Спосіб одержання 2-сульфініл-1н-бензоімідазолів
Номер патенту: 105479
Опубліковано: 26.05.2014
Автори: Рузіц Мілош, Смодіш Янез, Котар-Йордан Берта, Іскра Юрней, Ставбер Стоян, Зупет Рок
















Формула / Реферат
1. Спосіб одержання сполуки формули (І)
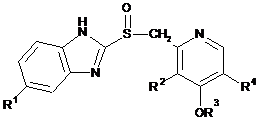 (I)
(I)
або її солі або сольвату, або гідрату, де
R1 вибраний з групи, що складається з водню, С1-4-алкілу і С1-4-алкокси, де С1-С4-алкіл і С1-С4-алкокси не заміщені або заміщені одним або більше ніж одним галогеном,
R2 вибраний з групи, що складається з водню, С1-С4-алкілу і С1-С4-алкокси, де С1-С4-алкіл і С1-С4-алкокси не заміщені або заміщені одним або більше ніж одним галогеном,
R3 є С1-С4-алкіл, не заміщений або заміщений одним або більше ніж одним галогеном або одним або більше ніж одним С1-С4-алкокси, і
R4 вибраний з групи, що складається з водню і С1-С4-алкілу, не заміщеного або заміщеного одним або більше ніж одним галогеном,
в якому здійснюють
(а) окислення сполуки формули (II)
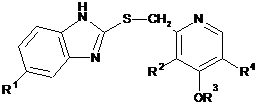 (II)
(II)
або її гідрату або сольвату, або солі, в якій R1, R2, R3 і R4 мають ті ж значення, як визначено для сполуки (І), з одержанням сполуки формули (І), де окислення проводять у присутності перекису водню або аддукту сечовини з перекисом водню, трифторетанолу і металевого каталізатора, вибираного з групи, що складається з CH3ReО3, C2H5ReО3, Re(PPh3)2OCl3, Na2MoO4, V2O5, VOCl3, VOF3, VO(OC2H5)3, VO(1-OC3H7)3, VO(2-OC3H7)3, VO(CH3COCHCOCH3)2, NaVO3, H2WO4, H4SiW12O40, (NH4)2Ce(NO3)6 і Yb(OSO2CF3)3,
(б) можливо, виділення сполуки формули (І) і
(в) можливо, очищення сполуки формули (І) і/або перетворення її в її сіль або сольват, або гідрат.
2. Спосіб за п. 1, який відрізняється тим, що R1 є дифторметокси, R2 є метокси, R3 є метилом і R4 є воднем.
3. Спосіб за п. 1, який відрізняється тим, що R1 є воднем, R2 є метилом, R3 є 2-трифторетилом і R4 є воднем.
4. Спосіб за п. 1, який відрізняється тим, що R1 є метокси, R2 є метилом, R3 є метилом і R4 є метилом.
5. Спосіб за п. 1, який відрізняється тим, що R1 є воднем, R2 є метилом, R3 є 3-метоксипропілом і R4 є воднем.
6. Спосіб за будь-яким з пп. 1-5, який відрізняється тим, що перекис водню використовують у формі водного розчину перекису водню.
7. Спосіб за будь-яким з пп. 1-6, який відрізняється тим, що перекис водню використовують в кількості від 0,5 до 3,0 еквівалентів щодо сполуки (II).
8. Спосіб за п. 1, який відрізняється тим, що металевий каталізатор використовують в кількості від 0,0001 до 0,1 еквівалента щодо сполуки (II).
9. Спосіб за будь-яким з пп. 1-8, який відрізняється тим, що стадію (а) здійснюють у присутності трифторетанолу і органічного розчинника.
10. Спосіб за п. 9, який відрізняється тим, що вказаний органічний розчинник вибирають з групи, що складається з метанолу, етанолу, ацетону, ацетонітрилу, C6H5CF3, простих ефірів, неполярних розчинників і їх сумішей.
11. Спосіб за п. 10, який відрізняється тим, що вказаний простий ефір є тетрагідрофураном і дані неполярні розчинники є дихлорметанолами або ізоалканами.
12. Спосіб за будь-яким з пп. 1-11, який відрізняється тим, що сполуку (II) використовують на стадії (а) в концентрації від 0,1 до 5 моль/л.
13. Спосіб за будь-яким з пп. 1-12, який відрізняється тим, що стадію (а) здійснюють при температурі, що знаходиться в діапазоні від -30 до 30 °C.
14. Спосіб за будь-яким з пп. 8-13, який відрізняється тим, що перекис водню або аддукт сечовини з перекисом водню додають до розчиненої сполуки формули (II) або її гідрату, сольвату або солі і потім реакцію ініціюють шляхом додавання металевого каталізатора.
15. Спосіб за п. 1, який відрізняється тим, що стадію (б) здійснюють, проводячи наступні операції:
(і) додавання ацетону або розчину тіосульфатної солі і, можливо, основи в реакційну суміш, одержану на стадії (а),
(іі) додавання води до суміші із операції (і) для осадження твердої сполуки (І), і
(ііі) виділення сполуки (І).
16. Спосіб за п. 1, який відрізняється тим, що стадію (б) здійснюють, проводячи наступні операції:
(і) додавання до реакційної суміші, одержаної на стадії (а), дихлорметану, розчину солі тіосульфату і, можливо, основи,
(іі) повне видалення розчинника з суміші із операції (і) з одержанням неочищеної сполуки (І),
(ііі) додавання етилацетату до одержаної в результаті операції (іі) неочищеної сполуки (І), і
(iv) видалення етилацетату з одержанням сполуки (І).
17. Спосіб за п. 1, який відрізняється тим, що стадію (б) здійснюють, проводячи наступні операції:
(і) додавання розчину солі тіосульфату і, можливо, основи до реакційної суміші, одержаної на стадії (а),
(іі) повне видалення розчинника з суміші із операції (і) з одержанням неочищеної сполуки (І),
(ііі) змішування неочищеної сполуки (І), одержаної в результаті операції (іі), з дихлорметаном, і
(iv) видалення дихлорметану з одержанням сполуки (І).
18. Спосіб за п. 1, який відрізняється тим, що стадію (б) здійснюють, проводячи наступні операції:
(і) виділення твердого продукту реакції з реакційної суміші, одержаної на стадії (а),
(іі) додавання дихлорметану до твердого продукту реакції, одержаного в результаті операції (і), і
(ііі) видалення дихлорметану з одержанням сполуки (І).
19. Спосіб за п. 1, який відрізняється тим, що стадію (б) здійснюють, проводячи наступні операції:
(і) додавання солі тіосульфату і, можливо, основи до реакційної суміші, одержаної на стадії (а),
(іі) видалення всього або щонайменше частини розчинника в суміші із операції (і) з одержанням неочищеної сполуки (І) або концентрованої суміші, що містить сполуку (І),
(ііі) змішування неочищеної сполуки (І) або концентрованої суміші, одержаної в результаті операції (іі), з розчинником, і
(iv) виділення сполуки (І) з суміші, одержаної в результаті операції (ііі).
Текст
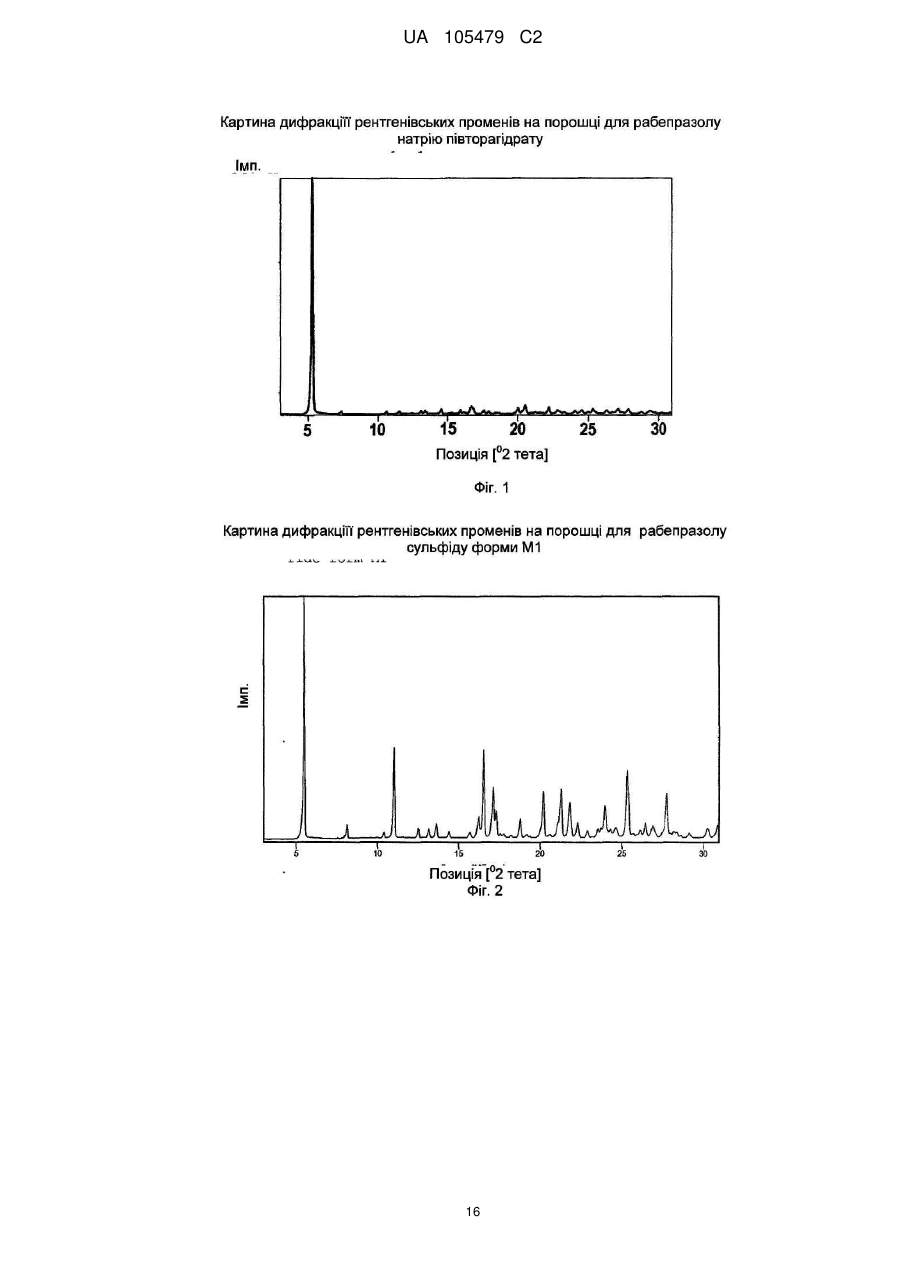
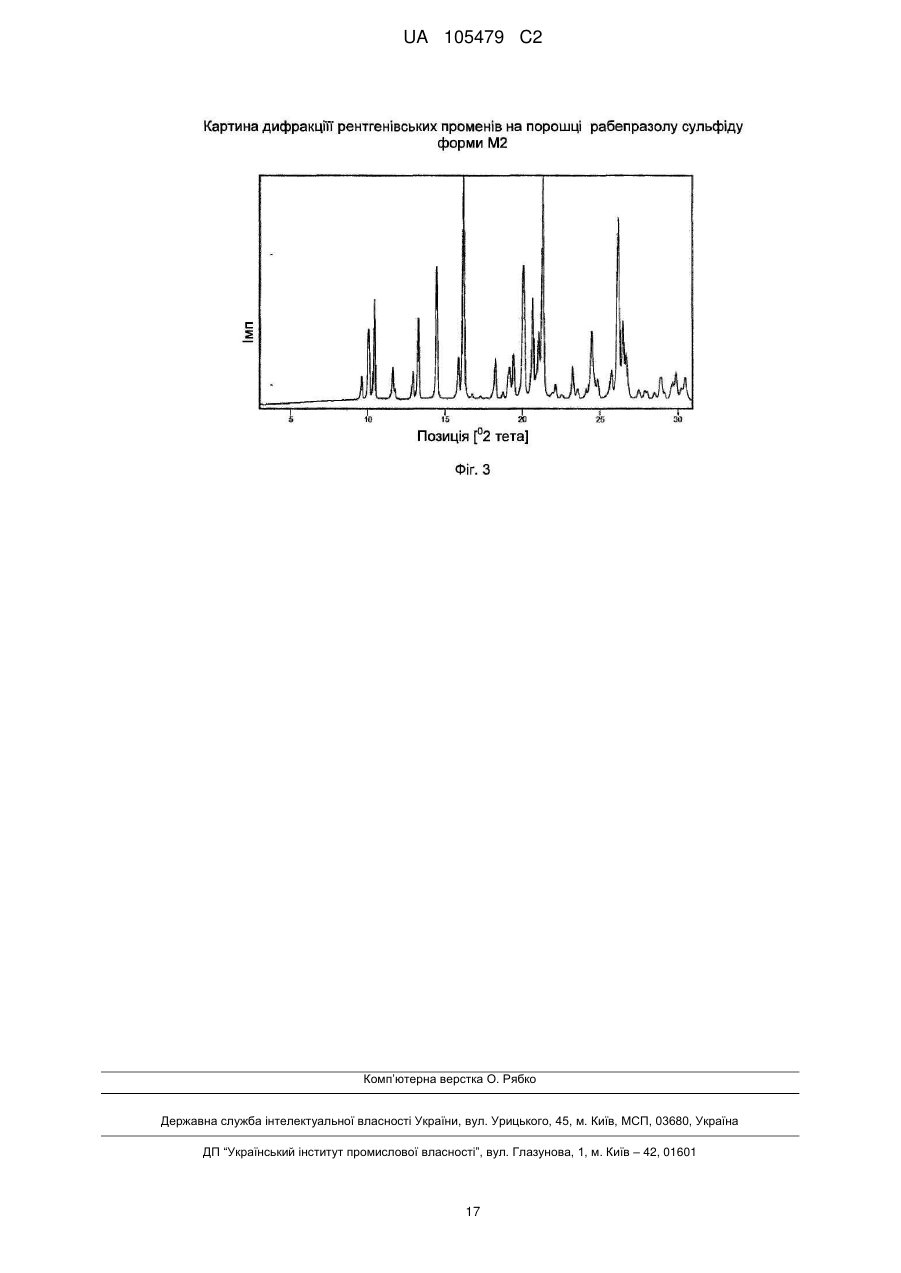
Реферат: Даний винахід бензоімідазолів стосується способу O H N S CH2 N R R N 2 R 1 OR 4 3 шляхом окисления тіоефірного попередника H N S CH2 N R R N 2 R 1 у присутності трифторетанолу. OR 3 4 одержання 2-(2-піридинілметилсульфініл)-1Н UA 105479 C2 5 ОБЛАСТЬ ВИНАХОДУ Даний винахід відноситься до способу одержання 2-(2-піридинілметилсульфініл)-1Нбензоімідазолів, таких як пантопразол, лансопразол, омепразол і рабепразол. ПОПЕРЕДНІЙ РІВЕНЬ ТЕХНІКИ Заміщені сполуки 2-(2-піридинілметилсульфініл)-1Н-бензимідазолу є добре відомими інгібіторами шлункового протонного насоса. Ці сполуки можуть інгібувати секрецію кислоти шлункового соку і застосовуються як противиразковий агент. їх можна представити наступною 1 4 загальною формулою, де R -R мають однакові значення, як описано нижче: 10 15 20 25 30 35 40 45 50 Приклади таких бензимідазолових сполук включають 2-[[[3-метил-4-(2,2,2-трифторетокси)-2піридиніл]метил]сульфініл]-1Н-бензимідазол (лансопразол), 5-(дифторметокси)-2-[[[3,4диметокси-2-піридиніл]метил] сульфініл]-1Нбензи мідазол (пантоп разол), 5-метокси-2-[[[4-метокси-3,5-д и мети л-2піридиніл]метил]сульфініл]-1Н-бензимідазол (омепразол) і 2-[[[4-(3-метоксипропокси)-3метил-2-піридиніл]метил]сульфініл]-1/-/-бензимідазол (рабепразол).· Способи одержання бензимідазолу відомі. Деякі способи включають застосування попередника, що має тіоефірну групу, яку піддають окисленню. У EP 0 174 726 В1 описане застосування перкислоти, такої як хлорпербензойна кислота, броміт натрію, гіпохлорит натрію або перекис водню, як окисляючий агент. Окислення проводили в галогенованих вуглеводнях, амідах, спиртах або їх сумішах. У EP 0 302 720 А1 описаний спосіб одержання 2-сульфінілбензимідазолів з використанням перекису водню у присутності сполуки ванадію, такої як пентоксид ванадію, метаванадат натрію або ацетилацетонат ванадію. У WO 02/062786 А1 окислення попередника тіоефіру проводили з використанням третбутилгідропероксиду (TBHP) у присутності пентоксиду ванадію, метаванадату натрію або ацетилацетонату ванадію. Переважно, окислення проводили в толуолі або ізопропанолі. У WO-2004/011455 А1 описаний спосіб одержання ланзопразола з використанням третбутилгідропероксиду (TBHP) у присутності окситрихлориду ванадію як каталізатора, де реакцію проводили в розчиннику, такому як С1-5-спирт, декан, нонан, толуол або їх суміш з водою. Крім того, реакцію переважно проводили у присутності слабкої основи. WO 03/008406 А1 відноситься до способу одержання сполук типу бензоімідазолів шляхом реакції відповідного попередника з окислювачем у відповідному розчиннику, з екстракцією сульфонових побічних продуктів і виділенням цільового продукту. Переважно, як окислювач застосовують хлорпербензойну кислоту. У EP 0 997 461 А1 описане окислення тіоефірного бензимідазолу відповідною сульфоксидною сполукою з використанням N-галосукциніміду, 1,3-дигало-5,5-диметилгідантоіну або дихлорізоціанурату у присутності основи. Альтернативно, як окислювач можна застосовувати перборатну сполуку у присутності кислого ангідриду або металевого каталізатора. У WO 01/21617 А1 запропонований спосіб одержання лансопразола з використанням перекису водню у присутності ренієвого каталізатора. Переважно, як каталізатор застосовують метилтріоксорений. Окислення проводять в етанолі. Селективність, досягнута на стадії окислення, як повідомляється, є прийнятною, якщо реакцію проводять при температурі від -20 0 до -3O C, і кількість сполуки ренію складає від 1 до 5 моль% по відношенню до початкової речовини. При вищих температурах реакції і нижчих концентраціях каталізатора підвищувалося утворення домішок. Нарешті, в WO 2004/056803 А1 описується спосіб одержання сульфінільних похідних з використанням перекису водню як окислювача у присутності сполуки ренію. В протилежність WO 01/21617 А1, згідно цього документу пропонується використовувати ренієвий каталізатор в кількості від 0,01 до 0,5 моль% по відношенню до початкової сполуки - сульфіду, і підтримувати температуру реакції в діапазоні від 0˚C до кімнатної температури. Як розчинник під час стадії 1 UA 105479 C2 5 10 15 окислення можна застосовувати лінійні або розгалужені С 1-С6-спирти, кетон, ефіри, складні ефіри або аміди самі по собі або в суміші з водою. Переважно застосовують метанол. При температурі реакції 5 °C, концентрації каталізатора 0,1 моль% і часу реакції 4 години, лансопразол утворюється з виходом 75 %. Способи одержання заміщених сполук 2-(2-піридинілметилсульфініл)-1/-/-бензимідазолу відповідно до попереднього рівня техніки все ж таки мають недоліки, що полягають в низькому виході одержаного бензимідазолу, високого вмісту домішок бензимідазолу і/або застосуванні неекономічних умов реакції. Це може відбуватися через те, що зокрема, при вищих температурах реакції селективність на стадії окислення нижча, і, таким чином, можуть утворюватися побічні продукти, такі як N-оксид відповідного бензимідазолу і відповідний сульфон, шляхом окислення азоту і надокислення сульфіду, відповідно. Отже, до цих пір існує необхідність в створенні покращуваного способу одержання заміщених сполук 2-(2-піридинілметилсульфініл)-1Н-бензимідазолу, що не має вищезазначених недоліків. Зокрема, бажаний спосіб, який забезпечує покращуваний баланс між селективністю реакції окислення, виходом продукту і економічною ефективністю умов реакції. Ця проблема несподівано вирішується за допомогою способів одержання лансопразолу за пп. 1-24. ДОКЛАДНИЙ ОПИС ВИНАХОДУ Згідно першого аспекту винаходу, спосіб одержання сполуки формули (І) 20 25 30 або його солі, сольвату або гідрату, де 1 R вибраний з групи, що складається з водню, С 1-4-алкілу і С1-4-алкокси, де С1-С4-алкіл і С1С4-алкокси не заміщені або заміщені одним або більш, ніж одним галогеном, 2 R вибраний з групи, що складається з водню, С 1-С4-алкіла і C1-C4-алкокси, де С1-С4-алкіл і С1-С4-алкокси не заміщені або заміщені одним або більш ніж одним галогеном, 3 R є С1-С4-алкіл, не заміщений або заміщений одним або більше, ніж одним галогеном або одним або більш ніж одним С1-С4-алкокси, і 4 R вибраний з групи, що складається з водню і С 1-С4-алкіла, не заміщеного або заміщеного одним або більше, ніж одним галогеном включає (а) окислення сполуки формули (II) 35 1 40 45 2 3 4 або його гідрата, сольвата або солі, де R , R , R і R мають ті ж значення, як визначено вище, з одержанням сполуки формули (І), де окислення проводять у присутності трифторетанола (б) можливо, виділення сполуки формули (І) і (в) можливо, очищення сполуки формули (І) і/або перетворення її в її сіль, сольват або гідрат. Несподівано було виявлено, що присутність трифторетанолу дає можливість одержання заміщених 2-(2-піридинілметилсульфініл)-1Н-бензимідазольних сполук з високим виходом і 2 UA 105479 C2 5 10 15 20 25 30 35 40 45 50 55 приводить до утворення заміщених 2-(2-піридинілметилсульфініл)-1Н-бензимідазольних сполук, які містять лише невеликі кількості домішок. 1 4 Переважно, одержують таку сполуку формули (І), в якій R -R мають незалежно один від одного наступні значення: 1 R вибраний з групи, що складається з водню і С 1-С4-алкокси, який не заміщений або заміщений одним або більш, ніж одним галогеном 2 R вибраний з групи, що складаєпься з незаміщеного С1-С4-алкіла і незаміщеного С1-С4алкокси 3 R є С1-С4-алкіл, не заміщений або заміщений одним або більше, ніж одним галогеном або одним або більш ніж одним С1-С4-алкокси, і 4 R вибраний з групи, що складається з водню і незаміщеного C1-C4-алкіла. 1 4 Переважніше, одержують сполуку формули (І), в якій R -R незалежно один від одного мають наступні значення: 1 R вибраний з групи, що складається з водню, метокси і дифторметокси, 2 R вибраний з групи, що складається з метилу і метокси, 3 R вибраний з групи, що складається з метилу, 2-трифторетилу і 3-метоксипропілу, і 4 R вибраний з групи, що складається з водню і метилу. 1 2 3 Найпереважніше, сполукою формули (І) є пантопразол (R = -OCHF2, R = -OCH3, R = -CH3 4 1 2 3 4 1 2 hR = -H)1 лансопразол (R = -H, R = -CH3, R = -CH2F3 и R = -H)1 омепразол (R = -OCH3, R = 3 4 1 2 3 4 CH3, R = -CH3 и R = -CH3) або рабепразол (R = -H1 R = -CH3, R = -CH2CH2CH2OCH3 и R = -H). На стадії (а) способу згідно винаходу реакції піддають сполуки формули (II) або її похідні. Переважно, застосовують сполуки (II), або сольват, або гідрат сполуки (II). Зазвичай застосовують гідрат сполуки (II). У переважних втіленнях способу згідно винаходу, значення 1 4 1 радикалів R -R сполуки (II) відповідають вище згаданим переважним значенням радикалів R 4 R сполуки (І). Переважно, концентрація сполуки (II) в реакційній суміші складає від 0,1 до 5,0 моль/л, переважніше, від 0,2 до 2,0 моль/л, і найпереважніше, від 0,3 до 1,2 моль/л. Переважно, в способі згідно винаходу окислення тіоефірної групи сполуки формули (II), гідрата, сольвата або її солі здійснюють перекисом водню як окислювач. Найпереважніше, перекис водню застосовують у формі водного розчину. Крім того, переважно, щоб концентрація водного розчину перекису водню варіювала від 10 до 70 мас-%, переважніше, від 20 до 50 мас%. Найбільш переважна концентрація складає приблизно від 30 до 35 мас-%. Також можливе застосування такого джерела перекису водню, як аддукт сечовини з перекисом водню. Вживана кількість перекису водню, загалом, складає приблизно від 0,5 до 3,0 еквівалентів, переважно, від 0,7 до 2,0 еквівалентів, і найпереважніше, від 0,9 до 1,5 еквівалентів згідно співвідношення до сполуки (II). Крім того, переважно проводити окислення на стадії (а) у присутності металевого каталізатора. Переважно, метал або металевий каталізатор вибирають з групи, що складається з ренію, ванадію, молібдену, вольфраму, церію і ітербію. Переважніше, металевий каталізатор вибирають з групи, що складається з CH3ReO3, C2H5ReO3, Re (PPh3) 2ОС13, Na2MoO4, V2O5, VOCI3, VOF3, VO(OC2Hs)3, VO(I-OC3H7)S, VO (2-ОС3Н7)3, VO(CH3COCHCOCH3)2, NaVO3, H2WO4, H4SiW 12O4O, (NH4)2Ce(NO3)6, Yb(OSO2CF3)3. Найпереважніше застосовують метилтріоксорений. Звичайна кількість металевого каталізатора складає приблизно від 0,0001 до 0,1 еквівалентів, переважно, від 0,0002 до 0,01 еквівалентів, і найпереважніше, від 0,0005 до 0,0015 еквівалентів згідно відношення до сполуки (II). Якщо металевий каталізатор є метилтріоксорений, то особливо переважно застосовувати його в кількості приблизного від 0,0005 до 0,0015 еквівалентів згідно відношення до сполуки (II) (відповідає 0,05-0,15 моль%). Інші сполуки металів зазвичай застосовують в концентрації від 0,005 до 0,015 еквівалентів згідно відношення до сполуки (II) (відповідає 0,5-1,5 моль%). Стадію" (а) проводять у присутності трифторетанолу (CF3CH2OH). Крім того, стадію (а) можна проводити у присутності трифторетанолу і органічного розчинника. Цей органічний розчинник може бути вибраний з групи, що складається з метанолу, етанолу, ацетону, ацетонітрилу, C6H5CF3, ефірів, таких як тетрагідрофуран (THF), неполярних розчинників, таких як дихлорметан і ізоалкани (наприклад, ізооктан), і їх сумішей. Відповідно до особливо переважного втілення стадію (а) проводять в розчиннику, що містить трифторетанол. Розчинник є або тільки трифторетанол, або суміш трифторетанолу з другим органічним розчинником. Переважно, другий органічний розчинник вибраний з групи, що містить метанол, етанол, ацетон, ацетонитрил, . CeH5CF31 ефіри, такі як THF, неполярні розчинники, такі як дихлорметан і ізоалкани (наприклад, ізооктан), і їх суміші. Якщо застосовують суміш трифторетанолу з другим органічним розчинником, то співвідношення між трифторетанолом і вказаним другим 3 UA 105479 C2 5 10 15 20 25 30 35 40 45 50 55 60 розчинником переважно варіює від 1:1 до 1:5, переважніше, від 1:2 до 1:4 за об'ємом розчинників. Зокрема, співвідношення між трифторетанолом і вказаним другим розчинником складає приблизно 1:3 за об'ємом розчинників. У переважному втіленні даного винаходу реакцію на стадії (а) проводять при температурі від -30 до 30˚C, переважно, від -10 до 30˚C, і переважніше, від 0 до 30˚C. · Якщо як розчинник застосовують тільки трифторетанол, то температура реакції переважно складає від -20 до 20˚C, переважніше, від -10 до 10˚C і найпереважніше приблизно 0˚C. Якщо застосовують суміш трифторетанолу і другого органічного розчинника, то переважна температура збільшується залежно від природи вказаного другого розчинника і пропорцій суміші. Наприклад, при співвідношенні трифторетанолу і другого розчинника 1:3, переважна температура реакції в цілому варіює від 15 до 30˚C. Несподівано було виявлено, що застосування трифторетанолу на стадії (а) робить можливим окислення сполуки (II) з одержанням сполуки (І) з високим виходом і високою селективністю. Крім того, використання суміші трифторетанолу з другим органічним розчинником навіть робить можливим виборче одержання сполуки (І) при вищих температурах реакції. Таким чином, можна одержати дуже чисту сполуку (І) в звичайних реакторах без спеціального низькотемпературного устаткування, що також істотно для застосування способу в промисловому масштабі. Більш того, сполука формули (І) добре розчинна в трифторетанолі, так що реакційний об'єм можна зменшити, що є додатковою перевагою. Показано, що реакція перетворення сполуки (II) або її гідрата, сольвата або солі в сполуку формули (І) може відбуватися при будь-якій послідовності додавання, тобто при первинному додаванні металевого каталізатора в реакційну суміш з подальшим додаванням перекису водню або її джерела, або, альтернативно, при первинному додаванні перекису водню або її джерела в реакційну суміш з подальшим додаванням металевого каталізатора. У переважному втіленні перекис водню або його джерело додають до розчиненої сполуки формули (II) або її гідрату, сольвату або солі, а потім реакцію ініціюють шляхом додавання металевого каталізатора. Переважно, реакцію на стадії (а) проводять протягом 1-10 годин. На стадії (б) способу згідно винаходу одержану сполуку (І) можуть видаляти з реакційної суміші із стадії (а). Це переважно здійснюють за допомогою одного з наступних способів. У одному втіленні стадію (б) здійснюють, проводячи, щонайменше, одну з наступних стадій: (і) додавання ацетону або розчину тіосульфатної солі і, можливо, основи в реакційну суміш, одержану на стадії (а) (іі) додавання води до суміші із стадії (і) для осадження твердої сполуки (І), і (ііі) виділення сполуки (І). На стадії (і) описаної процедури виділення в реакційну суміш стадії (а) додають ацетон або тіосульфатну сіль, переважно, тіосульфат натрію, розчинений у воді, щоб викликати повне розкладання надлишку перекису водню після реакції. Окрім цього, можуть додавати неорганічну сполуку, таку як гідроксид натрію або гідроксид калію, або органічну сполуку, таку як триетиламін. На стадії (іі) додають воду до суміші із стадії (а) для осадження сполуки (І), яку можна виділити традиційним способом на стадії (ііі). У переважному втіленні на стадії (ііі) сполуку (І), одержану на стадії (іі), фільтрують і, можливо, перекристалізовують. Переважно, перекристалізацію проводять в суміші води і органічного розчинника. Можна застосовувати органічний розчинник - алканол, такий як етанол, 1-метил-2-піролідон, 1-етил-2піролідон, Ν,Ν-диметилацетамід, N1N-диметилформамід або їх суміш. Зокрема, застосовують суміш води і 1-метил-2-піролідона в співвідношенні від 9:1 до 1:3 (о./об.). Також переважно, щоб перекристалізацію проводили в суміші води і етанолу у присутності слабкої основи, такої як триетиламін або аміак. Крім того, на стадії (ііі) перекристалізовану сполуку (І) можуть суспендувати у воді, переважно при температурі від 15 до 20˚C, і перемішувати протягом деякого часу, наприклад, двох годин. Одержаний продукт можуть збирати шляхом фільтрації і сушки, наприклад при зниженому тиску при температурі 40˚C. У переважному втіленні вода, використовувана для суспендування сполуки (І), має значення рН від 8 до 11, яке може бути скоректоване шляхом додавання основи, такої як гідроксид натрію, гідроксид калію, триетиламін або аміак. Крім того, переважно, щоб суспензію сполуки (І) у воді поволі охолоджували до 5 °C, а потім фільтрували. Також 4 UA 105479 C2 5 10 15 20 25 30 35 40 45 50 55 переважно, щоб стадію вилуговування здійснювали кілька разів для збільшення чистоти сполуки (І). Відповідно до ще одного втілення стадію (б) здійснюють, проводячи, щонайменше, одну з наступних стадій: (і) додавання до реакційної суміші, одержаної на стадії (а), дихлорметану, розчину солі тіосульфату і, можливо, основи (іі) видалення розчинника з суміші із стадії (і) з одержанням неочищеної сполуки (І) (ііі) додавання етилацетату до одержаної на стадії (іі) неочищеної сполуки (І) і (iv) видалення етилацетату з одержанням сполуки (І). На стадії (і) цього способу додають дихлорметан. Далі додають розчин солі тіосульфату, переважно тіосульфату натрію, у воді для повного руйнування надлишку перекису водню. Крім того, можуть додавати неорганічну основу, таку як гідрокарбонат натрію. Після цього реакційну суміш переважно сушать, наприклад, приводячи її в контакт з сульфатом натрію. На стадії (іі) розчинник реакційної суміші із стадії (і) видаляють, наприклад шляхом випаровування. Унаслідок реакцій комплексоутворення із сполуки (І) складно повністю видалити тритрифторетанол. Таким чином, на стадії (іі) одержують неочищену сполуку (І). Перед стадією (ііі) неочищену сполуку (І), одержану на стадії (іі), можуть очищати, наприклад, пропускаючи через хроматографічну колонку (наприклад, наступного складу: нерухома фаза: SiO2, рухома фаза: метанол/дихлорметан 1:9). На стадії (ііі) до неочищеної сполуки додають етилацетат (І), і суміш, що виходить в результаті, потім концентрують, наприклад, шляхом упарювання на стадії (iv). Переважно, стадії (ііі) і (iv) здійснюють кілька разів для збільшення чистоти сполуки (І). Крім того, стадію (б) можуть здійснювати, проводячи, щонайменше, одну з наступних стадій: (і) додавання розчину солі тіосульфату і, можливо, основи до реакційної суміші, одержаної на стадії (а) (іі) повне видалення розчинника з суміші із стадії (і) з одержанням неочищеної сполуки (І) (ііі) змішування неочищеної сполуки (І), одержаної на стадії (іі), з дихлорметаном, і (iv) видалення дихлорметану з одержанням сполуки (І). Відповідно до цього способу на стадії (і) розчин солі тіосульфату, переважно тіосульфату натрію, у воді, додають до реакційної суміші на стадії (а) для повного руйнування надлишку перекису водню. Крім того, можуть додавати неорганічну основу, таку як гідрокарбонат натрію. На стадії (іі) розчинник суміші, одержаної на стадії (і), видаляють, наприклад, шляхом упарювання з одержанням неочищеної сполуки (І). Цей неочищений продукт на наступній стадії розчиняють в дихлорметані, який потім видаляють на стадії (iv). Після стадії (iv) сполука (І) може бути перекристалізована, наприклад, з етилацетату. Крім того, стадію (б) можуть здійснювати шляхом, щонайменше, однієї з наступних стадій: (і) додавання солі тіосульфату і, можливо, основи до реакційної суміші, одержаної на.стадії (а), (іі) видалення всього або, щонайменше, частини розчинника з суміші на стадії (і) з одержанням неочищеної сполуки (І) або концентрованої суміші, що містить сполуку (І), (ііі) змішування неочищеної сполуки (І) або концентрованої суміші, одержаної на стадії (іі), з розчинником і (iv) виділення сполуки (І) з суміші, одержаної на стадії (ііі). На стадії (іі) цього способу розчинник суміші на стадії (і) повністю або частково видаляють, наприклад, за допомогою вакууму. Переважно, приблизно від 30 до 70 про. % і, переважніше, приблизно від 40 до 60 про. % розчинника видаляють з одержанням концентрованої суміші, що містить сполуку (І). На стадії (ііі) неочищену сполуку (І) або концентровану суміш, одержану на стадії (іі), змішують з розчинником. Переважно, цей розчинник вибраний з групи, що складається з ацетону, етанолу, ацентонітрилу, етилацетату або суміші цих розчинників з водою. Можливо, після стадії (ііі) розчинник суміші із стадії (ііі) повністю або частково видаляють, наприклад, шляхом упарювання, і знов додають розчинник, такий, як використовуваний на стадії (ііі). Дана необов'язкова стадія може бути повторена кілька разів. На стадії (iv) сполуку (І) виділяють з суміші із стадії (ііі) або суміші, одержаної після можливого видалення і додавання розчинника. Таке розділення можуть здійснювати, наприклад, шляхом кристалізації і подальшого збору, наприклад шляхом охолоджування суміші до температури приблизно від 5 до 15 °C, гомогенізації охолодженої суміші і фільтрації продукту, що кристалізується. 5 UA 105479 C2 5 10 15 20 25 30 35 40 45 50 55 60 У разі, коли сполуки (І) утворюється у вигляді твердого продукту на стадії (а), стадію (б) можуть здійснювати шляхом, щонайменше, однієї з наступних стадій: (і) виділення твердого продукту реакції з реакційної суміші, одержаної на стадії (а) (іі) додавання дихлорметану до твердого продукту реакції, одержаного на стадії (і), і (ііі) видалення дихлорметану з одержанням сполуки(І). На стадії (і) цього способу твердий продукт, одержаний на стадії реакції (а), звичайним способом виділяють з реакційної суміші із стадії (а). Переважно, одержаний продукт фільтрують. Після виділення продукт можуть додатково очищати. Переважно, його промивають водою. На стадії (іі) до продукту, одержаного на стадії (і), додають дихлорметан з одержанням розчину сполуки (І). Переважно сушать цей розчин перед стадією (ііі), наприклад, шляхом приведення його в контакт з сульфатом натрію. На стадії (ііі) розчинник видаляють, наприклад, шляхом упарювання з одержанням сполуки (І). Як можна бачити з приведених вище конкретних втілень, стадію (б) загалом, можуть проводити шляхом відновлення надлишку перекису водню (наприклад, шляхом додавання розчину солі тіосульфату), додавання органічного розчинника, такого як дихлорметан, сушки органічної фази і видалення розчинника. Якщо твердий продукт не утворюється, то можуть додавати і видаляти етилацетат з одержанням чистої сполуки (І) у формі порошку. На стадії (в) способу згідно винаходу одержану сполуку (І) можуть додатково очищати і/або, можливо, перетворювати на її сіль, або сольват, або гідрат. Загалом, сполуки формули (І), такі як пантопразол, можуть бути перекристалізовані з використанням етилацетату або суміші етилацетату і води. Наприклад, очищення сполуки (І) може бути здійснене шляхом (і) додавання (а) етилацетату або суміші етилацетату і води, і (б) можливо основи до сполуки (І), одержаної на стадії (б) (іі) охолоджування суміші, що виходить в результаті (ііі) виділення осаду сполуки, що виходить в результаті (І) і (iv) можливо, промивання і сушки сполуки (І) з одержанням чистої сполуки (І). На стадії (і) цього способу очищення до сполуки (І), одержаної на стадії (б) способу згідно винаходу додають етилацетат або суміш етилацетату і води. Якщо використовують суміш етилацетату і води, то відношення між етилацетатом і водою може знаходитися в діапазоні від 1:100 до 100:1, переважно від 10:1 до 1:50, переважніше від 1:1 до 1:20, і найпереважніше, складає приблизно 1:10 за об'ємом розчинників. Окрім етилацетату і/або води до сполуки (І) можуть додавати основу, таке як гідроксид натрію. Переважно, використовують водний розчин гідроксиду натрію. Молярне відношення між сполукою (І) ι гідроксидом натрію може знаходитися в діапазоні від 200:1 до 1:10, переважно від 150:1 до 10:1,переважніше від 60:1 до 20:1, і найпереважніше від 40:1 до 30:1. На стадії (іі) суміш, що виходить в результаті, переважно охолоджують до температури, що знаходиться в діапазоні від -20 до 25 °C, переважніше, від 0 до 20˚C і найпереважніше, від 5 до 15˚C, такий як від 10 до 15 °C для того, щоб забезпечити можливість осадження сполуки (І). На стадії (ііі) виділення твердої сполуки (І), що виходить, наприклад кристалічної сполуки (І), можуть здійснювати способами, відомими в області техніки, такими як фільтрування. Нарешті, виділену сполуку (І) можуть промивати з використанням, наприклад, охолодженого етилацетату і/або послідовної сушки з одержанням чистої сполуки формули (І). У ще одному втіленні сполуки (І) може бути очищена шляхом: (і) додавання етилацетату або суміші етилацетату і води до сполуки (І), одержаної на стадії (б) (іі) доведення рН до 10-15, переважно 11-14, переважніше, 12-14, наприклад, приблизно 13 (ііі) екстракції суміші, що виходить в результаті, один або кілька разів за допомогою метиленхлориду (iv) можливо, промивання об'єднаних органічних шарів водою (ν) доведення рН об'єднаних водних шарів із стадії (ііі) і, можливо, (iv) до 6-9, переважно 7-9, переважніше 7,5-8,5, наприклад, приблизно 8 (vi) виділення осаду сполуки, що виходить в результаті (І), і (vii) можливо, промивання і сушки твердої сполуки (І) з одержанням чистої сполуки (І). На стадії (і) цього способу очищення до сполуки (І), одержаної на стадії (б) способу згідно винаходу, додають етилацетат або суміш етилацетату і води. Якщо використовують суміш 6 UA 105479 C2 5 10 15 20 25 30 35 40 45 50 55 етилацетату і води, то відношення між етилацетатом і водою може знаходитися в діапазоні від 1:100 до 100:1, переважно, від 10:1 до 1:50, переважніше, від 1:1 до 1:20, і, найпереважніше, складає, приблизно, 1:10 за об'ємом розчинників. На наступній стадії (іі) рН суміші, що виходить в результаті, доводять до 10-15, переважно, 11-14, переважніше, 12-14, наприклад, приблизно, 13. На рН, наприклад, може впливати додавання водного розчину неорганічної основи, такої як гідроксид натрію. На стадії (3) суміш, що виходить, екстрагують один або більше, ніж один раз метиленхлоридом. Переважно, екстракцію здійснюють двічі. Після можливого промивання об'єднаних органічних шарів на стадії (iv), об'єднані водні шари обробляють кислотою на стадії (ν) для доведення рН до 69, переважно, 7-9, переважніше, 7,5-8,5, наприклад, приблизно, 8. Переважно, використовують розчин неорганічної або органічної кислоти, такий як оцетова кислота. Крім того, переважно охолоджувати об'єднані водні шари, що виходять в результаті, до температури, що знаходиться в діапазоні від -20 до 25 °C, переважніше, від 0 до 20˚C, і найпереважніше від 5 до 15˚C, наприклад, від 10 до 15 °C для того, щоб дати можливість сполуці (І) осісти. На стадії (vi) виділення осаду сполуки (І), що виходить, наприклад кристалів сполуки (І), можуть здійснювати за допомогою способів, відомих в області техніки, таких як фільтрування. Нарешті, виділена сполука (І) може бути промита з використанням, наприклад води, і/або подальшої сушки з одержанням чистого зразка сполуки (I). Додатково або альтернативно до очищення, сполуку формули (І) можуть перетворювати на її сіль або сольват, або в гідрат. Наприклад, пантопразол, одержаний відповідно до способу згідно винаходу, можуть перетворювати на солі пантопразолу, наприклад, пантопразолу натрієву сіль або пантопразолу магнієву сіль. Загалом, будь-які форми сполуки (II), такі як будь-які кристалічні форми пантопразолу сульфіду або лансопразола сульфіду, одержані відповідно до будь-яких способів синтезу і одержувані за допомогою будь-яких способів кристалізації, відомі в області техніки, можуть бути використані в способі згідно винаходу. Відповідні приклади різних кристалічних форм пантопразолу сульфіду, а також відповідні приклади способів очищення пантопразолу, і утворення солей пантопразолу, таких, як пантопразолу натрієва сіль, розкриті в J. Med. Chemistry 1992, 35, 1049-1057, Anal. Profiles of Drug Substances-Pantoprazole Sodium, Vol. 29, 213-259, Chin. Pharm, August 1999, Vol. 34, No. 8, 564-565, IPCOM000016610D від ip.com, Inc., WO 2004/111029, WO 2004/080961, WO 2004/063188, WO 2004/056804, EP 1 300 406, WO 2007/017244, WO 2006/040778, US 2004/018 6139, WO 2007/068925, WO 2007/026188, WO 03/097606. У другому аспекті винахід відноситься до способів одержання сполуки, що 1 2 3 4 має формулу (І) і замінники R , R , R і R · такі, як визначені вище, або її солі або сольвата, або гідрата, що включає: 1 2 3 4 (а) окислення сполуки, що має формулу формули (II) і замінники R , R , R і R , такі, як визначені вище, з одержанням реакційної суміші, що містить сполуку формули (І), (б) виділення сполуки формули (І), і (в) можливо очищення сполуки формули (І) і/або її перетворення на сіль або сольват, або гідрат, де стадію (б) здійснюють за допомогою будь-якого із способів виділення, описаних вище у зв'язку з першим аспектом винаходу. Винахід додатково проілюстрований наступними прикладами. У прикладах для визначення чистоти сполуки (І), вираженої у вигляді % площі, використовували високоефективну рідинну хроматографію (ВЕРХ) високого дозволу. Тести здійснювали з використанням Zorbax XDB С18, 1,8 мкм, 50 χ 4,6 мм. Рухома фаза була градієнтом води і води/ТЕА/ацетонітрилу (40/0,1/160), рН 7. Хроматограф обладнаний УФ (ультрафіолетовим) детектором, встановленим на 285 нм. Швидкість потоку складала 0,7 мл/хв при 40˚C. Приклади 1 і.-у.: Одержання 5-(дифторметокси)-2-[[[3,4-диметокси-2піридиніл]метил]сульфініл]-1 Н-бензимідазолу (пантопразолу) 3,67 г (10 моль) 5-дифторметокси-2-[[(3,4-диметокси-2-піридиніл]метил]тіо]-1Нбензимідазолу (пантопразолу сульфіду, безводого, кристалічної форми C відповідно до IPCOM000016610D) розчиняли в 10 мл трифторетанолу. Потім до розчину пантопразолу сульфіду додавали при кімнатній температурі 0,908 мл водного розчину Н2О2 (35 %). Потім розчин охолоджували до температури, вказаної в приведеній нижче таблиці, і додавали 2,492 міліграмів каталізатора CH3ReOs двома або 7 UA 105479 C2 чотирма порціями (інтервали по 10 хв). Реакційну суміш перемішували протягом однієї години при вказаній температурі і реакцію контролювали за допомогою аналізу ВЕРХ. Приклад і. іі. ііі. iv. 5 10 15 20 25 30 35 40 45 50 55 Температура реакції (°С) -10 0 -10 0 Кількість доданих порцій 2 2 4 4 Приклад 2: Одержання 5-(дифторметокси)-2-[[[3.4-диметокси-2піридиніл]мєтил]сульФініл] 1Н-бензимідазолу (пантопразолу) а) Окислення пантопразолу сульфіду: 1,473 г (4,0 моль) 5-(дифторметокси)-2-[[[3,4-диметокси-2-піридиніл]метил]тіо]-1Нбензимідазолу (пантопразолу сульфіду, безводого, кристалічна форма 3 відповідно до ІРСОМ000016610D) розчиняли в 4 мл трифторетанолу. Додавали 1 міліграм (0,004 моль) CHaReO3, і суміш перемішували при 0˚C. 1,5 еквів. 30 % водного розчину Н2О2 (0,60 мл) додавали до охолодженого розчину, і реакційну суміш перемішували при 0 °C протягом 2 годин. б) Виділення пантопразолу 10 мл ацетону додавали до реакційної суміші. Потім продукт осаджували шляхом додавання 0 10 мл води, фільтрували, промивали водою і сушили при 40 C у вакуумній сушці з одержанням 1,062 г (72 %) пантопразолу, що має чистоту 99,26 %; [PN 1 (пантопразолу сульфід): 0,07 %, PN2 (пантопразолу сульфон): 0,38 %, PN3 (пантопразолу сульфону N-оксид): 0,06 % і PN4 (пантопразолу N-оксид): 0,04 %]. Приклад 3: Одержання 5-(дифторметокси)-2-[[[З, 4-диметокси-2-піридиніл]метил]сульфініл]1Н-бензимідазолу (пантопразолу) Повторювали окислення пантопразолу сульфіду, описане в Прикладі 2а) і за винятком того, що використовували 1,469 г пантопразолу сульфіду. 20 мл CH2Cb додавали до реакційної суміші, а потім додавали 0,5 мл 1М розчину Na2S2O3 для руйнування надлишку перекису водню. Додавали 0,2 г твердого Na2S2O3, і суміш, що виходить в результаті, перемішували протягом 5 хв. Потім для сушки реакційної суміші додавали Na2SO4. Після фільтрування розчинник суміші упарювали і одержували 2,646 г неочищеного пантопразолу. Цей продукт очищали шляхом його пропускання через коротку колонку (2 г SiO2, метанол/дихлорметан 1:9) з одержанням 1,850 г маслянистого продукту. 10 мл етилацетату додавали для ініціації кристалізації і проводили упарювання. Повторно додавали 10 мл етилацетату і знову проводили упарювання. Одержували 1,550 г (100 %) кристалічного пантопразолу. Приклад 4: Одержання 5-(диФторметокси)-2-[[[3.4-диметокси-2-піридиніл]метил]сульфініл]1Н-бензимідазолу (пантопразолу) Повторювали окислення пантопразолу сульфіду, описане в Прикладі 2а), за винятком того, що використовували 1,471 г пантопразолу сульфіду. До реакційної суміші додавали 0,5 мл 1М розчину Na2S2Os, а потім 0,2 г твердого NaHCO3. Розчинник видаляли при зниженому тиску. Неочищений продукт виливали у воду. Проте, продукт не осідав. Додавали 20 мл дихлорметану, і водні і органічні фази розділяли. Органічну фазу сушили над Na2SO4, і після упарювання розчинника одержували 1,956 г неочищеного пантопразолу. Цей продукт кристалізували з 5 мл етилацетату, і одержували 761 мг (49 %) пантопразолу у формі білого порошку. Продукт (796 мг, 51 %), що залишився, одержували в чистій формі після видалення розчинника. Приклад 5: Одержання і виділення 5-(диФторметокси)-2-[ГГ3.4-диметокси-2піридинілімєтилісульФінілМ Н-бензимідазолу (пантопразолу) Окислення пантопразолу сульфіду здійснювали тим самим чином, як в Прикладі 1, за винятком того, що спочатку до розчину пантопразолу сульфіду при кімнатній температурі додавали каталізатор СНзКеОз (2,492 мг), потім реакційну суміш охолоджували до температури 0 -10 C і, нарешті, до розчину додавали 35 % водного розчину H2O2 (0,908 мл). Реакцію здійснювали протягом однієї години при вказаній температурі і контролювали за допомогою аналізу ВЕРХ. В кінці реакції додавали розчин 0,56 г тіосульфату натрію в 4 мл води при температурі від 5 0 до 10 C. рН одержаної суміші доводили до 7,5 з використанням водного NaOH, а потім шляхом перегонки трифторетанолу. 20 мл суміші ацетону і води (1:1) додавали до суміші, що 8 UA 105479 C2 0 5 10 залишилася, яку потім перемішували протягом двох годин при 5-1O C. Продукт фільтрували, промивали водою і сушили при температурі менше 45 °C. Одержували 2,8 г кристалічного пантопразолу, що має чистоту 99,23 % (PN2: 0,11 %, PN3: 0,05 %, PN4: 0,05 %). Приклади 6 і.-у.: Одержання 5-(дифторметокси)-2-[[[3,4-диметокси-2піридиніл]метил]сульфініл]-1 Н-бензимідазолу (пантопразолу) 1,473. г (4,0 моль) 5-(дифторметокси)-2-[[[3,4-диметокси-2-піридиніл]метил]тіо]-1Нбензимідазолу (пантопразолу сульфіду) розчиняли в 8 мл розчинника, вказаного в нижчеприведеній таблиці, одержуючи в результаті 0,5М розчину пантопразолу сульфіду. Додавали 1 мг (0,004 моль) CH3ReO3, і суміш перемішували. Потім температуру реакції доводили до значень, вказаних в нижчеприведеній таблиці, і до розчину додавали 1,2 еквів. 30 % водного розчину Η2О2 (0,48 мл). Реакційну суміш перемішували при приведеній температурі протягом двох годин (Приклад 6 і.) або 6 годин (Приклади 6 ii.-v.), відповідно. Таблиця: Одержання пантопразолу з використанням різних розчинників Приклад і. іі. ііі. IV. V. Розчинник CF3CH2OH CH3CN + CF3CH2OH (3:1 об./об.) CH3CH2OH + CF3CH2OH (3:1 об./об.) CH2CI2+ CF3CH2OH (3:1 об./об.) ізо-С8Н18+ CF3CH2OH (3:1 об./об.) Умови реакції 0°C, 2 год 22˚C, 6 год 22˚C, 6 год 22˚C, 6 год 22˚C, 6 год 15 20 25 Приклади 7 i.-vi.: Одержання 5-(дифторметокси)-2-[[[3,4-диметокси-2піридиніл]метил]сульфініл]-1 Н-бензимідазолу (пантопразолу) 1,473 г (4,0 моль) 5-(дифторметокси)-2-[[[3,4-диметокси-2-піридиніл]метил]тіо]-1/-/бензимідазолу (пантопразолу сульфіду) розчиняли в 8 мл трифторетанолу, одержуючи в результаті 0,5М розчину пантопразолу сульфіду. Потім додавали металевий каталізатор, вказаний в нижчеприведеній таблиці, в кількості 0,1 моль% (відповідає 0,001 еквів.; Приклад 7 і.), 1,0 моль% (відповідає 0,01 еквів.; Приклади 7 N.-7 ν.) або 5,0 моль% (відповідає 0,05 еквів.; Приклад 7 vi.) відносно пантопразолу сульфіду, і суміш перемішували. Потім реакційну суміш охолоджували до 0˚C і до розчину додавали 1,2 еквів. 30 % водним розчином H2O2 (0,48 мл). Реакційну суміш перемішували при вказаній температурі або діапазоні температур (градієнт від 0˚C до 22˚C протягом вказаного періоду часу). Закінчення реакції контролювали за допомогою тонкошарової хроматографії. Таблиця Одержання пантопразолу з використанням різних металевих каталізаторів Приклад і. іі. ііі. iv. V. vi. 30 35 Металевий каталізатор CH3ReO3(0,1 моль%) Na2MoO4 (1,0 моль%) V2O5 (1,0 моль%) VO(2-OC3H7)3, (1,0 моль%) VO(CH3COCHCOCH3)2 (1,0 моль%) VO(CH3COCHCOCHs)2 (5,0 моль%) Умови реакції 0˚C, 2 год. 0-22˚C, 2 год. 0-22 °C, 2 год. 0-22˚C, 2 год. 0-22 °C, 2 год. 0 °C, 4 год. Приклад 8: Очищення пантопразолу і утворення натрієвої солі пантопразолу і. Очищення пантопразолу 60,0 г пантопразолу суспендували в 210 мл етилацетату і 0,77 мл 6 M водного гідроксиду натрію при температурі приблизно 30˚C протягом 0,5 год. Потім суспензію поступово охолоджували до 10-15˚C і перемішували протягом ще двох годин. Продукт збирали шляхом фільтрації і тричі промивали 30 мл холодного етилацетату. Продукт потім сушили при температурі нижче 450C. Одержували 53,0 г кристалічного пантопразолу (99,60 % пантопразолу, 0,28 % PN2, 0,03 % PN3, 0,02 % PN4, кількість PN1 нижча за рівень виявлення). іі. Очищення пантопразолу 9 UA 105479 C2 5 10 15 20 25 30 35 40 45 50 55 60,0 г пантопразолу суспендували в 210 мл водного етилацетату (10 %) і 0,77 мл 6М водного гідроксиду натрію при, приблизно, 30 °C протягом 0,5 год. Потім суспензію поступово 0 охолоджували до 10-15 C і перемішували протягом ще двох годин. Продукт збирали шляхом фільтрації і тричі промивали 30 мл холодного етилацетату. Продукт потім сушили при температурі нижче 45 °C. Одержували 50,0 г кристалічного пантопразолу (99,65 % пантопразолу, 0,19 % PN2, 0,04 % PN3, 0,03 % PN4, прибл. PN1). ііі. Очищення пантопразолу 68,0 г пантопразолу суспендували в 750 мл суміші етанол/вода (1:10), що має рН, доведений до 13 за допомогою 10 % розчину гідроксиду натрію. Реакційну суміш двічі екстрагували метиленхлоридом. Об'єднані органічні шари промивали водою, і водний шар очищали активованим вуглецем. рН розчину доводили до 8±0,5 з використанням 17 мл 50 % оцетової кислоти при приблизно 15 °C для здійснення кристалізації пантопразолу. Кристалізований продукт збирали шляхом фільтрації і промивали 50 мл води. Продукт сушили при температурі менше 45 °C. Одержували 61,1 г кристалічного пантопразолу (99,57 % пантопразолу, 0,15 % PN2, 0,03 % PN3, 0,03 % PN4, 0,02 % PN1). iv. Одержання пантопразолу натрію півторагідрату 50,0 г пантопразолу розчиняли в 250 мл метиленхлориде і 15 мл етанолу при кімнатній температурі. Суміш, що виходить в результаті, фільтрували для видалення яких-небудь нерозчинних частинок. Розчин пантопразолу в метиленхлориді і етанолі обробляли 25 мл 6М водного розчину гідроксиду натрію до досягнення рН 12,5±0,5 при 20±3 °C. 500 мл диізопропілового ефіру поволі додавали до розчину пантопразолу натрію. Кристалізацію здійснювали при 20 ± 3°С. Продукт збирали шляхом фільтрації і промивали 50 мл диізопропілового ефіру. Продукт сушили в сушарці при температурі нижче 45˚C. Під час сушки продукт просівали для руйнування агломератів. Одержували 51,5 г кристалічного пантопразолу натрію півторагідрату (чистота: 99,70 %, 0,15 % PN2, 0,02 % PN3, 0,03 % PN4, 0,02 % PN1). Картина дифракції рентгенівських променів на порошці одержаного пантопразолу натрію півторагідрату (одержана на дифрактометрі Phillips PW3040/60 X'Pert PRO з використанням CuKa випромінювання, фільтр: Ni; напруга: 45 кВ; сила струму: 40 мА) представлена на Фіг. 1. Вміст води, виміряний відповідно до Ph. Eur. 2,5,12. Спосіб А, складало приблизно від 6,0 до 8,0 мас. %. ν. Виділення пантопразолу магнію дигідрату Після завершення реакції окислення до реакційної суміші додавали 5 мл 5 % розчину тіосульфату натрію і 5 мл води, що містить 4 моль пантопразолу, при 10-15˚C. Потім рН суміші, що виходить в результаті, доводили до 13 з використанням 10 % розчину гідроксиду натрію. Реакційну суміш двічі екстрагували метиленхлоридом. Водний шар очищали активованим вуглецем. 2 мл водного розчину хлориду магнію (2 моль) додавали до водного шару пантопразолу натрію при перемішуванні при 20-25˚C. Після осадження тверду речовину фільтрували, ще раз суспендували в 10 мл води і перемішували протягом двох годин. Продукт фільтрували, промивали 5 мл води і сушили при температурі нижче 50˚C до досягнення вмісту води 5,0 % (KF - вміст води, визначений по методу Карла Фішера). Одержували 1,1 г пантопразолу магнію дигідрату. vi. Одержання пантопразолу натрію півтора гідрату 43,0 г (100 моль) пантопразолу магнію дигідрату суспендували в 400 мл етанолу при 20-25˚C, потім додавали 200 мл води і рН доводили до 7,5-8,0 з використанням 6 % оцетової кислоти. Пантопразол екстрагували метиленхлоридом і освітлювали за допомогою активованого вуглецю. Зібрані органічні шари концентрували до 200 мл. Суміш, що виходить в результаті, необов'язково фільтрували для видалення яких-небудь нерозчинних частинок. Розчин пантопразолу в метиленхлориді і в етанолі обробляли 23 мл 6М водного розчину гідррксиду натрію до рН 12,5±0,5 при 20±3 °C. 450 мл диізопропілового ефіру поступово додавали до розчину пантопразолу натрію. Кристалізацію здійснювали при 20 ± 3°С. Продукт збирали шляхом фільтрації і промивали 50 мл диізопропілового ефіру. Продукт сушили в сушарці при температурі нижче 45 °C. Під час сушки продукт просівали для руйнування агрегатів. Одержували 38,5 г кристалічного пантопразолу натрію півторагідрату. Приклад 9: Одержан ня 2-[[[3-мети л-4-(2,2,2-трифторетокси)-2-піридиніл]метил]сульфініл]-1 Н-бензимідазолу (лансопразола) а) Окислення лансопразола сульфіду: 372 мг (1,0 моль) 2-[[[3-метил-4-(2,2,2-трифторетокси)-2-піридиніл]метил]тіо]-1/-/бензимідазолу (лансопразола сульфіду) розчиняли в 1 мл трифторетанолу. Додавали 0,25 мг (0,001 моль) CH3ReO3, і суміш перемішували при 0˚C. До охолодженого розчину додавали 1,2 10 UA 105479 C2 5 10 15 20 25 30 35 40 45 50 55 60 еквів. 30 % водного розчину H2O2 (0,12 мл), і реакційну суміш перемішували при 0 °C протягом однієї години. В результаті утворювався твердий продукт. б) Виділення лансопразола Твердий продукт фільтрували і промивали водою. Одержували 1,51 г білої твердої речовини, яку складно було сушити. Тверду речовину розчиняли в 15 мл дихлорметану. Розчин сушили над Na2SO4 і розчинником видаляли. Одержували 361 мг (98 %) білого лансопразола. Приклад 10: Одержання 2-[[[3-метил-4-(2,2,2-трифторетокси)-2-піридиніл]метил]сульфініл]1Н-бензимідазолу (лансопразола) Повторювали окислення лансопразола сульфіду, описане в Прикладі 9 а). До реакційної суміші додавали 10 мл дихлорметану, а потім 0,3 мл 1М розчину Na2S2O3. Суміш, що виходить в результаті, сушили над Na2SO4 і видаляли розчинник. Одержували 666 мг маслянистого продукту. 10 мл етилацетату додавали до продукту, і суміш концентрували шляхом упарювання. Одержували 371 мг (100 %) лансопразола, що має чистоту 97,56 %. Приклад 11: Очищення лансопразола і.) 48 г вологого лансопразола розчиняли в 80 мл 1-метил-2-піролідона (NMP) і 5 мл триетиламіну, охолоджували до 15 °C, і продукт осаджували за допомогою 240 мл води. Суспензію перемішували протягом 4-Ю годин і фільтрували з одержанням 34,9 г вологого лансопразола. Вологий кристалізований продукт суспендували в 200 мл води, обробляли триетиламіном, доводячи таким чином величину рН до приблизно 9-10. Суспензію охолоджували до 15 °C, перемішували при температурі від 15 до 20˚C протягом 2 годин, і потім охолоджували до температури від 0 до 5 °C. Одержані кристали збирали шляхом центрифужної фільтрації і сушили при зниженому тиску при 40˚C з одержанням 26,6 г форми А лансопразола. іі.) Приклад 11 і. повторювали за винятком того, що рН суспензії вологого лансопразола доводили гідроксидом натрію замість триетиламіну. iii.) Приклад 11 і. повторювали за винятком того, що вологий кристалізовані лансопразол суспендували в питній воді замість води, рН, що має, від 9 до 10. iv.) 48 г вологого лансопразола розчиняли в суміші 110 мл 1-метил-2-піролідона (NMP), 2,67 г гідроксиду натрію і 75 мл ізопропілацетату при 20±2 °C. Шари, що виходять в результаті, розділяли, водну фазу охолоджували до 10˚C, і продукт осаджували шляхом додавання 8,3 мл 50 % оцетової кислоти з одержанням величини рН, що знаходиться в діапазоні від 9,8 до 10,1 при 5-10˚C. Суспензію перемішували протягом двох годин при 5-10˚C, потім фільтрували і промивали 20 мл води з одержанням 36,8 г вологого лансопразола. Вологий кристалізований продукт суспендували в 228 мл води, що має величину рН приблизно 9-10 (обробляли гідроксидом натрію) при 16-18 °C, перемішували протягом двох годин і охолоджували до 5 °C. Продукт фільтрували, промивали 10 мл води і сушили при зниженому тиску при 40˚C з одержанням 26,7 г форми А лансопразола. ν.) Приклад 11 iv. повторювали за винятком того, що замість оцетової кислоти використовували мурашину кислоту. vi.) Приклад 11 iv. повторювали за винятком того, що замість оцетової кислоти використовували соляну кислоту. vii.) 1,58 кг вологого неочищених лансопразола суспендували в суміші етанол/вода/аміак (5,2: 0,6: 0,006. 1), і суміш нагрівали до 50˚C. Нерозчинну речовину фільтрували, фільтрат поступово охолоджували, і з початком кристалізації (32 °C) суспензію інтенсивно перемішували і охолоджували до 0˚C. Суспензію перемішували протягом ще півгодини. Тверду речовину фільтрували, промивали 1,0 л охолодженої суміші етанол/вода (9:1) з одержанням 1,19 кг вологого продукту, який суспендували при 22 °C в 6,65 л води, при доведенні рН до 9,66 триетиламіном. Суспензію перемішували протягом двох годин при 20±2˚C, потім охолоджували протягом однієї години до 5 °C, фільтрували і промивали 1,0 л води з рН 9,0 з одержанням 1,55 кг вологого продукту. Знов повторювали мацерацію у воді. Вологий продукт (1,48 кг) сушили у вакуумі при 40˚C з одержанням 0,71 кг форми А лансопразола, що має площу поверхні, 2 визначеної згідно методу Брунауера, Еммета і Теллера (метод БЕТ) 4,67 м /г. Приклад 12: Одержання 5-(дифторметокси)-2-[[[3, 4-диметокси-2-піридиніл]метил]сульфініл]1Н-бензимідазолу (пантопразолу) а) Окислення пантопразолу сульфіду: У інертизованому реакторі розчиняли 15 кг (40,83 моль) 5-дифторметокси-2-((3,4-диметокси2-піридиніл)метил) сульфініл)-1Нбензимідазолу (пантопразолу сульфіду) у 36 л 2,2,2-трифторетанолу при кімнатній температурі, і розчин, що виходить в результаті, охолоджували до 15-20˚C. Потім до розчину додавали 3,4 л водного розчину перекису водню (35 %). Потім реакційну суміш охолоджували в інертній атмосфері до -20-15˚C і протягом двох годин додавали 10,5 г каталізатора CH3ReO3, 11 UA 105479 C2 5 10 15 20 25 30 35 40 45 50 55 60 розчиненого в 5,5 л 2,2,2-трифторетанолу. Реакцію контролювали за допомогою ВЕРХ, і температуру підтримували при -15-20˚C. Нарешті, реакцію зупиняли шляхом додавання тіосульфату натрію пентагідрату (1,2 кг, розчиненого в 15,5 л дистильованої води 5 % водний розчин) при -10-0˚C для руйнування надлишку перекису водню. рН реакційної суміші доводили до 7,1-7,5 шляхом додавання 10 % водного розчину гідроксиду натрію. б) Виділення пантопразолу з використанням ацетону: Реакційну суміш, одержану на наведеній вище стадії а), перегонили у вакуумі при температурі 40˚C. Вакуумну перегонку зупиняли, коли залишалося приблизно, 28 кг реакційної суміші. Потім до реакційної суміші додавали 28 л ацетону протягом 1 години при температурі від 25 до 30˚C. Потім, суспензію охолоджували до 15 °C протягом 30 хв і гомогенізували при цій температурі протягом двох годин. Твердий продукт виділяли, промивали 8,4 л ацетону і 42 л води, і сушили при 40˚C у вакуумній сушці з одержанням 10,48 кг пантопразолу, що має чистоту 99,93 % (PN1: 0,02 %, PN2: 0,03 %, PN4: 0,02 %). Загальний вихід пантопразолу складав 66 % (вихід на стадії виділення: 70 %). Приклад 13: Одержання 5-(диФторметокси)-2-[[[3, 4-диметокси-2-піридиніл]метил]сульфініл]1Н-бензимідазолу (пантопразолу) а) Окислення пантопразолу сульфіду: У інертизованому реакторі розчиняли 272 кг (740 моль) 5-дифторметокси-2-((3,4-диметокси2-піридиніл)метил) сульфініл)-1 Н-бензимідазолу (пантопразолу сульфіду) у 900 кг 2,2,2-трифторетанолу при кімнатній температурі, і розчин, що виходить в результаті, охолоджували до 15-20˚C. Потім до розчину додавали 78 кг водного розчину перекису водню (35 %). Потім реакційну суміш охолоджували в інертній атмосфері до 20-15˚C і протягом проміжку часу до 5 годин додавали 220 г СН 3RеО3 каталізатора, розчиненого в 160 кг 2,2,2-трифторетанолу. Реакцію контролювали за допомогою ВЕРХ, і температуру підтримували при -15-20˚C. Нарешті, реакцію зупиняли, додаючи тіосульфату натрію пентагідрат (24 кг, розчинений в 295 кг дистилюваної води, 5 % водний розчин) при -10-0˚C для руйнування надлишку перекису водню. рН реакційної суміші доводили до 7,3-7,8, додаючи 10 % водний розчин гідроксиду натрію. б і.) Виділення пантопразолу з використанням етанолу: 1 л реакційної суміші, одержаної в наведеній вище стадії а) і що має концентрацію пантопразолу 190 мгм/мл, перегонили у вакуумі при температурі 40˚C до тих пір, поки не залишалося 400 мл реакційної суміші. Потім додавали 600 мл етанолу, і суміш, що виходить в результаті, знов перегонили до тих пір, поки не залишиться 600 мл реакційної суміші. Цю стадію повторювали двічі. Нарешті, додавали 300 мл етанолу, і суспензію, що виходить в результаті, охолоджували до 10˚C протягом однієї години і гомогенізували при цій температурі протягом ще однієї години. Одержаний продукт виділяли і промивали 300 мл етанолу і 1 л води з одержанням 167 г пантопразолу, що має чистоту 99,70 % (PN1: 0,11 %, PN2: 0,11 %, PN4: 0,04 %). Вихід пантопразолу на стадії виділення склав 85 %. б іі.) Виділення пантопразолу з використанням ацетонітрилу: 100 мл реакційної суміші, одержаної на наведеній вище стадії а), і що має концентрацію пантопразолу 190 мг/мл, 0 перегонили у вакуумі при температурі 40 C до тих пір, поки не залишалося 50 мл реакційної суміші. Потім додавали 50 мл ацетонітрилу, і суміш, що виходить в результаті, перегонили до тих пір, поки не залишиться 50 мл реакційної суміші. Цю стадію повторювали двічі. Нарешті, одержану суспензію охолоджували до 5 °C протягом однієї години і гомогенізували при цій температурі протягом наступних двох годин. Одержаний продукт виділяли і промивали 50 мл охолодженого на льоду ацетонітрилу з одержанням 12,8 г пантопразолу, що має чистоту 99,80 % (PN1: 0,03 %, PN2: 0,11 %, PN4: 0,04 %). Вихід пантопразолу на стадії виділення складав 68 %. б ііі.) Виділення пантопразолу з використанням етилацетату: Приклад 13 6 іі.) повторювали за винятком того, що замість ацентонітрилу використовували етилацетат. Одержували 13,95 г пантопразолу, що має чистоту 99,75 % (PN1: 0,05 %, PN2: 0,13 %, PN4: 0,06 %). Вихід пантопразолу на стадії виділення склав 73 %. б iv.) Виділення пантопразолу з використанням ацетону: Реакційну суміш, що залишилася, з наведеної вище стадії а) перегонили у вакуумі при температурі 40˚C. Вакуумну перегонку зупиняли, коли залишалося приблизно 60. % початкового об'єму реакційної суміші. Потім до реакційної суміші протягом однієї години додавали 280 кг ацетону при 30-35˚C. Потім, суспензію нагрівали до 45 °C, потім поступово (5 °C/год.) охолоджували до 15-5˚C і підтримували при цій температурі протягом двох годин. Продукт виділяли при 5˚C, промивали 60 кг охолодженого ацетону і 500 кг води, і сушили при 40˚C у вакуумній сушці з одержанням 221 кг пантопразолу, що має чистоту 99,7 % (PN1: нижче за 12 UA 105479 C2 5 10 15 20 25 30 35 40 45 50 55 рівень виявлення, PN2: 0,2 %, PN4: 0,02 %). Загальний вихід пантопразолу склав 78 % (вихід після стадії виділення: 82 %). Приклад 14: Одержання 2-[[[4-(3-метоксипропокси)-3-метил-2-піридиніл]мєтил]сульфініл]-1Нбензимідазолу (рабепразолу) а) Окислення рабепразола сульфіду: 34,3 г (0,1 моль) 2-[[[4-(3-метоксипропокси)-3-метил-2-піридиніл]метил]тіо]-1Н-бензимідазолу (рабпразолу сульфіду) розчиняли в 100 мл 2,2,2-трифторетанолу при кімнатній температурі. Потім при температурі від 15 до 20˚C додавали 9,41 мл 35 % водного розчину перекису водню і одержану суміш охолоджували до -20˚C. Розчин 75,25 мг (0,3 моль) метилтриоксоренію (VII) в 5 мл "2,2,2-трифторетанолу поступово додавали до реакційної суміші протягом однієї години при -20-10˚C. Реакцію здійснювали при тій же самій температурі і контролювали за допомогою аналізу ВЕРХ. До кінця реакції при -10-0˚C додавали розчин 3,2 г натрію тіосульфату в 40 мл води. рН реакційної суміші доводили до 11 за допомогою 10 % розчину гідроксиду натрію при 010 °C. Рабепразолу сульфід, використовуваний на цій стадії, може знаходитися в аморфній або кристалічній формі, таких як кристалічні форми, зображені на Фіг. 2 і 3. Картини дифракції рентгенівських променів на пороші на Фіг. 2 і З одержували на порошковому дифрактометрі Philips PW3040/60 X'Pert, детектор X'celerator з випромінюванням CuKa, 1,54178 Ǻ, 3°
ДивитисяДодаткова інформація
Автори англійськоюIskra, Jernei, Stavber, Stojan, Kotar Jordan, Berta, Ruzic, Milos, Smodis, Janez, Zupet, Rok
Автори російськоюЗупет Рок
МПК / Мітки
МПК: C07D 401/12
Мітки: спосіб, одержання, 2-сульфініл-1н-бензоімідазолів
Код посилання
<a href="https://ua.patents.su/19-105479-sposib-oderzhannya-2-sulfinil-1n-benzoimidazoliv.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання 2-сульфініл-1н-бензоімідазолів</a>
Фармацевтичний препарат, що містить 2-[[(2-піридиніл)метил]сульфініл]бензімідазол, та спосіб виготовлення такого препарату

Номер патенту: 63992
Опубліковано: 16.02.2004
Автори: Педерсен Сьорен Больс, Канн Хелле, Хенріксен Крістіан Лунд, Сьоренсен Карен Ейхштедт
МПК: A61P 1/04, A61K 9/22, A61K 31/4439
Мітки: 2-[[(2-піридиніл)метил]сульфініл]бензімідазол, містить, такого, спосіб, фармацевтичний, препарат, препарату, виготовлення
Формула / Реферат:
1. Фармацевтичний препарат для перорального вживання, який містить як активний інгредієнт 2-[[(2-піридиніл)метил]сульфініл]бензімідазол, що має противиразкову дію, причому цей препарат має форму гранул, які мають суттєво інертну серцевину, покриту 1) внутрішнім шаром покриття, що містить бензімідазол, роздрібнювальну речовину та поверхнево-активну речовину в основі, яка являє собою речовину для покривання з розплаву, яка, в основному,...
Оптично чисті солі (-)-5-метокси-2-[[(4-метокси-3,5-диметил-2-піридиніл)метил]сульфініл]-1н-бензімідазолу, спосіб їх одержання, проміжні сполуки та фармацевтична композиція

Номер патенту: 60289
Опубліковано: 15.10.2003
Автори: Вон Унге Свєркєр, Ліндберг Пер Леннарт
МПК: A61K 31/4427, A61K 31/4439, A61P 1/00, A61P 31/04, A61P 1/04, A61P 29/00, A61P 19/06, A61K 31/44, A61P 17/06, C07D 401/12
Мітки: солі, чисті, оптично, сполуки, 5-метокси-2-[[(4-метокси-3,5-диметил-2-піридиніл)метил]сульфініл]-1н-бензімідазолу, композиція, спосіб, одержання, фармацевтична, проміжні
Формула / Реферат:
1. Оптически чистые Na+, Мg2+, Li+, Κ+ или Са2+ соли (-)-5-метокси-2-[[(4-метокси-3,5-диметил-2-пиридинил)метил]сульфинил]-1Н-бензимидазола.2. Соединение по пункту 1, представляющее собой Na+, Мg2+ или Са2+ соль (-)-5-метокси-2-[[(4-метокси-3,5-диметил-2-пиридинил)метил]сульфинил]-1Н-бензимидазола.3. Соединение по пункту 1, представляющее собой Мg2+ соль...
Спосіб одержання 1-(тіометил)циклопропаноцтової кислоти та проміжних сполук для її одержання

Номер патенту: 50730
Опубліковано: 15.11.2002
Автори: Піпік Бренда, Конлон Девід Е., Кінг Стівен
МПК: C07C 253/16, C07C 319/00, C07D 327/00, C07C 327/00
Мітки: 1-(тіометил)циклопропаноцтової, спосіб, одержання, сполук, проміжних, кислоти
Формула / Реферат:
1. Спосіб одержання циклічного сульфіту 1,1-циклопропандиметанолу формули,який передбачає:а) взаємодію 1,1-циклопропандиметанолу з діалкілсульфітом в присутності кислоти або основи таb) вилучення з реакційної суміші спиртового побічного продукту реакції.2. Спосіб за п. 1, за яким реакцію проводять в присутності основи.3. Спосіб за...
Спосіб одержання сполуки фенілпіразолу, проміжні сполуки та спосіб їх одержання

Номер патенту: 79295
Опубліковано: 11.06.2007
Автори: Ансель Жан-Ерік, Відаль Жоель
МПК: C07D 231/44, C07C 323/54, C07D 231/38, C07C 323/58, C07B 43/00, C07C 319/00, C07B 45/00
Мітки: спосіб, одержання, проміжні, фенілпіразолу, сполуки
Формула / Реферат:
1. Спосіб одержання сполуки (III), який відрізняється тим, що проводять реакцію між сполукою загальної формули (V) і диціаноацетиленом (IV) у присутності води, ,де R вибраний з CF3 або С1-С6-алкілу; таМ вибраний з лужного або лужноземельного металу або срібла.2. Спосіб за п. 1, який відрізняється тим, що R означає CF3, a M...
Похідні октагідро-6,10-діоксо-6н-піридазино[1,2-а][1,2]діазепін-1- карбонової кислоти, спосіб їх одержання та одержання терапевтично активних сполук

Номер патенту: 71913
Опубліковано: 17.01.2005
Автори: Руссель Патрік, КОЛЛАДАН Колетт, Крок Веронік, Ларкін Джон Патрік
МПК: C07K 5/06, C07D 487/04, C07K 5/078
Мітки: кислоти, сполук, карбонової, октагідро-6,10-діоксо-6н-піридазино[1,2-а][1,2]діазепін-1, активних, похідні, спосіб, терапевтичної, одержання
Формула / Реферат:
1. Сполуки загальної формули (IA1):,де R являє собою С1-4 алкіл, що мають конфігурацію SR або що знаходяться у вигляді суміші SR+SS, які являють собою рацемічну суміш.2. Сполуки загальної формули (ІА1) за п.1, де R являє собою метил.3. Сполуки загальної формули (IA1) за п.1, 2, які являють собою рацемічну суміш, що складається...
Попередній патент: Настінний світильник
Наступний патент: Спосіб одержання сполук, які застосовують як інгібітори натрійзалежного переносника глюкози
Випадковий патент: Похідні діоксабіцикло[3.2.1]октан-2,3,4-тріолу