Спосіб одержання похідних сартану і проміжних сполук, придатних для цього способу
Номер патенту: 94591
Опубліковано: 25.05.2011
Автори: Жупанчіч Сілво, Путала Мартін, Веверка Мірослав, Брат Хейнріч






Формула / Реферат
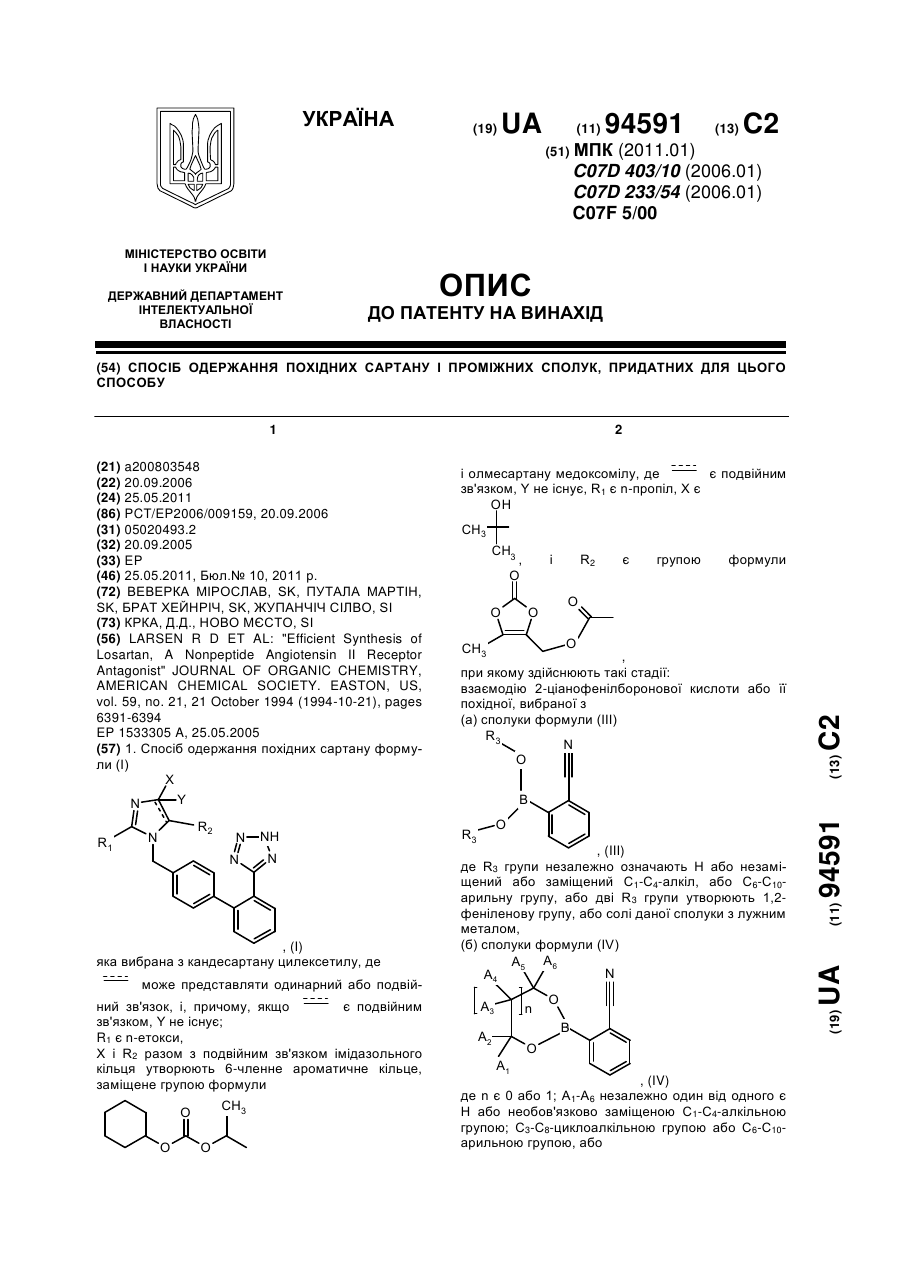
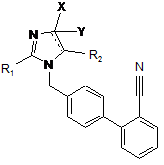
1. Спосіб одержання похідних сартану формули (І)
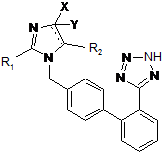 , (I)
, (I)
яка вибрана з кандесартану цилексетилу, де
![]() може представляти одинарний або подвійний зв'язок, і, причому, якщо
може представляти одинарний або подвійний зв'язок, і, причому, якщо ![]() є подвійним зв'язком, Y не існує;
є подвійним зв'язком, Y не існує;
R1 є n-етокси,
X і R2 разом з подвійним зв'язком імідазольного кільця утворюють 6-членне ароматичне кільце, заміщене групою формули

і олмесартану медоксомілу, де ![]() є подвійним зв'язком, Y не існує, R1 є n-пропіл, X є
є подвійним зв'язком, Y не існує, R1 є n-пропіл, X є
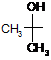 , і R2 є групою формули
, і R2 є групою формули  ,
,
при якому здійснюють такі стадії:
взаємодію 2-ціанофенілборонової кислоти або її похідної, вибраної з
(а) сполуки формули (III)
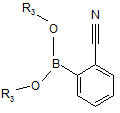 , (III)
, (III)
де R3 групи незалежно означають Н або незаміщений або заміщений С1-С4-алкіл, або С6-С10-арильну групу, або дві R3 групи утворюють 1,2-феніленову групу, або солі даної сполуки з лужним металом,
(б) сполуки формули (IV)
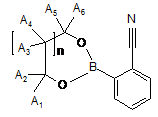 , (IV)
, (IV)
де n є 0 або 1; А1-А6 незалежно один від одного є Н або необов'язково заміщеною С1-С4-алкільною групою; С3-С8-циклоалкільною групою або С6-С10-арильною групою, або
(в) сполуки формули (V)
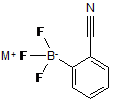 , (V)
, (V)
де М є лужним металом або групою NR4R5R6R7, де R4-R7 незалежно один від одного є Н або незаміщеною або заміщеною С1-С18-алкільною групою,
у реакції перехресного поєднання з похідним п-галогенобензил-1Н-імідазолу формули (VI)
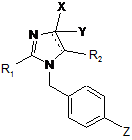 , (VI)
, (VI)
де![]() , X, Y, R1 і R2 визначені вище, Z є І, Вr або Сl;
, X, Y, R1 і R2 визначені вище, Z є І, Вr або Сl;
у присутності каталізатора на основі перехідного металу і органічної основи з утворенням заміщеної біфенілімідазольної сполуки формули (II)
 , (II)
, (II)
де ![]() , X, Y, R1 і R2 визначені вище;
, X, Y, R1 і R2 визначені вище;
перетворення сполуки формули (II) у відповідну 2-тетразол похідну з отриманням похідної сартану формули (І).
2. Спосіб одержання сполуки формули (II)
, (II)
де ![]() , R1, R2, X і Y визначені в будь-якій з попередніх формул, або її фармацевтично прийнятної солі, при якому здійснюють такі стадії взаємодії 2-ціанофенілборонової кислоти або її похідної, де вказану 2-ціанофенілборонову кислоту або її похідну вибирають із сполуки формули (III)
, R1, R2, X і Y визначені в будь-якій з попередніх формул, або її фармацевтично прийнятної солі, при якому здійснюють такі стадії взаємодії 2-ціанофенілборонової кислоти або її похідної, де вказану 2-ціанофенілборонову кислоту або її похідну вибирають із сполуки формули (III)
, (III)
де R3 визначено в п. 1, або її солі лужного металу,
сполуки формули (IV)
, (IV)
де n і А1-А6 визначені в п. 1,
або сполуки формули (V)
, (V)
де М визначено в п. 1;
в реакції перехресного поєднання з похідним п-галогенобензил-1Н-імідазолу формули (VI)
, (VI)
де ![]() , R1, R2, X, Y і Z визначені в будь-якій з попередніх формул;
, R1, R2, X, Y і Z визначені в будь-якій з попередніх формул;
у присутності каталізатора на основі перехідного металу і органічної основи з утворенням заміщеної біфенілімідазольної сполуки формули (II).
3. Спосіб за будь-яким із пп. 1, 2, який відрізняється тим, що основу вибирають з триетиламіну або діізопропілетиламіну.
4. Спосіб за будь-яким із пп. 1-3, який відрізняється тим, що розчинник вибирають з диметилформаміду, тетрагідрофурану, толуолу, толуолметанолу, метанолу або етанолу, або їх сумішій з водою.
5. Спосіб за будь-яким із пп. 1-4, який відрізняється тим, що каталізатор на основі металу є комплексом паладію, вибраним з тетракис(трифенілфосфін)паладію (0), тетракис(три-о-толілфосфін)паладію (0), 6ic[1,1'-біс(дифенілфосфіно)фероцен]паладію (0) або фосфінових комплексів паладію (II).
6. Спосіб за будь-яким із пп. 1-5, при якому здійснюють стадії:
а) приготування суміші із сполуки формули (III), (IV) або (V), сполуки формули (VI), додавання в суміш основи і попередників для синтезу каталізатора,
б) додавання розчинника і змішування,
в) нагрівання суміші і кип'ятіння протягом 3-12 годин з отриманням сполуки формули (II),
г) очищення реакційної суміші, що містить сполуку формули (II), і
д) необов'язкового перетворення сполуки формули (II) в сполуку формули (І).
7. Спосіб за будь-яким із пп. 1-6, при якому додатково здійснюють стадії:
взаємодії з літієм або магнієм 2-галогенобензонітрилу, де галоген означає І, Вr, Сl, і
взаємодії одержаного продукту з ефіром бору формули B(OR)3, де R є С1-С4-алкілом, і
взаємодії одержаної сполуки з неорганічним або органічним воденьдифторидом або комбінацією неорганічної або органічної основи з фторводневою кислотою, або з воденьдифторидом калію, далі необов'язково заміни катіона калію іншим катіоном шляхом реакції з органічною або неорганічною основою з отриманням сполуки формули (V)
, (V)
де М визначено вище.
8. Спосіб за будь-яким із пп. 1-6, при якому додатково здійснюють стадії:
взаємодії з перехідним металом 2-галогенобензонітрилу, причому галоген означає І, Вr, Сl, з сіллю цинку або міді,
взаємодії одержаного продукту з тетрафторборатом або BF3 і неорганічною або органічною основою з одержанням сполуки формули (V)
, (V)
де М визначено вище.
9. Спосіб за будь-яким із пп. 1-6, при якому додатково здійснюють стадію взаємодії 2-ціанофенілборонової кислоти з неорганічним або органічним воденьдифторидом або сумішшю органічної або неорганічної основи з фторводневою кислотою або з воденьдифторидом калію з отриманням сполуки формули (V).
10. Спосіб за п. 8 або 9, який додатково містить стадії заміни катіона калію іншим неорганічним або органічним катіоном шляхом реакції з органічною або неорганічною основою.
Текст
1. Спосіб одержання похідних сартану формули (І) X 2 3 94591 (в) сполуки формули (V) N вказану 2-ціанофенілборонову кислоту або її похідну вибирають із сполуки формули (III) R3 N O F M+ F 4 B B F , (V) де М є лужним металом або групою NR4R5R6R7, де R4-R7 незалежно один від одного є Н або незаміщеною або заміщеною С1-С18-алкільною групою, у реакції перехресного поєднання з похідним пгалогенобензил-1Н-імідазолу формули (VI) X A3 R2 N R1 , (III) де R3 визначено в п. 1, або її солі лужного металу, сполуки формули (IV) A5 A6 N A4 Y N O R3 n O B A2 O A1 Z , (VI) де , X, Y, R1 і R2 визначені вище, Z є І, Вr або Сl; у присутності каталізатора на основі перехідного металу і органічної основи з утворенням заміщеної біфенілімідазольної сполуки формули (II) X Y N R2 N R1 , (IV) де n і А1-А6 визначені в п. 1, або сполуки формули (V) N N F M+ F BF , (V) де М визначено в п. 1; в реакції перехресного поєднання з похідним пгалогенобензил-1Н-імідазолу формули (VI) X Y N R1 N R2 , (II) де , X, Y, R1 і R2 визначені вище; перетворення сполуки формули (II) у відповідну 2тетразол похідну з отриманням похідної сартану формули (І). 2. Спосіб одержання сполуки формули (II) X Y N R1 N R2 N , (II) де , R1, R2, X і Y визначені в будь-якій з попередніх формул, або її фармацевтично прийнятної солі, при якому здійснюють такі стадії взаємодії 2-ціанофенілборонової кислоти або її похідної, де Z , (VI) де , R1, R2, X, Y і Z визначені в будь-якій з попередніх формул; у присутності каталізатора на основі перехідного металу і органічної основи з утворенням заміщеної біфенілімідазольної сполуки формули (II). 3. Спосіб за будь-яким із пп. 1, 2, який відрізняється тим, що основу вибирають з триетиламіну або діізопропілетиламіну. 4. Спосіб за будь-яким із пп. 1-3, який відрізняється тим, що розчинник вибирають з диметилформаміду, тетрагідрофурану, толуолу, толуолметанолу, метанолу або етанолу, або їх сумішій з водою. 5. Спосіб за будь-яким із пп. 1-4, який відрізняється тим, що каталізатор на основі металу є комплексом паладію, вибраним з тетракис(трифенілфосфін)паладію (0), тетракис(три-отолілфосфін)паладію (0), 6ic[1,1'біс(дифенілфосфіно)фероцен]паладію (0) або фосфінових комплексів паладію (II). 5 6. Спосіб за будь-яким із пп. 1-5, при якому здійснюють стадії: а) приготування суміші із сполуки формули (III), (IV) або (V), сполуки формули (VI), додавання в суміш основи і попередників для синтезу каталізатора, б) додавання розчинника і змішування, в) нагрівання суміші і кип'ятіння протягом 3-12 годин з отриманням сполуки формули (II), г) очищення реакційної суміші, що містить сполуку формули (II), і д) необов'язкового перетворення сполуки формули (II) в сполуку формули (І). 7. Спосіб за будь-яким із пп. 1-6, при якому додатково здійснюють стадії: взаємодії з літієм або магнієм 2галогенобензонітрилу, де галоген означає І, Вr, Сl, і взаємодії одержаного продукту з ефіром бору формули B(OR)3, де R є С1-С4-алкілом, і взаємодії одержаної сполуки з неорганічним або органічним воденьдифторидом або комбінацією неорганічної або органічної основи з фторводневою кислотою, або з воденьдифторидом калію, далі необов'язково заміни катіона калію іншим катіоном шляхом реакції з органічною або неорганічною основою з отриманням сполуки формули (V) N 94591 6 де М визначено вище. 8. Спосіб за будь-яким із пп. 1-6, при якому додатково здійснюють стадії: взаємодії з перехідним металом 2галогенобензонітрилу, причому галоген означає І, Вr, Сl, з сіллю цинку або міді, взаємодії одержаного продукту з тетрафторборатом або BF3 і неорганічною або органічною основою з одержанням сполуки формули (V) N F M+ F BF , (V) де М визначено вище. 9. Спосіб за будь-яким із пп. 1-6, при якому додатково здійснюють стадію взаємодії 2ціанофенілборонової кислоти з неорганічним або органічним воденьдифторидом або сумішшю органічної або неорганічної основи з фторводневою кислотою або з воденьдифторидом калію з отриманням сполуки формули (V). 10. Спосіб за п. 8 або 9, який додатково містить стадії заміни катіона калію іншим неорганічним або органічним катіоном шляхом реакції з органічною або неорганічною основою. F M+ F BF , (V) Даний винахід відноситься до способу одержання заміщених сполук біфенілімідазолу в якості придатних проміжних сполук при синтезі певних похідних сартану, що зв'язуються з ангіотензином, зокрема лосартану, олмесартану медоксомілу, канденсартану цилексетилу або ірбесартану. Речовини, що зв'язуються з ангіотензином II, такі як лосартан, олмесартан медоксоміл (ЕР 0 503 785; ЕР 0 545 912), кандесартан цилексетил (ЕР 0 459 136; ЕР 0 720 982), та ірбесартан (ЕР 0 454 511) є ефективними інгібіторами ангіотензинперетворюючого ферменту і використовуються для лікування гіпертензії, ниркової недостатності та глаукоми. Повідомлялося про деякі непептидні аналоги, що володіють властивостями рецепторів ангіотензину II (U.S. 4 355 040, Wong P.C., J. Pharm. Exp. Then, 1990, 255 (2), 584). Більшість рецепторів ангіотензину II володіють загальною структурною особливістю біфенільного компоненту, з гетероциклом в 4 позиції. Наприклад, лосартан має наступну формулу: У контексті даного винаходу, термін «лосартан» також включає фармацевтично прийнятні гідрати і сольвати речовин формули (І). То ж відноситься і до інших похідних сартану, згаданих в данній заявці. Відомі різні підходи, які описують синтез заміщених сполук біфенілімідазолу, що є придатними для синтезу 1,2,4,5-заміщених імідазолів, що представляють цінні проміжні речовини при синтезі лосартану. Калій лосартан є першою сполукою нового класу ліків, отриманих в ключовій стадії, за допомогою гетерогенного перехресного скріплення 7 Сузукі між тритилтетразолфенілборонової кислоти і 1-(4-бромобензил)-2-н-бутил-4-хлор-1 Н-імідазол5-іл похідного (Larsen, D.R., et.al. J. Org. Chem. 1994, 59, 6391, US 5 130 439, US 5 310 928). Ларсен Д.Ρ, J. Org. Chem. 1994, 59, 6391 розкриває спосіб прямого N-алкілування похідних 1Німідазолу з отриманням бензилованого імідазолу. Дана сполука реагує з тритил-захищеною фенілтетразолбороновою кислотою за допомогою перехресного зв'язування Сузукі з отриманням лосартану. US 5 310 928 розкриває нові тетразолфенілборонові кислоти та їхні похідні, способи їхнього отримання, а також їхнє застосування в способах отримання антагоністів рецепторів ангіотензину II за допомогою перехресного скріплення Сузукі. Як у способі Ларсона, так і в US 5 310 928 потрібний захист атома азоту в положенні 2 тетразольної частини, оскільки незахищений тетразол забруднює каталізатор [Smith, G.B.; Dezeny, G: С; Hughes, D.L.; King, A.O.; Verhoeven, T.R. J. Org. Chem. 1994, 59, 8151.] Захист тетразольної частини виконується за допомогою тритильної групи [a) Larsen, R.D.; King, О.; Chen, С; Υ.; Corley, E.G.; Foster, В. S.; Roberts, F.E.; Yang, C; Lieberman, D.R.; Remwr, R.A.; Tschaen, D.M.; Verhoven, T.R.; Reider, P.J. J.Org.Chem. 1994, 59, 6391. b) PCT Int. Appl., 9310106, 1993. з) Ger. Often., 4313747, 1994.]. Проте, дані способи не є дуже ефективними, оскільки тритильна група є досить лабільною, і навіть кількість слідів дезтритил тетразолборонової кислоти істотно знижують вихід продукту перехресного скріплення [Smith, G.B.; Dezeny, G: С; Hughes, D.L.; King, A.O.; Verhoeven, T.R. J. Org. Chem. 1994, 59, 8151] Найбільш поширеним способом одержання тетразольної частини є перегрупування ціаногрупи. В Hird, Μ., J.C.S. Perkin. Trans. I. 1998, 20 3479, Norman H.M., J. Med. Chem. 1995, 38, описана реакція Сузукі-Міяура, де 2-бромбензонітрил застосовується в якості електрофілу для одержання 2ціанобіфенілу. Проте, проблема застосування 2ціанофенілборонової кислоти полягає в її низькій стабільності, оскільки зазнає екзотермічного розкладання, особливо при температурах вище 90°С [Urawa, Y.; Naka, H.; Miyazawa, M.; Souda, S.; Ogura, К. J. Organomet. Chem. 2002, 653, 269.] Отже, існує дуже невелика кількість повідомлень, що описують використання 2-ціанофенілборонової кислоти в реакціях перехресного зшивання, з одержанням сполук 2-ціанобіфенілу з середніми виходами (45-67%) [a) Thomas, А.Р. et. al., Bioorg. Med. Chem. Lett. 1994, 4, 2615; b) Yang, G.X. et. ΑΘ., Bioorg. Med. Chem. Lett. 2002, 12, 1497; з) Wu, T. Y. H. et. Al. Org. Lett. 2001, 3, 3827]. Основні способи для одержання, властивостей і використання боронових кислот, а також їхніх похідних приведені в "Metal-Organic Compounds", Advances in Chemistry Series, No. 23, American Chemical Society, 1959). У наших власних дослідженнях ми виявили, що з'єднання ефірів 2-ціанофенілборонової кислоти приводить практично до повної кількісної конверсії субстрату 1-пара-галогенобензил-1Німідазолу в продукт реакції, за умови правильно підібраних умов реакції. Проте відповідні боронати 94591 8 практично отримують тільки з виходами середніх значень, оскільки для їхнього синтезу з боронової кислоти потрібне нагрівання і декілька стадій кристалізації для отримання бажаного ефіру певної композиції. Даний факт є істотним, враховуючи чималу вартість о-ціанофенілборонової кислоти. Недавно повідомили про можливість використання арилтрифторборонатів замість арилборонових кислот в якості субстратів в реакціях перехресного зшивання Сузукі для одержання заміщених біарильних сполук з галогенарилами [a) Molander, G.A. et.al., J. Org. Chem. 2003, 68, 4302. b) Molander, G.A. et.al., Org. Lett. 2002, 4, 1867]. Проте невідомо, чи можуть заважати ціано замісники, і яким саме чином, цій реакції. Оскільки добре відомо, що гідроліз ціаногруп приводить до утворення відповідних карбонових кислот, можна припустити щонайменше часткове утворення небажаних побічних продуктів у водному середовищі (Advanced Organic Chemistry, J March, 4th Edition, p.888). Несподівано нами було виявлено, що нові (2ціанофеніл) трифторборонати можна дуже ефективно отримувати з о-ціанофенілборонової кислоти або похідних кислоти. Подібні сполуки є придатними проміжними речовинами для отримання похідних сартану. Так, даний винахід пропонує спосіб одержання похідних сартана формули (І) або їхніх фармацевтично прийнятних солей, зокрема лосартану, олмесартану медоксомілу, кандесартану цилексетилу, та ірбесартану: Причому може бути одинарним або подвійним зв'язком і причому якщо є подвійним зв'язком, то Υ не іс нує. R1 є С1-С7 лінійним або розгалуженим алкілом, переважно С3-С5 алкілом, найпереважніше нпропілом або н-бутилом. С2-С7 лінійну або розгалужену алкокси групу, переважно лінійну С2-С5 аклкокси, найпереважніше н-етокси, або С3-C9 циклоалкіл, наприклад циклопропіл, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил і циклононіл, найпереважніше циклопентил. R2 є гідроксиметил групою, формілом або необов'язково заміщеною карбоксигрупою, причому замісником може бути лінійна або розгалужена С1С10 акільна група, С3-С8 циклоалкільна група, С6С10 арильна група або С7-С13 аралкільна група, або R2 є групою формули 9 94591 10 де n є 0 або 1; Α1-Α6 є незалежно Η або необов'язково заміщеними С1-С4 алкіл, С3-С8 циклоалкіл або С6-С10 арил групами або (в) сполуки, представленої формулою (V) X є Н, СІ або , або X, a R2, разом з подвійним зв'язком імідазольного кільця вони утворюють 6-членне ароматичне кільце, яке можна замістити карбоксильною групою, яку далі можна замістити лінійною або розгалуженою С1-С10 алкільною групою, С3-C6 циклоалкільною групою, С6С10 арильною групою, С7-С13 аралкільною групою, або групою формули де Μ є лужним металом або групою NR4R5R6R7, причому R4-R7 незалежно є Η або незаміщеною або заміщеною С1-С18 алкіл групою у реакції перехресного зшивання з похідним пгалогенобензил-1Н-імідазолу формули (VI) у разі, коли є одинарним зв'язком, тоді R1 є С2-С7 лінійним або розгалуженим алкілом, переважно лінійним алкілом С3-С5, найпереважніше н-бутилом R2 є =0, а X і Υ утворюють С4-С7 циклоалкільну групу, переважно С5-С6 циклоалкільну групу, найпереважніше циклопентильну групу що включає стадії: взаємодія похідної 2-ціанофенілборонової кислоти, вибраної з (а) сполуки, представленої формулою (III) де , X, Y, R1 і R2 визначені вище, a Z є І, Вr або СІ у присутності каталізатора на основі перехідного металу і неорганічної або органічної основи, з утворенням заміщеної сполуки біфенілімідазолу формули (II) де R3 незалежно позначає Η або незаміщений або заміщений С1-С4 алкіл або С6-С10 арил групу, або дві R3 групи утворюють 1,2-фенільну групу (термін «незалежно», що тут використовується означає, що R3 групи можуть бути однаковими або різними, наприклад R3 групи можуть бути заміщеними алкіл групами або одна з них може бути заміщеною алкіл групою, а інша може бути незаміщеною алкіл групою або арил групою, і т.д.; те ж застосовуватиметься і згодом), або їхньою лужною сіллю де лужною сіллю переважно є натрій або калій, найпереважніше калій (б) сполуки, представленої формулою (IV) де , Χ, Υ, R1 i R2 визначені вище далі переведення сполуки формули (II) у відповідну 2-тетразол похідну з одержанням похідної сартану формули (І), переважно лосартану, олмесартану і їхнього ефіру медоксомілу, кандесартану і його ефіру цилексетилу, а також ірбесартану, і необов'язково переведення вказаної похідної сартану в одну з його фармацевтично прийнятних 11 94591 12 солей або ефірів, наприклад, лосартан в лосартан калій. Наступним аспектом винаходу є спосіб, описаний вище, який, як попередня стадія, додатково включає синтез сполуки формули (V) де Μ є лужним металом, переважно калієм, або NR4R5R6R7 групою, де R4-R7 незалежно є Н, С1-С18 незаміщеним або заміщеним алкілом, переважно тетра-н-бутиламонієм, що включає стадії взаємодії з літієм або магнієм 2галогенобензонітрилу, причому галогено означає І, Вr, СІ, і взаємодії отриманого продукту з ефіром бору формули B(OR)3, де R є С1-С4 алкілом, далі взаємодія отриманої сполуки з неорганічним або органічним воденьдифторидом або сумішшю неорганічної або органічної основи з фторводневою кислотою або з калієм воденьдифторидом, потім необов'язково слідує заміна катіону калію на інший катіон шляхом реакції з органічною або неорганічною основою з одержанням сполуки формули (V). Наступним аспектом винаходу є спосіб, описаний вище, який як попередня стадія додатково включає синтез сполуки формули (V), де Μ визначено вище, що включає стадії взаємодії з перехідним металом 2-галогенобензонітрилу, де галогено означає І, Вr, СІ, із сіллю цинку або міді, взаємодія отриманого продукту з тетрафторборатом або BF3 і неорганічною або органічною основою з одержанням сполуки формули (V). Альтернативно, 2-ціанофенілборонова кислота може реагувати з неорганічним або органічним воденьдифторидом або сумішшю неорганічної або органічної основи з фторводневою кислотою або фторводнем калію з отриманням сполуки формули (V). У додатковій стадії будь-якого з описаних вище способів катіон калію можна замінити на іншій, неорганічний або органічний катіон шляхом реакції з органічною або неорганічною основою. Винахід також надає нові проміжні сполуки формули (II) і (V), описані вище, і їхнє застосування при отриманні похідних сартану, зокрема лосартану, олмесартану медоксомілу, кандесартану цилексетилу, а також ірбесартану. Тут будуть детальніше описані численні реакційні стадії і проміжні сполуки, які можна використовувати для одержання важливих проміжних речовин при синтезі похідних сартану, таких як лосартан, олмесартан медоксоміл, кандесартан цилексетил та ірбесартан, з посиланнями на переважні втілення. У одному переважному втіленні даного винаходу пропонується спосіб одержання певних похідних сартану формули (II) де може бути одинарним або подвійним зв'язком, причому якщо є подвійним зв'язком, Υ не існує, R1, R2, X і Υ визначені вище, R1 є переважно н-пропілом, н-бутилом або н-етокси групою, R2 переважно є гідроксиметилом, або і X переважно є СІ, , або X і R2 разом з подвійним зв'язком імідазольного кільця утворюють 6-членне ароматичне кільце, яке переважно є заміщеним групою формули і якщо є одинарним зв'язком, то R1 переважно є н-бутилом, R2 є =0, а X і Υ утворюють переважно циклопентильну групу за допомогою реакції похідного 1Н-імідазолу формули (VI) Де , X, Y, R1 і R2 визначені вище, Ζ переважно є Вr і похідної 2-ціанофенілборонової кислоти формули (III), або її сіллю лужного металу, (IV) і (V) у присутності каталізатора на основі перехідного металу і основи, в органічному або водному розчиннику, або суміші розчинників з одержанням сполуки формули (II). Новий (2-ціанофеніл)-трифторборонат формули (V) характеризується дивно високою стабільністю і тому підходить для реакції перехресного зшивання Сузукі у водних умовах. Якщо, в одному з приведених вище визначень вказано, що група може бути «заміщеною», це означає, що група може містити 1-3 атоми галогену, переважно СІ або F, або одну, дві або три С 1 13 C3 алкоксигрупи, С1-С3 алкіл групи або С6-С10 арил групи. Для одержання лосартану синтез переважно виконують за допомогою реакції похідного 2-нбутил-4-хлор-1-п-бромбензил-1Н-імідазолу формули (V) (R1 = н-бутил, R2 = СН2ОН, X = СІ, Y= Br) з похідним 2-ціанофенілборонової кислоти у присутності каталізатора на основі металу і основи, такого як неорганічна основа, наприклад, карбонат натрію або калію, K3РО4·nН2О, KF·2H2O і алкоксиди лужних металів, або органічна основа, таких як триетиламін або диізопропілетиламін. Реакцію виконують в органічному розчиннику, такому як диметилформамід, диметилацетамід, НМП, диметилсульфоксид, ацетонітрил, С1-С4 спиртах, таких як метанол, етанол, н- і ізо-пропанол, або нбутанол, толуол, тетрагідрофуран, діоксан, ДМЕ, або в їхніх сумішах, або в їхніх сумішах з водою. Особливо переважною основою є диізопропіламін, іншим переважним розчинником є 95% водний етанол; диізопропіламін в технічному етанолі є переважною сумішшю основа/розчинник. Ще переважнішими є безводнні спирти, особливо безводнний етанол. Також, для одержання кандесартана цилексетилу, синтез переважно виконувати реакцією (±)1[[(циклогексилокси)-карбоніл]окси]етил-1-(4бромбензил)-2-етокси-1Н-бензімідазол-7карбоксилату з (2-ціанофеніл) тетрафторборонатом калію у присутності каталізатора на основі металу і основи, такої як неорганічна основа, наприклад карбонат натрію або калію, K3РО4·nН2О, KF·2H2O і алкоксиди лужних металів або органічна основа, такого як триетиламін або диізопропілетиламін. Реакцію виконують в органічному розчиннику, такому як диметилформамід, диметилацетамід, НМП, диметилсульфоксид, ацетонітрил, С1-С4 спиртах, таких як метанол, етанол, н- і ізопропанол, або н-бутанол, толуол, тетрагідрофуран, діоксан, ДМЕ, і в їхніх сумішах або в їхніх сумішах з водою. Особливо переважною основою є диізопропіламін, іншим переважним розчинником є 95% водний етанол; диізопропіламін в технічному етанолі є переважною сумішшю основа/розчинник. Ще переважнішими є безводні спирти, особливо безводний етанол. Також, для одержання ірбесартану синтез переважно виконують реакцією 1 -(4-бромбензил)-2н-бутил-4-спіроциклопентан-2-імідазол-5-ону з (2ціанофеніл)-тетрафторборонатом калію у присутності каталізатора на основі металу і основи, такої як неорганічна основа, наприклад карбонат натрію або калію, K3РО4·nН2О, KF·2H2O і алкоксиди лужних металів, або органічна основа, така як триетиламін або диізопропілетиламін. Реакцію проводять в органічному розчиннику, наприклад в диметилформаміді, диметилацетаміді, НМП, диметилсульфоксиді, ацетонітрилі, С1-С4 спиртах, таких як метанол, етанол, н- і ізо-пропанол, або н-бутанол; у толуолі, тетрагідрофурані, діоксані, ДМЕ, а також в їхніх сумішах або в їхніх сумішах з водою. Особливо переважною основою є диізопропіламін, іншим переважним розчинником є 95% вод 94591 14 ний етанол; диізопропіламін в технічному етанолі є переважною сумішшю основа/розчинник. Ще переважнішими є безводні спирти, особливо безводний етанол. Також, для одержання олмесартану синтез переважно виконують шляхом реакції етил-4-(1гідрокси-1-метилетил)-2-пропіл-1-(4-бромбензил)імідазол-5-карбоксилу з (2-ціанофеніл)тетрафторборонатом калію у присутності каталізатора на основі металу і основи, такого як неорганічна основа, наприклад карбонат натрію або калію, K3РО4·nН2О, KF·2H2O і алкоксиди лужних металів, або органічна основа, такого як триетиламін або диізопропілетиламін. Реакцію проводять в органічному розчиннику, наприклад в диметилформаміді, диметилацетаміді, НМП, диметилсульфоксиді, ацетонітрилі, С1-С4 спиртах, таких як метанол, етанол, н- та ізо-пропанол, або н-бутанол; у толуолі, тетрагідрофурані, діоксані, ДМЕ, а також в їхніх сумішах або в їхніх сумішах з водою. Особливо переважною основою є диізопропіламін, іншим переважним розчинником є 95% водний етанол; диізопропіламін в технічному етанолі є переважною сумішшю основа/розчинник. Ще переважнішими є безводні спирти, особливо безводний етанол. Продукт взаємодії даної реакції можна перевести в етиловий ефір олмесартану, переважно циклоприєднанням, далі етиловий ефір можна перетворити з отриманням іншого ефіру олмесартану, або можна піддати гідролізу з отриманням олмесартану. Альтернативно, продукт взаємодії можна гідролізувати до стадії циклоприєднання. Реакцію приєднання проводять при температурі від 25°С до 180°С, переважно від 50 до 130°С, найпереважніше від 70 до 110°С. Каталізатором на основі металу, використовуваним в реакції, є комплекс нікелю, паладію або платини, переважно комплекс паладію, наприклад біс(ацетонітрил) паладій дихлорид, тетракис(трифенілфосфін) паладій, біс(дибензиліденацетон) паладій, трис(дибензиліденацетон)дипаладій, фосфінові комплекси паладію II, вибрані з групи, що складається: біс(трифенілфосфін) паладій хлорид, біс(трифенілфосфін) паладій бромід, біс(трифенілфосфін) паладій ацетат, біс(триізопропілфосфіт) паладій хлорид, біс(триізопропілфосфіт) паладій бромід, біс(триізопропілфосфіт)паладій ацетат, [1,2біс(дифенілфосфіно)етан]паладій хлорид, [1,2біс(дифенілфосфіно) етан]паладій бромід, [1,2біс(дифенілфосфіно)етан] паладій ацетат, [1,3біс(дифенілфосфіно)пропан]паладій хлорид, [1,3біс(дифенілфосфіно) пропан]паладій бромід, [1,3біс(дифенілфосфіно)пропан]паладій ацетат, [1,4біс(дифенілфосфіно)бутан]паладій хлорид, [1,4біс(дифенілфосфіно)бутан]паладій бромід, [1,4біс(дифенілфосфіно)бутан]паладій ацетат і [1`1біс(дифенілфосфіно)фероцен]паладій хлорид. Активний каталізатор можна отримати завчасно, або синтезувати безпосередньо в реакційній суміші in situ. Активний каталізатор також можна отримати з солі Pd (II), наприклад хлориду паладію, броміду 15 паладію або ацетату паладію з фосфіном, зазвичай трифенілфосфіном або тритолуолфосфіном під дією відновника, такого як діалкілцинк, алкілцинкгалогенід, діалкілмагній, алкілмагнійгалогенід, триалкілалюміній, диалкілалюмінійгідрид, боргідрид натрію, гідразин або арилборонова кислота у відповідному розчиннику. У переважному втіленні у якості відновника використовують диетилцинк. За певних умов стадію відновлення можна замінити, для того, щоб відновився попередник паладію і в реакційній суміші утворився активний каталізатор. Реакцію можна проводити, використовуючи каталізатор з фосфіновими лігандами або без них. Проте, в переважному втіленні використовують фосфін як ліганд в співвідношенні Рd:фосфін 1:4, оскільки це збільшує стабільність активного каталітичного комплексу паладію. Металевий атом, іон або початкову металеву сполуку можна використовувати з носієм або без. Носії можуть бути органічної або неорганічної природи. У іншому втіленні, носій може не бути частиною початкової сполуки металу, відповідні носії включають кременеві, алюмінієві носії, цеоліти, поліетиленгліколи, полістирени, поліефіри, поліаміди, пептиди і тому подібне Особливі приклади Pd на носієві включають Pd/C, Pd/SiO2, Pd/CaCO3, Pd/ВаСО3, Pd/алюміній, Pd/оксид алюмінію, Pd/полістирен. Будь-який з вищеперелічених металів може замінювати Pd в даному списку, наприклад, Ni/C і так далі. В цілому, розчинники для реакції можна вибирати з безлічі відомих розчинників. Прикладами розчинників, які можна використовувати як окремо, так і в суміші, є бензол, толуол, етиловий ефір, тетрагідрофуран, діоксан, НМП, ацетонітрил, диметилформамід, диметилацетамід, диметилсульфоксид, етанол, метанол, пропанол, ізопропіловий спирт, вода, 2-метилтетрагідрофуран або диетоксіметан. Переважним розчинником є водний етанол, тетрагідрофуран або толуол, переважніший безводний етанол. Зручно використовувати дегазовані розчинники. Для виконання реакції(й) можна використовувати велику кількість основ. Ілюстративними прикладами є органічні четвертинні не-нуклеофільні основи, такі як триетиламін або диізопропілетиламін, неорганічні основи, такі як карбонат калію, карбонат натрію, карбонат цезію, фторид цезію, фторид калію, фосфат калію, гідроксид калію, гідроксид натрію або алкоксиди цих лужних металів. Коли використовують неорганічну основу, нерозчинну в органічному розчиннику, може бути потрібно розчинення у воді; застосування міжфазних каталізаторів, таких як тетра-н-бутиламоній бромід або краун-ефір також може сприяти реакції. Основи, розчинні в органічних розчинниках, такі як тетра-н-бутиламоній карбонат або тетра-нбутиламоній гідроксид, бензилтриметиламоній карбонат, бензилтриметиламоній метилкарбонат, бензилтриметиламоній метоксид або бензилтриметиламоній гідроксид, а також інші основні сполуки тетраалкіламонію можна застосовувати в певних випадках. Основи, розчинні в органічних розчинниках можна приготувати заздалегідь або 94591 16 отримати безпосередньо в реакційній суміші. Наприклад, отримати бензилтриметиламоній карбонат можна реакцією розчину гідроксиду бензилтриметиламонію з карбонатом амонію. Основи переважно використовувати в способі за винаходом в кількості приблизно від 1 до 1000 моль%, переважніше від приблизно 50 до 500 моль%, найпереважніше від приблизно 100 до 400 моль%, зокрема від 150 до приблизно 300 моль%, виходячи з похідної боронової кислоти. Через 2 - 24 години від початку реакції можна виділити продукт реакції, сполуку формули (II), способом, відомим фахівцям даної області техніки, але переважно осадженням з реакційного середовища за допомогою додавання води. Доцільно, щоб молярне співвідношення (III), (IV) або (V), відповідно, у відношенні до похідних формули (VI) складало між 1 і 1,5, зокрема, якщо сполука формули (VI) є 2-н-бутил-1-пгалогенобензил-1Н-імідазолом. У особливо переважному втіленні даний винахід включає умови використання 1% Pd(OAc)2 + 4% Р(о-С6Н4СН3) або 2% Pd(OAc)2 + 8% PPh3, 4 екв. i-Pr2NEt в 95% водному етанолі при реакції перехресного зшивання (2-ціанофеніл)трифторбороната калію з 2-н-бутил-1-пбромбензил-4-хлор-1Н-імідазол-5-ілметанолом або подібною сполукою, попередником сартана формули (VI), як вказано вище. За цих переважних реакційних умов похідна 4`-(2-бутил-4-хлор-5гідроксиметил-1Н-імідазол-1-іл)-1,1`-біфеніл-2карбонітрилу формули (II) отримують практично з високими виходами. Проте, використання інших каталізаторів паладію (без лігандів або з іншими фосфіновими лігандами), розчинників і основ також приводить до утворення бажаного субстрата формули (VI) з хорошими виходами. Нові похідні (2-ціанофеніл)-трифторбороната формули (V) отримують реакцією 2ціанофенілборонової кислоти з неорганічним або органічним фторводнем або із сумішшю органічної або неорганічної основи з фторводневою кислотою або з гідрофторидом калію, після цього проводять заміну катіона калію іншим органічним або неорганічним катіоном шляхом реакції з органічною або неорганічною основою. Основу вибирають з органічних четвертинних не-нуклеофільних основ, причому органічну не-нуклеофільну основу вибирають з групи, що складається з триетиламіну або диізопропілетиламіну, або використовують неорганічну основу, причому неорганічну основу вибирають з карбонату калію, карбонату натрію, карбонату цезію, фосфату калію, фториду калію, гідрофториду калію, алкоксиду калію або алкоксиду натрію, а також основ, розчинних в органічних розчинниках, причому основи вибирають з групи, що складається з тетра-н-бутиламоній карбонату, тетра-н-бутиламоній гідроксиду, бензилтриметиламоній карбонату, бензилтриметиламоній метилкарбонату, бензилтриметиламоній метоксиду або бензилтриметиламоній гідроксиду. Альтернативно, похідні (2ціанофеніл)трифторбороната формули (V) отримують взаємодією з літієм або магнієм 2галогенобензонітрилу, причому галогено означає І, 17 Вr, СІ і взаємодією отриманого продукту з ефіром бору формули B(OR)3, де R означає С1-С4 алкіл групу, далі взаємодією отриманої сполуки з органічним або неорганічним фторводнем або сумішшю органічної або неорганічної основи з фторводневою кислотою або з гідрофторидом калію, далі необов'язково замінюють катіон калію на інший катіон реакцією з органічною або неорганічною основою, отримуючи сполуку формули (V). Одержання проміжних сполук виконують способами, добре відомими фахівцям в даній області техніки. Так, наприклад, введення літію в орто-положення, описане в US 5 039 814. Проте, реакційні стадії в синтезі мають бути сумісними з функціональними групами імідазольного угрупування й іншими частинами молекули. У другому варіанті, похідні (2-ціанофеніл) трифторборонату формули (V) отримують взаємодією з перехідними металами 2-галогенобензонітрилу, причому галогено означає І, Вr, СІ, з сіллю цинку або магнію, далі реакцією отриманого продукту з тетрафторборонатом BF3 і неорганічною або органічною основою, отримуючи сполуку формули (V). Отримана 2-ціанофенілборонова кислота далі реагує з органічним або неорганічним воденьдифторидом або сумішшю неорганічної або органічної основи з фторводневою кислотою або з гідрофторидом калію. У наступній стадії можна замінити катіон калію на інший неорганічний або органічний катіон, реакцією з органічною або неорганічною основою. Переведення сполуки формули (II) в сполуку формули (І) можна виконувати будь-якими способами, відомими фахівцям в даній області техніки. Особливо переважна взаємодія ціаногрупи з азидом, особливо з азидом натрію. Альтернативно, похідні сартану формули (І) можна переводити в одну з фармацевтично прийнятних солей будьякими способами, відомими на сучасному рівні техніки. Переважним прикладом є реакція з гідроксидом калію або натрію. У переважному втіленні похідні сартану формули (І) переводять у відповідні солі калію. У разі ірбесартану і кандесартану, продукт взаємодії формули (І) можна далі перевести в ірбесартан і кандесартан цилексетил, відповідно, і очистити, переводячи дані сполуки у відповідну похідну з тетразольним захистом, наприклад, в похідну тритилу, а потім зняти захист, отримавши бажану похідну сартану формули (І). У разі олмесартану, продукт взаємодії формули (І) можна перевести в етиловий ефір олмесартану з тетразольним захистом, потім за допомогою транс-етерифікації отримати олмесартану медоксоміл ефір з тетразольним захистом, потім зняти захист і отримати олмесартану медоксоміл ефір. У кожному з цих випадків переважною захисною групою є тритил. Іншим аспектом винаходу є спосіб одержання сполуки формули (VI), як описано вище, що характеризується тим, що похідна імідазолу формули (VII) 94591 18 де X, Y, R1 і R2 визначені вище, реагує з 4галогенобензилгалогенідом, причому галогеном є СІ, Вг або І, переважно Вr, у присутності основи при дефлегмації. У переважному втіленні, 2-нбутил-4-спіроциклопентан-2-імідазолін-5-он гідрохлорид взаємодіє з 4 бромбензилбромідом у присутності тетрабутиламонію броміду, і гідроксиду калію. Винахід проілюстрований приведеними нижче прикладами. Приклади не обмежують об'єм даного винаходу, визначений формулою винаходу нижче. ПРИКЛАДИ Приклад 1 Одержання (2-ціанофеніл) трифторборонату калію 2,50 грам 2-ціанофенілборонової кислоти розчинили в 100 мл МеОН і додали розчин 4,40 грам KHF2 (3,30 екв.) в 50 мл Н2О. Реакційну суміш нагрівали до кипіння і розчинник відігнали у вакуумі. Білий залишок тричі екстрагували по 50 мл теплим осушеним ацетоном. Розчин концентрували до об'єму близько 50 мл і потім повільно 400 мл діетилового ефіру. Осаджені кристали (2ціанофеніл)трифторборонату калію відфільтрували, промивали ефіром і осушували. Вихід: 2,90 грам (82%) білого кристалічного осаду (т.пл. 1691 171°С). Н ЯМР (300 МГц, ДМСО, δ): 7,52 d (1H), 7,50 d (1H), 7,39 dd (1H), 7,23 ddd(1H). Приклад 2 А) Одержання 4`-(2-бутил-4-хлор-5гідроксиметил-1Н-імідазол-1-іл)-1`1-біфеніл-2карбонітрилу 0,50 грам (1,4 ммоль) (2ціанофеніл)трифторбороната калію, 0,35 грам (1,68 ммоль, 1,2 екв) 2-н-бутил-1-п-бромбензил-4хлор-1Н-імідазол-5-іл-метанолу (чистота 93 %), 3,00 міліграм (0,014 ммоль, 0,01 екв.) ацетату паладію, 17 міліграм (0,056 ммоль, 0,04 екв.) три-отолілфосфіну помістили в суху колбу з дефлегматором. Колбу закрили діафрагмою і заповнили аргоном в три етапи. За допомогою шприца в колбу додали 10 мл 95% водного етанолу, насиченого аргоном, і 0,50 мл (4 екв.) диізопропілетиламіну, насиченого аргоном. Реакційну суміш нагрівали і кип'ятили протягом 12 годин. Колір мінявся від жовтуватого (ацетат паладію) через червонокоричневий (активні частинки паладію) до осадження неактивного чорного паладію. Осадження чорного неактивного паладію відбулося через 6 годин. Далі, реакційну суміш відкрили і розчинник відігнали у вакуумі. До залишку додали 1,00 грам силікагелю, зволоженого 5,00 мл етилового ацетату і розчинник упарювали. Реакційну суміш, осаджену на силікагель, нанесли на кінець короткої колонки, заповненої силікагелем (10 грам силікагеля з етилацетат-гексаном 1:1 (об/об)), колонку елюювали сумішшю етилацетат-гексан 1:1 (об/об). Після упарювання розчинника отримали 0,50 грам 19 (94%, аналіз: 90%) неочищеного жовтуватого твердого продукту. Отриманий матеріал кристалізували з суміші етилацетат-гексану, отримавши 0,45 грам (84%) 4`-(2-бутил-4-хлор-5-гідроксиметил-1Німідазол1-іл)-1,1`-біфеніл-2-карбонітрилу у вигляді білого кристалічного осаду (т.пл. 154,1 - 155,5°С). 1 Н ЯМР (300 МГц, CDCI3, δ): 7,77 dd (1H), 7,65 ddd (1H), 7,53 d (2H), 7,42-7,50 m (2H) 7,12 d (2H), 5,29 s (2H), 4,53 d (2H), 2,60 t (2H), 1,68 m (2H), 1,36 m (2H), 0,891 (3Н). Б) Одержання 4`-(2-бутил-4-хлор-5гідроксиметил-1Н-імідазол1-іл)-1,1`-біфеніл-2карбонітрилу Аналогічно прикладу А, але замість 3 міліграм ацетату паладію і 23 міліграми три-о-толілфосфіну додали 6,00 міліграм (0,03 ммоль, 0,02 екв.) ацетату паладію і 29 міліграм (0,11 ммоль, 0,08 екв.) трифенілфосфіну. В) Одержання 4`-(2-бутил-4-хлор-5гідроксиметил-1Н-імідазол1-іл)-1,1`-біфеніл-2карбонітрилу 8,00 грам (22,5 ммоль) 2-н-бутил-1-пбромбензил-4-хлор-1Н-імідазол-5-іл-метанолу, 5,64 грам (27,0 ммоль, 1,2 екв.) (2ціанофеніл)трифторборонату калію, 50,0 міліграм (0,23 ммоль, 0,01 екв.) ацетату паладію, 273 міліграми (0,90 ммоль, 0,04 екв.) три-о-толілфосфіну помістили в суху колбу з дефлегматором. Колбу закрили діафрагмою і наповнили аргоном в три етапи. За допомогою шприца в колбу додали 150 мл 95% водного етанолу, насиченого аргоном, і 15,3 мл (0,90 ммоль, 4 екв.) диізопропілетиламіну, насиченого аргоном. Реакційну суміш нагрівали до кипіння і перемішували при цій температурі протягом 18 годин. Далі, реакційну суміш відкривали і розчинник упарювали у вакуумі. Реакційну суміш очищали хроматографічно (етилацетат-гексан 1:1). Розчинник видаляли і осад екстрагували теплим циклогексаном (для того, щоб видалити залишковий фосфін), а потім кристалізували з етилацетату-гексану (1:1), отримавши першу порцію продукту. Матковий розчин очистили хроматографічно і кристалізували, отримавши другу порцію продукту. Продукт отримали з виходом 89% (7,54 грам), у вигляді білого кристалічного порошку з т.пл. 154-156°С. 1 Н ЯМР бензил метиленовий сигнал 96,5 моль% (5,29 м.д.) (ізомерна сполука 1,2 моль% (5,25 м.д.) і 2-н-бутил-1-п-бромбензил-4-хлор-1Німідазол-5-іл-метанол 2,3 моль% (5,22 м.д.); ВЕРХ (фенільна зворотнофазова колонка, 40% водного ацетонітрилу) 254 нм: 98,2% (11,95 хв) + полярні домішки 1,8% (3,64 хв); 235 нм: 97,7% (11,96 хв) + полярні домішки 2,3% (3,70 хв). 0,60 грам (7%) 4`-(2-бутил-4-хлор-5гідроксиметил-1Н-імідазол1-іл)-1,1`-біфеніл-2карбонітрилу отримали у вигляді брудно-білого кристалічного порошку, т.пл. 152 - 154°С; Аналіз: 1Н ЯМР 93,5% (ізомерна сполука 2,8 моль% (5,25 м.д.) і 2-н-бутил-1-р-бромбензил-4-хлор-1Німідазол-5-іл-метанол 3,7 моль% (5,22 м.д.); ВЕРХ (фенільна зворотньофазова колонка, 40% водний ацетонітрил) 235 нм: 95,6% (12,00 хв) + полярні домішки 4,4% (3,69 хв). 94591 20 Загальний вихід: 8,14 грам (96 %). Приклад 3 Одержання 4`-(2-бутил-4-хлор-5гідроксиметил-1Н-імідазол-1-іл)-1,1`-біфеніл-2карбонітрилу Аналогічно прикладу 2Б, але каталізатор готували окремо. Трі-о-толілфосфін (17,0 міліграм) розчиняли в ТГФ (20 мл) і розчин дегазували вакуум/продування азотом (3 рази). Ацетат паладію (3,00 міліграм, 0,25 ммоль) додали до розчину і знову дегазували (3 рази). Отриманий розчин нагрівали до 60°С протягом 30 хв, потім охолодили до 25°С і застосовували для реакції. Приклад 4 Одержання 4`-(2-бутил-4-хлор-5гідроксиметил-1Н-імідазол-1-іл)-1,1`-біфеніл-2карбонітрилу Аналогічно прикладу 2, але каталізатор готували окремо. Суміш хлориду паладію (50,0 міліграм) і трифенілфосфіну (0,70 грам) додавали до безводного ТГФ (20 мл). Гетерогенний розчин дегазували вакуум/продуванням азотом (3 рази) і потім додавали однією порцією триізопропілфосфіт (0,30 мл). Суміш залишали при кімнатній температурі при перемішуванні до повного розчинення хлориду паладію і отримання гомогенного розчину. Приклад 5 Одержання 4`-(2-бутил-4-хлор-5-форміл-1Німідазол-1-іл)-1,1`-біфеніл-2-карбонітрилу Аналогічно прикладу 2Б, але замість 2-нбутил-1-п-бромбензил-4-хлор-1Н-імідазол-5-ілметанолу використовували 2-н-бутил-1-пбромбензил-4-хлор-1Н-імідазол-5-карбальдегід. Приклад 6 Одержання тетра-н-бутиламоній (2ціанофеніл) трифторборонату Колбу, що містить 2,00 грам 2бромбензонітрилу (11,0 ммоль) і закриту діафрагмою, заповнювали аргоном і додавали 30 мл сухого дегазованого ТГФ. Розчин охолоджували до 94°С і через 10 хв поволі додавали 10 мл 1,60 Μ розчину н-бутиллітію в гексані (16,0 ммоль, 1,5 екв). Отриманий розчин перемішували протягом 20 хв при даній температурі. 33,0 мл 1,5 Μ розчину хлориду цинку (22,0 ммоль, 2,00 екв.) додавали до реакційної суміші і перемішували 20 хв. Далі до реакційної суміші поволі додавали розчин, що містить 7,20 грам (22,0 ммоль, 2,00 екв.) тетра-нбутиламонію тетрафторборонату в 20 мл ТГФ, і перемішували протягом 1 години при -94°С. Реакційну суміш залишали нагріватися протягом ночі до кімнатної температури. Далі колбу відкривали і розчинник упарювали у вакуумі. Залишок промивали діетиловим ефіром і тричі екстрагували 50 мл теплого сухого ацетону. Отриманий розчин концентрували до об'єму приблизно 50 мл і додавали 400 мл діетилового ефіру. Осад фільтрували, промивали ефіром і просушували. Отримували 2,50 грам (56 %) тетра-н-бутиламонію (2ціанофеніл) трифторборонату у вигляді білого 1 кристалічного осаду. Аналіз: Н ЯМР > 98%. 21 Приклад 7 Одержання (2-ціанофеніл) трифторборонату калію Колбу, що містить 2,00 грам 2бромбензонітрилу (11,0 ммоль) і закриту діафрагмою, заповнювали аргоном і додавали 30 мл сухого дегазованого ТГФ. Розчин охолоджували до 94°С і протягом 10 хв поволі додавали 10 мл 1,60 Μ розчину н-бутиллітію в гексані (16,0 ммоль, 1,5 екв). Отриманий розчин перемішували протягом 20 хв при даній температурі. До реакційної суміші поволі додавали 5,00 мл триметилборату (45 ммоль, 4,0 екв.) і перемішували протягом 1 години при температурі -94°С. Реакційну суміш залишали нагріватися до кімнатної температури протягом ночі. Далі колбу відкривали і розчинник упарювали у вакуумі. Додавали 100 мл метанолу і розчин, що містить 2,10 грам KHF2 в 50 мл води. Отриману суміш нагрівали до кипіння і розчинник упарювали. Залишок промивали діетиловим ефіром і тричі екстрагували 50 мл сухого теплого ацетону. Розчин сконцентрували до об'єму приблизно 50 мл і додавали 400 мл діетилового ефіру. Осад відфільтрували, промивали ефіром і просушували. Отримували 1,58 грам (69 %) (2-ціанофеніл) три1 фторборонату калію. Аналіз: Н ЯМР > 98%. Приклад 8 Одержання лосартану 0,381 грам (1,00 ммоль) 4`-(2-бутил-4-хлор-5гідроксиметил-1Н-імідазол-1-іл)-1,1`-біфеніл-2карбонітрилу і 0,82 мл (3,00 ммоль) трибутилтин азиду розмішали в 6 мл толуолу і нагрівали до температури кипіння. Реакційну суміш перемішували при цій температурі протягом 96 год. Після завершення реакції суспензію охолоджували до кімнатної температури і додавали 4 мл 2М КОН. Фази розділили і водну фазу підкислювали до рН близько 3. Осаджений продукт фільтрували і просушували. Виділяли 0,33 грам продукту. 1 Н ЯМР (300 МГц, ДМСО-d6, δ): 7,63 - 7,72 m (2H), 7,50-7,60 m (2H), 7,05 m (4Н), 5,25 s (2Н), 4,33 bs (2Н), @,50 t (2Н), 1,45 m (2Н), 1,23 m (2Н), 0,80 t (3Н), ОН і NH обмінені. Приклад 9 Одержання кандесартана цилексетилу А) Одержання ((±)1[[(циклогексилокси)карбоніл]окси]етил-2-етокси-1[(2`-ціанобіфеніл-4-іл)-метил]-1Н-бензімідазол-7карбоксилату 0,06 грам (0,26 ммоль) (2ціанофеніл)тетрафторбороната калію, 0,11 грам (0,2 ммоль) (±)1[[(циклогексилокси)карбоніл]окси]етил-1-(4бромбензил)-2-етокси-1Н-бензімідазол-7карбоксилату, 1 міліграм (0,004 ммоль) ацетату паладію, 2 міліграми (0,007 ммоль) три-отолілфосфіну поміщали в суху колбу, забезпечену дефлегматором. Колбу закривали діафрагмою і заповнювали аргоном в три етапи. За допомогою шприца в колбу додавали 1,2 мл етанолу, насиченого аргоном, і 0,06 мл диізопропілетиламіну, насиченого аргоном. Реакційну суміш нагрівали і кип'ятили протягом 20 годин. Далі, реакційну суміш відкривали на повітрі, охолоджували, відфільтрували і розчинник упарювали у вакуумі. До залишку 94591 22 додавали 5 мл ізопропілацетату і 5 мл води. Суміш перемішували і потім розділяли. Органічну фазу двічі промивали 5 мл води, сушили над Na2SO4 і упарювали з одержанням 150 міліграм маслянистого осаду. 1 Н ЯМР (300 МГц, ДМСО, δ): 7,9 m (1Н), 7,75 m (2H), 7,45-7,60 m (5H), 7,22 m (1H), 7,10 d (2H), 6,80 m (1H), 5,60 d (2H), 4,46-4,68 m (3Н), 1,15-1,80 m (16H). Б) Одержання ((±)1[[(циклогексилокси)карбоніл]окси]етил-2-етокси-1[(2`-(1Н-тетразол-5-іл)[1`1-біфеніл]-4-іл]метил]-1Нбензімідазол-7-карбоксилату Суміш 0,057 грам (0,1 ммоль) (±)1[[(циклогексилокси)карбоніл]окси]етил-2-етокси-1[(2`-ціанобіфеніл-4-іл)-метил]-1 Н-бензімідазол-7карбоксилату, 2 мл толуолу, 0,054 мл (0,3 ммоль) трибутилтин хлориду і 20 міліграм (0,3 ммоль) NaN3 кип'ятили протягом 72 годин. Реакційну суміш охолоджували і концентрували. Залишок очищали колоночною хроматографією на силікагелі, отримавши 70 міліграм кандесартана цилексетилу. Приклад 10 А) Одержання 1-(4-бромбензил)-2-н-бутил-4спіроциклопентан-2-імідазолин-5-ону Суміш 50 мл ацетонітрилу, 2,76 (12 ммоль) 2н-бутил-4-спіроциклопентан-2-імідазолин-5-он гідрохлориду, 2,48 грам (10 ммоль) 4бромбензилбромиду, 1,39 грам (4,3 ммоль) тетрабутиламоній броміду і 3,67 грам (65,5 ммоль) КОН кип'ятили протягом 4 годин. Суспензію охолоджували і сконцентрували при зниженому тиску. До залишку додавали 50 мл води і далі суміш нейтралізовували додаванням приблизно 31 мл 1М НСІ до рН 6. Продукт екстрагували 80 мл СН2СІ2 і органічну фазу двічі промивали водою, просушували над Na2SO4, фільтрували і упарювали. Отримували 2,67 грам масляного осаду. Зразок для аналітичних цілей отримували, очищаючи неочищений продукт флешхроматографією (гексан:етилацетат:триетиламін 2:1:0,1, об/об/об). Одержання 1-(4-бромбензил)-2-н-бутил-4спіроциклопентан-2-імідазолін-5-ону виконували за способом, описаним в WO 2006/073376. 1 Н ЯМР (300 Мгц, ДМСО δ): 7,55 d (2H), 7,11 d (2H), 4,65 s (2H), 3,3 t (2H), 1,58-1,94 m (8Н), 1,46 m (2Н), 1,24 m (2Н), 0,791 (3Н). Б1) Одержання ірбесартану 0,712 грам (3,12 ммоль) (2-ціанофеніл) тетрафторборонату калію, 0,92 грам (2,53 ммоль) 1-(4бромбензил)-2-н-бутил-4-спіроциклопентан-2імідазолін-5-ону, 12 міліграм (0,05 ммоль) ацетату паладію, 22 міліграми (0,07 ммоль) три-отолілфосфіну поміщали в суху колбу з дефлегматором. Колбу закривали діафрагмою і заповнювали аргоном в три етапи. За допомогою шприца в колбу додали 14 мл етанолу, насиченого аргоном, і 0,72 мл диізопропілетиламіну, насиченого аргоном. Реакційну суміш кип'ятили протягом 20 годин. Далі, реакційну суміш охолоджували, фільтрували і розчинник упарювали у вакуумі. До залишку додавали 20 мл ксилену і 21 мл 0,05 Μ НСІ. Суміш перемішували і потім розділяли. Органічну фазу двічі промивали 5 мл води, висушували над 23 Na2SO4 і упарювали, отримуючи 10 мл розчину, що містить продукт. До цього розчину додавали 1,3 мл (7,2 ммоль) трибутилтин хлориду і 468 міліграм (7,2 ммоль) NaN3 і кип'ятили протягом 42 год. Суміш охолоджували і додавали 16 мл 0,2 Μ ΝΑΟΗ. Після перемішування фази розділяли і водну фазу екстрагували 20 мл терт-бутил метилового ефіру. Водну фазу підкисляли до рН 4-5 і охолоджували. Осаджений продукт фільтрували і виділяли 0,84 грам ірбесартану. Б2) Одержання ірбесартану 0,074 грам (0,5 ммоль) 2-ціанофенілборонової кислоти, 0,225 грам (0,6 ммоль) 1-(4-бромбензил)2-н-бутил-4-спіроциклопентан-2-імідазолин-5-ону, З міліграми (0,01 ммоль) ацетату паладію, 6 міліграм (0,02 ммоль) три-о-толілфосфіну поміщали в суху колбу з дефлегматором. Колбу закривали діафрагмою і заповнювали аргоном в три етапи. За допомогою шприца в колбу додавали 3,6 мл етанолу, насиченого аргоном, і 0,18 мл диізопропілетиламіну, насиченого аргоном. Реакційну суміш кип'ятили протягом 22 годин. Далі реакційну суміш відкривали на повітрі, охолоджували, фільтрували і розчинник упарювали при зниженому тиску. До залишку додавали 5 мл ксилолу і 5,3 мл 0,05 Μ НСІ. Суміш перемішували і розділяли. Органічну фазу двічі промили 5 мл води, просушували над Na2SO4 і упарювали, отримуючи 3 мл розчину, що містить ціано продукт. До цього розчину додали 0,33 грам (1 ммоль) трибутилтин азиду і кип'ятили протягом 42 годин. Ксилол упарювали і додавали 5 мл СН2СІ2 і 5 мл води. Фази розділяли і потім до органічної фази додавали 10 мл 0,2 Μ ΝΑΟΗ. Після перемішування фази розділяли і водну фазу екстрагували 10 мл трет-бутилметилового ефіру. Водну фазу підкисляли до рН 4-5 і охолоджували, осаджений продукт фільтрували. Виділяли 0,13 грам ірбесартану. Приклад 11 Одержання олмесартану А) Одержання етил-4-(1-гідрокси-1метилетил)-2-пропіл-1-(4-бромбензил)імідазол-5карбоксилату (VII) Суміш 240 мл ацетонітрилу, 20,7 грам (150 ммоль) K2СО3, 18 грам (75 ммоль) етил-4-(1гідрокси-1-метилетил)-2-пропілімідазол-5карбоксилату і 20,4 грам (81,6 ммоль) 4бромбензилбромиду кип'ятили протягом 7 годин. Суспензію охолоджували, фільтрували і сконцентрували при зниженому тиску приблизно до 1/3 від початкового об'єму, далі перемішували при біля 0°С протягом 1 години. Осад фільтрували і висушували при 35°С протягом години, потім розмішували в 218 мл води. Суміш перемішували протягом 2 год, фільтрували і знову сушили. Виділяли 21,56 грам неочищеного продукту. Неочищений продукт перекристалізовували з ацетонітрилу (87%, 55% за синтезом). 94591 24 Т=84 - 85°С ІК (основні піки): 3371, 2961, 1666, 1529, 1404, 1176, 1009, 780, 632 1 Н ЯМР (300 МГц, ДМСО, δ): 7,54 d (2Н), 6,90 d (2Н), 5,42 s (2Н), 5,39 s 1 Η), 4,14 q (2H), 2,60 t (2H), 1,59 m (2H), 1,50 s (6H), 1,07 t (3Н), 0,87 t (3Н). Б) Одержання етил-4-(1-гідрокси-1метилетил)-2-пропіл-1-[(2-ціанобіфеніл-4-іл)метил]імідазол-5-карбоксилату 0,89 грам (3,9 ммоль) (2-ціанофеніл) тетрафторбороната калію, 1,35 грам (3,3 ммоль)етил-4(1-гідрокси-1-метилетил)-2-пропіл-1-(4бромбензил)імідазол-5-карбоксилату, 15 міліграм (0,07 ммоль) ацетату паладію, 30 міліграм (0,1 ммоль) три-о-толілфосфіну поміщали в суху колбу з дефлегматором. Колбу закрили діафрагмою і заповнювали аргоном в три етапи. За допомогою шприца в колбу додавали 18 мл етанолу, насиченого аргоном, і 0,9 мл диізопропілетиламіну, насиченого аргоном. Реакційну суміш кип'ятили протягом 20 годин. Далі, реакційну суміш охолоджували, фільтрували і розчинник упарювали у вакуумі. До залишку додавали 40 мл ізопропілацетату і 42 мл 0,05 мл НСІ. Суміш перемішували і потім розділяли. Органічну фазу двічі промивали 40 мл води, просушували над Na2SO4 і упарювали, отримавши 1,76 грам маслянистого осаду. Зразок продукту для аналітичних цілей очищали за допомогою флеш-хроматографії (Мфз: гексан : етил ацетат 1:1 об/об). 1 Н ЯМР (300 МГц, ДМСО, δ): 7,93 ddd (1H), 7,78 ddd (1H), 7,61-7,54 m (4H), 7,11 d (2Н), 5,55 s 2Н), 5,42 s (1Н), 4,16 q (2Н), 2,65 t (2Н), 1,63 m (2Н), 1,49 s (6Н), 1,07 t (З Η), 0,90 t (3H). В) Одержання етил-4-(1-гідрокси-1метилетил)-2-пропіл-1-[[2`-(1Н-тетразол-5-іл)[1,1`біфеніл]-4-іл]метил]-імідазол-5-карбоксилату Суміш 0,56 грам (0,8 ммоль) етил-4-(1гідрокси-1-метилетил)-2-пропіл-1-[(2`ціанобіфеніл-4-іл)-метил]-імідазол-5-карбоксилату, 3 мл толуолу, 0,65 мл (2,1 ммоль) трибутилтин хлориду і 0,13 міліграм (2 ммоль) NaN3 кип'ятили протягом 42 годин. Реакційну суміш упарювали при зниженому тиску і залишок розчиняли в 5,5 мл 2,5 Μ розчину НСІ в етанолі. Розчин перемішували протягом 18 годин і потім сконцентрували. Залишок перетирали в диізопропіловому ефірі, отримуючи 0,51 грам кінцевого продукту у вигляді солі гідрохлориду. Т=100 - 103°С 1 Н ЯМР (300 МГц, CD3OD, δ): 6,9-7,8 m (8H), 5,70 s (2H), 4,30 q (2H), 3,00 t (2H), 1,70 s (6H), 1,57 m (2H), 1,24 t (3Н), 0,97 t (3Н). На наступній стадії етил-4-(1-гідрокси-1метилетил)-2-пропіл-1-[[2`-(1Н-тетразол-5-іл)[1,1`біфеніл]-4-іл]метил]-імідазол-5-карбоксилат можна гідролізувати, отримуючи олмесартан. 25 Комп’ютерна верстка Т. Чепелева 94591 Підписне 26 Тираж 24 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for the preparation of sartan derivates and intermediates useful in such process
Автори англійськоюVeverka Miroslav, Putala Martin, Brath Heinrich, Zuppancic Silvo
Назва патенту російськоюСпособ получения производных сартана и промежуточных соединений, пригодных для этого способа
Автори російськоюВеверка Мирослав, Путала Мартин, Брат Хейнрич, Жупанчич Силво
МПК / Мітки
МПК: C07D 233/54, C07D 403/10, C07F 5/00
Мітки: одержання, сполук, сартану, проміжних, придатних, цього, способу, похідних, спосіб
Код посилання
<a href="https://ua.patents.su/13-94591-sposib-oderzhannya-pokhidnikh-sartanu-i-promizhnikh-spoluk-pridatnikh-dlya-cogo-sposobu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання похідних сартану і проміжних сполук, придатних для цього способу</a>
Спосіб одержання похідних 4-трифтрометилсульфінілпіразолу та способи одержання проміжних сполук

Номер патенту: 73752
Опубліковано: 15.09.2005
Автори: Шарро Філіпп, Пельта Ізабелль, Клавель Жан-Луї, Ле Бар Сільві
МПК: C07D 401/04, C07D 231/44
Мітки: способи, спосіб, похідних, сполук, проміжних, одержання, 4-трифтрометилсульфінілпіразолу
Формула / Реферат:
1. Спосіб (А) одержання сполуки формули (І):, (I)в якій W означає азот або -CR3; R1 означає галоген, галоалкіл, галоалкокси, R4S(O)n- або -SF5; R2 означає водень або галоген; R3 означає галоген; R4 означає алкіл або галоалкіл і n означає 0, 1 або 2, що включає окислення сполуки формули (II):
Азольні сполуки з протигрибковою активністю, спосіб одержання цих сполук, спосіб одержання проміжних сполук та фармацевтична композиція

Номер патенту: 55405
Опубліковано: 15.04.2003
Автори: Альбіні Енріко, Фраіре Крістіна, Скіоппакассі Джованна, Наполетано Мауро
МПК: A61K 31/00, A61P 31/10, C07D 521/00, A61K 31/41, A61P 31/00, A61K 31/4196, C07D 249/08, C07D 233/60, A61K 31/4164, A61K 31/415
Мітки: фармацевтична, сполук, сполуки, протигрибковою, спосіб, композиція, активністю, проміжних, одержання, азольні, цих
Формула / Реферат:
1. Сполука формули (ІІ-А), (ІІ-А)де:R1 являє собою хлор, фтор, бром або трифторметил;R2 являє собою водень, хлор, фтор, бром або трифторметил;Z являє собою СН або N;R3, R4, R5, які можуть бути однаковими або різними, являють собою водень або С1-С4 алкіл при умові, що R4 відрізняється від R5, якщо R3 - водень;Х являє собою О, S, SO або SO2;R6 являє собою С1-С5 поліфторалкільну групу, яка...
Спосіб одержання 1-(тіометил)циклопропаноцтової кислоти та проміжних сполук для її одержання

Номер патенту: 50730
Опубліковано: 15.11.2002
Автори: Кінг Стівен, Піпік Бренда, Конлон Девід Е.
МПК: C07C 253/16, C07C 319/00, C07C 327/00, C07D 327/00
Мітки: проміжних, 1-(тіометил)циклопропаноцтової, спосіб, кислоти, одержання, сполук
Формула / Реферат:
1. Спосіб одержання циклічного сульфіту 1,1-циклопропандиметанолу формули,який передбачає:а) взаємодію 1,1-циклопропандиметанолу з діалкілсульфітом в присутності кислоти або основи таb) вилучення з реакційної суміші спиртового побічного продукту реакції.2. Спосіб за п. 1, за яким реакцію проводять в присутності основи.3. Спосіб за...
Спосіб одержання 3-(3-фторо-4-гідроксифеніл)-7-гідроксинафтонітрилу, способи одержання проміжних сполук, а також проміжна сполука

Номер патенту: 84166
Опубліковано: 25.09.2008
Автори: Геуш Моузумі, Равеендранат Паноліл, Сузерленд Карен Вігінз, Ву Янжонг, Рен Жянксін, Левент Махмут
МПК: C07C 39/00, C07C 255/53, C07C 41/00, C07C 37/00, C07C 255/00, C07C 253/30, C07C 255/54
Мітки: способи, також, проміжних, сполук, 3-(3-фторо-4-гідроксифеніл)-7-гідроксинафтонітрилу, одержання, сполука, спосіб, проміжна
Формула / Реферат:
1. Спосіб одержання сполуки формули V, Vде R1 являє собою CN, F або Сl;R2 являє собою Н або Вr;a R3 й R4 незалежно являють собою Н або F, який відрізняється тим, що включає реакцію сполуки формули (II), IIде R1 й R2 - як визначені вище, зі сполукою...
Спосіб одержання 2′,2′-дифторнуклеозиду та його проміжних сполук, а також сполуки, одержані цим способом

Номер патенту: 87637
Опубліковано: 27.07.2009
Автори: Ча Даі-Вон, Чой Жан-Хо, Лім Хонг-Гу, Кім Мун-Санг, Кім Йонг-Жік
МПК: C07D 307/32
Мітки: способом, одержання, проміжних, одержані, цим, сполук, сполуки, 2',2'-дифторнуклеозиду, також, спосіб
Формула / Реферат:
1. Спосіб одержання суміші енантіомерів еритро- і трео-лактонів формули 5, який відрізняється тим, що суміш 3-R та 3-S-енантіомерів та їх захищене похідне алкіл-2,2-дифтор-3-гідрокси-3-(2,2-діалкілдіоксолан-4-іл)пропіонат формули 4 піддають гідролізу в присутності реагентів для гідролізу, вибраних із групи, що включає оцтову кислоту або хлороцтову кислоту, воду та суміш органічних розчинників, вибраних із групи, що включає ацетонітрил,...
Попередній патент: Надульний пристрій зброї
Наступний патент: Установка для одержання, видалення та переміщення високов’язкої суспензії
Випадковий патент: Спосіб прогнозування незрощення перелому