Способи одержання комбретастатину та його солей, проміжна сполука
Номер патенту: 76079
Опубліковано: 15.06.2006
Автори: Малєжонок Іріна, Лавінь Мішель, Казімір Жан-Поль, Мютті Стефан







Формула / Реферат
1. Спосіб одержання комбретастатину наступної загальної формули (І):
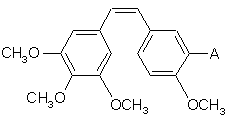 , (I)
, (I)
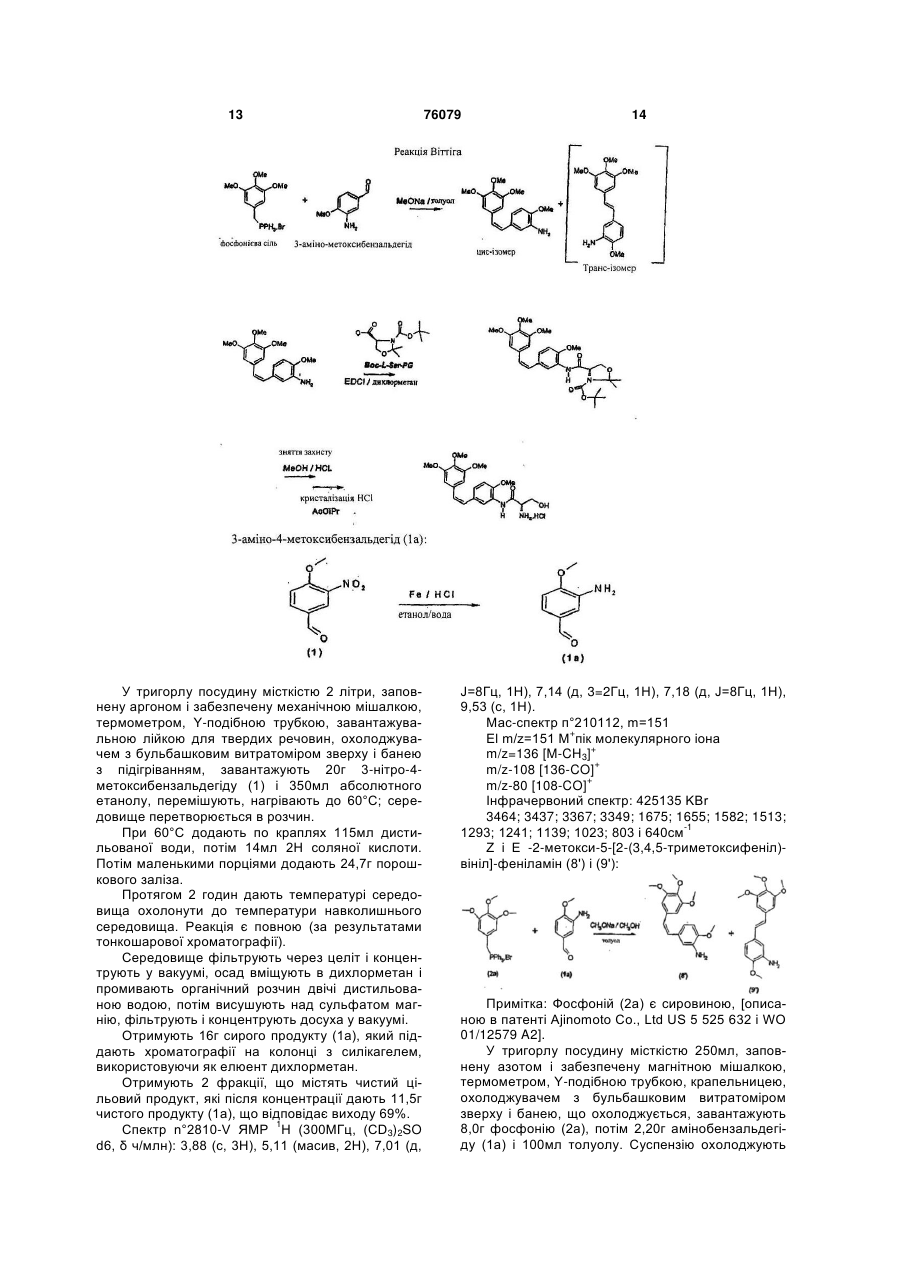
в якій А означає аміногрупу, який відрізняється тим, що після конденсації за методом Віттіга в присутності броміду або хлориду (3,4,5-триметоксибензил)-трифенілфосфонію і 3-нітро-4-метоксибензальдегіду або 3,4,5-триметоксибензальдегіду з сіллю (4-метокси-3-нітробензил)-трифенілфосфонію проводять відновлення нітрогрупи в присутності заліза.
2. Спосіб за п. 1, який відрізняється тим, що залізо використовують в надлишку.
3. Спосіб за п. 2, який відрізняється тим, що вказаний надлишок перевищує 2 еквіваленти на моль вихідного нітропохідного.
4. Спосіб одержання комбретастатину загальної формули (І):
, (I)
в якій А означає аміногрупу, який відрізняється тим, що конденсують сіль (3,4,5-триметоксибензил)-трифенілфосфонію з 3-аміно-4-метоксибензальдегідом або сіль (3-аміно-4-метоксибензил)-трифенілфосфонію з 3,4,5-триметоксибензальдегідом, переважно в присутності основи.
5. Спосіб за п. 4, який відрізняється тим, що конденсують сіль (3,4,5-триметоксибензил)-трифенілфосфонію з 3-аміно-4-метоксибензальдегідом.
6. Спосіб за пп. 4 або 5, який відрізняється тим, що реакцію проводять переважно в присутності основи, вибраної з трет-бутилату калію, трет-амілату натрію, гідриду натрію, бутиллітію, літійдіізопропіламіну, метилату натрію, карбонату калію або лужних похідних гексаметилдисиланів.
7. Спосіб за пп. 4 або 5, який відрізняється тим, що використовують метилат натрію.
8. Спосіб за пп. 4 або 5, який відрізняється тим, що реакційний розчинник, інертний відносно реакції, вибирають з простих ефірів, зокрема, тетрагідрофурану, полярних апротонних розчинників, зокрема, ацетонітрилу, N-метилпіролідону, диметилформаміду, диметилсульфоксиду, спиртів, ароматичних розчинників або води.
9. Спосіб за п. 7, який відрізняється тим, що температура реакції переважно складає від 0 до 10°C.
10. Спосіб одержання сполуки формули (І) в формі солі серину шляхом сполучення похідних формули (ІІа)
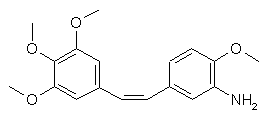 , (IIa)
, (IIa)
з циклічним похідним захищеного серину формули (IIb)
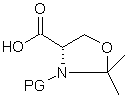 , (IIb)
, (IIb)
де PG означає захисну групу аміногрупи з одержанням нової проміжної сполуки наступної загальної формули:
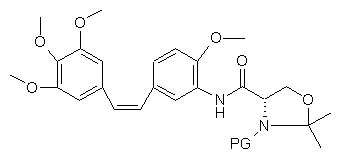 , (III)
, (III)
яку потім піддають зняттю захисту.
11. Проміжна сполука, утворена поєднанням амінокомбретастатину і циклічного похідного захищеного серину, яка відрізняється тим, що вона має формулу:
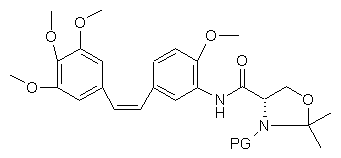 , (III)
, (III)
в якій PG означає захисну групу.
12. Проміжна сполука за п. 11, яка відрізняється тим, що PG означає захисну групу, вибрану з наступних груп: трет-бутоксикарбоніл, бензилоксикарбоніл або 9-флуоренілметилоксикарбоніл.
Текст
1. Спосіб одержання комбретастатину наступної загальної формули (І): 2 (11) 1 3 76079 4 ного серину, яка відрізняється тим, що вона має 12. Проміжна сполука за п. 11, яка відрізняється формулу: тим, що PG означає захисну групу, вибрану з наступних груп: трет-бутоксикарбоніл, бензилоксикаO O O рбоніл або 9-флуоренілметилоксикарбоніл. O O N H O N PG , (III) в якій PG означає захисну групу. Даний винахід відноситься до нового способу отримання комбретастатинів і їх похідних. Під комбретастатинами, або похідними стильбену, розуміють похідні наступної загальної формули: (I) в якій А означає гідроксильну групу або аміногрупу, а також їх фармацевтично прийнятні солі. З цих солей можна назвати гідрохлорид, ацетат, фосфат, метансульфонат. Коли сполука або А означає аміногрупу, вона може бути також зв'язана з амінокислотами з утворенням амідів, а також їх фармацевтично прийнятних солей. Синтез похідних стильбену, або комбретастатинів, які можуть знаходитися в формі фармацевтично прийнятної солі, а також фармацевтичні композиції, що їх містять, [описані в патентах US 4 996 237; US 5 525 632; US 5 731 353 і US 5 674 906]. У цих патентах описані комбретастатини, а також їх метаболіти і описана їх онкологічна активність in vitro. Згідно з цими патентами, комбретастатини отримують з солей (3,4,5-триметоксибензил)трифенілфосфонію, який конденсують з 3-нітроабо 3-гідрокси- (де гідроксильна група захищена) 4-метоксибензальдегідом в присутності гідриду натрію або похідних літію, після чого отримане похідне, якщо воно є нітропохідним, відновлюють в присутності цинку. Потім отримують ізомер в цисконфігурації дією світла або хроматографічним розділенням суміші. Даний винахід відноситься до нових способів отримання комбретастатинів або їх похідних, а також до удосконалень відомих способів. Передусім, був відкритий перший спосіб за шляхом V0 1 отримання похідних формули (І), в яких А означає аміногрупу, який являє собою вдосконалений спосіб, описаний у перелічених вище патентах, який полягає в тому, що після конденсації по Віттігу в присутності броміду або хлориду (3,4,5-триметоксибензил)-трифенілфосфонію і 3нітро-4-метоксибензальдегіду проводять відновлення в присутності заліза замість цинку, який використовується у відомому способі, що дозволяє отримати загальний вихід реакції, по відношенню до використаного альдегіду, який складає 60% (вихід по відношенню до використаного альдегіду [в патенті US 5 525 632] складає від 21% до 33%). Перший спосіб за шляхом V0 2 полягає в кон денсації 3,4,5-триметоксибензальдегіду з бромідом або хлоридом (4-метокси-3-нітробензил)трифенілфосфонію. В обох перших способах за шляхом V0 1 і V0 2 реакцію проводять в присутності основи, вибраної, зокрема, з трет-бутилату калію, трет-амілату натрію, гідриду натрію, бутиллітію, літійдіізопропіламіну (LDA), метилату натрію, карбонату калію або лужних похідних гексаметилдисиланів. Цю реакцію здійснюють в різних розчинниках, таких як простий ефір (ТГФ), полярні апротонні розчинники (ацетонітрил, NMP, DMF, DMSO...), спирти, ароматичні розчинники або вода, при температурі, вибраній фахівцем в залежності від основи і розчинника, що використовуються. Ця реакція, в тому, що стосується першого способу за шляхом V0 2, описана, зокрема, [в публікації K.G. Piney, Bioorg. Med. Chem. 8(2000) 2417-2425]. 2-метокси-5-[2-(3,4,5-триметоксифеніл)-вініл]нітрофеніл відновлюють згідно з поліпшеним способом згідно з винаходом дією заліза. Переважно використовують надлишок заліза, якщо потрібне повне перетворення вихідного продукту. Цей надлишок переважно перевищує 2 еквіваленти на моль вихідного нітропохідного. Було доведено, що та ж стадія, здійснена в присутності цинку в оцтовій кислоті, традиційному розчиннику, що використовується при відновленні за допомогою цинку, не дозволяє здійснити повну реакцію, [в патенті US 5 525 632] вихід реакції відновлення, здійснюваної на чистому ізомері Z, складає від 46 до 66%, і, з іншого боку, кількості цинку, що використовуються, непомірно великі, що приводить до великого об'єму промислових відходів; крім того, такий спосіб приводить до утворення великої кількості «азо»-сполук, що походять від злиття аміно, що утворюється, і нітрозо, що є проміжним продуктом реакції відновлення. Відновлення за рахунок водню, що утворюється форміатом амонію в присутності класичних каталізаторів, таких як паладій або платина, приводить до сильної ізомеризації подвійного зв'язку з утворенням небажаного ізомеру Е, а також до часткового насичення подвійного зв'язку. У згаданій вище публікації Ріnеу описане відновлення за допомогою гідросульфіту натрію чистого ізомеру Ζ нітро, отриманого хроматографією і перекристалізацією, з отриманням ізомеру Ζ аміно з виходом, що складає усього-навсього 37%. Гідрування молекулярним воднем, що каталізуються платиною або паладієм, рідко виявляють 5 76079 6 ся повними і, що гірше, приводять до насичення ном кислоти, органічну фазу промивають, конценетиленового подвійного зв'язку. трують і, в результаті хроматографії сирого концеТакож був знайдений другий спосіб, що дозвонтрату, отримують цільовий продукт. ляє уникнути стадії проміжного відновлення, яка є Похідне, що отримується згідно з другим спообов'язковою при використанні нітропохідного як собом за шляхом V0 3 або V0 4 або на другій ставихідного продукту. Дійсно, значно більш економідії першого способу за шляхом V0 1 або V0 2, має чним є перший метод здійснення цього другого наступну формулу: способу, в якому конденсують бромід або хлориди (3,4,5-триметоксибензил)-трифенілфосфонію з 3(IIa) аміно-4-метоксибензальдегідом, або другий метод здійснення цього другого способу, в якому конденсують 3,4,5-триметоксибензальдегід з сіллю (3Коли потрібно поєднати серин із сполукою фоаміно-4-метоксибензил)-трифенілфосфонію. рмули (llа), переважно використати L-серин, який Цей другий спосіб згідно з обома варіантами двічі захищений по атому азоту серину і по гідроквимагає на одну стадію менше з числа таких, на сильній групі і має загальну формулу (llb) яких використовують продукти CMR (канцерогени, мутагени, сполуки, токсичні відносно репродуктив(IIb) ної функції) в порівнянні з першими способами за шляхом V0 1 і V0 2, що в промисловому масштабі вважається відчутною перевагою з точки зору безде PG означає захисну групу аміногрупи, відопеки і виробничих витрат. му будь-якому фахівцеві, з отриманням нового Згідно з другим способом за шляхом V0 3 проміжного продукту наступної загальної формуздійснення винаходу вводять в реакцію сіль (3,4,5ли: триметоксибензил)-трифенілфосфонію і 3-аміно-4метоксибензальдегід, причому реакцію здійснюють переважно в присутності основи, вибраної, зокре(III) ма, з трет-бутилату калію, трет-амілату натрію, гідриду натрію, бутиллітію, LDA, метилату натрію, карбонату калію або лужних похідних гексаметилдисиланів. Переважно використовують метилат яку потім розщеплюють, переважно одночасно натрію. з розкриттям циклу шляхом кислотного гідролізу, в Цю реакцію проводять в різних розчинниках, реакції видалення захисної групи, відомій будьтаких як простий ефір (ТГФ), полярні апротонні якому фахівцеві. Переважно, група PG в формурозчинники (ацетонітрил, NMP, DMF, DMSO...), лах (llb) або (III) являє собою захисну групу, вибспирти, ароматичні розчинники або вода, при темрану з наступних груп: трет-бутоксикарбоніл, бенпературі, вибираній фахівцем в залежності від зилоксикарбоніл (CBZ) або 9основи і розчинника, що використовуються. флуоренілметилоксикарбоніл (FMOC). Температура реакції вибирається фахівцем в Сполука наведеної вище формули (III) є новим залежності від основи, що використовується; при і заявлена як така. використанні метилату вона складає переважно Конденсацію переважно здійснюють в присутвід 0 до 10°С. Після завершення реакції основу, ності EDCI (хлориду 1-етил-3-(3що використовується, нейтралізують водним роздиметиламінопропіл)карбодііміду) або в присутночином кислоти, органічну фазу промивають, консті DCC (дициклогексилкарбодііміду) і НОВТ (гідцентрують і в результаті хроматографії сирого роксибензотриазолу), або в присутності DCC (диконцентрату отримують цільовий продукт. циклогексилкарбодііміду) і HOSU (NЗгідно з другим способом за шляхом V0 4 гідроксисукциніміду), або, нарешті, в присутності здійснення винаходу, в якому вводять в реакцію TOTU (O-[(етоксикарбоніл)ціанометиленаміно]сіль (3-аміно-4-метоксибензил)-трифенілфосфонію N,N,N',N'-тетраметилуронійтетрафторборату) або і 3,4,5-триметоксибензальдегід, реакцію переважHBTU (О-бензотриазол-і-іл-Ν,Ν,Ν',Ν'но здійснюють в присутності органічної основи, тетраметилуронійгексафторфосфату) або Ν,Νвибраної, зокрема, з трет-бутилату калію, треткарбонілдіімідазолу. Реакцію переважно провоамілату натрію, гідриду натрію, бутиллітію, LDA, дять в розчиннику, інертному по відношенню до метилату натрію, карбонату калію або лужних пореакції, вибраному, зокрема, з полярних апротонхідних гексаметилдисиланів. Переважно викорисних розчинників, таких як ацетонітрил, диметилтовують метилат натрію. формамід, тетрагідрофуран, або хлоровмісних Цю реакцію проводять в різних розчинниках, аліфатичних розчинників, таких як дихлорметан, таких як простий ефір (ТГФ), полярні апротонні або з складних ефірів. розчинники (ацетонітрил, NMP, DMF, DMSO...), Зв'язування з похідним формули (IIа) може буспирти, ароматичні розчинники або вода, при темти також здійснене дією змішаного ангідриду, синпературі, вибираній фахівцем в залежності від тезованого in situ на основі хлорформіату або хлооснови і розчинника, що використовуються. рангідриду карбонової кислоти, наприклад, Температура реакції вибирається фахівцем в пивалоїлу, і двічі захищеного L-серину формули залежності від основи, що використовується; при (lIb) в присутності третинної основи типу NMM (Nвикористанні метилату вона складає переважно метилморфоліну) в різних розчинниках, інертних від 0 до 10°С. Після завершенні реакції основу, що по відношенню до реакції: складних ефірах, просвикористовується, нейтралізують водним розчитих ефірах, хлоровмісних розчинниках, ацетоніт 7 76079 8 рилі і т.д. Змішаний ангідрид отримують переважкції складає від 50 до 70°С. но при температурі від 0 до 10°С, потім реакцію Винахід буде більш повно розкритий за допопроводять при температурі навколишнього серемогою наступних прикладів, які не повинні вважадовища. Після завершення реакції реакційне сетися такими, що мають обмежувальний характер. редовище гідролізують за допомогою водного розСклад сумішей, послідовність і хід реакцій, а чину, потім середовище декантують і отриману також вихід не виділених продуктів/проміжних споорганічну фазу промивають гідроксильованою ослук і їх титри визначали шляхом ВЕРХ (високоеновою. фективної рідинної хроматографії). Зняття подвійного захисту із сполуки формули Приклад 1 - перший спосіб за шляхом V0 2 згі(III) здійснюють дією органічної або неорганічної дно з винаходом кислоти; переважно, використовують концентроГідрохлорид (Z)-N-[2-метокси-5-[2-(3,4,5ваний водний розчин соляної кислоти в спиртово(триметоксифеніл)вініл]феніл]-L-серинаміду му середовищі. Згідно з найбільш переважним Загальна схема синтезу варіантом здійснення винаходу, температура реа Новий спосіб, т.зв. «зворотний Віттіг», виходячи з броміду (4-метокси-3-нітробензил)трифенілфосфонію і 3,4,5триметоксибензальдегіду дозволяє отримати суміш ізомерів Ζ і Ε 2-метокси-5-[2-(3,4,5триметоксифеніл)-вініл]-нітрофенілу з відношенням Ζ/Ε=75/25. Це відношення відображає досить високу частку ізомеру Ζ для того, щоб суміш Ζ/Ε можна було безпосередньо піддати відновленню з отриманням шляхом кристалізації гідрохлориду ізомеру Ζ аміно з титрів ВЕРХ 97% (внутрішній стандарт). Отримання броміду (4-метокси-3-нітробензил)трифенілфосфонію (4) здійснюють згідно з наступним прикладом: 3-нітро-4-метоксибензиловий спирт (2): У тригорлу посудину місткістю 2 літри, забез печену механічною мішалкою, термометром, Yподібною трубкою, завантажувальною лійкою для твердих речовин і охолоджувачем з бульбашковим витратоміром зверху, завантажують 90,5г 3-нітро4-метоксибензальдегіду (1), потім 450мл ТГФ і 90мл етанолу. Отриманий блідо-жовтий розчин охолоджують до 10°С, потім вносять 10г боргідриду натрію протягом 40хв. при 10/15°С (ця реакція є сильно екзотермічною і температуру необхідно підтримувати за допомогою крижаної бані з ацетоном); в кінці додання розчин змінює колір з коричневого на колір морської хвилі. Розчин перемішують протягом 30хв. при 10°С, закінчення реакції відстежують за допомогою тонкошарової хроматографії і перемішують ще 1 годину при 10°С, потім температурі дають піднятися до температури навколишнього середовища. Завантажувальну лійку замінюють ізобарною крапельницею місткістю 500мл, через яку по краплях підливають 300мл дистильованої води протягом 30 хв., підтримуючи температуру середовища 20°С. На початку доливання спостерігають виділення газу. 9 76079 10 Середовище концентрують до 2/3 в ротацій30°С, фільтрують через скляний фільтр, промиваному випарнику (50°С/20ммHg); у водному конценють на фільтрі 2 рази за допомогою 300мл толуотраті кристалізується продукт білого кольору в лу, віджимають і висушують в сушильній шафі формі блоків. (35°С/20ммHg/20год.). Охолоджену водну фазу екстрагують за допоОтримують 217г броміду (4-метокси-3могою 250мл, потім 150мл дихлорметану, об'єднанітробензил)-трифенілфосфонію з хімічним вихоні органічні фази промивають 250мл дистильовадом 88%. ної води, потім висушують над сульфатом магнію. Синтез описаний в публікації: (розчинник, що Після фільтрації хлорметиленовий розчин вивикористовується: дихлорметан) [К. G. Piney et al. користовують таким, який він є в наступній реакції Bioorg. Med. Chem. 8(2000) 2417-2425]. бромування. Спектр n°=4 865-V 1 Вихід цієї стадії вважають таким, що дорівнює Н ЯМР (300МГц, (CD3)2SO d6, δ ч/млн): 3,90 100%. (с, 3Н), 5,26 (д, J=15Гц, 2Н), 7,33 (м, 2Н), 7,41 (м, Примітка: спирт (2) є промисловим, але мало1Н), 7,65-8,05 (м, 15Н). доступним продуктом. Мас-спектр n° 212217, m=428 3-нітро-4-метоксибромбензил (3): El m/z=262 [PPh3]+ пік молекулярного іона У тригорлу посудину місткістю 1 літр, забезпеDCI m/z=445 MNH3+ чену механічною мішалкою, термометром, Ym/z=428 M+ подібною трубкою, крапельницею і охолоджувачем m/z=263 [PPh3H]+ пік молекулярного іона з бульбашковим витратоміром зверху, завантажуІнфрачервоний спектр 426469 КВr ють хлорметиленовий розчин 3-нітро-42869; 2843; 2776; 1619; 1527; 1438; 1362; 1287; метоксибензилового спирту (2) і додають 100мл 1270; 1111; 752; 692 і 502 см-1 дихлорметану. Розчин при перемішуванні охолоСуміш Ζ і Ε 2-метокси-5-[2-(3,4,5джують до 5°С, потім підливають по краплях, підттриметоксифеніл)-вініл]-нітрофенілу (6) і (7): римуючи температуру середовища рівною 5°С, 135,4г триброміду фосфору. Розчин перемішують протягом 1год. 30хв. при 5°С; закінчення реакції відстежують за допомогою тонкошарової хроматографії, потім підливають по краплях, підтримуючи температуру 15°С, 250мл насиченого розчину гідрокарбонату натрію; в процесі додання з невеликою затримкою відбувається У тригорлу посудину місткістю 2 літри, забездуже інтенсивне виділення газу. печену механічною мішалкою, термометром, YСередовище декантують в колбі і промивають подібною трубкою, крапельницею і охолоджувачем послідовно за допомогою 250мл дистильованої з бульбашковим витратоміром зверху, завантажуводи і 200мл насиченого розчину гідрокарбонату ють при 20°С і в атмосфері азоту 54,7г 3,4,5натрію. Органічну фазу висушують над сульфатом триметоксибензальдегіду (5), 148,6г броміду (4магнію, фільтрують і концентрують в ротаційному метокси-3-нітробензил)-трифенілфосфонію (4) і випарнику (50°С/20ммHg). 1300мл толуолу. Отримують 119г твердої речовини в формі воСуспензію при перемішуванні охолоджують до рсистих голок жовто-зеленого кольору з хімічним 5°С на крижаній бані, потім за 40хв. при 5°С підливиходом на 2 стадіях 97%. вають 63,2г 25%-ого (по масі) розчину метилату Примітка: Цей продукт (3) може також бути натрію в метанолі. отриманий за наступною схемою, [описаною в пуУ міру доливання колір суспензії міняється з блікації K.G. Piney et al. Bioorg. Med. Chem. 8(2000) брудно-білого на жовтий і далі на коричневий. 2417-2425]. Перемішують 1 годину при 5°С, закінчення реакції відстежують за допомогою ВЕРХ (загальне споживання альдегіду), після чого додають 3г (0,05моль) оцтової кислоти. Суспензію нагрівають до 40°С і підтримують 30хв. при 40°С; при цій температурі нерозчинними Бромід (3-нітро-4-метоксибензил)-трифеніл залишаються тільки солі; середовище фільтрують фосфонію (4): при 40°С через скляний фільтр №3 і солі промиУ тригорлу посудину місткістю 2 літри, забезвають на фільтрі 3 рази за допомогою 100мл топечену механічною мішалкою, термометром, Yлуолу. подібною трубкою, завантажувальною лійкою для Фільтрат завантажують назад в посудину з твердих речовин і охолоджувачем з бульбашковим 250мл дистильованої води, цю двофазну суміш витратоміром зверху, завантажують 119г 3-нітро-4перемішують 20хв. при 40°С, потім декантують в метоксибромбензилу (3) на перемішуваний шар колбі. Фазу, що містить толуол, промивають двічі з 1000мл толуолу; при легкому нагріванні до 25°С 250мл дистильованої води, потім концентрують середовище стає розчином. Додають 126,5г тридосуха в ротаційному випарнику. фенілфосфіну, отриманий розчин поступово нагріОсад вміщують в 600мл ізопропілового спирту вають до 60°С; починаючи з температури 30°С, і 12мл толуолу з температурою 40°С, починається формується осад. Середовище витримують протякристалізація, при повільному перемішуванні дагом 4 годин при 60/65°С, потім охолоджують до ють температурі опуститися до кімнатної і зали 11 76079 12 шають до наступного ранку. зникнення азеотропу починається осадження масСуспензію при перемішуванні охолоджують і ла в залишковій водній фазі. витримують 1 годину при 5°С, потім фільтрують Цю водну фазу екстрагують в колбі двічі за через скляний фільтр, осад промивають двічі з допомогою 300мл дихлорметану, потім промива125мл ізопропілового спирту, віджимають і висують органічну фазу двічі з 300мл напівнасиченого шують в сушильній шафі у вакуумі водного розчину хлориду натрію і з 300мл дисти(35°С/30ммHg/18годин). льованої води. Отримують 91,7г суміші ізомерів Ζ і Ε (6) і (7) в Органічну фазу концентрують досуха в ротаспіввідношенні Ζ/Ε=75/25 (ВЕРХ внутрішній станційному випарнику; отримують 76г олії, що має дарт) і з виходом 95%. співвідношення Z/E=80/20 згідно з даними ВЕРХ. Синтез описаний в публікації: (розчинник, що Цю олію розчиняють в 591мл метанолу і переливикористовується: дихлорметан: основа, що виковають в посудину місткістю 1 літр при перемішуристовується: NaH) [К. G. Piney et al. Bioorg. Med. ванні, потім додають 100мл 2,32Η розчину соляної Chem. 8(2000)2417-2425]. кислоти в метанолі, запускають реакцію і залишаПримітка: Були випробувані різноманітні робоють на ніч при перемішуванні для осадження. чі умови, такі як: Кількість метанол + розчин соляної кислоти в Розчинники: ТГФ, ацетонітрил, метанол і інші метанолі таке, що кінцева концентрація ізомеру Ζ спирти, дихлорметан, NMP, DMF, DMSO і т.д. (визначена за допомогою ВЕРХ) становить 8,8% Основи: трет-бутилат калію, трет-амілат навага/об'єм. трію, гідроксид натрію. NaH, Buli/LDA, карбонат На наступний ранок середовище фільтрують калію і т.д. через скляний фільтр; висушений осад має масу Температури: від -10°С до температури кипін8,2г і складається виключно з ізомеру Ε (ВЕРХ). ня деяких розчинників Фільтрат (693г) з співвідношенням Ζ/Ε=86/14 Гідрохлорид 2-2-метокси-5-[2-(3,4,5(ВЕРХ, внутрішній стандарт) концентрують напотриметоксифеніл)-вініл]-феніламіну (8): ловину в ротаційному випарнику, додають до 347г концентрату 400мл ацетонітрилу і знов концентрують до отримання маси концентрату 347г. Додають 1000мл ацетонітрилу і концентрують до початку кристалізації, потім концентрат переливають в посудину місткістю 4 літри, що містить 1500мл ацетонітрилу, при перемішуванні; при 60°С спостерігається рясне випадання осаду з середовища. У тригорлу посудину місткістю 2 літри, забезСередовище підтримують 2,5 години при 60°С печену механічною мішалкою, термометром, Yі при перемішуванні, дають охолонути до 30°С подібною трубкою, завантажувальною лійкою для приблизно за 1 годину, кашку фільтрують через твердих речовин, охолоджувачем з бульбашковим скляний фільтр, ізомер Ε (9) розчинений в фільтвитратоміром зверху і банею з підігріванням, завараті, осад промивають двічі з 200мл ацетонітрилу і нтажують при 20°С і в атмосфері азоту 80г суміші висушують в сушильній шафі Ζ і Ε 75/25 2-метокси-5-[2-(3,4,5-триметоксифеніл)(35°С/30ммHg/18год.). вініл]-нітрофенілу (6) і (7), 640мл абсолютного Отримують 45,7г Е-2-метокси-5[2-(3,4,5етанолу і 160мл дистильованої води. триметоксифеніл)-вініл]-феніламіну (8) з титром по Швидко перемішують і нагрівають на масляній ВЕРХ, внутрішній стандарт, складаючий 97% і вибані, при 50°С до суспензії додають 7,8мл 6Η соходом тель-кель 56%, що відповідає виходу ізомеляної кислоти, потім температуру середовища ру Ζ отриманого по відношенню до ізомеру Ζ вихіпідвищують до 77±2°С; середовище стає практичдному 72%. но повністю розчинним. ПРИКЛАД 2 - Синтез згідно з другим способом Протягом 5 хвилин порціями додають 52г поза шляхом V0 3 згідно з винаходом рошкового заліза. Починаючи з першого додання Перевагою другого способу за шляхом V0 3 в середовище перетворюється в розчин, потім на порівнянні з першим способом за шляхом V0 2, стінках посудини формується чорнуватий осад. який називається «зворотний Віттіг», є проведення Середовище витримують 2 години при 77±2°С, реакції Віттіга між вже відновленим продуктом, за допомогою ВЕРХ контролюють зникнення вихіаміноальдегідом (1а) і фосфонієм (2а) і, отже, видних нітропохідних (6) і (7). ключення хімічної стадії з використанням продуктів Середовищу дають охолонути до 40°С і фільCMR. трують через скляний фільтр з фільтруючим матеГідро хлорид (Z)-N-[2-метокси-5-[2-(3,4,5ріалом Clarcel, осад промивають двічі з 160мл сутриметоксифеніл)вініл]феніл]-L-серинаміду міші етанол/вода 80/20. Загальна схема синтезу Фільтрат, маточні і промивальні розчини концентрують в ротаційному випарнику; з моменту 13 У тригорлу посудину місткістю 2 літри, заповнену аргоном і забезпечену механічною мішалкою, термометром, Y-подібною трубкою, завантажувальною лійкою для твердих речовин, охолоджувачем з бульбашковим витратоміром зверху і банею з підігріванням, завантажують 20г 3-нітро-4метоксибензальдегіду (1) і 350мл абсолютного етанолу, перемішують, нагрівають до 60°C; середовище перетворюється в розчин. При 60°С додають по краплях 115мл дистильованої води, потім 14мл 2Η соляної кислоти. Потім маленькими порціями додають 24,7г порошкового заліза. Протягом 2 годин дають температурі середовища охолонути до температури навколишнього середовища. Реакція є повною (за результатами тонкошарової хроматографії). Середовище фільтрують через целіт і концентрують у вакуумі, осад вміщують в дихлорметан і промивають органічний розчин двічі дистильованою водою, потім висушують над сульфатом магнію, фільтрують і концентрують досуха у вакуумі. Отримують 16г сирого продукту (1а), який піддають хроматографії на колонці з силікагелем, використовуючи як елюент дихлорметан. Отримують 2 фракції, що містять чистий цільовий продукт, які після концентрації дають 11,5г чистого продукту (1а), що відповідає виходу 69%. Спектр n°2810-V ЯМР 1H (300МГц, (CD3)2SO d6, δ ч/млн): 3,88 (с, 3Н), 5,11 (масив, 2Н), 7,01 (д, 76079 14 J=8Гц, 1Н), 7,14 (д, 3=2Гц, 1Н), 7,18 (д, J=8Гц, 1H), 9,53 (с, 1Н). Мас-спектр п°210112, m=151 El m/z=151 М+пік молекулярного іона m/z=136 [M-CH3]+ + m/z-108 [136-CO] + m/z-80 [108-CO] Інфрачервоний спектр: 425135 KBr 3464; 3437; 3367; 3349; 1675; 1655; 1582; 1513; 1293; 1241; 1139; 1023; 803 і 640см-1 Ζ і Е -2-метокси-5-[2-(3,4,5-триметоксифеніл)вініл]-феніламін (8') і (9'): Примітка: Фосфоній (2а) є сировиною, [описаною в патенті Ajinomoto Co., Ltd US 5 525 632 і WO 01/12579 A2]. У тригорлу посудину місткістю 250мл, заповнену азотом і забезпечену магнітною мішалкою, термометром, Y-подібною трубкою, крапельницею, охолоджувачем з бульбашковим витратоміром зверху і банею, що охолоджується, завантажують 8,0г фосфонію (2а), потім 2,20г амінобензальдегіду (1а) і 100мл толуолу. Суспензію охолоджують 15 76079 16 при перемішуванні до 5°С і підливають за 15хв. 93% ізомери Ζ+ неідентифіковані домішки, другий, 3,51мл 25%-ого (по вазі) розчину метилату натрію масою 2,09г, містить вихідний альдегід і суміш Ζ/Ε, в метанолі. Після витримування протягом 2,5год. що складають 39 і 37,5% за результатами ВЕРХ при 5°C реакція залишається неповною ((коефіці(внутрішній стандарт). єнт перетворення: 45%), але далі не розвивається Підсумкова кількість ізомеру Ζ (8'), що визнаВЕРХ) і відношення Z/E становить 61/39. Підливачається за допомогою ВЕРХ (внутрішній станють 0,2мл оцтової кислоти, розбавленої 50 мл водарт), становить 1,15г на 2,20г використаного альди, підвищують температуру до 13°С, перемішудегіду, що відповідає виходу 24%. ють 30хв., потім переливають в колбу, ПРИКЛАД 3 - Синтез згідно з другим способом концентрують органічну фазу у вакуумі в ротаційза шляхом V0 3 згідно з винаходом ному випарнику і отримують 8г олії жовтого кольоЯк і для шляху V0 2, перевагою шляху V0 3 в ру. порівнянні з першим способом за шляхом V0 2, За результатами ВЕРХ, ця олія містить вихідякий називається «зворотний Віттіг», є проведення ний альдегід, фосфіноксид і цільову суміш Z/E з реакції Віттіга між вже відновленим продуктом, співвідношенням 61/39. бромідом (3-аміно-4-метоксибензил)Олію хроматографують на колонці з силікагетрифенілфосфонієм (1b) і 3,4,5лем (40 частин мас/мас). використовуючи як елютриметоксибензальдегідом (5) і, отже, відсутність ент суміш циклогексан/етилацетат/триетиламін хімічної стадії з використанням продуктів CMR. (50/50/2). Гідрохлорид (Z)-N-[2-метокси-5-[2-(3,4,52 серії фракцій об'єднують і концентрують дотриметоксифеніл)віеіл]феніл]-L-серинаміду суха, перший сухий екстракт масою 360мг містить Загальна схема синтезу Бромід (4-метокси-3-амінобензил)трифенілфосфонію (1b) У тригорлу посудину місткістю 1 літр, забезпечену механічною мішалкою, термометром, Yподібною трубкою, завантажувальною лійкою для твердих речовин, охолоджувачем з бульбашковим витратоміром зверху і банею з підігріванням, завантажують 30г продукту (4), 240мл етанолу і 60мл дистильованої води. До суспензії додають при перемішуванні 1,76мл 6Η соляної кислоти і нагрівають до 70°С. Потім за 15 хвилин додають маленькими порціями 9,9г порошкового заліза; середовище залишається нерозчинним. Середовище витримують 2 години при 15°С, органічні речовини повільно розчиняються, паралельно формується коричнюватий осад заліза і оксиду заліза. За результатами ВЕРХ залишається ще 5% вихідних сполук; знов додають 2г заліза і продовжують нагрівання протягом 1год.; перетворення стає повним. Середовище охолоджують до 40°С і фільтрують через Clarcel, промивають за допомогою 100мл 20%-ого водного розчину етанолу і концентрують фільтрат досуха у вакуумі в ротаційному випарнику. Осад вміщують в 300мл ізопропілового спирту і кристалізують з середовища, перемішують і нагрівають до 50°С; при цьому середовище знов перетворюється в розчин. Потім підливають 14мл 5Η 17 76079 18 розчину соляної кислоти в ізопропіловому спирті; тить 86% ізомеру Ε і 7% Ζ. відбувається випадання осаду з середовища; сеПідсумкова кількість ізомеру Ζ (8'), визначена редовище витримують 1 годину при 50°С, потім за допомогою ВЕРХ (внутрішній стандарт), станодають охолонути до температури навколишнього вить 0,725г на 4г .використаного альдегіду, що середовища. відповідає як такому виходу 11,3%. Кашку фільтрують через скляний фільтр, осад Трет-бутиловий ефір Z-4-{2-метокси-5-[2промивають за допомогою 50мл ізопропілового (3,4,5-триметоксифеніл)-вініл]-фенілкарбамоїл}спирту, ретельно віджимають і висушують в су2,2-диметилоксазолідин-3-карбонової кислоти (11): шильній шафі у вакуумі. Отримують 27,3г (1b) як такої (не очищеної) з виходом 89,9%. Спектр n° 4584-V ЯМР 1H (300МГц, (CD3)2SO d6, δ ч/млн): 3,78 (с, 3Н), 5,03 (душ., J=15Гц, 2Н), 6,43 (масив, 1H), 6,62 (суш., 1H), 6,82 (д уш., J=8Гц, 1Н), 7,60-8,00 (м, 15Н). Мас-спектр n°211915, m=397 El m/z=397 M+ Вивільнення основи (8') з гідрохлориду (8): m/z=382 [M-CH3]+ У колбу Ерленмейєра місткістю 1 літр заванm/z=262 [PPh3]+ пік молекулярного іона + тажують 44г продукту (8), 16г гідрокарбонату наDCI m/z=398 MNH4 трію, потім 200мл дистильованої води і 375мл диm/z=263 [PPh3H]+ пік молекулярного іона хлорметану. Перемішують протягом 20хв. при Інфрачервоний спектр 426386 КВr температурі навколишнього середовища і отри3254; 2474; 1920; 1628; 1520; 1439; 1433; 1279; мують дві прозорі фази. 1110; 736; 690; 527 і 511см-1 Органічну фазу відділяють декантацією, висуΖ і Ε -2-метокси-5-[2-(3,4,5-триметоксифеніл)шують над сульфатом натрію, потім фільтрують. вініл]-феніламін (8') і (9') Отримують приблизно 400мл хлорметиленового розчину, що містить продукт (8'). Отримання 3-трет-бутилового ефіру 2,2диметилоксазолідин-3,4-дикарбонової кислоти (10) Цей продукт, хоча і випускається серійно, зустрічається дуже рідко; його отримували омилюванням літином його метилового складного ефіру [згідно з J. ORG. СНЕМ. 63(12) Р. 3983(1998)]. Спектр ЯМР 1H (300МГц, (CD3)2SO d6, δ ч/млн): 1,38 (с, 3Н), 1,45 (с, 9Н), 1,55 (с, 3Н), 3,95 У тригорлу посудину місткістю 250мл, запов(м, 1H), 4,16 (м, 1Н), 4,31 (м, 1H), 12,50-13,10 (манену азотом і забезпечену магнітною мішалкою, сив уш., 1Н). термометром, Y-подібною трубкою, крапельницею, Маса-спектр: n°213135, m=245 охолоджувачем з бульбашковим витратоміром DCI m/z=263 MNH4+ зверху і банею, що охолоджується, завантажують m/z=246 MH+ 11,02г продукту (1b), 4г продукти (5) і 70мл толуоm/z=207 [MNH4-tBu]+ пік молекулярного іона лу. m/z=146 [MH-BOC]+ Суспензію охолоджують при перемішуванні до Інфрачервоний спектр: 426759 КВr 5°С і підливають за 15хв. 4,92мл 25%-ого (по масі) 1744;170/; 1638;1407; 1368; 1164; 1104; 856; розчину метилату натрію в метанолі. Суспензію 836 і 623см-1 перемішують протягом 2,5год. при 5°С, потім підСполука ливають 0,2мл оцтової кислоти, розбавленої 50мл У тригорлу посудину місткістю 2 літри, забезводи, підвищують температуру до 14°С; середопечену механічною мішалкою, термометром, Yвище стає дуже густим, його розбавляють 10мл подібною трубкою, завантажувальною лійкою для толуолу і 10мл води; залишається нерозчинна твердих речовин, охолоджувачем з бульбашковим частина коричневого кольору. витратоміром зверху і крижаною банею, завантаСуміш фільтрують через Clarcel, осад промижують розчин продукту (8'), додають 600мл дихловають тричі за допомогою 50мл толуолу (промирметану і охолоджують при перемішуванні. вальні розчини містять майже виключно вихідний При 5°С додають 42,9г 3-трет-бутилового ефіальдегід і їх не з'єднують з двофазним фільтрару 2,2-диметилоксазолідин-3,4-дикарбонової кистом), прозорий фільтрат (рН12) переливають в лоти (10), який розчиняється, потім додають порколбу і органічну фазу концентрують досуха у ваціями при температурі від 5 до 10°С 48г куумі при 40°С; відношення Z/E, визначене за догідрохлориду 1-етил-3-(3-диметиламінопропіл)помогою ВЕРХ, становить 43/57. карбодііміду (EDCI). Отриману олію коричневого кольору піддають Середовищу дають повільно нагрітися до темхроматографії на колонці з силікагелем (100 часператури навколишнього середовища в процесі тин ваг./ваг.), використовуючи як елюент суміш танення льоду у водяній бані протягом ночі. циклогексан/етилацетат/триетиламін (50/50/2). На наступний ранок додають 330мл дистильо2 серії фракцій об'єднують і концентрують дованої води і інтенсивно перемішують. Протягом суха; перший сухий екстракт масою 1,1г містить 30хв. середовище стає каламутним (гідроліз 14% ізомеру Ε і 59% Ζ, другий, масою 1,08г, міс 19 76079 20 EDCI); перемішування продовжують ще 30хв. Середовище переливають в колбу і промивають органічну фазу послідовно двічі за допомогою 280мл 0,55Η гідроксиду натрію, потім за допомогою 300мл дистильованої води. Органічну фазу концентрують досуха в ротаційному випарнику (50°С/50ммHg). Отримують 79,4г клейкої олії (11), що твердне У тригорлу посудину місткістю 1 літр, забезпепри 20°С, з виходом по вазі по відношенню до вичену механічною мішалкою, термометром, Yкористаного продукту (8) 117%. подібною трубкою, охолоджувачем з бульбашкоСпектр n°=5 578-V 1 вим витратоміром зверху і банею з підігріванням, 'H ЯМР (400МГц, (CD3)2SO d6, при темперазавантажують при 20°С 61,8г продукту (11) в розтурі 373 К, 6 ч/млн): 1,41 (с, 9Н), 1,53 (с, 3Н), 1,64 чині в 54мл метанолу, додають 150мл ізопропіла(с, 3Н), 3,64 (с, 6Н), 3,71 (с, 3Н), 3,86 (с, 3Н), 3,99 цетату, 99мл 2,3Η розчину соляної кислоти в ме(д, J=9 і 3Гц, 1Н), 4,19 (дд, J=9 і 7Гц, 1Н), 4,52 (дд, танолі і 8,2мл дистильованої води. Перемішують і J=7 і 3Гц, 1Н), 6,48 (д, J=12,5Гц, 1H), 6,55 (д, нагрівають до 60°С протягом 3 годин. Розчин охоJ=12,5 Гц, 1H), 6,58 (с, 2H), 7,02 (м, 2Н), 8,13 (с лоджують до 40°С і очищають фільтрацією через уш., 1Н), 8,82 (с уш., 1Н). скляний фільтр n°4, потім промивають фільтр Мас-спектр n°213565, m=542 40мл метанолу. Фільтрат знов вміщують в тригорDCI m/z=560 MNH4+ пік молекулярного іона лу посудину при перемішуванні і додають 194мл m/z=543 MH+ ізопропілацетату, нагрівають до 40°С, додають m/z=504 [MNH4-tBu]+ 0,2г продукту (12), потім по краплях підливають m/z=443 [MH-BOC]+ протягом 1год. 194мл ізопропілацетату; в процесі Інфрачервоний спектр 425857 ССl4 доливання середовище повільно кристалізується. 3409; 2982; 2938; 2837; 1712; 1698; 1534; 1363; Середовищу дають вихолонути до кімнатної 1249; 1133; 1092 і 851см-1 температури, потім охолоджують до 5°С і залишаБули використані інші умови зв'язування, такі ють при цій температурі протягом ночі. як: На наступний ранок кашку фільтрують через - Змішаний ангідрид (пивалоїлхлорид/(10)) скляний фільтр, осад віджимають і промивають 4 ОСС/НОВТ-ОСС/НОSU-ТОТU-N-Nрази по 50мл ізопропілацетату, ретельно віджикарбонілдіімідазол і т.д. мають, потім висушують в сушильному Шафі до - В ацетонітрилі, DMF, ТГФ, дихлорметані, постійної маси (35°С/10ммHg). складному ефірі і т.д. EDCI, HCl в дихлорметані Отримують 28г продукту (12) з виходом за 2 дає найкращий результат. стадії (сполука, потім зняття захисту) 56% і титром Гідрохлорид (Z)-N-[2-метокси-5-[2-(3,4,5по ВЕРХ (внутрішній стандарт) >98%. триметоксифеніл)вініл]феніл]-L-серинаміду Цей показник відповідає загальному виходу як такому для синтезу, здійсненого згідно з першим способом за шляхом V0 2, що становить 30% (отриманий продукт (12) по відношенню до використаного продукту (5)). Комп’ютерна верстка Т. Чепелева Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethods for preparing combretastatin and salts thereof, an intermediary compound
Назва патенту російськоюСпособы получения комбретастатина и его солей, промужуточное соединение
Автори російськоюMutti, Stephane
МПК / Мітки
МПК: C07D 263/06, C07C 213/00
Мітки: способи, солей, одержання, сполука, проміжна, комбретастатину
Код посилання
<a href="https://ua.patents.su/10-76079-sposobi-oderzhannya-kombretastatinu-ta-jjogo-solejj-promizhna-spoluka.html" target="_blank" rel="follow" title="База патентів України">Способи одержання комбретастатину та його солей, проміжна сполука</a>
Похідні n-триазолілметилпіперазину, способи їх одержання, лікарський засіб та проміжна сполука

Номер патенту: 73728
Опубліковано: 15.09.2005
Автори: Брюкнер Райнхард, Екхоут Крістіан, Занн, Хольгер, Шьон, Уве, Яссеранд Даніель
МПК: A61P 29/00, C07D 413/14, C07D 491/10, A61P 25/00, A61P 43/00, A61P 1/00, A61K 31/497, C07D 405/14, A61K 31/5377, A61P 1/04, C07D 403/06, A61K 31/506, C07D 403/14, A61K 31/496
Мітки: проміжна, сполука, n-триазолілметилпіперазину, похідні, засіб, лікарський, одержання, способи
Формула / Реферат:
1. Сполуки загальної формули І, Iв якійR1 означає водень або (нижч.)алкіл, де (нижч.) означає нижчий,R2 означає (нижч.)алкіл, ди(нижч.)алкіламіно(нижч.)алкіл, (нижч.)алкоксикарбоніл(нижч.)алкіл, необов'язково одно- або двозаміщений (нижч.)алкілом цикло(гетеро)алкіл з 5-6 атомами циклу, який містить необов'язково 1-2 подвійні зв'язки,...
Спосіб одержання циклопропілацетилену та проміжна сполука для його здійснення

Номер патенту: 58470
Опубліковано: 15.08.2003
Автори: Йін Жангюо, Ванг Зе, Фортунак Джозеф М.
МПК: C07C 5/00, B01J 31/04, C07B 61/00, C07C 22/00, C07C 1/00, C07C 13/00, B01J 31/02, C07C 17/093, C07C 51/347
Мітки: проміжна, одержання, здійснення, спосіб, сполука, циклопропілацетилену
Формула / Реферат:
1. Спосіб одержання циклопропілацетилену, що включає:взаємодію циклопропанкарбоксальдегіду з малоновою кислотою або заміщеною малоновою кислотою в присутності основного каталізатора з утворенням 3-циклопропілакрилової кислоти,взаємодію 3-циклопропілакрилової кислоти з металовмісним каталізатором та галогенуючим агентом з утворенням (E,Z)-1-галоген-2-циклопропілетилену та взаємодію (Е,Z)-1-галоген-2-циклопропілетилену з...
Спосіб одержання моносахариду 25-циклогексил-22,23-дигідро-5-гідроксііміноавермектину в1, проміжна сполука та його кристалічні сольвати

Номер патенту: 56244
Опубліковано: 15.05.2003
Автори: Волш Найджел Дерек Артур, Кемберс Селена Джейн
МПК: A61K 31/7048, A61P 33/10, A61P 33/00, C07H 17/08
Мітки: одержання, проміжна, моносахариду, сольвати, спосіб, сполука, 25-циклогексил-22,23-дигідро-5-гідроксііміноавермектину, кристалічні
Формула / Реферат:
1. Спосіб одержання моносахариду 25-циклогексил-22,23-дигідро-5-гідроксііміноавермектину В1, що включає взаємодію 25-циклогексил-22,23-дигідро-5-оксоавермектину В1 з гідрохлоридом гідроксиламіну у водному органічному розчиннику.2. Спосіб згідно з п. 1, у якому розчинник є водним ізопропіловим спиртом.3. Спосіб згідно з п. 2, у якому процес здійснюють при температурі від 40-50 °С.4. Спосіб згідно з будь-яким з пп. 1-3, у...
Біциклічна сполука, способи її одержання, фармацевтичні композиції, що її містять,способи лікування та попередження різних захворювань, проміжні сполуки та способи їх одержання

Номер патенту: 72883
Опубліковано: 16.05.2005
Автори: Лохрей Брадж Бхушан, Калчар Шіварамаййа, Лохрей Відіа Бхушан, Баджі Ашок Чаннавеераппа, Чакрабарті Ранджан, Рамануджам Раджагопалан
МПК: A61K 9/20, A61P 3/06, A61P 19/10, A61P 25/28, A61K 9/08, A61P 13/12, A61K 9/14, A61P 3/04, A61P 17/06, A61P 3/10, A61K 9/10, A61P 9/10, A61K 31/5415, A61K 9/48, C07D 413/06, A61K 31/538, C07D 279/00, C07D 265/36
Мітки: біциклічна, містять,способи, захворювань, способи, сполуки, лікування, фармацевтичні, різних, композиції, одержання, сполука, попередження, проміжні
Формула / Реферат:
1. Сполука формули (І), (I)де групи R1, R2, R3, R4 і групи R5 і R6, коли вони приєднані до атома вуглецю, можуть бути однаковими або різними і означають водень, галоген, гідрокси або необов'язково заміщену групу, вибрану з алкілу, алкокси, фенілу, карбонової кислоти або сульфонової кислоти ; один або обидва замісники R5 і R6 можуть також означати оксогрупу,...
Спосіб одержання клавуланової кислоти або її фармацевтично прийнятих солей або ефірів, сіль клавуланової кислоти з алкіламіном як проміжна сполука для одержання клавуланової кислоти

Номер патенту: 22115
Опубліковано: 30.04.1998
Автори: Уілкінс Роберт Беннет, Кук Майкл Аллен
МПК: C07B 63/00, A61P 31/04, C07D 503/00, C12P 17/18, A61K 31/42
Мітки: спосіб, сполука, одержання, проміжна, сіль, кислоти, клавуланової, прийнятих, алкіламіном, солей, ефірів, фармацевтично
Формула / Реферат:
1. Способ получения клавулановой кислоты или ее фармацевтически приемлемых солей или эфиров, включающий контактирование неочищенной клавулановой кислоты в органическом растворителе с алкиламином, выделение полученной соли клавулановой. кислоты с алкиламином и превращение ее в клавулановую кислоту или ее фармацевтически приемлемую соль или эфир, отличающийся тем, что в качестве алкиламина используют трет-октиламин или амин общей...
Попередній патент: Споруда-рибохід
Наступний патент: Інтегральний тензоперетворювач
Випадковий патент: Високовольтне герконове реле перевантаження