Спосіб одержання дабігатрану









Формула / Реферат
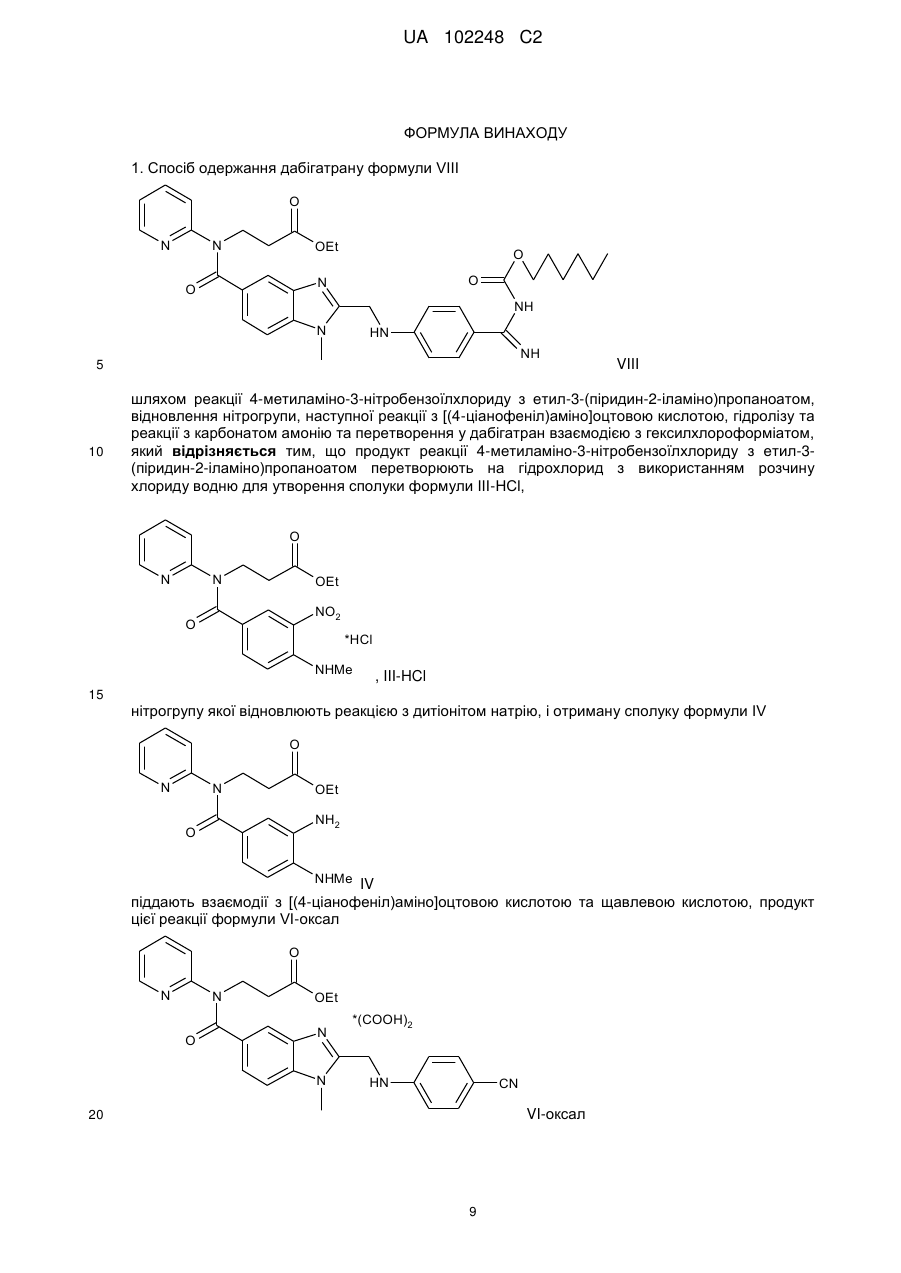
1. Спосіб одержання дабігатрану формули VIII
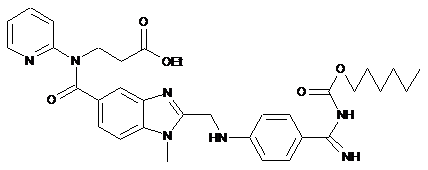 VIII
VIII
шляхом реакції 4-метиламіно-3-нітробензоїлхлориду з етил-3-(піридин-2-іламіно)пропаноатом, відновлення нітрогрупи, наступної реакції з [(4-ціанофеніл)аміно]оцтовою кислотою, гідролізу та реакції з карбонатом амонію та перетворення у дабігатран взаємодією з гексилхлороформіатом, який відрізняється тим, що продукт реакції 4-метиламіно-3-нітробензоїлхлориду з етил-3-(піридин-2-іламіно)пропаноатом перетворюють на гідрохлорид з використанням розчину хлориду водню для утворення сполуки формули ІІІ-НСl,
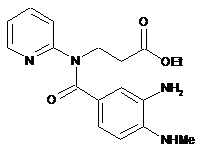
 , III-HCl
, III-HCl
нітрогрупу якої відновлюють реакцією з дитіонітом натрію, і отриману сполуку формули IV
 IV
IV
піддають взаємодії з [(4-ціанофеніл)аміно]оцтовою кислотою та щавлевою кислотою, продукт цієї реакції формули VІ-оксал
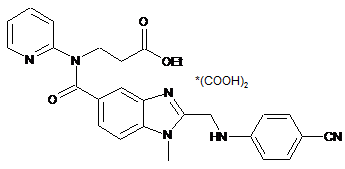 VI-оксал
VI-оксал
потім піддають гідролізу та взаємодії з карбонатом амонію для одержання проміжної сполуки формули VII-HCl,
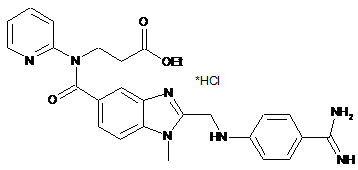 , VII-HCl
, VII-HCl
яку далі перетворюють на дабігатран реакцією з гексилхлорформіатом.
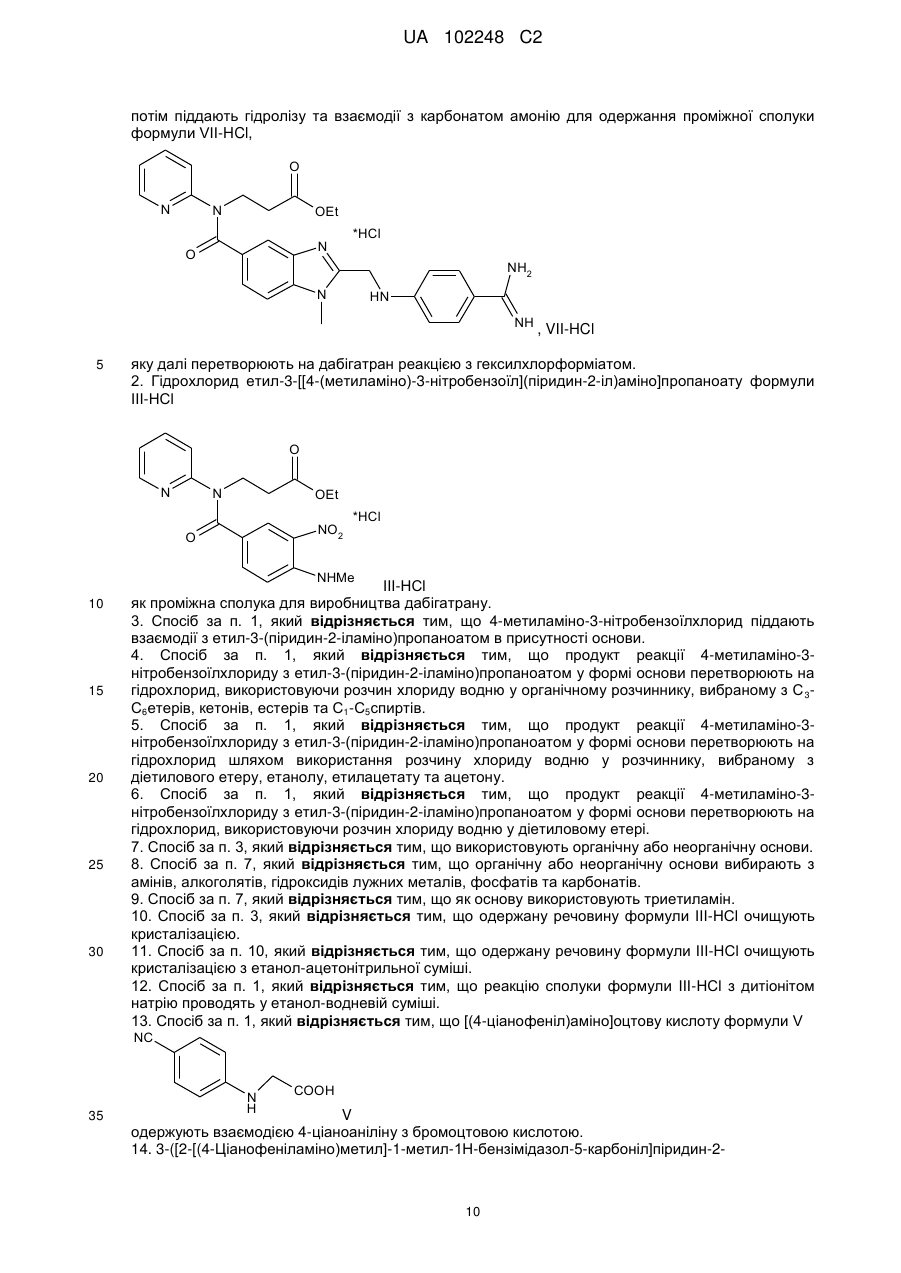
2. Гідрохлорид етил-3-[[4-(метиламіно)-3-нітробензоїл](піридин-2-іл)аміно]пропаноату формули ІІІ-НСl
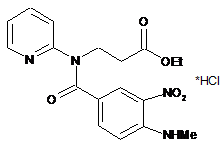 ІІІ-НСl
ІІІ-НСl
як проміжна сполука для виробництва дабігатрану.
3. Спосіб за п. 1, який відрізняється тим, що 4-метиламіно-3-нітробензоїлхлорид піддають взаємодії з етил-3-(піридин-2-іламіно)пропаноатом в присутності основи.
4. Спосіб за п. 1, який відрізняється тим, що продукт реакції 4-метиламіно-3-нітробензоїлхлориду з етил-3-(піридин-2-іламіно)пропаноатом у формі основи перетворюють на гідрохлорид, використовуючи розчин хлориду водню у органічному розчиннику, вибраному з С3-С6етерів, кетонів, естерів та С1-С5спиртів.
5. Спосіб за п. 1, який відрізняється тим, що продукт реакції 4-метиламіно-3-нітробензоїлхлориду з етил-3-(піридин-2-іламіно)пропаноатом у формі основи перетворюють на гідрохлорид шляхом використання розчину хлориду водню у розчиннику, вибраному з діетилового етеру, етанолу, етилацетату та ацетону.
6. Спосіб за п. 1, який відрізняється тим, що продукт реакції 4-метиламіно-3-нітробензоїлхлориду з етил-3-(піридин-2-іламіно)пропаноатом у формі основи перетворюють на гідрохлорид, використовуючи розчин хлориду водню у діетиловому етері.
7. Спосіб за п. 3, який відрізняється тим, що використовують органічну або неорганічну основи.
8. Спосіб за п. 7, який відрізняється тим, що органічну або неорганічну основи вибирають з амінів, алкоголятів, гідроксидів лужних металів, фосфатів та карбонатів.
9. Спосіб за п. 7, який відрізняється тим, що як основу використовують триетиламін.
10. Спосіб за п. 3, який відрізняється тим, що одержану речовину формули III-НСl очищують кристалізацією.
11. Спосіб за п. 10, який відрізняється тим, що одержану речовину формули ІІІ-НСl очищують кристалізацією з етанол-ацетонітрильної суміші.
12. Спосіб за п. 1, який відрізняється тим, що реакцію сполуки формули ІІІ-НСl з дитіонітом натрію проводять у етанол-водневій суміші.
13. Спосіб за п. 1, який відрізняється тим, що [(4-ціанофеніл)аміно]оцтову кислоту формули V
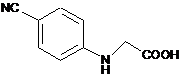 V
V
одержують взаємодією 4-ціаноаніліну з бромоцтовою кислотою.
14. 3-([2-[(4-Ціанофеніламіно)метил]-1-метил-1Н-бензімідазол-5-карбоніл]піридин-2-іламіно)етилпропіонат оксалат формули VI-оксал
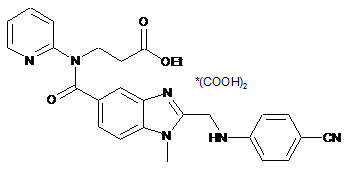 VI-оксал
VI-оксал
як проміжна сполука для одержання дабігатрану.
15. Спосіб за п. 1, який відрізняється тим, що використовують безводну форму або один з гідратів щавлевої кислоти.
16. Спосіб за п. 1, який відрізняється тим, що продукт формули VI-оксал очищують кристалізацією.
17. Спосіб за п. 16, який відрізняється тим, що продукт очищують кристалізацією, використовуючи розчинник, вибраний з етанолу та етилацетату.
18. Спосіб одержання дабігатрану за п. 1, який відрізняється тим, що реакцію 4-метиламіно-3-нітробензоїлхлориду з етил-3-(піридин-2-іламіно)пропаноатом проводять у присутності основи при температурі 40-120 °C, після чого продукт перетворюють на хлорангідрид, використовуючи розчин соляної кислоти для одержання сполуки формули ІІІ-НСl.
19. Спосіб одержання дабігатрану за п. 1, який відрізняється тим, що реакцію сполуки формули ІІІ-НСl з дитіонітом натрію, в процесі якої утворюється сполука формули IV, проводять при температурі 20-100 °C.
20. Спосіб одержання дабігатрану за п. 1, який відрізняється тим, що реакцію сполуки формули IV з 4-ціанофенілгліцином та щавлевою кислотою проводять при температурі 40-120 °C для одержання сполуки формули VI-оксал.
Текст
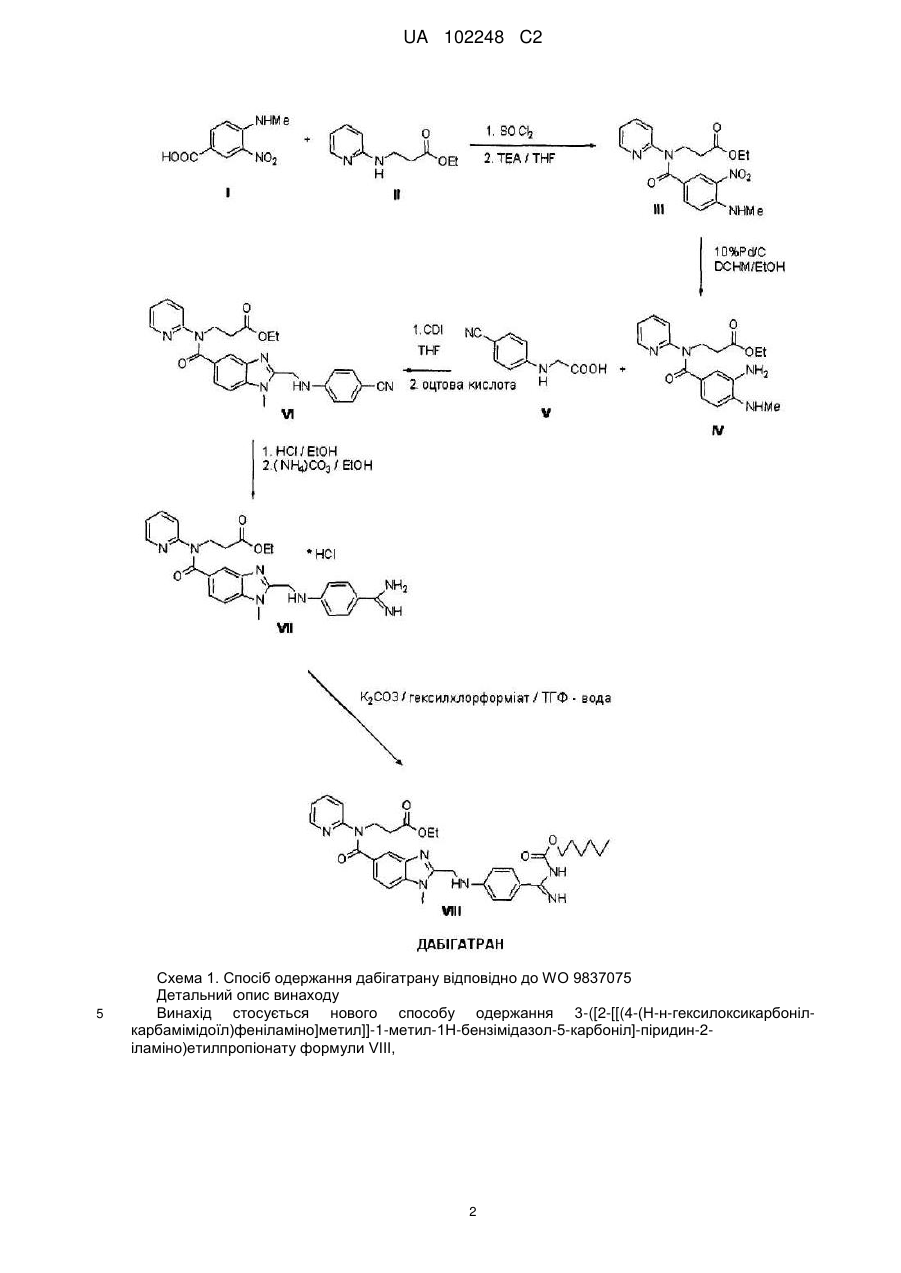
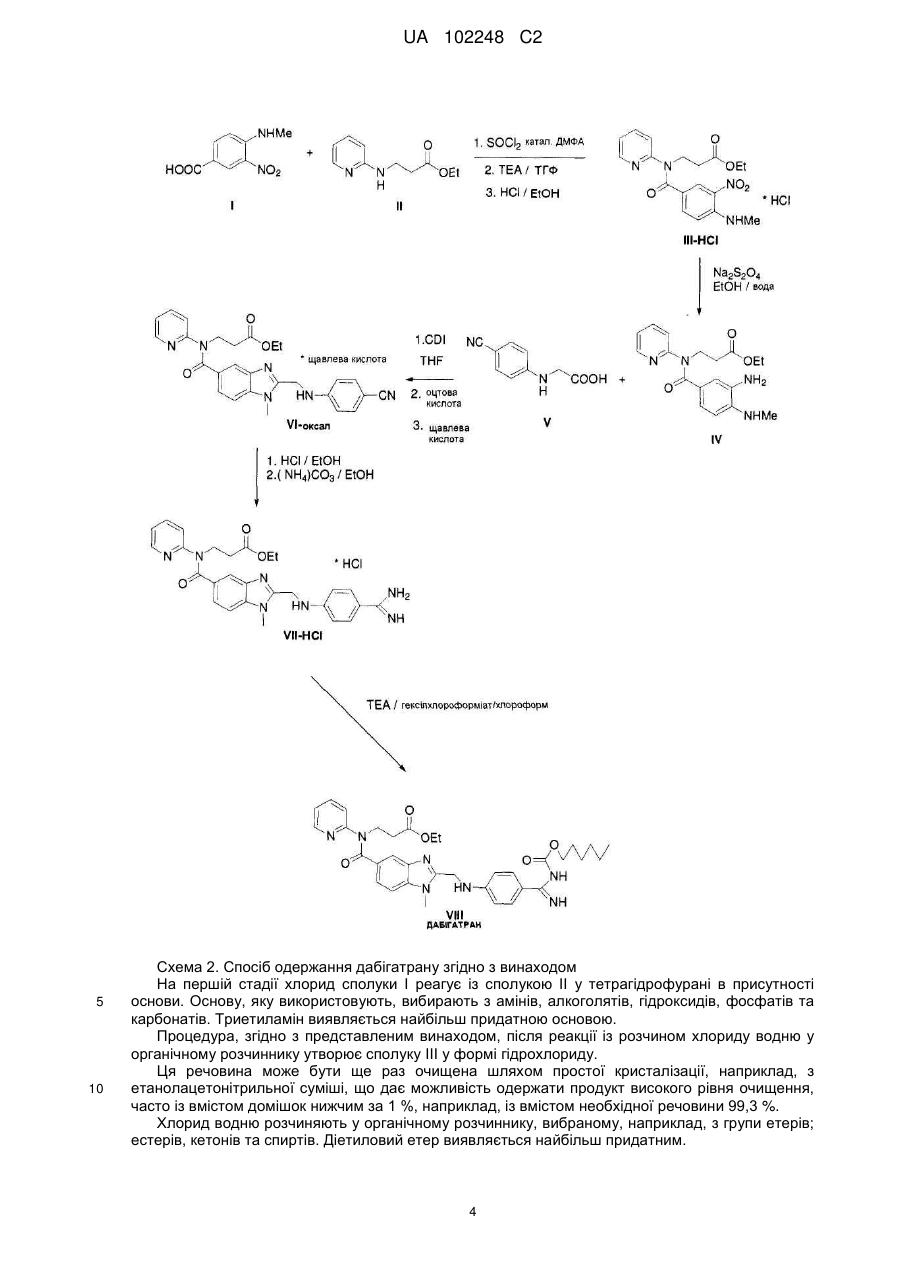
Реферат: Спосіб одержання дабігатрану формули VIII, в якому продукт реакції хлориду 4-етиламіно-3нітробензоїлхлориду з етил-3-(піридин-2-іламіно)пропаноатом перетворюють на гідрохлорид, використовуючи розчин хлориду водню для утворення сполуки формули ІІІ-НСl, в якій нітрогрупу відновлюють шляхом реакції із дитіонітом натрію, і одержану сполуку формули IV піддають взаємодії з [(4-ціанофеніл)аміно]оцтовою кислотою та щавлевою кислотою, продукт цієї взаємодії VІ-оксал піддають гідролізу та взаємодії з карбонатом амонію для утворення проміжної сполуки формули VII-HCl, яку далі перетворюють на дабігатран реакцією з гексилхлорформіатом. UA 102248 C2 (12) UA 102248 C2 UA 102248 C2 5 10 15 20 25 30 35 Галузь винаходу Винахід стосується нового способу виробництва 3-([2-[[(4-(N-н-гексилоксикарбонілкарбамімідоїл)феніламіно]метил]]-1-метил-1Н-бензімідазол-5-карбоніл-піридин-2іламіноетилпропіонату, відомого під міжнародною непатентованою назвою дабігатран. Дабігатран є антикоагулянтною сполукою та використовується для лікування тромбозів, серцево-судинних захворювань та таке інше. Передумови створення винаходу Речовина вперше була описана у документі WO 9837075. Спосіб її одержання згідно вищевказаного документу надано на Схемі 1. На першій стадії виробництва хлорид сполуки І взаємодіє зі сполукою II у тетрагідрофурані, використовуючи триетиламін як основу. Речовина III є продуктом реакції. Під час проведення вищевказаної процедури речовину III одержують у формі основи. Однак, згідно досвіду та у відповідності з WO 9837075, ця речовина потребує складних операцій з очищення, таких, як хроматографія, з метою її використання для виготовлення високоякісних активних фармацевтичних інгредієнтів. Наступна стадія виробництва - це відновлення нітрогрупи до аміногрупи. У документі WO 9837075 цю реакцію здійснюють за допомогою каталітичного гідрування з використанням паладію на активному вугіллі під високим тиском. Однак, цей процес є високотехнологічним та пов'язаний із ризиком для безпеки та навколишнього середовища. Він потребує використання високого тиску, тому необхідне спеціальне обладнання. Каталізатори, які при цьому використовують, є токсичними, ціна на них у поєднанні із ціною на обладнання негативно відбивається на економічній вартості всього процесу. Ще один недолік процесу полягає у низькому рівні якості продукту формули IV, одержаному у такий спосіб. Таким чином, продукт одержують у масляній формі зі вмістом домішок 20-40 %. Продукт IV, якнайменше згідно з відомим процесом, повинен переходити до наступної стадії у забрудненій формі. Винахід альтернативного методу проведення цієї реакції вочевидь робить процес виробництва дабігатрану ефективнішим. Під час третьої стадії речовина IV реагує із речовиною V, утворюючи речовину VI. Згідно з WO 9837075 сполуку V одержують шляхом реакції 4-ціанофеніланіліну із хлороцтовою кислотою в присутності основи. Під час відтворення процедури WO 9837075, ми з'ясували, що реакція дає низький вихід (близько 50 %). Продукт способу згідно WO 9837075 є сполукою VI в основній або ацетатній формі. Обидва ці продукти потребують хроматографічного очищення, що дуже складно здійснити в промисловому масштабі. На наступній стадії здійснюють кислотний гідроліз нітрильної функціональної групи сполуки VI та взаємодію з карбонатом амонію з утворенням сполуки VII. Завершальною стадією є реакція проміжної сполуки VII з гексилхлорформіатом з утворенням дабігатрану. 1 UA 102248 C2 5 Схема 1. Спосіб одержання дабігатрану відповідно до WO 9837075 Детальний опис винаходу Винахід стосується нового способу одержання 3-([2-[[(4-(Н-н-гексилоксикарбонілкарбамімідоїл)феніламіно]метил]]-1-метил-1Н-бензімідазол-5-карбоніл]-піридин-2іламіно)етилпропіонату формули VIII, 2 UA 102248 C2 5 відомого під міжнародною непатентованою назвою дабігатран. Дабігатран є антикоагулянтною сполукою та вживається для лікування тромбозів, серцево-судинних захворювань та таке інше. Спосіб одержання відповідно представленого винаходу поданий на схемі 2. 3 UA 102248 C2 5 10 Схема 2. Спосіб одержання дабігатрану згідно з винаходом На першій стадії хлорид сполуки І реагує із сполукою II у тетрагідрофурані в присутності основи. Основу, яку використовують, вибирають з амінів, алкоголятів, гідроксидів, фосфатів та карбонатів. Триетиламін виявляється найбільш придатною основою. Процедура, згідно з представленим винаходом, після реакції із розчином хлориду водню у органічному розчиннику утворює сполуку III у формі гідрохлориду. Ця речовина може бути ще раз очищена шляхом простої кристалізації, наприклад, з етанолацетонітрильної суміші, що дає можливість одержати продукт високого рівня очищення, часто із вмістом домішок нижчим за 1 %, наприклад, із вмістом необхідної речовини 99,3 %. Хлорид водню розчиняють у органічному розчиннику, вибраному, наприклад, з групи етерів; естерів, кетонів та спиртів. Діетиловий етер виявляється найбільш придатним. 4 UA 102248 C2 5 10 15 20 25 30 Наступна стадія одержання - це відновлення нітрогрупи до аміногрупи. Спосіб одержання, відповідно до представленого винаходу, проводиться у суміші розчинників етанолу та води. Діючою речовиною є дитіоніт натрію. Немає необхідності застосовувати підвищений тиск або дуже високу температуру. Цей спосіб набагато менш витратний та більш вигідний з економічної точки зору. Речовина III реагує у формі гідрохлориду, що позитивно впливає на протікання реакції та чистоту продукту. Використовуючи поєднання зміни якості, складу вихідної речовини та способу відновлення нітрогрупи, можливо одержати продукт IV із мінімальним вмістом домішок, наприклад, менш ніж 5 %, переважно, менш ніж 1 %. На третій стадії речовина IV реагує із речовиною формули V, утворюючи сполуку формули VI. В межах способу, що базується на представленому винаході, сполуку VI одержують у формі солі з щавлевою кислотою. Цю сіль просто вдруге очищують шляхом подальшої кристалізації, що забезпечує покращену проміжну сполуку VI із виходом, прийнятним для промислового виробництва, приблизно 80-90 %. Ця кристалізація може здійснюватися з полярного протонного органічного розчинника, бажано з нижчих С1 - С5 спиртів, наприклад, з етилового спирту. В способі, який представлено у винаході, 4-ціанофеніланілін реагує із бромоцтовою кислотою. Використання цього відмінного реагенту приводить до значно вищого виходу, приблизно 85-90 %, порівнюючи із 49 % у випадку реакції з хлороцтовою кислотою. Це буде, можливо, спричинено більш високою здатністю бромідного іону існувати у нуклеофільних реакціях, порівняно із хлоридним аніоном, що суттєво позначається на виході реакції у випадку реакції 4-хлороцтової або 4-бромоцтової кислоти відповідно, з порівняно слабким нуклеофілом, таким як 4-ціаноанілін. Вся процедура одержання зробить можливим промислове виробництво дабігатрану. Речовина VI, одержана шляхом, вказаним вище, у подальшому може перероблятися способом згідно з WO 9837075. На наступній стадії здійснюють кислотний гідроліз нітрильної функціональної групи сполуки VI та реакцію із карбонатом амонію з утворенням речовини фopмyлиVII. 35 5 UA 102248 C2 5 10 15 Використання вхідної речовини VI у формі оксалату, згідно з представленим винаходом, утворює сполуку VII у формі гідрохлориду. При цьому досягається значний очищувальний ефект із виходом проміжної сполуки VII високого рівня очищення. Останньою стадією є взаємодія проміжної сполуки VII з гексилхлорформіатом, продуктом якої є дабігатран. Спосіб згідно з представленим винаходом, робить можливим одержання високоякісного продукту з низьким вмістом домішок та відносно високим виходом. Одержання проміжних сполук III та VI у формі солей значно спрощує операції з очищення у процесі виробництва дабігатрану; хроматографічне очищення, зазначене у WO 9837075, не може використовуватися у промисловому масштабі. Така процедура значно збільшить вихід процесу. Відновлення нітрогрупи речовини III із дитіонітом замість описаного вище каталітичного гідрування з жорсткими вимогами щодо технології та матеріалів, забезпечує значне спрощення та позитивно відображається на економічній складовій всього процесу. Винахід відображено у поданих нижче прикладах: Приклад 1: Одержання гідрохлориду етил 3-[[4-(метиламіно)-3-нітробензоїл](піридин-2іл)аміно)]пропаноату (III) 20 25 30 35 40 45 Інгредієнти: Проміжна сполука І: 100 г - 0,51 моль Проміжна сполука II: 94 г - 0,48 моль Тіонілхлорид: 850 мл Сухий триетиламін: 99 мл Диметил формамід (сухий): 4 мл Тетрагідрофуран: 650 мл хлороформ: 500 мл 850 мл тіонілхлориду та 4 мл сухого диметилформаміду додавали до 100 г речовини І в нейтральній атмосфері. Суміш доводили до кипіння. У цей час речовина І розчинялась за декілька хвилин та утворювала розчин темно-коричневого кольору. Далі розчин кип'ятили впродовж наступних 40-45 хвилин. Після цього надмірний тіонілхлорид відганяли. До коричневого залишку додавали 200 мл зневодненого толуолу у нейтральній атмосфері та видаляли толуол шляхом дистилювання. Цю процедуру повторювали ще раз. Одержаний коричневий кристалічний залишок розчиняли у сухому тетрагідрофурані (600 мл у нейтральній атмосфері) при підвищеній температурі. Після охолодження розчину хлорангідриду І до температури 40 °C додавали сухий триетиламін. До цього розчину краплями додавали розчин речовини II у сухому тетрагідрофурані (50 мл). Час крапання становив 15 хвилин. Під час додавання речовини II реакційну суміш охолоджували на водяній бані. Це супроводжувалося падінням температури до 30 °С. Далі суміш реакції перемішували, не підігріваючи, протягом 2 годин. Після відгонки гідрохлориду триетіламіну у вакуумі, створюваному водоструминним насосом, тетрагідрофуран випарювали. Залишок випаровування розчиняли у 500 мл хлороформу та струшували з водою. Відокремлений органічний шар струшували із 2 М НСІ, і, після цього - з водою. Органічний шар висушували сульфатом натрію та випарювали хлороформ. Вихід: Сирий продукт: 200г (96 %); ВЕРХ: 88,2 %; коричневу медоподібну речовину Із цієї речовини одержували гідрохлорид. 6 UA 102248 C2 5 10 15 Одержання гідрохлориду: EtAc: 850 мл Соляна кислота/Et2O Первинний продукт розчинили у 850 мл EtAc при кімнатній температурі. До розчину при інтенсивному перемішуванні повільно додали краплями 95 мл розчину соляної кислоти в Et2O. Під час перемішування відокремився жовтий осад. Після охолодження у холодильнику його відфільтрували та висушили (протягом ночі при кімнатній температурі, після чого у вакуумі при температурі 55-75 °C, далі у вакуумі без нагрівання, впродовж вихідних (без зміни маси)). Вихід: Сирий гідрохлорид: 161,3 г (77,4 %); ВЕРХ: 93,3 % Кристалізація гідрохлориду: Сирий гідрохлорид кристалізували із суміші денатурованого незневодженого етанолу з ацетонітрилом у пропорції 9:1. Вихід: Кристалізований гідрохлорид: 120,2 г (61,6 %); ВЕРХ: 99,3 % Приклад 2: Одержання етил 3-[[3-аміно-4-(метиламіно)бензоїл](піридин-2-іл)аміно]-пропіонату (IV) 20 25 30 Інгредієнти Проміжна сполука III:120 г - 0,29 моль Дитіоніт натрію: 246 г - 1,2 моль Етанол:750 мл Етилацетат: 600 мл Речовину III поміщали у 1500 мл суміші етанолу з водою у пропорції 1:1 та підігрівали до температури 50 °C. Таким чином одержували розчин, до якого швидко та при інтенсивному перемішуванні додавали твердий дитіоніт натрію. Після реакції вихідної речовини, реакційну суміш концентрували у вакуумному випарнику. Після відокремлення масла концентрування завершували і продукт екстрагували етилацетатом. Після його висушування сульфатом натрію, розчинник випарювали. Одержаний продукт був коричневою, дуже в'язкою рідиною. Вихід: 82 г (81,3 %); ВЕРХ: 95 % Приклад 3: Одержання [(4-ціанофеніл)аміно]оцтової кислоти (V) 35 7 UA 102248 C2 5 10 15 Інгредієнти: F: 90 г - 0,75 моль G: 211,7 г - 1,5 моль Бікарбонат натрію: 35 г, 0,42 моль Вихідні речовини F та G змішували у 1250 мл води, і цю суспензію поміщали у баню, підігріту до температури 100-110 °C. Після трьох годин нагрівання лабораторний стакан вилучали із бані, охолоджували у холодильнику, і відокремлену речовину відсмоктували. Речовину висушували у вакуумній сушарці при температурі 100 °C. Вихід: Сирий продукт: 122 г (92,8 %), ВЕРХ: 97 % Сирий продукт очищали перетворенням у сіль натрію та повторним підкисленням з використанням водного розчину бікарбонату натрію. Кислоту вивільняли за допомогою розведеної соляної кислоти (1:1). Після відсмоктування, промивання водою, продукт висушували у вакуумній сушарці (105 °C). Вихід: Очищений продукт: 115 г (88 %), ВЕРХ: 99,1 %, вміст води: 0,13 %; сульфат (зола): 1,8 % Приклад 4: Одержання 3-([2-[(4-ціанофеніламіно)метил]-1-метил-1Н-бензімідазол-5-карбоніл]піридин-2іламіно)етилпропіонату оксалату (VI) 20 25 30 35 40 45 50 Інгредієнти: Проміжна сполука IV: 82,5 г - 0,24 моль Проміжна сполука V: 46,4 г - 0,26 моль 1,1'-карбонілдіімідазол КДІ: 42,2 г - 0,26 моль Методика Речовину V та 1,1'-карбонілдіімідазол поміщають у колбу з інертним газом. Додають 2000 мл сухого тетрагідрофурану і суміш кип'ятять у колбі зі зворотним холодильником з хлоркальцієвою трубкою протягом 40 хвилин. Через 40 хвилин до суміші додають розчин речовини IV у 330 мл сухого тетрагідрофурану і суміш кип'ятять протягом наступних 5 годин. Після цього реакцію суміші трохи охолоджують і тетрагідрофуран видаляють шляхом дистиляції у вакуумі. До медоподібного коричневого залишку додають 1000 мл льодяної оцтової кислоти, і одержаний розчин кип'ятять впродовж години. Кислоту відганяють, і утворений осад розчиняють у дихлорметані (850 мл) та струшують з водою. Органічний шар, що відокремлюється, висушують сульфатом натрію і випаровують розчинник. Отримують темно-коричневий медоподібний залишок. Вихід: 119 г (92 % у вигляді ацетату); ВЕРХ: 82 % Одержання оксалату речовини VI Сиру виділену речовину (119 г) розчиняють у 650 мл EtAc при кипінні. Розчин охолоджують до кімнатної температури, і розчин дигідрату щавлевої кислоти (27,1 г) додають краплями до 400 мл EtAc, інтенсивно перемішуючи. Швидко відокремлюється речовина світло-бежевого кольору. Кристали відсмоктують, промивають етанолом та висушують. Вихід сирого оксалату: 102 г (70 %); ВЕРХ: 90,2 % Кристалізація сирого оксалату речовини VI: 101 г сирого продукту розчиняють у 4100 мл етанолу при кипінні. Розчин охолоджують у водяній бані, перемішуючи. Суспензію кристалів охолоджують у холодильнику і відсмоктують кристали. Вихід кристалізованого оксалату: 82,5 г (57,2 %); ВЕРХ: 97,8 %. 8 UA 102248 C2 ФОРМУЛА ВИНАХОДУ 1. Спосіб одержання дабігатрану формули VIII O N N OEt O O N O NH N HN NH 5 10 VIII шляхом реакції 4-метиламіно-3-нітробензоїлхлориду з етил-3-(піридин-2-іламіно)пропаноатом, відновлення нітрогрупи, наступної реакції з [(4-ціанофеніл)аміно]оцтовою кислотою, гідролізу та реакції з карбонатом амонію та перетворення у дабігатран взаємодією з гексилхлороформіатом, який відрізняється тим, що продукт реакції 4-метиламіно-3-нітробензоїлхлориду з етил-3(піридин-2-іламіно)пропаноатом перетворюють на гідрохлорид з використанням розчину хлориду водню для утворення сполуки формули ІІІ-НСl, O N N OEt NO2 O *HCl NHMe , III-HCl 15 нітрогрупу якої відновлюють реакцією з дитіонітом натрію, і отриману сполуку формули IV O N N OEt NH2 O NHMe IV піддають взаємодії з [(4-ціанофеніл)аміно]оцтовою кислотою та щавлевою кислотою, продукт цієї реакції формули VІ-оксал O N N O OEt N N *(COOH)2 HN CN VI-оксал 20 9 UA 102248 C2 потім піддають гідролізу та взаємодії з карбонатом амонію для одержання проміжної сполуки формули VII-HCl, O N N OEt N O *HCl NH2 N HN NH , VII-HCl 5 яку далі перетворюють на дабігатран реакцією з гексилхлорформіатом. 2. Гідрохлорид етил-3-[[4-(метиламіно)-3-нітробензоїл](піридин-2-іл)аміно]пропаноату формули ІІІ-НСl O N N OEt NO2 O *HCl NHMe 10 15 20 25 30 ІІІ-НСl як проміжна сполука для виробництва дабігатрану. 3. Спосіб за п. 1, який відрізняється тим, що 4-метиламіно-3-нітробензоїлхлорид піддають взаємодії з етил-3-(піридин-2-іламіно)пропаноатом в присутності основи. 4. Спосіб за п. 1, який відрізняється тим, що продукт реакції 4-метиламіно-3нітробензоїлхлориду з етил-3-(піридин-2-іламіно)пропаноатом у формі основи перетворюють на гідрохлорид, використовуючи розчин хлориду водню у органічному розчиннику, вибраному з С 3С6етерів, кетонів, естерів та С1-С5спиртів. 5. Спосіб за п. 1, який відрізняється тим, що продукт реакції 4-метиламіно-3нітробензоїлхлориду з етил-3-(піридин-2-іламіно)пропаноатом у формі основи перетворюють на гідрохлорид шляхом використання розчину хлориду водню у розчиннику, вибраному з діетилового етеру, етанолу, етилацетату та ацетону. 6. Спосіб за п. 1, який відрізняється тим, що продукт реакції 4-метиламіно-3нітробензоїлхлориду з етил-3-(піридин-2-іламіно)пропаноатом у формі основи перетворюють на гідрохлорид, використовуючи розчин хлориду водню у діетиловому етері. 7. Спосіб за п. 3, який відрізняється тим, що використовують органічну або неорганічну основи. 8. Спосіб за п. 7, який відрізняється тим, що органічну або неорганічну основи вибирають з амінів, алкоголятів, гідроксидів лужних металів, фосфатів та карбонатів. 9. Спосіб за п. 7, який відрізняється тим, що як основу використовують триетиламін. 10. Спосіб за п. 3, який відрізняється тим, що одержану речовину формули III-НСl очищують кристалізацією. 11. Спосіб за п. 10, який відрізняється тим, що одержану речовину формули ІІІ-НСl очищують кристалізацією з етанол-ацетонітрильної суміші. 12. Спосіб за п. 1, який відрізняється тим, що реакцію сполуки формули ІІІ-НСl з дитіонітом натрію проводять у етанол-водневій суміші. 13. Спосіб за п. 1, який відрізняється тим, що [(4-ціанофеніл)аміно]оцтову кислоту формули V NC 35 N H COOH V одержують взаємодією 4-ціаноаніліну з бромоцтовою кислотою. 14. 3-([2-[(4-Ціанофеніламіно)метил]-1-метил-1Н-бензімідазол-5-карбоніл]піридин-2 10 UA 102248 C2 іламіно)етилпропіонат оксалат формули VI-оксал O N N O OEt N N 5 10 15 *(COOH)2 HN CN VI-оксал як проміжна сполука для одержання дабігатрану. 15. Спосіб за п. 1, який відрізняється тим, що використовують безводну форму або один з гідратів щавлевої кислоти. 16. Спосіб за п. 1, який відрізняється тим, що продукт формули VI-оксал очищують кристалізацією. 17. Спосіб за п. 16, який відрізняється тим, що продукт очищують кристалізацією, використовуючи розчинник, вибраний з етанолу та етилацетату. 18. Спосіб одержання дабігатрану за п. 1, який відрізняється тим, що реакцію 4-метиламіно-3нітробензоїлхлориду з етил-3-(піридин-2-іламіно)пропаноатом проводять у присутності основи при температурі 40-120 °C, після чого продукт перетворюють на хлорангідрид, використовуючи розчин соляної кислоти для одержання сполуки формули ІІІ-НСl. 19. Спосіб одержання дабігатрану за п. 1, який відрізняється тим, що реакцію сполуки формули ІІІ-НСl з дитіонітом натрію, в процесі якої утворюється сполука формули IV, проводять при температурі 20-100 °C. 20. Спосіб одержання дабігатрану за п. 1, який відрізняється тим, що реакцію сполуки формули IV з 4-ціанофенілгліцином та щавлевою кислотою проводять при температурі 40120 °C для одержання сполуки формули VI-оксал. 20 Комп’ютерна верстка Л. Литвиненко Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 11
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for the manufacture of dabigatran
Автори англійськоюJirman, Josef, Richter, Jindrich, Lustig, Petr
Назва патенту російськоюСпособ получения дабигатрана
Автори російськоюИрман Йосеф, Рихтер Индржих, Лустиг Петр
МПК / Мітки
МПК: C07D 401/12, C07D 213/74
Мітки: спосіб, одержання, дабігатрану
Код посилання
<a href="https://ua.patents.su/13-102248-sposib-oderzhannya-dabigatranu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання дабігатрану</a>
Похідні індолу, фармацевтична композиція та спосіб одержання сполук (варіанти)

Номер патенту: 68388
Опубліковано: 16.08.2004
Автори: Паллук Райнер, Грауерт Маттіас, Пшорн Уве, Хьонке Хрістоф, Вайзер Томас, Картер Адріан, Бєхтєль Вольф-Дітріх
МПК: A61P 25/28, C07D 209/04, C07D 401/04, A61P 9/10, A61P 23/02, C07D 209/60, A61K 31/404, A61P 9/06
Мітки: спосіб, похідні, композиція, індолу, одержання, сполук, фармацевтична, варіанти
Формула / Реферат:
1. Похідні індолу загальної формули 1, 1деХ означає простий зв'язок, -О-, С1-С4алкіл, С1-С3алкоксигрупу, -О-СН2-СН2-О- чи -O-CH2-CH2-NH-,R1 означає водень, метил, етил чи феніл, R2 означає водень або метил,R3 означає водень, F, Сl, Br, гідрокси- чи метоксигрупу, R4 означає водень, метил чи етил, R5 означає водень, метил чи етил, R6 означає водень, метил чи етил, R7 означає...
Спосіб одержання сполук, які посилюють секрецію гормону росту
Номер патенту: 70306
Опубліковано: 15.10.2004
Автори: Мелц Кліффорд Натаніель, Буш Френк Роберт, Чіу Чарльз Квок-Фунг, Пост Рональд Джеймс, Роуз Пітер Роберт
МПК: C07K 5/02, C07K 5/06, A61P 5/00, A61K 31/437, C07D 471/04, A61P 19/10, A61K 38/00, C07B 57/00
Мітки: сполук, спосіб, гормону, росту, секрецію, одержання, посилюють
Формула / Реферат:
1. Спосіб одержання сполуки формули II , (ІІ) в якійR1 є -(С1-С10)алкілом, необов'язково, заміщеним до трьох атомів фтору,R2 є фенілметилом або 2-піридилметилом,R3 є -(С1-С5)алкіл-О-(С0-С5)алкілфенілом, в якому фенільний замісник необов'язково заміщений до трьох атомів фтору, таPrt - амінозахисна...
Спосіб одержання похідної сполуки хіноліну та проміжна сполука

Номер патенту: 65627
Опубліковано: 15.04.2004
Автори: Такада Ясутака, Охара Йошіо, Янагава Йошінобу, Сузукі Мікіо
МПК: C07D 215/12, C07D 215/14
Мітки: сполуки, похідної, проміжна, хіноліну, одержання, сполука, спосіб
Формула / Реферат:
1. Проміжна сполука нітрилу формули (1). (1)2. Спосіб одержання похідного хіноліну (3), що проводять за допомогою сполуки нітрилу, одержуваної в результаті реакції сполуки альдегіду, представленої формулою (2), з діетилціанометилфосфонатом (2) (1) (3).
Тієнілазолілалкоксіетанаміни, спосіб їх одержання, проміжні продукти для їх одержання та фармацевтична композиція

Номер патенту: 58589
Опубліковано: 15.08.2003
Автори: МЕРСЕ-ВІДАЛЬ Рамон, АНДАЛУЗ-МАТАРО Блас, ФРІГОЛА КОНСТАНСА Хорді
МПК: A61K 31/381, A61K 31/4155
Мітки: фармацевтична, продукти, проміжні, спосіб, тієнілазолілалкоксіетанаміни, композиція, одержання
Формула / Реферат:
1. Похідна тієнілазолілалкоксіетанаміну загальної формули (І):, (І)деR1 - атом водню або атом галогену, або алкільний радикал з 1-4 атомами вуглецю;R2, R3 та R4, незалежно, - атом водню або алкільний радикал з 1-4 атомами вуглецю; таAz являє собою азотвмісне гетероциклічне ароматичне п'ятичленне N-метил-заміщене кільце, до складу якого входять від одного до трьох атомів азоту, загальної формули...
Спосіб одержання циталопраму (варіанти), s-циталопраму, проміжні кетони та спосіб одержання рацемічних сполук

Номер патенту: 72238
Опубліковано: 15.02.2005
Автори: Еллегор Петер, Петерсен Ханс, Рок Майкл Харольд
МПК: C07C 253/30, C07D 307/87, C07C 255/56
Мітки: одержання, варіанти, циталопраму, спосіб, s-циталопраму, проміжні, кетони, сполук, рацемічних
Формула / Реферат:
1. Спосіб одержання циталопраму, згідно з яким здійснюють реакцію сполуки формули IV, IVде R являє собою ацил, з 3-(N,N-диметиламіно)пропілмагнійгалогенідом, переважно з 3-(N,N-диметиламіно)пропілмагнійхлоридом, з одержанням циталопраму формули I, Iякий виділяють у вигляді основи або її фармацевтично прийнятної солі.2. Спосіб за п. 1, який відрізняється тим, що проміжну сполуку формули IV одержують...
Попередній патент: Імунологічні аналізи активності ботулінічного токсину серотипу а
Наступний патент: Спосіб виділення оцтової кислоти
Випадковий патент: Спосіб одержання спученого графіту