Заміщені карбоксаміди
Номер патенту: 95480
Опубліковано: 10.08.2011
Автори: Фішер Метью Джозеф, Бакер Райан Томас, Кукліш Стівен Лі, Голлінзхед Шон Патрік, Такеучі Куміко, Сміт Едвард К.Р.

















Формула / Реферат
1. Сполука формули І
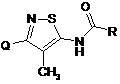 І
І
або фармацевтично прийнятна сіль такої сполуки, де
Q - фенільна група формули QA
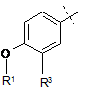 , QA
, QA
де R1 - метил або етил, та R3 - водень або фтор; або
Q - фенільна група формули QB
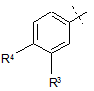 , QB
, QB
де
R3 - водень або фтор, та
R4 - водень, фтор, хлор або бром; або
кожний з R3 та R4 - хлор; або
R3 - водень та R4 - метилтіогрупа або 1,1-дифторетил; та
R-CO - (R-R)-транс-2-метилциклопропанкарбоніл.
2. Сполука за п. 1, де Q - 4-метоксифеніл, 3-фтор-4-метоксифеніл, 4-етоксифеніл, феніл, 4-фторфеніл, 4-хлорфеніл, 4-бромфеніл, 3,4-дихлорфеніл, 4-(метилтіо)феніл або 4-(1,1-дифторетил)феніл.
3. Сполука за п. 1, де Q - QA.
4. Сполука за п. 1, де Q - QB.
5. Сполука за п. 4, де R4 - хлор.
6. Сполука за будь-яким із пп. 1 та 3, 4, де R3 - водень.
7. Сполука за п. 1, яка являє собою (R-R)-N-[3-(4-хлорфеніл)-4-метилізотіазол-5-іл]-2-метилциклопропанкарбоксамід або його фармацевтично прийнятну сіль.
8. Фармацевтична композиція, яка містить сполуку формули І або фармацевтично прийнятну сіль такої сполуки за будь-яким із пп. 1-7 разом з фармацевтично прийнятним розріджувачем, наповнювачем або носієм.
9. Сполука за будь-яким із пп. 1-7 або фармацевтично прийнятна сіль такої сполуки для застосування як лікарського засобу.
10. Сполука формули І або фармацевтично прийнятна сіль такої сполуки за будь-яким із пп. 1-7 для застосування при лікуванні болю.
11. Застосування сполуки формули І або фармацевтично прийнятної солі такої сполуки за будь-яким із пп. 1-7 для виготовлення лікарського засобу для лікування болю.
Текст
1. Сполука формули І O N S R N Q H 2 3 Ця заявка претендує на пріоритет за попередньою заявкою № 60/811839 на патент США, поданою 8 червня 2006 р. Цей винахід стосується певних заміщених карбоксамідів, зокрема, певних N-ацилованих заміщених похідних 5-аміно-4-метилізотіазолу, а також способів їх одержання, фармацевтичних композицій, які містять заміщені карбоксаміди, та способів їх застосування. L-глутамат є основним збуджувальним нейротрансмітером у центральній нервовій системі та зветься збуджувальною амінокислотою. Рецептори глутамату складають два головні підтипи: керовані лігандами іонотропні рецептори іонних каналів та поєднані з G-протеїном метаботропні рецептори із сімома трансмембранними доменами (mGluRs). Метаботропне сімейство включає вісім членів та підрозділяється на три групи за ознаками подібності послідовностей, сигнальної трансдукції та фармакології. Рецептори Групи І (mGluR1 та mGluR5 а також їхні сплайсовані варіанти) позитивно пов'язані з гідролізом інозитфосфату та генерацією внутрішньоклітинного кальцієвого сигналу. Рецептори Групи II (mGluR2 та mGluR3) та рецептори Групи III (mGluR4, mGluR6, mGluR7 та mGluR8) негативно пов'язані з аденілілциклазою та регулюють рівні циклічного аденозинмонофосфату (AMP) шляхом непрямого інгібування активності аденілілциклази. Рецептори Групи І розташовані, головним чином, постсинаптично та посилюють нейронне збудження, тоді як рецептори Групи II та Групи III розташовані, головним чином, пресинаптично та діють як ауторецептори, знижуючи надлишкове вивільнення глутамату. Таким чином, підтипи рецепторів mGlu характеризуються унікальними схемами експресії у центральній нервовій системі, які можуть бути мішенями для нових та селективних засобів. Дивись, наприклад, публікацію Огеллі-Шафрана та Шварца (Augelli-Szafran C.E.; Schwarz R.D. Annual. Reports in Medicinal Chemistry (2003) 38, 21-30), де описані антагоністи mGluR1 як корисні нейропротекторні засоби, досліджені на тваринних моделях мозкового крововиливу, на моделях болю, в тому числі при лігуванні спинномозкового нерва, на моделі болю, індукованого формаліном, та на моделі мігрені. Корисність антагоністів mGluR1 показано також на моделях епілептичних нападів та на моделях заспокоювання. У тканинах, де знайдено рецептори mGluR1, вони можуть бути пов'язані з болями. Глутамат є головним збуджувальним нейротрансмітером, який під час стійких больових станів передає сенсорну інформацію нейронам спинного мозку та ЦНС. Припускається, що клінічні хронічні або стійкі болі принаймні частково залежать від тривалого посилення синаптичної ефективності глутаматергічних входів до соматосенсорних нейронів спинного мозку та супраспінальних чутливих до болю ділянок після інтенсивних периферичних збудників, пошкоджень тканини або нервів. Ця посилена синаптична передача викликає зниження 95480 4 больового порогу, посилення больових реакцій та поширення чутливості до болю на непошкоджені ділянки. Імуноцитохімічні дані показують, що рецептори mGlu1 експресуються у кількох ділянках висхідних глутаматергічних чутливих до болю сигнальних шляхів. Є відомості, що стимуляція рецепторів mGlu1 сприяє посиленню нейронного збудження та швидкій глутаматергічній синаптичній передачі. Тривалі впливи внутрішньоклітинних механізмів передачі сигналів, у яких бере участь стимуляція рецепторів mGlu1, підтримують ці рецептори у стані тривалої центральної сенсибілізації як на рівні спинного мозку, так і на супраспінальному рівні. Таким чином, можна очікувати, що послаблення збудження за допомогою антагоністів рецепторів mGlu1 забезпечить ефективну терапію для лікування стійких больових станів. Відома інша інформація, яка свідчить на користь застосування антагоністів рецепторів mGlu1 для полегшення больових реакцій, індукованих хронічними запальними та незапальними больовими збудниками при поведінкових дослідженнях. Селективний антагоніст рецептора mGlu1 LY456236 може послаблювати больову реакцію при формаліновому випробуванні та механічну алодинію у моделі невропатичного болю із застосуванням лігування спинномозкових нервів L5/L6. Дивись Варти та ін. (Varty G.B., et al., Psychopharmacology (Berl.). (2005), 179, 207-217). Сполуки за цим винаходом є селективними антагоністами метаботропних рецепторів Групи І, зокрема, рецептора mGluR1 (mGluR1), особливо у порівнянні з mGluR2, mGluR3 та mGluR4; вони також можуть бути селективними відносно mGluR5. Як такі, вони є корисними для лікування хворобливих станів, пов'язаних зі згаданими метаботропними рецепторами глутамату, наприклад, болю, зокрема, хронічного болю (або стійкого болю), наприклад, хронічного невропатичного болю, хронічного запального суглобного болю або хронічного незапального неневропатичного (NINN) болю, а також для лікування мігрені або епілепсії. Таким чином, згідно з цим винаходом, пропонується сполука формули І, або фармацевтично прийнятна сіль такої сполуки, де A Q - фенільна група формули Q , 5 1 95480 3 де R - метил або етил, та R - водень або фтор; або B Q - фенільна група формули Q дe 3 4 R - водень або фтор, та R - водень, фтор, 3 4 хлор або бром; або кожний з R та R - хлор; або 3 4 R - водень та R - метилтіогрупа або 1,1дифторетил; та R-CO (R,R)-транс-2метилциклопропанкарбоніл. У значенні, вживаному в цьому описі, вислів "сполука формули І" або вислів "сполука за цим винаходом" охоплює як сполуку, так і фармацевтично прийнятну сіль вказаної сполуки. При застосуванні у цьому описі подані нижче терміни мають такі значення (якщо не зазначено інше): "галоген" (halo) означає фтор, хлор, бром або йод, "алкіл", "алкокси[група]" тощо означають як лінійні, так і розгалужені групи; однак посилання на індивідуальний радикал, наприклад, "пропіл", охоплює тільки лінійний ("нормальний") радикал, а ізомер із розгалуженим ланцюгом, наприклад, "ізопропіл", має окреме позначення. Якщо група R-CO у сполуці за цим винаходом є хіральною, то сполука формули І може бути присутня у суміші з її енантіомером, наприклад, у рацемічній суміші, та/або з будь-яким із цисдіастереомерів. Відповідно до варіанта, якому віддається перевага, сполука за цим винаходом є практично чистим (R,R)-ізомером з енантіомерним надлишком, наприклад, 95% або більше. Як вказано нижче, сполука формули І або її фармацевтично прийнятна сіль може виявляти поліморфізм та/або утворювати сольват із водою або органічним розчинником. Цей винахід охоплює також будь-які такі поліморфні форми, будь-які сольвати або будь-які їх суміші. Конкретними значеннями Q є 4-метоксифеніл, 3-фтор-4-метоксифеніл, 4-етоксифеніл, феніл, 4фторфеніл, 4-хлорфеніл, 4-бромфеніл, 3,4дихлорфеніл, 4-(метилтіо)феніл або 4-(1,1дифторетил)феніл. Однією конкретною сполукою формули І є A сполука, де Q - Q . Іншою конкретною сполукою формули І є споB 4 лука, де Q - Q , і, більш конкретно, де R - хлор. У будь-якій з вищезазначених сполук конкрет3 ним значенням R є водень. B 4 3 Якщо Q - Q , R - хлор та R - водень, то сполукою формули І є (R,R)-N-[3-(4-хлорфеніл)-4метилізотіазол-5-іл]-2-метилциклопропанкарбоксамід (або його фармацевтично прийнятна сіль). Сполукою формули І, якій віддається перевага, є (R,R)-N-[3-(4-метоксифеніл)-4-метилізотіазол5-іл]-2-метилциклопропанкарбоксамід або його фармацевтично прийнятна сіль. 6 Фармацевтично прийнятною сіллю сполуки за цим винаходом є сіль сполуки формули 1 з органічною або неорганічною кислотою, яка має фізіологічно прийнятний аніон. Як ще один аспект цього винаходу пропонується фармацевтична композиція, яка містить сполуку формули І або її фармацевтично прийнятну сіль за будь-яким поданим нижче описом спільно з фармацевтично прийнятним розріджувачем, наповнювачем або носієм. Крім того, пропонується фармацевтична композиція для лікування болю, зокрема, хронічного болю, яка містить як активний інгредієнт сполуку формули І або її фармацевтично прийнятну сіль за будь-яким поданим нижче описом. Фармацевтична композиція, яка містить сполуку формули І, може бути виготовлена звичайним способом, який може включати стадію регулювання розмірів частинок, наприклад, подрібнення до мікронних розмірів або застосування нанодиспергування. Відповідно до варіанта, якому віддається перевага, фармацевтична композиція є композицією, придатною для перорального застосування. Сполуки формули І можна одержати способами, до яких належать відомі у галузі хімії процеси одержання структурно аналогічних сполук, або новими способами, описаними в цьому документі. Нові способи, описані в цьому документі, становлять ще один аспект цього винаходу. Способи одержання сполук формули І або їхніх фармацевтично прийнятних солей та нових проміжних продуктів для одержання сполук формули І є подальшими ознаками цього винаходу та ілюструються поданими нижче методиками, де значення основних радикалів відповідають поданим вище визначенням, якщо не зазначено інше. Таким чином, пропонується спосіб одержання сполуки формули І або її фармацевтично прийнятної солі за будь-яким поданим нижче описом, який включає стадію, вибрану з нижчезазначених стадій: (А) ацилювання аміну формули II, з використанням кислоти формули HOOC-R або її активованого похідного; (B) для одержання сполуки формули І, де Q A Q , алкілування фенольного атома кисню сполуки формули III , 7 95480 3 де R - водень або фтор, з використанням реа1 гента формули R -Y, де Υ - відома відщеплювана група для нуклеофільного заміщення; та (C) метилування відповідної сполуки формули VI , де X - бром або йод; після чого, при роботі за будь-якою з вищезазначених методик, в разі необхідності одержання фармацевтично прийнятної солі сполуки формули І, її виготовляють шляхом проведення реакції основної форми сполуки формули І з кислотою, яка постачає фізіологічно прийнятний протиіон, або будь-яким іншим відомим способом; та де, якщо вище не вказано інше, Q, R-CO та 1 R мають будь-яке з поданих вище значень. Таким чином, за одним аспектом цього винаходу, пропонується сполука, вибрана з групи, яку складають: (а) амін формули II , (b) сполука формули III , 8 алкілефіру або бензилового складного ефіру ацилування можна виконувати з використанням, наприклад, триметилалюмінію або трет-бутилату калію. У значенні, вживаному в цьому описі, відщеплювана група Υ означає групу, яка замінюється у реакції нуклеофільного заміщення, наприклад, галоген (наприклад, бром або йод), групу складного ефіру сульфонової кислоти (наприклад, метилсульфонілоксигрупу, n-толуїлсульфонілоксигрупу або трифторметилсульфонілоксигрупу, більш конкретно, у випадку метилування, метоксисульфонілоксигрупу), або групи, придатні для реакції Міцунобу, наприклад, групу, яка утворюється при обробленні спирту трифенілфосфіном, діетилазокарбоксилатом та триетиламіном. Для метилування сполуки формули VI, де X бром або йод, можна застосувати методику сполучення за Штілле (Stille) з використанням, наприклад, тетраметилолова та каталізатора Штілле, наприклад хлориду біс(трифенілфосфін)паладію (II), у диметилформаміді; або, за альтернативним способом, методику транс-металування - метилування з використанням, наприклад, бутиллітію, а потім метилйодиду у тетрагідрофурані. Кислоту формули HOOC-R можна одержати за відомою методикою. Кислоту формули HOOC-R доцільно одержати за способами, описаними нижче у підготовчих синтезах. Доцільно виконувати розділення кислоти формули HOOC-R у формі солі, за варіантами, яким віддається перевага - у формі солі (R,R)-2-метилциклопропанкарбонової кислоти з (S)-2-аміно-3-феніл-1-пропанолом (1:1), причому більша перевага віддається кристалічній формі, яка є ще одним аспектом цього винаходу. Таким чином, пропонується також описаний вище спосіб, згідно з яким кислоту формули HOOC-R або її активоване похідне одержують за звичайною методикою із солі (R,R)-2метилциклопропанкарбонової кислоти з (S)-2аміно-3-феніл-1-пропанолом (1:1). Амін формули II, 3 де R - водень або фтор, та (c) сполука формули VI , де X - бром або йод, 1 де, якщо не вказано інше, Q, R-CO та R мають будь-яке з поданих вище значень. Щодо кислоти формули HOOC-R, до типових активованих похідних належать складні ефіри (зокрема, нижчі складні алкілефіри, наприклад, складний метиловий або складний етиловий ефір або складний бензиловий ефір), галогенангідриди кислот (зокрема, хлорангідрид) та активовані складні ефіри або ангідриди (в тому числі 4нітрофеніловий складний ефір та активований складний ефір або ангідрид, одержані з реагента сполучення). При застосуванні нижчого складного доцільно одержувати за методикою, описаною нижче. Загалом, бензонітрил формули Q-CN конденсують із пропіонітрилом, і одержують нітрил формули IV, , який перетворюють у тіоамід формули V 9 95480 10 зонітрилу формули Q-CN та ацетонітрилу для одержання нітрилу формули IX , доцільно із застосуванням тіоацетаміду. Окиснення тіоаміду формули V, яке доцільно виконувати із застосуванням пероксиду водню, дає 5аміноізотіазол формули II. Сполуку формули III , який перетворюють у тіоамід формули X , , де R-CO (R,R)-транс-23 метилциклопропанкарбоніл та R - водень або фтор, є іншим аспектом цього винаходу, і його можна одержати шляхом ацилування відповідного амінофенолу формули IIа та піддають окиснювальній циклізації з одержанням аміну формули VIII. За альтернативним варіантом тіоамід формули X можна обробити двома еквівалентами (або дещо більшою кількістю, наприклад, 2,2 екв.) брому для здійснення як замикання циклу, так і бромування з одержанням аміну формули VIlla , , застосовуючи кислоту формули HOOC-R або її активоване похідне. Сполуку формули IIа можна одержати шляхом відщеплення групи захисту кисню від відповідної сполуки, де фенольний атом кисню захищений згаданою групою. Доцільно одержувати сполуку формули IIа шляхом Одеметилування відповідної метоксизаміщеної сполуки формули II. Конкретною сполукою формули III є (R,R)-N-[3-(4-гідроксифеніл)-4-метилізотіазол-5іл]-2-метилциклопропанкарбоксамід. Сполуку формули VI, де X - бром або йод, можна одержати шляхом бромування або йодування відповідної сполуки формули VII . Сполуку формули VII доцільно одержувати шляхом ацилування відповідного аміну формули VIII , із застосуванням кислоти формули HOOC-R або її активованого похідного. Амін формули VIII можна одержати, застосовуючи методику, аналогічну описаній для одержання аміну формули II, але з використанням бен який ацилюють, застосовуючи кислоту формули HOOC-R або її активоване похідне, і одержують відповідну сполуку формули VI, де X - бром. За ще одним альтернативним варіантом, сполуку формули VI, де X - бром, одержують, як описано нижче. Шляхом ацилювання 3,4дибромізотіазол-5-іламіну формули XI , застосовуючи кислоту формули HOOC-R або її активоване похідне, одержують відповідну сполуку формули XII , яку піддають перехресному сполученню з борною кислотою формули Q-B(OH)2, і одержують сполуку формули VI, де X - бром. Відносну ефективність та селективність сполук за цим винаходом стосовно до рецепторів mGlu1 оцінюють, застосовуючи клоновані лінії клітин, які експресують рецептори mGlu1, mGlu2, mGlu3, mGlu4, mGlu5 або mGlu8, трансфіковані у лінії клітин AV12, які містять клітини транспортера глутамату ЕААТ1 пацюка (RGT). Наприклад, більш детально, сполуки за цим винаходом оцінюють за ефектами індукованих глутаматом реакцій кальцієвого потоку, застосовуючи лінію клітин AV-12, які експресують реком 11 бінантний протеїн рецептора mGlu1a людини (дивись Кінгстон та ін. - Kingston et al. Neuropharmacology. 37(1): 1-12, 1998). Опосередковані рецептором mGlu1 реакції визначають за змінами внутрішньоклітинних концентрацій кальцію, які вимірюють за допомогою чутливого до кальцію флуоресцентного барвника Fluo-3. Клітини збирають та висівають у 96-лункові мікротитрувальні планшети. Після 48 год інкубації у вологій атмосфері при 37°С клітини обробляють 10 мкМ розчином барвника Fluo-3 AM протягом 60 хв при 25°С. Незв'язаний позаклітинний барвник видаляють із лунок шляхом промивання буферним розчином, після чого планшети переносять у 96канальний планшетний сканер з флуориметричною візуалізацією (FLIPR - Molecular Devices Corporation, La Jolla, CA, USA). Сканують фонову флуоресценцію протягом 10 с, після чого додають випробовувані сполуки за допомогою автоматичного піпетувального пристрою, який є складовою частиною приладу FLIPR. Після 20 с витримування додають у лунки глутамат у концентрації ЕС90% (10 мкМ), та спостерігають зміни флуоресценції протягом 60 с. Інгібувальні ефекти сполук визначають шляхом порівняння пікового значення флуоресцентної реакції на глутамат у присутності та за відсутності випробовуваної сполуки. Значення ІС50 обчислюють, застосовуючи 4-параметрову логістичну програму апроксимації кривих (пакет програм GraphPad Prism V4 або Activity Base V5,3). Сполуки, описані у поданих прикладах, виявляють значення ІС50 нижче від 100 нМ. Наприклад, кожна зі сполук, описаних у Прикладі 1 та Прикладі 6, при випробуванні за описаною вище методикою має ІС50 нижче від 20 нМ. Аналогічне випробування із застосуванням рецептора mGlu! пацюка можна виконувати для подальшого визначення характеристик сполуки у сполученні зі скринінговими дослідженнями in vivo, виконуваними на пацюках. Сполуки за цим винаходом продемонстрували активність in vivo у моделях болю із застосуванням пацюків. Ці добре відомі моделі болю доцільно реалізувати, як коротко описано нижче. Наприклад, у дозах, при яких у випробуванні на обертовому стрижні (rotorod), яке можна виконувати, як описано нижче, не спостерігаються порушення поведінки, сполука за Прикладом 1 виявляє дозозалежну активність у голодних самців пацюків лінії Harlan Sprague Dawley (HSD, маса тіла 200-250 г) у моделях із застосуванням формаліну, карагінану, капсаїцину та подразнення хвоста, а також у неголодних самців пацюків HSD (маса тіла 300350 г) у моделі із застосуванням лігування спинномозкових нервів L5/L6. Усі дані аналізують методами аналізу дисперсії (ANOVA) та t-тестування за Даннетом (Dunnett) із використанням статистичного програмного забезпечення JMPv4.1 (SAS Institute Inc., Сагу, NC), якщо не вказано інше. Дані виражають як середні значення ± СKВ. Конкретні умови для кожної моделі коротко описано нижче. Формалінова модель: Лікарський засіб вводять тваринам перед введенням формаліну (50 мкл, 5%), впорскуваного підшкірно у задню бічну частину правої задньої кінцівки. В рамках автоматизо 95480 12 ваного випробування визначають характеристику больової реакції, що виражається через кількість випадків лизання кінцівки за період від 0 хв до 50 хв після введення формаліну. Дані виражають як кількість випадків лизання кінцівки на ранній фазі (0-5 хв) або у пізній фазі (15-40 хв). У цій моделі сполука за Прикладом 1 виявляє дозозалежне послаблення больової реакції пізньої фази у голодних пацюків при пероральному застосуванні у дозах від 3 мг/кг до 60 мг/кг за 1 год до введення формаліну, а сполука за Прикладом 6 виявляє дозозалежне послаблення при застосуванні у дозах від 1 мг/кг до 30 мг/кг. Накладають тугу лігатуру на спинномозкові нерви L5 та L6 (тільки з одного боку). Через два тижні визначають механічну алодинію із застосуванням волокон фон-Фрея при зростаючому зусиллі згинання (0,5-15 г) у різні моменти часу після введення лікарського засобу. Карагінанова модель: У підошву правої кінцівки впорскують карагінан (100 мкл, 3%), а через 2 год після введення карагінану застосовують лікарський засіб. Визначають термічну гіпералгезію із застосуванням джерела променистого тепла протягом 30 с для запобігання пошкодженню тканин. Вимірюють затримку реакції відсмикування кінцівки як різницю між реакцією нагріваної кінцівки та ненагріваної лівої кінцівки. Капсаїцинова модель: Лікарський засіб застосовують до введення капсаїцину. Капсаїцин (25 мкл, 30 мкг) в оливковій олії впорскують у підошву правої кінцівки. Визначають механічну алодинію із застосуванням волокон фон-Фрея (Frey) при зростаючому зусиллі згинання (0,5-15 г) через 15 хв та 1 год після введення капсаїцину. Модель подразнення хвоста: Для збудження ударної реакції хвоста застосовують джерело променистого тепла протягом 10 с у різні моменти часу після застосування лікарського засобу. Випробування на приладі Rotorod: Для цього випробування використовують самців пацюків лінії Sprague Dawley (маса тіла 180-230 г, Harlan labs, Indianapolis). Здатність антагоністів mGlu1 індукувати седативний ефект та атаксію випробовують, застосовуючи автоматизований прискорюваний прилад Rotorod (Omnitech Electronics Inc., Columbus, OH), з'єднаний з комп'ютером IBM PC, як описано раніше (Сіммонс та ін. - Simmons et al., Neuropharmacol. (1998) 37, 25-36). Випробування на приладі Rotorod виконують перед застосуванням лікарського засобу та повторно через 1 год, 2 год, 3 год та 4 год після перорального введення, наприклад, 60 мг/кг лікарського засобу голодним пацюкам. Вибрані моменти часу відповідають поведінковим випробуванням на моделях болю. Тварини, які зберігають позу та не падають з ротороду, дають максимальний показник 40 с. Одержані дані аналізують методами аналізу дисперсії (ANOVA) та t-тестування за Даннетом (Dunnett) із використанням статистичного програмного забезпечення JMPv4,1 (SAS Institute Inc., Сагу, NC). Дані виражають як середні значення ± СKВ. Таким чином, слід очікувати, що сполуки за цим винаходом можуть бути корисними у випадках, коли показаним є антагонізм до рецепторів 13 mGlu1. Зокрема, можна очікувати, що сполуки за цим винаходом можуть бути корисними для лікування болю, зокрема, хронічного болю (або стійкого болю), наприклад, хронічного невропатичного болю, хронічного запального болю або суглобного болю, або хронічного болю незапального та неневропатичного походження (NINN), а також для лікування мігрені та епілепсії. Відповідно, одним із конкретних аспектів цього винаходу є лікування хронічного невропатичного болю; іншим конкретним аспектом цього винаходу є лікування хронічного запального болю або суглобного болю; та подальшим конкретним аспектом цього винаходу є лікування хронічного болю незапального та неневропатичного походження. Невропатичний біль охоплює біль, пов'язаний з діабетичною периферичною невропатією та постгерпетичною невралгією. Крім того, сполуки за цим винаходом можуть бути корисними як засоби лікування епілептичних нападів або як засоби для лікування станів неспокою, страху або тривоги, а також як нейропротекторні засоби після мозкових крововиливів. Таким чином, як ще один аспект цього винаходу пропонується спосіб лікування болю, зокрема, хронічного болю, у ссавців, зокрема, у людей, які потребують такого лікування, який включає введення в організм ссавця ефективної кількості сполуки формули І або її фармацевтично прийнятної солі, де Q та R мають будь-яке з вищевказаних значень. Ссавцем, який потребує лікування, може бути також свійська тварина, наприклад, кінь, або хатня тварина, наприклад, кіт або собака. Пропонується також сполука формули І за будь-яким поданим у цьому описі визначенням або її фармацевтично прийнятна сіль для застосування як лікарський засіб. Крім того, пропонується сполука формули І або її фармацевтично прийнятна сіль, де Q та R мають будь-яке з вищевказаних значень, для застосування при лікуванні болю, зокрема, хронічного болю. Далі, пропонується застосування сполуки формули І або її фармацевтично прийнятної солі, де Q та R мають будь-яке з вищевказаних значень, для виготовлення лікарського засобу для лікування болю, зокрема, хронічного болю. У будь-якому з вищезазначених тверджень конкретною формою хронічного болю є невропатичний біль. Іншою конкретною формою хронічного болю є хронічний запальний або суглобний біль. Ще однією конкретною формою хронічного болю є хронічний біль незапального та неневропатичного походження (NINN). Конкретна доза сполуки за цим винаходом, яка має бути введена в організм пацієнта, звичайно, визначається обставинами конкретного випадку, в тому числі, наприклад, застосовуваною сполукою, режимом застосування та станом, який підлягає лікуванню. Типова добова доза для лікування хронічного болю може становити 1-300 мг на добу, більш конкретно 5-200 мг на добу, яка застосовується у формі одиничної дози або двох чи більше часткових доз, у варіанті, якому віддається перевага, шляхом перорального введення. Таким чином, сполуки за цим винаходом можна застосову 95480 14 вати при лікуванні наявного болю або при профілактичному лікуванні. У поданих нижче методиках підготовчих синтезів та прикладах застосовуються такі позначення та абревіатури: R-CO - (R,R)-транс-2-иетилциклопропанкарбоніл; DMF (ДМФ) - диметилформамід; DMSO (ДМСО) - диметилсульфоксид (для ЯМР дейтерований [-d6]); екв. - еквівалент (еквіваленти); ES-MS - мас-спектр з іонізацією електророзпиленням; EtOAc - етилацетат; FID (ЦІП) - детектування з іонізацією в полум'ї; GC (ГХ) - газова хроматографія; HPLC (РХВЕ) - рідинна хроматографія високої ефективності (під високим тиском); LCMS (РХ-МС) - рідинна хроматографія, поєднана з мас-спектром; МеОН - метанол; МТВЕ - метилтрет-бутиловий простий ефір; ЯМР (ЯМР) - спектроскопія (або спектр) ядерного магнітного резонансу; TEA - триетиламін; TFA - трифтороцтова кислота; THF (ТГФ) - тетрагідрофуран; TLC (ТШХ) хроматографія в тонкому шарі; UV (УФ) - ультрафіолетовий (детектор); са. - приблизно; ее - енантіомерний надлишок; Ph - феніл; нас. - насичений. Реагенти одержували з різних ринкових джерел. Розчинники, як правило, видаляли (випарювали) під зниженим тиском. У деяких методиках підготовчих синтезів значення виходу є типовими значеннями виходу неочищених продуктів, виділених шляхом випарювання або фільтрування та використаних безпосередньо без додаткового очищення. Одержання бензил-(R,R)-2метилциклопропанкарбоксилату Готують 2,0 Μ розчин оксалілхлориду в дихлорметані шляхом додання при перемішуванні 98% оксалілхлориду (110,0 мл) до безводного дихлорметану (600,0 мл). Одержаний розчин оксалілхлориду (1,20 моль) додають краплями на протязі 1 год при перемішуванні до розчину 2метилциклопропанкарбонової кислоти (наявного на ринку продукту, який являє собою суміш цис- та транс-ізомерів, 120,0 г, 1,20 моль) у толуолі (800,0 мл), який містить DMF (0,6 мл, 7,8 ммоль). Суміш перемішують 2 год при кімнатній температурі; потім її додають краплями протягом 1,5 год до суміші бензилового спирту (114,0 мл, 1,10 моль), безводного THF (800,0 мл) та піридину (194,0 мл, 2,41 моль). Перемішують суміш після додання ще протягом 1 год, розподіляють між етилацетатом (2 л) та 10% водним розчином карбонату калію (2 л); органічну фазу промивають (10% водним розчином карбонату калію (2 л) та розсолом (2 л)), сушать (MgSO4), та випарюють, одержуючи рідку речовину. Після хроматографування на силікагелі з елююванням 5% етилацетату в гексані одержують як основний продукт рацемічний бензилтранс-2-метил-циклопропанкарбоксилат у вигляді прозорої безбарвної рідини (193,8 г, 93%). 1 H ЯМР (DMSO-d6) 7,38 (m, 5Н), 5,05 (s, 2Н), 1,42 (m, 1Н), 1,30 (m, 1H), 1,06 (d, J=6,0 Гц, 3H), 1,04 (m, 1H), 0,74 (m, 1H). Рацемічний складний ефір (189 г) розділяють хіральною РХВЕ. Умови препаративного розділення: метод рециркуляції в усталеному режимі із застосуванням колонки Chiralcel OJ, 832 см; елюент: суміш ізопропанол/гептан, 10/90; швидкість 15 потоку: 375 мл/хв; детектування: УФ на довжині хвилі 220 нм; тривалість циклу приблизно 7,1 хв; дозування: приблизно 21 мл розчину в елюенті (0,5 г речовини) на одну дозу при концентрації 0,025 мг/мл. Одержують бензил-(R,R)-транс-2метилциклопропанкарбоксилат (73 г) з ее понад 99,7% за даними хіральної РХВЕ. Умови аналізу: Chiralcel OJ, 4,6250 мм; елюент: суміш ізопропанол/гептан, 10/90; швидкість потоку 1,0 мл/хв; детектування: УФ на довжині хвилі 220 нм; час затримання 6,5 хв та 7,2 хв. Альтернативна методика одержання бензил(R,R)-2-метилциклопропан-карбоксилату Суміш 2-метилциклопропанкарбонової кислоти (наявного на ринку продукту, який являє собою суміш цис- та транс-ізомерів, 75,0 г, 0,75 моль) та 1-н. розчин NaOH (900 мл, 0,90 моль) нагрівають та перемішують при 45°С. До розчину додають при перемішуванні бензилбромід (131,0 мл, 1,10 моль) та хлорид метилтриалкіл(С8-С10)амонію (Adogen 464, 37,5г). Суміш перемішують при 40-45°С протягом 4 год, охолоджують до кімнатної температури та екстрагують діетиловим ефіром (800 мл). Органічний шар сушать (MgSO4), та випарюють, одержуючи рідку речовину. Після хроматографування на силікагелі з елююванням гексаном при поступовому збільшенні вмісту етилацетату в елюенті до 5% одержують рацемічний бензилтранс-2-метилциклопропанкарбоксилат у вигляді 1 рідини (132,2 г, 93%); H ЯМР (DMSO-d6) 7,40 (m, 5Η), 5,05 (s, 2Н), 1,43 (m, 1Н), 1,30 (m, 1Н), 1,06 (d, J=6,0 Гц, 3Н), 1,02 (m, 2H), 0,74 (m, 1H). [Примітка: якщо ЯМР виявляє присутність незначної кількості цис-ізомеру, то його можна видалити шляхом розділення хіральною РХВЕ, яку можна виконувати у вищезазначених умовах]. Одержання (R,R)-2метилциклопропанкарбонової кислоти, описаної також під назвою (R,R)-транс-2метилциклопропанкарбонової кислоти А. Одержання рацемічної 2метилциклопропанкарбонової кислоти і. Метилід диметилоксосульфонію (розчин у DMSO): До суспензії йодиду триметилоксосульфонію (2,47 кг, 1,05 екв.) у DMSO (8,00 л) додають при перемішуванні в атмосфері азоту гідроксид калію (90% мас., 0,69 кг, 1,05 екв.) дозами по 100 г. (За альтернативною методикою, можна додавати одразу всю кількість, в такому разі вивільнюється тепло). Додають ще DMSO (4,00 л), і реакційну суміш перемішують при температурі навколишнього середовища до одержання однорідної суміші (за винятком певної кількості нерозчинених гранул KOH, які не додають на наступній стадії) та завершення утворення іліду (приблизно2-2,5 год). іі. Етил-2-метилциклопропанкарбоксилат До розчину етил-транс-кротонату (1,20 кг, 1,31 л, 1,00 екв.) у DMSO (3,00 л) додають при температурі навколишнього середовища протягом 30 хв розчин іліду, одержаний, як описано вище, підтримуючи при цьому температуру реакційної суміші в межах приблизно 15-20°С. Хід реакції контролюють шляхом газохроматографічного (ГХ) аналізу в умовах, вказаних нижче, до виявлення лише не 95480 16 значної залишкової кількості кротонату відносно 2метилциклопропанкарбоксилату (приблизно 20-24 год). Реакційну суміш розділяють для подальшого оброблення на дві рівні частини (по 8,5 л). Кожну частину обробляють, як описано нижче. Додають метил-трет-бутиловий простий ефір (МТВЕ, 6 л), і двофазову суміш охолоджують до 15°С, після чого додають краплями воду (6 л) протягом приблизно 45 хв, підтримуючи температуру нижче 23°С. Після розділення фаз органічну фазу двічі промивають 10% розсолом, і розчинник обережно видаляють у вакуумі (400 мбар (0,04 МПа), температура бані 35°С), одержуючи етил-2-метилциклопропанкарбоксилат (1,00 кг, 26,8%), який містить приблизно 3,3 екв. МТВЕ. Умови ГХ аналізу: колонка Varian VF-lms, довжина 60 м, діаметр 320 мкм; товщина шару стаціонарної фази 1 мкм; газ-носій: гелій; температура: від 80°С до 300°С протягом 35°хв, тривалість випробування 35 хв детектор: ДІП; проба безпосередньо розведена у метанолі. ііі. 2-метилциклопропанкарбонова кислота Описану вище суміш, яка містить етил-2метилциклопропанкарбоксилат (1,00 кг, 1,00 моль, 1,00 екв.), змішують із водою (4,00 л) та 10,4 Μ розчином гідроксиду натрію (0,32 л, 1,20 екв), і одержану суміш нагрівають при 46°С, причому МТВЕ поступово дистилюється. (Якщо ГХ аналіз виявляє присутність складного ефіру у фракції дистиляту МТВЕ, то її повертають у реакційну суміш і знов дистилюють МТВЕ.) Коли ГХ аналіз перестає виявляти залишковий складний ефір у реакційній суміші (приблизно через 1-4 год), її охолоджують до 20°С, додають дистилят та додаткову кількість МТВЕ (2 л), і розділяють шари. Водну фазу підкислюють 12,18 Μ хлористоводневою кислотою та екстрагують МТВЕ (34 л). МТВЕ обережно видаляють з об'єднаних органічних екстрактів у вакуумі (наприклад, під тиском 400 мбар (0,04 МПа), потім 200 мбар (0,02 МПа), температура бані 35°С), і одержують рацемічну 2метилциклопропанкарбонову кислоту з домішкою незначної кількості залишкового МТВЕ, яку безпосередньо використовують для розділення. (Аналіз типового продукту ГХ та ЯМР посвідчує вихід 0,99 екв. рацемічної 2-метилциклопропанкарбонової кислоти з домішкою 1,7% цис-ізомеру та 0,3 екв. МТВЕ). В. Одержання солі (R,R)-2метилциклопропанкарбонової кислоти з (S)-2аміно-3-феніл-1-пропанолом (1:1), описаної також як сіль (R,R)-2-метил-циклопропанкарбонової кислоти з (S)-фенілаланінолом (1:1) Рацемічну транс-2метилциклопропанкарбонову кислоту (20 г, 0,2 моль) розчиняють в етилацетаті (200 мл). Додають однією порцією (S)-2-аміно-3-феніл-1-пропанол [відомий також як (S)-фенілаланінол] (15,6 г, 0,103 моль, 0,51 екв.), і суміш нагрівають до 65-70°С. Після кристалізації, якій можна сприяти затравлюванням, суспензію перемішують при кімнатній температурі 20 год, потім фільтрують, і кристали промивають етилацетатом (215 мл). Кристали сушать при 40°С у вакуумі протягом 3 год. Маса кристалів 18,4 г (молярний вихід 37%, енантіомер 17 ний склад за даними хіральної ГХ 85/15, методика хіральної ГХ описана нижче). Кристали повторно суспендують у 370 мл етилацетату; суспензію нагрівають зі зворотним холодильником протягом 1 год, охолоджують до кімнатної температури протягом ночі, кристали відфільтровують, промивають та сушать, як вказано вище. Маса кристалів 16,7 г (вихід 91%, хіральний склад 96/4). Після другого очищення в етилацетаті (170 мл) за описаною вище методикою одержують сіль (R,R)-2метилциклопропанкарбонової кислоти з (S)-2аміно-3-феніл-1-пропанолом (1:1) (16,12 г, вихід 96,5%, хіральний склад 99/1=98% ее; загальний вихід в розрахунку на рацемічну транс-2-метилциклопропанкарбонову кислоту 32%). 1 H ЯМР (400 МГц, DMSO): 0,47 (m, 1Н), 0,84 (m, 1H), 1,01 (d, 3H), 1,07 (m, 2H), 2,5 (m, 1H), 2,7 (m, 1H), 3,0 (m, 1H), 3,25 (dd, 1H), 3,35 (d, 2H), 5,05,2 (br, 4H), 7,2 (m, 3H), 7,3 (dd, 2H); т.пл. солі (98% ее) 130-131°С. Методика хіральної ГХ: колонка Hydrodex BPM; газ-носій гелій; температура випарника: 200°С; тиск: 30 фунтів на кв. дюйм (900 кПа); відношення поділу проби 1/100; детектор ДІП, 230°С; швидкість потоку 50 мл/хв; об'єм проби 1 мкл; початкова температура 130°С. Час затримання (R,R)-енантіомеру 8,3 хв (для S,S- енантіомеру 8,08 хв). Підготовка проби: сіль (приблизно 10 мг) розчиняють в 1-н. HCl (приблизно 1 мл), і вільну кислоту екстрагують етилацетатом (приблизно 1 мл). Екстракт в етилацетаті безпосередньо вводять у газовий хроматограф. С. Одержання (R,R)-2метилциклопропанкарбонової кислоти До солі (R,R)-2-метилциклопропанкарбонової кислоти з (S)-2-аміно-3-феніл-1-пропанолом (1:1) (12,6 г, 0,05 моль) додають 1-н. водну HCl (100 мл, 0,1 моль). Після перемішування протягом 10 хв розчин екстрагують етилацетатом (250 мл). Органічні екстракти сушать (MgSO4) та концентрують у вакуумі (40-45°С/200 мбар), і одержують (R,R)-)2-метилциклопропанкарбонову кислоту (5 г, 100%) у вигляді безбарвного масла. 1 H ЯМР (400 МГц, CDCl3): 0,75 (m, 1Н); 1,1 (d, 3H); 1,2 (m, 1H); 1,3 (m, 1H); 1,42 (m, 1H); 11,0 (br, 1H). Прискорена методика одержання (R,R)-2метилциклопропанкарбонової кислоти для одержання солі (R,R)-2-метилциклопропанкарбонової кислоти з (S)-2-аміно-3-феніл-1-пропанолом (1:1) і. Металід диметилоксосульфонію (розчин у DMSO): До суспензії йодиду триметилоксосульфонію (1,18 екв.) у DMSO (приблизно 3,3 мл на 1 г йодиду) додають однією порцією при перемішуванні в атмосфері азоту трет-бутилат калію (1,05 екв.). Реакція протікає з вивільненням тепла. Реакційну суміш перемішують при 20-35°С до утворення однорідної суміші та завершення утворення іліду. іі. Етил-2-метилциклопропанкарбоксилат: Розчин етил-транс-кротонату (1,00 екв.) у DMSO (3 мл на 1 г складного ефіру) нагрівають до 80°С. До цього розчину повільно додають розчин іліду, одержаний, як описано вище, підтримуючи 95480 18 при цьому температуру реакційної суміші 80°С. По збіганні типового часу додавання та протікання реакції (1 год) газохроматографічний аналіз (в умовах, вказаних вище) посвідчує наявність лише незначної залишкової кількості кротонату відносно 2-метилциклопропанкарбоксилату. ііі. 2-метилциклопропанкарбонова кислота Вищезазначену реакційну суміш охолоджують до 20°С, і додають до неї водний розчин KOH (5% (мас.), приблизно 1,14 екв. KOH), протягом 15 хв, підтримуючи температуру реакційної суміші 2030°С. Перемішують реакційну суміш ще протягом 2-3 год (до відсутності залишкового складного ефіру за даними ГХ аналізу у вищезазначених умовах). Одержаний розчин (рН приблизно 12 за індикаторним папером на рН) підкислюють до рН 2-3 при 20-30°С шляхом повільного додавання 1,5н. HCl; потім екстрагують трьома порціями ізопропілацетату (кожна порція по 5 мл на 1 г вихідного етил-транс-кротонату). Об'єднані органічні фази промивають 15% розсолом і частково випарюють шляхом дистиляції у вакуумі 100-300 мбар (0,010,03 МПа) при температурі бані 45°С (з повторним розведенням ізопропілацетатом в разі необхідності), і одержують розчин у кількості приблизно 10 мл на 1 г розрахункової кількості 2метилциклопропанкарбонової кислоти, який піддають розділенню за методикою, аналогічною описаній вище. Альтернативна методика одержання (R,R)метилцииклопропанкарбонової кислоти В атмосфері азоту гексиллітій (2,3 Μ у гексані, 8 мл, 18,4 ммоль) додають краплями протягом 20 хв до триетилфосфоноацетату (4,5 г, 19,67 ммоль) у безводному 2-метилтетрагідрофурані (40 мл), підтримуючи температуру в межах від 19°С до 25°С. Через 30 хв додають (S)-оксид пропілену (1,17 г, 20,15 ммоль), і суміш переносять у реактор з нержавіючої сталі (реактор Парра) місткістю 160 мл. Суміш нагрівають до 150°С протягом 15 хв та перемішують при цій температурі протягом 16 год. (ЯМР аналіз неочищеноїсуміші посвідчує ступінь перетворення в етил-(R,R)-2-метилциклопропанкарбоксилат >95%). Додають до суміші воду (50 мл) та 30% водний розчин NaOH (25 мл), і двофазову суміш перемішують при нагріванні зі зворотним холодильником протягом 5 год. Розділяють шари, і органічну фазу відкидають. До водного шару додають 37% водний розчин HCl (25 мл), і екстрагують суміш ізопропілацетатом (250 мл). Органічний шар, який містить (R,R)-2-метилциклопропанкарбонову кислоту, промивають 10% водним розчином NaCl (325 мл) і частково випарюють у вакуумі до загальної маси 14,5 г, після чого додають однією порцією (S)-2аміно-3-феніл-1-пропанол [відомий також під назвою (S)-фенілаланінолу] (3,01 г, 19,91 ммоль), при цьому спонтанно кристалізується сіль (R,R)-2метилциклопропанкарбонової кислоти з (S)-2аміно-3-феніл-1-пропанолом (1:1). Одержану суспензію перемішують протягом ночі. Кристали відділяють фільтруванням, промивають ізопропілацетатом (4 мл) і сушать у вакуумі при 40°С, одержуючи сіль (R,R)-2метилциклопропанкарбонової кислоти з (S)-2 19 аміно-3-феніл-1-пропанолом (1:1) (3,4 г, загальний вихід 69%). Аналіз методом хіральної ГХ: ее >99,5%, діасетреомерний надлишок (de) 98%. (За альтернативним варіантом, кислоту можна легко виділяти у формі солі з дициклогексиламіном (1:1)). Сіль можна перетворити у (R,R)-2метилциклопропанкарбонову кислоту за методикою, аналогічною описаній вище. Одержання нітрилу формули IV Якщо не описано інше, нітрил формули IV, який має вказане значення Q, одержують, застосовуючи відповідний бензонітрил формули Q-CN та пропіонітрил і методику, аналогічну описаній нижче для Підготовчого синтезу IV-1. Підготовчий синтез IV-1, Q=4-метоксифеніл Змішують 4-метоксибензонітрил (50,0 г, 376 ммоль), трет-бутилат калію (84,2 г, 752 ммоль), пропіонітрил (62,0 г, 1,130 ммоль) та толуол (1,880 мл); перемішують суміш протягом 72 год. Розводять насиченим розчином NaHCO3 та екстрагують EtOAc. Випарюють органічний розчин, та кристалізують із суміші гексан/EtOAc, і одержують 3-аміно3-(4-метоксифеніл)-2-метилакрилонітрил. Вихід 35,1%. ES-MS: m/е 189,2 (m+1). Підготовчий синтез IV-6, Q=4-хлорфеніл У круглодонній колбі місткістю 2 л (обладнаній гумовою мембраною, стрижневою мішалкою та пристроєм для створення атмосфери азоту) змішують 4-хлорбензонітрил (60,0 г, 1,00 екв., 432 ммоль), пропіонітрил (61,2 мл, 2,00 екв., 864 ммоль), тетрагідрофуран (43,2 мл) та 1,0 Μ розчин трет-бутилату калію у трет-бутанолі (калієве похідне трет-бутилового спирту, 475 мл, 1,10 екв.; 475 ммоль); і перемішують протягом 24 год. Гасять реакцію водним розчином NaHCO3, та екстрагують етилацетатом. Промивають органічну фазу двічі розсолом, сушать (K2СО3), фільтрують, і концентрують досуха. Очищають флеш-хроматографією на діоксиді кремнію при елююванні сумішами етилацетату з гексаном (від 15:85 до 50:50), і одержують 3-аміно-3-(4-хлорфеніл)-2метилакрилонітрил (у вигляді суміші ізомерів E/Z невизначеного складу). Вихід 49,5%. LCMS: 193,0 (m+1). Підготовчий синтез IV-7, Q=4-бромфеніл: 3аміно-3-(4-бромфеніл)-2-метилакрилонітрил Одержання тіоаміду формули V Якщо не описано інше, тіоамід формули V, який має вказане значення Q, одержують, застосовуючи відповідний нітрил формули IV і методи 95480 20 ку, аналогічну описаній нижче для Підготовчого синтезу V-1. Підготовчий синтез V-1, Q=4-метоксифеніл Додають HCl (4-н. у діоксані, 659 мл, 2,640 ммоль) до розчину 3-аміно-3-(4-метоксифеніл)-2метилакрилонітрилу (24,8 г, 132 ммоль), тіоацетаміду (19,8 г, 264 ммоль) та діоксану (132 мл) в атмосфері сухого азоту. Перемішують суміш протягом 2 год. Випарюють, розводять твердий залишок діоксаном (20 мл), і додають безводний TEA (40 мл). Додають насичений розчин K2СО3, та екстрагують EtOAc. Випарюють органічний розчин, та кристалізують із суміші СНСІ3/гексан (95/5); потім перекристалізують із суміші МеОН/Н2О для видалення надлишку тіоацетаміду, і одержують 3аміно-3-(4-метоксифеніл)-2-метилтіоакриламід. Вихід 90,8%. Підготовчий синтез V-6, Q=4-хлорфеніл Додають хлороводень (4 Μ розчин у 1,4діоксані, 858 мл, 16,0 екв.; 3,43 моль) до суміші E/Z-ізомерів 3-аміно-3-(4-хлорфеніл)-2метилакрилонітрилу (41,3 г, 1,00 екв., 214,4 ммоль) та тіоацетаміду (32,71г, 2,00 екв.; 428,8 ммоль) у круглодонній колбі місткістю 2 л (обладнаній гумовою мембраною, стрижневою мішалкою, пристроєм для створення атмосфери азоту та охолоджувальною банею). Підтримують температуру розчину нижче 30°С за допомогою льодяної бані до завершення додавання. Відстороняють льодяну баню, перемішують суміш протягом 2 год та повільно додають її до 1,5 л 30% водного ΝΗ4ΟΗ на льодяній бані при перемішуванні. Екстрагують суміш двічі етилацетатом. Промивають органічну фазу двічі розсолом, сушать (K2СО3), фільтрують, і концентрують досуха. Кристалізують залишок із суміші гексану з хлороформом (10/90). Розтирають одержану тверду речовину з етанольно-водною сумішшю (10/90), і одержують 3-аміно3-(4-хлорфеніл)-2-метилтіоакриламід (у вигляді суміші ізомерів Ε/Ζ невизначеного складу). Вихід 82,3%. 1 H ЯМР (CD3OD) 9,95 (s, 2Н), 8,74 (s, 1Н), 8,29 (d, J=6,0 Гц, 2H), 8,25 (s, 1Н), 8,11 (d, J=6,0 Гц, 2H), 4,099 (s, 3Н). Підготовчий синтез V-7, Q=4-бромфеніл: 3аміно-3-(4-бромфеніл)-2-метил-тіоакриламід. Одержання аміну формули II Якщо не описано інше, амін формули II, який має вказане значення Q, одержують, застосовуючи відповідний тіоамід формули V і методику, аналогічну описаній нижче для Підготовчого синтезу II-1. Підготовчий синтез II-1, Q=4-метоксифеніл Додають пероксид водню (30% мас., 2,39 г, 70,3 ммоль) до розчину 3-аміно-3-(4метоксифеніл)-2-метил-тіоакриламіду (7,800 г, 35,135 ммоль) у метанолі (703 мл). Перемішують протягом 3 год. Гасять реакцію Na2S2O3 (20% у 21 95480 воді), та випарюють суміш до об'єму 10 мл. Розводять EtOAc (500 мл), і промивають органічну фазу розсолом (3100 мл), випарюють та кристалізують із суміші EtOAc/гексан, одержуючи 3-(4метоксифеніл)-4-метилізотіазол-5-іл-амін. Вихід 88,2%. ES-MS: m/e 221,0 (m+1). Підготовчий синтез ІІ-6, Q=4-хлорфеніл У круглодонній колбі місткістю 2 л (обладнаній стрижневою мішалкою) змішують (Е/Z)-3-аміно-3(4-хлорфеніл)-2-метил-тіоакриламід (40,0 г, 1,00 екв., 176 ммоль), метанол (882 мл, 21,80 моль), пероксид водню (30% мас., 14,2 мл, 1,40 екв., 247 ммоль). Перемішують протягом 2 год, після чого гасять реакцію Na2S2O3 (20% у воді), та розводять суміш водою (1 л). Концентрують водну суміш у вакуумі до об'єму 1 л. Додають суміш гексану з етилацетатом (95/5) (500 мл) і інтенсивно перемішують протягом 20 хв. Фільтрують, промивають водою, потім гексаном, і сушать одержаний продукт у вакуумі, одержуючи 3-(4-хлорфеніл)-4метилізотіазол-5-іл-амін. Вихід 64,1%. LCMS: 225,0 (m+1). Підготовчий синтез ІІ-7, Q=4-бромфеніл: 3-(4бромфеніл)-4-метилізотіазол-5-іл-амін. ES-MS: m/e 257,0 (m+1). Одержання фенолу формули III 3 Фенол формули III, де R має вказане значення та R-CO є (R,R)-2-метилциклопропанкарбоніл, одержують, застосовуючи відповідну сполуку фо1 рмули II, де R - метил, і методику, аналогічну описаній нижче для Підготовчого синтезу III-1. 3 Підготовчий синтез III-1, R =H: (R,R)-N-[3-(4гідроксифеніл)-4-метилізотіазол-5-іл]-2метилциклопропанкарбоксамід А. 4-(5-аміно-4-метилізотіазол-3-іл)фенол Додають трибромід бору (0,227 г, 0,909 ммоль) до розчину 3-(4-метокси-феніл)-4метилізотіазол-5-іламіну (0,100 г, 0,455 ммоль) у дихлорметані (5 мл) при -20°С. Перемішують суміш і дають нагрітися до кімнатної температури. Перемішують суміш протягом 4 год, і гасять реакцію 1-н. HCl. Екстрагують EtOAc (двічі по 50 мл). Сушать (MgSO4), і випарюють. Одержаний продукт використовують на наступній стадії без додаткового очищення. Вихід 106,8% ES-MS: m/e 207,0 (m+1). В. (R,R)-N-[3-(4-гідроксифеніл)-4метилізотіазол-5-іл]-2-метилциклопропанкарбоксамід Додають оксалілхлорид (2 Μ у дихлорметані, (0,291 мл, 0,583 ммоль) до перемішуваного розчину (R,R)-2-метилциклопропанкарбонової кислоти (0,06 г, 0,58 ммоль) та DMF (1 крапля, каталітична кількість) у толуолі (1 мл). Перемішують протягом 3 год, і додають утворений хлорангідрид кислоти до перемішуваного розчину 4-(5-аміно-4метилізотіазол-3-іл)фенолу (0,060 г, 0,291 ммоль) 22 та піридину (0,07 г, 0,87 ммоль) у THF (1 мл). Перемішують протягом 1 год і розводять EtOAc (300 мл), промивають 1-н. NaOH (двічі по 100 мл), потім водою (100 мл), сушать (MgSO4), і випарюють. Кристалізують із хлороформу та гексану. Вихід 64,6%. ES-MS: m/e 289,0 (m+1). Одержання нітрилу формули IX Якщо не описано інше, нітрил формули IX, який має вказане значення Q, одержують, застосовуючи відповідний бензонітрил формули Q-CN та ацетонітрил і методику, аналогічну описаній нижче для Підготовчого синтезу ІХ-2. Підготовчий синтез ІХ-2, Q=3-фтор-4метоксифеніл Змішують 3-фтор-4-метоксибензонітрил (25,000 г, 165,563 ммоль), ацетонітрил (13,576 г, 331,126 ммоль) та THF (33 мл). Додають при перемішуванні розчин трет-бутилату калію у THF (1 М, 182,1 мл, 182,1 ммоль). Перемішують протягом ночі. Розводять насиченим розчином NaHCO3 та екстрагують EtOAc. Випарюють органічний розчин, та кристалізують із суміші гексан/EtOAc, одержуючи 3-аміно-3-(3-фтор-4метоксифеніл)акрилонітрил. Вихід 78,6%. ES-MS: m/e 193,0 (m+1). Підготовчий синтез ІХ-4, Q=феніл: 3-аміно-3фенілакрилонітрил. Підготовчий синтез IX-8, Q=3,4-дихлорфеніл Застосовуючи методику, аналогічну описаній для Підготовчого синтезу ІХ-2, але з використанням 1,5 екв. трет-бутилату калію та 1,5 екв. ацетонітрилу на 1 екв. 3,4-дихлорбензонітрилу, одержують 3-аміно-3-(3,4-дихлорфеніл)акрилонітрил. Підготовчий синтез IX-9, Q=4-(метилтіо)феніл Застосовуючи методику, аналогічну описаній для Підготовчого синтезу ІХ-2, але з використанням 1,5 екв. трет-бутилату калію та 1,5 екв. ацетонітрилу на 1 екв. 4-(метилтіо)бензонітрилу, одержують 3-аміно-3-[4-(метилтіо)феніл]акрилонітрил. Підготовчий синтез IX-10, Q=4-(1,1дифторетил)феніл: 3-аміно-3-[4-(1,1дифторетил)феніл]акрилонітрил. Вихідний 4-(1,1-дифторетил)бензонітрил для Підготовчого синтезу IX-10 одержують, як описано нижче. Додають трифторид біс-(2метоксіетил)аміносірки (30,516 г, 137,931 ммоль) до 4-ацетилбензонітрилу (10,000 г, 68,966 ммоль), і перемішують в атмосфері азоту у тефлоновій посудині протягом 24 год. Розводять дихлорметаном, а потім додають надлишок насиченого розчину NaHCO3 для гасіння. Екстрагують EtOAc, сушать (MgSO4), і випарюють. Хроматографують на силікагелі, елююючи гексаном та EtOAc (градієнт від 3% до 30%), і одержують 4-(1,1дифторетил)бензонітрил. Вихід 68,6%. Одержання тіоаміду формули X 23 Якщо не описано інше, тіоамід формули X, який має вказане значення Q, одержують, застосовуючи відповідний нітрил формули IX і методику, аналогічну описаній нижче для Підготовчого синтезу Х-2. Підготовчий синтез Х-2, Q=3-фтор-4метоксифеніл Додають діоксан (65 мл) до 3-аміно-3-(3-фтор4-метоксифеніл)-акрилонітрилу (25,0 г, 130 ммоль) та тіоацетаміду (19,5 г, 260 ммоль); потім додають 4-н. розчин HCl у діоксані (650 мл, 2,600 ммоль), і перемішують реакційну суміш протягом 4-8 до завершення реакції за даними ХТШ. Випарюють реакційну суміш досуха, потім додають діоксан (200 мл); повільно додають TEA (висушену над K2СО3, 1000 мл); а потім додають насичений розчин K2СО3 (1000 мл), і екстрагують EtOAc (2000 мл). Сушать органічну фазу (K2СО3) і випарюють розчинник, одержуючи 3-аміно-3-(3-фтор-4метоксифеніл)-тіоакриламід. Вихід 98,5%. ES-MS: m/e 227,0 (m+1). Підготовчий синтез Х-4, Q=феніл: 3-аміно-3(феніл)-тіоакриламід. Підготовчий синтез Х-8, Q=3,4-дихлорфеніл: 3аміно-3-(3,4-дихлорфеніл)тіоакриламід. Підготовчий синтез Х-9, Q=4-(метилтіо)феніл: 3-аміно-3-[4-(метилтіо)феніл]-тіоакриламід. Підготовчий синтез Х-10, Q=4-(1,1дифторетил)феніл Додають дифенілфосфінодитіокислоту (13,2 г, 52,9 ммоль) до розчину 3-аміно-3-[4-(1,1дифторетил)феніл]акрилонітрилу (5,50 г, 26,4 ммоль) у пропан-2-олі (264 мл). Нагрівають при 45°С протягом 4 год. Розводять EtOAc (400 мл), промивають тричі розсолом (100 мл), і випарюють. Кристалізують із суміші EtOAc/гексан, і одержують 3-аміно-3-[4-(1,1-дифторетил)феніл]-тіоакриламід. Вихід 67,2%. ES-MS: m/e 243,0 (m+1). Одержання аміну формули VIII Якщо не описано інше, амін формули VIII, який має вказане значення Q, одержують, застосовуючи відповідний тіоамід формули X і методику, аналогічну описаній нижче для Підготовчого синтезу VIII-2. Підготовчий синтез VIII-2, Q=3-фтор-4метоксифеніл Додають пероксид водню (30% у воді, 8,73 г, 257 ммоль) до розчину 3-аміно-3-(3-фтор-4метоксифеніл)тіоакриламіду (29,0 г, 128 ммоль) у метанолі (1,283 мл), і перемішують реакційну суміш при кімнатній температурі. Гасять реакцію Na2S2O3 (20% у воді), і концентрують суміш досуха. Розводять EtOAc (900 мл) та водою (900 мл), відділяють органічну фазу, та випарюють розчинник. Кристалізують залишок із суміші EtOAc/гексан, одержуючи 3-(3-фтор-4-метоксифеніл)ізотіазол-5 95480 24 іламін (1 г). Хроматографуючи маточний розчин (25 г) на силікагелі з елююванням 25-50% EtOAc у гексані, одержують додаткову кількість продукту. [В одному з експериментів помилка при завантаженні колонки призвела до втрати 1/2 матеріалу; однак хроматографуванням було одержано додаткову кіькість чистого матеріалу (1 г).] Вихід 12,2%. ES-MS: m/e 225,0 (m+1). Підготовчий синтез VIII-4, Q=феніл: 3фенілізотіазол-5-іламін. ES-MS: m/e 177,2 (m+1). Підготовчий синтез VIII-8, Q=3,4-дихлорфеніл: 3-(3,4-дихлорфеніл)-ізотіазол-5-іламін. ES-MS: m/e 247,0 (m+1). Підготовчий синтез VIII-9, Q=4(метилтіо)феніл: 3-[4-(метилтіо)-феніл]ізотіазол-5іламін. ES-MS: m/e 222,3 (m+1). Підготовчий синтез VIII-10, Q=4-(1,1дифтopeтил)фeнiл: 3-[4-(1,1дифторетил)феніл]ізотіазол-5-іламін. ES-MS: m/e 241,2 (m+1). Одержання ізотіазолу формули VII Якщо не описано інше, амід формули VII, який має вказане значення Q, одержують, застосовуючи відповідний амін формули VIII і методику, аналогічну описаній нижче для Підготовчого синтезу VII-2. Підготовчий синтез VII-1, Q=4-метоксифеніл Застосування бензилового складного ефіру та триметилалюмінію: До суспензії 3-(4-метоксифеніл)ізотіазол-5іламіну (5,0 г, 24,3 ммоль) у безводному дихлорметані (230 мл), охолодженої до 0-5°С, додають при перемішуванні триметилалюміній (2,0 Μ у толуолі, 12,1 мл, 24,2 ммоль) за допомогою шприца. Перемішують розчин 5 хв, і додають бензил-(R,R)2-метилциклопропанкарбоксилат (4,6 г, 24,21 ммоль) у безводному дихлорметані (10 мл). Потім одержану суміш нагрівають до 40-45°С у потоці азоту (голчастий випускний вентиль) для повільного видалення розчинника. Через 2 год більша частина дихлорметану видаляється, і температура суміші підвищується до приблизно 50°С. Перемішують розчин ще протягом 3 год, охолоджують і обережно гасять шляхом додавання краплями води, а потім 0,1-н. HCl при інтенсивному продуванні азотом. Одержаний залишок потім розподіляють між етилацетатом (400 мл) та 0,1-н. HCl (400 мл). Органічний шар сушать над карбонатом калію, фільтрують, і випарюють до малого об'єму. Одержану суспензію розводять гексаном, і відділяють тверду речовину фільтруванням, одержуючи 5,9 г неочищеного продукту, який повторно суспендують в МТВЕ (100 мл), нагрівають при слабкому кипінні протягом 1 год і охолоджують до кімнатної температури. Після стояння протягом 1 год відділяють тверду речовину фільтруванням і сушать (20 мм рт. ст., 40°С), одержуючи (R,R)-N[3-(4-метоксифеніл)ізотіазол-5-іл]-2-метилциклопропанкарбоксамід (5,2 г, 75%). 25 H ЯМР (DMSO-d6) 7,86 (d, J=8,8 Гц, 2Н), 7,20 (s, 1Н), 7,00 (d, J=8,8 Гц, 2H), 3,78 (s, 3Н), 1,62 (m, 1H), 1,34 (m, 1H), 1,10 (d, J=6,0 Гц, 3Н), 1,09 (m, 1H), 0,80 (m, 1H); ES-MS m/e 289 (m+H). Підготовчий синтез VII-2, Q=3-фтор-4метоксифеніл Застосування бензилового складного ефіру та триметилалюмінію: Додають бензил-(R,R)-2метилциклопропанкарбоксилат (0,55 г, 2,89 ммоль) до охолодженого до 0°С розчину 3-(3фтор-4-метоксифеніл)-ізотіазол-5-іламіну (0,59 г, 2,63 ммоль), триметилалюмінію (2,0 Μ у толуолі, 5,26 ммоль) у дихлорметані (5 мл), і дають реакційній суміші спонтанно нагрітися до кімнатної температури. Нагрівають реакційну суміш до 40°С і залишають при перемішуванні протягом ночі. Розводять реакційну суміш EtOAc (100 мл) та 1-н. HCl (40 мл) і водою (100 мл), відділяють органічну фазу, і випарюють розчинник. Хроматографують на силікагелі з елююванням 25-50% EtOAc у гексані, і одержують (R,R)-N-[3-(3-фтop-4метоксифеніл)ізотіазол-5-іл]-2метилциклопропанкарбоксамід. Вихід 55,9%. ESMS: m/e 307,0 (m+1). Підготовчий синтез VII-4, q=феніл: (R,R)-2метил-N-(3-феніл-ізотіазол-5іл)циклопропанкарбоксамід. ES-MS: m/e 259,2 (m+1). Підготовчий синтез VII-8, Q=3,4-дихлорфеніл: (R,R)-N-[3-(3,4-дихлорфеніл)ізотіазол-5-іл]-2метилциклопропанкарбоксамід. ES-MS: m/e 328,0 (m+1). Підготовчий синтез VII-9, Q=4-(метилтio)феніл: (R,R)-N-[3-[4-(метилтіо)феніл]ізотіазол-5-іл]-2метилциклопропанкарбоксамід. ES-MS: m/e 305,2 (m+1). Підготовчий синтез VII-10, Q=4-(1,1дифторетил)феніл: Застосування бензилового складного ефіру та трет-бутилату калію: Перемішують 3-[4-(1,1дифторетил)феніл]ізотіазол-5-іламін (0,200 г, 0,833 ммоль), бензил-(R,R)-2метилциклопропанкарбоксилат (0,238 г, 1,250 ммоль) та трет-бутилат калію (0,190 г, 1,667 ммоль) протягом 1 год. Розводять суміш насиченим розчином NaHCO3 і екстрагують EtOAc. Промивають органічну фазу розсолом, сушать (MgSO4), і випарюють. Хроматографують на силікагелі з елююванням 10% EtOAc у СНСІ3, і одержують (R,R)-N-[3-[4-(1,1дифторетил)феніл]ізотіазол-5-іл]-2метилциклопропанкарбоксамід. Вихід 50,3%. ESMS: m/e 323,3 (m+1). Одержання 4-бромізотіазолу Формули VI, Х=Br 1 Якщо не описано інше, амід формули VI, де X - бром, який має вказане значення Q, одержують, 95480 26 застосовуючи відповідний амід формули VII і методику, аналогічну описаній нижче для Підготовчого синтезу VI-2. Підготовчий синтез VІ-2, Q=3-фтор-4метоксифеніл Додають краплями бром (0,15 мл, 0,47 г, 2,94 ммоль) до (R,R)-N-[3-(3-фтор-4метоксифеніл)ізотіазол-5-іл]-2метилциклопропанкарбоксаміду (0,45 г, 1,47 ммоль) у дихлорметані (3 мл). Контролюють хід реакції ХТШ, і припиняють додавання брому, коли реакція завершується. Розводять реакційну суміш EtOAc (100 мл) і виливають у водний розчин Na2S2O3 (1-н.). Відділяють органічну фазу, промивають розсолом (50 мл) та водою (80 мл), і випарюють розчинник. Хроматографують на силікагелі з елююванням 10-50% EtOAc у гексані, і одержують (R,R)-N-[4-бром-3-(3-фтор-4метоксифеніл)ізотіазол-5-іл]-2метилциклопропанкарбоксамід. Вихід 97,1%. ESMS: m/e 387,0 (m+1). Підготовчий синтез VI-4, Q=феніл: (R,R)-N-[4бром-3-фенілізотіазол-5-іл]-2метилциклопропанкарбоксамід. ES-MS: m/e 339,1 (m+1). Підготовчий синтез VI-8, Q=3,4-дихлорфеніл: (R,R)-N-[4-бром-3-(3,4-дихлорфеніл)-ізотіазол-5іл]-2-метилциклопропанкарбоксамід. ES-MS: m/e 406,8 (m+1). Підготовчий синтез VI-9, Q=4-(метилтіо)феніл: (R,R)-N-[4-бром-3-[4-(метилтіо)феніл]ізотіазол-5іл]-2-метилциклопропанкарбоксамід. ES-MS: m/e 385,0 (m+1). Підготовчий синтез VI-10, Q=4-(1,1дифторетил)феніл: (R,R)-N-[4-бром-3-[4-(1,1дифторетил)-феніл]ізотіазол-5-іл]-2метилциклопропанкарбоксамід. ES-MS: m/e 403,0 (m+1). Одержання 3,4-дибромізотіазолу формули XI До перемішуваного розчину 2ціанотіоацетаміду (39,5 г, 0,395 моль) у дихлорметані (700 мл) додають при 0°С льодяну оцтову кислоту (79 мл). Потім додають краплями розчин брому (43,0 мл, 0,840 моль) у дихлорметані (395 мл) протягом 2 год, підтримуючи температуру при 0°С. Перемішують суміш ще протягом 1 год в холодному стані, а потім гасять доданням 10% водного розчину бісульфіту натрію (300 мл). Водний шар обробляють 2-н. водним розчином карбонату натрію до рН 9, доводять до кімнатної температури, і двофазову суміш фільтрують через діатомову землю, яку промивають дихлорметаном. Після розділення шарів темний органічний шар сушать (Na2SO4), і випарюють розчинник, одержуючи темну тверду речовину, яку повторно розчиняютьу дихлорметані, вносять без додаткового оброблення у колонку із силікагелем і хроматографують при елююванні 20% етилацетату в гексані, одержуючи 27 3,4-дибром-ізотіазол-5-іламін у вигляді злегка забарвленої твердої речовини (12 г, 12%). ES-MS m/E 259 (m+1). Одержання 3.4-дибромізотіазолу формули XII До (R,R)-2-метилциклопропанкарбонової кислоти (5,4 г, 36 ммоль) у дихлорметані (100 мл) додають DMF (дві краплі), а потім 98% оксалілхлорид (4,8 мл, 54 ммоль). Одержану суміш перемішують протягом 3 год при кімнатній температурі. В окремій колбі розчиняють 3,4дибромізотіазол-5-іламін (9,3 г, 36,05 ммоль) у THF (200 мл), охолоджують одержаний розчин до 0°С, та розчиняють у ньому триетиламін (30 мл, 215 ммоль). Додають краплями при 0°С розчин хлорангідриду кислоти, одержаний, як описано вище, нагрівають суміш до кімнатної температури, і перемішують протягом ночі (16 год). Темний розчин розподіляють між розсолом (800 мл) та етилацетатом (800 мл). Органічний шар сушать (Na2SO4), і випарюють, одержуючи темне масло. Це масло хроматографують на силікагелі (флеш65, 10% етилацетату в толуолі), і одержують неочищений продукт, який перекристалізовують із суміші дихлорметан/гексан, і одержують (R,R)-N[3,4-дибромізотіазол-5-іл]-2-метилциклопропанкарбоксамід (4,0 г, 33%). 1 H ЯМР (DMSO-d6) 11,90 (s, 1Н), 2,09 (m, 1H), 1,40 (m, 1H), 1,12 (m, 1H), 1,11 (d, J=6,0 Гц, 3H), 0,85 (m, 1H); ES-MS m/e 339 (m-H). Альтернативна методика одержання 4бромізотіазолу Формули VI, Х=Br Якщо не описано інше, амід формули VI, де X - бром, який має вказане значення Q, одержують, застосовуючи відповідну борну кислоту Q-B(OH)2 та амід формули XII, (R,R)-N-[3,4-дибромізотіазол5-іл]-2-метилциклопропанкарбоксамід, і методику, аналогічну описаній нижче для альтернативного Підготовчого синтезу VI-3. Альтернативний Підготовчий синтез VІ-3, Q=4етоксифеніл Знегажують азотом суміш (R,R)-N-[3,4дибромізотіазол-5-іл]-2-метилциклопропанкарбоксаміду (0,500 г, 1,471 ммоль), (4етоксифеніл)борної кислоти (0,485 г, 2,941 ммоль), DMF (3 мл) та толуолу (29 мл). Додають карбонат натрію (2 М) (4,41 ммоль) та Рd(РРh3)4 (0,255 г, 0,221 ммоль); після чого герметично закривають в атмосфері азоту. Нагрівають при 60°С протягом ночі. Додають 100 мг Рd(РРh3)4, і нагрівають ще протягом 1 доби. Розводять суміш EtOAc, і промивають розсолом. Розділяють шари, та випарюють. 95480 28 Хроматографують на силікагелі з елююванням 1550% EtOAc у гексані, після чого кристалізують з EtOAc та гексану, і одержують (R,R)-N-[4-бром-3(4-етоксифеніл)ізотіазол-5-іл]-2метилциклопропан-карбоксамід. Вихід 53,5%. ESMS: 380,0 (m+1). Альтернативний Підготовчий синтез VI-5, Q=4фторфеніл Застосовують методику, аналогічну описаній для Підготовчого синтезу VI-3, але з використанням 1,3 екв. 4-фторфенілборної кислоти на 1 екв. (R,R)-N-[3,4-дибромізотіазол-5-іл]-2метилциклопропанкарбоксаміду та з перемішуванням реакційної суміш при 70°С протягом 1 доби, і одержують (R,R)-N-[4-бром-3-(4-фторфеніл)ізотіазол-5-іл]-2-метилциклопропан-карбоксамід. ES-MS: m/e 357,0 (m+1). Приклади формули І Якщо не описано інше, амід формули І, який має вказане значення Q, одержують, застосовуючи відповідний амін формули II і методику, аналогічну описаній нижче для Прикладу 1. Приклад 1, Q=4-метоксифеніл, застосування методики (А) Застосування бензилового складного ефіру та триметилалюмінію: Додають триметилалюміній (2 Μ розчин у толуолі, 3,59 г, 45,5 ммоль) до охолодженого до 0°С розчину 3-(4-метоксифеніл)-4-метилізотіазол-5іламіну (10,0 г, 45,5 ммоль) у дихлорметані (455 мл). Перемішують протягом 5 хв, і додають бензил-(R,R)-2-метилциклопропанкарбоксилат (8,64 г, 45,5 ммоль). Нагрівають при 50°С у потоці азоту для видалення розчинника. Одержане масло нагрівають при 50°С протягом 3 год. Гасять водою, і розводять EtOAc (300 мл). Промивають 0,1-н. HCl, сушать (K2СО3), випарюють, і кристалізують із суміші гексан/EtOAc, одержуючи (R,R)-N-[3-(4метоксифеніл)-4-метилізотіазол-5-іл]-2метилциклопропанкарбоксамід. Вихід 80,4%. ESMS: m/e 303,0 (m+1). (Маточний розчин можна використати для подальших перекристалізацій). Застосування хлорангідриду кислоти: Оксалілхлорид (170 г, 120 мл, 1,05 екв.) додають краплями при 21°С протягом приблизно 1,5 год до розчину (R,R)-2метилциклопропанкарбонової кислоти (150 г, 1,06 екв.) та DMF (каталітична кількість, 0,03 екв.) у дихлорметані (1,30 л), за цей час суміш охолоджується до приблизно 16°С внаслідок ендотермічної реакції. Реакційну суміш перемішують при температурі навколишнього середовища протягом 30 хв, потім нагрівають зі зворотним холодильником протягом 30 хв, і одержують розчин (R,R)-2метилциклопропанкарбонілхлориду, який охолоджують до 25°С для використання на наступній стадії. 29 До розчину 3-(4-метоксифеніл)-4метилізотіазол-5-іламіну (280 г, 1,00 екв.) та піридину (каталітична кількість, 0,03 екв.) у безводному THF (350 мл) додають краплями вищезгаданий розчин хлорангідриду кислоти протягом 30 хв при температурі від 6°С до 13°С, і перемішують реакційну суміш при кімнатній температурі протягом 1 год. (Хід реакції можна контролювати методом РХВЕ і припиняти додавання хлорангідриду, якщо аналіз показує повне використання аміну). Додають воду (2,5 л), розділяють фази, і водну фазу повторно екстрагують дихлорметаном. Об'єднані органічні фази промивають водним розчином NaOH (1л), потім водою (1л), частково випарюють до маси приблизно 1 кг, після чого розводять THF (2 л), одержуючи однорідний розчин, від якого відділяється певна кількість води. Цю суміш залишають відстоюватися протягом ночі при кімнатній температурі; за цей час відбувається розділення двох фаз, причому водна фаза розташовується над органічною фазою. Розділяють фази, додають до кожної фази THF, додають воду до верхньої (органічної) фази, і знов доводять кожний шар до рівноважного стану, при цьому відбувається дуже повільне розділення фаз з утворенням верхнього органічного шару. Об'єднані органічні фази завантажують у роторний випарник (місткість колби 10 л) через просвітлювальну мембрану (5 мкм), додають толуол (4 л), причому суміш стає каламутною, і випарюють THF та воду (160 мбар (0,016 МПа), температура бані 45°С). Після випарювання THF та води починається утворення кристалів. Після відгонки 2 л додають толуол (2 л), і продовжують дистиляцію (85 мбар (0,0085 МПа), температура бані 45°С). Після віддистилювання подальших 2 л розчинника тиск у системі підвищують до атмосферного, охолоджують суміш до температури навколишнього середовища, і через 1 год фільтрують одержану суспензію. Осад повторно суспендують у толуолі (1 л), фільтрують, і промивають толуолом (1 л), після чого сушать протягом ночі, і одержують (R,R)-N-[3-(4-мeтoкcифeнiл)-4мeтилiзoтiaзoл-5-ίл]-2-мeтилциклопропанкарбоксамід. Вихід 357 г, 92%. Умови контролю реакції РХВЕ: колонка ХТеrrа MS C18 2,5 мкм; 4,650 мм; елюент А 0,1% TFA у воді; елюент В ацетонітрил; швидкість потоку 1,50 мл/хв; приблизний градієнт А/В: 0-0,5 хв при 85/15; 1,5-7 хв від 85/15 до 5/95; 7-7,5 хв при 5/95; 7,5-8 хв від 5/95 до 85/15; 8-10 хв при 85/15; тривалість циклу 10 хв; детектування УФ, 210 нм; підготовка проби: розведення сумішшю ацетонітрил-водаTFA (70:30:1). Енантіомерний надлишок (ее) та стереохімічну чистоту карбоксаміду можна визначити методом РХВЕ у таких умовах: стаціонарна фаза (колонка) Chiralcel OJ (2404,6 мм внутрішній діаметр) з колонки Daicel; рухома фаза метанол:діетиламін (100:0,1 за об'ємом); детектування УФ на довжині хвилі 280 нм; об'єм проби 5 мкл; температура проби 20°С; температура колонки навколишня; тривалість аналізу 20 хв; розчинник для проби метанол; концентрація проби приблизно 5 мг/мл метанолу. На типовій хроматограмі (R,R)-N-[3-(4метоксифеніл)-4-метилізотіазол-5-іл]-2 95480 30 метилциклопропан-карбоксамід має час затримання 10,519 хв, (S,S)-ізомер має час затримання 12,981 хв, а залишкові кількості двох цис-2метилізомерів мають час затримання 14,189 хв та 14,980 хв. Значення ее та вміст цис-домішок у типовому продукті залежать від ее та вмісту цисдомішок у вихідній кислоті та ступеня подальшого очищення шляхом перекристалізації Типові методики, описані вище, забезпечують значення ее понад 97% та вміст цис-домішок від приблизно 0,5% до приблизно 1%. Як правило, сполука за Прикладом 1, одержана та виділена, як описано у прикладах, згідно з даними мікроскопічного аналізу, порошкової рентгенографії (XRPD) та/або диференційної сканувальної калориметрії (DSC), має форму кристалічної твердої речовини. Партії продукту, проаналізовані XRPD, характеризуються такими піками (2, відносна інтенсивність): 5,944, 1,00; 13,856, 0,01; 15,445, 0,01; 17,806, 0,06; 19,797, 0,02; 22,718, 0,02; 23,812, 0,01; і, більш конкретно, піками (2, відносна інтенсивність): 5,944, 1,00; 17,806, 0,06; 19,797, 0,02; 22,718, 0,02; та позначаються як безводна форма І. При диспергуванні сполуки у сумішах води з метанолом утворюється друга форма, що позначається як безводна форма II, яка характеризується піками (2, відносна інтенсивність): 6,727, 1,00; 11,371, 0,04; 18,159, 0,04; 20,220, 0,12; 22,782, 0,09; 30,026, 0,04; 36,818, 0,02; 25,482; 0,03; і, більш конкретно, піками (2, відносна інтенсивність): 6,727, 1,00; 20,220, 0,12; 22,782, 0,09; її одержують в умовах зниженої активності води (aw), наприклад, при aw 0,66 або нижче; і моногідратну форму, яка позначається як моногідрат І і характеризується піками (2, відносна інтенсивність): 5,193, 0,07; 10,336, 0,82; 14,005, 0,77; 20,686, 0,19; 22,907, 1,00; 24,716, 0,53; 26,375, 0,29; і, більш конкретно, піками (2, відносна інтенсивність): 10,336, 0,82; 14,005, 0,77; 22,907, 1,00; 24,716, 0,53, одержують в умовах підвищеної активності води, наприклад, при aw 0,91 або вище. Порошкові рентгенодифрактограми (XRPD) було одержано на порошковому дифрактометрі Bruker D8 Advance, обладнаному джерелом випромінення CuK source (=1,54056 Å) та електронним детектором Sol-X із мінімальною робочою напругою 30 кВ та струмом 40 мА. Кожну пробу сканували при кімнатній температурі (25°С) в межах кута 2 від 4° до 40° з кроком 2 0,02° при максимальній швидкості сканування 3 с/крок, контрольованому змінному (ν 12) розходженні та ширині щілини детектора 0,1 мм. Приклад 1. (2=4-метоксифеніл, застосування методики (В) У суху колбу, яка містить (R,R)-N-[3-(4гідроксифеніл)-4-метилізотіазол-5-іл]-2метилциклопропанкарбоксамід (100 мг, 0,347 ммоль), додають безводний ацетон (2 мл), K2СО3 (50 мг, 0,347 ммоль) та метилйодид (19 мкл, 0,313 ммоль). Реакційну суміш нагрівають при 45°С, і додають додаткову кількість метилйодиду (19 мкл, 0,313 ммоль). Після нагрівання ще протягом 6 год розчинник випарюють, і залишок розподіляють між етилацетатом та водою. Етилацетатний розчин промивають насиченим водним розчином K2СО3 31 (двічі) та розсолом, сушать (MgSO4), і розчинник випарюють. Хроматографують залишок на силікагелі з елююванням сумішшю EtOAc/гексан, і одержують (R,R)-N-[3-(4-метоксифеніл)-4метилізотіазол-5-іл]-2метилциклопропанкарбоксамід (35 мг, 33%) у вигляді білої твердої речовини. 1 H ЯМР ES-MS: m/e 303 (m+1). Приклад 2, Q=3-фтор-4-метоксифеніл, застосування методики (С) - сполучення зі сполукою формули VI, Х=Br: Додають PdCl2(PPh3)2 (0,15 г, 0,21 ммоль) та Sn(CH3)4 (0,79 мл, 1,02 г, 5,71 ммоль) до (R,R)-N[4-бром-3-(3-фтор-4-метоксифеніл)ізотіазол-5-іл]2-метилциклопропанкарбоксаміду (0,55 г, 1,43 ммоль) у DMF (3 мл), і перемішують реакційну суміш при 130°С у запаяній трубці протягом ночі. Розводять суміш EtOAc (100 мл) та розсолом (100 мл). Відділяють органічну фазу, сушать (K2СО3), і випарюють. Одержане масло розчиняють у суміші 1:1 MTBE:KF (водний розчин, 15%) (50 мл), і перемішують при нагріванні зі зворотним холодильником протягом 1 год. Виливають охолоджений розчин на діатомову землю, та фільтрують з МТВЕ (100 мл). Відділяють органічну фазу, сушать (K2СО3), і випарюють. Хроматографують на силікагелі з елююванням 10-30% THF у гексані, і одержують (R,R)-N-[3-(3-фтор-4-метоксифеніл)-4метилізотіазол-5-іл]-2метилциклопропанкарбоксамід. Вихід 49,2%. ESMS: m/e 321,0 (m+1). Приклад 3, Q=4-етоксифеніл, застосування методики (С) - сполучення зі сполукою формули VI, Х=Br: Додають PdCl2(PPh3)2 (0,07 г, 0,09 ммоль) та Sn(CH3)4 (0,34 мл, 0,44 г, 2,48 ммоль) до (R,R)-N[4-бром-3-(4-етоксифеніл)ізотіазол-5-іл]-2метилциклопропанкарбоксаміду (0,24 г, 0,62 ммоль) у DMF (1 мл), і перемішують реакційну суміш при 130°С у запаяній трубці протягом ночі. Розводять суміш EtOAc (100 мл) та розсолом (100 мл). Відділяють органічну фазу, сушать (MgSO4), і випарюють. Фільтрують через силікагель, і промивають EtOAc. Випарюють розчинник, і кристалізують із суміші EtOAc/гексан, одержуючи (R,R)-N-[3(4-етоксифеніл)-4-метилізотіазол-5-іл]-2метилциклопропанкарбоксамід. Вихід 81,7%. ESMS: m/e 317,0 (m+1). Приклад 4, Q=феніл, застосування методики (С) - металування-метилювання сполукою формули VI, Х=Br: До розчину (R,R)-N-[4-бром-3-фенілізотіазол5-іл]-2-метилциклопропанкарбоксаміду (0,50 г, 1,48 ммоль) у THF (3 мл), охолодженого до -78°С, додають 1,1 екв. н-бутиллітію (n-BuLi) (1,6 Μ у гексані, 0,204 мл, 3,26 ммоль). Підтримують температуру в масі нижче від -68°С. Після додання реакційну суміш залишають при перемішуванні на 1 год. Потім додають ще 1,1 екв. n-BuLi (1,6 Μ у гексані, 0,204 мл, 3,26 ммоль), підтримуючи температуру в масі нижче від -66°С. Після перемішування протягом 2 год реакційну суміш підігрівають до -40°С протягом 15 хв, а потім знов охолоджують до 78°С. Потім додають метилйодид (0,10 мл, 1,63 ммоль). Дають реакційній суміші нагрітися до кім 95480 32 натної температури і перемішують протягом 2,5 діб, після чого гасять насиченим розчином NH4Cl, і розводять EtOAc. Органічну фазу промивають розсолом, сушать (K2СО3), і випарюють. Хроматографують на силікагелі з елююванням 5-35% EtOAc у гексані, і одержують (R,R)-2-метил-N-(4метил-3-фенілізотіазол-5-іл)циклопропанкарбоксамід. Вихід 16,4%. ES-MS: 273,2 (m+1). Приклад 5, Q=4-фторфеніл, застосування методики (С) - сполучення зі сполукою формули VI, Х=Br: За методикою, аналогічною описаній у Прикладі 3, використовуючи (R,R)-N-[4-бром-3-(4фторфеніл)ізотіазол-5-іл]-2метилциклопропанкарбоксамід, одержують (R,R)N-[3-(4-фторфеніл)-4-метилізотіазол-5-іл]-2метилциклопропанкарбоксамід. ES-MS: m/e 291,0 (m+1). Приклад 6, Q=4-хлорфеніл, застосування методики (A): (R,R)-N-[3-(4-хлорфеніл)-4метилізотіазол-5-іл] -2метилциклопропанкарбоксамід. ES-MS: m/e 307,0 (m+1). Використання хлорангідриду кислоти: До розчину (R,R)-2метилциклопропанкарбонової кислоти (7,65 мл, 7,83 г, 1,00 екв.) у дихлорметані (реактив для РХВЕ, 39,2 мл, 5 мл/г кислоти), додають диметилформамід (30 мкл, 390 мкмоль, 0,005 моль/моль кислоти), а потім повільно додають оксалілхлорид (6,85 мл, 77,4 ммоль, 0,99 моль/моль кислоти) при 0°С (льодо-водяна баня) в атмосфері азоту. Через 30 хв відстороняють льодо-водяну баню, і нагрівають суміш до 40°С за 30 хв. Дають розчину охолодитися до навколишньої температури і безпосередньо використовують на наступній стадії без додаткового оброблення. До розчину 3-(4-хлорфеніл)-4-метилізотіазол5-іламіну (17,1 г, 75,9 ммоль, 1 екв.), піридину (12,3 мл, 152 ммоль, 2 моль/моль аміну) та дихлорметану (75,9 мл для забезпечення концентрації аміну 1 М) у круглодонній колбі місткістю 500 мл (обладнаній пристроєм для створення атмосфери азоту, стрижневою мішалкою та охолоджувальною банею), додають вищезазначений розчин (R,R)-2метилциклопропанкарбонілхлориду (1,00 екв.; 75,9 ммоль). Перемішують протягом 30 хв, потім відстороняють льодяну баню, і перемішують протягом 3 год. Концентрують реакційну суміш у вакуумі, і розводять залишок етилацетатом. Промивають двічі розбавленою HCl і двічі водним розчином NaHCO3, сушать (K2СО3), фільтрують, і випарюють досуха. Кристалізують залишок із гексану та етилацетату, одержуючи білу тверду речовину, і одержують другу порцію шляхом повторення кристалізації; одержують (R,R)-N-[3-(4-хлорфеніл)-4метилізогіазол-5-іл]-2метилциклопропанкарбоксамід. Вихід 92,2%. LCMS: 307,0 (m+1). 1 H ЯМР (DMSO-d6, 300 МГц) 11,26 (s, 1Н), 7,62 (d, 2H, J=8,8 Гц), 7,50 (d, 2H, J=8,8 Гц), 2,28 (s, 3H), 1,90 (m, 1H), 1,32 (m, 1H), 1,11 (m, 4H), 0,79 (m, 1H). 33 95480 Приклад 7, Q=4-бромфеніл, застосування методики (A): (R,R)-N-[3-(4-бромфеніл)-4метилізотіазол-5-іл]-2метилциклопропанкарбоксамід. ES-MS: m/e 353,0 (m+1). Приклад 8, Q=3,4-дихлорфеніл, застосування методики (С) - сполучення зі сполукою формули VI, Х=Br: За методикою, аналогічною описаній у Прикладі 2, використовуючи (R,R)-N-[4-бром-3-(3,4діхлорфеніл)ізотіазол-5-іл]-2-метилциклопропанкарбоксамід, одержують (R,R)-N-[3-(3,4дихлорфеніл)-4-металізотіазол-5-іл]-2метилциклопропанкарбоксамід. ES-MS: m/e 341,0 (m+1). Приклад 9, Q=4-(метилтіо)феніл, застосування методики (С) - сполучення зі сполукою формули VI, Х=Br: Комп’ютерна верстка О. Гапоненко 34 За методикою, аналогічною описаній у Прикладі 2, використовуючи (R,R)-N-[4-бром-3-[4(метилтіо)феніл]ізотіазол-5-іл]-2метилциклопропан-карбоксамід, одержують (R,R)N-[3-[4-(метилтіо)феніл]-4-метилізотіазол-5-іл]-2метилциклопропанкарбоксамід. ES-MS: m/e 319 (m+1). Приклад 10, Q=4-(1,1-дифторетил)феніл, застосування методики (С) - сполучення зі сполукою формули VI, Х=Br: За методикою, аналогічною описаній у Прикладі 2, використовуючи (R,R)-N-[4-бром-3-[4-(1,1дифторетил)феніл]ізотіазол-5-іл]-2метилциклопропанкарбоксамід, одержують (R,R)N-[3-[4-(дифторетил)феніл]-4-метилізотіазол-5-іл]2-метилциклопропанкарбоксамід. ES-MS: m/e 337,3 (m+1). Підписне Тираж 23 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюSubstituted carboxamides
Автори англійськоюBacker Ryan Thomas, Fisher Matthew Joseph, Kuklish Steven Lee, Hollinshead, Sean Patrick, Smith Edward C. R., Takeuchi Kumiko
Назва патенту російськоюЗамещенные карбоксамиды
Автори російськоюБакер Райан Томас, Фишер Метью Джозеф, Куклиш Стивен Ли, Голлинзхед Шон Патрик, Смит Эдвард К.Р., Такеучи Кумико
МПК / Мітки
МПК: A61P 25/04, C07D 275/03, A61K 31/425
Мітки: карбоксаміди, заміщені
Код посилання
<a href="https://ua.patents.su/17-95480-zamishheni-karboksamidi.html" target="_blank" rel="follow" title="База патентів України">Заміщені карбоксаміди</a>
Заміщені 1-фенілпіразол-3-карбоксаміди, спосіб їх одержання та проміжні сполуки, фармацевтична композиція, що має спорідненість до рецепторів нейротензину

Номер патенту: 66750
Опубліковано: 15.06.2004
Автори: Жанжан Френсі, Молімар Жан-Шарль, Жиллі Даніель, Буажгрен Робер, Лабеу Бернар
МПК: C07C 243/00, A61K 31/445, C07D 401/10, C07D 453/00, A61K 31/495, A61K 31/4439, C07D 401/12, A61P 35/00, C07D 413/04, A61K 31/439, A61K 31/4427, A61P 25/00, A61K 31/496, A61K 31/5377, C07D 403/12, A61K 31/454, A61P 43/00, C07D 231/14, C07D 403/10, C07C 309/00, A61K 31/44, A61K 31/415, A61K 31/535, A61K 31/435, A61K 31/42, C07C 255/66, C07C 311/21
Мітки: заміщені, спосіб, 1-фенілпіразол-3-карбоксаміди, фармацевтична, композиція, рецепторів, спорідненість, нейротензину, сполуки, має, проміжні, одержання
Формула / Реферат:
1. Заміщений 1-фенілпіразол-3-карбоксамід загальної формули , (Ia)деR1x знаходиться у положенні 4 або5 і означає групу -T-CONRaRb, в якій Т - прямий зв’язок або С1-7алкілен;NRaRb означає групу, вибрану з: –NR9(CH2)sCR7R8(CH2)tNR5R6, ,
4-оксо-1-(3-заміщені феніл)-1,4-дигідро-1,8-нафтиридин-3-карбоксаміди як інгібітори фосфодіестерази-4

Номер патенту: 82208
Опубліковано: 25.03.2008
Автори: Лякомб Патрік, Жирар Ів, Аспіотіс Рене, Галлан Мішель, Дюб Даніель, Макдональд Дуайт, Дюб Лоранс
МПК: A61P 25/00, C07D 471/04
Мітки: 4-оксо-1-(3-заміщені, феніл)-1,4-дигідро-1,8-нафтиридин-3-карбоксаміди, інгібітори, фосфодіестерази-4
Формула / Реферат:
1. Сполука формули (І): (I)або її фармацевтично прийнятна сіль, деАr являє собою феніл, тієніл або його оксид;Y являє собою -COOR4, С1-6алкіл (С1-4алкіл)n-СООR4, -С3-4циклоалкіл (С1-4алкіл)m-СООR4, де -С1-6алкіл і С3-4циклоалкіл необов'язково заміщені галогеном, алкокси, гідрокси або нітрилом, і (С1-4алкіл) замісники необов'язково зв'язані з...
Заміщені 6-циклогексилалкілом заміщені 2-хінолінони та 2-хіноксалінони як інгібітори полі-(адф-рибоза)полімерази

Номер патенту: 91007
Опубліковано: 25.06.2010
Автори: Мабір Домінік Жан-П'єр, Сомерс Марія Вікторіна Франсіска, ван Дун Якобус Альфонсус Йозефус, Вутерс Вальтер Будевійн Леопольд
МПК: A61P 43/00, C07D 241/44, C07D 401/06, A61K 31/498
Мітки: інгібітори, 6-циклогексилалкілом, полі-(адф-рибоза)полімерази, 2-хіноксалінони, 2-хінолінони, заміщені
Формула / Реферат:
1. Сполука формули (І), (I)її N-оксидні форми, адитивні солі та стереохімічно ізомерні форми, деn означає 0 або 1;s означає 0 або 1;X являє собою -N= або -CR4=, де R4 являє собою водень, або, взятий разом з R1, може утворювати бівалентний радикал формули -СН=СН-СН=СН-;Y являє собою -N< або -СН<;Q являє собою -NH-, -О-,...
Заміщені b-дикетони і їх застосування для лікування респіраторних захворювань

Номер патенту: 64805
Опубліковано: 15.03.2004
Автори: Леннберг Карі, Лінден Інге-Брітт, Лотта Тімо, Похто Пентті, Копонен Аніта, Піппурі Айно, Ахо Пяйві, Бякстрем Рейо
МПК: A61K 31/12, C07C 317/24
Мітки: b-дикетони, заміщені, лікування, застосування, захворювань, респіраторних
Формула / Реферат:
1. Сполуки загальної формули І, (I)в якій один з Х1 і Х2 є MeSO2, а інший є галогеном, і Х3 є воднем або галогеном.2. Сполука формули І за п. 1, в якій Х1 є галогеном, Х2 є MeSO2 і Х3 є воднем.3. Сполука за п. 1, що являє собою 3-{[3-фтор-4-(метилсульфоніл)феніл]метилен)-2,4-пентандіон.4. Сполука за п. 1, яка являє собою 3-{[3-хлор-4-(метилсульфоніл)феніл]метилен}-2,4-пентандіон.5. Сполука за п. 1,...
Заміщені 1-гетероциклічні діариламіни

Номер патенту: 72239
Опубліковано: 15.02.2005
Автори: Теклі Хейл, Барретт Стівен Дуглас, Жанг Лу-Ян, Бріджіз Александер Джеймс
МПК: C07D 231/20, A61P 17/06, A61P 25/28, A61P 31/18, A61P 3/10, A61K 31/4196, A61P 9/00, A61K 31/421, C07D 257/00, C07D 249/06, C07D 249/14, A61P 9/10, C07D 271/10, A61K 31/4245, A61K 31/18, A61P 37/02, A61K 31/41, A61K 31/235, A61P 9/08, A61K 45/06, A61K 31/337, C07D 263/10, A61K 31/437, A61P 1/16, A61P 9/04, C07D 291/00, A61P 19/02, C07D 271/07, A61K 31/5517, A61P 29/00, C07D 231/46, A61P 31/12, C07D 285/08, C07D 275/00, C07D 249/12, A61P 35/00
Мітки: 1-гетероциклічні, заміщені, діариламіни
Формула / Реферат:
1. Сполука формули (І): (I),де X - -NH-,W - одна з наступних формул (і)-(хііі): (i) (ii) (iii) (iv) (v) (vi) (vii) (viii) (ix) (x) (xi) (xii) (xiii) (xiv) (xv) ...
Попередній патент: Двигун вихороімпульсний-2 (дві-2)
Випадковий патент: Програмований формувач імпульсів, тривалість яких визначається тривалістю вхідних, рівною або меншою встановленої