Похідні піразолів, спосіб їх одержання та застосування в терапії
Номер патенту: 106500
Опубліковано: 10.09.2014
Автори: Табар Мішель, Абекассі П'єр-Ів, Демазо Паскаль
















Формула / Реферат
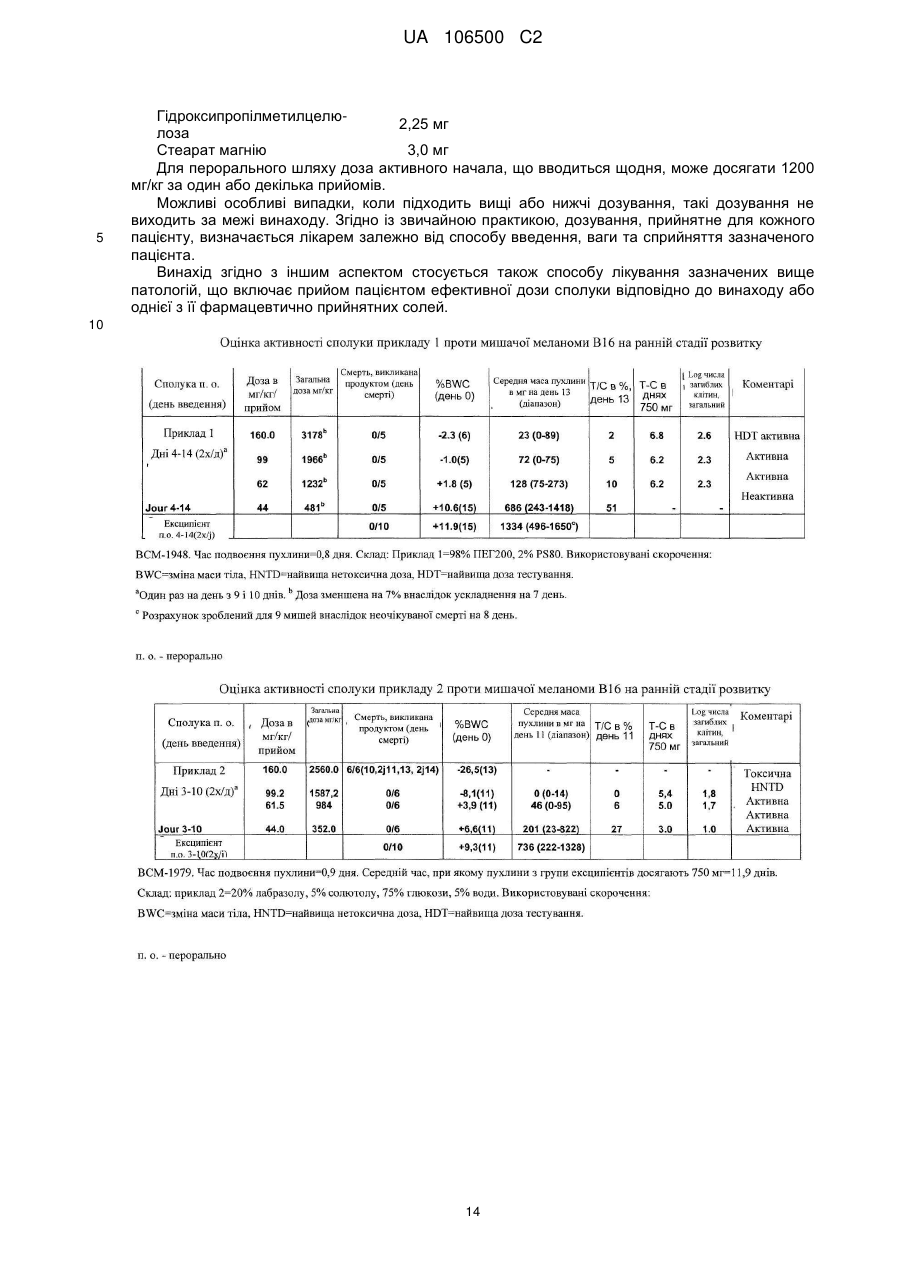
1. Сполука формули (І):
 ,
,
в якій:
X означає хлор або фтор,
у формі основи або кислотно-адитивної солі.
2. Сполука формули (І) за п. 1, де X означає хлор, у формі основи або кислотно-адитивної солі.
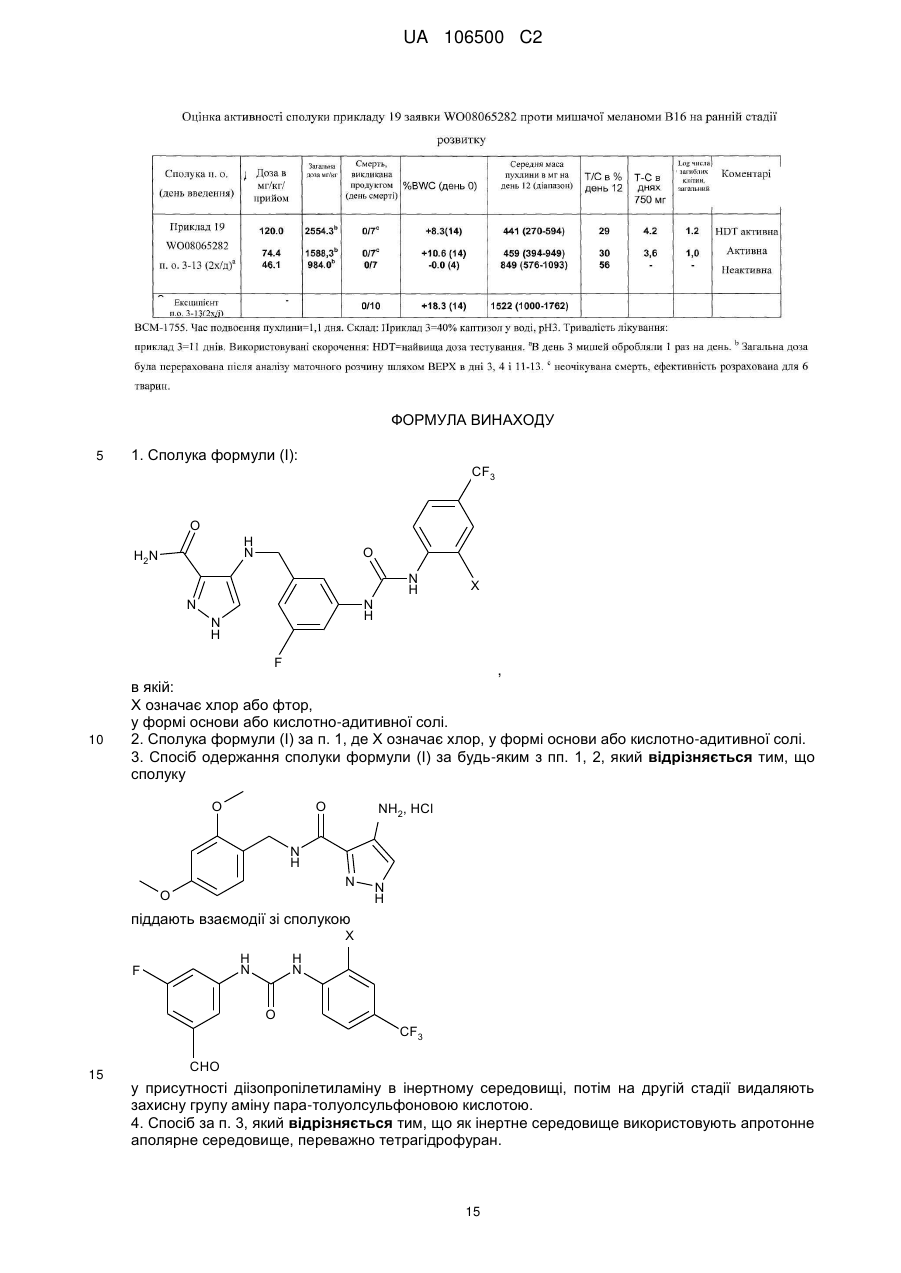
3. Спосіб одержання сполуки формули (І) за будь-яким з пп. 1, 2, який відрізняється тим, що сполуку
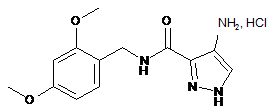
піддають взаємодії зі сполукою
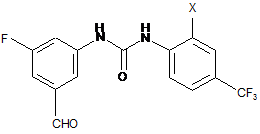
у присутності діізопропілетиламіну в інертному середовищі, потім на другій стадії видаляють захисну групу аміну пара-толуолсульфоновою кислотою.
4. Спосіб за п. 3, який відрізняється тим, що як інертне середовище використовують апротонне аполярне середовище, переважно тетрагідрофуран.
5. Сполука формули
,
де X означає хлор або фтор.
6. Сполука формули
 ,
,
де X означає хлор або фтор.
7. Лікарський засіб, який відрізняється тим, що містить сполуку формули (І) за будь-яким з пп. 1, 2 або адитивну сіль цієї сполуки з фармацевтично прийнятною кислотою.
8. Фармацевтична композиція, яка відрізняється тим, що містить сполуку формули (І) за будь-яким з пп. 1, 2 або фармацевтично прийнятну сіль цієї сполуки, а також щонайменше один фармацевтично прийнятний ексципієнт.
9. Сполука формули (І) за будь-яким з пп. 1, 2 для застосування як лікарського засобу при лікуванні раку.
Текст
Реферат: Винахід стосується похідних піразолів загальної формули (І), у якій X означає хлор або фтор. Винахід також стосується способу їх одержання і застосування в терапії. UA 106500 C2 (12) UA 106500 C2 UA 106500 C2 5 10 15 20 25 30 35 40 45 Даний винахід стосується способів одержання та використання похідних піразолу в терапії. Більш детально і згідно з першим аспектом винахід належить до нових замінних специфічних піразолів, які проявляють протиракову активність через модуляцію активності протеїнів, зокрема, протеїнкіназ. Протеїнкінази являють собою сімейство ферментів, що каталізують фосфорилювання гідроксильних груп специфічних залишків протеїнів, таких як залишки тирозину, серину або треоніну. Таке фосфорилування може сильно змінювати функцію протеїнів; таким чином, протеїнокінази відіграють важливу роль у регуляції різноманітних клітинних процесів, що включають в себе, зокрема, метаболізм, клітинну проліферацію, диференціювання, клітинну міграцію або живучість клітин. Серед різних клітинних функцій, в яких задіяна активність протеїнкінази, можна виділити такі процеси, використання яких є перспективним для лікування ракових захворювань та інших хвороб. Таким чином, однією із задач цього винаходу є надання такої композиції, що характеризуватиметься протираковою активністю, що буде виявлятися, зокрема, у впливі на кінази. Серед кіназ, відносно яких досліджується модуляція, слід назвати KDR, Tie2, VEGFR-1 (FLT1), VEGFR-3 (Flt4), PDGFR, FGFR. Переважними є кінази KDR і/або Tie2. Відомі сполуки із заявки на патент, що опублікований під номером WO08065282, які відповідають наступній загальній формулі (I): Формула (I) в якій: 1) A і Ar вибирають незалежно з групи, що складається з: арилу, гетероарилу, заміщеного арилу, заміщеного гетероарилу; 2) L вибирають із групи, що складається з: NH-CO-NH і O-CO-NH; 3) R1 вибирають із групи, що складається з: Н, R6, COR6, SO2R6, де R6 вибирають з Н, OR7, NR8R9, алкілу, циклоалкілу, гетероциклілу, заміщеного гетероциклілу, арилу, заміщеного арилу, гетероарилу, заміщеного гетероарилу, де R 7 вибирають з Н, фенілу, алкілу, і де R8 і R9 вибирають незалежно з групи, що складається з: Н, алкілу, циклоалкілу, гетероциклілу, заміщеного гетероциклілу, арилу, заміщеного арилу, гетероарилу, заміщеного гетероарилу, або R8 і R9 з'єднані між собою, утворюють 5-8-членний насичений цикл, що містить 0-3 гетероатомів, вибраних з O, S і N; 4) Х вибирають із групи, що складається з O і NH; 5) R3 вибирають із групи, що складається з: Н, алкілу, заміщеного алкілу, циклоалкілу, заміщеного циклоалкілу; 6) R4a вибирають із групи, що складається з Н і (С1-С4)алкілу; 7) R4b вибирають із групи, що складається з Н і (С1-С4)алкілу; 8) R5 вибирають із групи, що складається з Н, галогену, R 10, CN, O(R10), °C(O)(R10), °C(O)N(R10)(R11), OS(O2)(R10), N(R10)(R11), N=C(R10)(R11), N(R10)C(O)(R11), N(R10)C(O)O(R11), N(R12)C(O)N(R10)(R11), N(R12)C(S)N(R10)(R11), N(R10)S(O2)(R11), C(O)(R10), C(O)O(R10), C(O)N(R10)(R11), C(=N(R11)(R10), C(=N(OR11)(R10), S(R10), S(O)(R10), S(O2)(R10), S(O2)O(R10), S(O2)N(R10)(R11); де R10, R11, R12 незалежно вибирають із групи, що складається з: Н, алкілу, алкілену, алкинілу, арилу, гетероарилу, циклоалкілу, гетероциклілу, заміщеного алкілу, заміщеного алкілену, заміщеного алкінілу, заміщеного арилу, заміщеного гетероарилу, заміщеного циклоалкілу, заміщеного гетероциклілу. У цьому патенті переважно: Х означає, R3 означає метил, R4a і R4b означають H; L означає NHCONH; A означає феніл; Ar означає феніл; але у цьому патенті жоден з прикладів не описує заміщення Ar і його вплив на фармакокінетику. Даний винахід стосується двох сполук, що включені у зазначений патент, та відповідають формулі (I): 1 UA 106500 C2 5 10 15 20 25 , в якій: Х означає Cl або F. Сполуки формули (I) можуть існувати у формі основ або адитивних солей з кислотами. Дані адитивні солі є частиною винаходу. Сполуки у своїх двох таутомерних формах, що зазначені нижче, також є частиною винаходу: Ці солі можна отримати з фармацевтично прийнятними кислотами, але солі інших кислот, наприклад, корисних для очищення або відокремлення сполук формули (I), також є частиною винаходу. Серед використовуваних солей можна назвати, зокрема, гідрохлорид. Для сполук формули (I) можливе існування також у формі гідратів або сольватів, а саме асоціацій або комбінацій з однією або декількома молекулами води або з розчинником. Такі гідрати і сольвати також є частиною винаходу. Серед сполук формули (I) згідно з винаходом можна назвати наступні сполуки: -4-{3-[3-(2-хлор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Н-піразол-3карбоксамід і його гідрохлорид; -4-{3-[3-(2-фтор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Н-піразол-3карбоксамід і його гідрохлорид. Згідно з винаходом, сполуку формули (I) можна одержати використовуючи наведений нижче спосіб. На схемах нижче представлені вихідні сполуки та реагенти, якщо спосіб їх одержання не підлягає опису, то вони є комерційно доступними або вже описані в літературі. Або їх можна отримати способами, які відомі фахівцю або описані в літературі. Згідно з другим із своїх аспектів, винахід стосується також сполук формул: . в яких Х означає F або Cl. Ці сполуки корисні як проміжні для отримання сполук формули (I). Нижче наведені приклади, що описують алгоритм отримання сполук згідно з винаходом. Дані приклади не є обмежуючими і призначені лише для ілюстрування даного винаходу. 2 UA 106500 C2 Спосіб синтезу згідно з прикладами Синтез діарил-сечовинної частини 5 10 15 20 25 Приклад 1 Гідрохлорид 4-{3-[3-(2-хлор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Н-піразол3-карбоксаміду Суспензію 1,64 г (3,48 ммоль) 4-{3-[3-(2-хлор-4-трифторметилфеніл)-уреїдо]-5фторбензиламіно}-1Н-піразол-3-карбоксаміду у 50 мл етанолу перемішують при кімнатній температурі в атмосфері аргону. Потім додають по краплях 35 мл (35 ммоль) розчину соляної кислоти в діетиловому ефірі (1 н.). Реакційне середовище стає прозорим розчином. Після 10 годин перемішування при кімнатній температурі, розчинники випарюють в роторному випарнику при зниженому тиску. Отриманий залишок перемішують у 50 мл діетилового ефіру протягом 30 хвилин. Після фільтрації і висушування в сушильній шафі одержують 1,8 г гідрохлориду 4-{3-[3-(2хлор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Н-піразол-3-карбоксаміду у вигляді блідо-жовтих кристалів. МС: Час утримування Tr (хв)=1,01; [M+H]+ m/z=471; [M-H]- m/z=469 1 Н ЯМР (400 МГц, ДМСО-d) δ м. ч.: 4,32 (с, 2Н); 6,89 (д шир., J=9,6 Гц, 1Н); 7,16 (с шир., 1Н); 7,18-7,61 (м, 4Н); 7,68 (д шир., J=8,9 Гц, 1Н); 7,86 (с шир., 1Н); 8,44 (д, J=8,9 Гц, 1Н); 8,81 (м шир., 1Н); 10,17 (м шир., 1Н). Температура плавлення (за Кофлером) = 177ºС 4-{3-[3-(2-хлор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Н-піразол-3карбоксамід 3 UA 106500 C2 5 10 15 20 25 30 35 40 Розчин 5,9 г (9,5 ммоль) 4-{3-[3-(2-хлор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}1Н-піразол-3-(2,4-диметоксибензиламіду) і 4,5 г (24 ммоль) пара-толуолсульфонової кислоти в 400 мл толуолу нагрівають зі зворотним холодильником протягом 2 годин Після декантації толуольний розчин відокремлюють від жовтої смоли. Смолу змішують з 150 мл метанолу та 500 мл етилацетату. Потім додають 500 мл води. Після чого цей розчин охолоджують, а потім підлуговують 100 мл водного розчину їдкого калію (10 н.). Після декантації водний шар екстрагують розчином 400 мл етилацетату і 100 мл метанолу. Органічні шари поєднують та промивають 100 мл насиченого розчину хлориду натрію, висушують над сульфатом магнію і концентрують при зниженому тиску до отримання твердої речовини блідо-жовтого кольору, яку потім очищують на шарі, що складається з 200 г діоксиду кремнію, елююючи розчином дихлорметану/метанолу/ацетонітрилу 80/10/10 (за об'ємом): одержують 1,77 г 4-{3-[3-(2-хлор-4трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Н-піразол-3-карбоксаміду у вигляді білої твердої речовини. МС: Час утримування Tr (хв)=4,29; [M+H]+ m/z=471; [M-H]- m/z=469 1 Н ЯМР (400 МГц, ДМСО-d) δ м. ч.: 4,19 (д, J=6,7 Гц, 2Н); 5,74 (м шир., 1Н); 6,80 (д шир., J=9,6 Гц, 1Н); 6,95-7,11 (м, 3Н); 7,24 (м уш., 1Н); 7,42 (дт, J=11,3; 2,3 Гц, 1Н); 7,67 (д шир., J=8,9 Гц, 1Н); 7,86 (с шир., 1Н); 8,44 (д, J=8,9 Гц, 1Н); 8,70 (с шир., 1Н); 9,92 (с шир., 1Н); 12,57 (с шир., 1Н). Температура плавлення (за Кофлером) = 220ºС 4-{3-[3-(2-хлор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Н-піразол-3-(2,4диметоксибензиламід) Розчин 10 г (32 ммоль) гідрохлориду 4-аміно-1Н-піразол-3-(2,4-диметоксибензиламіду) і 5,9 мл (35,2 ммоль) діізопропілетиламіну в 300 мл тетрагідрофурану змішують при кімнатній температурі в атмосфері аргону. Додають 3,85 г (35 ммоль) сульфату магнію і 12,70 г (35,2 ммоль) 1-(2-хлор-4-трифторметилфеніл)-3-(3-фтор-5-формілфеніл)-сечовини. Після цього реакційну суміш нагрівають зі зворотним холодильником протягом 14 годин. Потім суміш охолоджують до 25ºС і після цього повільно додають 10,08 г (160 ммоль) ціанборгідриду натрію. Після перемішування протягом 12 годин при кімнатній температурі суміш концентрують до повного висихання в ротаційному випарнику. Отриману смолу перемішують з 300 мл води і 500 мл водного розчину їдкого натру (1 н.). Цю суспензію перемішують протягом 30 хвилин з 1 л дихлорметану. Після фільтрації через фритоване скло №3 отриманий твердий продукт промивають водою 2 рази по 500 мл, потім висушують у сушильній шафі під вакуумом при 40 ºС. Твердий продукт перекристалізують з 800 мл метанолу в гарячому стані й одержують 8,3 г 4{3-[3-(2-хлор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Н-піразол-3-(2,4диметоксибензиламіду) у вигляді білого порошку. МС: Час утримування Tr (хв)=4,97; [M+H]+ m/z=621; [M-H]- m/z=619 1 Н ЯМР (400 МГц, ДМСО-d) δ м. ч.: 3,73 (с, 3Н); 3,81 (с, 3Н); 4,19 (д, J=6,7 Гц, 2Н); 4,33 (д, J=6,7 Гц, 2Н); 5,71 (т шир., J=6,7 Гц, 1Н); 6,46 (дд, J=8,3, 2,3 Гц, 1Н); 6,55 (д, J=2,3 Гц, 1Н); 6,80 (д шир., J=9,6 Гц, 1Н); 7,04 (с шир., 1Н); 7,08 (с, 1Н); 7,10 (д, J=8,9 Гц, 1Н); 7,43 (дт, J=11,4, 2,3 Гц, 1Н); 7,67 (д шир., J=8,9 Гц, 1Н); 7,86 (с шир., 1Н); 7,94 (т шир., J=6,7 Гц, 1Н); 8,45 (д, J=8,9 Гц, 1Н); 8,62 (м уш., 1Н); 9,79 (м уш., 1Н); 12,60 (м уш., 1Н). Температура плавлення (за Кофлером)=168ºС 4 UA 106500 C2 1-(2-хлор-4-трифторметилфеніл)-3-(3-фтор-5-формілфеніл)-сечовина 5 10 15 20 25 30 35 40 Розчин 33,15 г (92,68 ммоль) 1-(2-хлор-4-трифторметилфеніл)-3-(3-ціано-5-фторфеніл)сечовини в 600 мл тетрагідрофурану перемішують при -10ºС в атмосфері аргону. Потім рівномірно по краплях додають 230 мл 20 %-го розчину гідриду діізобутилалюмінію в толуолі. Реакційну суміш перемішують при кімнатній температурі протягом 12 годин. Потім додають при -10ºС додаткові 80 мл гідриду діізобутилалюмінію. Після перемішування протягом 14 годин при кімнатній температурі, реакційне середовище концентрують до повного висихання в роторному розчиннику з одержанням густого масла, до якого повільно додають при перемішуванні 500 г льоду і 100 мл 100 %-ої оцтової кислоти. Отриману суспензію фільтрують. Отриманий твердий продукт обробляють етилацетатом 2 рази по 800 мл і органічний розчин промивають 600 мл насиченого розчину хлориду натрію, висушують над сульфатом магнію, фільтрують, концентрують до повного висихання в ротаційному розчиннику і висушують у сушильній шафі у вакуумі при 40ºС з одержанням 29,57 г 1-(2-хлор-4-трифторметилфеніл)-3-(3-фтор-5формілфеніл)-сечовини у вигляді порошку блідо-жовтого кольору. МС: Час утримування Tr (хв)=4,86; [M+H]+ m/z=361; [M-H]- m/z=359 1 Н ЯМР (400 МГц, ДМСО-d) δ м. ч.: 7,35 (ддд, J=8,4, 2,3, 1,8 Гц, 1Н); 7,67-7,75 (м, 2Н); 7,78 (т, J=1,8 Гц, 1Н); 7,88 (д, J=1,8 Гц, 1Н); 8,45 (д, J=8,8 Гц, 1Н); 8,72 (с шир., 1Н); 9,98 (д, J=1,8 Гц, 1Н); 10,07 (с шир., 1Н). 1-(2-хлор-4-трифторметилфеніл)-3-(3-ціано-5-фторфеніл)-сечовина Розчин 15 г (110,2 ммоль) 5-фтор-3-ціананіліну в 150 мл тетрагідрофурану змішують при кімнатній температурі в атмосфері аргону. Потім додають 17,5 мл (121,22 ммоль) 2-хлор-4трифторметилфенілізоціанату. Реакційне середовище нагрівають зі зворотним холодильником протягом 3 годин, потім концентрують до повного висихання в ротаційному випарнику. Отриманий твердий залишок перекристалізують у гарячому стані з 60 мл етилацетату й одержують 33,25 г 1-(2-хлор-4-трифторметилфеніл)-3-(3-ціано-5-фторфеніл)-сечовини у вигляді білої твердої речовини. МС: Час утримування Tr (хв)=4,97; [M+H]+m/z=356 Температура плавлення (за Кофлером)=286ºС 5-фтор-3-ціананілін є комерційним продуктом. Гідрохлорид 4-аміно-1Н-піразол-3-(2,4-диметоксибензиламіду) Суспензію 6,12 г (20 ммоль) 4-нітро-1Н-піразол-3-(2,4-диметоксибензиламіду) в 340 мл етанолу перемішують при кімнатній температурі. Потім додають 15,8 г (70 ммоль) дигідрату хлориду олова протягом 5 хвилин. Реакційну суміш перемішують 14 годин при кімнатній температурі, потім концентрують до повного висихання в ротаційному випарнику. Отриманий залишок перемішують із 330 мл насиченого водного розчину гідрокарбонату натрію і 300 мл дихлорметану. Після декантації органічний шар екстрагують дихлорметаном 2 рази по 150 мл. Органічні шари поєднують, промивають 150 мл насиченого розчину хлориду натрію, висушують 5 UA 106500 C2 5 10 15 20 25 30 35 40 над сульфатом магнію. Після концентрування в ротаційному випарнику одержують 4,57 г гідрохлориду 4-аміно-1Н-піразол-3-(2,4-диметоксибензиламіду) у вигляді твердої речовини фіолетового кольору. МС: IE: [M]+. m/z=276; пік молекулярного іона m/z=151 1 Н ЯМР (400 МГц, ДМСО-d) δ м. ч.: 3,73 (с, 3Н); 3,81 (с, 3Н); 4,31 (д, J=6,2 Гц, 2Н); 4,56 (м уш., 2Н); 6,46 (дд, J=8,3, 2,9 Гц, 1Н); 6,55 (д, J=2,9 Гц, 1Н); 7,06-7,13 (м, 2Н); 7,84 (т, J=6,2 Гц, 1Н); 12,50 (м уш., 1Н). Температура плавлення (прилад Buchi): 186ºС 4-нітро-1Н-піразол-3-(2,4-диметоксибензиламід) Розчин 14,65 г (76,4 ммоль) дигідрату 1-(3-диметиламінопропіл)-3-етилкарбодііміду, 10,32 г (76,4 ммоль) 1-гідроксибензотріазолу в 50 мл диметилформаміду перемішують при кімнатній температурі. Додають 11,7 г (70,03 ммоль) 2,4-диметоксибензиламіну, потім додають маленькими порціями 10,2 г 4-нітро-3-піразолкарбонової кислоти. Після 16 годин перемішування при кімнатній температурі - реакційне середовище виливають у 500 мл води. Суспензію фільтрують, потім промивають водою 2 рази по 250 мл. Отриманий твердий продукт висушують у сушильній шафі у вакуумі при 40ºС з одержанням 18,58 г 4-нітро-1Н-піразол-3-(2,4диметоксибензиламіду) у вигляді білої твердої речовини. 1 Н ЯМР (400 МГц, ДМСО-d) δ м. ч.: 3,75 (с, 3Н); 3,80 (с, 3Н); 4,35 (д, J=5,9 Гц, 2Н); 6,50 (дд, J=8,3, 2,4 Гц, 1Н); 6,56 (д, J=2,4 Гц, 1Н); 7,21 (д, J=8,3 Гц, 1Н); 8,71 (с шир., 1Н); 8,88 (т шир., J=5,9 Гц, 1Н); 14,13 (м уш., 1Н). МС (ES+/-); Час утримування Tr (хв)=3,23; [M+H]+ m/z=307; [M-H]- m/z=305 Температура плавлення (за Кофлером)=192ºС 4-Нітро-3-піразолкарбонова кислота є комерційним продуктом Приклад 2 Гідрохлорид 4-{3-[3-(2-фтор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Н-піразол3-карбоксаміду Суспензію 1,85 г (0,40 ммоль) 4-{3-[3-(2-фтор-4-трифторметилфеніл)-уреїдо]-5фторбензиламіно}-1Н-піразол-3-карбоксаміду в 40 мл етанолу перемішують при кімнатній температурі в атмосфері аргону. Потім додають по краплі 20 мл (40 ммоль) розчину соляної кислоти в діетиловому ефірі (1 н.). Реакційне середовище стає прозорим розчином. Після 12 годин перемішування при кімнатній температурі розчинники випарюють у роторному випарнику при зниженому тиску. Отриманий залишок перемішують у 200 мл діетилового ефіру протягом 30 хвилин. Після фільтрації і висушування в сушильній шафі одержують 1,75 г гідрохлориду 4-{3-[3-(2фтор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Н-піразол-3-карбоксаміду у вигляді блідо-жовтих кристалів. 1 Н ЯМР (400 МГц, ДМСО-d6) δ м. ч.: 4,24 (с, 2Н); 6,83 (д, J=9,0 Гц, 1Н); 7,08 (с, 1Н); 7,18 (с шир., 1Н); 7,23 (с, 1Н); 7,35 (с шир., 2Н); 7,42 (дт, J=11,3, 2,0 Гц, 1Н); 7,54 (д, J=8,8 Гц, 1Н); 7,69 (дд, J=11,2, 1,5 Гц, 1Н); 8,41 (т, J=8,2 Гц, 1Н); 8,99 (с, 1Н); 9,54 (с, 1Н). МС: Час утримування Tr (хв)=0,97; [M+H]+ m/z=455 Температура плавлення (за Кофлером)=184ºС (з розкладанням) 6 UA 106500 C2 4-{3-[3-(2-фтор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Н-піразол-3карбоксамід 5 10 15 20 25 30 35 40 Розчин 22,3 г (36,89 ммоль) 4-{3-[3-(2-фтор-4-трифторметилфеніл)-уреїдо]-5фторбензиламіно}-1Н-піразол-3-(2,4-диметоксибензиламіду) і 17,54 г (92,22 ммоль) паратолуолсульфонової кислоти в 600 мл толуолу нагрівають зі зворотним холодильником протягом 14 годин. Після декантації толуольний розчин відокремлюють від жовтої смоли. Смолу перемішують протягом 2 годин у 280 мл води і 100 мл їдкого натру (10 н.). Суспензію фільтрують. Отриманий твердий продукт промивають водою 3 рази по 350 мл, потім висушують і одержують твердий продукт кремового кольору, що очищають на шарі з 350 г діоксиду кремнію, елююючи розчином дихлорметану/метанолу/ацетонітрилу 95/2,5/2,5 (за об'ємом): одержують 3,85 г 4-{3-[3-(2-фтор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Нпіразол-3-карбоксаміду у вигляді білої твердої речовини. 1 Н ЯМР (400 МГц, ДМСО-d6) δ м. ч.: 4,18 (д, J=5,9 Гц, 2Н); 5,68-5,77 (м, 1Н); 6,80 (д, J=8,8 Гц, 1Н); 7,01-7,08 (с шир., 1Н); 7,04 (с, 1Н); 7,06 (с, 1Н); 7,24 (с шир., 1Н); 7,40 (дт, J=11,4, 2,0 Гц, 1Н); 7,54 (д, J=8,3 Гц, 1Н); 7,69 (д, J=11,5 Гц, 1Н); 8,41 (т, J=8,4 Гц, 1Н); 8,92 (с шир., 1Н); 9,40 (с шир., 1Н); 12,55 (с шир., 1Н). МС: Час утримування Tr (хв)=4,09; [M+H]+ m/z=455; [M-H]- m/z=453 Температура плавлення (за Кофлером)=233ºС 4-{3-[3-(2-фтор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Н-піразол-3-(2,4диметоксибензиламід) Розчин 11,40 г (36,45 ммоль) гідрохлориду 4-аміно-1Н-піразол-3-(2,4-диметоксибензиламіду) і 6,65 мл (40,09 ммоль) діізопропілетиламіну в 360 мл тетрагідрофурані перемішують при кімнатній температурі в атмосфері аргону. Додають 4,35 г (36,45 ммоль) сульфату магнію і 13,80 г (35,2 ммоль) 1-(2-фтор-4-трифторметилфеніл)-3-(3-фтор-5-формілфеніл)-сечовини. Потім реакційне середовище нагрівають зі зворотним холодильником протягом 14 годин. Суміш охолоджують до 25ºС, потім повільно додають 11,46 г (182,25 ммоль) ціаноборгідриду натрію. Після 72 годин перемішування при кімнатній температурі суміш концентрують до повного висихання в роторному випарнику. Отриману смолу перемішують з 400 мл води і 500 мл водного розчину їдкого натру (1 н.). Цю суспензію перемішують протягом 1 години, потім фільтрують через фритоване скло №3, отриману тверду речовину промивають водою 3 рази по 500 мл, потім висушують у сушильній шафі у вакуумі при 40ºС з одержанням 22,45 г 4-{3-[3-(2фтор-4-трифторметилфеніл)-уреїдо]-5-фторбензиламіно}-1Н-піразол-3-(2,4диметоксибензиламіду) у вигляді твердої рожевої речовини. 1 Н ЯМР (500 МГц, ДМСО-d6) δ м. ч.: 3,73 (с, 3Н); 3,81 (с, 3Н); 4,18 (д, J=6,3 Гц, 2Н); 4,33 (с, 2Н); 5,70 (т, J=6,2 Гц, 1Н); 6,47 (д, J=8,2 Гц, 1Н); 6,55 (с, 1Н); 6,79 (д, J=9,1 Гц, 1Н); 7,05 (с, 1Н); 7,07-7,12 (м, 2Н); 7,41 (д, J=11,3 Гц, 1Н); 7,53 (д, J=8,2 Гц, 1Н); 7,68 (д, J=11,3 Гц, 1Н); 7,95 (с шир., 1Н); 8,40 (т, J=8,2 Гц, 1Н); 9,25 (с шир., 2Н); 12,52 (с шир., 1Н). МС: Час утримування Tr (хв)=4,79; [M+H]+ m/z=605; [M-H]- m/z=603 Температура плавлення (за Кофлером)=152ºС 1-(2-фтор-4-трифторметилфеніл)-3-(3-фтор-5-формілфеніл)-сечовина 7 UA 106500 C2 5 10 15 20 25 30 35 40 45 Розчин 11,90 г (34,87 ммоль) 1-(2-фтор-4-трифторметилфеніл)-3-(3-ціано-5-фторфеніл)сечовини в 150 мл тетрагідрофурану перемішують при -2ºС в атмосфері аргону. Потім додають рівномірно "крапля за краплею" 86 мл 20 %-го розчину гідриду діізобутилалюмінію в гексані. Реакційну суміш перемішують при кімнатній температурі протягом 12 годин. Потім додають при -2ºС додаткові 60 мл гідриду діізобутилалюмінію. Після перемішування протягом 3 годин, при кімнатній температурі, реакційне середовище концентрують до повного висихання в роторному розчиннику з одержанням густого масла, до якого повільно додають при перемішуванні 500 г льоду і 300 мл 100 %-ої оцтової кислоти. Отриману суспензію фільтрують. Отриманий твердий продукт промивають водою 4 рази по 150 мл, центрифугують і висушують у сушильній шафі у вакуумі при 40ºС з одержанням 13,95 г 1-(2-фтор-4-трифторметилфеніл)-3-(3-фтор-5формілфеніл)-сечовини у вигляді твердої речовини блідо-жовтого кольору. 1 Н ЯМР (400 МГц, ДМСО-d6) δ м. ч.: 7,33-7,38 (м, 1Н); 7,57 (д, J=8,8 Гц, 1Н); 7,69-7,75 (м, 2Н); 7,79 (с, 1Н); 8,41 (т, J=8,3 Гц, 1Н); 9,05 (с шир., 1Н); 9,65 (с, 1Н); 9,98 (д, J=1,7 Гц, 1Н). МС: Час утримування Tr (хв)=4,62; [M+H]+ m/z=345; [M-H]- m/z=343 Температура плавлення (за Кофлером): 247ºС 1-(2-фтор-4-трифторметилфеніл)-3-(3-ціано-5-фторфеніл)-сечовина Розчин 6,15 г (45,18 ммоль) 5-фтор-3-ціананіліну в 90 мл тетрагідрофурану перемішують при кімнатній температурі в атмосфері аргону. Потім по краплях додають 4,3 мл (36,14 ммоль) дифосгену, потім 30 мл (135,54 ммоль) триетиламіну. Після нагрівання реакційного середовища зі зворотним холодильником протягом 3 годин повільно додають розчин 7,10 г (39,64 ммоль) 4аміно-3-фтортрифторметилбензолу в 10 мл тетрагідрофурану. Нагрівання зі зворотним холодильником підтримують більше 2 годин. Потім середовище перемішують у 100 мл води, після цього екстрагують 100 мл етилацетату. Органічний шар промивають 100 мл насиченого розчину хлориду натрію, висушують над сульфатом магнію і концентрують до повного висихання в ротаційному випарнику. Отриманий твердий продукт перекристалізують у жаркому стані з 90 мл ацетонітрилу й одержують 10,35 г 1-(2-фтор-4-трифторметилфеніл)-3-(3-ціано-5фторфеніл)-сечовини у вигляді твердої речовини кремового кольору. 1 Н ЯМР (400 МГц, ДМСО-d6) δ м. ч.: 7,47 (д, J=8,3 Гц, 1Н); 7,57 (д, J=8,6 Гц, 1Н); 7,69 (с, 1Н); 7,70-7,75 (м, 2Н); 8,37 (т, J=8,4 Гц, 1Н); 9,17 (с шир., 1Н); 9,63 (с шир., 1Н). + МС: Час утримування Tr (хв)=1,1; [M+H] : m/z=341 Температура плавлення (за Кофлером): 253ºС Продукти, згідно з винаходом, корисні як агенти, що здатні інгібувати одну або декілька реакцій, що каталізуються кіназою. KDR і/або Tie2 є кіназами, у зв'язку із якими продукти, згідно з винаходом, особливо корисні як інгібітори. Причини, з яких саме ці кінази були вибрані, викладені нижче: KDR KDR (Kinase insert Domain Receptor), що називається також VEGF-R2 (Vascular Endothelial Growth Factor Receptor 2) експресується головним чином у ендотеліальних клітинах. Такий рецептор має зв'язок із проангіогенним фактором росту VEGF, слугуючи, таким чином, медіатором трансдукційного сигналу шляхом активації свого внутрішньоклітинного домену кінази. Пряме інгібування активності кінази VEGF-R2 дозволяє зменшити явище ангіогенезу в присутності екзогенного VEGF (Vascular Endothelial Growth Factor: васкулярного ендотеліального фактора росту) (Strawn et al., Cancer Research, 1996, vol.56, p. 3540-3545). 8 UA 106500 C2 5 10 15 20 25 30 35 40 45 50 55 60 Такий процес було доведено, зокрема, за допомогою мутантних VEGF-R2 (Millauer et al., Cancer Research, 1996, vol.56, p. 1615-1620). Рецептор VEGF-R2, здається, не має жодної функції в дорослої людини, крім функції, зв'язаної з ангіогенною активністю VEGF. Доповненням до центральної ролі в динамічному ангіогенному процесі, згідно з отриманими нещодавно результатами, є те, що експресія VEGF сприяє виживанню клітин пухлини після хіміо- та радіотерапії, підкреслюючи потенційний синергізм інгібіторів KDR з іншими агентами (Lee et al., Cancer Research, 2000, vol.60, p. 5565-5570). Tie2 Tie-2 (TEK) являє собою член сімейства рецепторів тирозинкінази, специфічної до ендотелільних клітин. Tie2 є першим рецептором із тирозинкіназовою активністю, одночасно відомим як агоніст (ангіопоетин 1 або Ang1), що стимулює аутофосфорилування рецептора та клітинну сигналізацію [S. Davis et al. (1996) Cell 87, 1161-1169], і антагоніст (ангіопоетин 2 або Ang2) [P.C. Maisonpierre et al. (1997) Science 277, 55-60]. Ангіопоетин 1 може виявляти синергізм із VEGF на останніх стадіях неоангіогенезу [Asahara T. Circ. Res. (1998) 233-240]. Досліди з нокаутом генів і трансгенні маніпуляції експресії Tie2 або Ang1 приводять до тварин, які проявляють недостатність васкуляризації [D.J.Dumont et al (1994) Genes Dev. 8, 1897-1909 et C. Suri (1996) Cell 87, 1171-1180]. Зв'язок Ang1 із його рецептором призводить до аутофосфорилування домену кінази Tie2, що є суттєвою для неоваскуляризіції, а також для рекрутирування і взаємодії судин з перицитами і гладкими м'язовими клітинами; такі явища сприяють дозріванню та стабільності новоутворених судин [P.C. Maisonpierre et al.; (1997) Science 277, 55-60]. Автори Lin et al (1997) J. Clin. Invest. 100, 8; 2072-2078 et Lin P. (1998) PNAS 95, 8829-8834 показали інгібування росту і пухлинної васкуляризації, а також зменшення метастазів у легенях при інфікуванні аденовірусами або вакцинації позаклітинного домену Tie2 (Tek) в моделях ксенотрансплантів пухлини молочної залози та меланоми. З наведених нижче причин, за яких інгібітори Tie2 можуть застосовуватися у випадках, коли неоваскуляризація або ангіогенез відбуваються невідповідним чином, тобто при раку взагалі, а також при конкретних видах раку, таких як саркома Капоші або інфантильна гемоангіома, при ревматоїдному артриті, остеоартриті та/або при асоційованих із ними болях, запальних хворобах кишечнику, таких як геморагічний ректоколіт або хвороба Крона, патології очей, таких як м'язова дистрофія, що пов'язана з віком, діабетична ретинопатія, хронічне запалення, псоріаз. Ангіогенез являє собою процес утворення нових капілярних судин від вже існуючих судин. Пухлинний ангіогенез (формування нової кровоносної сітки), небажаний під час росту пухлини, є також одним із суттєвих факторів метастатичного обсіменіння (Oncogene. 2003 May 19:22(20):3172-9; Nat Med. 1995 Jan; 1(1): 27-31). Така неоваскуляризація обумовлена міграцією, а потім проліферацією і диференціюванням ендотеліальних клітин під впливом ангіогенних факторів, що секретуються раковими клітинами і стовбуровими клітинами (Recent Prog Horm Res.2000; 55:15-35; 35-6). Система ангіопоетин 1/рецептор Tie2 відіграє провідну роль у дозріванні судин, забезпечуючи рекрутирування пери-ендотеліальних клітин для стабілізації судинної структури (Cell. 1996 Dec. 27; 87(7):1161-9. Recent Prog Horm Res. 2004; 59:51-71). Таким чином, було доведено, що введення в розчинній рекомбінантній формі позаклітинного домену рецептора Tie-2 (exTek) інгібує ангіогенез у пухлині на моделях мишачих пухлин, а також поширення метастазів (Pengnian Lin, Jake A. Buxton, Ann Acheson, Czeslaw Radziejewski, Peter C. Mainsonpierre, George D. Yancopoulos, Keith M. Channon, Laura P. Hale, Mark W. Dewhirst, Samuel E. George, and Kevin G.Peters, Proc Natl Acad Sci USA. 1998 Jul 21; 95(15): 8829-34; Cecilia Melani, Antonelle Stoppacciaro, Chiara Foroni, Federica Felicetti, Alessandra Care et Mario. Colombo, Cancer immunol Immunother. 2004 Jul; 53(7):600-8). У культурі ендотеліальних клітин стимуляція Tie2 активує PI3-кіназовий шлях і р42/р44-кіназові шляхи, задіяні у проліферації і міграції клітин; шлях синтезу PAF (Cell Signal. 2006 Apr 14; ahead of print), залучений до прозапальної активності. Стимуляція Tie2 стимулює Akt-шлях та інгібує апоптоз (Laura M DeBusk, Dennis E Hallahan, Pengnian Chorles Lin, Exp Cell Res. 2004 Aug 1; 298(1): 167-77), сигнальний трансдукційний шлях, відомий своєю значущістю для виживання клітин. Додавання Extek (розчинний рецептор Tie2) інгібує утворення псевдотрубочок ендотеліальних клітин на Матригелі (Cecilla Melani, Antonelle Stoppacciaro, Chiara Foroni, Federica Felicetti, Alessandra Care et Mario. Colombo, Cancer immunol Immunother. 2004 Jul; 53(7):600-8). Ці дослідження вказують на те, що система Tie2/Ангіопоетин необхідна на перших стадіях утворення судинних вузлів у тканинах дорослої людини, і що функцією рецептору Tie2 є збільшення тривалості життя ендотеліальних клітин у процесі формування мережі кровоносних судин. Крім того, ангіопоетин-1 стимулює проліферацію лімфатичних ендотеліальних клітин, а 9 UA 106500 C2 5 10 15 20 25 30 35 40 45 50 55 також лімфоангіогенез (розвиток нових лімфатичних судин), переважний шлях метастазування. (Tohru… Morisada, Yuichi Oike, Yoshihiro Yamada, Takashi Urano, Masaki Akao, Yoshiaki Kubota, Hiromitsu Maekawa, Yoshishige Kimura, Masako Ohmura, Takeshi Miyamoto, Shiro Nozawa, Gou Young Koh, Kari Alitalo and Toshio Suda, Blood. 2005 Jun 15; 105(12): 4649-56. Серед ферментів, що грають певну роль в ангіогенезі, можна назвати також PDGFRβ (Guzman and Laurence H. Hurley, Univ of Arizona Molecular Cloning of the Human PDGFR-beta Promoter and Targeting the G-Quadruplex-Forming Region to Control gene Expression; Biomedical Drug Discovery, Genomics/Gentics, Therapeutic), FGFR1 (Somaia Elbauomy Elsheikh, Andrew R 1 3 1 Green, Maryou BK Lambros, Nicholas C Turner, Matthew J Graing, Des Powe, lan O Ellis and Jorge S Reis-Filho, FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis; FLT1 (Shibuya M (2007). "Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): a dual regulator for angiogenesis.” Angiogenesis 9(4): 225-30; discussion 231 et VEGFR3 (Tamela, T. et al Blocking VEGFR-3 supresses angiogenic sprouting and vascular network formation, Nature 454, 656-660 (20058). Процеси ангіогенезу відіграють провідну роль у розвитку численних солідних пухлин. Крім того, було доведено, що вірогідність появи метастазів сильно збільшується при підсилюванні васкуляризації первинної пухлини (Br J Cancer. 2002 May 20; 86(10): 1566-77). Нещодавно потенційна роль проангіогенних агентів при лейкеміях і в лімфомах також була підтверджена документально. Загалом, було показано, що клітинні клони за таких патологій можуть бути або зруйновані природним шляхом самою імунною системою, або перетворюються в ангіогенний фенотип, який сприяє виживанню клітин та їх проліферації. Така зміна фенотипу зумовлена суперекспресією ангіогенних факторів, зокрема, макрофагами, та/або мобілізацією цих факторів із позаклітинної матриці (Thomas DA, Giles FJ, Cortes J, Albitar M, Kantarjian HM., Acta Haematol, (2001), vol.207, pp 106-190). Існує кореляція між процесом ангіогенезу кісткового мозку та "екстрамедулярними ураженнями" при CML (хронічний мієломоноцитарний лейкоз). Різні дослідження показують, що інгібування ангіогенезу могло б слугувати терапією вибору для лікування даної патології (Leuk Res. 2006 Jan; 30(1): 54-9; Histol Histopathol. 2004 oct.; 19(4): 1245-60. Крім того, існує стійке припущення, що активація системи Tie2/ангіопоетин задіяна в розвитку ангіогенезу кісткового мозку у пацієнтів, хворих на множинну мієлому (Blood. 2003 Jul 15; 102(2): 638-45. Визначення активності сполук - Протоколи випробувань 1. KDR Інгібуючий ефект сполук визначали у тесті in vitro на фосфорилування субстрату методом сцинтиляції (96 ямковий планшет basic Flash Plate). Цитоплазматичний домен (залишок 790-1356) людського ферменту KDR був клонований у формі злитого GTS у вектор експресії бакуловірусу pFastBac. Білок був експресований у клітинах SF21, очищений та активований аутофосфорилуванням. Субстрат складався з залишків 658-850 PLCγ, експресованої і очищеної у формі злитого білка GST. Кіназну активність KDR вимірювали в буфері, що містить 20 мМ MOPS, 10 мМ MgCl 2, 10 мМ MnCl2, 1 мМ DTT, рН=7,4. Сполуки спочатку розбавляли в 100 % ДМСО, потім готували розчин 10Х в суміші ДМСО 30 % - буфер 70 %. Вносили 10 мкл розчину 10Х, потім 70 мкл буфера, що містить 150 нг (1,6 пмоль) ферменту KDR, при 4ºС. Реакцію запускали додаванням 20 мкл 33 розчину, що містить 2 мкг (41 пмоль) PLCγ-субстрату, 0,5 мкCi γ P[ATP] і 2 мкМ неміченого АТР. Планшет струшували. Після 30 хвилин інкубування при 37ºС інкубаційний буфер видаляли і ямки промивали три рази 300 мкл PBS. Радіоактивність вимірювали в кожній ямці, використовуючи лічильник радіоактивності Trilux-βWallac. Фоновий шум визначали вимірюванням радіоактивності в чотирьох різних ямках, що містять АТР (немічений і радіоактивно мічений) і субстрат, за відсутності ферменту і сполуки. Контрольну активність вимірювали в чотирьох різних ямках, що містили усі реагенти, але за відсутності сполуки. Інгібування активності KDR за допомогою сполуки за винаходом виражали у відсотках інгібування стосовно контрольної активності, що визначалась при відсутності сполуки. 2. Tie2 Інгібуючий ефект сполук визначали в тесті in vitro на фосфорилування субстрату методом сцинтиляції (96 ямковий планшет basic Flash Plate). Послідовність кодування людського Tie2, що відповідає амінокислотам внутрішньоклітинного домену 774-1124 була введена у вектор експресії бакуловірусу pFastBac у формі злитого білка GST. GST-Tie2 очищали й активували аутофосфорилуванням. Субстрат складався з залишків 658-850 PLCγ, експресованої і очищеної у формі злитого білка GST. 10 UA 106500 C2 5 10 15 20 25 30 35 40 Кіназну активність Tie2 вимірювали в буфері MOPS 20 мМ, рН=7,4, що містить 10 мМ MgCl 2, 10 мМ MnCl2, 1 мМ DTT. Сполуки спочатку розбавляли в 100 % ДМСО, потім готували розчин 10Х в суміші ДМСО 30 % - буфер 70 %. Вносили 10 мкл розчину 10Х, потім 70 мкл буфера, що містить 100 нг (1,5 пмоль) ферменту Tie2, при 4ºС. Реакцію запускали додаванням 20 мкл 33 розчину, що містить 2 мкг (41 пмоль) PLCγ-субстрату, 0,5 мкКю γ P[ATP] і 2 мкМ холодного АТР. Планшет струшували. Після 30 хвилин інкубування при 37ºС інкубаційний буфер видаляли і ямки промивали три рази 300 мкл PBS. Радіоактивність вимірювали в кожній ямці, використовуючи лічильник радіоактивності Trilux-βWallac. Фоновий шум визначали виміром радіоактивності в чотирьох різних ямках, що містять АТР (холодний і радіоактивно мічений) і субстрат, за відсутності ферменту і сполуки. Контрольну активність вимірювали в чотирьох різних ямках, що містили усі реагенти, але за відсутності сполуки. Вимірювали величину інгібування активності Tie2 і виражали у відсотках інгібування від контрольної активності, що визначалась за відсутності сполуки. 3. Скринінг сполук шляхом вимірювання фосфорилуючої активності кінази FLTI за присутності субстрату PLCγ за допомогою радіоактивного мічення. Кіназну активність FLT1 вимірювали таким же способом, що і інгібування Tie2, використовуючи реакційну суміш, що складається з 70 мкл кіназного буфера, що містить 13 нм ферменту FLT1 на ямку. Розраховували величину інгібування активності FLT1 і виражали у відсотках інгібування стосовно контрольної активності, що визначалась за відсутності сполуки. Відсоток інгібування, що відповідає кожній концентрації сполук, розраховували в такий спосіб: % інгібування=(середнє значення СРМ оброблених ямок - середнє значення СРМ контрольних ямок)/(СРМ неопрацьованих ямок - середнє значення СРМ контрольних ямок) ×100 4. Скринінг сполук шляхом вимірювання фосфорилуючої активності кінази PDGFR у присутності субстрату PLCγ за допомогою радіоактивного мічення. Тест був здійснений аналогічно тесту, проведеному з ферментом FLT1, але замість 13 нм FLT1 використовували 16 нм ферменту PDGFR, і в 96-ямкових планшетах змішували 18 мкл ферменту GST-PDGFR у концентрації 4 мг/мл (при чистоті 80 %), партія VLT802. 5. Скринінг сполук шляхом вимірювання фосфорилуючої активності кінази FGFR у присутності субстрату PLCγ за допомогою радіоактивного мічення. Тест був здійснений аналогічно тесту, проведеному з ферментом FLT1, але замість 13 нМ FLT1 використовували 27 нМ ферменту FGFR, і в 96-ямкових планшетах змішували 18 мкл ферменту GST-FGFR у концентрації 1,1 мг/мл (при чистоті 100 %): (партія JCE3666). Результати: Сполуки відповідно до прикладів винаходу знаходяться в концентрації, інгібуючої активність кінази на 50 %, як правило, між 0,1 нм і 2 мкм відносно кінази KDR та/або Tie2, переважно, між 0,1 нм і 500 нм, переважно, між 0,1 нм і 50 нм. Величини концентрацій, наведені нижче в таблиці 1, зазначені як ілюстрації. 11 UA 106500 C2 5 10 15 20 25 30 35 40 45 50 55 Сполуки згідно з винаходом були об'єктом фармакологічних досліджень, що дозволили визначити їх печінковий кліренс. Оцінка внутрішнього кліренсу сполук з використанням гепатоцитів людини: протокол експерименту. NB: Тест in vitro, описаний нижче, здійснений із клітинами печінки людини, був використаний для попереднього виявлення важливого фармакокінетичного параметру: вплив на печінковий метаболізм даної сполуки після її введення в організм людини. Умови культивування: Інкубування кріо-консервованих людських гепатоцитів людини (IVT: IVT-TLN-180608, отриманих від In Vitro Technologies, inc, Baltimore, Maryland, США) і свіжих людських гепатоцитів (від Biopredic: HEP200239) проводили в планшетах, що містять 48 ямок, покритих колагеном. Умови експерименту: Кінетичні процеси запускали додаванням у культуральне середовище сполук у кінцевій концентрації 5 мкм за відсутності або присутності кетоконазолу 10 мкм (інгібітор CYP3A4). Інкубаційний об'єм складав 100 мкл, тривалість інкубування: 0-24 години (звичайні кінетичні точки: 0-0,5-1-2-4-6-8-24 години). Кінетичні процеси зупиняли додаванням ацетонітрилу/води з використанням кортикостерону як внутрішнього стандарту. Клітини відкріплювали, потім лізували. Внутрішньоклітинні і позаклітинні середовища з'єднували і заморожували (-20ºС) для збереження до аналізу РХМС/МС. Метод аналізу РХ-МС/МС Об'єднані внутрішньоклітинні і позаклітинні середовища відтавали, піддавали впливу ультразвуку, перемішували на вортексі і центрифугували при 3000g протягом 20 хвилин. Супернатанти інжектували й аналізували методом РХ-МС/МС. Обробка даних і "класифікація» - Максимальну початкову швидкість in vitro розраховували для метаболітів, специфічних для цитохром Р450, і виражали в нмоль/год./мільйон клітин. V=[концентрація в Т(n) - концентрація в Т(n-1)]/[T(n)-T(n-1)] Визначали внутрішній кліренс і виражали в мл/год./мільйон клітин Clint= доза/AUC0-24 год. Доза=кількість у Т(0)(нмоль/мільйон клітин) AUC0-24 год.: розраховано за допомогою програми WinNonlin, використовуючи некомпартментний аналіз з моделюванням внутрішньовенної болюсної ін'єкції. Класифікація внутрішнього кліренсу була представлена в такий спосіб: -1 -6 Clint.
ДивитисяДодаткова інформація
Назва патенту англійськоюPyrazole derivatives, preparation thereof, and therapeutic use thereof
Автори англійськоюAbecassis, Pierre-Yves, Desmazeau, Pascal, Tabart, Michel
Автори російськоюАбекасси Пьер-Ив, Дэмазо Паскаль, Табар Мишель
МПК / Мітки
МПК: C07C 275/30, A61P 35/00, C07D 231/38, A61K 31/415
Мітки: застосування, піразолів, спосіб, похідні, терапії, одержання
Код посилання
<a href="https://ua.patents.su/18-106500-pokhidni-pirazoliv-sposib-kh-oderzhannya-ta-zastosuvannya-v-terapi.html" target="_blank" rel="follow" title="База патентів України">Похідні піразолів, спосіб їх одержання та застосування в терапії</a>
Похідні азакарболінів, спосіб їх одержання і застосування в терапії

Номер патенту: 101668
Опубліковано: 25.04.2013
Автори: Бедель Олів'є, Гуйон Тьєррі, Муркрофт Нейл, Папен Давід, Міньяні Серж, Арендт Крістофер, Лі Жунхуа, Лєвіт Міхаїл, Бабен Дід'є
МПК: C07D 471/14, A61K 31/437, A61P 35/00
Мітки: спосіб, застосування, терапії, азакарболінів, похідні, одержання
Формула / Реферат:
1. Сполука загальної формули (І):, формула (І)в якій:- R3, R4 незалежно один від одного означають:1) Н;2) галоген;3) CF3;4) заміщений окси;5) необов'язково заміщений...
Спосіб одержання піразолів, спосіб регіоселективного алкілування та застосування фосфату або фосфонату у способі

Номер патенту: 93860
Опубліковано: 25.03.2011
Автори: Корсі Камілла, Вальтер Харальд, Ламберт Клеменс, Еренфройнд Йозеф, Тоблер Ханс
МПК: C07D 231/14
Мітки: алкілування, способи, фосфонату, застосування, піразолів, фосфату, одержання, регіоселективного, спосіб
Формула / Реферат:
1. Спосіб одержання заміщеного піразолу формули І, (I)у якій R1 означає С1-С4галогеналкіл; R2 означає С1-С6алкіл і R3 означає метил або етил, у якому заміщений піразол формули II, (II)у якій R1 і R2 є такими, як визначено для формули І, вводять у реакцію з фосфатом або...
Хіназоліндіонові похідні, їх одержання і їх застосування в терапії

Номер патенту: 99485
Опубліковано: 27.08.2012
Автори: Нав Жан-Франсуа, Гласс Крістоф, Клосс Анні, Сейєр Андре, Віве Бертран, Мюзе Ніколя, Марсіньяк Жільбер
МПК: A61K 31/517, C07D 403/04
Мітки: хіназоліндіонові, одержання, похідні, терапії, застосування
Формула / Реферат:
1. Сполука, яка відповідає загальній формулі (І): , (I)у якій:- А означає арильну або гетероарильну групу;- R1 означає:• атом водню,• -C(O)R, де R означає атом водню, (С1-С6)-алкоксильну групу, арильну групу, (С3-С6)-циклоалкільну групу або (С1-С6)-алкільну групу, причому вищевказаний алкіл можливо заміщений:•...
Похідні азабіциклічних карбоксамідів, їх одержання і їх застосування в терапії

Номер патенту: 102837
Опубліковано: 27.08.2013
Автори: Маланда Андре, Дюбуа Лоран, Еванно Яннік, Машнік Давід
МПК: A61K 31/437, A61P 11/00, A61P 31/00, C07D 471/04, A61P 29/00
Мітки: застосування, похідні, карбоксамідів, азабіциклічних, одержання, терапії
Формула / Реферат:
1. Сполука, що відповідає загальній формулі (І):, (I) в якій: або X1, Х2, Х3 і Х4 означають незалежно один від одного групу C-R1; або, з Х1, Х2, Х3 і Х4, один з Х3 і Х4 означає атом азоту, а інші незалежно один від одного означають групу C-R1; R1 вибирають з атома водню, атома...
Похідні 5-піридиніл-1-азабіцикло[3.2.1]октану, їх одержання і їх застосування у терапії

Номер патенту: 92917
Опубліковано: 27.12.2010
Автори: Локхед Алістер, Леклерк Оділь, Ваше Жюльєн, Галлі Фредерік
МПК: A61K 31/439, A61P 25/00, C07D 471/18
Мітки: 5-піридиніл-1-азабіцикло[3.2.1]октану, терапії, похідні, одержання, застосування
Формула / Реферат:
1. Сполука загальної формули (І), (I) в якій R означає групу, вибрану з піразолілу, імідазолілу, триазолілу, оксазолілу, оксадіазолілу, тіазолілу, ізотіазолілу, тіадіазолілу, тетразолілу, причому вказана група, можливо, заміщена однією або декількома групами, вибраними з галогенів, (С1-С6)алкілу, (С1-С6)алкокси,...
Наступний патент: Пристрій для дослідження процесу фарбопередачі у друкарському контакті
Випадковий патент: Каталізатор - фільтр