,2-піролідиндикарбоксилату брутто-формули C24H28N2O6 (М=440,50 г). Загальний вихід, отже, становить 85%. При перемішуванні 948 мг одержаної вище сполуки розчиняють в 2 мл етилацетату. Охолоджують до температури 0-5°С, потім додають за один раз 4,7 мл приблизно 4,6 Μ розчину газоподібного хлороводню в етилацетаті. Через 1 годину розчинник випаровують при зниженому тиску і продукт обробляють 3 рази діетиловим ефіром. Розчинник випаровують при зниженому тиску. Таким чином одержують 842 мг гідрохлориду фенілметил-транс-4-[(бензоїламіно)окси]-2-піролідинкарбоксилату у вигляді крихкої білої піни формули C19H20N2O4.HCI (М=376,84 г). Ви хід є кількісним. В атмосфері інертного газу 47 мг (0,125 ммоль) одержаного вище гідрохлориду розчиняють в 0,5 мл дихлорметану. Додають 25,2 мкл піридину, потім охолоджують до температури 0-5°С і додають 9,5 мкл дифосгену. Залишають температуру підвищуватися до 20°С, розбавляють дихлорметаном, потім реакційне середовище промивають 10%-ним водним розчином винної кислоти, потім водою. Органічну фазу декантують і сушать її над сульфатом натрію. Потім розчинник випаровують при зниженому тиску. Таким чином одержують 43,8 мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю ди хлорметану і етилацетату у співвідношенні 90:10. Одержують 34,9 мг фенілметил-транс-4-[(бензоїламіно}окси]1 -(хлоркарбоніл)-2-піролідинкарбоксилату брутто-формули C20H19CIN 2O5 (М=402,83 г). Відповідний вихід становить 69%. 13 мг (0,032 ммоль) одержаної вище сполуки розчиняють в 4 мл толуолу. Додають 9 мкл триетиламіну і 7,8 мг DMAP. Нагрівають при температурі 100°С протягом ночі. Розчинник випаровують при зниженому тиску, потім залишок очищають шляхом хроматографії, елююючи дихлорметаном. Таким чином одержують 4,3 мг цільової сполуки брутто-формули C20H18N2O 5 (M=336,37 г). Відповідний вихід становить 40%. 1 H -ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і мул ьтипл етність): 1,97 (ддд) і 2,85 (ддд): N-O-CH-CH 2-CH; 3,80 (дд) і 4,14 (дд): N-O-CH-CH 2-N; 4,75 (дд): N-CH-CH 2; 4,93 (τ): N-O-CH-CH2; 5,04 і 5,31 [АВ]: О-СН 2-С6Н5; 7,77: і 7,25-7,50 (м) СН2-С6Н5 і ОС-С 6Н5. ІЧ-спектр (СНСІ3): 1735; 1612, 1575, 1496 см -1. ПРИКЛАД 24 3-Бензоїл-1,3-діазабіцикло[2,2,2]октан-2-он В атмосфері азоту 2,4 г (10 ммоль) гідрохлориду №-(4-піперидиніл)бензаміду (описується в J. Med. Chem. EN., 17, 736-739 (1974)) брутто-формули C12H, 6N2O розчиняють в 30 мл дихлорметану. Охолоджують до температури 0°С, при перемішуванні додають 2,8 мл триетиламіну і 0,66 мл дифосгену. Через декілька хвилин розбавляють дихлорметаном, потім промивають 10%-ним водним розчином винної кислоти, потім водою. Органічну фазу декантують, сушать її над сульфатом магнію і розчинник випаровують при зниженому тиску. Залишок очищають на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 90:10. Одержують 1,62 г хлорангідриду 4-(бензоїламіно)-1-піперидинкарбонової кислоти брутто-формули C13H15CIN2O5 (М=266,5 г). Відповідний вихід становить 61%. В атмосфері азоту 1,21 г (48 ммоль) одержаної вище сполуки розчиняють в 37 мл тетрагідрофурану. Розчин охолоджують до температури -78°С, потім додають краплями 5 мл 1 Μ розчину біс(триметилсиліл)аміду літію в тетрагїдрофурані. Витримують при температурі -78°С протягом 15 хвилин, потім залишають для підвищення температури до кімнатної і продовжують реакцію ще протягом години. Розчин охолоджують до температури 0°С, додають 720 мкл оцтової кислоти. Випадає осад. Розбавляють етилацетатом, потім промивають 10%-ним водним розчином винної кислоти і розчином фосфатного буфера з рН=7,0. Органічну фазу декантують і сушать її над сульфатом магнію. Фільтрують, потім розчинник випаровують при зниженому тиску. Сирий продукт очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 90:10. Таким чином одержують 0,214 г цільової сполуки формули Ci8Hi4N2O2 (M=230 г), кристалізованої з діетилового ефіру. Відповідний вихід становить 20%. 1 H -ЯМР-спектр (250 МГц; диметилсульфоксид; хімічні зсуви піків в м.ч. і м ультиплетність): 1,71-2,02 (м): (CH2)2-CHN; 3,14 (τ): N-(CH 2)2; 4,84 (м): (CH 2)2-CHN; 7,39-7,65 (м): С6Н5· ІЧ-спектр(СНСІ3): 1735, 1682; 1618, 1602, 1582; 1488 см'1. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [2 M+Na]+=483; [M+Na+CH3CN]+=294; [M+Na]+=253. ПРИКЛАД 25 Дифенілметил-транс-7-оксо-6-окса-1-азабіцикло[3,2,1]октан-2-карбоксилат В інертній атмосфері змішують 15 мл дихлорметану і 197 мг (0,633 ммоль) дифенілметил-транс-5гідрокси-2-піперидинкарбоксилату (описується в Rec. Tra v. Chim., 78,648-658 (1959)) брутто-формули C19H21NO3 . Охолоджують до температури 0°С, потім послідовно додають 42мкл дифосгену, 177 мкл триетиламіну, потім 77 мг DMAP. Залишають реакцію протягом 4 годин при кімнатній температурі. Потім реакційну суміш промивають 10%-ним водним розчином винної кислоти, потім водним насиченим розчином хлориду натрію. Органічні фази об'єднують і суша ть їх над сульфатом магнію, розчинник випаровують при зниженому тиску і таким чином одержують 195 мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи дихлорметаном, що містить 0,1% води. Одержують масло, яке кристалізують з суміші пентану і діетилового ефіру. Таким чином виділяють 108 мг цільової сполуки у вигляді кристалів білого кольору, відповідної брутто-формулі C20H19NO 4 (N=337,338 г). Відповідний вихід становить 51%. 1 H -ЯМР-спектр (400 МГц; CDCl3; хімічні зсуви піків в м.ч. і мультиплетність): 1,86 (м) і 2,03 (м): N-CH-CH2-CH2-CO; 2,27 (м): N-CH-CH2-CH2-CO; 3,07 (д) і 3,29 (м): N-CH 2-CHO; 4,31 (дд): N-CH-CH2; 4,73 (м): N-CH 2-CHO; 6,93 (с): СО2-СН-(С6Н5)2; 727-7,41 (м): СН(С 6Н5)2. ІЧ-спектр (СНСl3): 1788, 1736; 1496 см"1. МС (SIMS) m/z: [M+Na]+=360; [M+Li]+=344; [M]+=337, 167. ПРИКЛАД 26а (4-Нітрофеніл)метил-транс-7-оксо-6-окса-1-азабіцикло[3,2,1]октан-2-карбоксилат В інертній атмосфері 66 мл дихлорметану змішують з 1 г (3,56 ммоль) (4-нітрофеніл)метил-транс-5гідрокси-2-піперидинкарбоксилату брутто-формули C13H16N2O5 (М=280,282 г). Охолоджують до температури 0°С і додають 0,24 мл дифосгену. Реакцію проводять при перемішуванні протягом 10 хвилин при температурі 0°С, потім залишають нагріватися до кімнатної температури. Розчинник випаровують при зниженому тиску. Залишок розчиняють в 66 мл толуолу і додають 0,99 мл триетиламіну. Колбу занурюють в масляну баню з температурою 110°С і витримують там протягом 15 хвилин. Потім доводять температур у до кімнатної. Промивають 10%-ним водним розчином винної кислоти, потім насиченим водним розчином хлориду натрію. Органічну фаз у сушать над сульфатом магнію, потім розчинник випаровують при зниженому тиску. Таким чином одержують 0,885 г сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю толуолу і етилацетату у співвідношенні 85:15. Таким чином одержують 0,184 г цільової сполуки брутто-формули C14H14N2O 6 (М=306,276 г) у вигляді масла жовтого кольору. Відповідний вихід становить 17%. 1 H -ЯМР-спектр (400 МГц; CDCl3; хімічні зсуви піків в м.ч. і мультиплетність): 1,92 (м) і 2,07 (м): N-CH-CH 2-CH2-CO; 2,22 (м) і 2,30 (м): N-CH-CH 2-CH2-CO; 3,17 (д) і 3,35 (дм): N-CH 2-CHO; 4,28 (дд): N-CH-CH 2; 4,79 (м): N-CH 2-CHO; 5,33 [АВ]: CO 2-CH2-C6H4NO2 ; 7,56 і 8,25 [АА'ВВ1 ]: CH2-C6H 4NO2. ІЧ-спектр (СНСl3): 1791, 1745; 1609, 1526, 1495 см -1. МС (ЕУ) m/z: [M]+=306, 262, 136, 126, 82, 55. ПРИКЛАД 26b транс-7-Оксо-6-окса-1-азабіцикло[3,2,1]октан-2-карбонова кислота Змішують 140 мг (0,457 ммоль) складного ефіру, одержаного в прикладі 26а, 7 мл ацетону і 28 мг каталізатора 20 мас.%-ного паладію-на-вугіллі. Потім реакцію проводять при перемішуванні протягом 25 хвилин в атмосфері водню при нормальному тиску. Каталізатор відфільтровують і розчинник потім випаровують при зниженому тиску. Таким чином одержують 137 мг цільової сполуки брутто-формули C 7H9NO4 (М=171,152 г) у вигляді масла, в суміші з одним молем п-толуідіну. Відповідний вихід становить 97%. 1 H -ЯМР-спектр (400 МГц; диметилсульфоксид; хімічні зсуви піків в м.ч. і м ультиплетність): 1,84 (м) і 1,95-2,05 (м): N-CH-CH2-CH2-CO; 3,13 (д) і 3,24 (дд): N-CH2-CHO; 4,02 (дд): N-CH-CH 2; 4,81 (дм): N-CH2-CHO. ПРИКЛАД 26с Метил-транс-7-оксо-6-окса-1 -азабіцикло[3,2,1]октан-2-карбоксилат 17,25 мг (0,1 ммоль) одержаної в прикладі 26Ь кислоти розчиняють в 3 мл дихлорметану. Обробляють надлишком діазометану у вигляді розчину в ди хлорметані, потім розчинник випаровують при зниженому тиску. Таким чином одержують ЗО мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю толуолу і етилацетату у співвідношенні 90:10. Одержують 6,7 мг цільової сполуки (М=485,187 г). Відповідний вихід становить 36%. ПРИКЛАД 27 (4-Нітрофеніл)метил-цис-7-оксо-6-окса-1-азабіцикло[3,2,1]октан-2-карбоксилат В атмосфері азоту в 40 мл дихлорметану вводять 0,802 г (2,034 ммоль) трифторацетату (4нітрофеніл)метил-цис-5-гідрокси-2-піперидинкарбоксилату (описується в Rec. Trav. Chim., 78,648-658 (1959)) брутто-формули C13H16N2 O5 . CF3CO2H (M=394, 303 г) і охолоджують до температури 0°С. Додають 0,135 мл дифосгену. Перемішують протягом 15 хвилин при температурі 0°С, температуру підвищують до кімнатної і продовжують перемішування протягом 35 хвилин. Розчинник випаровують при зниженому тиску. Цей продукт розчиняють в 40 мл толуолу і 1,1 мл триетиламіну. Реакційну суміш протягом 35 хвилин витримують при температурі 100°С, потім залишають охолоджуватися до кімнатної температури. Промивають водою, потім розчином фосфатного буфера з рН=7. Органічну фазу сушать над сульфатом натрію і розчинник випаровують при зниженому тиску. Таким чином одержують 0,56 г сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю ди хлорметану і ацетону у співвідношенні 95:5. Таким чином одержують ПО мг цільової сполуки брутто-формули C14H14N2O6 (М=306,275 г) у вигляді масла. Відповідний вихід становить 17%. 1 H -ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і мультиплетність): 1,80-1,94 і 2,10-2,45: N-CH-CH2-CH2-CO; 3,07 (д), 3,04 (дм) і 3,86 (дд): CH-N-СН2; 4,80 (т): О-С-О-СН; 5,28 і 5,43 [АВ]: О=С-О-СН 2-С6Н5 ; 7,61 і 8,24 [АА'ВВ 1] C6H 4NO2. ІЧ-спектр(СНСІ3): 1801, 1794, 1745, 1704; 1609, 1525, 1498 см"1. МС (ЕУ) m/z: [M]+=306, 262, 136, 126, 83, 55. ПРИКЛАД 28а 1 -Пропенілтрифенілфосфонієва сіль фенілметил-транс-7-оксо-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан2-карбоксилату Стадія А: Фенілметил-цис-5-гідрокси-1-(трифторацетил)-2-піперидинкарбоксилат В інертній атмосфері 6,19 г (22,77 ммоль) гідрохлориду фенілметил-5-гідрокси-2-піперидинкарбоксилату бруттоформули C13H18ClNO3 (M=271,746 г) (описується в Rec. Trav. Chim., 78,648-658 (1959))розчиняють в 80 мл безводного дихлорметану. Охолоджують до температури 5°С і додають 9,5 мл триетиламіну, потім, краплями, 6,46 мл трифтороцтового ангідриду. Реакцію ведуть при перемішуванні при температурі 5 °С протягом однієї години, потім розбавляють дихлорметаном, промивають послідовно 10%-ним розчином винної кислоти, водним розчином фосфа тного буфера з рН=7 і водним розчином хлориду натрію. Органічну фазу декантують і сушать її над сульфатом магнію. Потім розчинник випаровують при зниженому тиску. Таким чином одержують 10 г масла червоного кольору, які розчиняють в 100 мл метанолу. Охолоджують до температури близько 10°С і повільно, при температурі максимальне 20°С, додають 6,8 г (78 ммоль) гідрокарбонату натрію у вигляді розчину в 100 мл води. Реакцію ведуть при перемішуванні і при температурі 20°С протягом 30 хвилин, екстрагують дихлорметаном. Органічну фазу декантують, промивають її водним насиченим розчином хлориду натрію і сушать її над сульфатом магнію. Розчинник випаровують при зниженому тиску і таким чином одержують 7,6 г масла оранжевого кольору, які очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 95:5. Таким чином одержують 6 г цільової сполуки брутто-формули C15H16F 3NO4 (M=331,294 г). Відповідний вихід становить 68%. Стадія В: Фенілметил-транс-5-[(2-пропенілокси)аміно]-1-(трифторацетил)-2-піперидинкарбоксилат У 29 мл ацетонітрилу вводять 1,74 г (5,26 ммоль) одержаного вище спирту. Охолоджують до температури -40°С і при цій температурі додають 0,61 мл 2,6-лутидину (C5H3N(CH3)2), потім 0,91 мл ангідриду трифторметансульфокислоти. Реакцію ведуть при перемішуванні протягом 30 хвилин при температурі -40°С. Потім додають, весь час при температурі -40°С, протягом однієї хвилини 0,7 мл (10,52 ммоль) О-алілгідроксиламіну. Доводять температуру до 0°С, потім додають 0,61 мл 2,6-лутидину і ви тримують реакційну суміш протягом ночі (15 годин) при температурі близько 5°С, потім ще 2 години при температурі 20°С. Після цього розбавляють дихлорметаном, промивають водним розчином гідрокарбонату натрію, потім 10%-ним водним розчином винної кислоти і водним насиченим розчином хлориду натрію. Органічну фазу декантують, сушать її над сульфатом магнію і розчинник випаровують при зниженому тиску. Таким чином одержують 2,1 г масла жовтого кольору, які очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю толуолу і етилацетату у співвідношенні 90:10. Одержують 1,23 г цільової сполуки брутто-формули C18H21F3N2O4 (М=386,374 г). Відповідний вихід становить 61%. Стадія С: Фенілметил-транс-5-[(2-пропенілокси)аміно]-2-піперидинкарбоксилат В інертній атмосфері 1,41 г (3,65 ммоль) одержаної вище сполуки розчиняють в 25 мл безводного метанолу. Охолоджують до температури 0-5°С, потім здійснюють 3 додавання, з проміжками в 45 хвилин, 145 мг NaBH,4. Реакційне середовище потім підкисляють до рН=2 1н водним розчином соляної кислоти, заздалегідь охолодженої до 5°С. Екстрагують етилацетатом. Водну фазу о холоджують до температури 5 °С, додають 100 мл етилацетату і обробляють насиченим розчином карбонату натрію аж до досягнення рН, рівного 8,5-9. Потім амін екстрагують етилацетатом. Органічну фазу промивають водним насиченим розчином хлориду натрію, потім сушать над сульфатом магнію і концентрують шляхом випаровування розчинника при зниженому тиску. Таким чином одержують 0,628 г цільового продукту брутто-формули C 16H22N2O3 (M=290,364 г). Відповідний вихід становить 59%. Стадія D: Фенілметил-транс-7-оксо-6-(2-пропенілокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксилат В інертній атмосфері 103 мг (0,35 ммоль) одержаного вище аміну розчиняють в 35 мл безводного дихлорметану. Розчин охолоджують до температури близько 0-5 °С і при цій температурі додають краплями 0,1 мл триетиламіну, потім 21 мкл дифосгену. Реакцію ведуть при перемішуванні протягом 15 хвилин при температурі 0-5°С, потім залишають для підвищення до 20°С і додають 42 мг DMAP. Продовжують перемішування при температурі 20°С протягом приблизно 5 годин. Розбавляють дихлорметаном, промивають 10%-ним водним розчином винної кислоти, потім водою. Органічну фазу сушать над сульфатом магнію і концентрують шляхом випаровування розчинника при зниженому тиску. Таким чином одержують 70 мг сирого продукту, який очищають шляхом хроматографії на 5 г діоксиду кремнію, елююючи сумішшю дихлорметану і метанолу у співвідношенні 98:2. Одержують 48 мг цільового продукту формули Q7H20N2O4 (М=316,36 г). Відповідний вихід становить 43%. ІЧ-спектр(СНСІ3): 1750; 1642; 1600, 1496 см-1. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [M+Na+CH 3CN]+=380; [M+Na]+=339; [M+H]+=317. Стадія Ε: 1 -Пропенілтрифенілфосфонієва сіль фенілметил-транс-7-оксо-6-(сульфокси)-1,6-діазабіцикло[3,2,1 ]октан-2-карбоксилату В інертній атмосфері 202 мг (0,638 ммоль) одержаної на стадії D сполуки розчиняють в 5,5 мл безводного дихлорметану. До одержаного розчину при температурі 20°С додають 73 мкл оцтової кислоти, потім 369 мг Ра[Р(С6Н5)3] 4· Після перемішування протягом 30 хвилин при кімнатній температурі Ν-гідроксисечовину, що утворилася, обробляють за допомогою 5,5 мл піридину і 358 мг комплексу 8Оз-піридин. Реакцію ведуть при перемішуванні протягом 18 годин при температурі 20°С, потім реакційне середовище концентрують шляхом випаровування розчинника при зниженому тиску. Залишок обробляють за допомогою 50 мл дихлорметану і промивають водою. Органічну фазу сушать над сульфатом магнію і дихлорметан випаровують при зниженому тиску. Таким чином одержують 650мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і ацетону у співвідношенні 60:40, що містить 0,1 об'ємн.% триетиламіну. Таким чином одержують 280мг фосфонієвої солі цільової сполуки брутто-формули C35H35N2O7PS (М=646,705 г). Відповідний вихід становить 68%. 1 H-ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультиплетність): 2,05 (м), 2,22 (дм) і 2,33 (м): N-CH-CH 2Ob; 2,95 (д) і 3,30 (д); O=C-N-CH 2; 4,10 (м) і 4,32 (μ): OC-N-CH і 0=C-N-CH 2-CH; 5,12 (с): СОО-СН 2-С6Н5; 7,36: С 6Н5 і 2,30 (м): СН3-СН=СН; 6,65 і 7,20 СН3-СН=СН; 7,65-7,85 Р(С6Н5)3. ІЧ-спектр (СНСІ3): 1746; 1638, 1605, 1587, 1495 см-1. МС (іонізація електронним розпиленням з утворенням позитивних і негативних іонів) m/z: [Μ аніон]"=355; [Μ катіон]+=303. ПРИКЛАД 28b Фенілметил-транс-7-оксо-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксилат натрію 236 мг (0,364 ммоль) фосфонієвой солі, одержаної на стадії Ε прикладу 28а, розчиняють в 0,8 мл тетрагідрофурану і 4 краплях води. Одержаний розчин пропускають через колонку зі смолою DOWEX 50WX8 в Ма+формі, елююючи водою. Після ліофілізації одержують 127 мг цільової натрієвої солі брутто-формули C14H15N2 O7SNa (M=378,339 г). Відповідний вихід становить 92%. 1 H -ЯМР-спектр (300 МГц; диметилсульфоксид; хімічні зсуви піків в м.ч. і м ультиплетність): 1,65-2,02: N-CH-CH2-CH2; 2,91 (д) і 3,04 (д): O=C-N-CH 2; 4,00-4,05: OC-N-CH і O=C-N-CH2-CH; 5,20 [АВ]: СОО-СН2-С6Н5; 7,39 (м): С6Н5. ІЧ-спектр (вазелінове масло): 1744; 1495 см -1. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]+=355. ПРИКЛАД 28с Фенілметил-транс-7-оксо-6-[(фенілсульфоніл)окси]-1,6-діазабіцикло[3,2,1] октан-2-карбоксилат 48 мг (0,152 ммоль) похідного, одержаного на стадії D прикладу 28а, розчиняють в 1,2 мл дихлорметану. При температурі 20°С до одержаного розчину додають 26 мкл оцтового ангідриду, потім 88 мг Ра[Р(С6Н5)3]4 і реакцію ведуть протягом 2 годин при температурі 20°С і при перемішуванні. Розбавляють шляхом додавання толуолу і розчинники випаровують при зниженому тиску. До одержаного сирого продукту додають 1,5 мл дихлорметану, 25мкл піридину і 24мкл бензолсульфонілхлориду. Реакцію ведуть при температурі 20°С і при перемішуванні протягом 1 години, потім додають 12,5 мкл піридину і 10 мкл бензолсульфонілхлориду. Перемішують протягом 15 хвилин при температурі 20°С і розбавляють дихлорметаном. Потім послідовно промивають 10%-ним водним розчином винної кислоти, розчином фосфатного буфера з рН=7 і водним насиченим розчином хлориду натрію. Органічну фазу сушать над сульфа том магнію і розчинник випаровують при зниженому тиску. Одержують 180 мг масла жовтого кольору, яке очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю ди хлорметану і трет-бутилметилового ефіру у співвідношенні 95:5. Таким чином одержують 20 мг цільової сполуки брутто-формули C20H20N2O 6S (M=416,456 г). Відповідний вихід становить 31%. 1 H -ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і мультиплетність): 1.83 (м) і 2,00-2,25 (м): N-CH-CH2-CH2; 3,02 (д) і 3,16 (дм): OON-CI3; 4,04 (м) і 4,11 (дд): O=C-N-CH і O=C-NCH2-CH; 5,21 (с): СОО-СН2-С6Н5; 7,34 (м): Сб Н5; 7,56 (м), 7,70 (м) і 8,03 (м): O2S-C6H5. ІЧ-спектр(СНСІ3): 1780, 1738; 1600, 1585, 1498; 1386, 1193 см -1. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [2 M+Na]+=855; [M+Na+CH3CH]+=480; [M+Na]+=439; [MH]+=417. ПРИКЛАД 28d Фенілметил-транс-7-оксо-6-[(2-тієнілсульфоніл)окси]-1,6-діазабіцикло [3,2,1]октан-2-карбоксилат Виходячи з 100 мг (0,316 ммоль) сполуки, одержаної на стадії D прикладу 28а, слідують методиці, подібній щойно описаної, за винятком того, що замість бензолсульфонілхлориду використовують 2тиенилсульфонілхлорид. Таким чином одержують 8 мг цільової сполуки брутто-формули Ci8Hi8N2O6S2 (М=422,481 г). Відповідний вихід становить 30%. 1 H -ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і мультиплетність): 1.84 (м) і 2,10-2,25: N-CH-CH2-CH2; 3,02 (д) і 3,24 (д): O=C-N-CH2; 4,06 (м): O=C-N-CH 2-CH; 4,14 (дд): O=CN-CH; 5,22 (с): СОО-СН2-С6Н5; 7,17 (дд): SO3-C-S-СН=СН; 7,35 (уш.с): Сб Н5; 7,80 (дд): S03-OCH; 7,87 (м): SO3C-S-CH. ІЧ-спектр (СНСІ3): 1780, 1739; 1600, 1503, 1495 см'1 МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [M+Na+CH3CN]+=867; [2M+Na]+=445; 339, 298, 91. ПРИКЛАД 28е Фенілметил-транс-6-(2-гідрокси-2-оксоетокси)-7-оксо-1,6-діазабіцикло [3,2,1]октан-2-карбоксилат Стадія А: Фенілметил-транс-7-оксо-6-[2-оксо-2-(2-пропенілокси)етокси]-1,6-діазабіцикло[3,2,1]октан-2-карбоксилат В інертній атмосфері 48 мг (0,15 ммоль) сполуки, одержаної на стадії D прикладу 28а, розчиняють в 1,5 мл безводного дихлорметану. При температурі 20°С додають 18 мкл оцтової кислоти, потім 88 мг Рd[Р(С6Н5)3]4 і перемішують протягом 1 години при температурі 20°С. Хроматографують на діоксиді кремнію, елююючи сумішшю ди хлорметану і метил-трет-бутилового ефіру у співвідношенні 7:3. Розчинник випаровують при зниженому тиску і одержують 70мг гідроксисечовини, яку обробляють за допомогою 2 мл дихлорметану, потім додають 85 мкл триетиламіну і 64мкл алілбромацетату. Перемішують протягом трьох з половиною годин при температурі 20°С. Промивають послідовно 10%-ним водним розчином винної кислоти, водним розчином фосфатного буфера з рН=7 і водою. Органічну фазу сушать і розчинник випаровують при зниженому тиску. Таким чином одержують 60 мг сирого продукту, якого хроматографують на діоксиді кремнію, елююючи сумішшю дихлорметану і метил-трет-бутилового ефіру у співвідношенні 90:10, що містить 0,1% триетиламіну. Одержують 22 мг продукту брутто-формули C19H22N2O6 (M=374,396 г). Відповідний вихід становить 39%. Стадія В Фенілметил-транс-6-(2-гідрокси-2-оксоетокси)-7-оксо-1,6-діазабіцикло [3,2,1]октан-2-карбоксилат В інертній атмосфері 22 мг (0,0587 ммоль) одержаної вище сполуки розчиняють в 1 мл безводного дихлорметану. При температурі 20 °С додають 10 мкл оцтової кислоти і 34 мг Pd[P(C6H5)3]4 і реакцію ведуть при перемішуванні протягом 30 хвилин при температурі 20°С. Реакційне середовище концентрують і обробляють толуолом для видалення оцтової кислоти. Таким чином одержують 49 мг сирого продукту, до якого додають 2 мл фосфатного буфера з рН=7, потім промивають 2 рази за допомогою 1 мл дихлорметану. Розчинник випаровують і одержують 46 мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи спочатку сумішшю дихлорметану і метилом-трет-бутилового ефір у у співвідношенні 90:10, потім сумішшю дихлорметану і етанолу у співвідношенні 60:40. Таким чином одержують 4,5 мг цільової сполуки бруттоформули C37H37N2O 6P (М=636,691 г). Відповідний вихід становить 12%. ПРИКЛАД 29а (4-Нітрофеніл)метил-транс-6-бензоїл-7-оксо-1,6-діазабіцикло[3,2,1]октан-2-карбоксилат Стадія А: 1-(1,1-Диметилетил)- і 2-[(4-нітрофеніл)метил]-цис-5-(метилсульфоніл)окси-1,2-піперидиндикарбоксилат В інертній атмосфері в 112 мл дихлорметану розчиняють 11,25 г (29,5 ммоль) 1 -(1,1 -диметилетил)- і 2[(4-нітрофеніл)метил]-цис-5-гідрокси-1,2-піперидин-дикарбоксилату (описується в Rec. Tra v. Chim., 78,648-658 (1959)) брутто-формули C18H24N2O 7 (М=380,398 г). Охолоджують до температури 0-5°С, потім послідовно вводять 5 мл триетиламіну, потім 2,44 мл метансульфонілхлориду. Температуру доводять до 20°С при перемішуванні і реакцію ведуть таким чином протягом 1 години. Потім розбавляють дихлорметаном, промивають два рази водою, сушать над сульфатом натрію і розчинник випаровують при зниженому тиску. Таким чином одержують 16 г сирого масла, яке очищають шляхом хроматографії на діоксиді кремнію, елююючи дихлорметаном, що містить 2% етилацетату. Одержують 9,14 г цільового продукту брутто-формули C 19H26N2 O9S (М=458,491 г). Відповідний вихід становить 67%. Стадія В: 1-( 1,1 -Диметилетил)- і 2-[(4-нітрофеніл)метил]-транс-5-азидо-1,2-піперидиндикарбоксилат В інертній атмосфері 11,1 г (24,2 ммоль) одержаного вище мезилату розчиняють в 111 мл диметилформаміду. Потім додають 1,73 г азиду натрію NaN3. Нагрівають при перемішуванні до температури 80°С і витримують при цій температурі протягом 18 годин. Температуру доводять до 20°С, потім диметилформамід випаровують при зниженому тиску аж до одержання незначного об'єму, потім розбавляють етилацетатом і промивають 2н розчином гідроксиду натрію, потім водою. Сушать над сульфа том магнію, потім розчинники випаровують при зниженому тиску. Одержане сире масло очищають шляхом хроматографії на діоксиді кремнію, елююючи дихлорметаном, що містить 2% етилацетату. Таким чином одержують 7,34 г цільової сполуки брутто-формули C18H23N5O6 (М=405,413 г) у вигляді масла жовтого кольору, яке кристалізується. Відповідний вихід становить 75%. Стадія С: 1-(1,1-Диметилетил)- і 2-[(4-нітрофеніл)метил]-транс-5-аміно-1,2піперидиндикарбоксилат 7,34 г (18,1 ммоль) одержаного вище азиду вводять в 150 мл тетрагідрофурану і ЗО мл води. Додають 7,2 г трифенілфосфін у, потім реакцію ведуть при перемішуванні притемпературі 20°С протягом ночі. Після цього розчинник випаровують при зниженому тиску і здійснюють дві обробки з відганянням при використанні етилацетату. Таким чином одержують сухий екстракт, який очищають шляхом хроматографії на діоксиді кремнію, елююючи дихлорметаном, що містить 5% метанолу. Одержують 5,62 г цільової сполуки брутто-формули C18H25N3 O6 (M=379,416 г). Відповідний вихід становить 82%. Стадія D: 1 -(1,1 -Диметилетил)- і 2-[(4-нітрофеніл)метил]-транс-5-(бензоїламіно)-1,2-піперидиндикарбоксилат 700 мг (1,84 ммоль) одержаного вище аміну розчиняють в 8 мл дихлорметану. Охолоджують до температури 0°С, потім вводять 257 мкл триетиламіну, потім 214 мкл бензоїлхлориду. Температуру доводять до 20°С. Після протікання реакції протягом 40 хвилин розбавляють дихлорметаном, промивають насиченим розчином гідрокарбонату натрію, потім водою. Сушать над сульфатом натрію, розчинник випаровують при зниженому тиску. Таким чином одержують 867 мг цільової сполуки брутто-формули C25H29N3O7 (М=483,525 г). Відповідний вихід становить 97%. Стадія Е: Гідрохлорид (4-нітрофеніл)метил-транс-5-(бензоїламіно)-2-піперидинкарбоксилату Змішують 861мг (8ммоль) одержаного вище аміду, 9 мл метанолу і 2,3 мл розчину газоподібного хлороводню в метанолі з концентрацією 8 моль/л. Температуру доводять до 20°С і реакцію ведуть протягом 3 годин. Потім додають 1,15 мл розчину хлороводню в метанолі. Перемішують протягом 20 хвилин при температурі 20°С, потім розчинник випаровують при зниженому тиску. Після цього здійснюють два відганяння при використанні дихлорметану, потім два відганяння при використанні діетилового ефіру. Продукт кристалізують з діетилового ефіру. Таким чином одержують 715 мг цільової сполуки брутто-формули C 20H22CIN3O 5 (М=419,967 г). Відповідний вихід становить 96%. Стадія F: (4-Нітрофеніл)метил-транс-5-(бензоїламіно)-1-(хлоркарбоніл)-2-піперидинкарбоксилат Змішують 1,08 г (2,58 ммоль) одержаного, як указано вище, гідро хлориду і 11 мл ди хлорметану. Одержану суспензію охолоджують до температури близько 0-5°С і додають 791 мкл триетиламіну, потім до одержаного розчину додають 161 мкл дифосгену. Перемішують протягом 5 хвилин при температурі 0-5°С, потім температуру доводять до 20°С і перемішують ще протягом ЗО хвилин. Після цього розбавляють дихлорметаном, промивають 10%-ним водним розчином винної кислоти, потім водою. Сушать над сульфатом натрію і розчинник випаровують при зниженому тиску. Сирий продукт очищають шляхом хроматографії на діоксиді кремнію, елююючи дихлорметаном, що містить 5% ацетону. Одержують 969 мг цільової сполуки брутто-формули C21H20ClN3O6 (M=445,862 г). Відповідний вихід становить 84%. Стадія G: (4-Нітрофеніл)метил-транс-6-бензоїл-7-оксо-1,6-діазабіцикло[3,2,1]октан-2-карбоксилат В атмосфері інертного газу, змішують 928 мг (2,08 ммоль) одержаної вище сполуки і 27 мл тетрагідрофурану. Одержаний розчин охолоджують до температури -78°С при перемішуванні, потім вводять 2,1 мл 1 Μ розчину біс(триметилсиліл)аміду літію в тетрагідрофурані. Перемішують при температурі -78°С протягом 10 хвилин, потім додають 130мкл оцтової кислоти і перемішують при доведенні температури до 15°С. Розбавляють етилацетатом, потім промивають послідовно 10%-ним водним розчином винної кислоти, розчином фосфатного буфера з рН=7 і водою. Сушать над сульфатом магнію і розчинник випаровують при зниженому тиску. Таким чином одержують 1,6 г сухого екстракту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю ди хлорметану і ацетону у співвідношенні 98:2. Продукт потім кристалізують з діетилового ефіру, одержуючи 204 мг цільової сполуки брутто-формули C 21H19N 3O6 (М=409,441 г). Відповідний вихід становить 24%. 1 H -ЯМР-спектр (250 МГц; CDC13; хімічні зсуви піків в м.ч. і мул ьтипл етність): 1,98 (м), 2,22 (м) і 2,40 (м): N-CH-CH2-CH2; 3,08 (д) і 3,42 (д): O=C-N-CH2; 4,23 (дд): OC-N-CH; 4,53 (м): O=C-N-CH2-CH; 5,34 [АВ]: СОО-СН 2-С6Н5; 7,69 (м): 8,25 (м): 7,44 (м) і 7,56 (м): С6Н5 і C6H4NO2. ІЧ-спектр (СНСl3): 1763, 1744, 1676; 1609, 1603, 1583, 1526, 1492 см"1. МС (ЕУ) m/z: [M]+=409, 304, 273, 201, 105, 77. ПРИКЛАД 29b транс-6-Бензоїл-7-оксо-1,6-діазабіцикло[3,2,1]октан-2-карбонова кислота Змішують 89 мг одержаного в прикладі 29а складного ефіру, 4 мл ацетону і 6 мг каталізатора 10%-ного паладію-на-вугіллі. Реакцію ведуть при перемішуванні, при температурі 20°С і в атмосфері водню протягом 2 годин 45 хвилин, потім каталізатор відфільтровують і фільтрат випаровують при зниженому тиску. Таким чином одержують 88 мг смоли, які кристалізують з 0,5 мл діетилового ефіру. Таким чином одержують 54 мг цільової сполуки брутто-формули C14H 14N2 O4 (М=274,278 г). Відповідний вихід становить 91%. 1 H -ЯМР-спектр (250 МГц; CDCl3; хімічні зсуви піків в м.ч. і мультиплетність): 1,96 (м) 2,10 (м) і 2,37 (м): N-CH-CH2-CH2; 3,13 (д) і 3,41 (дм): O=C-N-CH 2; 4,10 (уш.д): OC-N-CH; 4,52 (м): O=C-N-CH2-CH; 7,44 (м): 7,56 (т) і 7,69 (дд) С6Н5. МС (ЕУ) m/z: [M]+=274, 229, 169, 105, 77. ПРИКЛАД 29с Метил-транс-6-бензоїл-7-оксо-1,6-діазабіцикло[3,2,1]октан-2-карбоксилат При перемішуванні до 28 мг (0,102 ммоль) одержаної в прикладі 29Ь кислоти додають 2 мл розчину діазометану в дихлорметані з концентрацією 12,7 г/л. Розчинник випаровують при зниженому тиску і залишок очищають шля хом хроматографії на діоксиді кремнію, елююючи сумішшю ди хлорметану і етилацетату у співвідношенні 98:2. Одержують 18,4 мг цільової сполуки брутто-формули C15H16N2O 4 (М=288,305 г). Відповідний вихід становить 63%. 1 H Н-ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і м ультиплетність): 1,90-2,42: N-CH-CH2-CH2; 3,12 (д) і 3,44 (д): O=C-N-CH2; 3,83 (с): СН3; 4,17 (уш.д): OC-N-CH; 4,54 (м): O=CN-CH2-CH; 7,44 (τ), 7,56 (τ) і 7,69 (д): С6Н5. МС (ЕУ) m/z: [M]+=288, 229, 183, 155, 105, 77. ПРИКЛАД 29d транс-6-Бензоїл-7-оксо-N-(фенілметил)-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід Змішують 30 мг (0,109 ммоль) одержаної в прикладі 29b транс-6-бензоїл-7-оксо-1,6діазабіцикло[3,2,1]октан-2-карбонової кислоти, 0,5 мл дихлорметану, 23 мг EDCI і 13 мкл бензиламіну. Реакцію ведуть протягом 30 хвилин при перемішуванні. Потім розбавляють дихлорметаном, промивають 10%-ним водним розчином винної кислоти, декантують і сушать органічну фазу над сульфатом натрію. Розчинник випаровують при зниженому тиску, одержуючи сирий продукт, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і ацетону у співвідношенні 98:2. Таким чином одержують 19,5 мг цільової сполуки брутто-формули C21H21N3O 3 (M=363,419 г). Відповідний вихід становить 49%. 1 H -ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і мультиплетність): 1,97 (м), 2,34 (м) і 2,59 (м): N-CH-CHrCH2; 2,90 (д), 3,33 (м), 3,99 (уш.д) і 4,50 (м): O=C-N-CH, O=C-N-CH 2CH, O=C-N-CH2, CO-NH-CH2-C6H5; 6,94 (уш.т): NH; 7,24-7,58 (м) і 7,68 (м): С6Н5-СО і С б Н5-СН2. ІЧ-спектр (СНСl3): 3411, 1763, 1680; 1603, 1583, 1519, 1498 см-1, ПРИКЛАД 29е 6-Бензоїл-1N-[метил(фенілметил)]-7-оксо-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід Слідують методиці, аналогічної такій прикладу 29d, виходячи з 50 мг (0,182 ммоль) одержаної в прикладі 29Ь кислоти і 45 мкл N-метилбензиламіну. Таким чином одержують 12 мг цільової сполуки брутто-формули C22H23N3O3 (М=377,45 г). Відповідний вихід становить 17%. МС (ЕУ) m/z: [М]+=377, 272, 105. ПРИКЛАД 29f 6-Бензоїл-2-(гідроксиметил)-1,6-діазабіцикло[3,2Д ]октан-7-он В інертній атмосфері в 3 мл тетрагідрофурану розчиняють 100 мг (364 ммоль) одержаної в прикладі 29Ь транс-6-бензоїл-7-оксо-1,6-діазабіцикло[3,2,1]октан-2-карбонової кислоти. Охолоджують до температури -10°С і додають 40 мкл метилморфоліну, потім 38 мкл етилхлорформіату. Реакцію ведуть протягом 15 хвилин при температурі -10°С, потім температуру доводять до 0°С і додають 27 мг NaBH4, потім, краплями, 1,5 мл метанолу. Перемішують при температурі 0°С протягом 2 годин, потім температуру доводять до кімнатної температури. Додають З мл води, перемішують протягом 15 хвилин, потім додають декілька крапель розчину хлориду амонію. Екстрагують етилацетатом, сушать над сульфа том магнію, фільтрують і розчинник випаровують при зниженому тиску. Таким чином одержують 85 мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і метанолу у співвідношенні 98:2. Таким чином одержують 25 мг цільової сполуки брутто-формули C14H16N2 O3 (М=260,3 г). Відповідний вихід становить 26%. 1 H -ЯМР-спектр (300 МГц; CDCl3, хімічні зсуви піків в м.ч. і мультиплетність): 1,61 (м, 1Н), 2,00 (м, 2Н), 2,30 (м, 1Н): СН-СН 2-СН2-СН; 2,19: 3,23 (д) і 3,26 (д): N-CH2; 3,60 (м): N-CH-CH2OH; 3,70 (м) і 3,77 (дд): СН-СН 2-0; 4,56 (м): N-CH-CH 2-N. МС (SIMS) m/z: [M+Na]+=283; [M+H]+=261; [M]+=260, 229, 105. ПРИКЛАД 30 (4-Нітрофеніл)метил-транс-6-ацетил-7-оксо-1,6-діазабіцикло[3,2Д ]октан-2-карбоксилат 1 г (2,63 ммоль) одержаного на стадії С прикладу 29 продукту розчиняють в 12 мл дихлорметану. Додають 250 мкл оцтового ангідриду, реакцію ведуть протягом 10 хвилин при перемішуванні, потім розбавляють дихлорметаном і промивають водним насиченим розчином гідрокарбонату натрію. Органічну фазу сушать над сульфатом натрію, випаровують досуха при зниженому тиску, одержуючи 1,2 г і-(ІД-диметилетил)- і 2-[(4нітрофеніл)метил]-транс-5-(ацетиламіно)-і ,2-піперидиндикарбоксилату брутто-формули C20H27N3 O7 (М=421,453 г). Цей продукт використовують без очищення на стадіях, подібних стадіям E-G прикладу 29, і таким чином одержують 14 мг цільової сполуки брутто-формули C16H17N3O 6 (M=347,330 г). Відповідний вихід становить 17%. 1 H -ЯМР-спектр (300 МГц; CDC13; хімічні зсуви піків в м.ч. і мул ьтипл етність): 1,87 (м), 2,00-2,30 (м): N-CH-CH2-CH2; 2,54 (с): N-CO-CH3; 2,95 (д) і 3,21 (м): O=C-N-CH2; 4,26 (уш.д): O=CN-CH; 4,55 (м): O=C-N-CH 2-CH; 5,3.4 [АВ]: СО 2-СН2-С6Н4; 7,57 і 8,25 [АА'ВВ 1]: C6H4-NO 2. МС (ЕУ) m/z: [М]+=347, 304, 211, 169, 125, 43. ПРИКЛАД 31 (4-Нітрофеніл)метил- і 2-пропеніл-транс-7-оксо-1,6-діазабіцикло[3,2,1]октан-2,6-дикарбоксилат В атмосфері азоту, в 8 мл дихлорметану розчиняють 1,24 г (3,278 ммоль) продукти, одержаного на стадії С прикладу 29а. Розчин охолоджують до температури 0°С, потім додають краплями 0,45 мл триетиламіну, потім 0,35 мл алілхлорформіату. Витримують при температурі 0°С протягом 15 хвилин, потім реакцію ведуть при перемішуванні протягом 1 години при кімнатній температурі. Після цього розбавляють за допомогою 20 мл дихлорметану, промивають водним розчином гідрокарбонату натрію і два рази водою. Сушать над сульфатом магнію і розчинник випаровують при зниженому тиску. Таким чином одержують 1,5 г 1-(1,1-диметилетил)- і 2[(4-нітрофеніл)метил]-транс-5-[(2-пропенілокси)[карбоніл]аміно]-1,2-піперидиндикарбоксилату брутто-формули C22H28N3 O8 (М=462,486 г). Відповідний вихід становить 99%. Цей продукт використовують на стадіях, подібних стадіям E-G прикладу 29а, і таким чином одержують 30,6 мг цільової сполуки брутто-формули C18H19N3 O7 (М=389,368 г) у вигляді твердої речовини білого кольору. Відповідний вихід становить 40%. 1 H -ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і мультиплетність): 1,91 (м), 2,00-2,29 (м): N-CH-CH 2-CH2; 2,98 (д) і 3,25 (уш.д): O=C-N-CH 2; 4,27 (τ) OC-N-CH; 4,37 (уш.с): O=CN-CH2-CH; 4,77 (уш.д): С00-СН2-СН=; 5,33 (с): СОО-СН2-С6Н4; 5,29-5,46: СН^СН; 5,98 (м): СН2=СН; 7,96 і 8,29 [АА'ВВ1]: С6Н4-NO 2. ІЧ-спектр (СНСl3): 1801, 1775, 1738, 1724; 1649; 1608, 1595, 1526 см -1. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [2 M+Na]+=801; [M+Na+CH3CN]+=453; [M+Na]+=412. ПРИКЛАД 3 Ibis Фенілметил-транс-6-бензоїл-7-оксо-1,6-діазабіцикло[3,2,1]октан-2-карбоксилат В інертній атмосфері змішують 200 мг фенілметил-транс-5-(бензоїламіно)-1-(хлоркарбоніл)-2піперидинкарбоксилату брутто-формули C21H21ClN2O4 (М=400,87 г), одержаного згідно з методиками, подібним таким стадій A-F прикладу 29а, і 6 мл безводного тетрагідрофурану і охолоджують до температури -78°С. Додають краплями 0,55 мл 1 Μ розчину біс(триметилсиліл)аміду літію в тетрагідрофурані. Реакцію ведуть при перемішуванні при температурі -78°С протягом 10 хвилин, потім додають 25 мкл оцтової кислоти. Температуру доводять до кімнатної, потім виливають в 10 мл 10%-ного водного розчину винної кислоти. Екстрагують етилацетатом, промивають водним розчином фосфатного буфера з рН=7, потім водою і сушать над сульфатом магнію. Доводять досуха шляхом випаровування розчинника при зниженому тиску. Таким чином одержують 158 мг сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і ацетону у співвідношенні 98:2. Таким чином одержують 70 мг цільової сполуки бруттоформули C21H20N2O 4 (M=364,40 г). Відповідний вихід становить 39%. 1 H -ЯМР-спектр (400 МГц; CDCl3; хімічні зсуви піків в м.ч. і мультиплетність): 2,15 (м) і 2,25 (м): NCH-CH 2-CH2-CH-CO2; 1,94 (м) і 2,36 (м): NCH-CH 2-CH2-СН-СО2; 4,20 (д) N-CH-CO2; 4,50 (квадруплет): NCH-CH 2-CH2-CH-CO2; 3,08 (д) і 3,40 (д): N-CH2; 5,25 [АВ]: СО 2-СН2-С6Н5; 7,38 (уш.с): СН2-С6Н5; 7,43 (уш.т) і 7,55 (уш.т) і 7,69 (уш.д) С6Н5-СО. ІЧ-спектр (СНСl3): 1764, 1744, 1675; 1602, 1584, 1498 см -1. MC (SIMS) m/z: [M+Na]+=387; [M+H]+=365, 259, 257, 229, 105, 91. ПРИКЛАД 31 ter Фенілметил-6-бензоїл-7-оксо-1,6-діазабіцикло[3,2,1]окт-2-ен-2-карбоксилат В атмосфері азоту змішують 46 мг (0,126 ммоль) одержаного в прикладі 3 Ibis продукту і 0,5 мл безводного тетрагідрофурану. Охолоджують до температури -70°С і додають 0,31 мл 1Μ розчину біс(триметилсиліл)аміду літію в те трагідрофурані. Реакцію ведуть протягом 2 годин при температурі -70°С, потім температуру доводять до -15°С і при цій температурі додають 0,41 мл розчину C 6H5SeCl в тетрагідрофурані з концентрацією 0,7 моль/л. Перемішують при температурі -15°С протягом 15 хвилин, потім температуру до водять до кімнатної температури протягом 15 хвилин і виливають в суміш води і льоду, що містить декілька крапель водного насиченого розчину гідрокарбонату натрію. Екстрагують етилацетатом, промивають водою, сушать і розчинник випаровують при зниженому тиску. Залишок очищають шля хом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і ацетону у співвідношенні 98:2, і таким чином одержують 15 мг фенілметил-6-бензоїл7-оксо-2-(фенілселеніл)-1,6-діазабіцикло[3,2,1]октан-2-карбоксилату брутто-формули C27H 24N2O4Se (M=519,46 г). Відповідний вихід становить 23%. Змішують 15 мг (0,029 ммоль) одержаної вище сполуки і 0,3 мл дихлорметану. Охолоджують до температури 0°С і додають 15 мг м-хлорнадбензойної кислоти у вигляді розчину в 0,15 мл дихлорметану. Перемішують при температурі 0°С протягом 15 хвилин, потім температуру доводять до кімнатної температури. Виливають приблизно в 20 мл води, екстрагують дихлорметаном і органічну фазу промивають водним розчином фосфа тного буфера з рН=7. Сушать над сульфатом магнію, фільтрують і розчинник випаровують при зниженому тиску. Таким чином одержують 15 мг сирого продукту, який очищають на діоксиді кремнію, елююючи сумішшю дихлорметану і ацетону у співвідношенні 98:2. Таким чином одержують 5 мг цільової сполуки бруттоформули C2iH18N2O4 (M=362,39 г). Відповідний вихід становить 48%. 1 H -ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і мультиплетність): 2,66 (тд) і 2,99 (тдд): N-CH-CH 2; 3,03 (д) і 3,77 (ддд): N-CH 2; 4,76 (τ): N-CH; 5,23 [AB]: CO 2-CH2-C6H5; 7,02 (д): N-C=CH; 7,30-7,38 (м): СН2-С6Н5; 7,42 (тм), 7,54 (тм) і 7,62 (дм); С6Н5-СО. ПРИКЛАД 31 quater 6-Бензоїл-7-оксо-1,6-діазабіцикло[3,2,1]окт-2-ен-2-карбонова кислота Змішують 20 мг (0,055 ммоль) одержаного в прикладі 31ter продукту, 0,4 мл ацетону і 4 мг каталізатора 10%-ного паладію-на-вугіллі. Вміщують в атмосферу водню і реакцію ведуть протягом 3 годин при інтенсивному перемішуванні. Відфільтровують і промивають каталізатор ацетоном, потім метанолом. Фільтрат випаровують при зниженому тиску. Таким чином одержують 14 мг цільової сполуки брутто-формули Ci4H12N2O4 (M=272,4 г). Відповідний вихід становить 93%. МС (ЕУ) m/z: [M]+: 272, 105. ПРИКЛАД 32а 2-Пропеніл-транс-7-оксо-6-(2-фенілметокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксилат Стадія А: 2-Пропеніл-цис-5-гідрокси-1-(трифторацетил)-2-піперидинкарбоксилат В 17 мл етилацетату розчиняють 17 г (0,059 моль) 1-(1,1-диметилетил)- і 2-(2-пропеніл)-цис-5-гідрокси-1,2піперидиндикарбоксилату (описується в Rec. Trav. Chim., 78,648-658 (1959)) брутто-формули C14H23NO5 (М=285,3431 г). При температурі 0°С додають 51 мл розчину хлороводню в етилацетаті з концентрацією 150 г/л. Температуру доводять до кімнатної температури і реакцію ведуть при перемішуванні протягом 1 години 30 хвилин. Етилацетат випаровують при зниженому тиску, потім залишок обробляють діетиловим ефіром, який в свою чергу видаляють при зниженому тиску. Таким чином одержують 12 г твердої речовини блідо-жовтого кольору, яку змішують з 200 мл тетрагідрофурану. Охолоджують до температури 0°С, потім додають 37,6 мл триетиламіну. Витримують при температурі 0°С, потім повільно додають 16,8 мл трифтороцтового ангідриду. Залишають температуру підвищуватися до 20°С і реакцію ведуть ще протягом 20 хвилин при перемішуванні. Після цього додають 20 мл води. Одержаний розчин перемішують протягом 1 години при кімнатній температурі і виливають в 300 мл води. Екстрагують етилацетатом, промивають водою, сушать над сульфатом натрію і розчинник випаровують при зниженому тиску. Одержують 15,7 г сирого продукту, який очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю дихлорметану і етилацетату у співвідношенні 90:10. Таким чином одержують 12,3 г цільової сполуки брутто-формули С 11Н14F 3NO4 (М=281,23 г) у вигляді масла жовтого кольору. Відповідний вихід становить 73%. Стадія В: 2-Пропеніл-транс-5-[(фенілметокси)аміно]-1-(трифторацетил)-2-піперидинкарбоксилат Змішують 10,9 г (38,7 ммоль) одержаної на стадії А сполуки і 150 мл ацетонітрилу. Одержаний розчин блідо-жовтого кольору охолоджують до температури -30°С, потім додають 4,94 мл 2,6-лутидину і 6,7 мл ангідриду трифторметансульфокислоти. Перемішують протягом 15 хвилин, потім додають, весь час при температурі ЗО°С, 9,57 г О-бензилгідроксиламіну. По закінченні додавання температуру доводять до 0°С і реакцію ведуть протягом 1 години при цій температурі. Після цього додають 4,9 мл 2,6-лутидину і залишають контактувати протягом 3 днів при температурі 0°С. Потім реакційну суміш виливають в 500 мл води і екстрагують етилацетатом. Промивають послідовно водою, водним розчином фосфатного буфера з рН=7,0, водним насиченим розчином хлориду натрію, потім знов водою. Сушать над сульфатом натрію і розчинник випаровують при зниженому тиску. Таким чином одержують 23 г сирого продукту, який розчиняють в 150 мл дихлорметану. Промивають 10%-ним водним розчином винної кислоти, потім сушать над сульфатом натрію, випаровують розчинник при зниженому тиску. Таким чином виділяють 16,1 г масла жовтого кольору, які очищають шляхом хроматографії на діоксиді кремнію. Одержують 12,1 г цільової сполуки брутто-формули C18H2iF3N2O4 (M=386,37 г) в кристалічній формі. Відповідний вихід становить 72%. Стадія С: 2-Пропеніл-транс-5-[(фенілметокси)аміно]-2-піперидинкарбоксилат Охолоджують до температури -10°С 80 мл метанолу, потім додають 4,15 г (37,8 ммоль) NaBH4. До цієї суміші при перемішуванні повільно додають, протягом 30 хвилин, при підтриманні температури -10°С, розчин 10,6 г (27,4 ммоль) одержаної вище сполуки в 80 мл метанолу. Залишають потім температуру підвищуватися до 0°С, потім підтримують цю температуру протягом 3 годин. Реакційну суміш виливають в 450 мл води з льодом і 150 мл етилацетату. Декантують, промивають водою, органічну фазу потім сушать над сульфатом натрію і після цього розчинник випаровують при зниженому тиску. Таким чином одержують 8,2 г масла жовтого кольору, які розчиняють в 80 мл тетрагідрофурану, додають розчин 2,43 г щавлевоїкислоти в 25 мл тетрагідрофурану. Викристалізуваний оксалат відфільтровують і промивають невеликою кількістю тетрагідрофурану, потім висушують при зниженому тиску і розчиняють в насиченому розчині гідрокарбонату натрію. Екстрагують етилацетатом, органічну фаз у промивають водою, суша ть над сульфатом натрію і розчинник випаровують при зниженому тиску. Таким чином одержують 4,39 г цільової сполуки брутто-формули C16H22N2O3 (М=290,36 г) у вигляді масла, яке кристалізується, коли температура нижче 20°С. Відповідний вихід становить 55%. Стадія D: 2-Пропеніл-транс-7-оксо-6-(2-фенілметокси)-1,6-діазабіцикло [3,2,1] октан-2-карбоксилат В атмосфері азоту 3,2 г (11 ммоль) одержаного вище масла розчиняють в 500 мл ацетонітрилу. Одержаний розчин охолоджують до температури 0°С за допомогою бані з льодом і додають 3,37 мл триетиламіну, потім 0,796 мл дифосгену і 1,48 г D MAP. Температуру доводять до 20°С і реакцію ведуть протягом 2 годин при перемішуванні. Після цього реакційну суміш виливають в 200 мл водного розчину соляної кислоти, додають 400 мл води, екстрагують дихлорметаном, промивають водою і сушать над сульфатом натрію. Потім розчин ник випаровують при зниженому тиску, одержуючи 3,1 г цільової сполуки брутто-формули C17H20N2O4 (М=316,36 г) у ви гляді кристалів. Відповідний вихід становить 89%. 1 H-ЯМР-спектр: 1,66 (м) і 2,00-2,16 (м) О=С-СН-СН 2-СН2; 2,94 (д) і 3,07 (д) N-СН2; 3,31 (м) N-СН2-СН; 4,14 (дд) О=С-СН , 4,68 (д) СН2-СН=СН2; 4,90 і 5,06 [АВ] СН2-С6Н5; 5,26 (д. квадруплет) і 5,34 (д. квадруплет) СН2-СН=СН2; 5,92 (м) СН2-СН=СН2; 7,37-7,42 (м) С6Н5. ІЧ-спектр (СНСl3): 1748; 1646; 1496 см -1. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [2 M+Na]+=655; [M+Na+CH3CN]+=380; [M+Na]+=339; [M+H]+=317, 289, 91. ПРИКЛАД 32b транс-7-Оксо-6-(фенілметокси)-1,6-діазабіцикло[3,2Д]октан-2-карбонова кислота і її сіль з циклогексиламіном В атмосфері азоту 2,21 г (6,98 ммоль) одержаної у прикладі 32а сполуки розчиняють в 44 мл дихлорметану. Додають 0,5 Μ розчин етилгексаноату натрію в етилацетаті. Потім за один раз додають 242 мг тетракистрифенілфосфінпаладію, після чого перемішують протягом 1 години. Розбавляють за допомогою 22 мл етилацетату і виливають в 75 мл насиченого розчину NaH2PO4· Потім екстрагують етилацетатом і органічну фазу сушать над сульфатом натрію. Розчинник випаровують при зниженому тиску, одержуючи 3,5 г залишку жовтого кольору, який розчиняють в суміші з 11 мл етилацетату і 0,8 мл циклогексиламіну. Кристалічну сіль циклогексиламіну відділяють шляхом відфільтровування і промивають діетиловим ефіром, потім розчинник випаровують при зниженому тиску. Таким чином одержують в цілому 2,51 г кристалічної солі, яку розчиняють в 25 мл водного насиченого розчину NaH2PO4. Екстрагують етилацетатом, органічні фази об'єднують і сушать їх над сульфатом натрію, потім розчинник випаровують при зниженому тиску. Таким чином одержують 1,82 г цільової сполуки брутто-формули C14H16N 2O4 (М=276,29 г) у кристалічній формі. Відповідний вихід становить 94%. 1 H -ЯМР-спектр (300 МГц; CDCl3; хімічні зсуви піків в м.ч. і мультиплетність): 1,68 (м) і - 2,20-2,22 (м): СН-СНг-СНз-СН; 2,89 (д) і 3,11 (ддд): N-CH2; 3,34 (дд): N-CH2-CH, 4,13 (уш.д): NCH-O0; 4,90 і 5,05 [АВ]: СН2-О; 7,32-7,43: С6Н5. МС (SIMS) m/z: [M+Na]+=299; [M+H]+=277,91. ПРИКЛАД 33а Піридинієва сіль транс-7-оксо-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксаміду Стадія А: транс-7-Оксо-6-(фенілметокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід У 30 мл дихлорметану розчиняють 1,1 г (4 ммоль) одержаної в прикладі 32b сполуки. До цього розчину додають 0,67 мл триетиламіну. Розчин охолоджують до температури 5°С і досить швидко додають 0,57 мл ізобутилхлорформіату. Перемішують протягом 20 хвилин при температурі 5°С, потім повільно, при інтенсивному перемішуванні, додають 3 мл концентрованого розчину аміаку. Продовжують перемішування протягом години при кімнатній температурі, реакційне середовище розбавляють за допомогою 30мл води, екстрагують дихлорметаном, промивають водою, сушать над сульфатом натрію і концентрують при зниженому тиску. Таким чином одержують 1,1 г цільового продукту бр утто-формули Q4H17N3O3 (М=275,31 г). Ви хід є кількісним. Стадія В: транс-6-Пдрокси-7-оксо-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід Змішують 1,1г одержаної на стадії А сполуки, ЗО мл метанолу і 300 мг 10%-ного паладію-на-вугіллі. Вміщують в атмосферу водню, потім суміш інтенсивно перемішують протягом 45 хвилин. Каталізатор після цього відфільтровують, промивають метанолом, потім сумішшю ди хлорметану і метанолу. Фільтрат випаровують при зниженому тиску. Таким чином одержують 800 мг цільового продукту бруттоформули C7H11N3 O3 (М= 185,18 г) у вигляді безбарвної піни. Стадія С: Піридинієва сіль транс-7-оксо-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксаміду В атмосфері азоту змішують 800 мг одержаної вище сполуки і 20 мл безводного піридину. Потім додають 1,91 г комплексу БОз-піридин. Суміш перемішують протягом 20 годин при кімнатній температурі. Реакційне середовище після цього відфільтровують і розчинник випаровують при зниженому тиску. Таким чином одержують цільовий продукт брутто-формули C12H16N4O 6S.C6H5N (М=344,35 г) у вигляді продукту жовтого кольору. ПРИКЛАД 33b Тетрабутиламонієва сіль транс-7-оксо-6-(сульфокси)-1,6-діазабіцикло [3,2,1]октан-2-карбоксаміду Одержаний вище продукт вводять в 40 мл водного концентрованого розчину NaH2PO4 так, щоб досягнути значення рН, рівного 4. Екстрагують етилацетатом, потім у водну фазу додають 1,01 г тетрабутиламонійгідросульфату. Перемішують протягом 10 хвилин при кімнатній температурі, екстрагують 4 рази за допомогою 300 мл етилацетату, органічну фазу сушать над сульфатом натрію і концентрують при зниженому тиску. Таким чином одержують 1,530 г безбарвної піни, яку очищають шляхом хроматографії на діоксиді кремнію, елююючи сумішшю ацетону, дихлорметану і триетиламіну у співвідношенні 50:48:2. Таким чином одержують 1,02 г цільового продукту бр утто-формули C23H 46N4O 6S (М=506,71 г) у вигляді безбарвної піни. Відповідний загальний вихід становить 50%. ПРИКЛАД 33с транс-7-Оксо-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксамід натрію Одержаний в прикладі ЗЗЬ продукт розчиняють в 7 мл суміші ацетону і води у співвідношенні 1:1, потім вносять в колонку з 180 г смоли DOWEX 50WX8 в Na+-формі і елююють водою. Після випаровування води при зниженому тиску продукт кристалізується. Таким чином одержують 542 мг цільової сполуки формули C7H10N3NaO6S (M=287,23 г). Відповідний вихід становить 94%. 1 H -ЯМР-спектр (300 МГц; диметилсульфоксид; хімічні зсуви піків в м.ч. і м ультиплетність): 1,55-2,10 (3Н): СН-СН2-СН2-СН; 2,91 (д) і 3,02 (уш.д): N-CH 2; 3,38 (уш.с): N-СН2-СН; 3,68 (д): N-CH-C=O; 7,23 і 7,44: NHj МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]"=264. ПРИКЛАДИ 34-47 Згідно з методикою, подібною такій, яку використовують в прикладі 33, виходячи з ПО мг одержаної в прикладі 32b кислоти, одержують нижченаведені карбоксаміди. Єдина відмінність полягає в тому, що на стадії 1 реагент, що використовується, тобто, розчин аміаку, замінюють розчином відповідного аміну. Таким чином, тільки змінюється група R1 яка описана у формулі (І). ПРИКЛАД 34 Виходячи з 49 мкл бензиламіну, одержують 64 мг транс-7-оксо-N-(фенілметил)-6-(сульфокси)-1,6діазабіцикло[3,2,1]октан-2-карбоксаміду натрію, або загальний вихід становить 38%. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [M+Na]+=400; [M+H]+=378. ПРИКЛАД 35 Виходячи з 43 мкл 2-піридинметанаміну, одержують 37 мг транс-7-оксо-М-(2-піридинілметил)-6(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксаміду натрію, або загальний вихід становить 14%. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [М+Н]+=379. ПРИКЛАД 36 Виходячи з 51,3 мг 3-піридинетанаміну, одержують 42 мг транс-7-оксо-ІЧ-[2-(3-піридиніл)етил]-6(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксаміду натрію, або загальний вихід становить 20%. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [М+Н]+=393. ПРИКЛАД 37 Виходячи з 51,3 мг 4-піридинетанаміну, одержують 40 мг транс-7-оксо-ІЧ-[2-(4-піридиніл)етил]-6(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксаміду натрію, або вихід становить 20%. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [M+Na]+=415; [M+H]+=393. ПРИКЛАД 38 Виходячи з 50,2 мг 2-піридинетанаміну, одержують 45 мг транс-7-оксо-ТЧ-[2-(2-піридиніл)етил]-6(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксаміду натрію, або вихід становить 23%. МС (іонізація електронним розпиленням з утворенням позитивних іонів) m/z: [М+Н]+=393. ПРИКЛАД 39 Виходячи з 58,3 мг 3-амінобензаміду, одержують 43 мг TpaHc-N-[3-(амінокарбоніл)феніл]-7-оксо-6(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксаміду натрію, або вихід становить 22%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [M]+=383. ПРИКЛАД 40 Виходячи з 58,3 мг 4-диметиламінобензоламіну, одержують 65,3 мг транс-N-[4-(диметиламіно)феніл]-7оксо-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксаміду натрію, або вихід становить 40%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]'=383. ПРИКЛАД 41 Виходячи з 58,3 мг 3-диметиламінобензоламіну, одержують 91 мг транс-N-[3-(диметиламіно)феніл]-7-оксо6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксаміду натрію, або вихід становить 54%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]+=383. ПРИКЛАД 42 Виходячи з 43 мкл 4-піридинметанаміну, одержують 24,6 мг транс-7-оксо-N-[(4-піридиніл)метил]-6(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксаміду натрію, або вихід становить 15%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]=355. ПРИКЛАД 43 Виходячи з 44 мкл 3-піридинметанаміну, одержують 44,7 мг транс-7-оксо-N-(3-піридинілметил)-6(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксаміду натрію, або вихід становить 26%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]+=355. ПРИКЛАД 44 Виходячи з 84 мг (±)-a-амінобензолпропанаміду, одержують 55 мг транс-N-(1-аміно-1-оксо-3-феніл-2пропіл)-7-оксо-6-(сульфокси)-1,6-діазабіцикло[3,2,1] октан-2-карбоксаміду натрію, або вихід становить 27%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]'=411,321. ПРИКЛАД 45 Виходячи з 46 мг гідрохлориду 2-аміноацетаміду і 61 мкл триетиламіну, одержують 25 мг транс-1чї-(2аміно-2-оксоетил)-7-оксо-6-(сульфокси)-1,6-діазабіцикло-[3,2,1]октан-2-карбоксаміду натрію, або вихід становить 13%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]+=321,249. ПРИКЛАД 46 Виходячи з 64 мг (З-амінофеніл)сечовини, одержують 43 мг транс-N-[3-[(амінокарбоніл)аміно]феніл]-7оксо-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксаміду натрію, або вихід становить 24%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]=398,153, 111. ПРИКЛАД 47 Виходячи з 63 мг (±)-а-амінобензолацетаміду, одержують 64 мг транс-]Ч-(2-аміно-2-оксо-1-фенілетил)-7оксо-6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-2-карбоксаміду натрію, або вихід становить 38%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]=397. ПРИКЛАДИ 48-51 Наступні сполуки одержують, виходячи з 110 мг сполуки, одержаної на стадії Ε прикладу 32, яку етерифікують кожний раз за допомогою відповідного спирту для одержання кінцевого продукту. Потім слідують методиці, подібній такій, яка описана у разі стадій Ε прикладу 33. ПРИКЛАД 48 Виходячи з 31,5 мг 2-гідроксиацетаміду, одержують 54 мг 2-аміно-2-оксоетил-транс-7-оксо-6-(сульфокси) 1,6-діазабіцикло[3,2,1]октан-2-карбоксилату натрію, або вихід становить 32%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]+=322. ПРИКЛАД 49 Виходячи з 51,7 мг 4-піридинетанолу, одержують 20 мг 2-(4-піридиніл)етил-транс-7-оксо-6-(сульфокси)1,6-діазабіцикло[3,2,1]октан-2-карбоксилату натрію, або вихід становить 8,5%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]-=370. ПРИКЛАД 50 Виходячи з 47,3 мг 2-піридинетанолу, одержують 47 мг 2-(2'-піридиніл)етил-транс-7-оксо-6-(сульфокси)1,6-діазабіцикло[3,2,1]октан-2-карбоксилату натрію, або вихід становить 23,4%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]-=370. ПРИКЛАД 51 Виходячи з 57,7 мг 3-піридинетанолу, одержують 50 мг 2-(3-піридиніл)етил-транс-7-оксо-6-(сульфокси)1,6-діазабіцикло[3,2,1]октан-2-карбоксилату натрію, або вихід становить 26%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]+=370. ПРИКЛАД 52 Натрієва сіль 3-метокси-6-(сульфокси)-1,6-діазабіцикло[3,2,1]окт-3-ен-7-ону Стадія А: Розчиняють 10 г (50 ммоль) 1,1-диметилетил-3,5-діоксо-1-піперидинкарбоксилату в 10 мл метанолу, потім додають 6 г (54 ммоль) О-алілгідроксиламінгідрохлориду. Перемішують протягом 3 годин, потім розчинник випаровують при зниженому тиску. Залишок обробляють водою, екстрагують дихлорметаном, органічну фазу промивають водою, потім сушать її над сульфатом натрію. Після фільтрації і випаровування розчинника при зниженому тиску одержують 10,6 г 1,1-диметилетил-5-метокси-3-[(2-пропенілокси)іміно]-3,6-дигідро-1(2Н)піридинкарбоксилату брутто-формули C14H22N 2O4 (М=282,342 г). Відповідний вихід становить 75%. Стадія В: У колбу вводять 10,6 г (37,6 ммоль) одержаного на стадії А продукту і 212 мл метанолу. Розчин охолоджують до температури -5°С, додають 37,8 г ціаноборгідриду натрію, потім 58,2 мл ефірату трифториду бору. Після цього розбавляють дихлорметаном, потім сумішшю води і 2н гідроксиду натрію, екстрагують дихлорметаном, органічну фазу промивають водою, сушать її над сульфатом натрію, фільтрують і розчинник випаровують при зниженому тиску. Одержаний продукт очищають шля хом хроматографії на діоксиді кремнію, елююючи сумішшю етилацетату і дихлорметану у співвідношенні 10:90. Таким чином одержують 5,5 г 1,1-диметилетил5-метокси-3-[(2-пропенілокси)аміно]-3,6-дигідро-1(2Н)-піридинкарбоксилату бр утто-формули (М=284,36 г). Відповідний вихід становить 51%. Стадія С: У колбу вводять 5,5 г (19,3 ммоль) одержаного на стадії В продукту, 27,5 мл дихлорметану і 4,2 мл анізолу. Потім додають 27,5 мл трифтороцтової кислоти. Трифтороцтову кислоту і дихлорметан видаляють при зниженому тиску. Залишок обробляють водою і екстрагують 3 рази етилацетатом. Водну фазу підлуговують шляхом додавання гідроксиду амонію, потім екстрагують етилацетатом. Органічні фази промивають водою, сушать їх над сульфатом натрію. Фільтрують, потім розчинник випаровують при зниженому тиску. Таким чином одержують 2,45 г 5-метокси-М-(2-пропенілокси)-1,2,3,6-тетрагідро-3-піридинаміну брутто-формули C9H16N2O 2 (M=l 84,24 г). Відповідний вихід становить 69%. Стадія D: В інертній атмосфері 2,45 г (0,0133 ммоль) одержаного на стадії С продукту розчиняють в 826 мл ацетонітрилу і розчин охолоджують до температури 0°С. Додають 0,778 мл дифосгену. Залишають для підвищення до кімнатної температури, потім додають 5,56 мл триетиламіну. Перемішують протягом ночі при кімнатній температурі, потім розчинник випаровують при зниженому тиску. Залишок обробляють водою, екстрагують етилацетатом, органічну фазу промивають водою, сушать її над сульфатом натрію, фільтрують, потім розчинник випаровують при зниженому тиску. Залишок очищають шля хом хроматографії на діоксиді кремнію, елююючи сумішшю етилацетату і дихлорметану у співвідношенні 1:9. Таким чином одержують 1,13 г 3-метокси-6-(2пропенілокси)-1,6-діазабіцикло[3,2,1]окт-3-ен-7-ону брутто-формули C10H14N2O 3 (М=210,23 г). Відповідний вихід становить 40,3%. Стадія Е: У колбі з інертною атмосферою 105 мг (0,5 ммоль) одержаного на стадії D продукту розчиняють в 1,1 мл дихлорметану, додають 57 мкл оцтової кислоти, потім 317 мг Рd[Р(С6Н5)3]4· Після протікання реакції протягом 1 години додають 1,1 мл піридину, потім 238 мг комплексу 8О3-піридин. Перемішують протягом ночі, потім розчинник випаровують при зниженому тиску. Залишок обробляють водою, екстрагують його дихлорметаном, екстракт промивають водою, органічну фазу суша ть над сульфатом натрію, фільтрують і розчинник випаровують при зниженому тиску. Залишок очищають шля хом хроматографії на діоксиді кремнію, елююючи сумішшю три хлорметану і ацетонітрилу у співвідношенні 50:50. Таким чином одержують 148 мг 1пропенілтрифенілфосфонієвої солі З-метокси-6-(сульфокси)-1,6-діазабіцикло[3,2,1]окт-3-ен-7-ону бруттоформули C28H29N2O 6PS. Відповідний вихід становить 53%. Стадія F: 148мг продукту, одержаного на стадії Е, розчиняють у воді, що містить 10% тетрагідрофурану. Одержаний розчин пропускають через колонку зі смолою DOWEX 50WX8 в Na+-формі, елююючи за допомогою води, що містить 10% тетрагідрофурана. Зібраний продукт ліофілізують, одержуючи 51 мг цільової натрієвої солі брутто-формули C7H9N2O 6SNa (M=272,21 г). Відповідний вихід становить 70%. 1Н-ЯМР-спектр: 3,04 (д) і 3,25 (дд): C=CH-CH-CH 2-N; 3,41 (д) і 3,71 (дд): N-CH2-C=CH; 3,47 (с): СН3-О; 4,20 (дд): C=CH-CHCH2-N; 5,19 (уш.д): C=CH-CH-CH2-N. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]+=249; [М-СН3]+=235. Приклад 53 Натрієва сіль 3-метокси-6-(сульфокси)-1,6-діазабіцикло[3,2,1]окт-3-ен-7-ону Стадія А: 1,03 г (5,2 ммоль) 1,1-диметилетил-3,6-дигідро-3-оксо-1(2Н)-піридинкарбоксилату брутто-формули C10H15NO 3 розчиняють в 15 мл етанолу. Додають 572 мг (5,2 ммоль) О-алілгідроксиламіну, потім 1,3 мл піридину. Перемішують протягом 15 хвилин, потім додають 100 мл дихлорметану, промивають 10%-ним водним розчином винної кислоти, після чого органічну фазу сушать над сульфатом магнію. Фільтрують і розчинник випаровують при зниженому тиску. Таким чином одержують 1,36 г 1,1-диметилетил-3,6-дигідро-3[(2-пропенілокси)имино]-1(2Н)-піридинкарбоксилату брутто-формули C13H20N2 O3 (М=252,32 г). Відповідний вихід є кількісним. Стадія В: Слідують методиці, вказаній у разі стадії А прикладу 52, ви ходячи з 1,38 г одержаного на стадії А продукту, 15,1 г ціаноборгідриду натрію і 8,3 мл ефірату трифториду бору. Таким чином, після очищення, одержують 0,99 г суміші з 2/3 1,1-диметилетил-3-[(2-пропенілокси)аміно]-1-піперидинкарбоксилату і 1/3 1,1-диметилетил3,6-дигідро-3 -[(2-пропенілокси)аміно]-1 (2Н)-піридинкарбоксилату брутто-формули C13H22N2O3 (М=254,33 г). Відповідний вихід становить 71%. Стадія С: 1,07 г (4,26 ммоль) одержаної на стадії В суміші розчиняють в 2 мл етилацетату. Охолоджують до температури 0°С, потім додають 5,8 мл 7,3 Μ розчину хлороводню в етилацетаті. Залишають реагувати протягом 2 годин ЗО хвилин при температурі 0°С. Розчинник випаровують при зниженому тиску, потім обробляють простим ефіром, осад відфільтровують і потім висушують при зниженому тиску. Таким чином одержують 560 мг дигідрохлориду N-(2-пропенілокси)-1,2,3,6-тетрагідро-3-піридинаміну брутто-формули С 8Н16СЫЧ2О (М=227,14 г). Відповідний вихід становить 57%. Стадія D: 560 мг (2,46 ммоль) одержаного на стадії С продукту розчиняють в 6 мл дихлорметану, потім додають 2,5 мл 2н розчину гідроксиду натрію. Декантують і водну фазу екстрагують етилацетатом. Органічні фази об'єднують, сушать їх над сульфатом магнію, фільтрують їх, потім розчинник випаровують при зниженому тиску. Таким чином одержують 278 мг 1-(2-пропенілокси)-1,2,3,6-тетрагідро-3-піридинаміну брутто-формули C8H14N2O (M=l 54,21 г). Відповідний вихід становить 73%. Стадія Е: В атмосфері аргону 270 мг (1,75 ммоль) одержаного на стадії D продукту розчиняють в 45 мл ацетонітрилу, додають 760 мкл триетиламіну і 105 мкл дифосгену. Залишають реагувати протягом 15 хвилин при температурі 0°С, потім температур у доводять до кімнатної температури і залишають реагувати ще протягом 2 годин. Після цього додають 213 мг DMAP і реакцію ведуть протягом ночі. Додають етилацетат, потім промивають 10%-ним водним розчином винної кислоти і водою. Органічну фазу сушать над сульфатом магнію, фільтрують її і розчинник випаровують при зниженому тиску. Одержаний сирий продукт очищають на діоксиді кремнію, елююючи сумішшю дихлорметану і ацетону у співвідношенні 95:5, що містить 0,1% триетиламіну. Таким чином одержують 36 мг 6-(2-пропенілокси)-1,6-діазабіцикло[3,2,1]окт-3-ен-7ону брутто-формули C8H12N2O 2 (M=l 80,21 г). Відповідний вихід становить 11%. Стадія F: Слідують методиці, аналогічній такій, описаній у випадку стадії Ε прикладу 52, виходячи з 51 мг (0,27 ммоль) одержаного на стадії Ε продукту, 33 мкл оцтової кислоти, 165 мг Ра[Р(С 6Н5)з]4 і 132 мг комплексу 8О3піридин. Таким чином одержують 29,6 мг 1-пропенілтрифенілфосфонієвої солі 6-(сульфокси)-1,6діазабіцикло[3,2Д]окт-3-ен-7-ону. Цю сіль пропускають через колонку зі смолою DOWEX 50WX8 в Ка+-формі, елююючи водою, що містить 10% тетрагідрофурану. Зібраний продукт ліофілізують, одержуючи 13 мг цільової натрієвої солі брутто-формули C6H7N2O5SNa (Μ=242,19 г). Відповідний вихід становить 20%. МС (іонізація електронним розпиленням з утворенням негативних іонів) m/z: [М]=219. ПРИКЛАД 54 Натрієва сіль 6-(сульфокси)-1,6-діазабіцикло[3,2,1]октан-7-ону Стадія А: Слідують методиці, вказаній у разі стадії А прикладу 53, виходячи з 12 г (0,061 моль) 1,1-диметилетил-3,6дигідро-3-оксо-1(2Н)-піридинкарбоксилату брутто-формули C10H15NO 3, 9,7 г гідрохлориду Обензилгідроксиламіну і 15 мл піридину. Таким чином одержують 19,4 г 1,1-диметилетил-3,6-дигідро-3[(фенілметокси)іміно]-1(2Н)-піридинкарбоксилату брутто-формули С 17Н22^Оз (М=302,38 г). Відповідний вихід є кількісним. Стадія В: Слідують методиці, вказаній у разі стадії В прикладу 53, виходячи з 14,9. г (0,0496 моль) одержаного на стадії А продукту, 12 г ціаноборгідриду натрію і 30 мл ефірату трифториду бору. Таким чином, після очищення, одержують 8,2 г суміші з 2/3 1,1-диметилетил-3,6-дигідро-3(фенілметокси)аміно]-1(2Н)-піридинкарбоксилату і 1/3 1/1-диметилетил-3-[(фенілметокси)асміно]-1-піперидинкарбоксилату брутто-формули C17H24N2 O3 (М=304,39 г). Відповідний вихід становить 55%. Стадія С: Слідують методиці, вказаній у разі стадії С прикладу 53, виходячи з 9,3 г (0,0306 моль) одержаної на стадії В суміші і 106 мл розчину хлороводню в етилацетаті з концентрацією 7 моль/л. Таким чином одержують 8,39 г суміші з 2/3 дигідрохлориду К-(фенілметокси)-1,2,3,6-тетрагідро-3-піридинаміну і 1/3 дигідрохлориду >«[(фенілметокси)-3-піперидинаміну брутто-формули C12H18Cl2N2O (M=277,20 г). Відповідний вихід становить 98%. Стадія D: Слідують методиці, вказаній у разі стадії D прикладу 53, виходячи з 8,30 г (0,0299 моль) одержаної на стадії С суміші і 30 мл 2н розчину гідроксиду натрію. Таким чином одержують 5,95 г суміші з 2/3 1чї(фенілметокси)-1,2,3,6-тетрагідро-3-піридинаміну і 1/3 N-(фенілметокси)-3-піперидинаміну брутто-формули
ДивитисяДодаткова інформація
Назва патенту англійськоюAzabicyclic compounds, a method for the preparation thereof and use thereof as medicines, in particular as antibacterial agents
Автори англійськоюAszodi Jozsef, Rowlands David Alan, Fromentin Claude
Назва патенту російськоюАзабициклические соединения, способ их получения и их применение в качестве лекарственных средств, в частности, в качестве антибактериальных средств
Автори російськоюАсзоди Жозеф, Роулендс Девид Ален, Фроментен Клод
МПК / Мітки
МПК: A61K 31/529, C07D 498/08, A61P 11/00, C07B 61/00, A61K 31/4188, A61P 9/10, A61P 31/00, A61P 31/04, A61K 31/5386, A61K 31/439, C07D 487/08, C07D 471/08, A61P 17/00
Мітки: лікарських, спосіб, застосування, антибактеріальних, сполуки, зокрема, азабіциклічні, одержання, засобів
Код посилання
<a href="https://ua.patents.su/32-73791-azabiciklichni-spoluki-sposib-kh-oderzhannya-i-kh-zastosuvannya-yak-likarskikh-zasobiv-zokrema-yak-antibakterialnikh-zasobiv.html" target="_blank" rel="follow" title="База патентів України">Азабіциклічні сполуки, спосіб їх одержання і їх застосування як лікарських засобів, зокрема, як антибактеріальних засобів</a>
Похідні фенілімідазолідинів, спосіб їх одержання (варіанти), фармацевтична композиція і їх застосування як лікарських засобів

Номер патенту: 64691
Опубліковано: 15.03.2004
Автори: ГУБЕ Франсуа, Клосснер Андре, Тетч Жан-Жорж
МПК: A61K 31/415, C07D 235/02, C07D 233/78
Мітки: композиція, фармацевтична, фенілімідазолідинів, одержання, варіанти, лікарських, застосування, похідні, засобів, спосіб
Формула / Реферат:
1. Производные фенилимидазолидинов общей формулы (I):, (I)в которой:Y означает атом кислорода или радикал NH, Z2 означает трифторметил и Z1 означает циано или нитро,Х означает атом кислорода или серы,R3 означает атом водорода или алкил, содержащий не более 4 атомов углерода, возможно замещенный одним или несколькими радикалами,...
Похідні ксантону, їх одержання та використання як лікарських засобів

Номер патенту: 72260
Опубліковано: 15.02.2005
Автори: Несснер Герхард, Сантісук Тайватчай, Нікель Бернд, Кленнер Томас, Шмідт Юрген, Хозе Себастьян, Реутракул Вічай
МПК: A61P 43/00, C07D 493/20, A61P 35/00, A61K 31/352, C07D 493/22
Мітки: засобів, ксантону, лікарських, використання, одержання, похідні
Формула / Реферат:
1. Сполука загальної формули І,(І)деR1 - атом водню; метил(-СН3), -С2-С6-алкіл, форміл(-СНО); ацетил(-СОСН3), -СО-С2-С6-алкіл, -СО-С3-С8-циклоалкіл, -СО-С6-С18-арил або -СО-С7-С24-аралкіл, кожен з яких факультативно несе один або декілька замісників, вибраних із групи, яка складається з таких радикалів: -ОН, -SH, -NH2, -NHC1-С6-алкіл, -N(С1-С6-алкіл)2, -NНС6-С14-арил, -N(С6-С14-арил)2, -N(С1-С6-алкіл)(С6-С14-арил),...
Аміди антранілової кислоти та їхнє застосування як лікарських засобів

Номер патенту: 71587
Опубліковано: 15.12.2004
Автори: Зайдельманн Дітер, Болд Гвідо, Фюре Паскаль, Шірнер Міхаель, Манлі Пол Вілліам, Оттов Екхард, Крюгер Мартін, Местан Юрген, Хут Андреас, Вуд Жанетт Маржорі, Тіраух Карл-Хайнц, Менрад Андреас, Феррарі Стефано, Брюгген Йозе
МПК: A61K 31/443, A61P 17/06, A61K 31/4436, C07D 213/36, A61K 31/166, A61P 13/12, A61K 31/444, C07D 265/26, C07D 403/12, C07D 213/75, A61K 31/4725, A61P 9/14, A61K 31/4433, A61P 35/00, C07C 237/30, A61P 43/00, C07D 401/14, A61K 31/4409, C07D 409/12, C07D 213/40, A61K 31/4709, A61P 19/02, A61K 31/506, A61K 31/517, C07D 209/04, C07D 401/12, C07C 237/32, A61P 27/06, A61K 31/404, C07D 417/12, A61K 31/536, A61P 3/10, C07D 405/12, A61P 9/10, A61K 31/4427, A61K 31/4439, A61P 27/02, A61P 1/16, A61K 31/403, C07D 413/12
Мітки: антранілової, лікарських, засобів, кислоти, застосування, аміди, їхнє
Формула / Реферат:
1. Сполуки загальної формули І (І),деА означає групу = NR2,W означає кисень, сірку, два атоми водню або групу = NR8,Ζ означає групу = NR10, =Ν- або -N(R10)-(CH2)q, розгалужений або нерозгалужений С1-С6алкіл або групу,або А, Ζ і R1 разом...
Похідні ехінокандину, спосіб їх одержання, їх застосування, проміжні сполуки та фармацевтична композиція

Номер патенту: 72200
Опубліковано: 15.02.2005
Автори: Фово Патрік, Шио Лоран, Мелон Мангер Домінік, Маркус Астрид, Куртен Олів'є, Мішель Жан-Марк
МПК: C07K 7/56, A61P 31/10, A61K 38/04
Мітки: одержання, похідні, фармацевтична, застосування, проміжні, сполуки, композиція, спосіб, ехінокандину
Формула / Реферат:
1. Похідні ехінокандину формули (І):(І),їх можливі ізомерні форми або їх суміші, в яких:або R1 і R2, однакові або відмінні один від одного, означають атом водню, гідроксил; лінійний, розгалужений або циклічний алкільний, алкенільний або алкінільний радикал, що містить до 8 атомів вуглецю, що можливо переривається атомом кисню, можливо заміщений атомом...
Спіро-азабіциклічні сполуки, способи їх одержання та фармацевтична композиція

Номер патенту: 54375
Опубліковано: 17.03.2003
Автори: Гріффіт Роналд Конрад, Балєстра Майкл, Мюррей Роберт Джон, Гордон Джон Чарлз
МПК: A61P 43/00, A61P 25/22, A61P 25/04, A61P 25/28, A61K 31/439, A61P 25/18, C07D 487/08, A61K 31/424, A61P 25/24, A61P 25/26, C07D 453/00, C07D 498/20, A61K 31/42
Мітки: спіро-азабіциклічні, одержання, способи, сполуки, композиція, фармацевтична
Формула / Реферат:
1. Спиро-азабициклические соединения формулы I , (I)гдеR представляет водород или метил, иn = 1 или 2; или их фармацевтически приемлемые соли присоединения кислоты или энантиомеры.2. Соединение по п. 1, в котором R представляет водород, или его фармацевтически приемлемая соль присоединения кислоты или...
Попередній патент: Спосіб виготовлення теплоізоляційного і/або звукоізоляційного виробу на основі мінеральної вати, теплоізоляційний і/або звукоізоляційний виріб, виготовлений цим способом
Наступний патент: Гідростатичний привід сільськогосподарської машини
Випадковий патент: Привід керування прохідного дискового крана