Спосіб стереоселективного одержання z-1,2-діарилалілхлоридів
Номер патенту: 37175
Опубліковано: 15.05.2001
Автори: Хайнц Ізак, Еккард Хікман, Райнер Кобер, Райнер Зееле, Томас Цирке, Норберт Гец







Формула / Реферат
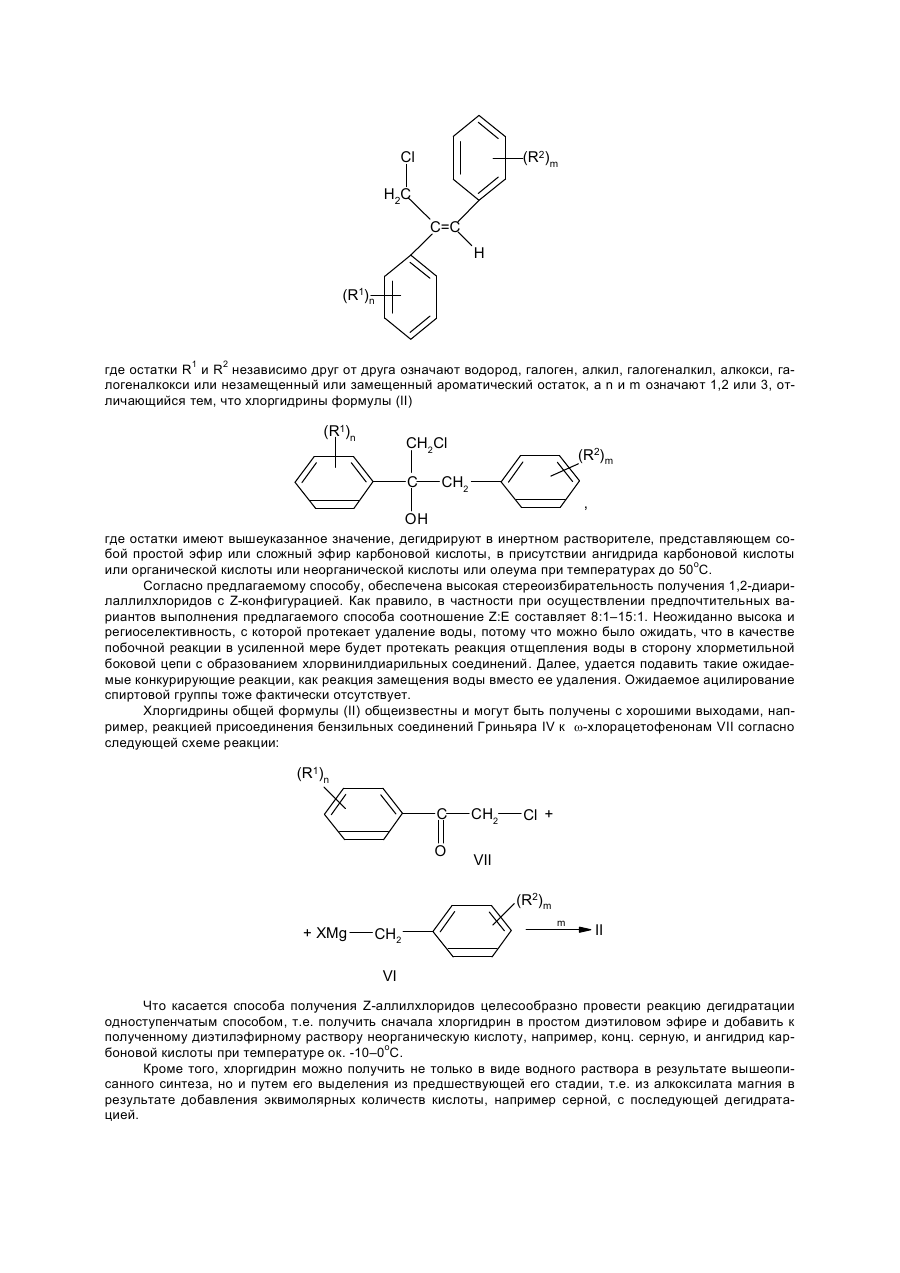
1. Способ стереоселективного получения Z -1,2-диарилаллилхлоридов общей формулы
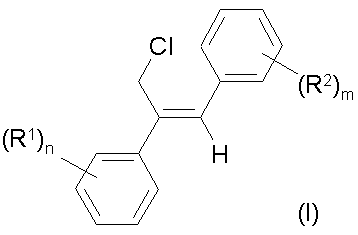
где R1 и R2 независимо друг от друга - водород, галоген, алкил, галогеналкил, алкокси, галогеналкокси или незамещенный или замещенный ароматический остаток,
n и m =1,2 или 3, отличающийся тем, что хлоргидрины общей формулы
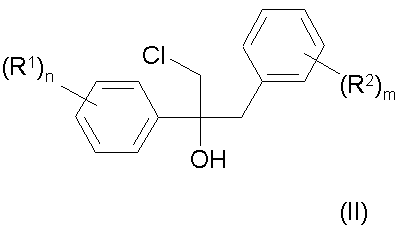
где R1 и R2 имеют указанное выше значение, дегидратируют в инертном простом эфире или сложном эфире карбоновой кислоты в качестве растворителя в присутствии ангидрида карбоновой кислоты или соответствующего кетена и органической или неорганической кислоты или олеума при температурах до 50 °С.
2. Способ по п. 1, отличающийся тем, что в качестве растворителя применяют простой циклический эфир или низкомолекулярный сложный эфир алифатической карбоновой кислоты.
3. Способ по п. 1, отличающийся тем, что в качестве ангидрида карбоновой кислоты берут ангидрид алифатических монокарбоновых кислот.
4. Способ по любому из пп. 1 - 3, отличающийся тем, что дегидратацию осуществляют в диоксане и/или тетрагидрофуране в качестве растворителя в присутствии ацетангидрида и серной кислоты.
5. Способ по любому из пп. 1-3, отличающийся тем, что дегидратацию осуществляют в этиловом эфире уксусной кислоты в качестве растворителя в присутствии ангидрида изомасляной кислоты.
6. Способ по п. 1, отличающийся тем, что используют кетен, если необходимо, в комбинации с органической карбоновой кислотой в количестве от каталитического до стехиометрического, считая на хлоргидрин указанной выше формулы.
7. Способ по п. 1, отличающийся тем, что дегидратацию осуществляют при температуре от -25 до +30 °С.
8. Способ по п. 1, отличающийся тем, что дегидратацию осуществляют в присутствии 0,01 - 4,0 моль серной кислоты и 0,5 - 3,0 моль ангидрида карбоновой кислоты.
9. Способ по п. 1, отличающийся тем, что дегидратацию осуществляют в присутствии 0,05 - 1,0 моль олеума в смеси с 1 - 2 моль ангидрида карбоновой кислоты.
Текст
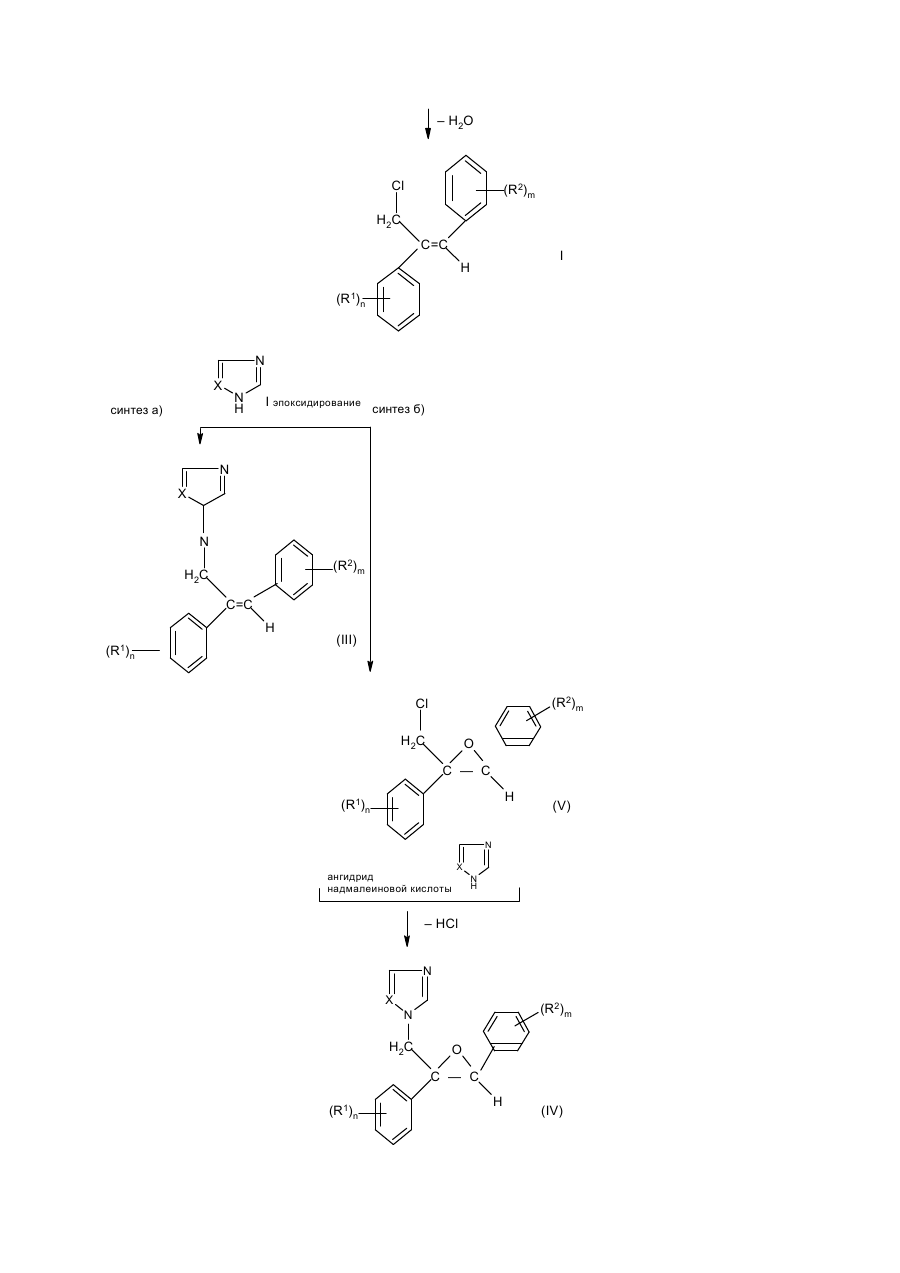
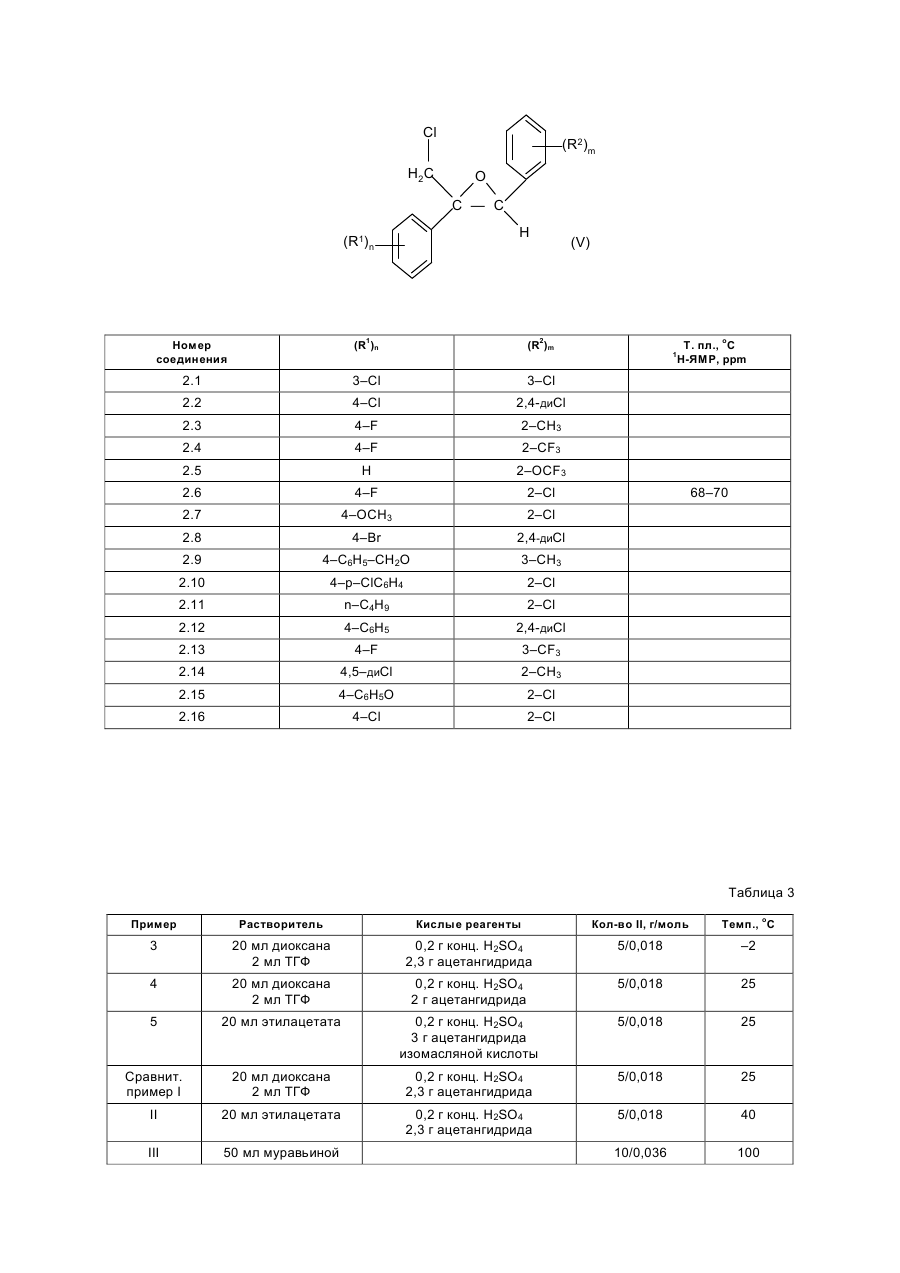
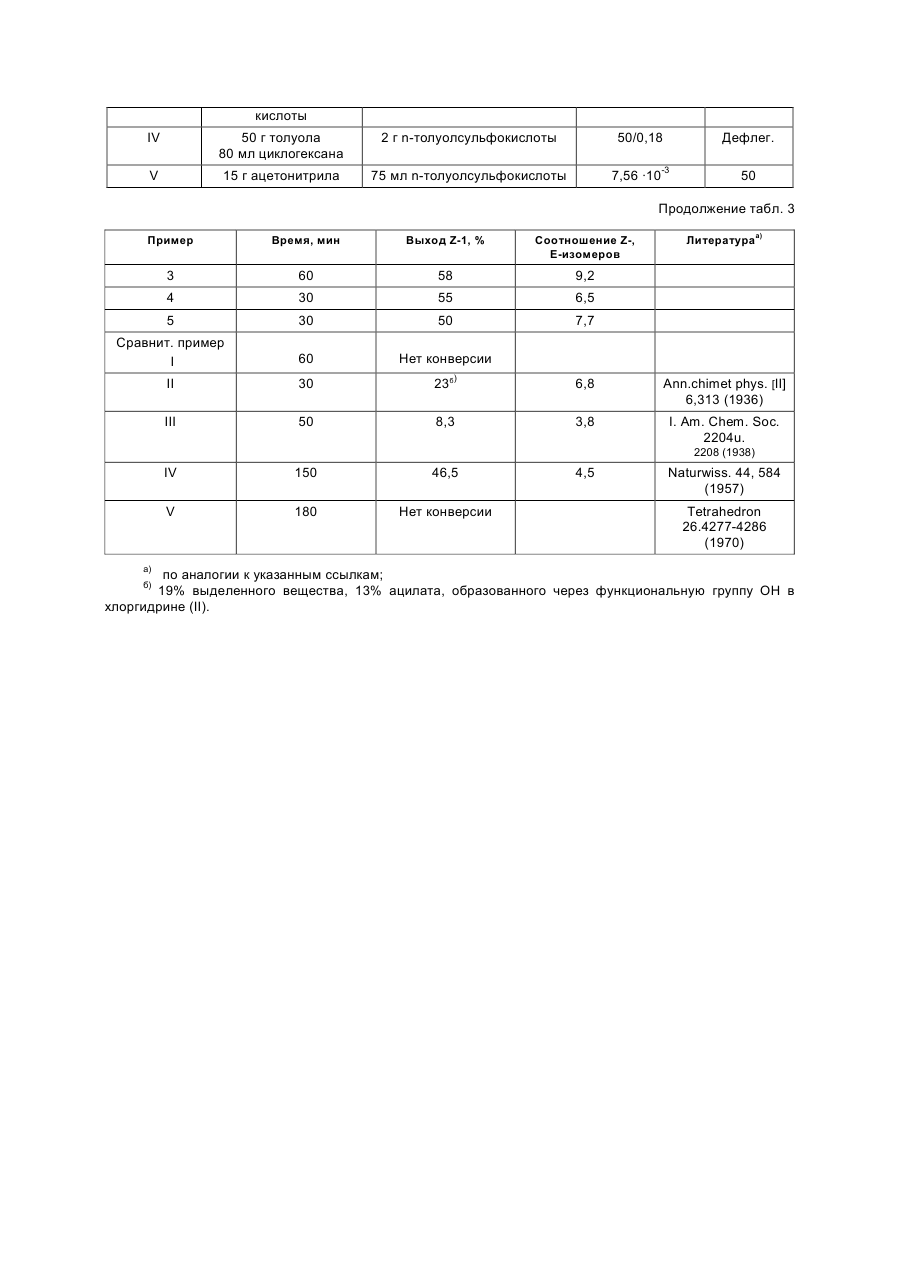
Изобретение относится к стереоизбирательному получению 1,2-диарилаллилхлоридов общей формулы Cl (R2)m H2C C=C H (R1)n , (I) где остатки R1 и R2 независимо друг от друга означают водород, галоген, алкил, галогеналкил, алкокси, галогеналкокси или незамещенный или замещенный ароматический остаток, а n и m означают 1, 2 или 3. Согласно выкладкам ФРГ №№ 3218129 и 3218130, а также Патенту Европы 196038 и Патенту США 3422153, соединения с типом структуры I представляют собой ценные промежуточные продукты в получении фармакологических, фунгицидных и противогрибковых действующих начал. До сих пор их получали путем радикального галогенирования соответствующих диарилпропеновых соединений или путем окисления с последующей реакцией замещения. Недостатком известных из данного уровня техники способов является применение дорогостоящих реагентов, например, таких дорогих реагентов галогенирования, как N-бромсукцинимид в реакции радикального бромирования, число ступеней реакции синтеза и, в частности, низкая стереоизбирательность реакции. Общеизвестно, что молекулы, проявляющие специфическое биологическое или фармакологическое действие, во многих случаях требуют определенного геометрического расположения определенных функциональных групп. Например, среди фунгицидных действующих начал особенно высокой эффективностью в качестве средств защиты растений обладают в первую очередь соединения с Z-конфигурацией (см. правило последовательности заместителей по Кану, Ингольду и Прелогу), т.е. соединения, в которых незамещенные или замещенные фенилы один относительно другого находятся в транс-положении. Поэтому задача настоящего изобретения заключалась в разработке способа, согласно которому удается получить с высоким выходом промежуточные продукты 1 с максимальной чистотой изомеров, т.е. с высокой избирательностью относительно Е- и транс-конфигурации фенильных остатков, присоединенных к двойной связи соединения. Другая задача заключалась в том, чтобы с применением выгодных промежуточных продуктов разработать способ получения фунгицидных азолилметилоксиранов IV, отличающийся высокими общими выходами и меньшим числом стадий реакций, чем у известных способов. Согласно уровню техники арилзамещенные спирты в кислых условиях реакции, например, с применением серной кислоты в органической фазе можно перевести в соответствующие арилзамещенные олефины или стиролы (см. например, Houber–Weyl. Methoden der organischen Chemie. 4-e изд., т. 5/1b Alkene, Cycloalkene, Arylalkene изд-во Georg Thleme Verlag г. Штуттгарт, 1972 г., стр. 62 и сл., в частности стр. 70 и 71: Tetrahedron т. 26, стр. 4277 и сл. (1790 г.)). Известно также, что подобного рода реакции можно осуществить с использованием акцепторов воды, например ацетатангидрида. Однако для осуществления этих реакций отщепления обычно требуются высокие температуры. В таких условиях реакции получают только недостаточное соотношение Е- и Z-изомеров относительно расположения арила по отношению к арилу. Теперь найден способ стереоизбирательного получения Z-1,2-диарил-аллилхлоридов общей формулы (I) Cl (R2)m H2C C=C H (R1)n где остатки R1 и R2 независимо друг от друга означают водород, галоген, алкил, галогеналкил, алкокси, галогеналкокси или незамещенный или замещенный ароматический остаток, а n и m означают 1,2 или 3, отличающийся тем, что хлоргидрины формулы (II) (R1)n CH2Cl C (R2)m CH2 , OH где остатки имеют вышеуказанное значение, дегидрируют в инертном растворителе, представляющем собой простой эфир или сложный эфир карбоновой кислоты, в присутствии ангидрида карбоновой кислоты или органической кислоты или неорганической кислоты или олеума при температурах до 50оС. Согласно предлагаемому способу, обеспечена высокая стереоизбирательность получения 1,2-диарилаллилхлоридов с Z-конфигурацией. Как правило, в частности при осуществлении предпочтительных вариантов выполнения предлагаемого способа соотношение Z:E составляет 8:1–15:1. Неожиданно высока и региоселективность, с которой протекает удаление воды, потому что можно было ожидать, что в качестве побочной реакции в усиленной мере будет протекать реакция отщепления воды в сторону хлорметильной боковой цепи с образованием хлорвинилдиарильных соединений. Далее, удается подавить такие ожидаемые конкурирующие реакции, как реакция замещения воды вместо ее удаления. Ожидаемое ацилирование спиртовой группы тоже фактически отсутствует. Хлоргидрины общей формулы (II) общеизвестны и могут быть получены с хорошими выходами, например, реакцией присоединения бензильных соединений Гриньяра IV к w-хлорацетофенонам VII согласно следующей схеме реакции: (R1)n C O CH2 Cl + VII (R2)m + XMg CH2 m II VI Что касается способа получения Z-аллилхлоридов целесообразно провести реакцию дегидратации одноступенчатым способом, т.е. получить сначала хлоргидрин в простом диэтиловом эфире и добавить к полученному диэтилэфирному раствору неорганическую кислоту, например, конц. серную, и ангидрид карбоновой кислоты при температуре ок. -10–0оС. Кроме того, хлоргидрин можно получить не только в виде водного раствора в результате вышеописанного синтеза, но и путем его выделения из предшествующей его стадии, т.е. из алкоксилата магния в результате добавления эквимолярных количеств кислоты, например серной, с последующей дегидратацией. Согласно изобретению, целесообразно постепенное добавление ангидрида карбоновой кислоты, причем процесс О-ацилирования хлоргидрина в значительной мере удается подавить в пользу процесса дегидратации. Предлагаемую реакцию дегидратации хлоргидринов (II) осуществляют в простом или сложном эфире в качестве растворителя. В случае использования нециклических простых эфиров предпочтение следует отдать эфирам с числом атомов кислорода не менее 2, например эфирам гликолей и низкомолекулярных алифатических спиртов, например, этиленгликольдиметиловому или диэтиловому эфиру. Особенно выгодным оказались циклические простые эфиры, например ТГФ и в частности диоксан. Для улучшения сольволиза при низких температурах, например, ниже ~10оС можно добавить незначительные количества апротонных растворителей, например этилацетата, галогенуглеводородов, таких как метиленхлорид или ТГФ, например к диоксану в качестве растворителя. Для предлагаемого способа особенно выгодными сложными эфирами оказались эфиры низкомолекулярных алифатических карбоновых кислот, в частности монокарбоновых, с низкомолекулярными алифатическими спиртами, причем понятие низкомолекулярных спиртов включает спирты с числом атомов С от одного до шести. В качестве примеров эфиров можно назвать этиловый эфир уксусной кислоты, этиловый эфир муравьиной кислоты, метиловый эфир пропионовой кислоты, метиловый эфир масляной кислоты, метиловый или этиловый эфир изомасляной кислоты, причем предпочтение отдается этилацетату. Количества растворителей не играют решающей роли и колеблются в широких пределах. Они обычно составляют 1–50 мас.%, в частности 2,5–10 мас.%, считая на хлоргидрин (II). Более высокие количества растворителей вполне возможны. Для дегидратации можно также использовать смеси растворителей, упомянутых, например, в п.1–5 формулы изобретения, причем соотношение компонентов может колебаться в широких пределах ~ от 10:1 до 1:10. В целях достижения высоких выходов за единицу времени и высоких долей Z-изомеров рекомендуется прибавить добавки в количествах 5–20 мас.%, считая на диоксан. В качестве акцептора кислоты к реакционной массе добавляют ангидрид карбоновой кислоты. В частности применяют такие ангидриды алифатических низкомолекулярных монокарбоновых кислот, как ацетангидрид, ангидриды пропионовой, масляной и изомасляной кислот. Но можно также использовать и ангидриды алифатических или ароматических дикарбоновых кислот, например малоновой, малеиновой, янтарной или фталевой кислот. В реакции дегидратации, как правило, используются 0,5–3, в частности 1–2 мольных эквивалента ангидрида, считая на хлоргидрин (II). Возможно также применение больших количеств, но они не дают какихлибо дополнительных выгод. Особенно выгодные результаты достигаются комбинацией диоксана и/или ТГФ в качестве растворителя и ацетангидрида и серной кислоты или в случае применения этилового эфира уксусной кислоты в качестве растворителя в комбинации с ангидридом изомасляной кислоты и серной кислотой. Дегидратация осуществляется в кислых реакционных условиях, для создания которых применяют обычные кислоты, например такие органические сульфокислоты, как трифторметансульфоновая, метансульфоновая, п-толуолсульфоновая или нафталинсульфоновая, и в частности такие концентрированные минеральные кислоты, как хлорная, фосфорная и в частности серная с концентрацией 30–99,9%, предпочтительно 50–99%, или олеум. В случае применения кислот с большим содержанием воды обычно применяют большее количество ангидрида карбоновой кислоты. Кислоту применяют в каталитическом, стехиометрическом или избыточном количестве, считая на соединение (II). Предпочитаются количества около 0,01–4 мольн. эквивалента, считая на соединение (II). В случае применения олеума целесообразно использовать меньшие количества – 0,05–1 мольн. эквивалент, считая на соединение (II). Выгодный вариант выполнения предлагаемого изобретения заключается в том, что в качестве акцептора воды вместо ангидрида карбоновой кислоты применяют кетен как таковой или в сочетании, считая на соединение (II), со стехиометрическими или каталитическими количествами алифатической карбоновой кислоты. В этом случае целесообразно поместить в реактор карбоновую кислоту, например одну из вышеприведенных низкомолекулярных алифатических, и добавить в реакционную массу газообразный кетен или же в растворенный в растворителе хлоргидрин (II) добавляют кетен в газообразном виде без добавки карбоновой кислоты. Количество добавляемого кетена соответствует вышеуказанным количествам ангидрида карбоновой кислоты. Для достижения высоких долей Z-изомеров рекомендуется провести дегидратацию при минимальных температурах, т.е. при температурах до ~ 50оС, предпочтительно от -25 до +40оС, в частности от -25 до +30оС. Как правило, дегидратацию осуществляют под обычным давлением. Осуществление реакции под повышенным или пониженным давлением в некоторых случаях может привести к увеличению выхода за единицу времени. Таким образом, согласно предлагаемому способу, получают Z-1,2-диарилаллилхлориды формулы (I) Cl (R2)m H2C C=C H (R1)n где остатки R1 и R2 независимо друг от друга означают водород, галоген, алкил С1–С7, галогеналкил С1–С5 алкокси С1–С5, галогеналкокси С1–С5 или незамещенный или замещенный одним–тремя остатками, указанными для радикалов R1 и R2, ароматический остаток, n-1 или 2, или 3. В формуле (I) показатели n и m предпочтительно означают 1, а заместители R1 и R2 в этой формуле независимо друг от друга водород, галоген, например, фтор, хлор, бром, йод, предпочтительно хлор и фтор: разветвленный или неразветвленный алкил С1–С7, например метил, этил, пропил, 1-метилэтил, бутил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил, пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, гексил, 1-метилпентил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,2-диметилбутил, 2,3-диметилбутил, 3,3-диметилбутил, 1-этилбутил, 2-этилбутил, 1,1,2-триметилпропил, 1,2,2– триметилпропил, 1-этил-1-метилпропил, 1-этил-2-метилпропил; галогеналкил С1–С6, например фторметил, дифторметил, трифторметил, хлорфторметил, дихлорфторметил, трихлорметил, 1-фторэтил, 2-фторэтил, 2,2-дифторэтил, 2,2,2-трифторэтил, 2-хлор-2,2дифторэтил, 2,2-дихлор-2-фторэтил, 2,2, 2-трихлорэтил, пентафторэтил, предпочтительно трифторметил; алкокси С1–С5, например, метокси, этокси, пропокси, 1-метилэтокси, бутокси, 1-метилпропокси, 2-метилпропокси, 1,1-диметилокси, предпочтительно метокси, этокси и пропокси; галогеналкокси С1–С5, например дифторметокси, трифторметокси, хлордифторметокси, дихлорфторметокси, 1-фторэтокси, 2-фторэтокси, 2,2-дифторэтокси, 1,1,2,2-тетрафторэтокси, 2,2,2-трифторэтокси, 2хлор-1,1,2-трифторэтокси, пентафторэтокси, предпочтительно трифторметокси; ароматический остаток, например фенил, который незамещен или одно-, двух- или трехкратно замещен остатком R3, имеющий предпочтительное значение, указанное для R1 или R2, т.е. который означает водород, галоген, разветвленный или неразветвленный алкил С1–С7, галогеналкил С1–С6, алкокси С1–С5 или галогеналкокси С1–С5. Предпочитаются остатки R1–2-F и R2–2-Се. По сравнению с известными из патента ДЕ–А 32 18 129 Z-1,2-диарилаллилбромидами, Z-1,2-диарилаллилхлориды общей формулы (I) обладают неожиданными преимуществами. Помимо очень простого их эпоксидирования до диарилоксиранов следует назвать его то преимущество, что благодаря стереоизбирательности эпоксидирования получают не смеси изомеров оксиранов, что имеет место, когда исходным материалом являются известные Z-1,2-диарилаллилбромиды, а оксираны, у которых арилы имеют трансоидное расположение. Могут присутствовать, например, указанные в табл. 1 типы замещения. Определение соотношения изомеров Z:E в диарилаллилхлоридах (I) проводится известным образом, например ВСЖХ (высокоскоростной жидкостной хроматографией), газовой хроматографией или методами 1 Н-ЯМР-спектрометрии с применением чистых Z- и E-изомеров для сравнения и стандартизацией соответствующих соотношений изомерных компонентов смеси. Получение фунгицидных действующих начал III и IV на основе исходных материалов – диарилаллилхлоридов I и хлоргидринов II изображено в нижеследующей схеме реакций. CH2Cl C (R1)n (R2)m CH2 II OH – H2O – H2O Cl (R2)m H2 C C=C I H (R1)n N X N H синтез а) I эпоксидирование синтез б) N X N (R2)m H2C C=C H (R1) (III) n (R2)m Cl H2C O C — C H (R1)n (V) N ангидрид надмалеиновой кислоты X N H – HCl N X (R2)m N H2C O C — C (R1)n H (IV) Последовательность реакций согласно синтезу б) можно осуществить известным образом, например, по методу, принцип которого описан в патенте ДЕ–А 32 18 129. Реакция замещения хлорного атома азольной или имидазольной группой в соединении V обычно осуществляется в инертном растворителе, например диметилформамиде или N-метилпирролидоне, в присутствии неорганического или органического основания, такого как гидроксид натрия или калия, карбоната натрия или калия, дициклогексиламина, диметилциклогексиламина. Промежуточные продукты (V) являются новыми соединениями. Предпочтительные остатки R1 и R2, а также показатели n и m имеют значение, аналогичное определениям, изложенным в связи с описанием соединения 1 (см. табл.2). В случае синтеза а) первая стадия, т.е. стадия замещения соответствует последней стадии синтеза б). Целесообразно осуществить процесс дегидратации и последующей процесс замещения одностадийным способом без выделения и очистки промежуточного продукта (II). В целях эпоксидирования соединений (III) целесообразно использовать высокий избыток надмалеиновой кислоты, которую получают "ин ситу" путем взаимодействия 5–30 мольн. экв., в частности 5–10 мольн. экв. ангидрида малеиновой кислоты, считая на соединение (III), с раствором перекиси водорода в количествах ниже стехиометрических, считая на ангидрид малеиновой кислоты. Обычно используют мольное соотношение ангидрида с пероксидом водорода 1,5–10, в частности 2–4. Предпочтительно используют 30–50%-ный водный раствор пероксида водорода. Температура реакции для процесса эпоксидирования 0–100оС, в частности 20–80оС. Эпоксидирование осуществляют в присутствии апротонного полярного растворителя. В качестве таких растворителей можно использовать, например, такие галогенуглеводороды, как дихлорметан, дихлорэтан, хлорбензол или хлортолуол, или такие ароматические углеводороды, как бензол, толуол или ксилол. Количество растворителя не имеет решающего значения. Оно обычно составляет 5–50, в частности 10–20 мас.%, считая на олефин. Согласно этому методу эпоксидирования, можно получить намного более высокие выходы азолилметилоксиранов (IV), чем по способу, описанному в Патенте DЕ–А 3218129. Отдельные стадии синтеза описаны в последующих примерах. Пример 1. Получение исходных веществ (II). 1-Хлор-2-(4-хлорфенил)-3-(2-хлорфенил)-пропан-2-ол. К 9,7 г (0,404 моль) магниевой стружки в 20 мл абс. диэтилового эфира при 24–36оС в течение 5 мин добавляют 5,0 г (0,031 моль) 2-хлорбензилхлорида. После того, как начнется реакция, по каплям добавляют раствор 200 мл абс. диэтилового эфира и 50,2 г (0,31 моль) 2-хлорбензилхлорида. Затем продолжают нагревать массу при температуре дефлегмации в течение еще 10 мин. В атмосфере азота декантируют избыточный магний. Полученный в результате раствор Гриньяра помещают в реактор при 0оС. Затем к нему по каплям добавляют 55,7 г (0,3 моль) п-хлор-w-хлорацетофенона, растворенного в 350 мл толуола, и продолжают перемешивать массу при 0оС в течение еще 1,5 ч. При температуре ~ 2–6оС реакционную массу по каплям добавляют в 1,5 л конц. раствора хлористого аммония. После экстрагирования метил-трет-бутиловым эфиром с последующей обычной переработкой получают 92,9 г (выход 99%, степень чистоты по данным ВСЖХ – 68,2 %) 1-хлор-2-(4-хлорфенил)-3-(2-хлорфенил)пропан-2-ол в виде сырого масла, которое подвергается последующей реакции. Для определения продукта проводили перекристаллизацию из нгексана; т.пл. 64–69оС. Примеры 2–5 и сравнительные примеры I–IV. Дегидратация хлоргидринов (II). Z-3-хлор-2-(4-хлорфенил)-1-(2-хлорфенил)- пропен. (Соединение № 1.16 в табл. 1). При -2оС к 60 г (0,2 моль) хлорного спирта, описанного в примере 1, в 230 мл диоксана и 23 мл ТГФ добавляют 24,5 г (0,24 моль) ацетангидрида. Затем к массе по каплям добавляют 2,36 г (0,024 моль) конц. серной кислоты. После перемешивания в течение 3 ч при 0о по данным ВСЖХ фактически весь исходный материал прореагировал. Затем при 0оС в течение 30 мин добавляют смесь из полунасыщенного раствора хлористого натрия и 50%-ного раствора едкого натра для того, чтобы установилось значение рН 8–9. Наконец, высушивают органическую фазу и концентрируют ее в вакууме, после чего она может применяться для последующих реакций без какой-либо дополнительной очистки. Выход 55,7 г (ZE=9,1/1) сырого масла, которое после перекристаллизации из н-гексана дает чистый Z-изомер с т.пл. 79–82оС. Аналогично можно получить Z-1,2-диарилаллилхлориды (cм. табл. 1). Z-3-хлор-2-(4-фторфенил)-1-(2-хлорфенил)-пропен. (примеры № 1.6 в табл. 1). 1-хлор-2-(4-фторфенил)-3-(2-хлорфенил)-пропен-2-ол, полученный реакцией присоединения по Гриньяру хлористого 2-хлорбензилмагния к п-фтор-w-хлорацетофенону и применяемого в качестве сырого материала со степенью чистоты по данным ВСЖХ 78–87%, в условиях реакции, описанных в табл. 2, подвергали реакции, как описано в примере 2. Долю Z- и E-изомеров определяли методом высокоскоростной жидкостной хроматографии (нескорректированный относительный процент площади) (см. табл. 3). Дегидратация 1-хлор-2-(4-фторфенил)пропан-2-ола. CH2Cl C F OH CH2 Cl II CH2Cl Cl C=C H I (Z= конфигурация) Пример 6. Получение хлоргидрина и процесс дегидратации "ин ситу" 1-хлор-2-(4-хлорфенил)-3-(2хлорфенил)пропан-2-ол. К 36,0 г (1,5 моль) магниевой стружки в 200 мл простого диэтилового эфира по каплям добавляют раствор 170 г (1,0 моль) 2-хлорбензилхлорида в 400 мл диэтилового эфира. Затем при -10оС по каплям добавляют раствор 155 г (0,9 моль) п-фтор-w-хлорацетофенона в 450 мл диэтилового эфира. Затем еще 2 ч перемешивают при 25оС. Потом при -10оС по каплям добавляют 49,0 г (0,5 моль) конц. серной кислоты в 300 мл диэтилового эфира. Потом нагревают до 25оС и отсасывают от выпавшей соли. Полученный сырой эфирный раствор хлоргидрина используют для нижеуказанной реакции. Z-3-хлор-2-(4-фторфенил)-1-(2-хлорфенил) пропен. К 525 мл вышеописанного сырого раствора, содержащего ок. 134,5 г хлоргидрина (0,45 моль), при 10оС добавляют 8,0 г (0,08 моль) конц. серной кислоты, после чего в течение 2 ч добавляют 57,1 г (0,58 моль) ацетангидрида. Затем выпавшую соль вновь отделяют фильтрацией. Полученный в результате выпаривания растворителя из фильтрата сырой аллилхлорид используют для тризолового замещения или для эпоксидирования. Пример 7. Кетеновый вариант. Z-3-хлор-2(4-фторфенил)-1-(2-хлорфенил)- пропен. При 0оС через исходную смесь 250 мл диоксана, 25 мл ТГФ, 12,4 г уксусной кислоты (0,2 моль) и 69 г (0,23 моль) сырого 1-хлор-2-(4-фторфенил)-3-(2-хлорфенил)пропан-2-ола, полученного в результате реакции Гриньяра по примеру 1, в течение ~ 1 ч пропускают 43 г (1,02 моль) кетена. После обычной переработки продукта на основе данных ВСЖХ получают выход, который практически равняется выходу, получаемому в случае применения ацетангидрида в соответствии с примером 2. Соотношение Z- и E-изомеров для данного типа ведения реакции составляет 11:1. Примеры 8 и 9. Получение азолилметилоксиранов (IV) по синтезу (а). Z-3-(1,2,4-триазол-1-ил)-2-(4-хлорфенил)-1-(2-хлорфенил)пропен. К раствору 11,5 г (0,17 моль) триазола в 150 мл диметилформамида добавляют 6,6 г гидрооксида натрия и с перемешиванием нагревают до температуры ~ 70о до тех пор, пока не образуется прозрачный раствор. Затем охлаждают до 10оС, после чего в течение 1 ч по каплям добавляют 49,5 г полученного по примеру 2 Z-3-хлор-2-(4-хлорфенил)-1-(2-хлорфенил)пропен в виде сырого продукта в 50 мл диметилформамида. Затем массу перемешивают еще 4 ч при комнатной температуре. Затем добавляют 200 мл воды и неоднократно экстрагируют простым метил-трет-бутиловым эфиром. Собранные органические фазы промывают, высушивают и концентрируют в вакууме. После перекристаллизации из метил-трет-бутилового эфира и н-гексана получают 24,4 г Z-3-(1,2,4-триазол-1-ил)-2-(4-хлорфенил)-1-(2-хлорфенил)про-пена с т.пл. 106–110оС. Цис-2-(1,2,4-триазол-1-илметил)-2-(4-фторфенил)-3-(2-хлорфенил)-оксиран. 84 г (0,9 моль) ангидрида малеиновой кислоты и 6 капель конц. серной кислоты в 90 мл дихлорэтана вместе с 22 г 50%-ной перекиси водорода нагревают до 50оС. Затем по каплям добавляют 28 г (0,089 моль) Z-3-(1,2,4-триазол-1-ил)-2-(4-фторфенил)-1-(2-хлорфенил)пропена в 75 мл дихлорэтана. Массу перемешивают 3 ч при указанной температуре и затем еще 2,5 ч при 70оС. После охлаждения реакционной массы отсасывают продукт от выпавшей малеиновой кислоты и встряхивают его с раствором тиосульфата и разбавленным раствором едкого натра. Высушенная и максимально выпаренная в вакууме при температуре около 50оС органическая фаза после охлаждения и повторного выпаривания маточного раствора дает 14 г целевого продукта (что равняется выходу в 50%). Примеры 10 и 11. Получение азолилметилоксиранов (IV) согласно синтезу б). Цис-1-хлорметил-2-(2-хлорфенил)-1-(4-фторфенил)оксиран (соединение № 2.6 по табл. 2). Исходят из 56,2 г (0,2 моль) Z-3-хлор-2-(4-фторфенил)-1-(2-хлорфенил)пропена в 530 мл ледяной уксусной кислоты и добавляют 196 г (2 моль) ангидрида малеиновой кислоты. Затем в течение 1 ч при 25оС добавляют 68 г (1 моль) 50%-ного раствора перекиси водорода. Массу перемешивают в течение еще 3–4 ч при 40оС, затем в течение 10 ч при 25оС. Наконец, реакционную массу с перемешиванием вводят в 3 л воды и 50 мл 10%-ного раствора тиосульфата натрия и потом, если необходимо, вновь добавляют раствор тиосульфата натрия до исчезновения перекиси. Получаемый в результате бесцветный осадок отсасывают и высушивают. Сырое вещество, полученное в результате перекристаллизации из п-гексана можно использовать без очистки (т.пл. 68– 70оС). Цис-2-(1,2,4-триазол-1-илметил)-2-(4-фторфенил)-3-(2-хлорфенил) оксиран. 1,5 г (5 ммоль) цис-1-хлорметил-2-(2-хлорфенил)-1-(4-фторфенил)оксирана и 0,69 г (7,5 ммоль) 1,2,4-триазолида натрия перемешивают в течение 5 ч при 75оС в 7 мл ДМФ. После охлаждения реакционную массу нейтрализуют путем добавления незначительного количества уксусной кислоты, после чего добавляют около 10 мл воды, в результате чего выпадает кристаллический продукт (выход 1,4 г). Полученный продукт отсасывают, промывают водой и высушивают в вакууме. Таблица 1 Cl (R2)m H2C C=C H (R1) n (R1)n 1 (R2)m 2 Номер соединения (R )n (R )m 1 2 3 1.1 3–Cl 3–Cl 1.2 4–Cl 2,4-диCl 1.3 4–F 2–CH3 1.4 4–F 2–CF3 1.5 H 2–OCF3 1.6 4–F 2–Cl 1.7 4–OCH3 2–Cl 1.8 4–Br 2,4-диCl 1.9 4–C6H5–CH2O о 3–CH3 1 Т. пл., С Н-ЯМР, ppm 4 66 Продолжение табл. 1 1 2 3 1.10 4–p–ClC6H4 2–Cl 1.11 n–C4H9 2–Cl 1.12 4–C6H5 2,4-диCl 1.13 4–F 3–CF3 1.14 4,5–диCl 2–CH3 1.15 4–C6H5O 2–Cl 1.16 4–Cl 2–Cl 4 79–82 Таблица 2 Cl H2C (R2)m O C — C H (R1)n 1 (V) 2 Номер соединения (R )n 2.1 3–Cl 3–Cl 2.2 4–Cl 2,4-диCl 2.3 4–F 2–CH3 2.4 4–F 2–CF3 2.5 H о (R )m 2–OCF3 2.6 4–F 4–OCH3 2–Cl 2.8 4–Br 2,4-диCl 2.9 4–C6H5–CH2O 3–CH3 2.10 4–p–ClC6H4 2–Cl 2.11 n–C4H9 2–Cl 2.12 4–C6H5 Т. пл., С Н-ЯМР, ppm 2–Cl 2.7 1 2,4-диCl 2.13 4–F 3–CF3 2.14 4,5–диCl 2–CH3 2.15 4–C6H5O 2–Cl 2.16 4–Cl 68–70 2–Cl Таблица 3 о Пример Растворитель Кислые реагенты Кол-во ІІ, г/моль Темп., С 3 20 мл диоксана 2 мл ТГФ 0,2 г конц. Н2SO4 2,3 г ацетангидрида 5/0,018 –2 4 20 мл диоксана 2 мл ТГФ 0,2 г конц. Н2SO4 2 г ацетангидрида 5/0,018 25 5 20 мл этилацетата 0,2 г конц. Н2SO4 3 г ацетангидрида изомасляной кислоты 5/0,018 25 Сравнит. пример I 20 мл диоксана 2 мл ТГФ 0,2 г конц. Н2SO4 2,3 г ацетангидрида 5/0,018 25 II 20 мл этилацетата 0,2 г конц. Н2SO4 2,3 г ацетангидрида 5/0,018 40 III 50 мл муравьиной 10/0,036 100 кислоты IV 50 г толуола 80 мл циклогексана 2 г n-толуолсульфокислоты 50/0,18 Дефлег. V 15 г ацетонитрила 75 мл n-толуолсульфокислоты 7,56 ·10-3 50 Продолжение табл. 3 а) Пример Время, мин Выход Z-1, % Соотношение Z -, E-изомеров Литература 3 60 58 9,2 4 30 55 6,5 5 30 50 7,7 Сравнит. пример I 60 Нет конверсии ІІ 30 23б ) 6,8 Ann.chimet phys. [II] 6,313 (1936) ІІІ 50 8,3 3,8 I. Am. Chem. Soc. 2204u. 2208 (1938) IV 46,5 V а) 150 180 Нет конверсии 4,5 Naturwiss. 44, 584 (1957) Tetrahedron 26.4277-4286 (1970) по аналогии к указанным ссылкам; 19% выделенного вещества, 13% ацилата, образованного через функциональную группу ОН в хлоргидрине (II). б) Тираж 50 екз. Відкрите акціонерне товариство «Патент» Україна, 88000, м. Ужгород, вул. Гагаріна, 101 (03122) 3 – 72 – 89 (03122) 2 – 57 – 03
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for stereoselective preparation of z -1,2-diarylallylchlorides
Автори англійськоюReiner Kober, Reiner Seele, Heinz Isak, Eckhard Hickmann, Norbert Goetz, Thomas Zierke
Назва патенту російськоюСпособ стереоселективного получения z-1,2-диарилаллилхлоридов
Автори російськоюРайнер Кобер, Райнер Зееле, Хайнц Изак, Эккард Хикман, Норберт Гец, Томас Цирке
МПК / Мітки
МПК: C07C 25/00, C07C 17/35, C07D 521/00, C07D 249/08, C07C 43/29, C07D 233/58, C07D 303/00, C07D 233/56, C07C 43/225
Мітки: спосіб, стереоселективного, одержання, z-1,2-діарилалілхлоридів
Код посилання
<a href="https://ua.patents.su/11-37175-sposib-stereoselektivnogo-oderzhannya-z-12-diarilalilkhloridiv.html" target="_blank" rel="follow" title="База патентів України">Спосіб стереоселективного одержання z-1,2-діарилалілхлоридів</a>
Спосіб одержання змішаних ангідридів хінолінкарбонової кислоти та борної кислоти

Номер патенту: 19065
Опубліковано: 25.12.1997
Автори: Марія Балог, Агнеш Хорват, Лелле Вашварі, Геза Керестурі, Петер Рітлі, Іштван Хермец
МПК: C07D 215/22
Мітки: хінолінкарбонової, кислоти, борної, спосіб, ангідридів, одержання, змішаних
Формула / Реферат:
1. Способ получения смешанных ангидридов хинолинкарбоновой кислоты и борной кислоты общей формулыгде R1 и R2 - галоген или алифатическая ацилоксигруппа, содержащая 2-6 атомов углерода, R3 - хлор или фтор; R4 - циклопропил, этил или фенил, возможно замещенный одним или двумя галогенами; R5 - водород или фтор, отличающий-с я тем, что соединение общей формулыгде R3, R4 и R5 имеют указанные значения;R6 -...
Спосіб одержання 4-(4-n,n-диметиламінофеніл) піридину

Номер патенту: 29574
Опубліковано: 15.11.2000
Автори: Лисенко Юрій Олексійович, Старовойтова Ірина Юріївна, Ютілов Михайло Юрійович, Щербина Любов Іванівна, Ютілов Юрій Михайлович
МПК: C07D 213/24, C07D 213/127
Мітки: спосіб, 4-(4-n,n-диметиламінофеніл, одержання, піридину
Текст:
...воронкой, загружают 24 мл (23,45 г, 0,297 моль) пиридина и, охлаждая водяной баней, быстро прикапывают при температуре 20-25° 20,8 мл (25,35 г; 0,18 моль) хлористого бензоила. Реакционную смесь нагревают при температуре в бане 100-105°С в течение 1 часа, затем охлаждают и прикапывают при температуре 2022°С 22,7 мл (21,7 г; 0,179 моль) N,N-диметиланилина. При интенсивном перемешивании и охлаждении ледяной водой прибавляют порциями 21,1 г...
Спосіб одержання похідних n-(2-оксо-3-оксазолідініл)ацетамідів

Номер патенту: 7033
Опубліковано: 31.03.1995
Автори: Рудольф Зандмайєр, Йост Харр, Ханспетер Шеллінг
МПК: A01N 43/76, C07D 263/26, C07D 263/22, C07D 413/12
Мітки: похідних, спосіб, одержання, n-(2-оксо-3-оксазолідініл)ацетамідів
Формула / Реферат:
(57) Способ получения производных N-{2-оксо-3-оксазолидинил)ацетамидов, общей формулыгдеотличающийся тем, что проводят внутримолекулярную конденсацию соединения общей формулы
Спосіб одержання n-карбоксифенілімидів 4-діметиламінонафталін-1, 8-дікарбонової кислоти

Номер патенту: 16696
Опубліковано: 29.08.1997
Автори: Кормилова Людмила Іванівна, Сердечна Тамара Андріївна, Єрмоленко Інна Григорівна, Переяслова Дія Георгіївна
МПК: C07D 221/14
Мітки: кислоти, спосіб, одержання, 4-діметиламінонафталін-1, n-карбоксифенілімидів, 8-дікарбонової
Формула / Реферат:
Способ получения N-карбоксифенилимидов 4- диметиламинонафталин-1,8-дикарбоновой кислоты взаимодействием соответствующего N-карбоксифепилимида 4-галоі'еннафталин-1,8- дикарбоновой кислоты с амином в среде растворителя при повышенной температуре, отличающийся тем, что, с целью упрощения процесса, в качестве N-карбоксифенилимида 4-галогеннафталин-1,8-дикарбоновой кислоты используют N-карбоксифенилимид 4-хлорнафталин-1,8-дикарбоновой кислоты или...
Спосіб одержання похідних хінолінкарбонової кислоти або її фармацевтично придатних солей та проміжні сполуки для їх одержання
Номер патенту: 26568
Опубліковано: 11.10.1999
Автори: Вашварі Лелле, Керестурі Геза, Шіпош Юдіт, Рітлі Петер, Балог Марія, Хермец Іштван, Хорват Агнеш, Пайор Аніко
МПК: C07D 401/04, C07D 215/56, C07F 5/00, A61K 31/495, A61P 31/04
Мітки: одержання, спосіб, фармацевтично, проміжні, похідних, хінолінкарбонової, придатних, сполуки, солей, кислоти
Формула / Реферат:
1. Способ получения производных хинолинкарбоновой кислоты общей формулы lгде R1 и R3 - водород или C1 - 4-алкил;R2 - C1 - 4-алкил;R4, R5 и R6 - каждый - водород или галоген,или ее фармацевтически приемлемых солей, взаимодействием производного хинолина с производным пиперазина, отличающийся тем, что в качестве производного хинолина используют соединение общей формулы llгде R - галоген,...
Попередній патент: Система цифрової передачі та використовувані в системі передавач і приймач.
Наступний патент: Спосіб вилучення корозійних металевих забруднень із рідкої композиції і спосіб одержання карбонової кислоти і/або її ангідриду
Випадковий патент: Спосіб нагрівання хвостового газу у виробництві азотної кислоти