Спосіб одержання 2-(6-фтор-1н-індол-3-іл)етиламіну
Номер патенту: 111584
Опубліковано: 25.05.2016
Автори: Теркельсен Франс, Рок Майкл Харольд, Треппендахль Свенн

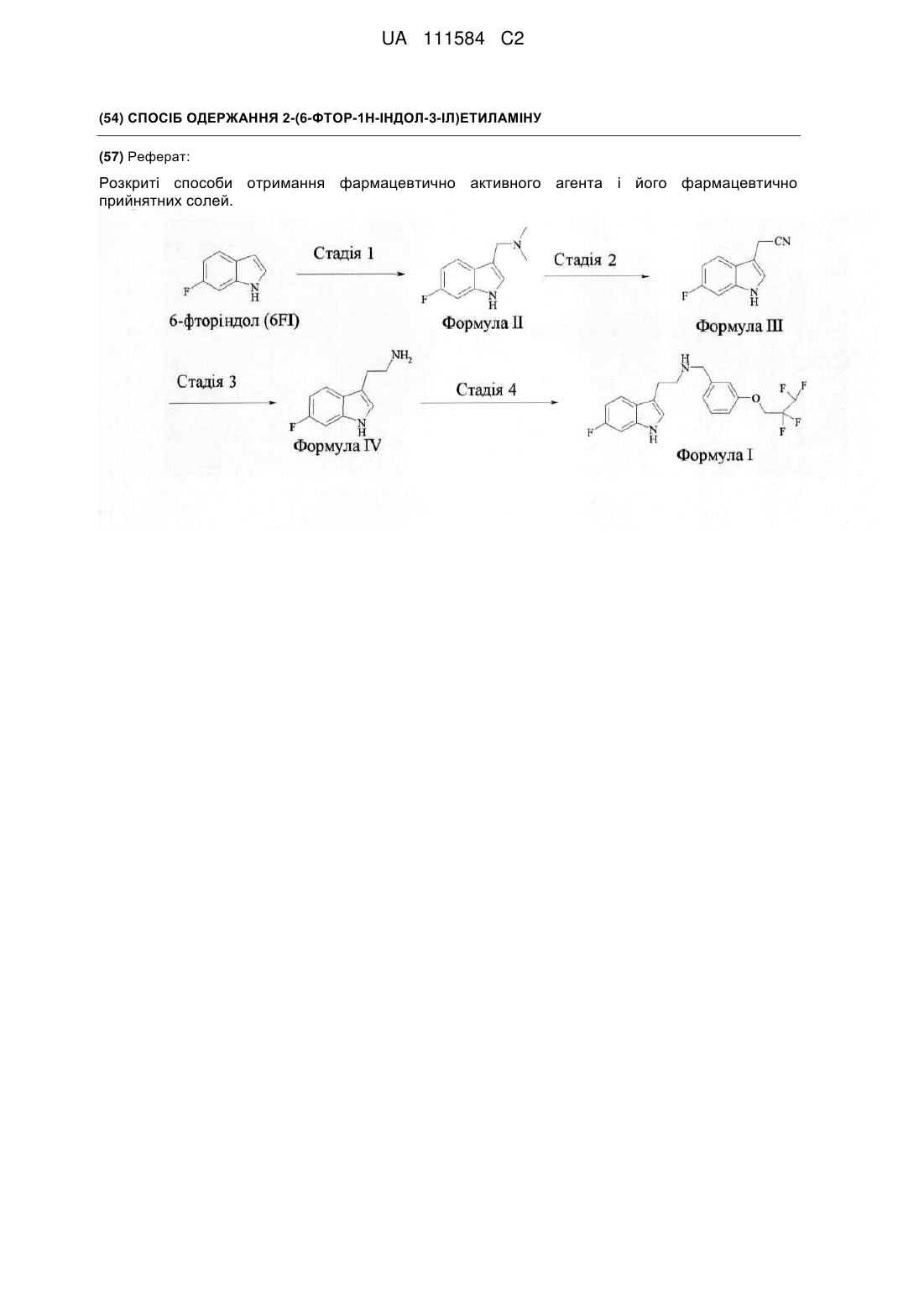








Формула / Реферат
1. Спосіб одержання 2-(6-фтор-1H-індол-3-іл)етиламіну формули IV
 ,
,
при якому здійснюють:
(a) змішування (6-фтор-1H-індол-3-іл)ацетонітрилу, водного розчину NH3 і каталізатора на основі перехідного металу в спиртовому розчиннику і
(b) гідрування отриманої суміші воднем.
2. Спосіб за п. 1, який відрізняється тим, що каталізатором на основі перехідного металу є RaNi (нікель Ренея).
3. Спосіб за п. 1 або 2, який відрізняється тим, що спиртовим розчинником є метанол.
4. Спосіб за будь-яким з пп. 1-3, який відрізняється тим, що гідрування здійснюють при тиску приблизно 2,5 бар (0,25 МПа) протягом приблизно 16 годин.
5. Спосіб за будь-яким з пп. 1-4, який відрізняється тим, що гідрування здійснюють при температурі від приблизно 55 °C до приблизно 65 °C.
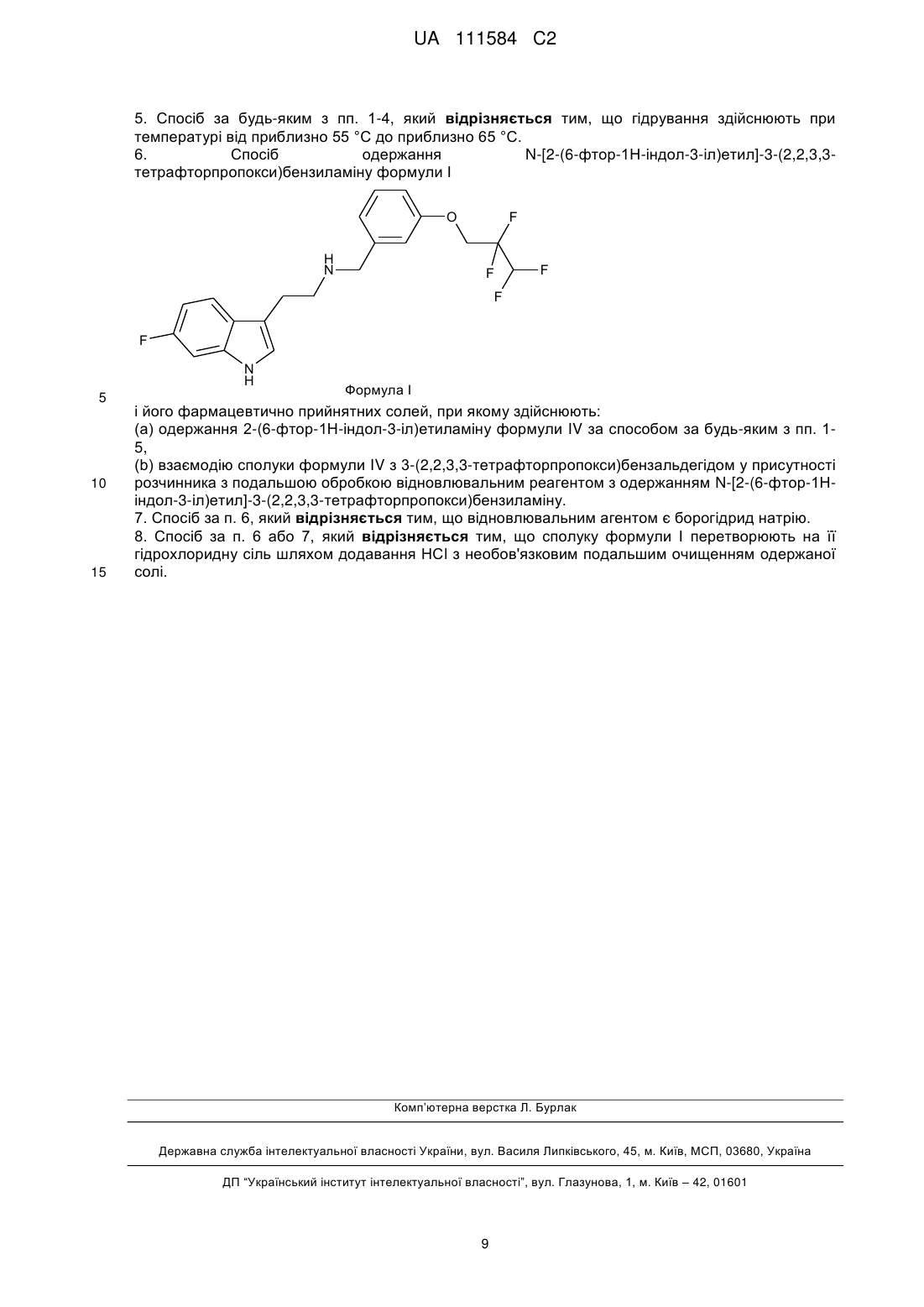
6. Спосіб одержання N-[2-(6-фтор-1Н-індол-3-іл)етил]-3-(2,2,3,3-тетрафторпропокси)бензиламіну формули І

і його фармацевтично прийнятних солей, при якому здійснюють:
(a) одержання 2-(6-фтор-1Н-індол-3-іл)етиламіну формули IV за способом за будь-яким з пп. 1-5,
(b) взаємодію сполуки формули IV з 3-(2,2,3,3-тетрафторпропокси)бензальдегідом у присутності розчинника з подальшою обробкою відновлювальним реагентом з одержанням N-[2-(6-фтор-1Н-індол-3-іл)етил]-3-(2,2,3,3-тетрафторпропокси)бензиламіну.
7. Спосіб за п. 6, який відрізняється тим, що відновлювальним агентом є борогідрид натрію.
8. Спосіб за п. 6 або 7, який відрізняється тим, що сполуку формули І перетворюють на її гідрохлоридну сіль шляхом додавання НСl з необов'язковим подальшим очищенням одержаної солі.
Текст
Реферат: Розкриті способи отримання фармацевтично активного агента і його фармацевтично прийнятних солей. UA 111584 C2 5 10 15 20 25 30 35 40 45 50 Галузь техніки, до якої відноситься винахід Цей винахід відноситься до одержання 2-(6-фтор-1Н-індол-3-іл)етиламіну, придатного для отримання N-[2-(6-фтор-1Н-індол-3-іл)етил]-3-(2,2,3,3-тетрафторпропокси)бензиламіну і його фармацевтично прийнятних солей. Попередній рівень техніки Рецептор 5-HT6 є представником суперродини пов'язаних з G-протеїном рецепторів і, так само як рецептори 5-HT4 і 5-HT7, позитивно пов'язаний з аденілатциклазою (Monsma, F. et al. Mol. Pharmacol. 1993, 43, 3, 320-327). Рецептор 5-HT6 щурів був вперше клонований в 1993 році, а про клонування людського гомолога, ідентичність послідовності якого складає 89 %, було повідомлено в 1996 році (Kohen, R. et al. J Neurochem. 1996, 66, 1, 47-56). Локалізація рецепторів 5-HT6 в головному мозку щурів досліджувалася з використанням кількісного аналізу матричних РНК шляхом нозерн-аналізу та полімеразної ланцюгової реакції із зворотною транскриптазою (RT-PCR), імуногістохімічного і авторадіографічного аналізів (Ward, R., et al. J. Comp Neurol. 1996, 370, 3, 405-414; і Ward, R. et al. Neuroscience 1995, 64, 4, 1105-1111). Високі рівні цих рецепторів були знайдені за усіма вказаними методами в ділянці нюхового горбика, гіпокампа, смугастого тіла, прилеглого ядра і кори мозку. Рецептори 5-HT6 або відсутні, або присутні на дуже низьких рівнях в периферійних тканинах. В значній мірі первинний інтерес до рецепторів 5-HT6 був пов'язаний із спостереженням, що деякі психотропні препарати є високоефективними антагоністами людських рецепторів 5-HT6. Ці препарати включають амітриптилін (Ki=65 нМ) і атипові нейролептики: клозапін (Ki=9,5 нМ), оланзапін (Ki=10 нМ) і кветіапін (33 нМ). Дивися: Roth, B. L., et al. J. Pharmacol. Exp. Ther. 1994, 268, 3, 1403-1410. Використання селективних антагоністів рецепторів 5-HT6 при лікуванні когнітивної дисфункції широко прийнято і засновано на низці логічних міркувань. Наприклад, селективні антагоністи рецептора 5-HT6 модулюють холінергічну і глутаматергічну нейрональну функцію. Холінергічна і глутаматергічна нейрональні системи відіграють важливу роль в когнітивній функції. Холінергічний нейрональний метаболічний шлях, як відомо, є важливим для формування пам'яті і запам'ятовування. Як показано в дослідженнях на тваринах і клінічних випробуваннях, антихолінергічні препарати центральної дії послаблюють когнітивну функцію, а втрата холінергічних нейронів є однією з ознак хвороби Альцгеймера. З іншого боку, стимулювання холінергічної функції, як відомо, покращує когнітивну діяльність, і два препарати, схвалені на даний час для лікування когнітивних розладів, пов'язаних з хворобою Альцгеймера, галантамін і донепезил, є інгібіторами ацетилхолінестерази. Також відомо, що глутаматергічна система в префронтальній корі мозку пов'язана з когнітивною функцією (Dudkin, K.N., et al. Neurosci. Behav. Physiol. 1996, 26, 6, 545-551). Активність селективних антагоністів рецепторів 5-HT6 також показана на тваринних моделях когнітивної функції. Починаючи з відкриття перших селективних антагоністів рецептора 5-HT6, було декілька повідомлень про активність цих селективних препаратів в моделях когнітивної функції. Наприклад, селективний антагоніст рецепторів 5-HT6, препарат SB-271046, покращує показники в тесті водного лабіринту Моріса (Morris) (Rogers, D. et al. Br. J. Pharamcol. 1999, 127 (suppl): 22P). Ці результати узгоджувалися з відкриттям, що постійне введення в шлуночок головного мозку (церебро-вентрикулярне введення (i.c.v.)) антисмислових олігонуклеотидів, спрямованих на послідовність рецепторів 5-HT6, приводить до деякого поліпшення при виконанні тесту водного лабіринту Моріса (Bentley, J. et al. Br. J. Pharmacol. 1999, 126, 7, 153742). Лікування препаратом SB-271046 також приводить до поліпшення у старих щурів при проходженні тесту просторових функціональних здібностей. В даний час декілька антагоністів рецепторів 5-HT6 проходять клінічні випробування як потенційні ліки для хвороб, пов'язаних з когнітивною дисфункцією. Перше повідомлення про те, що антагоніст рецептора 5-HT6, препарат SB-742457, приводить до клінічного поліпшення у пацієнтів, страждаючих хворобою Альцгеймера, додатково підтверджує терапевтичний потенціал даного методу лікування. N-[2-(6-фтор-1Н-індол-3-іл)етил]-3-(2,2,3,3-тетрафторпропокси)бензиламін є ефективним і селективним антагоністом рецепторів 5-HT6, що проходить в даний час клінічні випробування. Його хімічна структура зображена нижче як структурна формула I. 55 1 UA 111584 C2 5 10 15 20 25 30 35 40 Синтез N-[2-(6-фтор-1Н-індол-3-іл)етил]-3-(2,2,3,3-тетрафторпропокси)бензиламіну, його використання при лікуванні захворювань, таких як розлади когнітивної функції, і фармацевтичні композиції, що містять цю речовину, розкриті в патенті США № 7157488 (далі "патент "488"). Також в патенті "488 описується одержання відповідної солі у вигляді моногідрохлориду. Хоча методи синтезу, розкриті у вищезгаданому патенті, достатні для отримання невеликих кількостей препарату, вони мають різні недоліки, пов'язані з проблемами безпеки, низькими виходами або неможливістю застосувати дані методи у виробництві великих кількостей. Тому існує актуальна потреба в пошуку способу отримання сполуки формули I. Таким чином, цей винахід описує ефективний і економічний спосіб отримання сполуки формули I, що є застосовним для виробництва кілограмових кількостей речовини для доклінічних і клінічних випробувань, а також для комерційного використання. Зокрема, автори цього винаходу несподівано встановили роль аміаку, яка полягає в запобіганні димеризації при проведенні відновлення нітрил-вмісної проміжної сполуки у відповідний амін. Суть винаходу Цей винахід відноситися до способу отримання N-[2-(6-фтор-1Н-індол-3-іл)етил]-3-(2,2,3,3тетрафторпропокси)бензиламіну і його фармацевтично прийнятних солей, що включає стадії: (а) взаємодії 6-фторіндолу з різновидом імінієвого іона, що генерується in-situ з формальдегіду і диметиламіну у присутності водного розчину кислоти, що приводить до одержання сполуки формули II (b) взаємодії сполуки формули II з KCN у присутності суміші диметилформамід/вода, що приводить до одержання сполуки формули III (c) гідрування сполуки формули III воднем у присутності аміаку, використовуючи каталіз перехідним металом, що приводить до одержання сполуки формули IV і (d) взаємодії сполуки формули IV з 3-(2,2,3,3-тетрафторпропокси)бензальдегідом у присутності розчинника з подальшою обробкою відновлювальним реагентом. Окремий аспект винаходу відноситися до способу отримання сполуки формули II, що включає стадії: (a) змішування розчину діетоксиметану, води і мурашиної кислоти; (b) додавання розчину, отриманого на стадії (a), до суміші 6-фторіндолу, метиламіну і оцтової кислоти і (c) додавання водного лужного розчину. У одному з варіантів здійснення розчин, отриманий на стадії (a), змішують при температурі від приблизно 75 °C до приблизно 85 °C. У іншому варіанті здійснення розчин, отриманий на стадії (a), перемішують менше приблизно 2 годин. Ще в іншому варіанті здійснення розчин, отриманий на стадії (a), додають до суміші 6 2 UA 111584 C2 5 10 15 20 25 30 35 40 45 фторіндолу і оцтової кислоти при температурі приблизно 2-8 °C. У іншому варіанті здійснення водним лужним розчином є водний розчин NaOH. У одному з варіантів здійснення вихід продукту реакції становить більше 90 %. У одному з варіантів здійснення вихід продукту реакції становить більше 95 %. У окремому варіанті здійснення вихід продукту реакції становить більше 98 %. Інший аспект винаходу відноситься до способу отримання сполуки формули IV, що включає наступні стадії: (a) змішування (6-фтор-1Н-індол-3-іл)ацетонітрилу, 25 % водного розчину NH3 і каталізатора на основі перехідного металу в спиртовому розчиннику і (b) гідрування отриманої суміші воднем. У одному варіанті здійснення каталізатором на основі перехідного металу є RaNi (нікель Ренея). У одному варіанті здійснення спиртовим розчинником є метанол. У іншому варіанті здійснення гідрування проводять при тиску приблизно 2,5 бар (0,25 МПа) протягом приблизно 16 годин. У одному варіанті здійснення гідрування проводять при температурі від приблизно 55 °C до приблизно 65 °C. Інший аспект винаходу відноситься до способу очищення 2-(6-фтор-1Н-індол-3-іл)етиламіну, що включає наступні стадії: (a) розчинення 2-(6-фтор-1Н-індол-3-іл)етиламіну в спиртовому розчиннику; (b) додавання розчину L(+)-винної кислоти і (c) відділення солі винної кислоти у вигляді осаду. У одному варіанті здійснення спиртовим розчинником є метанол. У одному з варіантів здійснення разом із спиртовим розчинником використовують етилацетат. Докладний опис винаходу Як показано вище, цей винахід заснований на відкритті відповідного ефективного і економічного способу отримання N-[2-(6-фтор-1Н-індол-3-іл)етил]-3-(2,2,3,3-тетрафторпропокси)бензиламіну і його фармацевтично прийнятних солей. Нижче винахід описується докладніше, але не мається на увазі, що цей опис є обмежуючим переліком всіх шляхів, за якими може бути здійснений винахід, або всіх ознак, які можуть бути додані до цього винаходу. Відповідно, цей винахід був досягнутий розробкою нового способу, що описується Схемою I Схема I Спосіб, що починається з комерційно доступного 6-фторіндолу, може бути охарактеризований таким чином. • На першій стадії комерційно доступний 6-фторіндол перетворюють на (6-фтор-1Н-індол-3ілметил)диметиламін. Дане перетворення включає реакцію Манніха, в якій генерується різновид імінієвого іона in-situ. У одному варіанті здійснення цей різновид імінієвого іона генерують in-situ з діетоксиметану і диметиламіну. У іншому варіанті здійснення цей різновид імінієвого іона генерують in-situ з формальдегіду і диметиламіну. У іншому варіанті здійснення реакцію проводять у водному розчиннику. • На другій стадії (6-фтор-1Н-індол-3-ілметил)диметиламін перетворюють на (6-фтор-1Ніндол-3-іл)ацетонітрил реакцією з ціанідом калію у присутності суміші диметилформаміду і води 3 UA 111584 C2 5 10 15 20 25 30 35 40 45 50 55 60 при підвищених температурах. У іншому варіанті здійснення підвищеною температурою є температура, близька до температури кипіння реакційної суміші із зворотним холодильником. • На третій стадії (6-фтор-1Н-індол-3-іл)ацетонітрил перетворюють на 2-(6-фтор-1Н-індол-3іл)етиламін. Це перетворення включає відновлення нітрилу до первинного аміну з використанням водню і каталізатора на основі перехідного металу у присутності аміаку. У одному варіанті здійснення каталізатором на основі перехідного металу є нікель Ренея. • На четвертій стадії 2-(6-фтор-1Н-індол-3-іл)етиламін перетворюють на N-[2-(6-фтор-1Ніндол-3-іл)етил]-3-(2,2,3,3-тетрафторпропокси)бензиламін поєднанням аміну з 3-(2,2,3,3тетрафторпропокси)бензальдегідом у присутності розчинника з подальшим відновленням імінного зв'язку під дією відновлювального реагенту. Дане перетворення є реакцією відновного амінування. У одному варіанті здійснення відновлювальним реагентом є борогідрид натрію. Істотна перевага даного способу відповідно до даного винаходу полягає в тому, що використання аміаку на третій стадії несподівано запобігає небажаній димеризації (6-фтор-1Ніндол-3-іл)ацетонітрилу, забезпечуючи чистоту реакції і високий вихід. Хоча гідрування основного нітрилу на нікелі Ренея було відоме протягом деякого часу (Huber, W. JACS 1944, 66, 876-879), використання тільки нікелю Ренея в синтезі сполуки формули I не може бути практичним методом. Використання аміаку як реакційної добавки, яка діє узгоджено з каталітичним промотором, таким як нікель Ренея, вже було розкрито (Robinson and Snyder, Organic Syntheses Collective Volume 3, 720-722). Проте, попередній рівень техніки указує на той факт, що використання аміаку зменшує загальну активність (Thomas-Pryor, et al. Chem. Ind. 1998, 17, 195, Viullemin, et al. Chem. Eng. Sci. 1994, 49, 4839-4849; і Fouilloux, New Frontiers in Catalysis-Proceedings of the th 10 International Congress on Catalysis, 1992, Elsevier Science, Amsterdam, 255-2558). Як додаткові приклади див. EP 0913388, WO 00/27526, WO 99/22561, US 5777166 і US 5801286. Таким чином, в попередньому рівні техніки не розкривається і не пропонується використовувати аміак у відновленні нітрилів з використанням нікелю Ренея через те, що спостерігається зменшення загальної активності. Тому, автори винаходу несподівано відкрили, що використання аміаку в даному способі дозволяє проводити реакцію без зменшення загальної активності і запобігає протіканню небажаної димеризації. Нижче наводяться визначення різних скорочень, які використовуються в даному описі: "DEM" ("ДЕМ") – діетоксиметан, "DMF" ("ДМФА") – N, N-диметилформамід, "MeOH" – метанол, "TGF" ("ТГФ") – тетрагідрофуран, "6FI" – 6-фторіндол, "RaNi" – це активований нікелевий каталізатор, який може містити домішки заліза і хрому і який зустрічається у вигляді частинок різного розміру. У одному варіанті здійснення використовуваний RaNi є губчастим металевим каталізатором, комерційно вироблюваним компанією Fluka. У іншому варіанті здійснення використовуваний RaNi є каталізатором A5009 (5 %, 33 мікрони) фірми Johnson Matthey. У іншому варіанті здійснення використовуваний RaNi є каталізатором B111 W фірми Degussa; "Джерело ціаніду" – це KCN, NaCN або інший реагент, який вивільняє аніон CN . "aq" ("водн.") – водний, "DI" – перегнаний або особливо чистий, "RT" – кімнатна температура, "екв" – еквівалент, "г" – грам, "мл" – мілілітр, "л" – літр, "кг" – кілограм, "М" – молярність, "мас. %" – масові відсотки, "ВЕРХ" – високоефективна рідинна хроматографія. Сполука формули I утворює фармацевтично прийнятні кислотно-адитивні солі з різними органічними і неорганічними кислотами, включаючи фізіологічно прийнятні солі, які часто застосовуються у фармацевтичній хімії. Такого роду солі також є частиною цього винаходу. Такі солі включають фармацевтично прийнятні солі, перелічені в Journal of Pharmaceutical Science, 66, 2-19 (1977), які відомі фахівцям в даній галузі. Типові неорганічні кислоти, використовувані для отримання таких солей, включають хлористоводневу, бромистоводневу, йодистоводневу, 4 UA 111584 C2 5 10 15 20 25 30 35 40 45 50 азотну, сірчану, фосфорну, гіпофосфорну, метафосфорну, пірофосфорну і аналогічні кислоти. Солі, що походять з органічних кислот, таких як аліфатичні моно- і дикарбонові кислоти, фенілзаміщені алканові кислоти, гідроксіалканові кислоти і гідроксіалкандикислоти, ароматичні карбонові кислоти, аліфатичні і ароматичні сульфонові кислоти, також можуть бути використані. Такі фармацевтично прийнятні солі включають хлорид, бромід, йодид, нітрат, ацетат, фенілацетат, трифторацетат, акрилат, аскорбат, бензоат, хлорбензоат, динітробензоат, гідроксибензоат, метоксибензоат, метилбензоат, о-ацетоксибензоат, ізобутират, фенілбутират, α-гідроксибутират, бутин-1,4-дикарбоксилат, гексин-1,4-дикарбоксилат, сіль капринової кислоти, сіль каприлової кислоти, цинамат, цитрат, форміат, фумарат, сіль гліколевої кислоти, сіль гептанової кислоти, гіппурат, лактат, малат, малеат, гідроксималеат, малонат, сіль мигдалевої кислоти, мезилат, нікотинат, ізо-нікотинат, оксалат, фталат, терефталат, пропіолат, пропіонат, фенілпропіонат, саліцилат, себакат, сукцинат, суберат, бензолсульфонат, пбромбензолсульфонат, хлорбензолсульфонат, етилсульфонат, 2-гідроксіетилсульфонат, метилсульфонат, нафталін-1-сульфонат, нафталін-2-сульфонат, нафталін-1,5-сульфонат, птолуолсульфонат, ксилолсульфонат, тартрат тощо. Експериментальна частина Опис ВЕРХ Аналіз шляхом ВЕРХ проводився в наступних хроматографічних умовах: колонка: Xterra RP18 (100 мм х 4,6 мм, 3,5 мкм), рухома фаза: 10 мМ карбонат амонію (pH 8,5) / ацетонітрил, від 86/14 до 14/86 (об'ємні відсотки), витрата потоку: 2 мл/хв., температура колонки: приблизно 45 °C, детектування: УФ при 280 нм. Приклад 1. Синтез сполуки формули II Нижче описується докладний синтез сполуки формули II з комерційно доступного 6фторіндолу. На Схемі II використовується діетоксиметан і диметиламін для генерування "різновиду імінієвого іона". Нижче також наводиться альтернативна методика, в якій замість діетоксиметану застосовується формальдегід. Схема II Методика синтезу Отримання формальдегіду було виконане в реакторі A. Синтез сполуки формули II був здійснений в реакторі B. Осадження кінцевого продукту реакції було виконане в реакторі C. Методика У реактор A поміщали діетокиметан (65 мл/54 г), воду (50 мл) і мурашину кислоту (39 мл/47 г). Суміш нагрівали до приблизно 80 °C/кип'ятили із зворотним холодильником протягом 2 годин, а потім охолоджували до приблизно 20 °C. У реактор B поміщали 6-фторіндол (50 г) і 80 % оцтову кислоту (66 мл/70 г, 2,5 екв. по відношенню до 6-фторіндолу). Отриману суспензію охолоджували до 2-5 °C. 40 % Диметиламін (водн.) (103 мл/92 г, 2,2 екв. по відношенню до 6фторіндолу) додавали по краплях в реактор B, підтримуючи температуру нижче приблизно 15 °C. Реакційну суміш перемішували протягом приблизно 20 хвилин, підтримуючи протягом цього часу температуру в діапазоні 2-4 °C. Суміш, отриману в реакторі A (ДЕМ, вода, мурашина кислота, формальдегід і етанол при температурі приблизно 20 °C), додавали по краплях в реактор B, підтримуючи температуру 28 °C. Реакційну суміш перемішували протягом додаткових 10 хвилин при температурі 2-8 °C. Потім реакційну суміш поволі, протягом 1 години, нагрівали приблизно до 40 °C. Реакційну суміш додатково перемішували при температурі приблизно 40 °C протягом 1 години. Реакційну суміш охолоджували приблизно до 20 °C. У реактор C поміщали 3М розчин NaOH (800 мл, 1,24 екв. по відношенню до оцтової кислоти + мурашиної кислоти) і розчин охолоджували приблизно до 10 °C. Реакційну суміш, отриману в реакторі B, додавали по краплях до розчину NaOH в реакторі C, підтримуючи температуру 1015 °C (рН>14). Суспензію перемішували протягом 40 хвилин при 5-20 °C (рН>14). Продукт реакції відокремлювали фільтруванням, двічі промивали на фільтрі водою (2 × 250 мл). Продукт реакції сушили у вакуумі при температурі приблизно 60 °C протягом 16 годин. Вихід: 95 %. Чистота за даними ВЕРХ (280 нм): 98 % площі. 5 UA 111584 C2 5 10 15 20 25 30 35 40 45 Методика з використанням формальдегіду замість діетоксиметану У 250-літровий реактор в атмосфері азоту поміщали приблизно 40 %-вий водний розчин диметиламіну (35,68 кг, 1,0 екв.) при температурі приблизно 17 °C. Суміш охолоджували до приблизно 4,5 °C і протягом 140 хвилин додавали по краплях льодяну оцтову кислоту (43,4 кг, 2,5 екв.), підтримуючи температуру нижче 15 °C. Після перемішування протягом 20 хвилин при температурі приблизно 3 °C поволі протягом приблизно 20 хвилин додавали 37 % водний розчин формальдегіду (25,9 кг, 1,1 екв.), підтримуючи температуру між приблизно 0 °C та приблизно 10 °C. Потім додавали 6-фторіндол (39,2 кг). Спостерігалася екзотермічна реакція, і досягалася кінцева температура приблизно 40 °C, яку потім знижували до приблизно 20 °C. Реакційний розчин поволі протягом приблизно 40 хвилин додавали в 650-літровий реактор, що містить 3М водний розчин NaOH. Суспензію перемішували протягом приблизно 40 хвилин, підтримуючи температуру між приблизно 5 і 20 °C. Продукт реакції відфільтровували, промивали DI водою (120 кг) і сушили при температурі приблизно 50 °C, отримуючи сполуку формули II (45,4 кг). Вихід: 85 %. Приклад 2. Синтез сполуки формули III На Схемі III наводиться докладний синтез сполуки формули III із сполуки формули II. Схема III Покрокова методика У реактор поміщали (6-фтор-1H-індол-3-ілметил)диметиламін (65 г), KCN (31 г), DMF (195 мл) і воду (104 мл). Реакційну суміш нагрівали до приблизно 100-105 °C (інтенсивне кип'ятіння із зворотним холодильником) протягом 5-8 годин. Реакційну суміш охолоджували до 20-25 °C. У реактор поміщали воду (780 мл) і толуол (435 мл) і суміш енергійно перемішували протягом >2 годин. Водний і органічний шари розділяли. Органічний шар послідовно промивали 5 % NaHCO3 (6 × 260 мл), 2М HCl (260 мл), 5 % NaHCO3 (260 мл) і 5 % NaCl (260 мл). Органічний шар фільтрували і упарювали насухо. Додавали MeOH (260 мл) і розчин випарювали насухо. Сполуку формули III виділяли у вигляді оливи коричневого кольору. Вихід: 90 %. Чистота за + даними ВЕРХ (280 нм): 95 %. MS m/z: 193 (М+H) . Приклад 3. Синтез сполуки формули IV Нижче на Схемі IV наводиться докладний синтез сполуки формули IV. Схема IV Методика синтезу Відновлення сполуки формули III до сполуки формули IV воднем виконувалося в автоклаві. Для отримання суспензії RaNi і розчину реагентів, які переносили в автоклав, використовували реактор A і реактор B. Реактори C і D використовувалися для обробки реакційної суміші, а реактори E і F – для виділення сполуки формули IV у вигляді тартрату. Методика У реактор A поміщали RaNi (66 г, вологий) і метанол (600 мл). У автоклав поміщали (використовуючи вакуумну лінію) 25 % NH3 в H2O (375 мл). Суспензію RaNi в метанолі переміщали (використовуючи вакуумну лінію) з реактора A в автоклав. У реактор A поміщали 25 % NH3 в H2O (200 мл) і потім переміщали (використовуючи вакуумну лінію) в автоклав. У реактор B поміщали сполуку формули III (211 г) і метанол (500 мл), які потім переміщали (використовуючи вакуумну лінію) в автоклав. MeOH (600 мл) поміщали в реактор B, а потім переміщали (використовуючи вакуумну лінію) в автоклав. 25 % NH3 в H2O (175 мл) поміщали в 6 UA 111584 C2 5 10 15 20 25 30 35 40 45 50 реактор B, а потім переміщали (використовуючи вакуумну лінію) в автоклав. Реакційну суміш продували азотом (3 х N2 при тиску приблизно 2-3 бар (0,2-0,3 МПа)). Потім реакційну суміш продували воднем (4 х H2 при тиску 2 бар (0,2 МПа)). Тиск водню доводили до приблизно 2 бар (0,2 МПа). Реакційну суміш нагрівали до 60 °C. Тиск водню доводили до 2,5 бар (0,25 МПа). Після витримки протягом приблизно 16 годин при 60 °C і тиску (Н2) у 2,5 бар (0,25 МПа) реакційну суміш охолоджували до кімнатної температури. Реакційну суміш продували азотом (3 х N2 при тиску 2-3 бар (0,2-0,3 МПа)). Реакційну суміш переміщали з автоклава в реактор C. Автоклав промивали MeOH (500 мл). Метанол переносили в реактор С. Реакційну суміш залишали без перемішування протягом 2-16 годин. Надосадову рідину збирали в реактор D. Метанол (350 мл) поміщали в реактор D. Суміш поволі перемішували протягом 5 хвилин, а потім залишали без перемішування протягом 2-16 годин. Надосадову рідину збирали в реактор D. Після розкладання залишки RaNi збирали як відходи. Надосадову рідину в реакторі D фільтрували в атмосфері азоту через celite. Додаткову порцію MeOH (350 мл) фільтрували через celite, отримуючи об'єднаний фільтрат. Цей фільтрат переміщають в реактор E і концентрують при зниженому тиску приблизно до 2 об'ємів (~400-450 мл). Поміщають MeOH (600 мл). Отриману суміш концентрують при зниженому тиску приблизно до 2 об'ємів (~400-450 мл). Поміщають MeOH (600 мл). Суміш концентрують при зниженому тиску приблизно до 2 об'ємів (~400-450 мл). Поміщають MeOH (600 мл). Суміш концентрують при зниженому тиску приблизно до 2 об'ємів (~400-450 мл). Поміщають MeOH (1420 мл), етилацетат (1135 мл) і воду (190 мл). Отриманий в реакторі E розчин нагрівають до кип'ятіння із зворотним холодильником. У реактор F поміщали L(+)-винну кислоту (163,6 г) і MeOH (1135 мл). Розчин з реактора F переносили протягом 5-10 хвилин до розчину в реакторі E, що приводило до утворення осаду необхідного продукту у вигляді тартратної солі. Суміш перемішували протягом приблизно 15 хвилин при кип'ятінні із зворотним холодильником, а потім охолоджували протягом 1 години до температури 5-10 °C. Потім суміш перемішували протягом 1 години при температурі 5-10 °C. Продукт відокремлювали фільтруванням. Осад на фільтрі промивали холодною сумішшю етилацетат:MeOH (1:2, 380:760 мл). Білий продукт сушили у вакуумі при температурі приблизно 40-45 °C протягом 16 годин. Вихід: 82 %. Чистота за даними ВЕРХ (280 нм): 99-100 % площі. MS + m/z: 179 (М+H) . Методика з використанням BH3-ТГФ. Замість гідрування було також досліджено відновлення нітрильної групи сполуки формули III комплексом BH3-ТГФ до відповідного аміну. У 1600-літровий реактор в атмосфері азоту при кімнатній температурі поміщали толуольний розчин, що містить сполуку формули III (18,46 кг). Поволі, протягом приблизно 133 хвилин, до цього розчину додавали 1М розчин комплексу боран-ТГФ (211 кг, 2,2 екв.), підтримуючи температуру між 15 і 25 °C. Отриманий розчин жовтуватого кольору нагрівали приблизно до 65 °C і перемішували при цій температурі протягом приблизно 1 години. Після охолоджування до приблизно 21 °C реакційну суміш додавали по краплях в струмі азоту протягом приблизно 80 хвилин до добре перемішаного 15 % водного розчину NaOH. Двофазну суміш поволі нагрівали до приблизно 50 °C, перемішували в інтервалі приблизно 50-60 °C, нагрівали до приблизно 65 °C і перемішували при цій температурі протягом 1 години. Після охолоджування до приблизно 25 °C, лужний водний шар збирали і відкидали. Потім, для того, щоб відігнати ТГФ при зниженому тиску (0,2 бар (0,02 МПа)), реакційну суміш нагрівали до приблизно 50 °C. До водної фази, що залишилася, додавали дихлорметан (93 л) і при температурі приблизно 22 °C поволі протягом приблизно 30 хвилин додавали водну HCl (18,8 кг 37 % водн. HCl і 22 кг DI води). Цю реакційну суміш залишали перемішуватися при кімнатній температурі протягом 2 годин перед фільтруванням, двічі промивали дихлорметаном (2 × 19 л) і сушили при зниженому тиску протягом ночі, отримуючи сполуку формули IV у вигляді моногідрохлоридної солі. Вихід: 72 %, 17,3 кг. Приклад 4. Синтез сполуки формули I На Схемі V наводиться докладний синтез N-[2-(6-фтор-1Н-індол-3-іл)етил]-3-(2,2,3,3тетрафторпропокси)бензиламіну у вигляді моногідрохлоридної солі. Схема V 7 UA 111584 C2 5 10 15 20 25 Методика Сіль винної кислоти 2-(6-фтор-1H-індол-3-іл)етиламіну (49,3 г) перемішували в суміші толуолу (270 мл), ТГФ (100 мл), 2M NaOH (200 мл) і 15 % NaCl (65 мл). Фази розділяли. Органічну фазу промивали 5 % NaCl (200 мл). Органічну фазу випарювали при зниженому тиску насухо, а залишок розчиняли в ізопропанолі (400 мл). До реакційної суміші додавали 3-(2,2,3,3-тетрафторпропокси)бензальдегід (39 г) і ізопропанол (200 мл). Реакційну суміш нагрівали протягом 2,5 години при 60 °C, а потім охолоджували до приблизно 55 °C. До гарячої реакційної суміші додавали суспензію NaBH4 (7,4 г) в ізопропанолі (100+50 мл). Реакційну суміш нагрівали протягом 2,5 години при 55 °C, а потім охолоджували до приблизно 15-20 °C. Протягом приблизно 30 хвилин додавали по краплях 2М HCl (80 мл). Протягом 15 хвилин додавали по краплях 2М HCl (140 мл). Суміш енергійно перемішували протягом 15 хвилин. Суміш концентрувала до половини об'єму, після чого додавали 6M NaOH (83 мл) до рН ≥ 14. Додавали толуол (400 мл). Фази розділяли і органічну фазу послідовно промивали 2M NaOH (200 мл), 3 % NH4Cl (200 мл) і водою (200 мл). Органічну фазу профільтрували і випарювали насухо. Залишок розчиняли в суміші толуолу (550 мл) і ацетонітрилу (50 мл). По краплях додавали 6М HCl (33 мл). Отриману суспензію перемішували протягом 2-4 годин і потім фільтрували. Осад на фільтрі послідовно промивали сумішшю толуол/ацетонітрил (9:1, 2 × 75 мл) і 0,1М HCl (2 × 75 мл). Сирий гідрохлорид сполуки формули I сушили у вакуумі протягом приблизно 16 годин при температурі приблизно 45 °C. Для остаточного очищення гідрохлоридної солі сполуки формули I, виділену спочатку сіль HCl розчиняли в ацетоні (300 мл). Розчин фільтрували і концентрували до об'єму приблизно 90120 мл. Протягом 30 хвилин додавали по краплях фільтровану 0,1M HCl (1900 мл). Отриману суспензію перемішували при температурі 20-25 °C протягом 16 годин, а потім фільтрували. Осад на фільтрі послідовно промивали фільтрованою 0,1М HCl, (200 мл) і фільтрованою водою (150 мл). Очищену гідрохлоридну сіль сушили у вакуумі протягом 16 годин при 40 °C і виділяли у вигляді твердої речовини білого кольору. Вихід: 80 %. Чистота за даними ВЕРХ (280 нм): + >99,5 %. MS m/z: 399 (М+H) . 30 ФОРМУЛА ВИНАХОДУ 1. Спосіб одержання 2-(6-фтор-1H-індол-3-іл)етиламіну формули IV NH2 F N H Формула IV 35 40 , при якому здійснюють: (a) змішування (6-фтор-1H-індол-3-іл)ацетонітрилу, водного розчину NH3 і каталізатора на основі перехідного металу в спиртовому розчиннику і (b) гідрування отриманої суміші воднем. 2. Спосіб за п. 1, який відрізняється тим, що каталізатором на основі перехідного металу є RaNi (нікель Ренея). 3. Спосіб за п. 1 або 2, який відрізняється тим, що спиртовим розчинником є метанол. 4. Спосіб за будь-яким з пп. 1-3, який відрізняється тим, що гідрування здійснюють при тиску приблизно 2,5 бар (0,25 МПа) протягом приблизно 16 годин. 8 UA 111584 C2 5. Спосіб за будь-яким з пп. 1-4, який відрізняється тим, що гідрування здійснюють при температурі від приблизно 55 °C до приблизно 65 °C. 6. Спосіб одержання N-[2-(6-фтор-1Н-індол-3-іл)етил]-3-(2,2,3,3тетрафторпропокси)бензиламіну формули І F O H N F F F F N H 5 10 15 Формула I і його фармацевтично прийнятних солей, при якому здійснюють: (a) одержання 2-(6-фтор-1Н-індол-3-іл)етиламіну формули IV за способом за будь-яким з пп. 15, (b) взаємодію сполуки формули IV з 3-(2,2,3,3-тетрафторпропокси)бензальдегідом у присутності розчинника з подальшою обробкою відновлювальним реагентом з одержанням N-[2-(6-фтор-1Ніндол-3-іл)етил]-3-(2,2,3,3-тетрафторпропокси)бензиламіну. 7. Спосіб за п. 6, який відрізняється тим, що відновлювальним агентом є борогідрид натрію. 8. Спосіб за п. 6 або 7, який відрізняється тим, що сполуку формули І перетворюють на її гідрохлоридну сіль шляхом додавання НСl з необов'язковим подальшим очищенням одержаної солі. Комп’ютерна верстка Л. Бурлак Державна служба інтелектуальної власності України, вул. Василя Липківського, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 9
ДивитисяДодаткова інформація
Назва патенту англійськоюProcesses for the manufacture of a pharmaceutically active agent
Автори англійськоюTherkelsen, Frans, Rock, Michael, Harold, Treppendahl, Svend
Автори російськоюТеркельсен Франс, Рок Майкл Харольд, Треппендахль Свэнн
МПК / Мітки
МПК: C07D 209/14, C07D 209/16, C07D 209/10
Мітки: 2-(6-фтор-1н-індол-3-іл)етиламіну, одержання, спосіб
Код посилання
<a href="https://ua.patents.su/11-111584-sposib-oderzhannya-2-6-ftor-1n-indol-3-iletilaminu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання 2-(6-фтор-1н-індол-3-іл)етиламіну</a>
Спосіб одержання n,n-диметил-2-[5-(1,2,4-тріазол-1-ілметил)-1н-індол-3-іл]-етиламіну та 2-[5-(1,2,4-тріазол-1-ілметил)-1н-індол-3-іл]-етиловий спирт

Номер патенту: 52586
Опубліковано: 15.01.2003
Автори: Чен Ченг И., Верхоевен Томас Р., Ларсен Роберт Д.
МПК: A61P 9/00, A61K 31/41, A61P 43/00, C07D 521/00, C07D 403/06, A61P 25/04, A61K 31/415
Мітки: спосіб, одержання, n,n-диметил-2-[5-(1,2,4-тріазол-1-ілметил)-1н-індол-3-іл]-етиламіну, спирт, 2-[5-(1,2,4-тріазол-1-ілметил)-1н-індол-3-іл]-етиловий
Формула / Реферат:
1. Способ получения N,N-диметил-2-[5-(1,2,4-триазол-1-илметил)-1Н-индол-3-ил]-этиламина формулы Vотличающийся тем, что он включает стадию обработки 2-[5-(1,2,4-триазол-1-илметил)-1Н-индол-3-ил]-этилового спирта формулы IVмезилхлоридом при -20°С в сухом тетрагидрофуране в...
Похідні 1,4-діарил-2-фтор-1-бутен-3-олу, спосіб їх одержання та способи одержання похідних 1,4-діарил-2-фтор-1,3-бутадієну і 1,4-діарил-2-фтор-2-бутену

Номер патенту: 70975
Опубліковано: 15.11.2004
Автори: Ху Юлін, Хант Девід Аллен
МПК: C07C 41/00, C07C 43/295, C07C 43/29, C07C 33/00
Мітки: 1,4-діарил-2-фтор-2-бутену, похідні, одержання, способи, 1,4-діарил-2-фтор-1-бутен-3-олу, спосіб, похідних, 1,4-діарил-2-фтор-1,3-бутадієну
Формула / Реферат:
1. Сполука структурної формули (І)(I),деR означає водень, С1-С4алкіл, С1-С4галоалкіл, С3-С6циклоалкіл або С3-С6галоциклоалкіл;Аr означає феніл, необов'язково заміщений будь-якою комбінацією замісників, вибраних із ряду, який містить:один(ну)-три атом(и) галогену, С1-С4алкільну(і), С1-С4галоалкільну(і), С1-С4алкокси- або С1-С4галоалкоксигрупу(и);1- або 2-нафтил, необов'язково заміщений будь-якою...
Кристалічна форма солі яблучної кислоти n-[2-(діетиламіно)етил]-5-[(5-фтор-2-оксо-зн-індол-3-іліден) метил]-2,4-диметил-1н-пірол-3-карбоксаміду (варіанти), спосіб їх одержання і їх композиції

Номер патенту: 76483
Опубліковано: 15.08.2006
Автори: Прескотт Стівен П., Хоулі Майкл, Мелоні Марк Т., Флек Томас Дж.
МПК: C07D 403/06
Мітки: метил]-2,4-диметил-1н-пірол-3-карбоксаміду, композиції, n-[2-(діетиламіно)етил]-5-[(5-фтор-2-оксо-зн-індол-3-іліден, спосіб, яблучної, одержання, солі, форма, кристалічна, кислоти, варіанти
Формула / Реферат:
1. Безводна кристалічна форма солі яблучної кислоти сполуки, яка має структуру:.2. Кристалічна форма згідно з пунктом 1, де яблучною кислотою є L-яблучна кислота.3. Безводна кристалічна форма солі яблучної кислоти N-[2-(діетиламіно)етил]-5-[(5-фтор-1,2-дигіро-2-оксо-3Н-індол-3-іліден)метил]-2,4-диметил-1Н-пірол-3-карбоксаміду.4. Кристалічна форма солі яблучної кислоти...
Сульфатна сіль n,n-диметил-2-[5-(1,2,4-триазол-1ілметил)-1н-індол-3-іл]етиламіну та/або її сольвати, фармацевтична композиція на її основі, спосіб одержання фармацевтичної композиції і спосіб лікування

Номер патенту: 27908
Опубліковано: 16.10.2000
Автори: Олів Кароль, Стріт Леслі Джозеф, Бейкер Раймонд, Гіблін Александр Річард, Матасса Віктор Гіліо, Сторі Девід Едвард, Пітт Кендал Джордж
МПК: A61K 31/415, A61P 25/04, C07D 403/06, A61K 9/08, A61K 9/20, A61P 25/06, A61K 31/00, A61K 31/4196, A61P 43/00, A61K 31/41, C07D 521/00
Мітки: сіль, основі, n,n-диметил-2-[5-(1,2,4-триазол-1ілметил)-1н-індол-3-іл]етиламіну, композиції, одержання, фармацевтично, лікування, композиція, фармацевтична, сольвати, спосіб, сульфатна
Текст:
...Композиции для интраназального введения в общем случае могут быть представлены в форме жидкости или сухого порошка. Хорошие композиции для интраназального введения должны быть достаточно стабильными - химически и физически, быть равномерно распределены в точно измеренных дозах даже после длительного хранения с возможными колебаниями температур в пределах от 0 до 40°С. Соответственно активный ингредиент должен быть совместим с...
Спосіб одержання 8-метил-8-азабіцикло (3,2,1) окт-3-илового ефіру індол-3-карбонової кислоти

Номер патенту: 33170
Опубліковано: 15.02.2001
Автори: Хабаров Костянтин Михайлович, Забудкін Олександр Фрідріхович, Попов Анатолій Федорович, Дуленко Володимир Іванович, Донець Володимир Федорович, Щербина Любов Іванівна
МПК: C07D 451/12, C07D 451/04
Мітки: кислоти, спосіб, ефіру, окт-3-илового, індол-3-карбонової, 3.2.1, 8-метил-8-азабіцикло, одержання
Текст:
...за слідуючую схемою иж Механізм складається за галоформеного реакції схемою розпаду шляхом взаємодії алкоголяту тропіну з 3 трихлорацетиліндолом. Така реакція добре проходить при синтезі ефірів індол3-карбонової кислоти та різних первинних спиртів. Нами знайдено умови, при яких ця реакція з успіхом протікає з вторинними спиртами, представником якого є М метил-8-азабіцикло [3,2,1] октан-3-о (тропін). Приклад 1. У 250 мл...
Попередній патент: Зубний імплантат, абатмент для зубного імплантата та їх комбінація та імплантаційний комплект
Наступний патент: Перетворення високоенергетичних фотонів в електрику
Випадковий патент: Гідробіологічний спосіб боротьби із забрудненням морських акваторій