Спосіб отримання 2-арил-3-арил-5-галогенпіридинів, що використовуються як інгібітори сох-2 (варіанти)
Номер патенту: 66792
Опубліковано: 15.06.2004
Автори: Сейджер Джесс, Маліакал Ашок, Россен Кай, Воланте Ральф П., Пайє Філіп Дж.








Формула / Реферат
1. Спосіб одержання сполук формули I
, І
де R1 вибирають з групи, що складається з
(a) СН3,
(b) NH2,
(с) NНС(О)СF3,
(d) NНСН3;
Аr являє собою моно-, ди- або тризаміщений феніл або піридиніл (або його N-оксид), де замісники вибирають з групи, що складається з
(a) водню,
(b) галогену,
(с) С1-4алкокси,
(d) С1-4алкілтіо,
(е) CN,
(f) С1-4алкілу,
(g) С1-4фторалкілу;
R2 вибирають з групи, що складається з
(a) F, Cl, Br, I,
(b) CN,
(с) азиду;
при якому
сполуку формули А1
А1
конденсують в кислих умовах і, необов'язково, в присутності нереакційноздатного розчинника, і в присутності амонієвого реагенту, із сполукою А2
з одержанням сполуки формули I.
2. Спосіб за п.1, в якому нереакційноздатний розчинник являє собою оцтову кислоту.
3. Спосіб за п.1, в якому Аr являє собою моно-, ди- або тризаміщений 3-піридиніл.
4. Спосіб за п.1, в якому R1 являє собою СН3 або NH2.
5. Спосіб за п.1, в якому Аr являє собою моно- або дизаміщений 3-піридиніл, і замісники вибрані з групи, що складається з
(a) водню,
(b) галогену,
(с) C1-3алкокси,
(d) C1-3алкілтіо,
(е) C1-3алкілу,
(f) СF3 і
(g) CN.
6. Спосіб за п.1, в якому R1 являє собою СН3 або NH2; і Аr являє собою моно- або дизаміщений 3-піридиніл, і замісники вибрані з групи, що складається з
(a) водню,
(b) галогену,
(с) C1-3алкілу,
(d) СF3 і
(е) CN.
7. Спосіб за п.1, в якому R2 являє собою Сl; R1 являє собою СН3 або NH2; Аr являє собою монозаміщений 3-піридиніл, і замісники вибрані з групи, що складається з водню і C1-3алкілу.
8. Спосіб за п.1, в якому R2 являє собою хлор.
9. Спосіб за п.1, в якому амонієвий реагент вибирають з аміаку і ацетату амонію.
10. Спосіб одержання сполук формули I, що використовуються для лікування запальних та інших захворювань, опосередкованих циклооксигеназою-2
, I
де
R1 вибирають з групи, що складається з
(a) СН3,
(b) NH2,
(с) NНС(О)СF3,
(d) NНСН3;
Аr являє собою моно-, ди- або тризаміщений феніл або піридиніл (або його N-оксид), де замісники вибирають з групи, що складається з
(a) водню,
(b) галогену,
(с) С1-4алкокси,
(d) С1-4алкілтіо,
(е) CN,
(f) C1-4алкілу,
(g) C1-4фторалкілу;
R2 вибирають з групи, що складається з
(a) F, Cl, Вr, I,
(b) CN,
(с) азиду;
що включає стадії:
(а) сполуку формули А2
піддають взаємодії в присутності другого нереакційноздатного розчинника з сильною основою з одержанням еноляту формули В1
, В1
де М являє собою калій, літій або натрій, і
(b) сполуку формули В1 піддають взаємодії в присутності третього нереакційноздатного розчинника із сполукою формули В2
, B2
де R3 являє собою групу, що відходить, таку як тозил, мезил або галоген, яка після нагрівання в присутності амонієвого реагенту дає сполуку формули I.
11. Спосіб за п.10, в якому Аr являє собою моно- або дизаміщений 3-піридиніл.
12. Спосіб за п.10, в якому R1 являє собою СН3 або NH2.
13. Спосіб за п.10, в якому Аr являє собою моно- або дизаміщений 3-піридиніл, і замісники вибрані з групи, що складається з
(a) водню,
(b) галогену,
(с) C1-3алкокси,
(d) C1-3алкілтіо,
(е) C1-3алкілу,
(f) СF3 і
(g) CN.
14. Спосіб за п.10, в якому R1 являє собою СН3 або NH2; і Ar являє собою моно- або дизаміщений 3-піридиніл, і замісники вибрані з групи, що складається з
(a) водню,
(b) галогену,
(с) C1-3алкілу,
(d) СF3 і
(е) CN.
15. Спосіб за п.10, в якому R2 являє собою Сl; R1 являє собою СН3 або NH2; Ar являє собою монозаміщений 3-піридиніл, і замісники вибрані з групи, що складається з водню і C1-3алкілу.
16. Спосіб за п.10, в якому R3 являє собою хлор.
17. Спосіб за п.10, в якому амонієвий реагент вибирають з аміаку і ацетату амонію.
18. Спосіб за п.10, в якому сильна основа являє собою біс(триметилсиліл)амід літію.
19. Спосіб за п.10, в якому третій нереакційноздатний розчинник являє собою толуол.
Текст
Даний винахід відноситься до способу одержання деяких протизапальних сполук. Зокрема, заявка стосується способу одержання сполук формули І, як описано далі, які є сильнодіючими інгібіторами циклооксигенази-2. Нестероїдні протизапальні лікарські засоби чинять протизапальну, болезаспокійливу та жарознижуючу дію та інгібують гормон-індуковані скорочення матки і зростання деяких видів ракових пухлин за рахунок інгібування простагландин G/H синтази, також відомої як циклооксигеназа. Донедавна була охарактеризована тільки одна форма циклооксигенази, яка відповідала циклооксигеназі-1 або конститутивному ферменту, спочатку ідентифікованому в бичачих сім'яних пухирцях. Нещодавно був клонований, секвенований та охарактеризований ген другої індукуємої форми циклооксигенази (циклооксигеназа-2) з джерел, отриманих від курчати, миші та людини. Цей фермент відрізняється від циклооксигенази-1, яка в цей час також була клонована, секвенована та охарактеризована з джерел, отриманих від вівці, миші та людини. Друга форма циклооксигенази, циклооксигеназа-2, швидко та легко індукується рядом агентів, включаючи мітогени, ендотоксин, гормони, цитокіни та фактори росту. Оскільки простагландини грають як фізіологічну, так та патологічну ролі, автори даного винаходу зробили висновок, що конститутивний фермент, циклооксигеназа-1, є відповідальним, переважно, за ендогенне базальне вивільнення простагландинів, і, отже, він важливий для їх фізіологічних функцій, таких як підтримка працездатності шлунково-кишкового тракту та ниркоподібного кровотоку. У протилежність цьому, авторами зроблений висновок про те, що індукуєма форма циклооксигеназа-2, головним чином є відповідальною за патологічну дію простагландинів, коли відбувається швидке індукування ферменту у відповідь на такі агенти, як запальні агенти, гормони, фактори росту і цитокіни. Таким чином, селективний інгібітор циклооксигенази-2 буде володіти аналогічними протизапальними, жарознижуючими і болезаспокійливими властивостями, що і звичайні нестероїдні протизапальні лікарські засоби, і, крім того, він буде інгібувати гормон-індуковані скорочення матки і володіти потенційною протираковою дією, але буде мати знижену здатність індукувати деякі засновані на механізмі побічні ефекти. Зокрема, така сполука повинна володіти зниженим потенціалом відносно шлунково-кишкової токсичності, зниженої можливістю надання побічної дії на нирки, зниженою дією на час кровотечі і, можливо, зниженою здатністю індукувати приступи астми у аспірин-чутливих суб'єктів-астматиків. У W0 96/24585, опублікованому 15 серпня 1996 року, і W0 96/10012, опублікованому 4 квітня 1996 року, описані способи одержання 2-арил-3-арилпіридинів. У винаході, розкритому нижче, 2-арил-3-арилпіридини одержують простою для здійснення одностадійною конденсацією з легко доступних вихідних матеріалів. Таким чином, цей спосіб виявився несподівано зручним і більш ефективним, ніж описаний раніше спосіб, в якому 2-арил-З-арилпіридин одержували послідовним постадійним введенням арильних груп в центральне піридинове кільце. Більш того спосіб за даним винаходом також має ту несподівану перевагу, що не вимагає ні дорогих паладієвих реагентів, ні проведення ряду послідовних реакцій введення захисних груп/зняття захисних груп, як в способі, описаному в попередньому рівні техніки. Одержання 2-хлормалондіальдегіду було вперше здійснене Дікманном в 1904 році (W. Dieckmann, L. Platz, Ber. Deut. Chem. Ges. 1904, 37, 4 638). З хімії 2-галогенмалондіальдегідів є докладний огляд 1975 року (С. Reichardt and К. Halbritter, Angew. Chem. Int. Ed. 1975, 14, 86). У цьому огляді не вказаний синтез піридину з використанням таких реагентів. Єдине застосування 2-хлормалондіальдегіду для одержання піридину, що згадується - це нещодавня патентна заявка (F.J. Urban, US 5206367, Pfizer and Brackeen Μ., і Howard, H.R., Заявка на Європейський патент номер 89307339.5 (ЕР 0352959), Pfizer), де хлормалондіальдегід спочатку перетворюють в 2,3-дихлоракролеїн, який потім конденсують з енаміном, одержаним з 1,3-циклогександіону, з утворенням циклічного піридину з 28% виходом. У нещодавньому вичерпному огляді з синтезу і реакційної здатності піридину (D. Spitzner, в Methoden der Organischen Chemie (Houben-Weyl), стор.286-686, тому Е 7b/ Editor R.P.Kreher, 1992, Georg Thieme Verlag) не наводиться прикладів застосування галогенмалондіальдегідів для синтезу піридину. Нітромалондіальдегід конденсували з етил-2-амінокротонатом, одержуючи 5-нітропіридин, хоч і з невисоким виходом (35-50%) (J.M.Hoffman et al., J.Org. Chem., 1984, 49, 193 і P.Ε.Fanta, J.Am. Chem. Soc, 1953, 75, 737). Застосування похідних етоксикарбонілмалондіальдегіду дає5-етоксикарбонілпіридини (S. Torii et al., Synthesis, 1986, 400). Короткий виклад суті винаходу Винахід включає спосіб одержання сполук формули І, що використовуються для лікування запальних захворювань та інших захворювань, опосередкованих циклооксигеназою-2. Докладний опис винаходу У одному з своїх аспектів винахід включає спосіб одержання сполук формули І, що використовуються для лікування запальних захворювань та інших захворювань, опосередкованих циклооксигеназою-2. де R1 вибирають з групи, що складається з (a) СН3, (b) NH2, (c) NHC(O)CF3, (d) NHCH3; Аr являє собою моно-, ди- або тризаміщений феніл або піридиніл (або його N-оксид), де замісники вибирають з групи, що складається з (a) водню, (b) галогену, (c) С1-4алкоксі, (d) С1-4алкілтіо, (e) CN, (f) С1-4алкілу, (g) С1-4фторалкілу, R2 вибирають з групи, що складається з (a) F, СІ, Br, I (b) CN, (c) азиду, який включає: конденсацію сполуки формули А1 в кислих умовах і, необов'язково, в присутності нереакційноздатного розчинника і в присутності амонієвого реагенту, із сполукою А2 з отриманням сполуки формули І. Як очевидно для фахівців в даній області, в загальному випадку реагенти самі по собі забезпечують кислі умови. Таким чином, застосування не вступаючої в реакцію кислоти не є необхідним. Однак, додавання кислоти, такої як оцтова або пропіонова, або іншої карбонової кислоти, входить в об'єм даного винаходу. У даному описі термін нереакційноздатний розчинник включає тетрагідрофуран, діоксан, С1-6алканол і толуол. У даному описі термін амонієвий реагент включає аміак і солі амонію, такі як ацетат амонію і пропіонат амонію. Більш того термін амонієвий реагент включає суміш різних амонієвих реагентів. Молярне співвідношення сполук А1 і А2 звичайно змінюється від 2:1 до 1:2, переважно 1:1 до 1,5. Звичайно використовують надлишок сполуки А1. Молярне співвідношення сполуки А1 і амонієвого реагенту звичайно змінюється від 1:1 до 1:10. Взаємодію звичайно можна провести при температурі в діапазоні від 40 до 180°С, переважно від 80 до 140°С, і часі, що дозволяє провести взаємодію практично до повного завершення, що складає від 2 до 18 годин, звичайно від 6 до 12 годин. У іншому аспекті винахід включає спосіб одержання сполук формули І, що використовуються для лікування запальних та інших захворювань, опосередкованих циклооксигеназою-2. де R1 вибирають з групи, що складається з (a) СН3, (b) NH2, (c) NHC(O)CF3, (d) NHCH3; Аr являє собою моно-, ди- або тризаміщений феніл або піридиніл (або його N-оксид) , де замісники вибирають з групи, що складається з (a) водню, (b) галогену, (c) С1-4алкоксі, (d) С1-4алкілтіо, (e) CN, (f) С1-4алкілу, (g) С1-4фторалкілу, R2 вибирають з групи, що складається з (a) F, СІ, Br, I (b) CN, (c) азиду, який включає: (а) взаємодію сполуки формули А2 в присутності другого нереакційноздатного розчинника з сильною основою з одержанням еноляту формули В1 де Μ являє собою калій, літій або натрій. У даному описі термін сильна основа включає диізопропіламід літію, калію або натрію, біс(триметилсиліл)амід літію, калію або натрію, гідрид літію, калію або натрію та амід літію, калію або натрію. У даному описі термін другий нереакційноздатний розчинник включає тетрагідрофуран, діоксан, толуол і простий ефір. Молярне співвідношення сполуки А2 і основи звичайно можна змінювати від 1:1 до 1:1,5. Звичайно використовують надлишок основи. Реакційну стадію звичайно можна провести при температурі в діапазоні від -80 до 40°С; переважно від -10 до 20°С, і часу, який дозволяє провести реакцію до практично повного завершення, що складає від 1 до 3 годин; звичайно протягом від 1 до 2 годин. (b) взаємодію сполуки формули В1 в присутності третього нереакційноздатного розчинника зі сполукою формули В2 де R3 являє собою групу, що відходить, таку як тозил, мезил або галоген, яка після нагрівання в присутності амонієвого реагенту дає сполуку формули І. Для використання в даній реакції третій нереакційноздатний розчинник являє собою тетрагідрофуран, толуол і діоксан. Молярне співвідношення сполуки В1 і 2,3-дихлоракролеїну звичайно може змінюватися від 1:1,5 до 1,5:1, переважно від 1:1 до 1,5. Звичайно використовують надлишок 2,3-дихлоракролеїну. Реакційну стадію звичайно можна провести при температурі в діапазоні від 0 до 80°С; переважно від 20 до 50°С, і часі, що дозволяє провести взаємодію практично до повного завершення, що складає від 2 до 18 годин; звичайно від 4 до 12 годин. У обох варіантах винаходу R2 переважно являє собою галоген, найбільш переважно F або СІ, найбільш переважно СІ. Переважно, щоб R3 був таким же, як і R2. У обох варіантах винаходу переважним підкласом сполук формули І є той, в якому Аг являє собою моноабо дизаміщений піридиніл. У рамках цього підкласу особливо переважні ізомери 3-піридинілу. Знову ж, в обох варіантах винаходу іншим переважним підкласом сполук формули І є той, в якому R1 являє собою СН3 або NH2. Звичайно, СН3 є переважним для специфічності відносно СОХ-2, a NH2 є переважним для сили дії. Знову ж, в обох варіантах винаходу іншим переважним підкласом сполук формули І є той, в якому Аr є незаміщеним або заміщеним СН3 . Сполуки формули І використовується для полегшення болю, жару і запалення при безлічі хворобливих станів, включаючи ревматичну лихоманку, симптоми, пов'язані з грипом або іншими вірусними інфекціями, загальну простуду, біль в поперековій області і в області шиї, дисменорею, головний біль, зубний біль, різні види розтягнення, міозит, невралгію, синовіт, артрит, включаючи дегенеративні захворювання суглобів при ревматоїдному артриті (остеоартрит), подагру і анкілозуючий спондиліт, бурсит, опіки, рани після хірургічного втручання і стоматологічних процедур. Крім того, така сполука може інгібувати пухлинні переродження кліток і метастазне зростання пухлини і, отже, його можна використати при лікуванні раку. Сполуки формули І також можуть використовуватися для лікування слабоумства, включаючи пресенильне і старече недоумство і, зокрема, недоумство, пов'язане з хворобою Альцгеймера (тобто недоумство Альцгеймера). За рахунок високої активності у відношенні циклооксигенази-2 (СОХ-1) і/або селективності до циклооксигенази-2 в порівнянні з циклооксигеназою-1 (СОХ-1), як вказано вище, сполуки формули І є корисними як альтернатива звичайним нестероїдним протизапальним лікарським засобам (НСПВС), особливо коли є протипоказання до таких нестероїдних протизапальних лікарських засобів, як, наприклад, для пацієнтів з пептичними виразками, гастритом, регіональним ентеритом, виразковим колітом, дивертикулезом або з рецидивуючими шлунково-кишковими виразками; при шлунково-кишковій кровотечі, захворюваннях здатності скипатися крові, включаючи анемію, таку як гіпопротромбінова анемія, гемофілія, або при інших проблемах кровообігу (включаючи ті, які відносяться до пониження або порушення функції тромбоцитів); при ниркоподібних захворюваннях (наприклад, ниркоподібна недостатність); перед хірургічним втручанням або прийомі антикоагулянтів; і для пацієнтів з НСПВС-індукованою астмою. Сполуки за даним винаходом є інгібіторами циклооксигенази-2 і тому корисні для лікування захворювань, опосередкованих циклооксигеназою-2, перерахованих вище. Така активність проілюстрована їх здатністю селективно інгібувати циклооксигеназу-2 в порівнянні з циклооксигеназою-1. Відповідно в одному з аналізів здатність сполук за даним винаходом до лікування захворювань, опосередкованих циклооксигеназою, може бути продемонстрована шляхом вимірювання кількості простагландину Е2 (PGE2), синтезованого в присутності арахідонової кислоти, циклооксигенази-1 або циклооксигенази-2 і сполуки формули І. Значення ІС50 показують концентрацію інгібітора, необхідну для звертання синтезу PGE2 на 50% в порівнянні з контрольним значенням, отриманим без інгібування. Ілюструючи цей аспект, встановили, що сполуки з прикладів ефективні більш ніж в 100 разів при інгібуванні СОХ-2, ніж при інгібуванні СОХ-1. Крім того, для всіх з них значення ІС50 для СОХ-2 складає від 1нМ до 1мМ. Для порівняння, Ібупрофен має значення ІС50 для СОХ-2, рівне 1мМ, а Індометацин має ІС50 для СОХ-2 приблизно 100нМ. Для лікування будь-яких захворювань, опосередкованих циклооксигеназою, сполуки формули І можуть вводитися перорально, місцево, парентерально, шляхом інгаляційного розпилення або ректально у вигляді препаративних форм з одиничною дозою, що містить звичайні нетоксичні фармацевтично прийнятні носії, ад'юванти і розчинники. Як використано в даному описі, термін "парентеральне введення" включає підшкірні ін'єкції, внутрішньовенні, внутрішньом1язові, внутрішньогрудинні ін'єкції або інфузійні методики. Крім лікування теплокровних тварин, таких як миші, пацюки, коні, велика рогата худоба, вівці, собаки, кішки і т.д., сполука за даним винаходом ефективна для лікування людей. Винахід далі проілюстрований наступними необмежуючими прикладами, в яких, якщо не вказано інакше: (і) всі операції проводили при кімнатній температурі або температурі навколишнього середовища, тобто при температурі в діапазоні 18-25°С; випаровування розчинника здійснювали з використанням роторного випарника при зниженому тиску (600-4000 паскаль: 4/5-30мм рт.ст.) при температурі бані до 60°С; протікання реакцій контролювали за допомогою тонкошарової хроматографії (ТШХ) або високоефективної рідинної хроматографії (ВЕЖХ); час реакції приведений тільки для ілюстрації; температури плавлення не коректували, і "різн." вказує розкладання; приведені температури плавлення дані для речовин, одержаних описаним способом; в деяких способах одержання поліморфізм може привести до виділення речовин, отриманих за тими ж методиками, з іншими температурами плавлення; будова і чистота одержаних продуктів були підтверджені, принаймні, одним з наступних способів: ТШХ, мас-спектроскопія, спектроскопія ядерного магнітного резонансу (ЯМР) або дані мікроаналізу; виходи приведені тільки для ілюстрації; коли вони приводяться, дані ЯМР представлені у вигляді значень дельта (d) для основних тестових протонів в мільйонних частках (м.ч.) відносно тетраметилсилану (ТМС) як внутрішній стандарт, які зареєстровані при 300МГц або 400МГц з використанням вказаного розчинника; для позначення форми сигналу використали стандартні абревіатури: с - синглет; д - дублет; τ - триплет; м - мультиплет; роз. розширений сигнал; і т.д., крім того, "Аr" означає ароматичні сигнали; хімічні символи мають звичайні значення; використані наступні абревіатури: об. (об'єм), вага. (вага), т. кип. (температура кипіння), т. пл. (температура плавлення), л (літр(и)), мл (мілілітри), г (грам(и)), мг (міліграм(и)), моль (молі), ммоль (мілімолі), екв. (еквівалент(и)). Наступні абревіатури мають вказані значення: Абревіатури алкільних груп: Me = метил Et = етил n-Рr = нормальний пропіл і-Рr = ізопропіл n-Bu = нормальний бутил і-Bu = ізобутил s-Bu = вторинний бутил t-Bu = третинний бутил с-Рr = циклопропіл с-Bu = циклобутил с-Реn = циклопентил с-Нех = циклогексил Приклад 1 5- Хлор-3-(метилсульфоніл)феніл-2-(3-піридил)піридину; 2- Хлормалондіальдегід 4,8г (0,045моль) Кетон В 5,0г (0,018моль) Пропіонова кислота 30мл Ацетат амонію 8,4г (0,11моль) Суміш кетону В (5,0г), 2-хлормалондіальдегіду (4,8г) і ацетату амонію нагрівали до 130°С. Оцтову кислоту, що утворюється, відганяли і нагрівання продовжували при 136°С протягом 15 годин. Реакційну суміш підлуговували карбонатом натрію, додавали воду і продукт екстрагували дихлорметаном (2x150мл). Органічні шари обробляли активованим вугіллям (Dowex), сушили (MgSO4) і видаляли розчинник, одержуючи сполуку 1 у вигляді білої твердої речовини (3,4г, 55% вихід). 2- Хлормалондіальдегід 220мг (2,1ммоль) Оксалілхлорид 180мл (2,1ммоль) Толуол 3мл N/N-Диметилформамід 20мл Ν,Ν-Диметилформамід додавали до суспензії 2-хлор-малондіальдегіду (220мг) в толуолі. Додавали оксалілхлорид і реакційну суміш перемішували до повного розчинення. Кетон В 500мг (1,8ммоль) Біс(триметилсиліл)амід літію (1M в ТГФ) 1,8мл (1,8ммоль) Тетрагідрофуран 15мл 2,3-Дихлоракролеїн в толуолі 2,1ммоль в 3мл толуолу Ацетат амонію 1,0г Біс(триметилсиліл)амід літію (1,8мл; 1M в ТГФ) додавали по краплях до кетону В (500мг) в ТГФ (15мл) при 78°С. Реакційну суміш нагрівали до температури навколишнього середовища протягом 1 години з одержанням літієвого еноляту В (див. загальну формулу В1), потім охолоджували до -78°С. Додавали розчин 2,3 дихлоракролеїну і температуру доводили до кімнатної. Через 1 годину через розчин пропускали газоподібний аміак і через 30 хвилин додавали ацетат амонію (1г). Реакційну суміш нагрівали при 60°С протягом 1 години і виливали у водний розчин гідроксиду натрію (2М; 100мл). Продукт екстрагували дихлорметаном (2х150мл), сушили (MgSO4) і видаляли розчинник, отримуючи 1 (500мг; 80%). Одержання вихідних речовин Приклад одержання 1 Синтез 4-метилсульфонілфенілоцтової кислоти Тіоанізол 2(ΜΒ=124,2; d=1,058) Етілоксалілхлорид (МВ=136,5;d=1,222) 50,00г(0,403моль, 47,3мл) 82,43г (0,604моль, 67,5мл) Хлорид алюмінію (МВ=133,3) 75,13г (0,564моль) Діхлорбензол (ОДХБ) 112мл Етилоксалілхлорид і ОДХБ завантажували в колбу, обладнану підвісною механічною мішалкою, і охолоджували до 0°С. Повільно додавали АlСl3. Додавання АlСl3 являло собою екзотермічний процес. Через краплинну воронку протягом 1,5 годин додавали по краплях тіоанізол 2. Реакційна суміш швидко набувала темно-фіолетового забарвлення. Це додавання також було екзотермічним. Через 1 год за даними ВЕЖХ реакція завершилася. Реакцію зупиняли повільним додаванням 300 мл 1н НСl при 0°С. Після нагрівання до кімнатної температури додавали воду і ОДХБ (по 50мл кожного). Шари змішували і розділяли. Органічну (нижню) фазу промивали 1x250мл води і потім сушили над MgSO4. Зупинка реакції також була екзотермічною. Під час зупинки колір реакційної суміші змінювався від темнофіолетового до блідо-зеленого. Висушений розчин ОДХБ завантажували в колбу Нортона, обладнану механічною мішалкою. Додавали 1н розчин NaOH (800мл) . Двофазну суміш інтенсивно перемішували і нагрівали до 50°С. Гідроліз з утворенням сполучення завершувався за даними ВЕЖХ за 2-3 години. Водну фазу, що містить продукт, безпосередньо вводили в реакцію Вольфа-Кишнера. 4-(Метилтіо)фенілоцтова кислота 3 (в 1н розчині NaOH) (0,402моль) Гідразин (МВ=32,1,35вага. 206/14г % у воді) (2252моль204мл) NaOH (5н розчин) 5мл Гідразин і NaOH завантажували в колбу Нортона, обладнану механічною мішалкою. Після нагрівання розчину гідразину до 75°С протягом 35-40 хвилин додавали розчин 3 в NaOH. Після завершення додавання реакційну суміш кип'ятили із зворотним холодильником протягом 5 днів. За даними ВЕЖХ до цього моменту реакція протікала приблизно на 95%. Вихідна речовина переважно витрачалася протягом 24 годин, однак третій пік вимагає декількох днів для перетворення в 4. Реакцію підкисляли концентрованою НСl до рН=1,5 і екстрагували EtOAc (1x750мл і 1x250мл) . Об'єднані, утримуючі продукт органічні фази промивали 2x250мл 1н НСl. При підкисленні реакційна суміш набувала яскраво-жовте забарвлення. 4-(Метансульфоніл)фенілоцтова кислота 4-(Метилтіо)фенілоцтова кислота 4 (МВ=182,3) (0,402моль) Na2W04·Н2O(МВ=329,9) 2,64г (0,008моль) Aliquat 336 (МВ=404) 8,08г (0,020моль) Перекис водню (МВ=34,0, 136г (1,200моль, 30вага.% у воді) 123мл) У колбу, обладнану механічною мішалкою, завантажували сполуку 3 (з реакції вище в EtOAc), Aliquat 336 і Na2W04·2H2O (розчинений приблизно в 15мл Н2О) . Повільно через краплинну воронку додавали перекис водню протягом приблизно 30хв. Завершення реакції контролювали ВЕЖХ. Реакційну суміш промивали 2x400мл Н2О і сушили над MgSO4. За кількісною оцінкою в органічному шарі містилося 61,29г сполуки 5 (71% вихід по тіоанізолу). При концентруванні розчину випадає осад білого твердого продукту. Суспензію відфільтровували і промивали гексанами. Виділено 49,02г 5 (57% по тіоанізолу). Конденсація Іванова-Кляйзена для одержання 1-(3-піридил)-2-(4-метилсульфонілфеніл)етан-1-ону 4-Метилсульфонілбензил-З-піридилкетон, одержаний з етилнікотинату і 4-метилсульфонілфенілоцтової кислоти 4Метилсульфонілфенілоцтова кислота (МВ=217) 10г (46,7ммоль) трет-Бутилмагнійхлорид 128,11мл (1н/ТГФ) (128,11ммоль) Етілнікотинат (МВ=151,2; 5,54мл d=1, 107) (39,4ммоль) ТГФ 400мл Фенілоцтову кислоту розчиняли в ТГФ в атмосфері азоту. Протягом 5 хвилин до розчину додавали 1,9 еквівалента (88,73мл) трет-бутилмагнійхлориду. Реакція була екзотермічною. Температура підіймалася від 20°С до 50°С. Після додавання першого еквівалента трет-бутилмагнійхлориду розчин набував червоного забарвлення. Температуру реакції підтримували рівною 50°С. Через 1 годину додавали 0,5 еквівалента етилнікотинату. Розчин набував жовтого забарвлення і утворювався білий осад. Через одну годину при 50°С додавали 0,5 еквівалента трет-бутилмагнійхлориду. Розчин набував червоного забарвлення. Послідовність додавань повторювали, використовуючи 0,25екв., 0,125екв., 0,0625екв. етилнікотинату і трет-бутилмагнійхлориду. Реакційну суміш витримували протягом 1 години між кожним додаванням. Після останнього додавання реакцію гасили, додаючи в реакційну суміш при інтенсивному перемішуванні 2н соляну кислоту (100 мл). Тверді речовини на дні реакційної суміші розчинялися з бурхливим виділенням газу при перемішуванні в соляній кислоті. рН водної фази реакційної суміші доводили до 10 карбонатом натрію. За даними ЖХ-аналізу вихід кетону становив 91%. Одержання 4-метилсульфонілбензальдегіду Одержання проводили за способом Ulman JOC, pp. 4691 (1989). 4-Метилсульфонілбензальдегід (2) з 4фторбензальдегіду 4-Фторбензальдегід 23,3мл МВ=124,11; d=1,157) (217ммоль) Метансульфінова кислота, 24,23г натрієва сіль (МВ=102,09) (237ммоль) Метилсульфоксид 170мл Реагенти додавали до метилсульфоксиду і нагрівали при 130°С протягом 18 годин. Метансульфінат натрію був частково нерозчинний при кімнатній температурі, але переходив в розчин при 130°С. З розчину випадав осад фториду натрію. Реакційну суміш виливали в 300мл води. Продукт випадав в осад у вигляді білої твердої речовини. Реакційну суміш відфільтровували. Виділений продукт промивали 100мл води і 2х50мл метанолу для видалення метилсульфоксиду. Розчинник випаровували з продукту при зниженому тиску, одержуючи 39,9г ,2 у вигляді білого порошку (вихід виділеного продукту 86%). 13С-ЯМР (CDCl3): 44,33, 128,25, 130,43, 139,70, 145,38, 190,72. 4-Метилсульфонілбензальдегід (2) з 4-хлорбензальдегіду 4-Хлорбензальдегід (МВ=140,57) Метансульфінова кислота, натрієва сіль (МВ=102,09) 6,31г (45ммоль) 7,5г (74ммоль) Метилсульфоксид 50мл Реагенти додавали до метилсульфоксиду і нагрівали при 130°С протягом 18 годин. Метансульфінат натрію був частково нерозчинний при кімнатній температурі, але переходив в розчин при 130°С. З розчину випадав осад хлориду натрію. Реакційну суміш виливали в 100мл води. Продукт випадав в осад у вигляді білої твердої речовини. Реакційну суміш відфільтровували. Виділений продукт промивали 50мл води і 2x25мл метанолу для видалення метилсульфоксиду. Розчинник випаровували з продукту при зниженому тиску, одержуючи 5,1г 4-метилсульфонілбензальдегіду у вигляді білого порошку (вихід виділеного продукту 62%). Спосіб Хорнера/Віттіга для одержанняя 1-(3-піридил)-2-(4-метилсульфонілфеніл)етан-1-ону Посилання: Н. Zimmer, J.P. Bercz, Liebigs Ann. Chem. 1965, 686, 107-114. Анілін 89,4г (0,96моль) 3- піридинкарбоксальдегід 102,8г (0,96моль Етанол 150мл Дифенілфосфіт 224,7г (0,96моль Розчин аніліну в етанолі (50мл) додавали до розчину 3-піридинкарбоксальдегіду в етанолі (100мл) при 0°С. Через 2 години додавали дифенілфосфіт і продовжували перемішування при кімнатній температурі протягом 18 годин. Для додаткового осадження продукту додавали метил-трет-бутиловий простий ефір (МТБЕ) (400мл), продукт відфільтровували, промивали (МТБЕ) і сушили у вакуумі, отримуючи 320г (80%) піридиламінодифенілфосфонату у вигляді білої твердої речовини. 13С-ЯМР (CDCl3): Піридиламінодифенілфосфонат 14,0г (0,034моль 10% КОН в МеОН 23мл (0,04моль) Тетрагідрофуран 150мл 4- Метансульфонілбензальдегід 5,6г (0,03моль) До розчину фосфонату (14,0г) в тетрагідрофурані при -45°С протягом 10 хвилин додавали 10% КОН/МеОН (23мл). Через 10 хвилин додавали однією порцією бензальдегід і через 1 годину нагрівали реакційну суміш до температури навколишнього середовища. Додавали водну соляну кислоту (2н, 100мл) і розчин залишали на 18 годин. Додавали EtOAc (200мл) і воду (200мл) і органічний шар відкидали. Кислий шар підлуговували (рН=9) карбонатом натрію і екстрагували дихлорметаном (2x150мл). Органічні шари об'єднували, сушили (MgSO4) і концентрували. Після розтирання з гексанами одержували 4-метилсульфонілбензил-3-піридилкетон у вигляді блідо-жовтої твердої речовини (6,3г; 76%). 13С-ЯМР (ДМСО-d6):196,4, 153,6, 149,4, 140,8, 139,1, 135,7, 131,5, 130,9, 126,8, 123,9, 44,6 і 43,5 м.ч. Анілін 4,47г (0,05моль) 3- Піридинкарбоксальдегід 5,36г (0,05моль) Метанол 10мл Дифенілфосфіт 11,2г (0,05моль) 10% КОН в МеОН 28мл (0,05моль) 4- Метансульфонілбензальдегід 8,3г (0,45моль) Розчин аніліну в метанолі (5мл) додавали до розчину 3-піридинкарбоксальдегіду в метанолі (5мл) при 0°С. Через 2 години додавали дифенілфосфіт і продовжували перемішування при кімнатній температурі протягом 18 годин. Додавали ТГФ (100мл) і реакційну суміш охолоджували до -40°С. Додавали 10% КОН/метанол (28мл) і через 30 хвилин додавали 4-метансульфонілбензальдегід. Реакційній суміші давали нагрітися до кімнатної температури і перемішували протягом 18 годин. Додавали EtOAc (200мл) і воду (200мл), органічний шар відкидали. Кислий шар підлуговували (рН=9) карбонатом натрію і екстрагували дихлорметаном (2х150мл). Органічні шари об'єднували, сушили (MgSO4) і концентрували. Розтирання з гексанами давало 4метилсульфонілбензил-З-піридилкетон у вигляді блідо-жовтої твердої речовини (9,7г; 71%). Одержання хлормалондіальдегіду Одержання хлормалондіальдегіду можна здійснювати різними шляхами. Одержання з 1,1,2,3,3пентахлорпропану Докладний опис експерименте опублікований в Houben-Weyl-Muller: Methoden der Organischen Chemie, 4th Edit., Vol.7/1, Thieme Verlag, Stuttgart, 1954, page 119. Вихідна речовина -1,1,2,3,3-пентахлорпропан, комерційно доступний від Pfaltz and Bauer. Одержання з мукохлорної кислоти Наступний спосіб являє собою невелику зміну вихідної методики Дікманна (Ber. Deut. Chem. Ges.r 1904, 37, 4638). Мукохлорна кислота 50,0г (0,30моль) Анілін 54мл (0,60моль) Вода 1000мл До розчину аніліну у воді при 85°С в 2л колбі при інтенсивному перемішуванні додавали невеликими порціями протягом 30 хвилин мукохлорну кислоту. При додаванні мукохлорної кислоти з'являлося жовте забарвлення, яке швидко зникало. Реакційна суміш залишалася гетерогенною, і фільтрування аліквоти через 30 хвилин нагрівання показало завершення реакції. Реакційну суміш нагрівали при 90°С протягом 60хв, охолоджували до 50°С і відфільтровували. Осад на фільтрі промивали 50мл 2н НСl і 100мл Н2О. Продукт сушили в потоці N2 , одержуючи 57г (100% вихід) 3анілідо-2-хлоракролеїну у вигляді сірої твердої речовини. 13С-ЯМР (ДМСО-d6 в м.ч.): 108, 117, 124, 129, 140, 147, 182. 3-Анілідо-2-хлоракролеїн 57г (0,30моль) 5н розчин NaOH 120мл (0,6моль) Розчин 3-анілідо-2-хлоракролеїну в 120мл 5н NaOH нагрівали при 100°С протягом 90 хвилин. Темний чорний розчин екстрагували двічі по 50мл МТБЕ. Перша органічна промивка видаляла більшу частину темного кольору розчину, і друга органічна промивка була лише злегка забарвлена. При охолоджуванні водної фази утворювався кристалічний осад. Продукт являв собою натрієву сіль 3хлормалондіальдегіду. Водну фазу підкислювали, додаючи 60мл 37% розчину НСl. Водну фазу екстрагували (МТБЕ/ТГФ 50/50, всього 400мл) і об'єднані органічні фази сушили над MgSO4. Після обробки Darco G60 і фільтрування через шар SiO2 розчин упарювали, одержуючи 19,6г (загальний вихід 62%) хлормалондіальдегіду у вигляді темної твердої речовини. Перекристалізація з приблизно 10мл МТБЕ давала 11,13г чистого хлормалондіальдегіду у вигляді коричнюватої твердої речовини. 13С-ЯМР (ДМСО-d6 в м.ч.): 113,175 (розширений). Одержання з хлорацетилхлориду Arnold (Collect. Czech. Chem. Commun., 1961, 26, 3051) відмічає утворення 3-диметиламіно-2хлоракролеїну при взаємодії хлороцтової кислоти з реагентом Вільсмейера, отриманим з РОСl3 і ДМФ. Зміна і розширення його способу приводить до одержання хлормалондіальдегіду у вигляді Na солі. Оксалілхлорид (280мл, 3,2моль) додавали при 10°С до 1000мл ДМФ. Реакція була дуже екзотермічною і утворювався важкий осад. Через 2 години витримування додавали хлорацетилхлорид (110мл, 1,4моль) і реакційну суміш нагрівали при 75°С протягом 3 годин. Аналіз аліквоти методом 1Н-ЯМР показав повне зникнення хлорацетилхлориду, і реакційну суміш гасили, додаючи її в 1л Н2O. До охолодженого розчину додавали 500мл 50% розчину NaOH. Реакційну суміш кип'ятили із зворотним холодильником протягом 5 годин. При охолоджуванні утворювався осад, який відфільтровували і промивали водою. Рудувато-коричневу тверду речовину сушили в потоці N2, одержуючи рудувато-коричневу тверду речовину (54% вихід).
ДивитисяДодаткова інформація
Назва патенту англійськоюA method for the preparation of 2-aryl-3-aryl-5-halogenopyridines being used as сох-2 inhibitors
Назва патенту російськоюСпособ получения 2-арил-3-арил-5-галогенпиридинов, которые используются как ингибиторы сох-2
МПК / Мітки
МПК: C07D 213/50, C07D 213/34, C07D 213/61, A61K 31/44, A61P 43/00, C07D 213/85, C07B 43/00, A61P 29/00
Мітки: сох-2, 2-арил-3-арил-5-галогенпіридинів, отримання, використовуються, варіанти, спосіб, інгібітори
Код посилання
<a href="https://ua.patents.su/10-66792-sposib-otrimannya-2-aril-3-aril-5-galogenpiridiniv-shho-vikoristovuyutsya-yak-ingibitori-sokh-2-varianti.html" target="_blank" rel="follow" title="База патентів України">Спосіб отримання 2-арил-3-арил-5-галогенпіридинів, що використовуються як інгібітори сох-2 (варіанти)</a>
Похідні нітрометилтіобензолу як інгібітори альдегідредуктази, спосіб їх отримання (варіанти) та фармацевтична композиція

Номер патенту: 56205
Опубліковано: 15.05.2003
Автори: Дюрбен Філіп, Дюмаз Іве, Коллопжес Франсуа, Ларді Клод
МПК: A61K 31/381, A61P 3/10, C07C 323/49, A61K 31/167, C07D 333/34, C07D 213/71, A61P 13/12, C07D 307/91, A61P 25/00, C07C 317/40, A61P 9/00, A61P 27/00, A61K 31/4406, A61K 31/343, A61K 31/18, A61P 43/00, C07D 307/82, C07C 317/42, C07C 317/34
Мітки: інгібітори, фармацевтична, похідні, варіанти, отримання, композиція, нітрометилтіобензолу, альдегідредуктази, спосіб
Формула / Реферат:
1. Похідні нітрометилтіобензолу загальної формули 1 (1) ,у якій:Р означаєрадикал (і): -(CO-NH)m-SO2-R;радикал (іі):або радикал (ііі):,R означає радикал, обраний з фенілу, бензилу, дифенілметилу, нафтилу, циклоалкілалкілу, у якому алкільна частина є С1-С4, а циклоалкільна частина є С3-С7, та стирилу, причому зазначений радикал є необов'язково заміщений однією чи кількома...
N-гідрокси-2-(алкіл-, арил-, або гетероарилсульфаніл, -сульфініл або -сульфоніл)-3-заміщені алкіл-, арил- або гетероариламіди як інгібітори матричних металопротеїназ

Номер патенту: 58543
Опубліковано: 15.08.2003
Автори: Бейкер Жанні Леа, Венкатесан Аранапакам Мудумбай, Гросу Джордж Теодор, Девіс Жамі Марі
МПК: A61K 31/4035, C07C 317/44, C07D 233/54, C07D 213/89, A61K 31/41, A61K 31/423, A61P 29/00, A61P 35/04, A61P 17/02, C07C 317/46, A61K 31/4164, C07D 215/38, A61K 31/445, A61K 31/433, A61K 31/415, A61P 19/08, A61K 31/341, A61K 31/351, C07D 285/14, C07D 309/12, A61K 31/5375, C07D 233/64, A61K 31/4409, C07D 213/30, A61K 31/165, C07D 215/12, A61K 31/55, C07D 213/54, A61K 31/4402, A61P 1/02, C07D 409/04, C07D 333/28, A61K 31/40, C07D 233/84, C07D 207/12, A61P 19/02, A61K 31/4436, A61P 35/00, A61P 3/10, C07D 233/02, C07D 307/64, C07D 213/56, C07D 215/36, C07D 233/76, C07D 213/71, C07D 209/48, C07D 307/30, C07D 277/20, A61K 31/4184, A61K 31/4535, C07D 277/36, A61K 31/495, C07D 231/18, A61K 31/426, A61K 31/381, C07D 213/74, A61K 31/16, C07D 295/08, C07C 317/40, C07D 295/092, C07D 263/58, A61P 1/14, C07D 211/54, C07D 333/34, A61P 31/18, C07D 215/48, A61K 31/47, A61K 31/4166, C07D 257/00, C07C 323/60, C07D 241/12, C07D 213/70, C07D 277/76, C07D 235/28, A61K 31/4406, C07D 277/74, A61P 1/04, A61K 31/4965, C07D 307/38, A61K 31/4453, C07D 211/66, A61K 31/428, A61P 9/02, A61P 37/06, A61P 9/04, C07D 213/32, C07D 277/70
Мітки: n-гідрокси-2-(алкіл, гетероарилсульфаніл, гетероариламіди, металопротеїназ, інгібітори, матричних, сульфініл, сульфоніл)-3-заміщені, арил, алкіл
Формула / Реферат:
1. Сполука формули І, Ів якій:R1 являє собою алкіл із 1-18 атомами вуглецю, необов'язково заміщений однією або двома групами, незалежно вибраними з R5;алкеніл, що містить 3-18 атомів вуглецю із 1-3 подвійними зв'язками, необов'язково заміщений однією або двома групами, незалежно вибраними з R5;алкініл, що містить 3-18 атомів вуглецю із...
Інгібітори металопротеіназ, спосіб їх отримання (варіанти), спосіб лікування, спосіб отримання засобу для лікування та фармацевтична композиція

Номер патенту: 43358
Опубліковано: 17.12.2001
Автори: Мартін Фіонна Мітчелл, Уіттакер Марк, Бекетт Раймонд Паул, Міллер Ендрю
МПК: C07D 285/135, A61P 1/02, C07D 401/12, A61K 31/341, A61K 31/496, A61K 31/215, C07D 409/12, A61K 31/4406, C07D 213/89, C07D 277/82, A61K 31/41, C07D 249/18, A61K 31/165, A61K 31/4409, A61P 29/00, C07D 277/44, A61K 31/4427, C07D 295/22, C07C 323/50, C07D 277/46, A61K 31/428, C07C 317/44, A61K 31/415, C07D 307/52, C07D 277/20, A61P 43/00, A61K 31/425, A61K 31/4245, C07D 233/88, C07D 271/10, A61P 37/08, A61K 31/495, A61P 9/00, A61K 31/44, A61P 27/14, C07D 213/75, A61K 31/395, A61K 31/426, C07C 259/00, A61K 31/195, A61K 31/433, A61P 35/00, A61P 17/00, C07D 271/06, A61K 31/34, A61K 31/4402, C07D 277/56, C07D 333/34, A61K 31/42, A61P 27/02, C07D 417/12, C07D 285/08, C07C 237/22, A61K 31/4433, C07D 261/14
Мітки: варіанти, лікування, спосіб, засобу, фармацевтична, інгібітори, композиція, металопротеіназ, отримання
Формула / Реферат:
1. Соединение формулы (I):гдеX означает -СО2Н или -CONHOH,R1 означает водород, (С1-С6)алкил, (С2-С6)алкенил, фенил, замещенный фенил, фенил-(С1-С6)алкил; замещенный фенил-(С1-С6)алкил; гетероцикл; замещенный гетероцикл, гетероцикл-(С1-С6)алкил, группу BSOnA, где n равно 0,1 или 2 и В означает водород или (С1-С6)алкил, фенил, замещенный фенил, гетероцикл, (С1-С6)ацил, фенацил, или замещенный фенацил, и А означает...
Спосіб одержання фенільних гетероциклів, які використовуються як інгібітори цог-2, а також сполуки, одержані цим способом
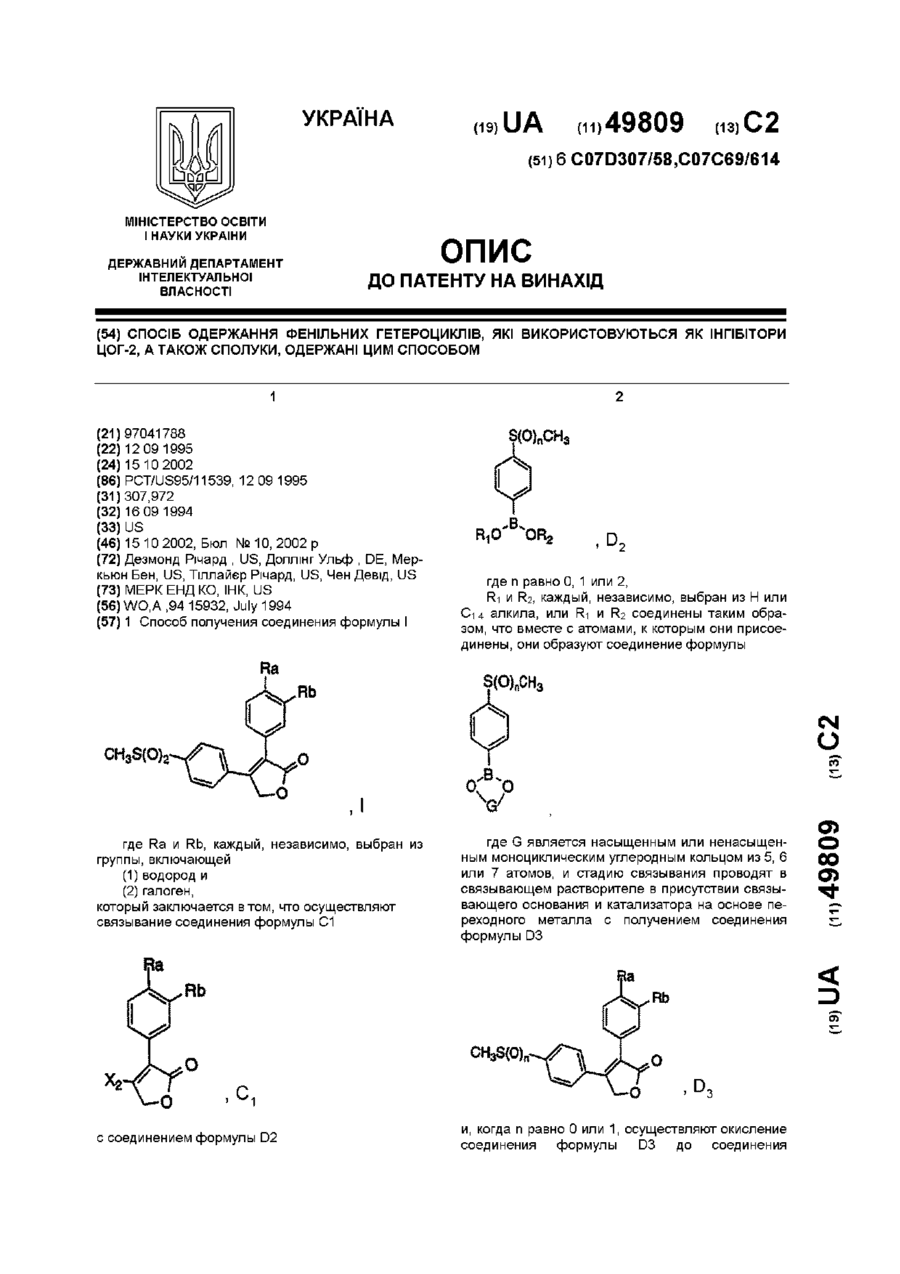
Номер патенту: 49809
Опубліковано: 15.10.2002
Автори: Тіллайєр Річард, Чен Девід, Доллінг Ульф, Меркьюн Бен, Дезмонд Річард
МПК: C07C 69/675, C07D 307/58
Мітки: цог-2, способом, також, гетероциклів, цим, інгібітори, одержання, одержані, використовуються, фенільних, сполуки, спосіб
Формула / Реферат:
1. Способ получения соединения формулы Iгде Ra и Rb, каждый, независимо, выбран из группы, включающей(1) водород и(2) галоген;который заключается в том, что осуществляют:связывание соединения формулы С1с соединением формулы D2где n равно 0, 1 или 2;R1 и R2, каждый, независимо, выбран из Н или C1-4 алкила, или R1 и R2 соединены таким образом, что вместе с атомами, к...
Похідні фенілоцтової кислоти, спосіб їх одержання (варіанти), проміжні продукти, що використовуються для їх одержання та засоби боротьби з шкідниками

Номер патенту: 48991
Опубліковано: 16.09.2002
Автори: Байер Херберт, Штратманн Зігфрид, Гроте Томас, Харріес Фолькер, Аммерманн Еберхард, Саутер Хуберт, Кірстген Райнхард, Лоренц Гізела, Мюллер Рут
МПК: A01N 37/50, A01N 35/10, C07C 251/60, C07C 249/00
Мітки: варіанти, фенілоцтової, продукти, кислоти, шкідниками, похідні, одержання, засоби, боротьби, спосіб, використовуються, проміжні
Формула / Реферат:
1. Похідні фенілоцтової кислоти формули Ів якій замісники та індекс мають наступне значення:Х означає NОСН3, СНОСН3, СНСН3;Y означає О, NR;R1, R незалежно один від одного означають водень і С1-С4алкіл;R2 означає ціано, нітро, трифторметил, галоген, С1-С4алкіл і, С1-С4алкокси;m означає 0,1 або 2, причому радикали R2 можуть бути різними, якщо m дорівнює 2;R3 означає водень, ціано,...
Попередній патент: Резервована система обробки інформації
Наступний патент: Спосіб гідрування альфа-, омега-динітрилів та каталізатор гідрування
Випадковий патент: Установка для визначення вмісту вуглецю в золі винесення пиловугільних котлоагрегатів теплових електростанцій