Похідні лептоміцину








Формула / Реферат
1. Похідна лептоміцину формули (І):
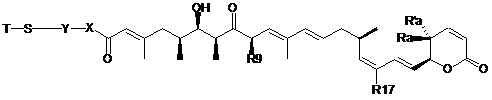 , (I)
, (I)
де:
- Ra і R'a являють собою Н або лінійний або розгалужений С1-С20алкіл;
- R17 являє собою алкіл, необов'язково заміщений OR, CN, NRR', перфторалкілом;
- R9 являє собою алкіл, необов'язково заміщений OR, CN, NRR', перфторалкілом;
- X означає -NR-;
- Y являє собою лінійний або розгалужений С1-С20алкіл;
- Т являє собою Н, Ac, R1 або SR1 де R1 являє собою Н, метил, лінійний або розгалужений С1-С20алкіл, циклоалкіл, арил або гетероцикліл;
- R, R', однакові або різні, являють собою Н або лінійний або розгалужений С1-С20алкіл;
або фармацевтично прийнятні солі, гідрати або гідратовані солі, або поліморфні кристалічні структури вказаної похідної лептоміцину, або їх оптичні ізомери, рацемати, діастереомери або енантіомери.
2. Похідна лептоміцину за п, 1, в якій Ra є лінійним або розгалуженим С1-С20алкілом і R'a є Н.
3. Похідна лептоміцину за п. 1 або 2, в якій R17 є лінійним або розгалуженим С1-С20алкілом.
4. Похідна лептоміцину за пп. 1-3, в якій R9 є лінійним або розгалуженим С1-С20алкілом.
5. Похідна лептоміцину за пп. 1-4, в якій Т являє собою Н або SR1 де R1 є лінійним або розгалуженим С1-С20алкілом.
6. Похідна лептоміцину, вибрана з групи, що містить:
- (2-метилсульфанілетил)амід (2Е,10Е,12Е,16Z,18Е)-(R)-6-гідрокси-3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метал-6-оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадека-2,10,12,16,18-пентаєнової кислоти,
- біс-[(2-меркаптоетил)амід (2E,10E,12E,16Z,18E)-(R)-6-гідрокси-3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадека-2,10,12,16,18-пентаєнової кислоти],
- (2-меркаптоетил)амід (2E,10E,12E,16Z,18E)-(R)-6-гідрокси-3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадека-2,10,12,16,18-пентаєнової кислоти;
(2-метилдисульфанілетил)амід (2Е,10Е,12Е,16Z,18Е)-(R)-6-гідрокси-3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадека-2,10,12,16,18-пентаєнової кислоти;
(2-метил-2-метилдисульфанілпропіл)амід (2Е,10Е,12Е,16Z,18E)-(R)-6-гідрокси-3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадека-2,10,12,16,18-пентаєнової кислоти;
(2-меркапто-2-метилпропіл)амід (2Е,10Е,12Е,16Z,18E)-(R)-6-гідрокси-3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадека-2,10,12,16,18-пентаєнової кислоти
або їх фармацевтично прийнятні солі, гідрати або гідратовані солі, або поліморфні кристалічні структури цих сполук, або їх оптичні ізомери, рацемати, діастереомери або енантіомери.
7. Фармацевтична композиція, що містить похідну лептоміцину за пп. 1-6 і фармацевтично прийнятний носій.
8. Застосування похідної лемтоміцину за пп. 1-6 для отримання лікарського засобу для лікування раку.
9. Спосіб отримання сполуки за пп. 1-6, що включає стадію взаємодії відповідних сполук формули (II) і (III):
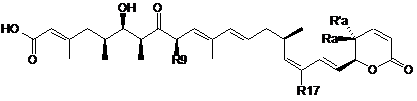 ,
,
(II)
T-S-Y-NHR,
(III)
де Ra, R'a, R17, R9, Y, T визначені в пп. 1-6.
10. Спосіб отримання сполуки за будь-яким з пп. 1-6, де в формулі (І) Т є Н, що включає взаємодію відповідної сполуки формули (І), де Т є S-R1, в присутності засобу, який відновлює дисульфідні зв'язки.
11. Спосіб отримання кон'югата, що включає стадію взаємодії похідної лептоміцину за будь-яким з пп. 1-6 з модифікованим засобом, що зв'язується з клітиною, який містить функціональну групу, реакційноздатну відносно зв'язувальної групи похідної лептоміцину, так що дана похідна і засіб, що зв'язується з клітиною, з'єднані один з одним через вказаний лінкер, включаючи сіль зв'язувальної речовини.
12. Спосіб за п. 11, за яким вказаний засіб, що зв'язується з клітиною, який містить функціональну групу, реакційноздатну відносно вказаної зв'язувальної групи даної похідної, отримують шляхом взаємодії вказаного засобу, що зв'язується з клітиною з реагентом, вибраним з SMCC, SSNPB, LC-SMCC, SIAB, SIA, SBA і SPAP.
13. Спосіб одержання сполуки через тіоефір кон'югата, який включає стадію взаємодії тіолвмісної похідної лептоміцину за будь-яким з пп. 1-6 з антитілом або засобом, який зв'язується з клітиною, модифікованими сполукою, такою як N-сульфосукцинімідил-4-(5-нітро-2-піридилдитіо)бутаноат, N-сукцинімідил-4-(малеімідометил)циклогексанкарбоксилат, N-сукцинімідил-4-(N-малеімідометил)циклогексан-1-карбокси-(6-амідокапроат), N-сукцинімідил-4-(йодацетил)амінобензоат, N-сукцинімідилйодацетат, N-сукцинімідилбромацетат або N-сукцинімідил-3-(бромацетамідо)пропіонат.
14. Спосіб одержання кон'югата, який включає стадію взаємодії дисульфід- або тіолвмісної похідної лептоміцину за будь-яким з пп. 1-6 з пептидом або антитілом, модифікованим реагентом, вибраним з N-сукцинімідил-4-(2-піридилдитіо)пентаноату, 4-сукцинімідилоксикарбоніл-a-метил-a-(2-піридилдитіо)толуолу, N-сукцинімідил-3-(2-піридилдитіо)бутирату, сукцинімідилпіридилдитіопропіонату, складного ефіру N-гідросукциніміду 4-(2-піридилдитіо)бутанової кислоти, сукцинімідил-4-[N-малеімідометил]циклогексан-1-карбоксилату, N-сульфосукцинімідил-3-(2-(5-нітропіридилтіо)бутирату, 2-імінотіолану і S-ацетилянтарного ангідриду.
15. Терапевтичний засіб для пригнічення росту вибраних клітинних популяцій, який містить:
(a) цитотоксичну кількість похідного лептоміцину за будь-яким з пп. 1-6, поєднаних із засобом, який зв'язується з клітиною, і (b) фармацевтично прийнятний носій, розріджувач або ексципієнт.
16. Терапевтичний засіб за п. 15, призначений для лікування злоякісного захворювання.
17. Терапевтичний засіб за п. 15, призначений для лікування раку легень, молочних залоз, товстого кишечнику, простати, нирок, підшлункової залози, яєчників і лімфатичних органів або меланоми.
Текст
1. Похідна лептоміцину формули (І): 3 95959 8-оксононадека-2,10,12,16,18-пентаєнової кислоти], - (2-меркаптоетил)амід (2E,10E,12E,16Z,18E)-(R)6-гідрокси-3,5,7,9,11,15,17-гептаметил-19-((2S,3S)3-метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)-8оксононадека-2,10,12,16,18-пентаєнової кислоти; (2-метилдисульфанілетил)амід (2Е,10Е,12Е,16Z,18Е)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадека2,10,12,16,18-пентаєнової кислоти; (2-метил-2-метилдисульфанілпропіл)амід (2Е,10Е,12Е,16Z,18E)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадека2,10,12,16,18-пентаєнової кислоти; (2-меркапто-2-метилпропіл)амід (2Е,10Е,12Е,16Z,18E)-(R)-6-гідроксиOH 4 3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадека2,10,12,16,18-пентаєнової кислоти або їх фармацевтично прийнятні солі, гідрати або гідратовані солі, або поліморфні кристалічні структури цих сполук, або їх оптичні ізомери, рацемати, діастереомери або енантіомери. 7. Фармацевтична композиція, що містить похідну лептоміцину за пп. 1-6 і фармацевтично прийнятний носій. 8. Застосування похідної лемтоміцину за пп. 1-6 для отримання лікарського засобу для лікування раку. 9. Спосіб отримання сполуки за пп. 1-6, що включає стадію взаємодії відповідних сполук формули (II) і (III): O R'a Ra HO O R9 O R17 (II) T-S-Y-NHR, (III) де Ra, R'a, R17, R9, Y, T визначені в пп. 1-6. 10. Спосіб отримання сполуки за будь-яким з пп. 16, де в формулі (І) Т є Н, що включає взаємодію відповідної сполуки формули (І), де Т є S-R1, в присутності засобу, який відновлює дисульфідні зв'язки. 11. Спосіб отримання кон'югата, що включає стадію взаємодії похідної лептоміцину за будь-яким з пп. 1-6 з модифікованим засобом, що зв'язується з клітиною, який містить функціональну групу, реакційноздатну відносно зв'язувальної групи похідної лептоміцину, так що дана похідна і засіб, що зв'язується з клітиною, з'єднані один з одним через вказаний лінкер, включаючи сіль зв'язувальної речовини. 12. Спосіб за п. 11, за яким вказаний засіб, що зв'язується з клітиною, який містить функціональну групу, реакційноздатну відносно вказаної зв'язувальної групи даної похідної, отримують шляхом взаємодії вказаного засобу, що зв'язується з клітиною з реагентом, вибраним з SMCC, SSNPB, LCSMCC, SIAB, SIA, SBA і SPAP. 13. Спосіб одержання сполуки через тіоефір кон'югата, який включає стадію взаємодії тіолвмісної похідної лептоміцину за будь-яким з пп. 1-6 з антитілом або засобом, який зв'язується з клітиною, модифікованими сполукою, такою як Nсульфосукцинімідил-4-(5-нітро-2піридилдитіо)бутаноат, N-сукцинімідил-4(малеімідометил)циклогексанкарбоксилат, Nсукцинімідил-4-(N-малеімідометил)циклогексан-1 O , карбокси-(6-амідокапроат), N-сукцинімідил-4(йодацетил)амінобензоат, Nсукцинімідилйодацетат, Nсукцинімідилбромацетат або N-сукцинімідил-3(бромацетамідо)пропіонат. 14. Спосіб одержання кон'югата, який включає стадію взаємодії дисульфід- або тіолвмісної похідної лептоміцину за будь-яким з пп. 1-6 з пептидом або антитілом, модифікованим реагентом, вибраним з N-сукцинімідил-4-(2піридилдитіо)пентаноату, 4сукцинімідилоксикарбоніл--метил--(2піридилдитіо)толуолу, N-сукцинімідил-3-(2піридилдитіо)бутирату, сукцинімідилпіридилдитіопропіонату, складного ефіру N-гідросукциніміду 4(2-піридилдитіо)бутанової кислоти, сукцинімідил-4[N-малеімідометил]циклогексан-1-карбоксилату, Nсульфосукцинімідил-3-(2-(5нітропіридилтіо)бутирату, 2-імінотіолану і Sацетилянтарного ангідриду. 15. Терапевтичний засіб для пригнічення росту вибраних клітинних популяцій, який містить: (a) цитотоксичну кількість похідного лептоміцину за будь-яким з пп. 1-6, поєднаних із засобом, який зв'язується з клітиною, і (b) фармацевтично прийнятний носій, розріджувач або ексципієнт. 16. Терапевтичний засіб за п. 15, призначений для лікування злоякісного захворювання. 17. Терапевтичний засіб за п. 15, призначений для лікування раку легень, молочних залоз, товстого кишечнику, простати, нирок, підшлункової залози, яєчників і лімфатичних органів або меланоми. 5 95959 6 Галузь техніки Даний винахід належить до похідних лептоміцину і їх терапевтичного використання. Більш конкретно, даний винахід належить до нових похідних лептоміцину, які містять групу (зв'язувальну групу), яка може ковалентно зв'язуватися з речовиною, яка зв'язується з клітиною, і відповідні кон'югати, що містять вказану похідну лептоміцину, зв'язану через лінкер з вказаною речовиною, яка зв'язується з клітиною. Вказані кон'югати забезпечують терапевтичні засоби, які здатні активуватися і вивільнятися in vivo, і які доставляються до конкретних популяцій клітин цільовим чином. Передумови створення винаходу Є багато повідомлень, які стосуються спрямованої доставки до пухлинних клітин за допомогою кон'югатів моноклональне антитіло-лікарський засіб {Sela et al., in Immunoconjugates, pp. 189-216 (C. Vogel, ed. 1987); Ghose et al., in Targeted Drugs, pp. 1-22 (E. Goldberg, ed. 1983); Diener et al., in Antibody Mediated Delivery Systems, pp. 1-23 (J. Rodwell, ed. 1988); Pietersz et al., in Antibody Mediated Delivery Systems, pp. 25-53 (J. Rodwell, ed. 1988); Bumol et al., in Antibody Mediated Delivery Systems, pp. 55-79 (J. Rodwell, ed. 1988); G. A. Pietersz & K. Krauer, 2 J. Drug Targeting, 183-215 (1994); R. V. J. Chari, 31 Adv. Drug Delivery Revs., 89-104 (1998); W. A. Blattler & R. V. J. Chari, in Anticancer Agents, Frontiers in Cancer Chemotherapy, 317-338, ACS Symposium Series 796; and I. Ojima et al eds, American Chemical Society 2001}. Цитотоксичні лікарські засоби, такі як метотрексат, даунорубіцин, доксорубіцин, вінкристин, вінбластин, мелфалан, мітоміцин С, хлорамбуцил, каліхеаміцин і майтанзиноїди були кон'юговані з рядом мишачих моноклональних антитіл. У деяких випадках молекули ліків були зв'язані з молекулами антитіл за допомогою проміжної молекули-носія, такої як сироватковий альбумін {Garnett et al., 46 Cancer Res. 2407-2412 (1986); Ohkawa et al., 23 Cancer Immunol. Immunother. 81-86 (1986); Endo et al., 47 Cancer Res. 1076-1080 (1980)}, декстран {Hurwitz et al., 2 Appl. Biochem. 25-35 (1980); Manabi et al., 34 Biochem. Pharmacol. 289-291 (1985); Dillman et al., 46 Cancer Res. 4886-4891 (1986); і Shoval et al., 85 Proc. Natl. Acad. Sci. U. S. A. 8276-8280 (1988)}, або поліглутамінова кислота {Tsukada et al., 73 J. Natl. Cane. Inst. 721-729 (1984); Kato et al., 27 J. Med. Chem. 1602-1607 (1984); Tsukada et al., 52 Br. J. Cancer 111-116 (1985)}. Існує широкий вибір лінкерів з доступної в цей час номенклатури для отримання таких імунокон'югатів, включаючи як відщеплювані, так і не відщеплювані лінкери. Випробування на цитотоксичність in vitro, однак, виявили, що з кон'югатами антитіло-лікарська речовина рідко досягається такий же рівень цитотоксичної дії, що і з вільними некон'югованими лікарськими речовинами. Це свідчить про те, що механізми, за допомогою яких молекули лікарського засобу вивільняються з кон'югованих антитіл, є дуже неефективними. Рання робота в галузі імунотоксинів показала, що кон'югати, утворені за допомогою дисульфідних містків між моноклональними антитілами і каталітично активними білковими токсинами були більш цитотоксичними, ніж кон'югати, що містять інші лінкери {Lambert et al., 260 J. Biol. Chem. 1203512041 (1985); Lambert et al., in Immunotoxins 175209 (A. Frankel, ed. 1988); Ghetie et al., 48 Cancer Res. 2610-2617 (1988)}. Підвищена цитотоксичність була пов'язана з високою внутрішньоклітинною концентрацією відновленого глютатіону, сприяючого ефективному розщепленню дисульфідного зв'язку між молекулою антитіла і токсином. Майтанзиноїди і каліхеаміцин були першими прикладами високо цитотоксичних лікарських засобів, які були зв'язані з моноклональними антитілами за допомогою дисульфідних зв'язків. Кон'югати антитіл і цих лікарських засобів, як показано, мають високу активність in vitro і виняткову протипухлинну активність на моделях ксенотрансплантатів людських пухлин на мишах {R. V. J. Chad et al., 52 Cancer Res., 127-131 (1992); С. Liu et al., 93, Proc. Natl. Acad. Sci, 8618-8623 (1996); L. M. Hinman et al., 53, Cancer Res, 3536-3542 (1993); and P. R. Hamann et al., 13, BioConjugate Chem., 40-46 (2002)} Лептоміцин В: є природним продуктом, спочатку виділеним зі Streptomyces spp., про що повідомлено в US 4771070 і US 4792522. Спочатку він був ідентифікований в результаті масового тестування на антимікробну активність, а потім ідентифікований як протипухлинний засіб (Komiyama et al., J. Antibiotics 1985, 38(3), 427-429 і US 2003/0162740). На молекулярному рівні лептоміцин В діє як інгібітор рецепторів ядерного експорту CRM1, який зв'язується з «транспортними білками» і впливає на ядерне переміщування «транспортних білків». На клітинному рівні лептоміцин В діє шляхом зупинення клітин в кінці фаз G1 і G2 клітинного циклу (Kalesse et al., Synthesis 2002, 8, 981-1003). Однак його надзвичайна токсичність відносно клітин ссавців (Hamamoto et al., J. Antibiotics 1983, 36(6), 639645) зробила його клінічне застосування неможливим. Таким чином, дуже бажано знизити токсичність похідних лептоміцину відносно нецільових клітин. Терапевтична ефективність похідних лептоміцину могла бути значно підвищена шляхом зміни 7 розподілу in vivo шляхом спрямованої доставки в місце пухлини, що дає в результаті знижену токсичність відносно нецільових тканин, і таким чином, більш низьку системну токсичність. Щоб досягнути цієї мети, розглянуте отримання кон'югатів похідних лептоміцину В із засобами, що зв'язуються з клітиною, які специфічно спрямовуються до пухлинних клітин, маючи на увазі вияв високої цільової специфічної цитотоксичності. Короткий опис винаходу Задача, на рішення якої спрямований даний винахід, полягає в отриманні похідних лептоміцину, які містять зв'язувальну групу, яка може бути ковалентно зв'язана із засобом, що зв'язується з клітиною, і відповідні кон'югати, що містять вказану похідну лептоміцину, зв'язану через лінкер з вказаним засобом, що зв'язується з клітиною. Вказані кон'югати забезпечують терапевтичні засоби, здатні активуватися і вивільнятися in vivo і бути доставленими до конкретних популяцій клітин цільовим чином. Щоб додатково підвищити розчинність у воді, у разі необхідності в зв'язувальну групу може бути введена роздільна поліетиленгліколева група. Сполуки даного винаходу можна використовувати в цитотоксичних кон'югатах, в яких засіб, що зв'язується з клітиною, з'єднаний з однією або більше із сполук даного винаходу. Засоби, що зв'язуються з клітиною включають антитіла і їх фрагменти, інтерферони, лімфокіни, вітаміни, гормони і фактори росту. Представлені також фармацевтичні композиції, що містять такі кон'югати. Цитотоксичні кон'югати можна використовувати в способі лікування пацієнта шляхом введення ефективної кількості представленої вище фармацевтичної композиції. Відповідно до виду клітин, з яким зв'язується вибраний засіб, що зв'язується з клітинами, багато які захворювання можна лікувати або in vivo, ex vivo або in vitro. Такі захворювання включають, наприклад, лікування багатьох видів раку, включаючи лімфоми, лейкемії, рак легенів, молочних залоз, товстого кишечнику, простати, нирок, підшлункової залози і тому подібного. Таким чином, представлені похідні лептоміцину, які застосовні для спрямованої доставки до конкретних видів клітин за допомогою кон'югації зі специфічним засобом, що зв'язується з клітиною. Короткий опис креслення Фіг. представляє цитотоксичність in vitro і специфічність кон'югату huC242-SSNPB-noxiznry лептоміцину з прикладу 6. Докладний опис винаходу Були знайдені похідні лептоміцину, які здатні до з'єднання із засобами, що зв'язуються з клітиною, в результаті чого терапевтична ефективність таких похідних підвищується шляхом зміни розподілу in vivo за допомогою спрямованої доставки даних похідних в місце пухлини, що дає в результаті знижену токсичність для нецільових тканин і, отже, більш низьку системну токсичність. Щоб досягти цієї мети синтезовані зразки похідних лептоміцину, які містять зв'язувальну групу для кон'югації похідної лептоміцину із засобом, що зв'язується з клітиною. Зв'язувальна група може 95959 8 містити роздільну поліетиленгліколеву групу. Зв'язувальну групу використовують для з'єднання із засобами, що зв'язуються з клітиною, і вона переважно містить дисульфідний зв'язок або сульфідний (або, який називається тут тіоефірний) зв'язок. Раніше було показано, що цей зв'язок високотоксичних лікарських засобів з антитілами з використанням розщеплюваного зв'язку, такого як дисульфідний зв'язок, забезпечує повне вивільнення активної лікарської речовини в клітині, і що такі кон'югати є цитотоксичними в залежності від антигенної специфічності {R. V. J. Chari et al., 52 Cancer Res. 127-131 (1992); R. V. J. Chari et al., 55 Cancer Res. 4079-4084 (1995) і патенти США №№ 5208020 і 5475092}. У даному винаході описаний синтез похідних лептоміцину, способи їх з'єднання з моноклональними антитілами і визначення цитотоксичності in vitro і специфічності таких кон'югатів. Таким чином, даний винахід представляє сполуки, застосовні для отримання терапевтичних засобів, спрямовані на усунення хворих і аномальних клітин, які треба знищити або лізувати, таких як пухлинні клітини, інфіковані вірусом клітини, інфіковані мікроорганізмами клітини, інфіковані паразитами клітини, аутоімунні клітини (клітини, які продукують аутоантитіла), активовані клітини (ті, які беруть участь у відторгнені трансплантату або в реакції трансплантат проти хазяїна) або будь-який інший вид хворих або аномальних клітин, в той же час, які виявляють мінімальні побічні дії. Таким чином, в даному винаході представлений синтез похідних лептоміцину, які можуть бути хімічно з'єднані із засобом, що зв'язується з клітиною і показано, що вони зберігають високу цитотоксичність при звільненні від захисної групи вихідних похідних лептоміцину. Ці сполуки, коли вони з'єднані із засобом, що зв'язується з клітиною, є цитотоксичними для клітин, з якими зв'язуються засоби, що зв'язуються з клітиною, і значно менш токсичні для нецільових клітин. Похідні лептоміцину даного винаходу Похідні лептоміцину за даним винаходом містять зв'язувальну групу, здатну до з'єднання даної похідної із засобом, що зв'язується з клітиною. Відповідно до даного винаходу «похідні лептоміцину» належать до представників сімейства лептоміцину, як визначено Kalesse et al. in Synthesis 2002, 8, 981-1003, і включають: лептоміцини, такі як лептоміцин А і лептоміцин В, калістатини, рат'ядони, такі як рат'ядон А і рат'ядон В, ангуїноміцини, такі як ангуїноміцини А, В, С, D, казузаміцини, лептостатини, лептофураніни, такі як лептофураніни А, В, С, D. Похідні лептоміцину А і В є переважними. Щоб з'єднати дану похідну із засобом, що зв'язується з клітиною, дана похідна повинна включати групу (зв'язувальну групу), яка дає можливість приєднати дані похідні із засобом, що зв'язується з клітиною шляхом такого зв'язку, як дисульфідний зв'язок, сульфідний (або, який називається тут тіоефірний) зв'язок, за допомогою нестійкої в кислому середовищі групи, нестійкої до світла групи, нестійкої до пептидази групи або нестійкої до естерази групи. Дані похідні створюють так, що вони містять групу, необхідну для з'єднання похідної 9 95959 10 лептоміцину із засобом, що зв'язується з клітиною через, наприклад, дисульфід ний зв'язок, тіоефірний зв'язок, нестійку в кислому середовищі групу, нестійку до світла групу, нестійку до пептидази групу або нестійку до естерази групу. Щоб додатково підвищити розчинність у водних розчинах, зв'язувальна група може містити поліетиленгліколевий роздільник. Переважно використовують сульфідний або дисульфідний зв'язок, тому що відновлювальне середовище цільових клітин приводить до розщеплення сульфіду або дисульфіду і вивільнення даних похідних з підвищенням в результаті цього цитотоксичності. Відповідно до переважного аспекту даний винахід представляє похідні лептоміцину, причому кінцева карбонільна функціональна група являє собою групу, яка робить можливим з'єднання даної похідної із засобом, що зв'язується з клітиною. Зв'язувальна група може містити поліетиленгліколевий роздільник. Приклади включають групи, які роблять можливими з'єднання через дисульфідний зв'язок, тіоефірний зв'язок, нестійку в кислому середовищі групу, нестійку до світла групу, нестійку до пептидази групу або нестійку до естерази групу і добре відомі фахівцям в даній галузі {див., наприклад, патент США 5846545, який включений сюди у вигляді посилання}. Переважними групами є ті, які роблять можливим з'єднання через дисульфідний зв'язок, наприклад, тіол або дисульфід. Можна використовувати змішані дисульфіди, що містять будь-яку кінцеву групу, що видаляється, таку як глютатіон, алкілтіо, таку як метилтіо, піридилтіо, арилтіо, нітропіридилтіо, гідроксикарбонілпіридилтіо, (нітро)гідроксикарбонілпіридилтіо і тому подібне, за умови, що такі дисульфіди здатні зазнавати реакції дисульфідного обміну для з'єднання похідної із засобом, що зв'язується з клітиною. Більш конкретно, похідні даного винаходу мають формулу (І): дe Ra і R'a представляють Η або -Alk; переважно Ra представляє -Alk, переважно метил, a R'a є Η; R17 є алкіл, необов'язково заміщений OR, CN, NRR', перфторалкіл; переважно, R17 є алкіл, більш переважно, метил або етил; R9 є алкіл, необов'язково заміщений OR, CN, NRR', перфторалкіл; переважно, R9 є алкіл, більш переважно, метил; X представляє -О- або -NR-; переважно, X є -NR-; Υ представляє -U-, -NR-U-, -O-U-, -NR-CO-U-, U-NR-CO-, -U-CO-, -CO-U-; переважно, коли X є О-, Υ представляє -U-, -NR-U-, -U-NR-CO-; де U вибрана з лінійних або розгалужених Alk-, -Alk(OCH2CH2)m-, -(OCH2CH2)m-Alk-, Alk(OCH2CH2)m-Alk-, -(OCH2CH2)m-, -циклоалкіл-, гетероцикліл-, -циклоалкіл-Alk-, -Alk-циклоалкіл-, гетероцикліл-Alk-, -Alk-гетероцикліл-; де m є ціле число від 1 до 2000; переважно U є лінійний або розгалужений -Alk-, Ζ є -Alk-; n дорівнює 0 або 1; переважно n дорівнює 0; Τ представляє Η, захисну тіол групу, таку як Ас, R1 або SR1, де R1 представляє Н, метил, Alk, циклоалкіл, необов'язково заміщений арил або гетероцикліл, або Τ представляє де Ra, R'a, R17, R9, Φ, Υ, Ζ, n дане визначення ви ставляє ціле число від 0 до 2; переважно, Alk представляє -(СН2)- або -С(СН3)2-; або представляють їх фармацевтично прийнятні солі, гідрати або гідратовані солі, або поліморфні кристалічні структури цих сполук, або їх оптичні ізомери, рацемати, діастереомери або енантіомери. Переважні сполуки можуть бути вибрані з: (2-метилсульфанілетил)аміду (2Е,10Е,12Е,16Z,18Е)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6 ще; переважно Т є Η або SR1, де R1 представляє Alk, більш переважно, метил; R, R' є ідентичні або різні і представляють Η або алкіл; Alk представляє лінійний або розгалужений алкіл; переважно, Alk представляє -(CH2-q(CH3)q)p-, де p представляє ціле число від 1 до 10; і q пред 11 оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти, біс-[(2-меркаптоетил)аміду (2Е,10Е,12Е,16Z,18Е)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти], (2-меркаптоетил)аміду (2Е,10Е,12Е,16Z,18Е)(R)-6-гідрокси-3,5,7,9,11,15,17-гептаметил-19((2S,3S)-3-метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)8-оксононадеку-2,10,12,16,18-пентаєнової кислоти; (2-метилдисульфаніл етил)аміду (2Е,10Е,12Е,16Z,18Е)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти; (2-метил-2-метилдисульфанілпропіл)аміду (2E,10E,12E,16Z,18E)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти; (2-меркапто-2-метилпропіл)аміду (2Е, 10Е, 12Е, 16Z, 18Е)-(R)-6-гідрокси-3,5,7,9,11,15,17гептаметил-19-((2S,3S)-3-метил-6-оксо-3,6дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти або їх фармацевтично прийнятних солей, гідратів або гідратованих солей, або поліморфних кристалічних структур цих сполук, або їх оптичних ізомерів, рацематів, діастереомерів або енантіомерів. Відповідно до опису алкіл включає лінійні або розгалужені С1-С20 алкіли. Приклади лінійних алкілів включають метил, етил, пропіл, бутил, пентил і гексил. Приклади розгалужених алкілів включають ізопропіл, ізобутил, вторинний бутил, трет-бутил, ізопентил і 1-етилпропіл. Приклади циклоалкілів, тобто циклічних алкілів, включають циклопропіл, циклобутил, циклопентил і циклогексил. Приклади арилів включають феніл і нафтил. Приклади заміщених арилів включають арили, такі як феніл або нафтил, заміщені алкільними групами, галогенами, такими як СІ, Br, F, нітрогрупами, аміногрупами, групами сульфонової кислоти, групами карбонової кислоти, гідроксильними групами або алкоксигрупами. Гетероцикліли належать до, необов'язково, ароматичних кілець, що містять один або більше гетероатомів, вибраних з О, N і S, і їх приклади включають фурил, піроліл, піридил (наприклад, 2заміщену піримідинову групу) і тіофен. Сульфідо- або дисульфідовмісні і меркаптовмісні похідні даного винаходу можна оцінювати за їх здатністю придушувати проліферацію ліній різних небажаних клітин in vitro в умовах інкубації. Лінії клітин, такі як, наприклад, лінії клітин Ramos і HL60, можна легко використовувати для оцінки цитотоксичності цих сполук. Клітини, які треба оцінити, можуть бути піддані впливу даних сполук протягом 24 годин, і частину клітин, що вижила, оцінювали в прямих дослідженнях відомими методами. Значення ІС50 потім можна вирахувати за результатами цих досліджень. Відповідно до опису вираз «з'єднана із засобом, який зв'язується з клітиною» належить до похідних лептоміцину, що містить щонайменше 95959 12 одну зв'язувальну групу або її попередник, прийнятні для з'єднання вказаних похідних із засобом, що зв'язується з клітиною; переважними зв'язувальними групами є тіольні, сульфідні або дисульфідні зв'язувальні групи або їх попередники. Відповідно до опису вираз «з'єднана із засобом, який зв'язується з клітиною» належить до кон'югатної молекули, що містить щонайменше одну похідну лептоміцину, з'єднану із засобом, що зв'язується з клітиною через прийнятну зв'язувальну групу або її попередник; переважними зв'язувальними групами є тіольні, сульфідні або дисульфідні зв'язувальні групи або їх попередники. Відповідно до опису термін «пацієнт» належить або до тварини, такої як тварина, цінна для розведення, або переважно, людини або дитини, які уражені одним або більше із захворювань і патологічних станів, описаних тут, або мають потенційну загрозу їх вияву. Відповідно до опису «терапевтично ефективна кількість» належить до кількості сполуки даного винаходу, яка ефективна для запобігання, зниження, усунення, лікування або контролю симптомів описаних тут захворювань і патологічних станів. Термін «контроль», як мається на увазі, належить до всіх процесів, за якими може бути сповільнення, переривання, припинення або зупинка прогресування захворювань і патологічних станів, описаних тут, але немає необхідності показувати повне усунення симптомів захворювання і патологічного стану, і, як мається на увазі, включає профілактичне лікування. Відповідно до опису термін «фармацевтично прийнятний» належить до тих сполук, речовин, ексципієнтів, композицій або дозованих форм, які в рамках здорової медичної думки придатні для контакту з тканинами людини і тварин без зайвої токсичності, подразнення, алергійних реакцій або інших проблемних ускладнень, відповідних розумному співвідношенню благотворна дія/ризик несприятливої дії. Відповідно до опису «фармацевтично прийнятні солі» належать до похідних описаних сполук, для яких вихідна сполука модифікується шляхом отримання їх солей з кислотами або основами. Фармацевтично прийнятні солі включають звичайні нетоксичні солі або солі четвертинного амонію вихідної сполуки, що утворюються, наприклад, з нетоксичними неорганічними або органічними кислотами. Наприклад, такі загальноприйняті нетоксичні солі включають ті, які отримують з неорганічними кислотами, такими як соляна, бромистоводнева, сірчана, сульфамінова, фосфорна, азотна і тому подібне; і солі, отримувані з органічними кислотами, такими як оцтова, пропіонова, янтарна, виннокам'яна, лимонна, метансульфонова, бензолсульфонова, глюкуронова, глютамінова, бензойна, саліцилова, толуолсульфонова, щавлева, фумарова, малеїнова, молочна і тому подібне. Крім того, солі приєднання включають амонієві солі, такі як трометамінова, меглумінова, еполамінова і т. д., солі металів, такі як солі натрію, калію, кальцію, цинку або магнію. Фармацевтично прийнятні солі даного винаходу можна синтезувати з вихідної сполуки, яка міс 13 тить лужну або кислотну групу, звичайними хімічними методами. Звичайно такі солі можна отримати за реакцією форм вільних кислоти або основи цих сполук зі стехіометричною кількістю відповідної основи або кислоти у воді або в органічному розчиннику, або в суміші цих двох розчинників. Звичайно переважні неводні середовища, подібні до ефіру, етилацетату, етанолу, ізопропанолу або ацетонітрилу. Списки прийнятних солей представth лені в Remington's Pharmaceutical Sciences, 17 ed., Mack Publishing Company, Easton, PA, 1985, p. 1418, опис якого включений сюди у вигляді посилання. Відповідно до ще одного об'єкта, даний винахід належить до способу отримання сполук даного винаходу. Сполуки і спосіб даного винаходу можна здійснити різними шляхами, добре відомими фахівцям в даній галузі. Дані сполуки можна синтезувати, наприклад, шляхом застосування або адаптації способів, описаних нижче, або їх варіантів, що зрозуміло фахівцеві в даній галузі. Відповідні модифікації і заміни легко побачити, і вони добре відомі, або їх легко знайти в науковій літературі фахівцям в даній галузі. Зокрема, такі методи можна знайти в R. С. Larock, Comprehensive Organic Transformations, Wiley-VCH Publishers, 1999. Буде зрозуміло, що сполуки даного винаходу можуть містити один або більше асиметрично заміщені атоми вуглецю, і їх можна виділити в оптично активній або рацемічній формі. Таким чином, якщо конкретно не вказана специфічна стереохімія або ізомерна форма, то маються на увазі хіральні, діастереомерні, рацемічні форми і всі геометричні ізомерні форми структури. Фахівцям в даній галузі добре відомо, як отримати і виділити такі оптично активні форми. Наприклад, суміші стереоізомерів можна поділити за стандартними методиками, включаючи, але не обмежуючи цим, розділення рацемічних форм, нормальну, із зворотною фазою і хіральну хроматографію, переважне утворення солі, перекристалізацію і тому подібне, або шляхом хірального синтезу з хіральних вихідних речовин або шляхом спрямованого синтезу цільових хіральних центрів. Сполуки даного винаходу можна отримати з використанням різних методик синтезу. Реагенти і вихідні матеріали доступні для придбання або їх легко синтезувати добре відомими методами фахівцеві в даній галузі. Всі заміни, якщо не указано інакше, визначені раніше. У ході реакцій, описаних тут далі, може виникнути необхідність захистити реакційноздатні функціональні групи, наприклад, гідроксильні, аміно-, іміно-, тіо- або карбоксильні групи, коли вони бажані в кінцевому продукті, щоб уникнути їх небажаної участі в реакціях. Відповідно до стандартної 95959 14 практики можна використовувати звичайні захисні групи, наприклад, див., Т. W. Greene and P. G. Μ. rd Wuts in Protective Groups in Organic Chemistry, 3 ed., John Wiley and Sons, 1999; J. F. WmcOmie in Protective Groups in Organic Synthesis, Plenum Press, 1973. Деякі реакції можна проводити в присутності основи. Не існує конкретного обмеження основи, яке можна використовувати в даній реакції, і будьяку основу, звичайно використовувану при реакціях цього типу, однаково можна використовувати тут за умови, що вона не має побічної дії на інші частини молекули. Приклади прийнятних основ включають: гідроксид натрію, карбонат калію, триетиламін, гідриди лужних металів, такі як гідрид натрію і гідрид калію; сполуки алкіллітію, такі як метиллітій і бутиллітій; і алкоксиди лужних металів, такі як метоксид натрію і етоксид натрію. Звичайно реакції здійснюють у прийнятному розчиннику. Можна використовувати ряд розчинників, за умови, що він не створює побічної дії на реакцію або на реагенти, які беруть участь в ній. Приклади прийнятних розчинників включають: вуглеводні, які можуть бути ароматичними, аліфатичними або циклоаліфатичними вуглеводнями, такими як гексан, циклогексан, бензол, толуол і ксилол; аміди, такі як диметилформамід; спирти, такі як етанол і метанол, і ефір, такий як діетиловий ефір і тетрагідрофуран. Реакції можуть відбуватися в широкому інтервалі температур. Загалом виявлено, що зручно проводити реакцію при температурі від 0°С до 150°С (більш переважно, від приблизно кімнатної температури до 100°С). Час, необхідний для реакції, може також коливатися в широких межах, в залежності від багатьох факторів, особливо від температури реакції і природи реагентів. Однак за умови, що реакцію здійснюють в переважних умовах, описаних вище, звичайно вистачить період від 3 годин до 20 годин. Дану сполуку, отриману таким чином, можна виділити з реакційної суміші звичайними способами. Наприклад, дані сполуки можна виділити шляхом відгонки розчинника з реакційної суміші або, якщо необхідно, після відгонки розчинника з реакційної суміші виливання осаду в воду з подальшим екстрагуванням з органічним розчинником, який не змішується з водою, і відгонки розчинника з екстракту. Крім того, даний продукт можна, якщо бажано, додатково очистити різноманітними добре відомими методами, такими як перекристалізація, переосадження або різні хроматографічні методики, особливо колонковою хроматографією або препаративною тонкошаровою хроматографією. Спосіб отримання сполук формули (І) включає стадію взаємодії відповідних сполук формули (II) і (III): 15 95959 де Ra, R'a, R17, R9, Φ, Υ, Ζ, Τ, n мають значення, вказані до формули (І). В основному цю реакцію можна провести в присутності звичайних зв'язувальних реагентів, включаючи реагенти, які придушують рацемізацію, таких як НОВТ і/або дегідратуючі засоби, використовувані для активації карбонової кислоти відносно утворення аміду або складного ефіру, такого як DIC, DCC. Звичайно цю реакцію можна здійснити у прийнятному органічному розчиннику, такому як дихлорметан. Коли у формулі (І) Τ є Н, реакцію альтернативно можна здійснити з Ν-ацілюючою речовиною, такою як півалоїлхлорид, в присутності основи, включаючи органічні основи, такі як триетиламін. Альтернативно, коли в формулі (І) Τ є Н, сполуки формули (І) можна отримати з відповідних сполук формули (І), де Τ є S-R1, в присутності речовини, яка відновлює дисульфідні зв'язки, такої як триалкілфосфіни, і більш конкретно, ТСЕР. Цю реакцію можна звичайно провести у водному середовищі, такому як суміші органічного розчинника і води, наприклад, ТГФ/води. Димерні сполуки формули (І) можуть бути отримані за реакцією відповідної сполуки формули (II) з відповідною сполукою формули (IV): HX-Y-(Z)n-S-S-(Z)n-Y-XH (IV) в присутності звичайних зв'язувальних реагентів, включаючи реагенти для придушення рацемізації, таких як НОВТ, і/або засобів дегідратації, використовуваних для активації карбонової кислоти відносно утворення аміду і складного ефіру, таких як DIC, DCC. Звичайно цю реакцію можна здійснити у прийнятному органічному розчиннику, такому як дихлорметан. Даний спосіб може також включати додаткову стадію виділення отриманого продукту. Даний винахід належить також до кон'югату похідної лептоміцину, що включає засіб, який зв'язується з клітиною, з'єднаний з однією або більше з похідних лептоміцину за даним винаходом за допомогою лінкеру, що містить вказану зв'язувальну групу. Переважно, засоби, які зв'язуються з клітиною, є антитілами або їх фрагментами. 16 Переважно лінкер містить групу -S- або -S-S-. Отримання засобів, що зв'язуються з клітиною Засоби, що зв'язуються з клітиною можуть представляти собою будь-які відомі в цей час засоби такого виду або які стануть відомими і включають пептиди і не пептиди. В основному цими засобами можуть бути антитіла (зокрема, моноклональні антитіла) або фрагменти антитіл, які містять щонайменше один зв'язувальний сайт, лімфокіни, гормони, фактори росту, молекули транспорту поживних речовин (такі як трансферин) або будь-які інші молекули або речовини, що зв'язуються з клітиною. Більш конкретні приклади речовин, що зв'язуються з клітиною, які можна використовувати, включають: - моноклональні антитіла; - одноланцюгові антитіла; - фрагменти антитіл, такі як Fab, Fab', F(ab')2 і Fv {Parham, 131 J. Immunol. 2895-2902 (1983); Spring et al., 113 J. Immunol. 470-478 (1974); Nisonoff et al., 89 Arch. Biochem. Biophys. 230-244 (1960)}; - інтерферони; - пептиди; - лімфокіни, такі як IL-2, IL-3, IL-4, IL-6; - гормони, такі як інсулін, TRH (релізинггормони тиротропіну), MSH (меланоцитостимулюючий гормон), стероїдні гормони, такі як андрогени і естрогени; - фактори росту і колонієстимулюючі фактори, такі як EGF, TGF, інсуліноподібний фактор росту (IGF-I, IGF-II), G-CSF, M-CSF і GM-CSF {Burgess, 5 Immunology Today 155-158 (1984)}; вітаміни, такі як фолат і - трансфернії {O'Keefe et al., 260 J. Biol. Chem. 932-937 (1985)}. Технологія моноклональних антитіл дає можливість продукції надзвичайно селективних засобів, що зв'язуються з клітиною у вигляді специфічних моноклональних антитіл. Зокрема, фахівцям добре відомі методики створення моноклональних антитіл, продукованих імунізованими мишами, щурами, хом'ячками або будь-якими іншими ссавцями за допомогою цікавлячого антигену, такого як інтактні цільові клітини, антигени, виділені з цільових клітин, цілі віруси, ослаблені цілі віруси і вірусні білки, такі як білки вірусної оболонки. 17 Вибір відповідного засобу, що зв'язується з клітиною є справою переваги, яка залежить від конкретної популяції клітин, на які він повинний бути спрямований, але в основному переважні моноклональні антитіла, якщо доступні для придбання відповідні одні з них. Наприклад, моноклональні антитіла MY9 є мишачими антитілами IgG1, які специфічно зв'язуються з антигеном CD33 {J. D. Griffin et al., 8 Leukemia Res., 521 (1984)}, і їх можна використовувати, якщо цільові клітини експресують CD33, як при захворюванні гострою мієлогенною лейкемією (ГМЛ). Подібним же чином моноклональні антитіла до В4 є мишачим IgG1, який зв'язується з антигеном CD19 на В-клітинах {Nadler et al., 131 J. Immunol. 244-250 (1983)}, і його можна використовувати, якщо цільові клітини є В-клітинами або хворими клітинами, які експресують цей антиген, такі як лімфоми не-Ходжкіна або хронічної лімфобласної лейкемії. Крім того, GM-CSF, який зв'язується з мієлоїдними клітинами, можна використовувати як засіб, що зв'язується з хворими клітинами при гострій мієлогенній лейкемії. IL-2, який зв'язується з активованими Т-клітинами, можна використовувати для профілактики відторгнення пересадженого трансплантату, для терапії і попередження реакції трансплантат проти хазяїна і для лікування гострої Т-клітинної лейкемії. MSH, які зв'язуються з меланоцитами, можна використовувати для лікування меланоми. Отримання кон'югатів Кон'югати похідних і засобу, який зв'язується з клітиною можуть бути утворені з використанням будь-яких методик, відомих в цей час або які будуть розроблені пізніше. Звичайно спосіб отримання кон'югатів даного винаходу включає стадію взаємодії похідної лептоміцину даного винаходу із засобом, який зв'язується з клітиною в присутності реагенту, що містить функціональні групи, реакційноздатні відносно зв'язувальної групи похідної і засобу, який зв'язується з клітиною, так що дана похідна і засіб, який зв'язується з клітиною з'єднані один з одним через лінкер, що містить вказану зв'язувальну групу. Переважно, вказаний лінкер містить сульфідний або дисульфідний зв'язок. Можна отримати похідні, які містять вільну аміногрупу, а потім з'єднати з антитілами або іншим засобом, який зв'язується з клітиною через нестійкий в кислому середовищі лінкер або нестійкий до світла лінкер. Похідні можна конденсувати з пептидом, що має відповідну послідовність, а потім з'єднати із засобом, який зв'язується з клітиною з отриманням нестійкого до пептидази лінкеру. Можна отримати цитотоксичні сполуки, які містять первинну гідроксильну групу, яка може бути сукцинільована і з'єднана із засобом, що зв'язується з клітиною з отриманням кон'югату, який може бути розщеплений внутрішньоклітинними естеразами із звільненням вільної похідної лептоміцину. Переважно, похідні синтезують так, що вони містять вільну або захищену тіолову групу з ПЕГ-вмісною роздільною групою або без неї, і потім одна або більше з похідних, що містять сульфідну, дисульфідну або тіолову групу, кожна, ковалентно з'єд 95959 18 нана із засобом, що зв'язується з клітиною через дисульфідний або тіоефірний зв'язок. Типовими кон'югатами даного винаходу є кон'югати похідних лептоміцину з антитілами, фрагментами антитіл, епідермальним фактором росту (EGF), стимулюючим меланоцити гормоном (MSH), тиреоїдним стимулюючим гормоном (TSH), естрогеном, аналогами естрогену, андрогеном і аналогами андрогену. Типові приклади отримання різних кон'югатів похідних лептоміцину і речовин, що зв'язуються з клітиною, описані нижче. Дисульфідні лінкери. Антитіла huMy-9-б є генетично гуманізованою формою мишачих моноклональних антитіл Му-9-6, спрямовані проти CD33 антигену, виявленого на поверхні людських мієлоїдних клітин, включаючи більшість випадків гострої мієлоїдної лейкемії (ГМЛ) (Е. J. Favaloro, K. F. Bradstock, A. Kabral, P. Grimsley & M. С Berndt, Disease Markers, 5(4): 215 (1987); M. G. Hoffee, D. Tavares, R. J. Lutz, Robert J., PCT Int. Appl. (2004) WO 2004043344). Му-9-6 можна використовувати для отримання кон'югатів. Антитіла модифікують N-сукцинімідил-3-піридилдитіопропіонатом, як описано раніше {J. Carlsson, Η. Drevin & R. Axen, Biochem. J., 173-723 (1978)}, щоб ввести, в середньому 4 піридилдитіогрупи на молекулу антитіла. Модифіковані антитіла приводять у взаємодію з тіоловмісною похідною лептоміцину з отриманням з'єднаного через дисульфід кон'югату. Тіоефірні лінкери. Тіоловмісні похідні даного винаходу можуть бути з'єднані з антитілами і іншими засобами, які зв'язуються з клітиною через тіоефірний зв'язок, як описано раніше (патент США № 5208020). Антитіла або інші засоби, які зв'язуються з клітиною можуть бути модифіковані відомою або доступною для придбання сполукою, такою як N-сульфосукцинімідил-4-(5-нітро-2піридилдитіо)бутаноат (SSNPB), N-сукцинімідил-4(малеімідометил)циклогексанкарбоксилат (SMCC), N-сукцинімідил-4-(N-малеімідометил)циклогексан1-карбокси-(6-амідокапроат), яка є «довголанцюговим» аналогом SMCC (LC-SMCC). Ці зшивні реагенти утворюють нерозщеплювальні лінкери, які отримуються з малеімідовмісних груп. Зшивні реагенти, що містять групу на основі галогенацетилу, включають N-сукцинімідил-4(йодацетил)амінобензоат (SIAB), Nсукцинімідилйодацетат (SIA), Nсукцинімідилбромацетат (SBА) і N-сукцинімідил-3(бромацетамідо)пропіонат (SBAP). Ці зшивні реагенти утворюють нерозщеплювальні лінкери з груп на основі галогенацетилу. Модифікований засіб, який зв'язується з клітиною можна приводити у взаємодію з тіоловмісною лікарською речовиною з отриманням з'єднаного через тіоефір кон'югату. Нестійкі в кислоті лінкери. Похідні лептоміцину, які містять аміногрупу даного винаходу можуть бути з'єднані з антитілами і іншими засобами, які зв'язуються з клітиною через нестійкий до кислоти лінкер, як описано раніше {W. A. Blatter et al., Biochemistry 24, 1517-1524 (1985); патенти США №№ 4542225; 4569789; 4618492; 4764368}. Подібним же чином похідна лептоміцину даного винаходу, яка містить гідразидну групу, може 19 бути з'єднана з вуглеводною частиною антитіл і інших засобів, які зв'язуються з клітиною через нестійкий до кислоти гідразоновий лінкер {відносно прикладів гідразонових лінкерів див. В. С. Laguzza et al., J. Med. Chem, 32, 548-555(1989); R. S. Greenfield et al., Cancer Res., 50, 6600-6607 (1990)}. Нестійкі до світла лінкери. Похідні лептоміцину даного винаходу, які містять аміногрупу, можуть бути з'єднані з антитілами і іншими засобами, які зв'язуються з клітиною через нестійкий до світла лінкер, як описано раніше {Р. Senter et al., Photochemistry and Photobiology, 42, 231-237 (1985); патент США № 4625014}. Нестійкі до пептидази лінкери. Похідні лептоміцину даного винаходу, які містять аміногрупу, можуть бути з'єднані з антитілами і іншими засобами, що зв'язуються з клітиною через пептидні роздільні групи. Раніше показано, що короткі пептидні роздільні групи між лікарськими засобами і макромолекулярними білковими носіями стабільні в сироватці, але легко гідролізуються внутрішньоклітинними пептидазами {A. Trouet et al., Proc. Natl. Acad. Sci., 79, 626-629 (1982)}. Похідні лептоміцину даного винаходу, які містять аміногрупу можна конденсувати з пептидами, використовуючи конденсуючі засоби, такі як 1-етил-3-(3диметиламінопропіл)карбодіімід-НСl (EDC-HCl) з отриманням пептидної похідної, яка може бути з'єднана із засобом, який зв'язується з клітиною. Нестійкі до естерази лінкери. Похідні лептоміцину даного винаходу, які несуть гідроксіалкільну групу, можуть бути сукцинільовані янтарним ангідридом, а потім з'єднані із засобом, який зв'язується з клітиною з отриманням кон'югату, який може бути розщеплений внутрішньоклітинними естеразами зі звільненням вільної лікарської речовини {Відносно прикладів див.: Е. Aboud-Pirak et al., Biochem. Pharmacol, 38, 641-648 (1989)}. Кон'югати антитіл, фрагменти антитіл, білкових або пептидних гормонів, білкових або пептидних факторів росту і інших білків отримують таким же чином за відомими методиками. Наприклад, пептиди і антитіла можуть бути модифіковані за допомогою зшивних реагентів, таких як Nсукцинімідил-3-(2-піридилдитіо)пропіонат, Nсукцинімідил-4-(2-піридилдитіо)пентаноат (SPP), 4сукцинімідилоксикарбоніл--метил--(2піридилдитіо)толуол (SMPT), N-сукцинімідил-3-(2піридилдитіо)бутират (SDPB), сукцинімідилпіридилдитіопропіонат (SPDP), складний ефір Nгідросукциніміду4-(2-піридилдитіо)бутанової кислоти (SPDB), сукцинімідил-4-[Nмалеімідометил]циклогексан-1-карбоксилат (SMCC), N-сульфосукцинімідил-3-(2-(5нітропіридилтіо)бутират (SSNPB), 2-імінотіолан або S-ацетилянтарний ангідрид, відомими методами. Див., Carlsson et al., 173, Biochem. J. 723-737 (1978); Blattler et al., 24, Biochem. 1517-1524 (1985); Lambert et al., 22, Biochem. 3913-3920 (1983); Klotz et al., 96, Arch. Biochem. Biophys., 605 (1962); and Liu et al., 18, Biochem., 690 (1979), Blakey and Thorpe, 1 Antibody, Immunoconjugates & Radiopharmaceuticals, 1-16 (1988), Worrell et al. 1 AntiCancer Drug Design 179-184 (1986). Вільна або 95959 20 захищена речовина, яка містить тіол і зв'язується з клітиною, отримана таким чином, потім приводять у взаємодію з похідною лептоміцину, яка містить дисульфідну або тіолову групу, з отриманням кон'югатів. Кон'югати, отримані вказаними вище способами можуть бути очищені стандартною колонковою хроматографією або ВЕРХ. Переважними кон'югатами між моноклональними антитілами або засобами, що зв'язуються з клітиною і похідними лептоміцину даного винаходу є ті, які з'єднані через дисульфідний зв'язок або тіоефірний зв'язок, як обговорено вище. Такі кон'югати, які зв'язуються з клітиною, отримують відомими методами, такими як модифікація моноклональних антитіл сукцинімідилпіридилдитіопропіонатом (SPDP) {Carlsson et al., 173, Biochem. J., 723-737 (1978)}. Отримувану тіопіридильну групу потім переміщують шляхом обробки похідної лептоміцину, яка містить тіол, з отриманням кон'югатів, зв'язаних через дисульфід. Кон'югати, які містять від 1 до 10 похідних лептоміцину, приєднаних через дисульфідний місток, легко отримати цим способом. Кон'югація за цим способом повністю описана в патенті США № 5585499, який включений у вигляді посилання. Цитотоксичність in vitro кон'югатів між засобами, які зв'язуються з клітиною і похідними лептоміцину даного винаходу Цитотоксичність похідних лептоміцину даного винаходу і їх кон'югатів із засобами, що зв'язуються з клітиною, можна оцінити після відщеплення захисної групи і перетворення в активну лікарську речовину. Цитотоксичність відносно ліній клітин, які не зв'язуються, таких як Namalwa і HL60 може бути оцінена шляхом зворотної екстраполяції за кривими проліферації клітин, як описано у Goldmacher et al., 135, J. Immunol., 3648-3651 (1985). Цитотоксичність цих сполук відносно зв'язувальних ліній клітин, таких як А-375 і SCaBER, можна визначити за допомогою клоногенних досліджень, як описано у Goldmacher et al., 102 J. Cell Biol. 1312-1319(1986). Терапевтичний засіб і спосіб придушення росту вибраних клітинних популяцій Даний винахід представляє також терапевтичний засіб для придушення росту вибраних клітинних популяцій, що містить: (a) цитотоксичну кількість однієї або більше з описаних вище похідних лептоміцину, з'єднаних із засобом, що зв'язується з клітиною, і (b) фармацевтично прийнятний носій, розріджувач або ексципієнт. Подібним же чином даний винахід представляє спосіб придушення росту вибраних популяцій клітин, що включає контактування клітинної популяції або тканини, в якій передбачається вміст клітин з вказаної клітинної популяції з цитотоксичною кількістю цитотоксичного засобу, що містить одну або більше з описаних вище похідних лептоміцину, з'єднаних із засобом, що зв'язується з клітиною. Цитотоксичний засіб отримують, як описано вище. 21 Прийнятні фармацевтично прийнятні носії, розріджувачі і ексципієнти добре відомі і можуть бути встановлені фахівцями в даній галузі, як того вимагають клінічні обставини. Приклади прийнятних носіїв, розріджувачів і/або ексципієнтів включають: (1) фосфатнобуферний фізіологічний розчин Dulbecco, pH приблизно 7,4, який містить приблизно від 1 мг/мл до 25 мг/мл людського сироваткового альбуміну, (2) 0,9% фізіологічний розчин (0,9% в/о NaCl) і (3) 5% (в/о) розчин декстрози. Спосіб придушення росту вибраних клітинних популяцій може бути практично здійснений in vitro, in vivo або ex vivo. Приклади використання in vitro включають обробку клітинних культур, щоб знищити всі клітини за винятком бажаних варіантів, які не експресують антиген-мішень; або щоб знищити варіанти, які експресують небажаний антиген. Умови неклінічного використання in vitro легко визначать фахівці в даній галузі. Приклади використання ex vivo включають обробку аутологічного кісткового мозку перед їх трансплантацією тому ж самому пацієнту, щоб знищити хворі або злоякісні клітини: обробка кісткового мозку перед його трансплантацією, щоб знищити компетентні Т-клітини і запобігти реакції трансплантат проти хазяїна (GVHD). Клінічну обробку ex vivo для видалення пухлинних клітин або лімфоїдних клітин з кісткового мозку перед аутологічною трансплантацією при лікуванні раку або при лікуванні аутоімунного захворювання, або для видалення Т-клітин і інших лімфоїдних клітин з алогенного кісткового мозку або тканини перед трансплантацією, щоб запобігти GVHD можна здійснювати таким чином. Кістковий мозок беруть у пацієнта або іншої особи і потім інкубують в середовищі, що містить сироватку, в яке додають цитотоксичний засіб даного винаходу, концентрації в інтервалі від приблизно 10 мкМ до 1 пМ, протягом від приблизно 30 хвилин до приблизно 48 годин при приблизно 37°С. Точні умови концентрації і часу інкубації (=доза) легко визначить фахівець в даній галузі. Після інкубації клітини кісткового мозку промивають середовищем, що містить сироватку, і повертають пацієнту шляхом внутрішньовенного вливання відомими методами. При обставинах, коли пацієнт отримує інше лікування, таке як курс руйнуючої хіміотерапії або загального опромінення організму між часом забору кісткового мозку і зворотного вливання оброблених клітин, оброблені клітини кісткового мозку зберігають замороженими в рідкому азоті з використанням стандартного медичного обладнання. Для клінічного використання in vivo цитотоксичний засіб даного винаходу буде застосовуватися у вигляді розчинів, які випробовують на стерильність і рівні ендотоксину, або у вигляді ліофілізованого твердого препарату, який може бути знову розчинений в стерильній воді для ін'єкції. Приклади відповідних методик введення кон'югату представлені наступним. Кон'югати дають щотижня протягом 6 тижнів у вигляді швидкого в/м введення. Навантажувальні дози дають в 50-400 мл нормального фізіологічного розчину, до якого може 95959 22 бути доданий людський сироватковий альбумін (наприклад, 0,5-1 мл концентрованого розчину людського сироваткового альбуміну, 100 мг/мл). Дозування буде складати приблизно від 50 мкг до 10 мг/кг ваги тіла на тиждень, в/м (в інтервалі від 10 мкг до 100 мг/кг на ін'єкцію). Через шість тижнів після лікування пацієнт може отримати другий курс терапії. Конкретні клінічні режими і методики відносно шляху введення, ексципієнтів, розріджувачів, дозування, термінів і т. д. можуть бути визначені фахівцем в даній галузі, як того вимагає клінічна ситуація. Приклади патологічних станів, які можна лікувати за методами знищення вибраних клітинних популяцій in vivo або ex vivo, включають злоякісне захворювання будь-якого типу, включаючи, наприклад, рак легенів, молочних залоз, товстого кишечнику, простати, нирок, підшлункової залози, яєчників і лімфатичних органів; меланом; аутоімунних захворювань, таких як системний червоний вовчак, ревматоїдний артрит і розсіяний склероз; відторгнення трансплантату, таке як відторгнення пересадженої нирки, відторгнення трансплантату печінки, відторгнення трансплантату легенів, відторгнення трансплантату серця і відторгнення трансплантату кісткового мозку; реакцію трансплантату проти хазяїна; вірусні інфекції, такі як ЦМВ (цитомегаловірусна інфекція), інфікування ВІЛ, СНІД і т. д.; бактерійну інфекцію; і паразитарні інфекції, такі як лямбліоз, амебіаз, шистосоміаз і інші, що визначається фахівцем в даній галузі. Приклади Даний винахід тепер буде проілюстрований шляхом звернення до необмежуючих прикладів. Якщо не указано інакше, всі проценти, співвідношення, частини і т. д. дані по вазі. Матеріали і методи Температуру плавлення визначали, застосовуючи електротермічний прилад і не коректували. Спектри ЯМР реєстрували на спектрометрі Bruker AVANCE400 (400 МГц). Хімічні зсуви представлені в м. ч. по відношенню до ТМС як внутрішній стандарт. Мас спектри отримували, використовуючи систему Bruker Esquire 3000. Ультрафіолетові спектри реєстрували на спектрофотометрі Hitachi U1200. ВЕРХ виконували, використовуючи систему Beckman Coulter GOLD 125, обладнану системою детектора варіабельної довжини хвилі Backman Coulter GOLD 168 і Waters RADIALPAK (колонка С-18 із зворотною фазою). Тонкошарову хроматографію виконували на пластинках силікагелю для ТШХ Analtech GF. Силікагель для колонкової флеш-хроматографії отриманий від Baker. Тетрагідрофуран сушили відгонкою над металевим натрієм. Диметилактамід і диметилформамід сушили дистиляцією над гідридом кальцію під зниженим тиском. Всі використані розчинники були марки для реактивів або марки для ВЕРХ. Лінії ракових клітин людини HL60, Namalwa, A375, COLO205 і Ramos отримані з Американської колекції типових культур (АТСС). Kara являє собою лінію клітин мишачої пухлини, яка була стабільно трансфікована людським CD33 антигеном. Експериментальна частина 23 95959 24 Mac-спектрометричний аналіз проводили таким чином: ЕІ-СІ Аналіз: пряме введення (DCI = нанесення зразка на волокно). Мас-спектрометр Finnigan SSQ7000; масінтервал m/z=29-900; енергія електронів 70 еВ; температура джерела 70°С; реагентний газ СІ аміачний; ЕІ = іонізація за допомогою електронного удару; СІ = хімічна іонізація. Аналіз з електророзпиленням: (з реєстрацією + позитивних іонів: ES ; з реєстрацією негативних іонів: ES ) LC-MS-DAD-ELSD: МС: Waters-Micromass ZQ; РХ: Agilent HP 1100; Колонка РХ Xbridge Waters С18, 350 мм, 2,5 мкм; елюент: градієнт води (з 0,1% мурашиної кислоти) + ацетонітрил; УФ: DAD (=200-400 нм). Приклад 1 (2-Метилсульфанілетил)амід (2Е,10Е,12Е,16Ζ,18E)-(R)-6-гiдрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил~6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти У розчин 20 мг (2Е,10Е,12Е,16Z,18Е)-(R)-6гідрокси-3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)-8оксононадеку-2,10,12,16,18-пентаєнової кислоти і 5,6 мг 1-гідроксибензотриазолу в 0,3 мл дихлорметану вводять при температурі приблизно 20°С 7,65 мкл ДІК (Ν,Ν'-діізопропілкарбодіімід), потім 5,2 мг 2-(тіометил)етиламіну. Реакційну суміш перемішують при температурі близько 20°С протягом 20,5 годин, потім очищують прямим нанесенням на 2 пластини для препаративної ТШХ на силікагелі (товщина 0,5 мм, 2020 см). Пластини для препаративної ТШХ елююють сумішшю метанолу/дихлорметану (5/95 по об'єму), потім бажаний продукт екстрагують з силікагелю сумішшю метанолу/дихлорметану (15/85 по об'єму). Отримують 1,4 мг (2-метилсульфанілетил)аміду (2Е,10Е,12Е,16Z,18Е)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-(3-метил6-оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти у вигляді жовтої твердої речовини, характеристики якої є наступними: Мас-спектр: + + СІ: m/z=617: [M+NH4] ; m/z=600: [M+H] 1H ЯМР спектри при 600 МГц отримали на спектрометрі Bruker Avance DMX-600 з наступними хімічними зсувами ( в м.ч.) - в хлороформі як розчинник -d1(CDCl3-d1) еталон на 7,27 при температурі 303К: 0,79 (д, J=6,5 Гц, 3H); 0,97 (д, J=7,0 Гц, 3H); 1,07 (д, J=7,0 Гц, 3H); 1,12 (д, J=7,0 Гц, 3H); 1,15 (д, J=7,5 Гц, 3H); 1,71 (м, 1Н); 1,81 (с, 3H); 1,82 (м частково замаскований, 1Н); 1,83 (с, 3H); 2,08 (м, 2Н); 2,11 (с, 3H); 2,13 (с, 3H); 2,15 (дд, J=6,5 і 13,5 Гц, 1Н); 2,45 (м ушир., 1Н); 2,53 (м, 1Н); 2,66 (т, J=6,5 Гц, 2Н); 2,70 (м, 1Н); 2,82 (м, 1Н); 3,50 (кв, J=6,5 Гц, 2Н); 3,61 (м, 1Н); 3,65 (м, 1Н); 5,01 (дд, J=4,5 і 7,5 Гц, 1Н); 5,09 (д, J=10,0 Гц, 1Н); 5,26 (д, J=10,0 Гц, 1Н); від 5,55 до 5,66 (м, 2Н); 5, 69 (дд, J=7,5 і 16,0 Гц, 1Н); 5,97 (т ушир., J=6,5 Гц, 1Н); 6,00 (д, J=10,0 Гц, 1Н); 6,02 (д, J=15,5 Гц, 1Н); 6,75 (д, J=16,0 Гц, 1Н); 6,95 (дд, J=6,0 і 10,0 Гц, 1Н). Приклад 2 Біс-[(2-меркаптоетил)амід (2Е,10Е,12Е,16Z,18Е)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти] До розчину 20 мг (2E,10E,12E,16Z,18E)-(R)-6гідрокси-3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)-8оксононадєку-2,10,12,16,18-пентаєнової кислоти і 5,6 мг 1-гідроксибензотриазолу в 0,3 мл дихлорметану вводять при температурі приблизно 20°С 12,8 мг дихлоргідрату цистаміну, 7,65 мкл Ν,Ν'діізопропілкарбодііміду, потім 11,6 мкл триетила міну. Реакційну суміш перемішують при температурі близько 20°С протягом 22 годин, потім очищують прямим нанесенням на 2 пластини для препаративної ТШХ на силікагелі (товщина 0,5 мм, 2020 см). Пластини для препаративної ТШХ елююють сумішшю метанолу/дихлорметану (5/95 по об'єму), потім бажаний продукт екстрагують з силікагелю сумішшю метанолу/дихлорметану 25 95959 26 + + (15/85 по об'єму). Отримують 4,6 мг біс[(2тіоетил)аміду (2E,10E,12E,16Z,18E)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти] у вигляді білої чистої твердої речовини, характеристики якої є наступними: Мас-спектр: ES : m/z=1167: [M+H] ES: m/z=1211: [М-Н+НСООН] Приклад 3 (2-Меркаптоетил)амід (2Е,10Е,12Е,16Z,18Е)(R)-6-гідрокси-3,5,7,9,11,15,17-гептаметил-19((2S,3S)-3-метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)8-оксононадеку-2,10,12,16,18-пентаєнової кислоти До розчину 20 мг (2E,10E,12E,16Z,18E)-(R)-6гідрокси-3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)-8оксононадеку-2,10,12,16,18-пентаєнової кислоти і 8 мкл триетиламіну в 0,15 мл дихлорметану вводять при температурі близько 0°С 6,1 мкл півалоїлхлориду. Через 15 хвилин при температурі близько 0°С додають розчин 4,4 мг 2-аміноетантіолу в 0,15 мл дихлорметану і 0,05 мл етанолу. Реакційну суміш перемішують при температурі близько 20°С протягом 1 години, потім очищують прямим нанесенням на 2 пластини для препаративної ТШХ на силікагелі (товщина 0,5 мм, 2020 см). Пластини для препаративної ТШХ елююють сумішшю метанолу/дихлорметану (8/92 по об'єму), потім бажаний продукт екстрагують з силікагелю сумішшю метанолу/дихлорметану (15/85 по об'єму). Отримують 2,3 мг (2-меркаптоетил)аміду (2Е,10Е,12Е,16Z,18Е)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти у вигляді безбарвної скловидної речовини, характеристики якої є наступними: Мас-спектр: + + ES : m/z=586: [M+H] ES : m/z=630: [М-Н+НСООН] 1Н ЯМР спектри при 400 МГц отримали на спектрометрі Bruker Avance DRX-400 з наступними хімічними зсувами ( в м.ч.) - в хлороформі як роз чинник - d1(CDCl3-d1) еталон на 7,27 при температурі 303К: 0,80 (д, J=6,5 Гц, 3H); 0,98 (д, J=6,5 Гц, 3H); 1,08 (д, J=7,5 Гц, 3H); 1,14 (д, J=6,5 Гц, 3H); 1,16 (д, J=7,0 Гц, 3H); 1,72 (м, 1Н); від 1,80 до 1,87 (м замаскований, 1Н); 1,82 (с, 3H); 1,84 (с, 3H); 2,09 (м, 2Н); 2,11 (с, 3H); 2,16 (дд, J=6,5 і 13,5 Гц, 1Н); 2,54 (м, 1Н); від 2,65 до 2,74 (м, 3H); 2,83 (м, 1Н); 3,48 (кв, J=6,5 Гц, 2Н); від 3,60 до 3,70 (м, 2Н); 5,00 (дд, J=4,0 і 7,0 Гц, 1Н); 5,10 (д, J=10,5 Гц, 1Н); 5,27 (д, J=10,5 Гц, 1Н); 5,58 (с, 1Н); 5,60 (тд частково замаскований, J=7,5 і 15,5 Гц, 1Н); 5,70 (дд, J=7,0 і 15,5 Гц, 1Н); від 5,96 до 6,06 (м, 3H); 6,75 (д ушир., J=15,5 Гц, 1Н); 6,97 (дд, J=6,0 і 10,0 Гц, 1Н). Приклад 4 (2-Метилдисульфанілетил)амід (2Е,10Е,12Е,16Z,18Е)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти До розчину 20 мг (2Е,10Е,12Е,16Z,18Е)-(R)-6гідрокси-3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)-8оксононадеку-2,10,12,16,18-пентаєнової кислоти і 5,6 мг 1-гідроксибензотриазолу в 0,15 мл дихлорметану вводять при температурі близько 20°С 7,65 мкл Ν,Ν'-діізопропілкарбодііміду, потім розчин 8 мг 2-метилдитіоетиламіну в 0,15 мл дихлорметану. Реакційну суміш перемішують при температурі близько 20°С протягом 2 годин, потім очищують прямим нанесенням на 2 пластини для препаративної ТШХ на силікагелі (товщина 0,5 мм, 2020 см). Пластини для препаративної ТШХ елююють сумішшю метанолу/дихлорметану (7/93 по об'єму), потім бажаний продукт екстрагують з силікагелю сумішшю метанолу/дихлорметану (15/85 по об'єму). Отримують 3,4 мг (2метилдисульфанілетил)аміду (2Е,10Е,12Е,16Z,18E)-(R)-6-riдрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-(3-метил 27 95959 28 6-оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти у вигляді блідожовтої олії, характеристики якої є наступними: Мас-спектр: + + ES : m/z=632: [M+H] ES : m/z=630: [М-Н] ; m/z=676: [М-Н+НСООН] 1H ЯМР спектри при 400 МГц отримали на спектрометрі Bruker Avance DRX-400 з наступними хімічними зсувами ( в м.ч.) - в хлороформі як розчинник -d1(CDCl3-d1) еталон на 7,27 при температурі 303К: 0,81 (д, J=6,5 Гц, 3H); 0,98 (д, J=6,5 Гц, 3H); 1,08 (д, J=7,5 Гц, 3H); 1,14 (д, J=6,5 Гц, 3H); 1,16 (д, J=7,0 Гц, 3H); 1,72 (м, 1Н); від 1,79 до 1,86 (м, 1Н); 1,82 (с, 3H); 1,84 (с, 3H); 2,09 (м, 2Н); 2,11 (з ушир., 3H); 2,15 (дд, J=5,5 і 13,5 Гц, 1Н); 2,35 (з ушир., 1Н); 2,43 (м, 3H); 2,54 (м, 1Н); 2,70 (м, 1Н); 2,83 (м, 1Н); 2,86 (т, J=6,5 Гц, 2Н); від 3,59 до 3,70 (м, 4Н); 5,01 (дд, J=4,0 і 7,0 Гц, 1Н); 5,11 (д, J=10,0 Гц, 1Н); 5,27 (д, J=10,0 Гц, 1Н); 5,56 (с, 1H); 5,60 (тд частково замаскований, J=7,5 і 15,5 Гц, 1Н); 5,70 (дд, J=7,0 і 15,5 Гц, 1Н); 5,93 (т, J=6,0 Гц, 1Н); 6,00 (д, J=10,0 Гц, 1Н); 6,02 (д, J=15,5 Гц, 1Н); 6,75 (д, J=15,5 Гц, 1Н); 6,96 (дд, J=6,0 і 10,0 Гц, 1Н). Приклад 5 (2-Метил-2-метилдисульфанілпропіл)амід (2Е,10Е,12Е,16Z,18E)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти До розчину 225,4 мг (2Е,10Е,12Е,16Z,18Е)-(R)6-гідрокси-3,5,7,9,11,15,17-гептаметил-19-((2S,3S)3-метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)-8оксононадеку-2,10,12,16,18-пентаєнової кислоти в 1,5 мл дихлорметану вводять при температурі близько 0°С розчин 63,6 мг 1гідроксибензотриазолу і 110 мг 2-метил-2метилдисульфанілпропіламіну в 1,5 мл дихлорметану, потім 86,2 мкл Ν,Ν'-діізопропілкарбодііміду. Реакційну суміш перемішують при температурі близько 0°С протягом 15 годин, потім розріджують 30 мл дихлорметану. Органічну фазу промивають двічі 10 мл води, сушать над сульфатом натрію, фільтрують через скляний фільтр, потім концентрують під зниженим тиском при температурі близько 40°С. Отриманий таким чином залишок очищують колонковою хроматографією на силікагелі (20 г SiO2 15-35 мкм, елюювання градієнтом метанол/дихлорметан від 0/100 до 10/90 (по об'єму). Фракції, які містять бажаний продукт концентрують під зниженим тиском при температурі близько 40°С. Отримують 212,1 мг (2-метил-2метилдисульфанілпропіл)аміду (2Е,10Е,12Е,16Z,18Е)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти у вигляді жовтої скловидної речовини, характеристики якої є наступними: Мас-спектр: + + ES : m/z=660: [M+H] ES : m/z=658: [М-Н] ; m/z=704: [М-Н+НСООН] 1H ЯМР спектри при 400 МГц отримали на спектрометрі Bruker Avance DRX-400 з наступними хімічними зсувами ( в м.ч.) - в хлороформі як розчинник -d1(CDCl3-d1) еталон на 7,27 при температурі 303К: 0,80 (д, J=6,5 Гц, 3H); 0,97 (д, J=6,5 Гц, 3H); 1,08 (д, J=7,5 Гц, 3H); 1,14 (д, J=6,5 Гц, 3H); 1,16 (д, J=7,0 Гц, 3H); 1,32 (с, 6Н); 1,72 (м, 1Н); 1,82 (с, 3H); 1,83 (м замаскований, 1Н); 1,84 (с, 3H); 2,08 (м, 2Н); 2,11 (с ушир., 3H); 2,15 (дд, J=6,5 і 13,5 Гц, 1Н); 2,43 (с, 3H); 2,54 (м, 1Н); 2,70 (м, 1Н); 2,83 (м, 1Н): 3,45 (д, J=6,0 Гц, 2Н); від 3,59 до 3,69 (м, 2Н); 5,00 (дд, J=4,0 і 7,0 Гц, 1Н); 5,10 (д, J=10,5 Гц, 1Н); 5,26 (д, J=10,0 Гц, 1Н); від 5,54 до 5,64 (м, 2Н); 5,70 (дд, J=7,0 і 15,5 Гц, 1Н); 5,83 (т, J=6,0 Гц, 1Н); 6,00 (д, J=10,5 Гц, 1Н); 6,02 (д, J=15,5 Гц, 1Н); 6,75 (д, J=15,5 Гц, 1Н); 6,96 (дд, J=6,5 і 10,5 Гц, 1Н). Приклад 6 (2-Меркапто-2-метилпропіл)амід (2Е,10Е,12Е,16Z,18Е)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку2,10,12,16,18-пентаєнової кислоти До розчину 200 мг (2-метил-2метилдисульфанілпропіл)аміду (2Е,10Е,12Е,16Z,18Е)-(R)-6-гідрокси3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3-метил-6оксо-3,6-дигідро-2Н-піран-2-іл)-8-оксононадеку 2,10,12,16,18-пентаєнової кислоти в 7,7 мл тетрагідрофурану і 3,85 мл води при температурі близько 20°С додають 217,2 мг хлоргідрату трис(2карбоксіетил)фосфіну. Через 16 годин при температурі близько 20°С, реакційну суміш розріджують 29 95959 30 30 мл етилацетату, промивають двічі 15 мл води, 15 мл насиченого розчину солі, сушать над сульфатом магнію, фільтрують через скляний фільтр і концентрують під зниженим тиском при температурі близько 40°С. Отриману таким чином жовту олію очищують колонковою хроматографією на силікагелі (25 г SiO2 15-35 мкм, елюювання градієнтом метанолу/дихлорметану від 1/99 до 10/90 (по об'єму). Фракції, які містять бажаний продукт концентрують під зниженим тиском при температурі близько 40°С. Отримують 122,6 мг (2-меркапто-2метилпропіл)аміду (2Е,10Е,12Е,16Z,18Е)-(R)-6гідрокси-3,5,7,9,11,15,17-гептаметил-19-((2S,3S)-3метил-6-оксо-3,6-дигідро-2Н-піран-2-іл)-8оксононадеку-2,10,12,16,18-пентаєнової кислоти у вигляді жовтої скловидної речовини, характеристики якої є наступними: Мас-спектр: + + ES : m/z=614: [M+H] ES : m/z=612: [М-Н] ; m/z=658: [М-Н+НСООН] 1H ЯМР спектри при 500 МГц отримали на спектрометрі Bruker Avance DMX-500 з наступними хімічними зсувами ( в м.ч.) - в хлороформі як розчинник -d1(CDCI3-d1) еталон на 7,27 при температурі 303К: 0,80 (д, J=6,5 Гц, 3H); 0,97 (д, J=6,5 Гц, 3H); 1,07 (д, J=7,5 Гц, 3H); 1,13 (д, J=6,5 Гц, 3H); 1,16 (д, J=6,5 Гц, 3H); 1,38 (с, 6Н); 1,72 (м, 1Н); 1,82 (с, 3H); 1,84 (с, 3H); 1,85 (м частково замаскований, 1Н); 2,09 (м, 2Н); 2,11 (с, 3H); 2,17 (дд, J=6,5 і 13,5 Гц, 1Н); 2,54 (м, 1Н); 2,70 (м, 1Н); 2,83 (м, 1Н); 3,38 (д, J=6,5 Гц, 2Н); від 3,61 до 3,70 (м, 2Н); 5,01 (дд, J=4,0 і 7,0 Гц, 1Н); 5,09 (д, J=10,0 Гц, 1Н); 5,26 (д, J=10,0 Гц, 1Н); 5,59 (дт частково замаскований, J=7,5 і 15,5 Гц, 1Н); 5,63 (с, 1Н); 5,69 (дд, J=7,0 і 15,5 Гц, 1Н); 6,00 (д, J=10,0 Гц, 1Н); 6,02 (д, J=15,5 Гц, 1Н); 6,04 (т частково замаскований, J=6,5 Гц, 1Н); 6,75 (д, J=15,5 Гц, 1Н); 6,97 (дд, J=6,0 і 10,0 Гц, 1Н). Кон'югація антитіл з похідними лептоміцину Кон'югація антитіл huC242 до раку товстого кишечнику з похідними лептоміцину: Отримували зв'язані дисульфідом кон'югати гуманізованих антитіл (huC242) до пухлини товстого кишечнику із сполукою з прикладу 6 (які називаються тут huС242-SSNРВ-лептоміцин з прикладу 6). Антитіла huC242 приводили у взаємодію з 6кратним молярним надлишком модифікуючого антитіла засобу SSNPB (N-сульфосукцинімідил-4(5-нітро-2-піридилтіо)бутаноат) при концентрації антитіл, яка дорівнює 9 мг/мл в 50 мМ калійфосфатному буфері (рН 6,5, що містить 50 мМ NaCl, 2 мМ ЕДТА, 5% диметилацетаміду) протягом 90 хвилин при кімнатній температурі. Реакційну суміш очищують за допомогою гель-хроматографії на Sephadex G-25 з урівноваженням в 50 мМ калій-фосфатному буфері рН 6,5, який містить 50 мМ NaCl і 2 мМ ЕДТА. Зразок модифікованих антитіл аналізували з доданням -меркаптоетанолу або без нього, і визначено, що вони мають ~6 нітропіридилдитіогруп, включених в антитіло. До зразка модифікованих антитіл при концентрації 2 мг/мл в 50 мМ калій-фосфатному буфері (рН 6,5, що містить 50 мМ NaCl, 2 мМ ЕДТА, 10% диметилацетаміду) додавали 2-кратний молярний надлишок лікарської речовини лептоміцин-SH на зв'язувальну групу. Реакцію відстежували спектрофотометрично (394 нм) і за допомогою ВЕРХ у відношенні 5-нітропіридин-2-тіону. Після взаємодії при кімнатній температурі протягом приблизно 90 хв суміш очищували гель хроматографією на Sephadex G-25 в 50 мМ фосфатному буфері (рН 6,5, який містить 50 мМ NaCl, 2 мМ ЕДТА). Відношення поглинання при 250 нм/280 нм, що дорівнює 0,78 для кон'югату в порівнянні з 0,37 для немодифікованих антитіл показувало включення групи лептоміцину в кон'югат (що приводить до підвищеного поглинання при 250 нм). Mac-спектрометричний аналіз деглікозильованого кон'югату huC242-лептоміцину показав піки кон'югату при 147492, 148212, 148936 і 149660 дальтон, які відповідають 1, 2, 3 і 4 включеним молекулам лептоміцину на молекулу антитіла. Кон'югування анти-CD19 (huB4) антитіл з похідними лептоміцину: Кон'югати анти-CD19 антитіл (гуманізованих В4 антитіл) з лептоміцином отримували за допомогою дисульфідних і нерозщеплюваних тіоефірних лінкерів. Перший зразок (-S-S-лінкер) складався з анти-CD19 (huB4) антитіл, зв'язаних із сполукою з прикладу 6 через дисульфідний лінкер (SSNPB; N-сульфосукцинімідил-4-(5-нітро-2піридилдитіо)бутаноат). Другий зразок складався з huB4 антитіла, зв'язаного із сполукою з прикладу 6 через малеімідний лінкер (SMCC; N-сукцинімідил4-(мелеімідометил)циклогексанкарбоксилат). Кон'югат HuВ4-SSNPB-лептоміцин (HuВ4SSNPB-лептоміцин з прикладу 6): 4 мг антитіл huB4 приводили у взаємодію з 7,5-кратним молярним надлишком лінкеру SSNPB при концентрації антитіл, яка дорівнює 8 мг/мл в 50 мМ калій-фосфатному буфері (рН 6,5, що містить 50 мМ NaCl, 2 мМ ЕДТА, 5% диметилацетаміду) протягом 90 хвилин при кімнатній температурі. Реакційну суміш очищували гельхроматографією на Sephadex G-25 в 50 мМ калійфосфатному буфері (рН 6,5, що містить 50 мМ NaCl, 2 мМ ЕДТА). Зразок модифікованих антитіл аналізували з доданням -меркаптоетанолу або без нього, і визначено, що вони мають 5,3 нітропіридилдитіогрупи, включених в антитіло. До зразка модифікованих антитіл при концентрації 1 мг/мл в 50 мМ калій-фосфатному буфері (рН 6,5, що міс тить 50 мМ NaCl, 2 мМ ЕДТА, 10% диметилацетаміду) додавали 3-кратний молярний надлишок лікарської речовини з прикладу 6 на зв'язувальну групу. Реакцію відстежували спектрофотометрично (394 нм) і за допомогою ВЕРХ відносно виділення 5-нітропіридин-2-тіону. Після взаємодії при кімнатній температурі протягом ночі суміш очищували гель-хроматографією на Sephadex G-25 в 10 мМ цитратного буфера (рН 5,5, що містить 135 мМ NaCl). Зразок аналізували з доданням меркаптоетанолу або без нього, і визначено, що є 4,3 лінкери, які прореагували на антитіло. Аналіз гель-хроматографією (ГХ) показав 95% мономерних антитіл. Кон'югат HuВ4-SМСС-лептоміцин 4 мг антитіл huB4 приводили у взаємодію з 7,5-кратним молярним надлишком лінкеру СМЦК при концентрації антитіл, яка дорівнює 8 мг/мл в 50 мМ калій-фосфатному буфері (рН 6,5, що містить 50 мМ NaCl, 2 мМ ЕДТА, 5% диметилацетаміду) протягом 90 хвилин при кімнатній температурі. Реакційну суміш очищували гельхроматографією на Sephadex G-25 в 50 мМ калійфосфатному буфері (рН 6,5, що містить 50 мМ NaCl, 2 мМ ЕДТА). Кількість малеімідних груп, які включаються в зразок, встановлювали шляхом додання надлишку тіолу (цистеїну), і визначено, що є 3,3 лінкерних групи на антитіло. До зразка модифікованих антитіл при концентрації 1 мг/мл в 50 мМ калій-фосфатному буфері (рН 6,5, що містить 50 мМ NaCl, 2 мМ ЕДТА, 10% диметилацетаміду) додавали 3-кратний молярний надлишок лікарської речовини з прикладу 6 на малеімідну групу. Після взаємодії при кімнатній температурі протягом ночі суміш очищували гельхроматографією (ГХ) на Sephadex G-25 в 10 мМ цитратному буфері (рН 5,5, що містить 135 мМ NaCl). ГХ аналіз показав 98% мономерних антитіл. Mac-спектрометричний аналіз деглікозильованих кон'югатів HuB4-SMCC-лептоміцину показав піки кон'югату при 145138, 145860 і 146566 даль тон, що відповідають 1, 2 і 3 включеним молекулам лептоміцину на молекулу антитіла. Біологічні результати: Оцінка цитотоксичності HuВ4-SSNPBлептоміцину, кон'югату з прикладу 6, на ракових клітинах Ramos (позитивні на антиген CD19) і HL60 (антигенонегативні) за аналізом життєздатності WST показала значення ІС50, що дорівнює -9 -9 1,410 М і 4,210 М, відповідно, демонструючи таким чином антигеноспецифічну цитотоксичну активність кон'югату лептоміцин-антитіло. Оцінка цитотоксичності HuВ4-SSNPBлептоміцину, кон'югату з прикладу 6, на ракових клітинах COLO 205 (позитивних на антиген CanAg) і клітинах А375 показала значення ІС50, що дорів-10 -9 нює 1,310 Μ і >5,010 М, відповідно. (Фіг.). У даному описі зроблені посилання на деякі патенти і друкарські публікації, ідеї і вказівки яких тим самим, включені шляхом посилання у всій їх відповідній повноті. Хоч даний винахід був описаний в деталях і з посиланнями на його конкретні втілення, фахівцеві в даній галузі буде очевидно, що в ньому можуть бути зроблені різні зміни і модифікації без виходу з його суті і обсягу. 33 95959 34 В описі до патенту на винахід графічні зображення та текст подаються в редакції заявника Комп’ютерна верстка О. Гапоненко Підписне Тираж 23 прим. Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюLeptomycin derivatives
Автори англійськоюBouchard, Herve, Commercon Alain, Chari, Ravi V. J.
Назва патенту російськоюПроизводные лептомицина
Автори російськоюБушар Эрве, Коммерсон Ален, Чаре Рави В. Дж.
МПК / Мітки
МПК: A61P 35/00, A61K 47/48
Мітки: лептоміцину, похідні
Код посилання
<a href="https://ua.patents.su/17-95959-pokhidni-leptomicinu.html" target="_blank" rel="follow" title="База патентів України">Похідні лептоміцину</a>
Похідні морфоліну, спосіб їх одержання та фармацевтичні препарати, що містять вказані похідні

Номер патенту: 73931
Опубліковано: 17.10.2005
Автори: Гейоль Патрік, Проєтто Вінченцо, Дюку Жан Філіп, ЕМОН-АЛЬТ Ксав'є
МПК: C07D 413/06, A61P 1/04, A61P 25/24, A61P 13/02, A61P 1/08, A61P 25/10, A61P 29/00, C07D 265/30, A61P 25/00, A61K 31/5377, A61P 37/00, A61P 25/06, A61P 29/02, A61P 9/00, A61P 11/06, A61P 13/00, A61P 1/00, A61P 11/00, A61P 17/00, A61P 25/22
Мітки: морфоліну, одержання, фармацевтичні, містять, спосіб, вказані, препарати, похідні
Формула / Реферат:
1. Сполука формули (I): (I),в якій Аr являє собою феніл, монозаміщений або дизаміщений атомом галогену; (С1-С3)алкіл;Χ являє собою групу R2-N=; групу R2-CH=;R1 являє собою атом хлору, атом брому, (С1-С3)алкіл або трифлуорметил;R2 являє собою (С1-С6)алкіл; (С3-С6)циклоалкіл; групу –CR4R5CONR6R7;R3 являє собою групу...
Фармацевтичні композиції, що містять похідні 3-аміноазетидину, похідні і спосіб їхнього одержання

Номер патенту: 72319
Опубліковано: 15.02.2005
Автори: Ашард Даніель, Міерс Мішель, Боушард Херве, Філош Бруно, Букерель Жан, Грізоні Серж, Хіттінгер Огюстін
МПК: A61K 47/00, A61P 29/00, A61P 27/02, A61P 25/18, C07D 403/12, A61P 25/00, C07D 401/06, A61P 25/08, C07D 401/12, A61P 25/20, C07D 205/00, A61P 11/00, A61P 25/14, A61K 31/4427, A61K 31/4178, A61P 35/00, A61P 25/24, A61P 9/02, A61K 31/506, A61P 5/00, C07D 403/08, A61P 1/08, C07D 409/12, A61P 25/28, A61P 1/00, A61P 37/00, A61K 31/4025, A61P 25/16, A61P 9/08, A61K 31/397, A61P 23/00, A61P 1/14, A61P 9/00, A61P 25/02, A61P 11/06, A61K 31/4709, A61P 25/22, A61P 3/04
Мітки: спосіб, 3-аміноазетидину, композиції, фармацевтичні, їхнього, містять, похідні, одержання
Формула / Реферат:
1. Фармацевтична композиція, що містить як активний інгредієнт сполуку формули: , (І)у якійR1 означає радикал –NНСОR4 або -N(R5)-Y-R6,Υ означає CO або SO2,R2 і R3, однакові або різні, означають або ароматичний радикал, вибраний з фенілу, нафтилу і інденілу, що можуть бути незаміщеними або заміщеними одним або декількома замісниками:...
Похідні карбамату хінуклідину та лікарські композиції, що містять названі похідні

Номер патенту: 76131
Опубліковано: 17.07.2006
Автори: Фернандез Форнер Марія Долорс, Прат Квінонес Марія, Буіл Альберо Марія Антонія
МПК: C07D 209/00, A61P 13/00, A61K 31/439, C07D 203/00, C07D 453/00, A61P 11/00, C07D 471/08, A61P 1/00, C07D 221/00
Мітки: композиції, похідні, містять, названі, лікарські, карбамату, хінуклідину
Формула / Реферат:
1. Сполука, що являє собою карбамат формули (І): , (I)в якій:R1 являє собою,,,
Похідні ди- або трифторметансульфоніланіліду, спосіб їх одержання і гербіциди, що містять вказані похідні як активні інгредієнти

Номер патенту: 56338
Опубліковано: 15.05.2003
Автори: Оно Юкімаса, Тамару Масатосі, Янасігава Кацутада, Накатані Масао, Дандзо Такесі, Йосімура Такумі
МПК: A01N 41/06, A01N 43/54, C07C 317/36, C07D 239/52
Мітки: вказані, активні, похідні, гербіциди, одержання, дії, інгредієнти, спосіб, трифторметансульфоніланіліду, містять
Формула / Реферат:
1. Похідне ди- або трифторметансульфоніланіліду, представлене наступною загальною формулою:,де R1 являє атом водню, алкільну групу або алкоксіалкільну групу; і R2 являє атом водню, коли R1 являє атом водню або алкільну групу, і являє атом водню або атом фтору, коли R1 являє алкоксіалкільну групу, або його сіль.2. Похідне ди- або трифторметансульфоніланіліду або його сіль за п. 1, в якому R1 являє атом водню.3....
Похідні 2,3-бензодіазепіну і фармацевтичні композиції, що містять ці похідні як активний інгредієнт

Номер патенту: 74615
Опубліковано: 16.01.2006
Автори: Гіглер Габор, ШІМІГ Дьюла, Харшінг Ласло Габор, Вег Міклош, Лінг Іштван, Леваі Дьйордь, Сабо Геза, Мартонне Марко Бернадетт, Раткаі Зольтан, Сенаші Габор, БАРКОЦІ Йожеф, Грефф Зольтан
МПК: A61P 25/02, C07D 243/10, A61P 25/00, A61P 13/00, A61P 25/30, A61P 25/22, A61P 25/04, A61P 25/06, A61K 31/551, A61P 25/18, A61P 1/08, A61P 25/16, A61P 25/28, A61P 25/08, A61P 9/00, C07D 243/02
Мітки: інгредієнт, містять, фармацевтичні, похідні, 2,3-бензодіазепіну, активний, композиції
Формула / Реферат:
1. Похідне 2,3-бензодіазепіну формули І: (І),деΧ - водень, хлор або метоксигрупа,Y - водень або галоген,Ζ - метил або хлор,R – С1-4 алкіл або група формули –NR1R2, де R1 і R2, незалежно, являють собою водень, С1-4алкіл, С1-4алкоксил або С3-6циклоалкіл, та його фармацевтично прийнятні кислотно-адитивні солі.2....
Попередній патент: Антитіло, що зв’зується з cd22, імунокон’югат та їх застосування
Наступний патент: Водонепроникна та паропроникна підошва для взуття та взуття з такою підошвою
Випадковий патент: Механізм синхронного горизонтального переміщення вантажонесучих кареток кліті в багатоярусній механізованій автостоянці